
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Barium-doped iron nanoparticles supported on MgO as an efficient catalyst for ammonia synthesis under mild reaction conditions†
Kohei
Era
 a,
Katsutoshi
Sato
*ab,
Shin-ichiro
Miyahara
a,
Takahiro
Naito
c,
Kanishka
De Silva
c,
Saeid
Akrami
c,
Hiroshi
Yamada
a,
Takaaki
Toriyama
d,
Takehiro
Tamaoka
d,
Tomokazu
Yamamoto
d,
Yasukazu
Murakami
de,
Koji
Inazu
f and
Katsutoshi
Nagaoka
*ac
a,
Katsutoshi
Sato
*ab,
Shin-ichiro
Miyahara
a,
Takahiro
Naito
c,
Kanishka
De Silva
c,
Saeid
Akrami
c,
Hiroshi
Yamada
a,
Takaaki
Toriyama
d,
Takehiro
Tamaoka
d,
Tomokazu
Yamamoto
d,
Yasukazu
Murakami
de,
Koji
Inazu
f and
Katsutoshi
Nagaoka
*ac
aDepartment of Chemical Systems Engineering, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: nagaoka.katsutoshi.n2@f.mail.nagoya-u.ac.jp; satou.katsutoshi.j5@f.mail.nagoya-u.ac.jp
bInstitute for Advanced Research, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan
cInstitutes of Innovation for Future Society, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan
dThe Ultramicroscopy Research Center, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
eDepartment of Applied Quantum Physics and Nuclear Engineering, Kyushu University, Motooka 744, Nishi-ku, Fukuoka, 819-0395, Japan
fNational Institute of Technology, Numazu College, 3600 Ooka, Numazu, Shizuoka 410-8501, Japan
First published on 8th May 2024
Abstract
To realize a carbon-neutral society, developing highly active and inexpensive catalysts for ammonia (NH3) production working under moderate conditions (<400 °C, <10 MPa) using hydrogen fabricated from the electrolysis of water is demanded. Current industrial fused iron (Fe) catalysts show deficient activity under such conditions. Although Ru-based catalysts have been introduced as highly efficient catalysts working under desired conditions, they are scarce and consequently expensive. This work introduces Fe/Ba/MgO (reduced at 700 °C) as an active catalyst with a high ammonia production rate working at mild temperature and pressure (350 °C, 1.0 MPa). Ba doping to Fe nanoparticles supported on MgO and pre-reduction at high temperatures dramatically ameliorated the NH3 production rate. Due to the absence of hydrogen poisoning, the catalytic activity of the Fe/Ba/MgO increased gradually by raising the pressure from 0.1 to 3.0 MPa. The activity of this catalyst at 3.0 MPa was higher than that of two benchmark Ru catalysts. After pre-reduction at high temperature, electrons are donated from the BaO encapsulating the Fe0 nanoparticles to the N2 molecule, which promotes the rate-determining step of ammonia synthesis. We anticipate that these findings will contribute to developing inexpensive Fe catalysts for decarbonizing the ammonia synthesis process to achieve a carbon-neutral society.
Introduction
Ammonia (NH3) is a promising component for helping to achieve a carbon-neutral society.1–7 Today, most NH3 is produced by the industrial Haber–Bosch process using fused Fe-based catalysts. These conventional catalysts, which include Al2O3 as a structural promoter and K2O as a chemical promoter, are introduced as highly active catalysts for NH3 production at high temperatures of more than 450 °C and pressure higher than 20 MPa.8 Al2O3 and K2O promoters facilitate the cleavage of the nitrogen bond (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) with a high dissociation energy of 945 kJ mol−1.8 However, in its current form, the Haber–Bosch process is not suitable for achieving a carbon-neutral society for two reasons. First, the utilized H2 gas in the process is provided by fossil fuel steam reforming, which leads to the emission of huge amounts of CO2 (1.9 tons-CO2 per ton-NH3).9 Second, the NH3 yield of current Fe-based catalysts is significantly decreased at temperatures and pressures lower than 400 °C and 10 MPa, respectively.10 Thus, novel catalysts are needed that show high NH3 synthesis activity under mild conditions, which will allow for the use of H2 produced from clean processes and ultimately, decarbonization of the Haber–Bosch process.11
N) with a high dissociation energy of 945 kJ mol−1.8 However, in its current form, the Haber–Bosch process is not suitable for achieving a carbon-neutral society for two reasons. First, the utilized H2 gas in the process is provided by fossil fuel steam reforming, which leads to the emission of huge amounts of CO2 (1.9 tons-CO2 per ton-NH3).9 Second, the NH3 yield of current Fe-based catalysts is significantly decreased at temperatures and pressures lower than 400 °C and 10 MPa, respectively.10 Thus, novel catalysts are needed that show high NH3 synthesis activity under mild conditions, which will allow for the use of H2 produced from clean processes and ultimately, decarbonization of the Haber–Bosch process.11
Recently, various alternative NH3 synthesis catalysts have been developed.12 Reported Ru catalysts have demonstrated considerable NH3 yield under moderate conditions, and they are considered the most promising candidates for synthesizing decarbonized NH3.13–17 However, Ru utilization in large-scale commercial applications is limited due to its high cost and rarity, and transition elements, such as Mn,18 Fe,19 and Co,20 which are more accessible, have been proposed as catalysts for decarbonized NH3 synthesis. Fe is the most appropriate element in terms of cost and abundance, which makes the development of Fe-based catalysts for use in a decarbonized NH3 synthesis process highly desirable.
In our previous study, the performance of potassium-doped Fe nanoparticles supported on MgO was investigated for catalytic NH3 synthesis.21 Characterization of this catalyst revealed that use of the optimal reduction temperature and the optimal amount of K doping increased the number of Fe0 nanoparticles on the catalyst surface and the Fe turnover frequency. Moreover, it was reported that a Co/Ba/MgO catalyst prepared by impregnating cobalt(II) acetylacetonate into Ba-doped MgO followed by pre-reduction at high temperature (700 °C) contained BaO encapsulating Co nanoparticles and that this encapsulation dramatically improved the NH3 synthesis activity of Co.20 These findings motivated us to study the effects of Ba doping on the catalytic behaviour of the Fe/MgO catalyst.
