
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A first-in-class dual-chelator theranostic agent designed for use with imaging-therapy radiometal pairs of different elements†
James L.
Wood
 ab,
Saikat
Ghosh
b,
Zachary H.
Houston
b,
Nicholas L.
Fletcher
b,
James
Humphries
b,
Karine
Mardon
b,
Dewan T.
Akhter
b,
William
Tieu
c,
Alesia
Ivashkevich
d,
Michael P.
Wheatcroft
d,
Kristofer J.
Thurecht
b and
Rachel
Codd
*a
ab,
Saikat
Ghosh
b,
Zachary H.
Houston
b,
Nicholas L.
Fletcher
b,
James
Humphries
b,
Karine
Mardon
b,
Dewan T.
Akhter
b,
William
Tieu
c,
Alesia
Ivashkevich
d,
Michael P.
Wheatcroft
d,
Kristofer J.
Thurecht
b and
Rachel
Codd
*a
aThe University of Sydney, School of Medical Sciences, New South Wales 2006, Australia. E-mail: rachel.codd@sydney.edu.au
bCentre for Advanced Imaging (CAI), Australian Institute for Bioengineering and Nanotechnology (AIBN) and ARC Training Centre for Innovation in Biomedical Imaging Technology, The University of Queensland, Brisbane, Queensland 4072, Australia
cMolecular Imaging and Therapy Research Unit (MITRU), South Australian Health and Medical Research Institute (SAHMRI), Adelaide, Australia
dTelix Pharmaceuticals Limited, North Melbourne, Victoria 3051, Australia
First published on 3rd June 2024
Abstract
A covalent adduct of DFOB and DOTA separated by a L-lysine residue (DFOB-L-Lys-N6-DOTA) exhibited remarkable regioselective metal binding, with {1H}-13C NMR spectral shifts supporting Zr(IV) coordinating to the DFOB unit, and Lu(III) coordinating to the DOTA unit. This first-in-class, dual-chelator theranostic design could enable the use of imaging-therapy radiometal pairs of different elements, such as 89Zr for positron emission tomography (PET) imaging and 177Lu for low-energy β−-particle radiation therapy. DFOB-L-Lys-N6-DOTA was elaborated with an amine-terminated polyethylene glycol extender unit (PEG4) to give DFOB-N2-(PEG4)-L-Lys-N6-DOTA (compound D2) to enable installation of a phenyl-isothiocyanate group (Ph-NCS) for subsequent monoclonal antibody (mAb) conjugation (mAb = HuJ591). D2-mAb was radiolabeled with 89Zr or 177Lu to produce [89Zr]Zr-D2-mAb or [177Lu]Lu-D2-mAb, respectively, and in vivo PET/CT imaging and in vivo/ex vivo biodistribution properties measured with the matched controls [89Zr]Zr-DFOB-mAb or [177Lu]Lu-DOTA-mAb in a murine LNCaP prostate tumour xenograft model. The 89Zr-immuno-PET imaging function of [89Zr]Zr-D2-mAb and [89Zr]Zr-DFOB-mAb showed no significant difference in tumour accumulation at 48 or 120 h post injection. [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb showed similar ex vivo biodistribution properties at 120 h post-injection. Tumour uptake of [177Lu]Lu-D2-mAb shown by SPECT/CT imaging at 48 h and 120 h post-injection supported the therapeutic function of D2, which was corroborated by similar therapeutic efficacy between [177Lu]Lu-D2-mAb and [177Lu]Lu-DOTA-mAb, both showing a sustained reduction in tumour volume (>80% over 65 d) compared to vehicle. The work identifies D2 as a trifunctional chelator that could expand capabilities in mixed-element radiometal theranostics to improve dosimetry and the clinical outcomes of molecularly targeted radiation.
Introduction
The use of radiometals for targeted imaging and therapy in nuclear medicine is expanding,1–7 as evident by recent U.S. Food and Drug Administration approvals for 177Lu endo-radiotherapy agents Lutathera® for neuroendocrine tumours (NET)‡ and Pluvicto™ for metastatic castration-resistant prostate cancer (mCRPC). A major clinical advantage of these agents is afforded by the ability to individualise the dose of β−-particle radiation therapy by first measuring the tumour burden using an imaging agent containing the same targeting vector as the therapeutic agent (e.g., octreotate, which targets somatostatin receptors in NET; or vipivotide tetraxetan (PSMA-617), which targets prostate-specific membrane antigen (PSMA) in mCRPC). However, pairing metal ions of different elements with different coordination chemistries in a theranostic approach requires the use of different chelators (e.g., HBED-CC in 68Ga-gozetotide paired with DOTA in 177Lu-based Pluvicto™), which can result in different biodistribution properties between the imaging and therapy pair. Using different isotopes of the same element (e.g., 64Cu for imaging, 67Cu for therapy) is one solution to this problem, since the requirement for a unique chelator (e.g., sarcophagine)8,9 guarantees the properties of the imaging and therapy complexes are identical. Same-element imaging-therapy radiometal pairs, however, are few which limits opportunities to use mixed-element radiometal pairs with useful decay properties. Theranostic systems compatible with radiometals of different elements are being explored using discrete chelators with capacity for cross-metal binding.10–14We have taken a different approach by grafting together two chelators, desferrioxamine B (DFOB, 1) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, 2) (Chart 1), which have been established experimentally and/or clinically as useful chelators for 89Zr(IV) (positron-emission tomography (PET) imaging) or 177Lu(III) (β-radiation therapy), respectively.
 | ||
| Chart 1 Structure of desferrioxamine B (DFOB, 1) and DOTA (2). | ||
The risk of this approach is that most chelators have broad metal selectivity,1–4 forming complexes with a range of metal ions with variable affinities and stabilities, which could complicate robust verification and function of the radiolabelled product. The polyaminocarboxylic acid chelator 2 is a case in point, which can coordinate many radiometal ions, including but not limited to 68Ga, 177Lu, 44Sc, 212Pb, 225Ac and 211At.1–4,15,16 The hydroxamic acid chelator 1 can complex 89Zr, 68Ga, 90Nb, and others.1–4,17–22 Considering the metal selectivity of 1 and 2 as discrete compounds (Table 1) indicated the possibility that a covalent DFOB-DOTA adduct might have an unusual and useful regioselective metal binding function towards sets of imaging-therapy radiometal pairs of different elements, such as 89Zr (t½ = 3.3 d, E(βmax+) = 901 keV (22.8%), EC = 77%, Eγ = 909 keV (100%))18,23 and 177Lu (t½ = 6.6 d, E(βmax−) = 497 keV (78.6%), Eγ = 208 keV (11.1%), 113 keV (6.6%)).24
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K values of 1:1 complexes formed between DFOB (1) or DOTA (2) and selected metal ions with corresponding theranostic radioisotopes in use/development
K values of 1:1 complexes formed between DFOB (1) or DOTA (2) and selected metal ions with corresponding theranostic radioisotopes in use/development
| Chelator | LogK |
||||
|---|---|---|---|---|---|
| Zr(IV) | Lu(III) | Ga(III) | La(III) | Pb(II) | |
| a 25 °C, I = 1 M NaClO4. b NR = not reported. c 25 °C, I = 1 M KCl. d 25 °C, I = 0.1 M KCl. e 25 °C, I = 0.1 M NaCl. f 25 °C, I = 0.1 (CH3)4N+Cl−. g 25 °C, I = 0.1 M NaClO4. | |||||
| DFOB (1) | 40.0a38 | NRb | 27.6c39 | NRb | 10.0d40 |
| DOTA (2) | NRb | 25.4e41 | 26.1f42 | 22.9e41 | 24.3g43 |
This work describes the preparation and characterisation of the dual chelator DFOB-N2-(PEG4)-L-Lys-N6-DOTA (3) (named ‘D2’ = ![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) FOB and OTA, Scheme 1). Compound 3 (D2) has been designed for regioselective binding of nat/89Zr(IV) in the 1 region and nat/177Lu(III) in the 2 region, and includes an amine-terminated polyethylene glycol (PEG4) unit to enable further chemistry, including biomolecule conjugation. Radiolabelling D2-mAb (mAb = monoclonal antibody = HuJ591) with 89Zr(IV) or 177Lu(III) gave [89Zr]Zr-D2-mAb (4) or [177Lu]Lu-D2-mAb (5) complexes, respectively (Scheme 1). The in vitro and in vivo properties of [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb were evaluated together with the matched single chelator control compounds [89Zr]Zr-1-mAb and [177Lu]Lu-2-mAb in LNCaP cell-based assays and a murine LNCaP prostate cancer xenograft model. [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb showed similar PET/CT imaging function and tumour uptake (about 15% ID g−1) at 48 h and 120 h post injection, with SPECT/CT imaging showing tumour uptake of [177Lu]Lu-D2-mAb to support therapeutic function, which was corroborated by therapeutic efficacy data. The work identifies D2 as a trifunctional dual-chelator theranostic compound with potential for use with 89Zr for imaging or 177Lu for therapy, and other mixed-element radiometal pairs, to expand capabilities in nuclear medicine.
FOB and OTA, Scheme 1). Compound 3 (D2) has been designed for regioselective binding of nat/89Zr(IV) in the 1 region and nat/177Lu(III) in the 2 region, and includes an amine-terminated polyethylene glycol (PEG4) unit to enable further chemistry, including biomolecule conjugation. Radiolabelling D2-mAb (mAb = monoclonal antibody = HuJ591) with 89Zr(IV) or 177Lu(III) gave [89Zr]Zr-D2-mAb (4) or [177Lu]Lu-D2-mAb (5) complexes, respectively (Scheme 1). The in vitro and in vivo properties of [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb were evaluated together with the matched single chelator control compounds [89Zr]Zr-1-mAb and [177Lu]Lu-2-mAb in LNCaP cell-based assays and a murine LNCaP prostate cancer xenograft model. [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb showed similar PET/CT imaging function and tumour uptake (about 15% ID g−1) at 48 h and 120 h post injection, with SPECT/CT imaging showing tumour uptake of [177Lu]Lu-D2-mAb to support therapeutic function, which was corroborated by therapeutic efficacy data. The work identifies D2 as a trifunctional dual-chelator theranostic compound with potential for use with 89Zr for imaging or 177Lu for therapy, and other mixed-element radiometal pairs, to expand capabilities in nuclear medicine.
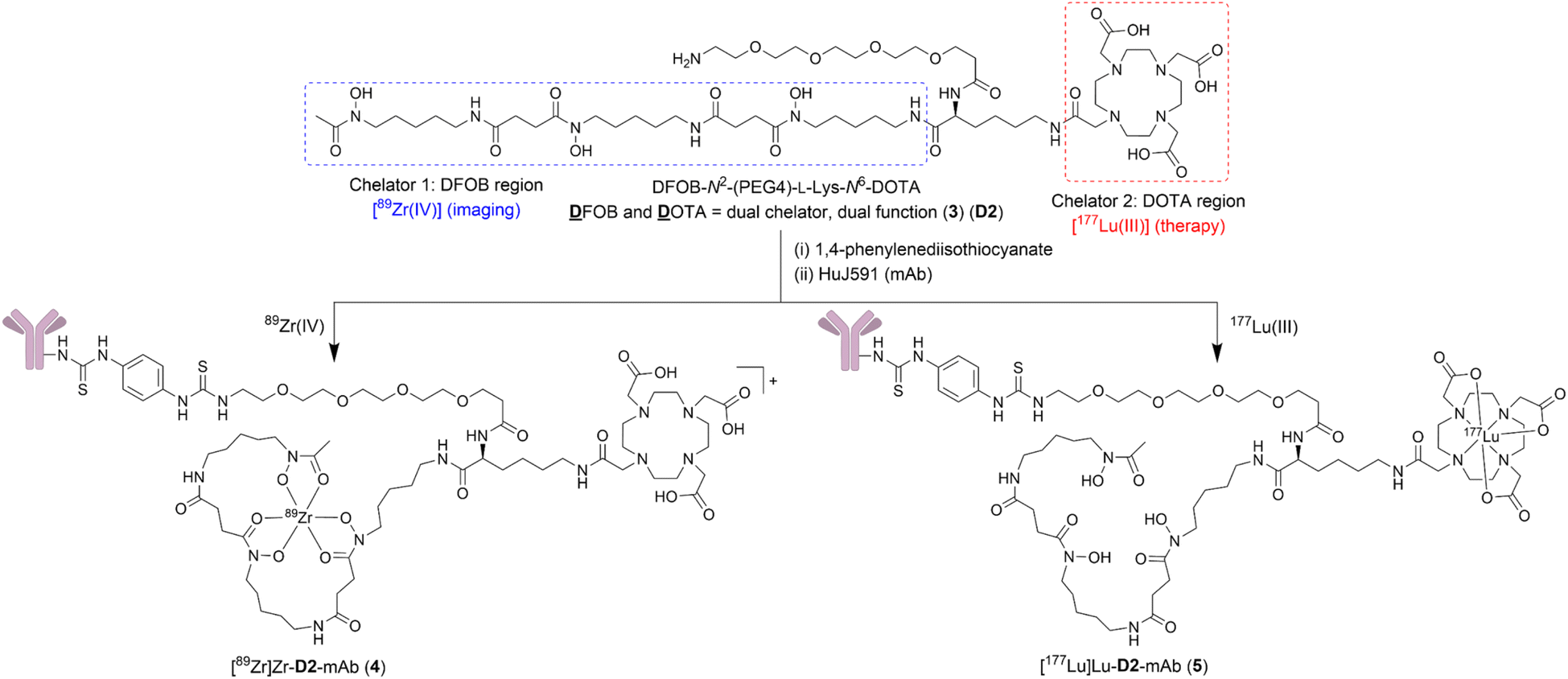 | ||
| Scheme 1 Structure of compound D2 (3), [89Zr]Zr-D2-mAb (4), and [177Lu]Lu-D2-mAb (5). | ||
Results and discussion
Metal selectivity of discrete chelators DFOB and DOTA
The ability of 1 to bind Zr(IV) and not Lu(III) was confirmed from {1H}–13C NMR spectroscopic measurements (Fig. 1) from D2O solutions of 1 alone, or in the presence of a stoichiometric excess of Zr(IV) or Lu(III). The diamagnetism of Zr(IV) and Lu(III) made this approach feasible with minimal metal-dependent line broadening.
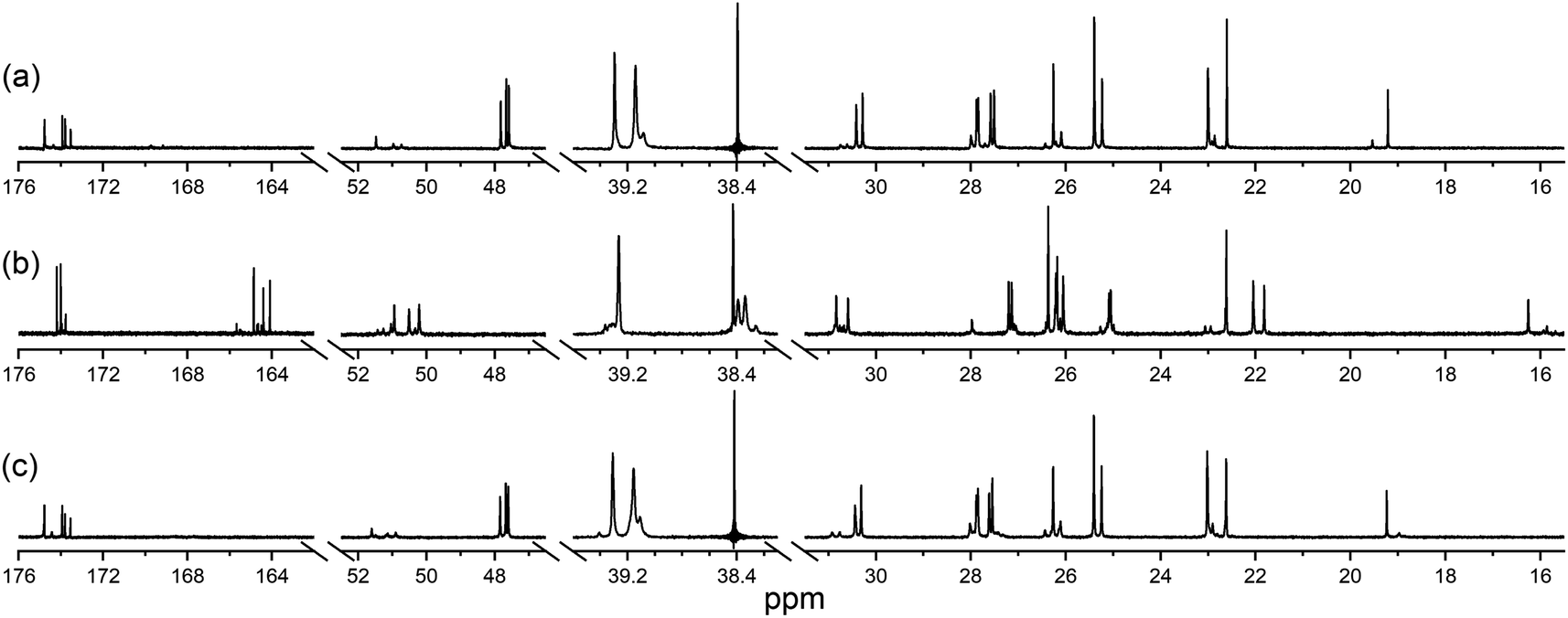 | ||
| Fig. 1 {1H}–13C NMR spectra from solutions (D2O) of (a) 1, (b) Zr(IV):1 (1.8:1), or (c) Lu(III):1 (1.3:1). The signal at 38.4 ppm in (a) and (c) is due to a decoupler zero frequency spike. | ||
Consistent with previous work on complexes between 1 and Al(III) or Ga(III),39 or Zr(IV),44 the cluster of signals between 173-175 ppm attributable to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (amide) and CO (hydroxamic acid) carbon atoms in the spectrum from 1 (Fig. 1a) were resolved in the presence of Zr(IV) (Fig. 1b) into two sets, with the CO (hydroxamate) signals involved in metal coordination appearing upfield at 164.1–164.9 ppm. The two CO signals in the Zr(IV)-1 complex at about 174 ppm that did not shift appreciably compared to 1 could be assigned as the two CO (amide) carbon atoms, since these would experience less change in chemical environment. The signals at 47.6–47.8 ppm attributable to the methylene groups adjacent to the hydroxamic acid N–OH in free 1 moved downfield to 50.2–50.9 ppm in the presence of Zr(IV). The signal attributable to the terminal CH3 group in 1 shifted upfield from 19.2 to 16.3 ppm in the presence of Zr(IV), supporting Zr(IV)-1 complexation. In addition to these readily assigned signals, there were other spectral shifts in the Zr(IV)-1 system that demonstrated complex formation, which would likely include the presence of isomers.39
O (amide) and CO (hydroxamic acid) carbon atoms in the spectrum from 1 (Fig. 1a) were resolved in the presence of Zr(IV) (Fig. 1b) into two sets, with the CO (hydroxamate) signals involved in metal coordination appearing upfield at 164.1–164.9 ppm. The two CO signals in the Zr(IV)-1 complex at about 174 ppm that did not shift appreciably compared to 1 could be assigned as the two CO (amide) carbon atoms, since these would experience less change in chemical environment. The signals at 47.6–47.8 ppm attributable to the methylene groups adjacent to the hydroxamic acid N–OH in free 1 moved downfield to 50.2–50.9 ppm in the presence of Zr(IV). The signal attributable to the terminal CH3 group in 1 shifted upfield from 19.2 to 16.3 ppm in the presence of Zr(IV), supporting Zr(IV)-1 complexation. In addition to these readily assigned signals, there were other spectral shifts in the Zr(IV)-1 system that demonstrated complex formation, which would likely include the presence of isomers.39
The spectra from free 1 or in solution with Lu(III) (Fig. 1c) were close to coincident, indicating the absence of Lu(III)-1 complexation. Metal complexation experiments were conducted at room temperature or at mildly elevated temperatures (≤37 °C) to mimic radiolabelling conditions and provide results relevant to procedures used for mAbs and other thermally labile biological vectors.