In this study, the Fe/MgO catalyst doped with barium was employed for the NH3 synthesis process under mild conditions, and its catalytic behaviour was evaluated. This catalyst showed a high NH3 production rate (13.8 mmol h−1 gcat−1) under moderate conditions. Moreover, raising the pressure from 1.0 MPa to 3.0 MPa resulted in improving the NH3 production activity to 26.1 mmol h−1 gcat−1 for the Fe/Ba(1)/MgO-700red (Ba/(Ba + Mg) = 0.01 mol mol−1, pre-reduced at 700![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C) catalyst. This value is more than the activity of two benchmark Ru catalysts (Ru/CeO2-500red22 and Cs+/Ru/MgO-500red23). Reduction of Fe/Ba(1)/MgO at 700 °C, a higher temperature than that used for conventional Fe-based catalysts, resulted in the decomposition of BaCO3 and the formation of a structure in which Fe0 nanoparticles were encapsulated within BaO. This encapsulation led to a significant enhancement of the NH3 synthesis activity and turnover frequency of Fe/Ba(1)/MgO-700red by electron donation to adsorbed N2 molecules from the BaO encapsulating Fe0 nanoparticles. We also discuss the difference in dopant effects between Ba and K.18
°C) catalyst. This value is more than the activity of two benchmark Ru catalysts (Ru/CeO2-500red22 and Cs+/Ru/MgO-500red23). Reduction of Fe/Ba(1)/MgO at 700 °C, a higher temperature than that used for conventional Fe-based catalysts, resulted in the decomposition of BaCO3 and the formation of a structure in which Fe0 nanoparticles were encapsulated within BaO. This encapsulation led to a significant enhancement of the NH3 synthesis activity and turnover frequency of Fe/Ba(1)/MgO-700red by electron donation to adsorbed N2 molecules from the BaO encapsulating Fe0 nanoparticles. We also discuss the difference in dopant effects between Ba and K.18
Experimental
Synthesis of the Ba-doped Fe/MgO catalyst
The stepwise impregnation method was employed to fabricate the Ba-doped Fe/MgO catalyst.20,21 In this regard, specified amounts of MgO (MgO-500A, Ube Material Industries, Ltd, Japan) and Ba(OH)2·8H2O (Fujifilm Wako Pure Chemicals, Co., Japan) solution were mixed for 1 h. It should be noted that different amounts of Ba(X) (X = 0.5, 1 and 3) were loaded for investigation of the influence of doping on catalytic behaviour (X is the mole percentage of Ba with respect to the total molar value of Ba plus Mg). After blending, the powder was separated using a rotary evaporator and treated in a muffle furnace at 700 °C for 5 h to form the Ba/MgO support. To deposit the Fe catalyst on the support, iron(III) acetylacetonate [Fe(acac)3] (Tokyo Chemical Industry Co., Ltd, Japan) was mixed with Ba/MgO in tetrahydrofuran (Fujifilm Wako, Japan). The Fe loading amount was considered to be 20 wt% of the catalyst. However, this value was 16.2 wt% according to the X-ray fluorescence analysis after the catalytic tests. The powder and solvent were separated using a rotary evaporator at 35 °C and calcined in a tube furnace in Ar at 500 °C for 5 h. The procedure to synthesize the catalyst without Ba doping (Fe/MgO) was the same as the mentioned route except for the Ba loading step. It should be noted that for this catalyst, MgO was heated in a muffle furnace at 700 °C for 5 h before loading the Fe catalyst.20 Ru/CeO2 and Cs+/Ru/MgO as benchmark catalysts were fabricated utilizing the same method reported before.20Catalytic NH3 synthesis test
For the catalytic NH3 synthesis test, 100 mg of catalyst powder was pressed to make a pellet and then was sieved (250–500 μm particle size).20,21 The pelleted catalyst was located in a tubular Inconel reactor (The Nilaco Corporation, Japan) and reduced at 500 or 700 °C with a 2 °C min−1 ramping rate.20,21 The reduction was performed under a mixture of H2 and N2 gas flow for 1 h. The H2 to N2 ratio, total stream rate, and pressure were 3, 240 mL min−1 and 0.1 MPa, respectively. After reduction, the reactor temperature was decreased under the same flow. Then, the H2–N2 stream with H2/N2 = 3 and total stream rate of 120 mL min−1 was fed to the reactor to obtain the space velocity of 72000 mL h−1 gcat−1 and the pressure increased to 1.0 or 3.0 MPa. The synthesis rate of the catalysts was measured by trapping the produced NH3 in 1–10 mM H2SO4 solution. The rate decrease in conductivity, measured using an electronic conductivity detector (CM-30R, DKK-TOA, Japan), is employed to calculate the NH3 synthesis rate based on a standard curve of electronic conductivity (Fig. S1†). All utilized gases in this work were obtained from high-pressure and high-grade (>99.9999%) cylinders and purified using an inline gas purifier (MicroTorr MC50-904FV, SAES Pure Gas, USA).
Kinetic calculations
Kinetic calculations were conducted using a similar procedure reported before at 350 °C and 0.1 MPa.24–26 Ammonia synthesis reaction rate was calculated using eqn (1) by considering that the synthesis rate of NH3 depends on the N2, H2 and NH3 pressures. More details regarding the kinetic calculations and retardation index can be found in the ESI.†| r = kPN2nPH2hPNH3a | (1) |
Characterization
It should be noted that all catalyst characterizations were performed after samples were passivated with CO2 (>99.95% purity with O2 as an impurity).Scanning transmission electron microscopy (STEM) with high-angle annular dark-field (HAADF) imaging and corresponding Energy dispersive X-ray spectroscopy (EDX) mapping were performed at 120 kV using a JEM-ARM200CF electron microscope (JEOL, Japan). The sample was prepared by crushing the catalysts in ethanol, dispersing them on a carbon-coated copper grid, and drying them at ambient temperature for 1 day in vacuum.
An EDXL 300 apparatus (Rigaku, Japan) was utilized for X-ray fluorescence analysis of the samples.
The crystal structure was examined by X-ray diffraction analysis using a SmartLab X-ray diffractometer (Rigaku) with a CuKα source. Analysis of X-ray diffraction profiles was conducted using the PDXL2 software (Rigaku) and three databases (ICDD, COD,27 AtomWork).28 The crystallite size of Fe0 was measured by Scherrer's equation using the Fe0 (110) peak located at 44.67° in the X-ray diffraction profile.
After pre-treatment under a N2 flow at 300 °C, the Brunauer–Emmett–Teller method was performed to measure the specific surface area of the catalysts using a BELSORP MINI X-instrument (Micro-Trac BEL, Japan).
The turnover frequencies of Fe/Ba(1)/MgO and Fe/MgO catalysts were determined by CO-chemisorption capacity measurement using a BEL-CAT-II apparatus (Micro-Trac BEL). To prepare the sample, 50 mg of the catalyst was exposed to a mixture flow of H2 and N2 (H2/N2 = 3, 120 mL min−1 total) for reduction. The temperature was increased to 500 or 700 °C with a 2 °C min−1 ramping rate and kept constant for 1 h. For pre-treatment, the sample was located under the He flow for 2 h. The temperature decreased to 50 °C, and the sample was flushed with the He flow for 10 min. Pulse chemisorption was utilized for CO-chemisorption measurement under the He flow (30 mL min−1) at 50 °C.