Synthesis of DFOB-L-Lys-N6-DOTA (3a) and regioselective coordination of Zr(IV) and Lu(III)
The metal selectivity results from the {1H}–13C NMR spectroscopic data together with established Zr(IV)-1 and Lu(III)-2 chemistry (Table 1) supported that under mild temperature conditions, a DFOB–DOTA covalent adduct would preferentially bind natZr(IV) or 89Zr(IV) at the DFOB region and not at the DOTA region, and bind natLu(III) or 177Lu(III) at the DOTA region and not at the DFOB region. A construct useful for targeted radiometal delivery requires a functional group to graft peptides and/or mAbs, which led to the design of DFOB-L-Lys-N6-DOTA (Scheme 2, 3a) as the preliminary target compound.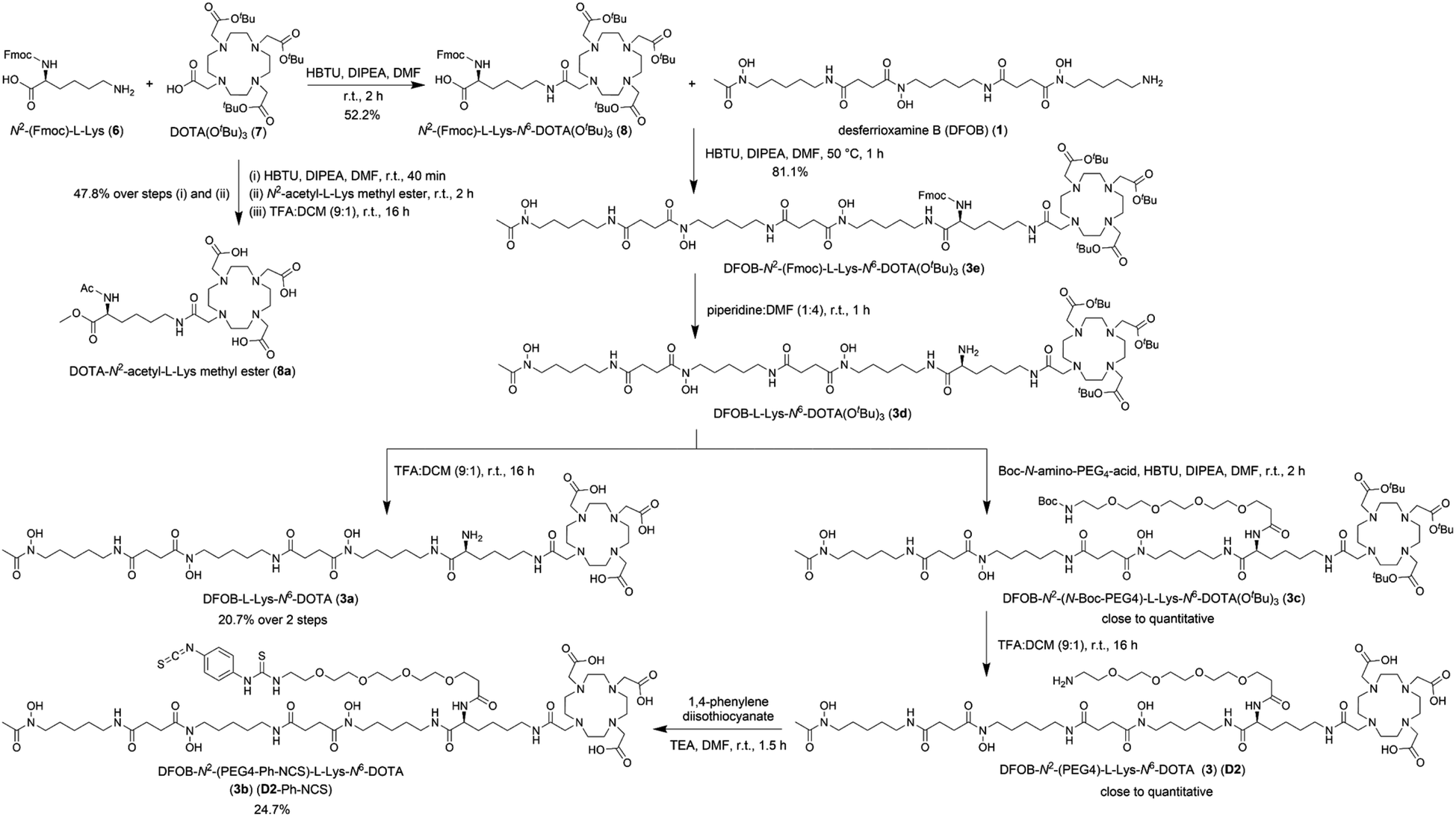 | ||
| Scheme 2 Synthesis of DFOB-N2-(PEG4)-L-Lys-N6-DOTA (3) (D2), DFOB-L-Lys-N6-DOTA (3a), and DFOB-N2-(PEG4-Ph-NCS)-L-Lys-N6-DOTA (3b) (D2-Ph-NCS). | ||
A protected precursor of 3a, DFOB-N2-(Fmoc)-L-Lys-N6-DOTA(OtBu)3 (3e), was prepared from an HBTU-mediated amide coupling reaction between 1 and N2-(Fmoc)-L-Lys-N6-DOTA(OtBu)3 (8), which itself was prepared from N2-(Fmoc)-L-Lys (6) and DOTA(OtBu)3 (7). Global deprotection of 3e generated 3a in an overall yield of 21% (Scheme 2). The {1H}–13C NMR spectrum of 3a (Fig. 2b), assigned using data from the composite fragments 1 (Fig. 2a)39 and 8a, showed signals between 47.9–52 ppm and 54.8–57.1 ppm ascribed to the endo- and exo-methylene carbon atoms of the DOTA region, respectively. The carbonyl carbon atoms in the DOTA region of 3a were resolved at 169.6–169.8 ppm (C45, C47) and 169.9 ppm (C46). Compared to 2 or 8a, the nature and conformation of the extended structure present in 3a modulated the molecular tumbling to enable resolution of the signals ascribable to the DOTA region at room temperature.
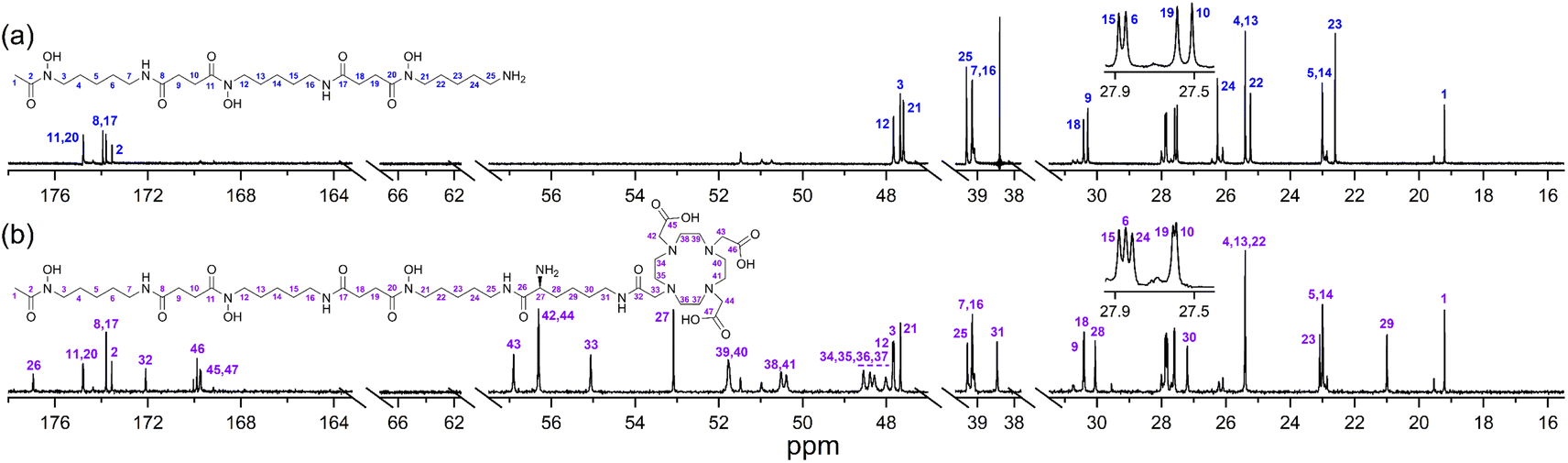 | ||
| Fig. 2 {1H}–13C NMR spectra from solutions (D2O) of (a) 1 or (b) 3a. | ||
The {1H}–13C NMR spectra from 3a alone (Fig. 3a) or in the presence of Zr(IV) or Lu(III) supported the regioselective coordination of Zr(IV) to the 1 region and Lu(III) to the 2 region. The spectral changes in the Zr(IV)-3a system (Fig. 3b) were similar to those observed in the Zr(IV)-1 system (Fig. 1b), with an upfield shift in the three signals ascribed to the carbonyl carbon atoms of the Zr(IV) coordinating hydroxamic acid groups (164.1–164.8 ppm) and the terminal methyl group (16.3 ppm), and a downfield shift (50.1–51.3 ppm) in the three signals attributable to the methylene carbon atoms adjacent to the N–OH groups. These latter sharp signals were overlaid with the signals for the endo-methylene carbon atoms in the 2 region, and although broadened, their appearance supported the non-coordinating role of the 2 region in solutions of Zr(IV) and 3a.
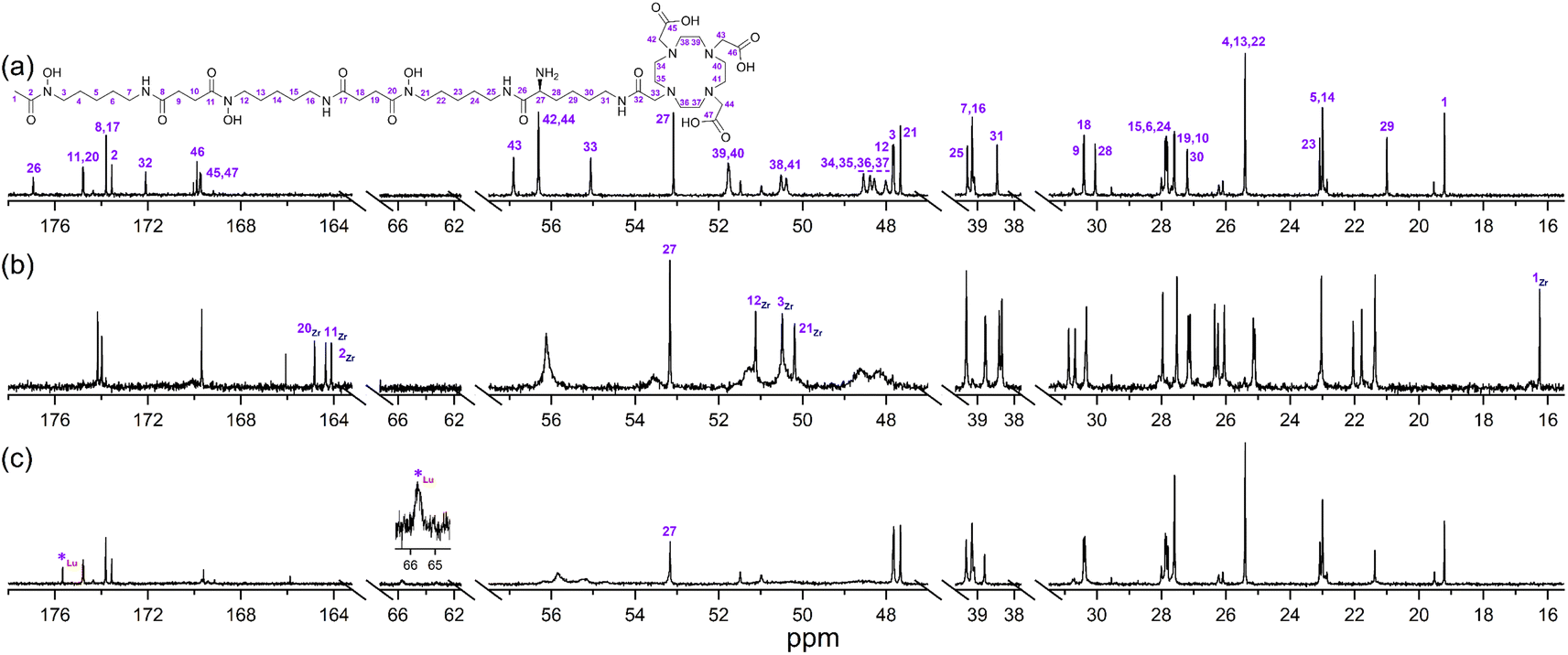 | ||
| Fig. 3 {1H}–13C NMR spectra from solutions (D2O) of (a) 3a, (b) Zr(IV):3a (0.8:1), or (c) Lu(III):3a (0.8:1). | ||
Signals ascribed to the 1 region in the spectrum from a solution of 3a and Lu(III) (Fig. 3c) mapped closely to the same ascribed signals in 3a alone, supporting the 1 region was non-coordinating towards Lu(III). In the presence of Lu(III), the signals for the endo- and exo-methylene carbon atoms of the 2 region in 3a were significantly broadened, with low intensity signals appearing at 63.3 ppm and 65.7 ppm (inset) and an upfield shift of a signal ascribed to one of the carbonyl carbon atoms in the 2 region in 3a. Together, these results supported that at room temperature, Zr(IV) preferentially coordinated the 1 region and not the 2 region in 3a, and Lu(III) preferentially coordinated the 2 region and not the 1 region in 3a.
Stoichiometry of complexes between Zr(IV), Lu(III) or Ga(III) and 3a
Having established the regioselectivity of Zr(IV)/Lu(III)-3a coordination from {1H}–13C NMR spectroscopy, complementary experiments to support selective binding were undertaken using liquid chromatography-mass spectrometry (LC-MS) to report metal:3a stoichiometry. In addition to Zr(IV) and Lu(III), these experiments included Ga(III), which has an affinity towards 1 and 2 (Table 1), and was useful in evaluating a potential case of cross-chelator coordination. Solutions of 3a were incubated with a molar equivalent (Fig. 4) of Zr(IV), Lu(III) or Ga(III) at 37 °C and analysed by LC-MS. The {1H}–13C NMR spectroscopic data predicted the formation of 1:1 Zr(IV):3a (Chart 2), [Zr(IV)-3a(DFOB)]+ (9), and not 2:1 Zr(IV):3a [(Zr(IV))2-3a(DFOB)(DOTA)]2+ (10). This expected trend in stoichiometry would be matched for Lu(III), albeit with binding in the 2 region, with the formation of 1:1 Lu(III):3a ([Lu(III)-3a(DOTA)] (11)) and not 2:1 Lu(IV):3a ([(Lu(III))2-3a(DFOB)(DOTA)] (12)). The affinity of Ga(III) for 1 and 2 could result in the formation of two 1:1 Ga(III):3a isomers indistinguishable by LC-MS ([Ga(III)-3a(DFOB)] (13a) and [Ga(III)-3a(DOTA)] (13b)), and the 2:1 Ga(III):3a complex ([(Ga(III))2-3a(DFOB)(DOTA)] (14)).
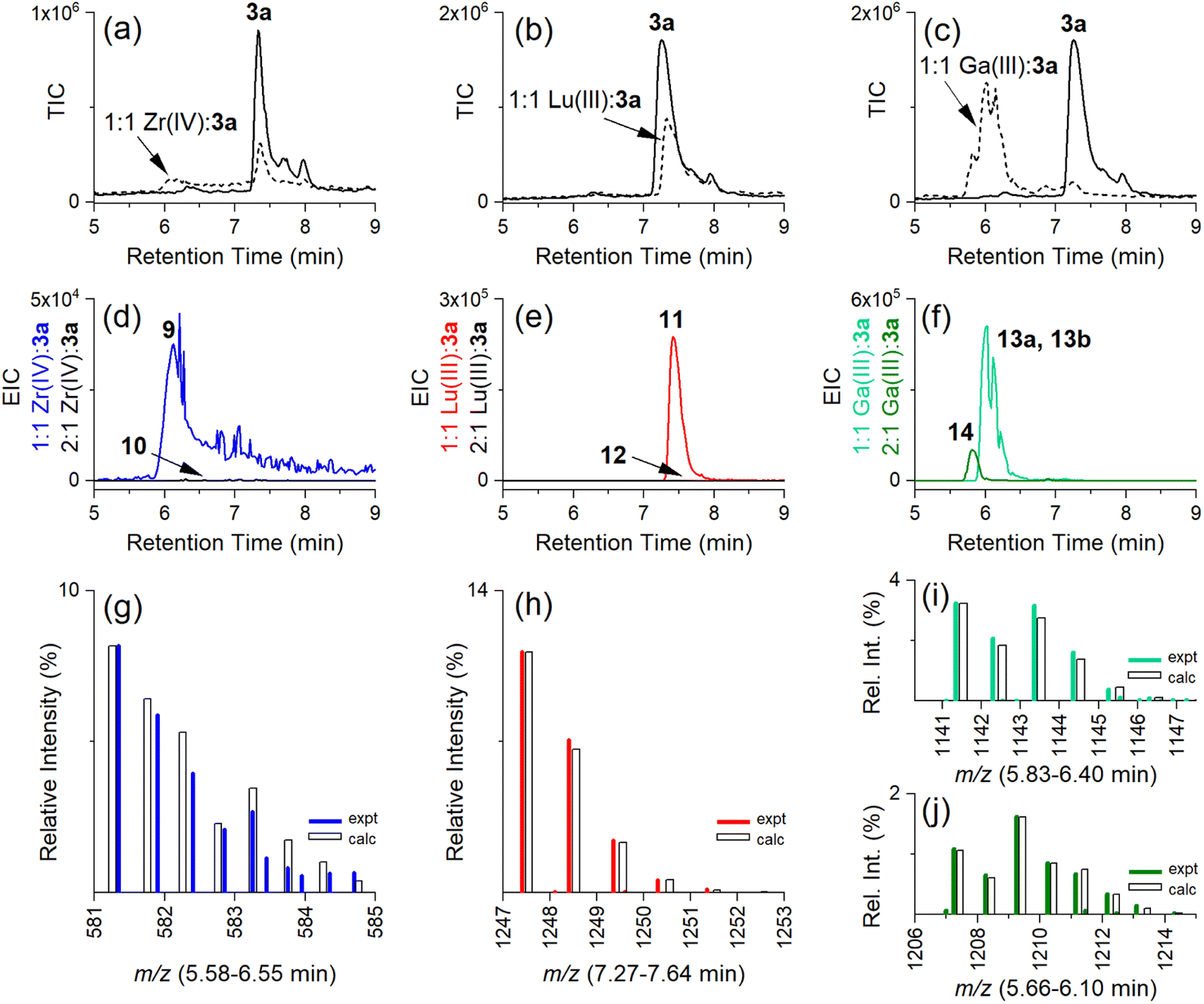 | ||
| Fig. 4 LC-MS traces (total ion current (TIC)) from solutions of 3a in the absence (a–c, solid line) or presence (broken line) of (a) Zr(IV), (b) Lu(III), or (c) Ga(III). EIC traces set to report protonated double-charged 1:1 or 2:1 metal:3a complexes for (d) Zr(IV) (9) (blue) or (10) (black), (e) Lu(III) (11) (red) or (12) (black), or (f) Ga(III) (13a, 13b) (green) or (14) (olive), with the isotope pattern experimental (coloured line) or calculated (white column) for (g) 1:1 Zr(IV):3a (9) ({[M+ + H]}2+ adduct), (h) 1:1 Lu(III):3a (11) ([M + H]+ adduct), (i) 1:1 Ga(III):3a (13a, 13b) ([M + H]+ adduct), or (j) 2:1 Ga(III):3a (14) ([M + H]+ adduct). | ||
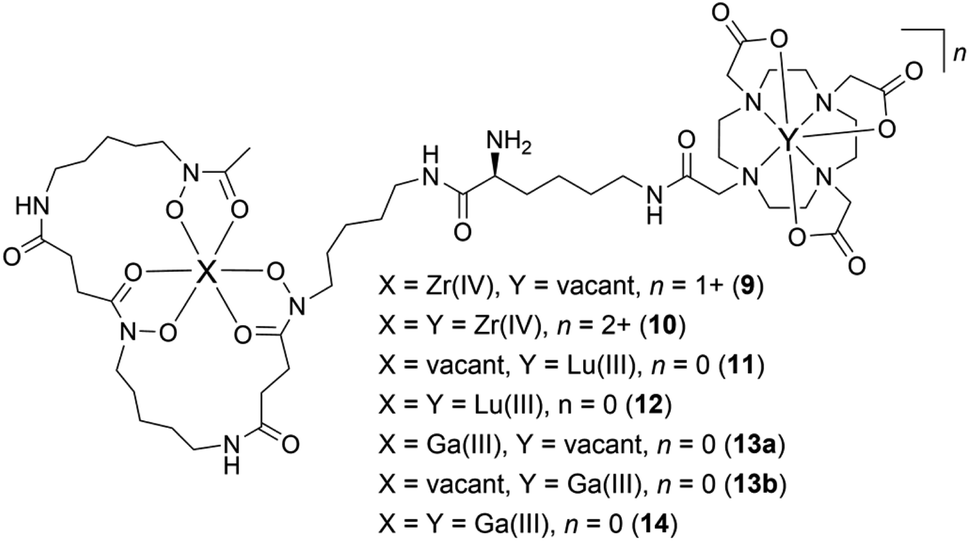 | ||
| Chart 2 Complexes between Zr(IV), Lu(III), or Ga(III), and 3a, with a 1:1 (9, 11, 13a, 13b) or 2:1 (10, 12, 14) metal:3a stoichiometry. | ||
The intensity of the signal at 7.3 min in the LC-MS traces of the free ligand 3a (Fig. 4a–c, solid line) (total ion chromatogram (TIC)) was reduced in the presence of Zr(IV), Lu(III), or almost completely in the presence of Ga(III) (Fig. 4a–c, respectively, broken line), as consistent with the formation of complexes between Zr(IV), Lu(III) or Ga(III) and 3a.
Signals at tR = 6.1 min or 7.4 min were detected from the extracted ion chromatogram (EIC) traces set to report 1:1 Zr(IV):3a (9) (Fig. 4d, blue) or Lu(III):3a (11) (Fig. 4e, red), respectively (Table 2). No signals were observed for the 2:1 Zr(IV):3a (10) (Fig. 4d, black) or 2:1 Lu(III):3a (12) (Fig. 4e, black) complexes. The experimental isotope patterns for 9 (Fig. 4g, blue) and 11 (Fig. 4h, red) were consistent with calculated patterns (white columns) in support of the assignment of these species.