X-ray Absorption Fine Structure (XAFS) analyses of the Fe K-edge were conducted using the BL01B1 beamline at Spring-8 (Hyogo, Japan) with Japan Synchrotron Radiation Research Institute permission. To avoid the natural oxidation of Fe upon exposure to air, the measurements were done using in situ XAFS techniques. Samples (50 mg) diluted with boron nitride were pressed into self-supporting disks with 7 mm diameter and located in a quartz in situ XAFS cell (Makuhari Rika Glass, Japan). Each disk was reduced by increasing the cell's temperature by 2 °C min−1 from room temperature to 500 or 700 °C under a mixed H2–N2 gas stream (H2/N2 = 3, 120 mL min−1 total) and held for 1 h. The spectra were then measured after cooling to 50 °C under the same stream. Measurements of the Fe K-edge were performed in transmission mode using two ion chambers. A Si (111) two-crystal monochromator was used for the monochromatization of X-rays. X-ray energy was calibrated against the spectrum of a standard Fe foil. Data reduction was carried out using the Athena program (ver. 0.9.26) included in the Demeter package.29
Infrared spectra of Fe/Ba(1)/MgO measured by in situ reduction under a gas flow of H2–N2 mixture (H2/N2 = 3) were obtained with a spectrometer (FT/IR-6600, JASCO, Japan) equipped with a mercury–cadmium–telluride detector at a resolution of 4 cm−1. The sample (about 10 mg) was pressed into the self-supporting disk (10 mm in diameter), placed in a silica-glass cell with a CaF2 window, and connected to a closed gas-circulation system (Makuhari). The disk was pre-treated with a H2–N2 mixture (H2/N2 = 3, about 80 kPa) that had been passed through a liquid N2 trap and reduced by increasing the temperature of the cell by 10 °C min−1 from room temperature to 700 °C. The infrared spectrum of air measured at room temperature was used as the background, and the difference spectrum was obtained by subtracting the background spectrum from the spectrum measured before reduction (fresh) or after reduction at 500 or 700 °C.
Results and discussion
Catalytic NH3 production
In the first step, the efficiency of the Fe/Ba(1)/MgO catalyst for NH3 production was evaluated. Since Fe is easily sintered at high temperatures, a low-temperature, slow reduction is generally used.19,30–32 A previous study on the Fe/K/MgO catalyst indicated that catalyst reduction at 500 °C results in the best NH3 production rate. However, because Co/Ba/MgO-700red has been previously reported to show high activity, we performed pre-reduction of Fe/Ba(1)/MgO at 700 °C.Fig. 1 shows the NH3 synthesis rates and yields versus reaction temperature for Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, and Fe/MgO-700red, compared with fused iron (wüstite-based) as the commercial catalyst at 1.0 MPa. It was observed that Fe/Ba(1)/MgO-700red activity was better than that of the other three catalysts at temperatures of 150 °C to 400 °C. As equilibrium NH3 yields at 350 °C and 400 °C are 14.8 and 7.91%, respectively, NH3 yields were far below the equilibrium value. Fe/Ba(1)/MgO-700red even showed NH3 synthesis activity at 150 °C (0.1 mmol h−1 gcat−1). From our previous work, it was realized that the K(3)-doped Fe catalyst supported on MgO and reduced at 500 °C cannot produce NH3 at 150 °C.21 This indicates that Ba can enhance the NH3 synthesis activity at lower temperatures. The activity of Fe/Ba (1)/MgO-700red for NH3 production at 350 °C was 13.8 mmol h−1 gcat−1, which is 5.3 times higher than that of Fe/Ba(1)/MgO-500red and Fe/MgO-700red, and 1.6 times higher than that of the commercial catalyst as shown in Table 1. Thus, Ba was found to be an effective additive for Fe nanoparticles, which is similar to what we reported previously for Co nanoparticles.20 Further analyses revealed that the highest NH3 synthesis rates were observed for the catalyst with a reduction temperature of 700 °C (Fig. S2†) and when the amount of Ba doping (Ba/(Ba + Mg)) was 0.01 mol mol−1 (Fig. S3†).
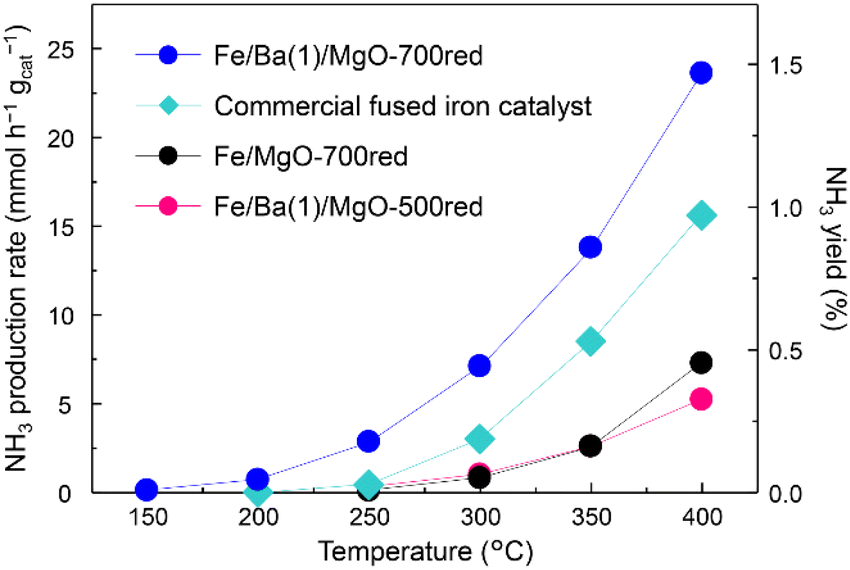 | ||
| Fig. 1 Ammonia production activity versus temperature at 1.0 MPa for Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, Fe/MgO-700red, and a commercial fused iron catalyst. xxxred stands for pre-reduction of the catalyst at xxx °C. Detailed information regarding the commercial catalysts can be found in the ESI.† | ||
| Entry | Catalyst | SSAa [m2 gcat−1] | Fe0 crystallite sizeb [nm] | CO-chemisorptionc [μmol gcat−1] | Rated [mmol h−1 gcat−1] | TOFe [s−1] |
|---|---|---|---|---|---|---|
| a BET specific surface area. b Measured by using Scherrer's equation. c Measured as CO-chemisorption capacity. d Ammonia production rate at 350 °C and 1.0 MPa. e Turnover frequency measured from the amount of CO-chemisorption and the ammonia production rate. | ||||||
| 1 | Fe/Ba(1)/MgO-700red | 64 | 10 | 11.9 | 13.8 | 0.322 |
| 2 | Fe/Ba(1)/MgO-500red | 110 | 7 | 11.4 | 2.6 | 0.063 |
| 3 | Fe/MgO-700red | 90 | 8 | 20.5 | 2.6 | 0.035 |
| 4 | Commercial fused iron catalyst | — | — | — | 8.6 | — |
The catalytic activity for NH3 production versus time at 350 °C and 1.0 MPa was observed in Fig. 2, indicating that the activity of Fe/Ba(1)/MgO-700red slightly decreased at the beginning and then stabilized. At lower amounts of weight hourly space velocity (36000 mL h−1 gcat−1), the NH3 production activity of the Fe/Ba(1)/MgO-700red catalyst at 350 °C and 1.0 MPa reached 8.8 mmol h−1 gcat−1. Even at this lower space velocity, the catalyst synthesized 0.05 mmol h−1 gcat−1 NH3 at 150 °C. Fe/Ba(1)/MgO-700red showed NH3 synthesis activity comparable to Fe-based catalysts, which have been introduced in the literature under mild conditions (Table S1†). Compared to our previously reported Fe/K(3)/MgO-500red, the NH3 synthesis rate for Fe/Ba(1)/MgO-700red was lower above 300 °C, but it was higher below 250 °C.21 This suggests that Ba and K have different effects on the NH3 synthesis activity of Fe0.