:ligand complexes in 1:1 or 2:1 stoichiometries between Zr(IV), Lu(III) or Ga(III) and 3a (observed and theoretical)
| Xa | Ya | n | Adduct (1+) m/z | Adduct (2+) m/z | Observed | |
|---|---|---|---|---|---|---|
| a Refer Chart 2. b [M]+. c {([M]+ + H)}2+. d [M]2+. e [M + H]+. f [M + 2H]2+. | ||||||
| 9 | Zr(IV) | Vacant | 1+ | 1161.5b | 581.3c | Y |
| 10 | Zr(IV) | Zr(IV) | 2+ | N/A | 624.2d | N |
| 11 | Vacant | Lu(III) | 0 | 1247.6e | 624.3f | Y |
| 12 | Lu(III) | Lu(III) | 0 | 1419.5e | 710.2f | N |
| 13a | Ga(III) | Vacant | 0 | 1141.5e | 571.3f | Y |
| 13b | Vacant | Ga(III) | 0 | 1141.5e | 571.3f | Y |
| 14 | Ga(III) | Ga(III) | 0 | 1207.4e | 604.2f | Y |
The solution of Ga(III) and 3a gave EIC signals at tR = 6.0 min and 5.8 min correlating with complexes with 1:1 (13a, 13b) (Fig. 4f, green) and 2:1 (14) (Fig. 4f, olive) stoichiometries, respectively, showing that, unlike Zr(IV) and Lu(III), Ga(III) was able to simultaneously coordinate to the 1 and 2 regions of 3a. The assignment of the 1:1 isomer pair (13a, 13b) (Fig. 4i) and the 2:1 complex (14) (Fig. 4j) was supported by the match between the experimental and theoretical isotope patterns.
The selectivity of coordination of 3a as established with natural abundance natZr(IV) and natLu(III) metal solutions supported progressing to radiolabelling studies and evaluating the potential of the D2 dual-chelator system to deliver molecularly targeted imaging/therapy radiation.
Synthesis and characterisation of DFOB-N2-(PEG4)-L-Lys-N6-DOTA (3) (D2) and DFOB-N2-(PEG4-Ph-NCS)-L-Lys-N6-DOTA (3b) (D2-Ph-NCS)
A polyethylene glycol unit (PEG4) was appended to the internal L-Lys residue in 3a to increase water solubility and to increase the distance between the dual-chelator scaffold and the mAb to maintain mAb-antigen recognition properties. PEG units and simpler ether-containing constructs have been incorporated in other 1- and 2-type chelators to improve water solubility and other performance measures.30,32,50–53 Compound 3 (D2) (Scheme 2) was prepared from 3e by removing the Fmoc group to give 3d followed by an amide coupling reaction with N-Boc-amine-PEG4-acid to give 3c, and a final global deprotection step to give 3 (D2). A sample of 3 (D2) was reacted with 1,4-phenylenediisothiocyanate and the resultant 3b (D2-Ph-NCS) purified by HPLC and lyophilised. Other types of activating groups§ would be compatible with D2,2,29,54,55 with Ph-NCS selected here as one in common use, and useful for benchmarking performance against the discrete chelators DFOB-Ph-NCS (1-Ph-NCS) and DOTA-Bn-NCS (2-Bn-NCS). The overall yield of 3b (D2-Ph-NCS) starting from the reaction between 6 and 7 was approximately 10.4%. The most significant loss of yield occurred from the reaction between 3 (D2) and 1,4-phenylene diisothiocyanate, due in part to the poor solubility of the latter reagent in organic solvents.Antibody conjugation with D2-Ph-NCS (3b) and radiolabelling D2-mAb
The monoclonal antibody HuJ591 (mAb) was conjugated to D2-Ph-NCS (3b), 1-Ph-NCS or 2-Bn-NCS, and after buffer exchange, the resulting D2-mAb, 1-mAb or 2-mAb complexes were radiolabelled with 89Zr or 177Lu. The chelator:mAb ratio determined from MALDI data (Fig. S26†) was about 2 for 1-mAb, 3 for 2-mAb and 4 for D2-mAb (Table S1†). HuJ591 is a de-immunised variant of the antibody J591 that targets PSMA expressed on LNCaP cells, which is a cell line established from metastatic prostate cancer.56–61
The radio-iTLC traces from D2-mAb radiolabelled with 89Zr or 177Lu gave well-resolved signals characteristic of [89Zr]Zr-D2-mAb (Fig. 5c) or [177Lu]Lu-D2-mAb (Fig. 5g) and correlated with the respective signals for [89Zr]Zr-1-mAb (Fig. 5a) or [177Lu]Lu-2-mAb (Fig. 5e) as matched controls. An earlier radiolabelling study using D2 conjugated to the mAb girentuximab showed similar results with D2-mAb and showed minimal (<1%) radiolabelling of 2 with 89Zr and no detectable radiolabelling of 1 with 177Lu at mild temperatures (Fig. S25†), in accord with the chelator-metal selectivity observed using natZr(IV) and natLu(III).