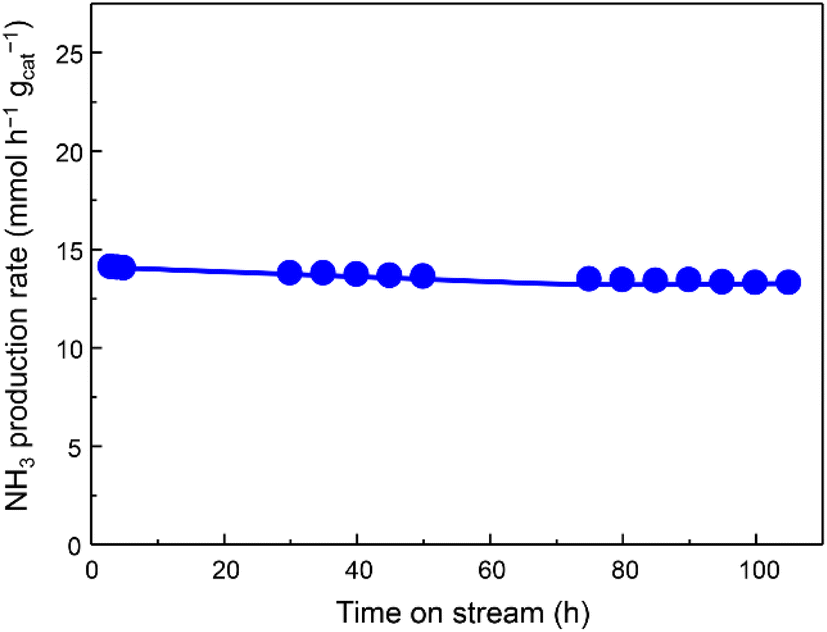 | ||
| Fig. 2 Ammonia production activity of Fe/Ba(1)/MgO-700red versus time course at 350 °C and 1.0 MPa. | ||
Arrhenius plots of ammonia production reactions using Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, Fe/MgO-700red, and the commercial catalyst are observed in Fig. 3. Following our previous report, the apparent activation energy was calculated separately because the slope changed above and below 300 °C.21 Here, it was confirmed that these differences were not due to internal diffusion, as the same activity was obtained when testing with smaller catalyst pellets. The different slopes can be attributed to the fact that the adsorbed species which strongly adsorb on the catalyst surface and retard the reaction vary depending on the temperature. Based on the idea that the rate-limiting step is the dissociative adsorption of dinitrogen, this step is retarded by adsorbed species such as NHx(a) (x = 0, 1, 2) and H(a), and these adsorbed species are in equilibrium with gaseous NH3 and H2. At lower temperatures, H(a) may be strongly adsorbed on the surface, while at higher temperatures NHx(a) replaces H(a) as discussed in ref. 33. The apparent activation energies which are equivalent to the slope of the curves were 57.3, 74.5, 111.5, and 85.5 kJ mol−1 for Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, Fe/MgO-700red, and the commercial catalyst at temperatures lower than 300 °C, respectively. On the other hand, for the higher temperature range (300–400 °C), it was 38.5, 53.3, 70.5 and 52.5 kJ mol−1, respectively. Thus, Fe/Ba(1)/MgO-700red showed the lowest apparent activation energy over the whole temperature range, suggesting that the high NH3 synthesis activity of Fe/Ba(1)/MgO-700red resulted from its low apparent activation energy.
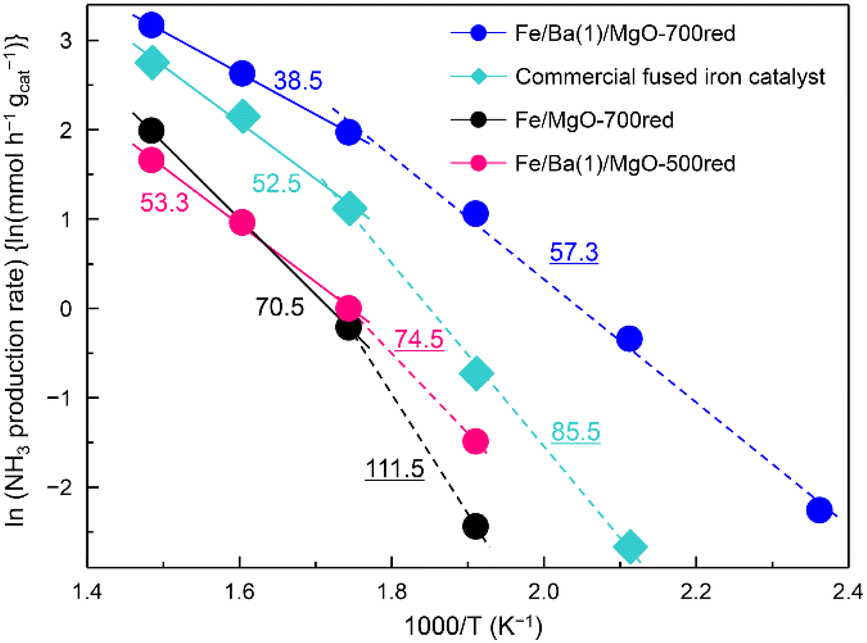 | ||
| Fig. 3 Arrhenius plots corresponding to ammonia production reactions over Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, Fe/MgO-700red, and a commercial fused iron catalyst at 1.0 MPa. The numbers in the figure denote the apparent activation energy (kJ mol−1) calculated from the slope. The dashed lines and underlined numbers are plots and apparent activation energies below 300 °C, and the solid lines and non-underlined numbers are the plots and apparent activation energies above 300 °C. xxxred stands for pre-reduction of the catalyst at xxx °C. | ||
The effect of pressure on the NH3 synthesis activity of Fe/Ba(1)/MgO-700red at 350 °C and also for Cs+/Ru/MgO-500red and Ru/CeO2-500red as benchmark Ru catalysts is observed in Fig. 4. Cs+/Ru/MgO and Ru/CeO2 are known catalysts due to their activity and for ammonia production by renewable energy utilization, respectively.22,23 Although the activity of the Fe/Ba(1)/MgO-700red catalyst at 0.1 MPa was lower than that of benchmark Ru catalysts, at 1.0 MPa it was higher than that of Cs+/Ru/MgO and at 3.0 MPa it improved significantly and reached a value higher than that of both benchmark Ru catalysts.
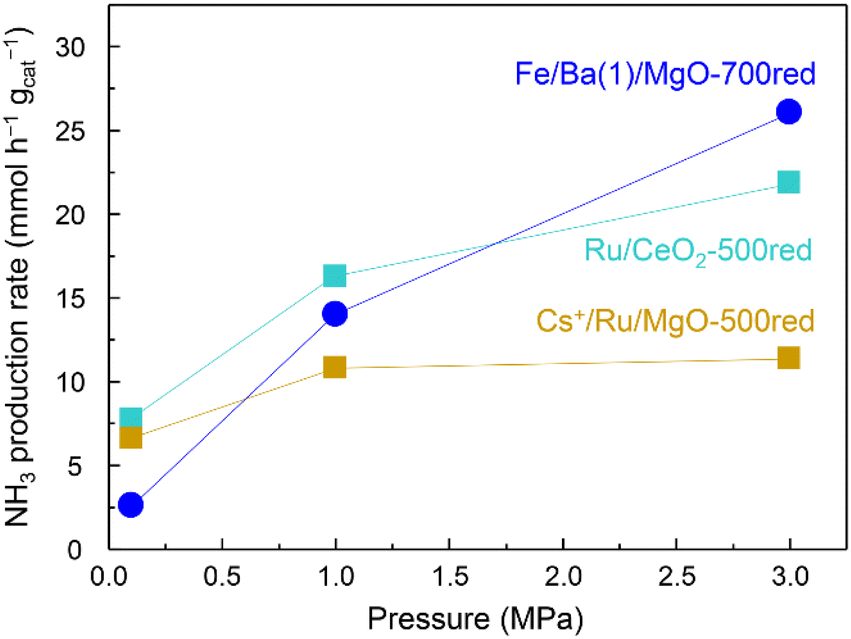 | ||
| Fig. 4 Ammonia production activity versus pressure at 350 °C for Fe/Ba(1)/MgO-700red and two benchmark Ru catalysts. xxxred stands for pre-reduction of the catalyst at xxx °C. | ||
To determine the reason for the different pressure dependencies of Fe/Ba(1)/MgO-700red and the two benchmark Ru catalysts, we performed kinetic analyses at 350 °C and 0.1 MPa (Table S2 and Fig. S4†). Orders of the reaction with respect to N2 for the three catalysts were almost unity (Table S2†), suggesting that dissociation of N2 is the rate-determining step. The reaction orders for Cs+/Ru/MgO-500red and Ru/CeO2-500red with respect to H2 were negative. It demonstrates the significant adsorption of hydrogen atoms on the surface of Ru catalysts. This hydrogen adsorption, which is known as hydrogen poisoning, is the main limitation of Ru catalyst utilization. However, the reaction order for Fe/Ba(1)/MgO-700red was positive. Thus, one of the reasons for the increase in NH3 synthesis rate over Fe/Ba(1)/MgO-700red in response to increased pressure is the absence of hydrogen poisoning. The reaction order for NH3 was negative for all three catalysts, this value for Fe/Ba(1)/MgO-700red was lower than −1.0, which is a larger negative amount compared to other catalysts. This result shows the significant adsorption of NHx species on the surface of the catalyst and the equilibrium formed between NHx and NH3 components, which prevent N2 adsorption and activation.
To further investigate the kinetics of Fe/Ba(1)/MgO-700red, the retardation index was considered following previous reports.21,34 We define the retardation index for NHx(a) and H(a) as y and z, respectively. A higher value of y indicates strong adsorption of NHx, while a higher value of z indicates strong adsorption of H. If the sum of y and z exceeds 1 when x = 0, then x = 0 is not suitable and x = 1 or 2 is considered. The calculated retardation index was (y, z) = (0.69, −0.16) when x = 1 (Table S3†), meaning that the main surface adsorbed species of Fe/Ba(1)/MgO-700red was NH. The main adsorbed species of Fe/K(3)/MgO-500red estimated in our previous study was N,21 suggesting that the kinetics and reaction pathways differ, depending on whether K or Ba is doped.
Effect of Ba on the ammonia synthesis activity of Fe/Ba/MgO-700red
The effect of barium doping on the efficiency of the catalysts for NH3 production, was investigated by evaluating their physicochemical properties. Comparison of these properties for Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, and Fe/MgO-700red is observed in Table 1. Comparison of the catalysts with and without Ba showed that the specific surface area of Fe/Ba(1)/MgO-700red (entry 1) is smaller, and it's Fe0 crystallite diameter (calculated with Scherrer's equation using the Fe0 (110) peak in the X-ray diffraction patterns (Fig. S5†)) is larger than that of Fe/MgO-700red (entry 3). For the Fe/Ba(1)/MgO-700red catalyst, the CO-chemisorption capacity, which is considered as an index of Fe dispersion, and the turnover frequency were approximately half and 9.2 times that of Fe/MgO-700red. This means that even though the ratio of surface Fe0 sites is greater on Fe/MgO-700red than on Fe/Ba(1)/MgO-700red, the latter has significantly more active Fe0 sites owing to the Ba doping. Here, it was also confirmed that the crystallite size of Fe0 of Fe/Ba(1)/MgO-700red was 10 nm and was not changed before and after the reaction (Fig. S6†).Comparison of the Ba-doped catalysts with different reduction treatments showed that Fe/Ba(1)/MgO-700red (entry 1) had about half the specific surface area of Fe/Ba(1)/MgO-500red (entry 2) and a larger Fe0 crystallite size (Fig. S7†). However, the CO-chemisorption capacity of Fe/Ba(1)/MgO-700red was comparable to that of Fe/Ba(1)/MgO-500red. This is strange from the viewpoint of the sintering of Fe0 and will be discussed in the section of Effect of Ba on catalyst surface morphology. The turnover frequency of Fe/Ba(1)/MgO-700red was about 5.1 times that of Fe/Ba(1)/MgO-500red, which means that the high-temperature reduction treatment enabled Ba to effectively activate Fe0.
In the X-ray diffraction pattern of Fe/Ba(1)/MgO-700red, the peaks attributed to BaCO3 that were observed for Fe/Ba(1)/MgO-fresh and Fe/Ba(1)/MgO-500red had disappeared (Fig. S7†). It should be noted that basicity and electron-donating decrease with the formation of carbonates in catalyst samples. Therefore, the significantly improved turnover frequency of Fe/Ba(1)/MgO-700red can be attributed to the decomposition of Ba carbonates during high-temperature reduction. We also performed Fourier transform infrared spectroscopy analyses of Fe/Ba(1)/MgO under an H2–N2 mixed gas (H2/N2 = 3) flow with heating (Fig. S8†). The infrared spectrum of fresh Fe/Ba(1)/MgO before reduction showed characteristic peaks attributed to MgCO3 (1555 cm−1) and BaCO3 (1443 cm−1).20,35 At 500 °C, the spectrum showed carbonate peaks, but these peaks were primarily absent at 700 °C. Together, these results indicate that higher temperature reduction is necessary to eliminate carbonates from the Ba-doped Fe/MgO catalyst.