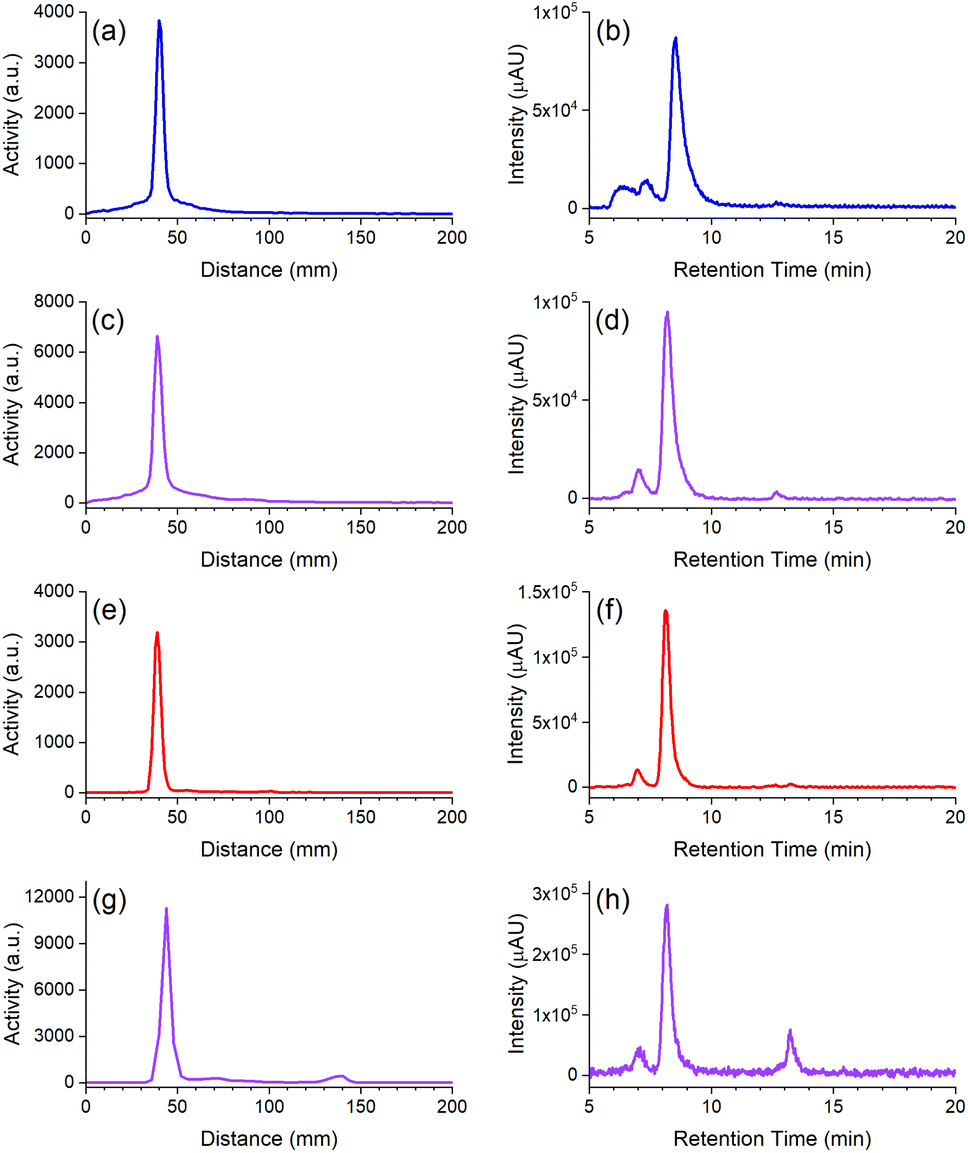 | ||
| Fig. 5 Radio-iTLC (left column) or radio-SEC-HPLC (right column) traces from 1-mAb (a and b) or D2-mAb (c and d) radiolabelled with 89Zr, and 2-mAb (e and f) or D2-mAb (g and h) radiolabelled with 177Lu. Radio-iTLC reaction mixtures were mixed with 1:1 5 mM EDTA (177Lu) or DTPA (89Zr) and developed in a 1:1 EtOH:H2O solvent system (Agilent iTLC-SG Glass microfiber chromatography paper impregnated with silica gel). | ||
In vitro properties and radiochemical stability of [89Zr]Zr-D2-mAb (4) and [177Lu]Lu-D2-mAb (5)
The in vitro cell membrane binding and internalisation of [89Zr]Zr-D2-mAb, [177Lu]Lu-D2-mAb, and the single-chelator controls [89Zr]Zr-1-mAb and [177Lu]Lu-2-mAb, were assessed in LNCaP prostate cancer cells, in which the binding specificity of HuJ591 and cell-surface PSMA is well established.58,59,62In vitro cell-associated activity at 1 h was similar between [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb (Fig. 6a), and between [177Lu]Lu-D2-mAb and [177Lu]Lu-2-mAb (Fig. 6c), showing the mAb was functional in D2-mAb. Levels of cell-membrane and internalised activity were similar between [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb (Fig. 6b), with a slight reduction in internalisation of [177Lu]Lu-D2-mAb compared to [177Lu]Lu-2-mAb (Fig. 6d). The capacity of radiolabelled antibodies to accumulate and remain intracellularly (residualisation) can be useful in maximising DNA radiation damage in endo-radiotherapy.63,64 The radiochemical stability of [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb was measured using radio-iTLC and radio-SEC-HPLC in human serum and in phosphate-buffered saline (Fig. S27–S30 and Table S3†), with sampling at day 0, 2, 4 and 7. [89Zr]Zr-D2-mAb maintained radiochemical purity in human serum at day 7 to 81% (radio-SEC-HPLC) or 88% (radio-iTLC) with the respective values for [89Zr]Zr-1-mAb at 61% or 76%.
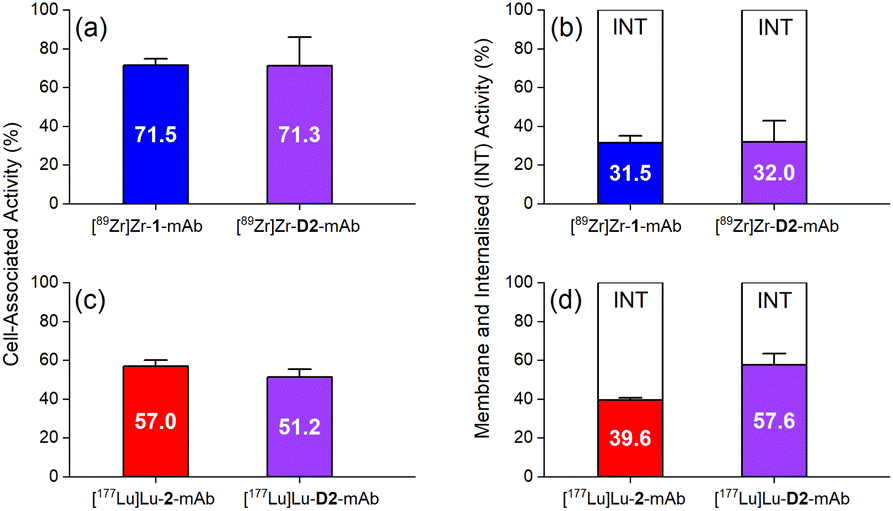 | ||
| Fig. 6 Average percentage of cell-associated activity (LNCaP cells) at 1 h of (a) [89Zr]Zr-1-mAb (blue), [89Zr]Zr-D2-mAb (purple), and (c) [177Lu]Lu-2-mAb (red), [177Lu]Lu-D2-mAb (purple); and membrane (coloured) or internalised (white) fractions of (b) [89Zr]Zr-1-mAb (blue), [89Zr]Zr-D2-mAb (purple), and (d) [177Lu]Lu-2-mAb (red), [177Lu]Lu-D2-mAb (purple). | ||
In vivo properties of [89Zr]Zr-D2-mAb (4) and [177Lu]Lu-D2-mAb (5)
The biodistribution in tumour and other tissues of [89Zr]Zr-D2-mAb, [177Lu]Lu-D2-mAb, and the single-chelator controls [89Zr]Zr-1-mAb and [177Lu]Lu-2-mAb, were assessed in vivo in a murine LNCaP xenograft model by PET/CT imaging and SPECT/CT imaging or ex vivo by gamma counting.¶ [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb showed similar tumour/tissue biodistribution at 4 h post-injection (Fig. 7a), with [89Zr]Zr-D2-mAb showing significantly higher accumulation in the tumour. This trend continued at 24 h post-injection (Fig. 7b).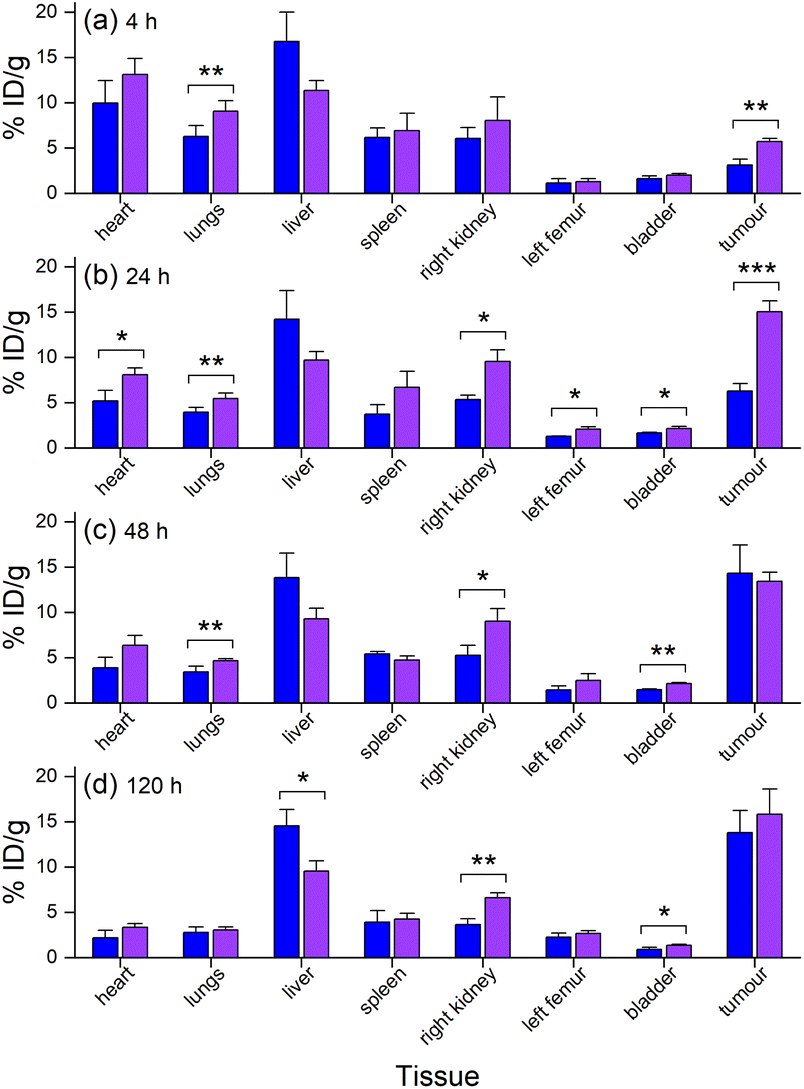 | ||
| Fig. 7 In vivo biodistribution (n = 3) of [89Zr]Zr-1-mAb (blue) and [89Zr]Zr-D2-mAb (purple) (n = 3) at (a) 4 h, (b) 24 h, (c) 48 h, or (d) 120 h post-injection as determined by ROI analysis of PET/CT images. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. | ||
From 48 h to 120 h post-injection, the level of accumulation in the tumour showed no difference between [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb (Fig. 7c and d). At 120 h post-injection, there was no statistical difference in bone uptake between [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb, suggesting the stability of 89Zr binding was similar between the two conjugates (Fig. 7d).