Next, we conducted in situ X-ray absorption fine structure (XAFS) measurements to investigate the changes induced by Ba doping in the state and local structure of Fe in the catalyst. Fig. 5 shows the Fe K-edge X-ray absorption near edge structure (XANES) spectra of Fe/Ba(1)/MgO and Fe/MgO, as well as spectra of several reference samples. The spectra of the fresh, non-reduced Fe/Ba(1)/MgO (spectrum e) and Fe/MgO (spectrum f) were comparable to that of Fe3O4 (spectrum c), suggesting that the valence of Fe in the catalyst was +2 or +3. In the spectra of the reduced samples (spectra g, h, and i), we observed that the intensity of the white line decreased, whereas the intensity of the pre-edge characteristic of the Fe0 (spectrum a) spectra increased. We examined the degree of reduction of each sample by calculating the fraction of Fe0 from the linear combination of the Fe K-edge XANES spectra of FeO and Fe foil. Comparing the catalysts with and without Ba doping, the reduction degree of oxidized Fe species to Fe0 in Fe/Ba(1)/MgO-700red and Fe/MgO-700red was calculated to be 100% and 85%, respectively; thus, Ba doping enhanced the reduction of oxidized Fe species to Fe0. Comparing the Ba-doped catalysts with different reduction treatments, the degree of reduction for Fe/Ba(1)/MgO-700red and Fe/Ba(1)/MgO-500red was calculated to be 100% and 54%, respectively, indicating that reduction to Fe0 is incomplete at 500 °C.
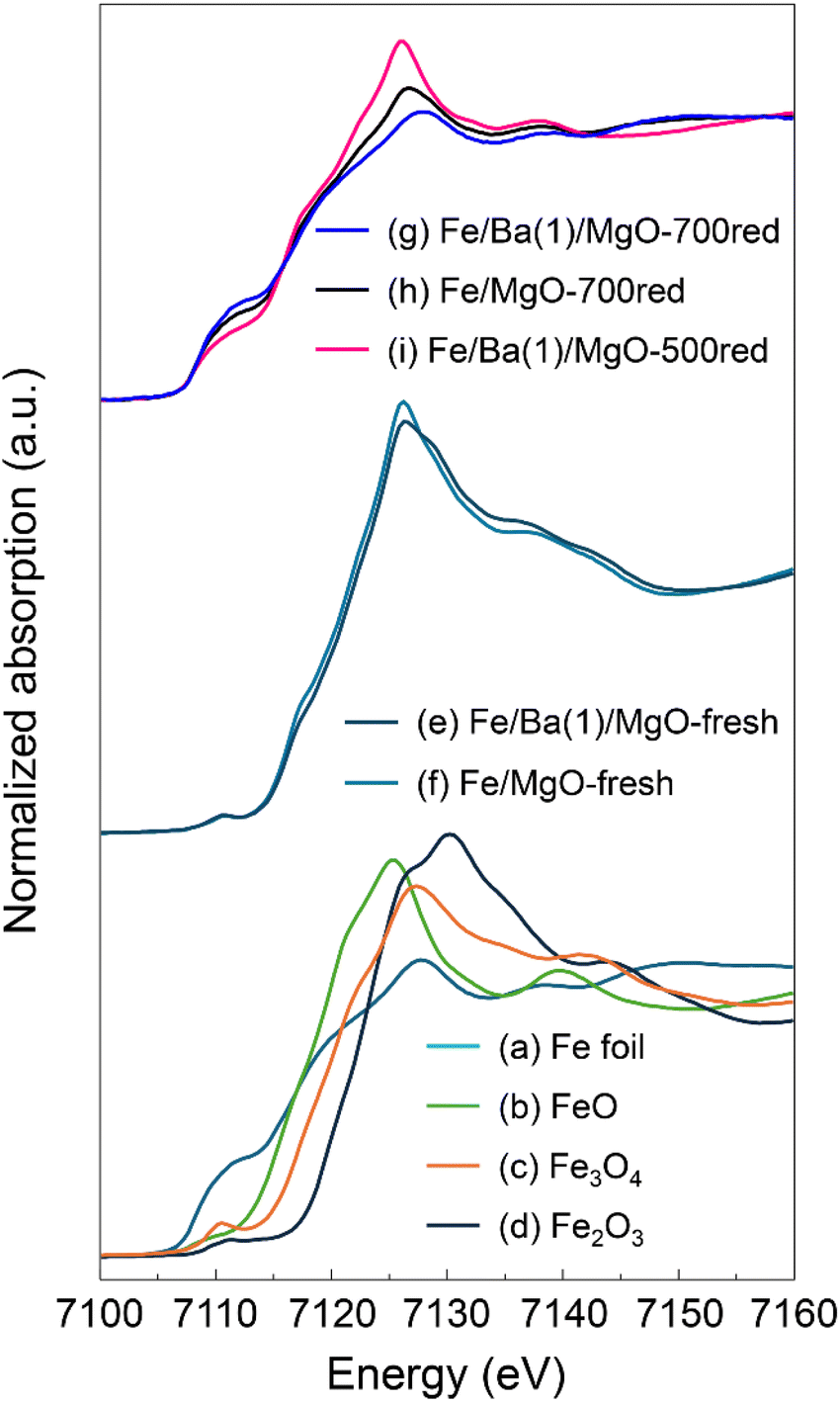 | ||
| Fig. 5 Normalized Fe K-edge XANES spectra for Fe/Ba(1)/MgO and Fe/MgO before (fresh) and after in situ reduction and for reference samples. | ||
Effect of Ba on catalyst surface morphology
Fig. 6 and S9–S11† show the scanning transmission electron microscopy (STEM) images and corresponding EDX mapping for Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, and Fe/MgO-700red after the catalytic ammonia production experiment as surface morphology evaluation. As shown in our previous report, Fe is oxidized rapidly by trace amounts of oxygen,21 and because it is challenging to prepare samples completely without exposure to oxygen, the samples were observed after exposed to air using high-magnification, high-angle annular dark-field (HAADF) STEM at atomic resolution. Thus, the contours of the Fe0 particles were not clear.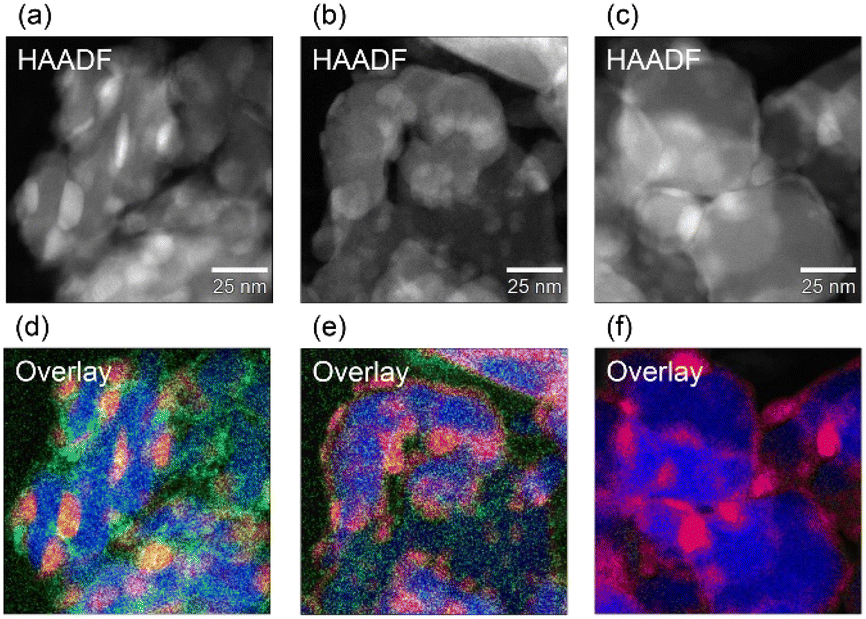 | ||
| Fig. 6 Comparison of the surface morphology of Fe/Ba(1)/MgO-700red (a and d), Fe/Ba(1)/MgO-500red (b and e), and Fe/MgO-700red (c and f) after the ammonia synthesis test. (a–c) High-angle annular dark-field scanning transmission electron microscopy images. (d–f) Overlay energy dispersive X-ray maps. (d and e) Overlay of Fe K (red), Ba L (green), and Mg K (blue). (f) Overlay of Fe K (red) and Mg K (blue). | ||
EDX mapping for Fe/Ba(1)/MgO-700red after the catalytic ammonia production experiment indicated that Ba was enriched near the Fe0 particles (Fig. 6a and d). This observation was also reported in our previous work on a barium doped Co catalyst supported on MgO.20
Fig. 6 shows the HAADF-STEM images of Fe/Ba(1)/MgO-700red, Fe/Ba(1)/MgO-500red, and Fe/MgO-700red (Fig. 6a–c), and overlay EDX maps of Fe K, Ba L, and Mg K (Fig. 6d and e) and Fe K and Mg K (Fig. 6f). The overlay EDX maps of Fe/Ba(1)/MgO-500red and Fe/MgO-700red indicate that Fe is distributed along the MgO edges. Together with the Fe0 ratios calculated from the XANES spectra shown in Fig. 5, it can be inferred that the Fe existing along the MgO edges is related to the unreduced Fe oxidized species. In contrast, no such distribution of Fe was observed for Fe/Ba(1)/MgO-700red. This supports our earlier calculation that the fraction of Fe0 in Fe/Ba(1)/MgO-700red is 100%. Comparison of Fig. 6d and e shows that Ba is enriched near Fe0 after reduction at 700 °C. This indicates that Fe0 particles and Ba species move across the MgO surface during reduction at 700 °C, and that the Ba species is enriched in the vicinity of the Fe0 nanoparticles, reducing the surface energy of Fe0.20
We use our present findings to summarize the influence of reduction temperature on the Fe0 active sites on a Ba-doped Fe/MgO catalyst. On Fe/Ba(1)/MgO-500red, the degree of reduction to Fe0 is low, and small Fe0 nanoparticles are formed. With increased reduction temperature, the Fe oxides on Fe/Ba(1)/MgO-700red are completely reduced, and the size of the Fe0 nanoparticles is increased. A high reduction temperature leads to decomposition of BaCO3 to Ba(OH)2 with a low melting point, 490 °C, migration of the melted Ba(OH)2 over Fe nanoparticles, decomposition of Ba(OH)2 to BaO, and coagulation of the BaO with a high melting point, resulting in the Fe nanoparticles enriched by BaO nano-fractions.20 Eventually, the CO-chemisorption of Fe/Ba(1)/MgO-700red is almost the same as that of Fe/Ba(1)/MgO-500red, but the turnover frequency of Fe/Ba(1)/MgO-700red is much higher than that of Fe/Ba(1)/MgO-500red. The very high turnover frequency of Fe/Ba(1)/MgO-700red can be attributed to the supply of electrons to the antibonding orbital of the nitrogen–nitrogen triple bond from the Fe0 nanoparticles encapsulated by BaO, which facilitates dissociation of the triple bond.20
Finally, we address the different doping effects of Ba and K on Fe nanoparticles in Fe/MgO. Unlike Fe/K(3)/MgO-500red, Fe/Ba(1)/MgO-700red demonstrated ammonia synthesis activity even at a low temperature of 150 °C. This can be attributed to the high N2 dissociation ability of Fe, which is caused by BaO encapsulation and its enrichment over Fe0 nanoparticles. K species are volatile and cannot encapsulate Fe0 nanoparticles, but instead appear to increase the number of Fe0 on the surface.21 The main adsorbed species of Fe/Ba(1)/MgO-700red was estimated to be NH at 350 °C, while that of Fe/K(3)/MgO-500red was N.21 This might be related to the affinity of these catalysts with hydrogen. Such different characteristics of the dopants bring about different behaviours of these catalysts; Fe/Ba(1)/MgO-700red shows higher NH3 synthesis activity than Fe/K(3)/MgO-500red at low temperatures and vice versa at high temperatures.21
Conclusions
We report here the catalytic behaviour of Fe/Ba(1)/MgO-700red for NH3 synthesis under mild reaction conditions. The Fe/Ba(1)/MgO catalyst pre-reduced at 700 °C was about 1.6 times more active per catalyst weight than a commercial fused iron catalyst at 350 °C and 1.0 MPa, and it was able to catalyze the synthesis of NH3 even at 150 °C. Because Fe/Ba(1)/MgO-700red suffered no hydrogen poisoning, catalyst efficiency improved gradually with raising the pressure from 0.1 to 3.0 MPa, and at 3.0 MPa, the catalyst showed higher activity than two benchmark Ru catalysts. The addition of Ba promoted the reduction of Fe oxides to Fe0. The reduction of the Fe/Ba(1)/MgO catalyst at 700 °C, a temperature far higher than that used for conventional Fe-based catalysts, caused BaCO3 to decompose and the resulting BaO to be enriched in the vicinity of the Fe0 nanoparticles. Subsequently, the donation of electrons from BaO encapsulating Fe0 nanoparticles to the adsorbed N2 molecules significantly improved the NH3 synthesis activity and turnover frequency of Fe/Ba(1)/MgO-700red. The dopant effects of K and Ba on Fe nanoparticles are related to their physicochemical properties. Some K is volatile but forms highly dispersed Fe0 sites, while Ba forms fewer but highly active Fe0 sites. Furthermore, the main adsorbed species on the catalyst surface were different: NH and N for the Ba-doped and K-doped catalysts, respectively. These differences led to different catalytic behaviours. Our findings are expected to encourage future research on multi-component doping, which promotes the development of inexpensive Fe catalysts to facilitate progress toward a carbon-neutral society.Author contributions
K. N. designed and he and K. S. coordinated this project. K. E. and S. Miyahara prepared the catalysts, performed the characterizations, and tested catalytic activities. K. E. and K. S. conducted XAFS measurements and analysed the data. T. Toriyama, T. Tamaoka, T. Y. and S. Murakami conducted HAADF-STEM observations and EDX mapping. K. I. performed the catalytic activity tests under high-pressure conditions. All the authors discussed the results and commented on the study. K. E., K.·S., and K. N. co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of the results presented in this article was obtained based on a project (JPNP21012) commissioned by the New Energy and Industrial Technology Development Organization (NEDO). STEM-EDX observations were supported by the “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) (Proposal Number JPMXP1223KU0012). K. Sato gratefully acknowledges the financial support from JST FOREST (JPMJFR223N). XAFS measurements were performed at the BL01B1 public beamline of SPring-8 with approval from the Japan Synchrotron Radiation Research Institute (JASRI; proposal no. 2022B1920 and 2023B1679). We thank Mr K. Kato (JASRI) and Dr Y. Nishida (Nagoya Institute of Technology) for their kind support with the XAFS measurements.References
- Ammonia: Zero-Carbon Fertiliser, Fuel and Energy Store, https://royalsociety.org/topics-policy/projects/low-carbon-energy-programme/green-ammonia/, accessed 19 January, 2024 Search PubMed.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- A. Valera-Medina, H. Xiao, M. Owen-Jones, W. I. F. David and P. J. Bowen, Prog. Energy Combust. Sci., 2018, 69, 63–102 CrossRef.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- K. Eguchi, in Energy Technology Roadmaps of Japan, ed. Y. Kato, M. Koyama, Y. Fukushima and T. Nakagaki, Springer Japan, 2016, pp. 167–181, DOI:10.1007/978-4-431-55951-1.