Tumour xenografted mice imaged by PET/CT using [89Zr]Zr-D2-mAb (upper) or [89Zr]Zr-1-mAb (lower) (Fig. 8) or by SPECT/CT using [177Lu]Lu-D2-mAb (Fig. 9) showed a discernible tumour at all timepoints, with signals isolated from background and able to be delineated without interference.
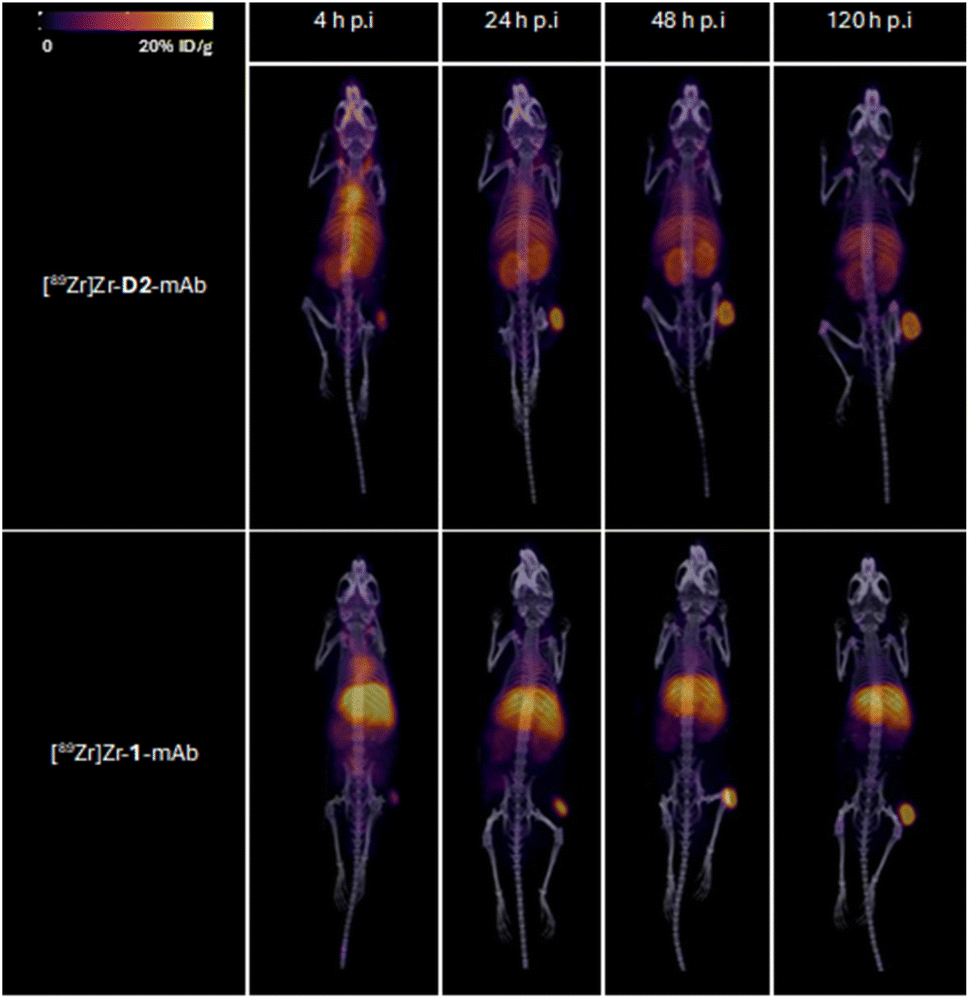 | ||
| Fig. 8 Representative 3D maximum intensity projections for all mice ([89Zr]Zr-D2-mAb upper; [89Zr]Zr-1-mAb, lower); in 89Zr PET/CT imaging cohort at 4, 24, 48 and 120 h post-injection. Signal normalised to same thresholding (% ID g−1). | ||
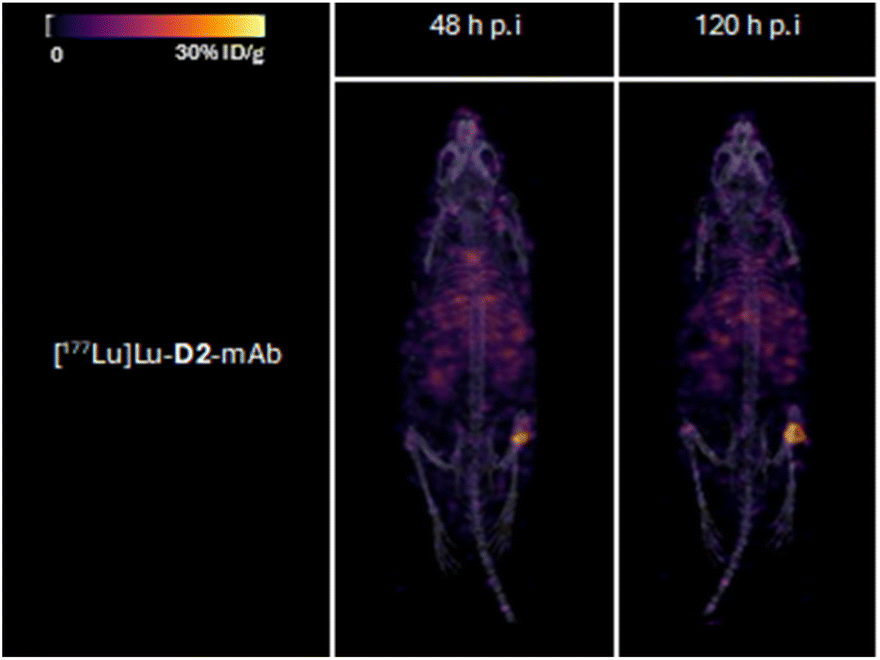 | ||
| Fig. 9 Representative 3D maximum intensity projections for all mice ([177Lu]Lu-D2-mAb) in 177Lu SPECT/CT imaging cohort at 48 and 120 h post-injection. Signal normalised to same thresholding (% ID g−1). | ||
Ex vivo measurements of [89Zr]Zr-D2-mAb or [89Zr]Zr-1-mAb correlated similarly to in vivo biodistribution and the PET/CT images at 48 h (Fig. 7c, 8 and 10a) and 120 h (Fig. 7d, 8 and 10b) post-injection. There was a significant difference in ex vivo biodistribution in the kidney and liver tissues at 48 h post-injection (Fig. 10a), with these differences reducing at 120 h post-injection (Fig. 10b). Most other collected tissues showed no significant difference in biodistribution between [89Zr]Zr-D2-mAb or [89Zr]Zr-1-mAb, with trends similar to in vivo biodistribution at the same time points (Fig. 7c, d; 10a and b).