- H. Kobayashi, A. Hayakawa, K. D. K. A. Somarathne and E. C. Okafor, Proc. Combust. Inst., 2019, 37, 109–133 CrossRef CAS.
- Y. Wang, Y. Tian, S. Y. Pan and S. W. Snyder, ChemSusChem, 2022, 15, e202201290 CrossRef CAS PubMed.
- A. Mittasch and W. Frankenburg, Adv. Catal., 1950, 2, 81–104 Search PubMed.
- S. L. Foster, S. I. P. Bakovic, R. D. Duda, S. Maheshwari, R. D. Milton, S. D. Minteer, M. J. Janik, J. N. Renner and L. F. Greenlee, Nat. Catal., 2018, 1, 490–500 CrossRef.
- K. Sato and K. Nagaoka, Chem. Lett., 2021, 50, 687–696 CrossRef CAS.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- H. Liu, Chin. J. Catal., 2014, 35, 1619–1640 CrossRef CAS.
- N. Saadatjou, A. Jafari and S. Sahebdelfar, Chem. Eng. Commun., 2014, 202, 420–448 CrossRef.
- K.-i. Aika, Catal. Today, 2017, 286, 14–20 CrossRef CAS.
- M. Hattori, S. Iijima, T. Nakao, H. Hosono and M. Hara, Nat. Commun., 2020, 11, 2001 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, M. Sasase, K. Kishida, Y. Kobayashi, K. Nishiyama, T. Tada, S. Kawamura, T. Yokoyama, M. Hara and H. Hosono, Angew. Chem., Int. Ed., 2018, 57, 2648–2652 CrossRef CAS PubMed.
- K. Sato, S.-i. Miyahara, Y. Ogura, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura and K. Nagaoka, ACS Sustainable Chem. Eng., 2020, 8, 2726–2734 CrossRef CAS.
- P. Wang, F. Chang, W. Gao, J. Guo, G. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 CrossRef CAS PubMed.
- W. Al Maksoud, R. K. Rai, N. Morlanés, M. Harb, R. Ahmad, S. Ould-Chikh, D. Anjum, M. N. Hedhili, B. E. Al-Sabban, K. Albahily, L. Cavallo and J.-M. Basset, J. Catal., 2021, 394, 353–365 CrossRef CAS.
- K. Sato, S.-i. Miyahara, K. Tsujimaru, Y. Wada, T. Toriyama, T. Yamamoto, S. Matsumura, K. Inazu, H. Mohri, T. Iwasa, T. Taketsugu and K. Nagaoka, ACS Catal., 2021, 11, 13050–13061 CrossRef CAS.
- K. Era, K. Sato, S. I. Miyahara, T. Naito, K. De Silva, S. Akrami, H. Yamada, T. Toriyama, T. Yamamoto, Y. Murakami, K. I. Aika, K. Inazu and K. Nagaoka, ChemSusChem, 2023, 16, e202300942 CrossRef CAS PubMed.
- T. Nanba, Y. Nagata, K. Kobayashi, R. Javaid, R. Atsumi, M. Nishi, T. Mochizuki, Y. Manaka, H. Kojima, T. Tsujimura, H. Matsumoto, T. Fujimoto, K. Suzuki, T. Oouchi, S. Kameda, Y. Hoshino, S. Fujimoto, M. Kai and Y. Fujimura, J. Jpn. Petrol. Inst., 2021, 64, 1–9 CrossRef CAS.
- R. Javaid, H. Matsumoto and T. Nanba, ChemistrySelect, 2019, 4, 2218–2224 CrossRef CAS.
- K. Aika, J. Kubota, Y. Kadowaki, Y. Niwa and Y. Izumi, Appl. Surf. Sci., 1997, 121–122, 488–491 CrossRef.
- R. Kojima and K.-i. Aika, Appl. Catal., A, 2001, 219, 157–170 CrossRef CAS.
- K. Imamura, S.-i. Miyahara, Y. Kawano, K. Sato, Y. Nakasaka and K. Nagaoka, J. Taiwan Inst. Chem. Eng., 2019, 105, 50–56 CrossRef CAS.
- S. Grazulis, D. Chateigner, R. T. Downs, A. F. Yokochi, M. Quiros, L. Lutterotti, E. Manakova, J. Butkus, P. Moeck and A. Le Bail, J. Appl. Crystallogr., 2009, 42, 726–729 CrossRef CAS PubMed.
- Y. Xu, M. Yamazaki and P. Villars, Jpn. J. Appl. Phys., 2011, 50, 11RH02 CrossRef.
- B. Ravel, M. Newville and J. Synchrotron, Radiat., 2005, 12, 537–541 CAS.
- H. Fan, X. Huang, K. Kahler, J. Folke, F. Girgsdies, D. Teschner, Y. Ding, K. Hermann, R. Schlogl and E. Frei, ACS Sustainable Chem. Eng., 2017, 5, 10900–10909 CrossRef CAS.
- P. Q. Yan, W. H. Guo, Z. B. Liang, W. Meng, Z. Yin, S. W. Li, M. Z. Li, M. T. Zhang, J. Yan, D. Q. Xiao, R. Q. Zou and D. Ma, Nano Res., 2019, 12, 2341–2347 CrossRef CAS.
- M. Hattori, N. Okuyama, H. Kurosawa and M. Hara, J. Am. Chem. Soc., 2023, 145, 7888–7897 CrossRef CAS PubMed.
- C. M. Goodwin, P. Lomker, D. Degerman, B. Davies, M. Shipilin, F. Garcia-Martinez, S. Koroidov, J. Katja Mathiesen, R. Rameshan, G. L. S. Rodrigues, C. Schlueter, P. Amann and A. Nilsson, Nature, 2024, 625, 282–286 CrossRef CAS PubMed.
- K. Aika, M. Kumasaka, T. Oma, O. Kato, H. Matsuda, N. Watanabe, K. Yamazaki, A. Ozaki and T. Onishi, Appl. Catal., 1986, 28, 57–68 CrossRef CAS.
- E. Roedel, A. Urakawa, S. Kureti and A. Baiker, Phys. Chem. Chem. Phys., 2008, 10, 6190–6198 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4se00411f |
| This journal is © The Royal Society of Chemistry 2024 |