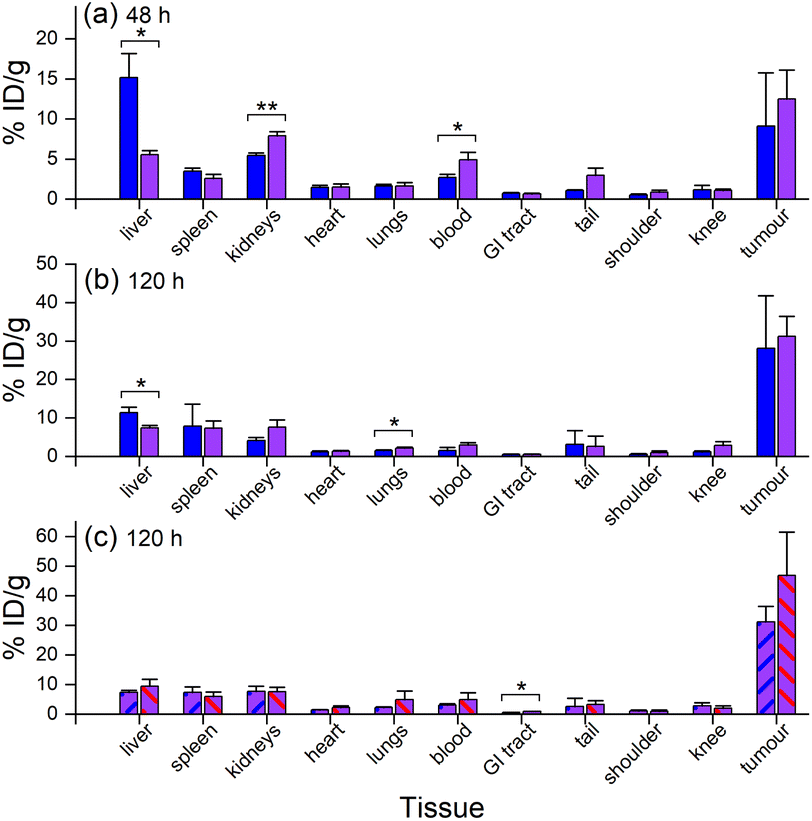 | ||
| Fig. 10 Ex vivo biodistribution from gamma counting (n = 3) of [89Zr]Zr-1-mAb (blue) and [89Zr]Zr-D2-mAb (purple) at (a) 48 h p. i. or (b) 120 h p. i., or (c) [89Zr]Zr-D2-mAb (purple, blue diagonal line) and [177Lu]Lu-D2-mAb (purple, red diagonal line) at 120 h p. i. *p ≤ 0.05, **p ≤ 0.01. | ||
Ex vivo biodistribution of [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb showed a significant difference in accumulation only within the gastrointestinal tract tissue, with all other tissues showing similar biodistribution (Fig. 10c). Both [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb showed minimal uptake into bone tissue (shoulder and knee) at 120 h post-injection, which correlated to the minimal uptake observed in the in vivo biodistribution (Fig. 8 and 9). This suggests that the [89Zr]Zr-D2-mAb complex remained stable up to 120 h in vivo, with bone tissue signals a typical marker of complex instability arising from the osteophilic nature of 89Zr(IV).35,65 The similar biodistribution properties of [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb indicate D2 could have potential utility for improved imaging-therapy dosimetry. The biodistribution of [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb, and [177Lu]Lu-D2-mAb and [177Lu]Lu-2-mAb, supported that D2-mAb formulated for either imaging or therapy could function similar to the matched single-chelator controls.66
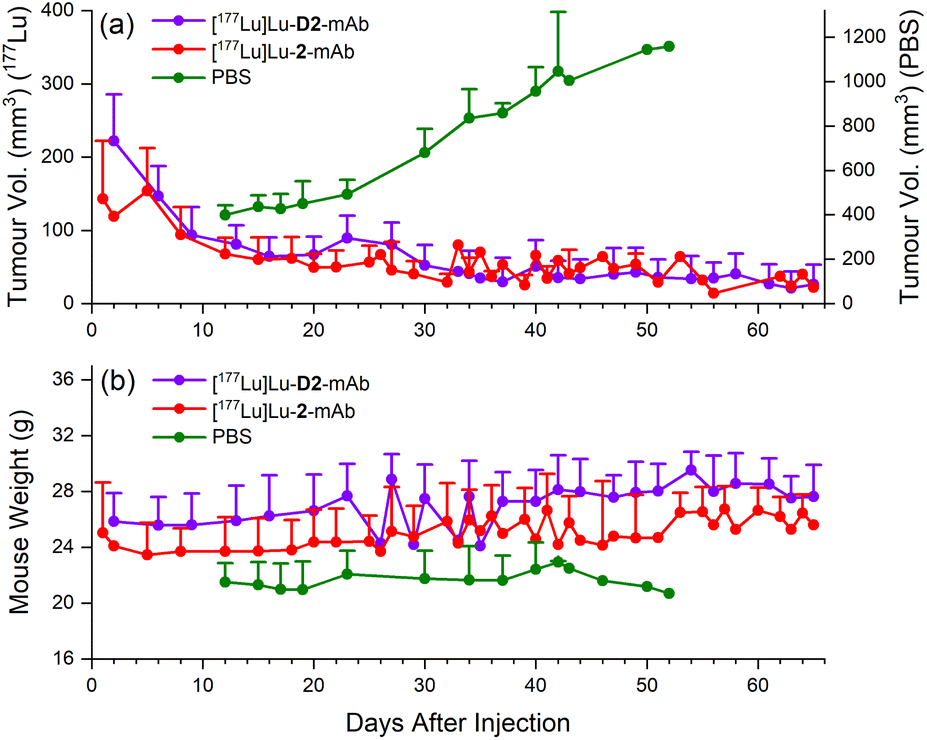 | ||
| Fig. 11 Therapeutic efficacy of [177Lu]Lu-D2-mAb (purple) and [177Lu]Lu-2-mAb (red) in a murine LNCaP xenograft model, monitored over a 65 day period, with vehicle only PBS as control (green). Therapeutic efficacy in each cohort was measured by changes in tumour volume (a) and weight (b). | ||
Therapeutic efficacy of [177Lu]Lu-D2-mAb was found to closely match that of [177Lu]Lu-2-mAb, with both compounds showing >80% reduction in tumour volume (<40 mm3 after 65 days, Fig. 11a) and a slight increase in weight over the 65 day monitoring period (Fig. 11b). Both treatment cohorts exhibited a sustained reduction in tumour volume and sustained increase in weight over the observation period. The vehicle only cohort displayed an increase in tumour volume until ethical cut-offs were reached and mice were euthanised in accordance with the ethics approval guidelines (Fig. 11a). A trend of increasing weight loss was exhibited in the vehicle only cohort (Fig. 11b). These efficacy data showed the therapeutic potential of [177Lu]Lu-D2-mAb, with similar performance to [177Lu]Lu-2-mAb.
Conclusions
A covalent adduct between desferrioxamine B (DFOB, 1) and 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA, 2) separated by an L-lysine unit (DFOB-L-Lys-N6-DOTA, 3a) and containing an amine-terminated PEG motif for further chemistry and biomolecule conjugation was prepared and named ‘D2’ (3). The dual chelator D2-mAb complex was selectively radiolabelled with 89Zr or 177Lu to form [89Zr]Zr-D2-mAb (4) or [177Lu]Lu-D2-mAb (5), respectively, with the regioselectivity of 89Zr(IV) binding to the 1 region or 177Lu(III) to the 2 region, consistent with {1H}–13C NMR spectroscopic data, and complementary LC-MS measurements supporting the 1:1 metal:ligand stoichiometry. [89Zr]Zr-D2-mAb and [89Zr]Zr-1-mAb showed similar PET/CT imaging function and [177Lu]Lu-D2-mAb and [177Lu]Lu-2-mAb showed similar therapeutic efficacy in a murine LNCaP prostate cancer xenograft model, with tumour localisation of [177Lu]Lu-D2-mAb shown by SPECT/CT imaging.
The functional properties of [89Zr]Zr-D2-mAb and [177Lu]Lu-D2-mAb demonstrates the potential of D2 as a single 89Zr–177Lu theranostic agent which could streamline development pathways and improve patient dosimetry. D2 could also open opportunities to use other mixed-element radiometal pairs, including 89Zr (imaging) and 225Ac (therapy) or 212Pb (therapy), which forms ongoing work, together with studies to examine properties of D2 co-labelled with 89Zr and natLu (imaging) or with 177Lu and natZr (therapy), with this iso-structural and iso-charge imaging-therapy pair predicted to have identical biodistribution properties.
Experimental
Materials and methods
O-(1H-Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU, 98%) was obtained from Alfa Aesar. 2,2-Dimethyl-4-oxo-3,8,11,14,17-pentaoxa-5-azaicosan-20-oic acid (N-Boc-PEG4-CO2H, 97%) was obtained from AmBeed. Ammonium acetate (97%) was obtained from APS Finechem. Sodium chloride (99.7%) was obtained from Chemsupply. HEPES (99.7%) was obtained from Formedium. DOTA-tri(tBu) ester (≥95%), DFOB-Ph-NCS (≥94%) and DOTA-Bn-NCS (≥94%) were obtained from Macrocyclics. Dichloromethane (≥99.9%), N-ethyldiisopropylamine (≥98%), sodium hydrogen carbonate (99.7%), formic acid (98–100%) and magnesium sulfate, anhydrous (≥98%) were obtained from Merck. Acetonitrile (99.9%) was obtained from RCI Labscan. Hydrochloric acid (36%) and toluene (99.5%) were obtained from Ajax Finechem. Desferrioxamine mesylate (≥92.5%), N,N-dimethylformamide, anhydrous (99.8%), piperidine (99%), sodium hydroxide, gallium(III) acetylacetonate (99.99%), lutetium(III) chloride (99.99%), zirconium(IV) chloride (≥99.9%), sodium hydroxide (≥98%), methanol, anhydrous (99.8%), triethylamine (≥99%), p-phenylene diisothiocyanate (98%), diethyl ether, anhydrous (≥99.7%) and trifluoroacetic acid (99%) were obtained from Sigma-Aldrich. Nα-[(9H-Fluoren-9-ylmethoxy)carbonyl]-L-lysine hydrochloride (>98%) was obtained from TCI. Milli-Q water was prepared using a Millipore Q-pod system and used as required. Healthy male Balb/c nude mice from 8 weeks old (weight ∼20 g) were obtained from Ozgene. Human serum used for stability studies was obtained from Sigma-Aldrich (product number H4522). Human LnCaP clone FGC cells were obtained from the American Type Culture Collection (ATCC, product number CRL-1740) and used for xenograft tumour growth and cell assays.Synthesis and characterisation
:piperidine (4 mL:1 mL) and the solution was stirred at r. t for 1 h, and the solvent removed in vacuo. Cold ether (approx. 10 mL) was added to the residue and after stirring for 15 min at 0 °C, the ether was decanted, and the residue dried in vacuo to yield 3d as a pale-yellow oil. LRMS (ESI) m/z: [M + H]+ calculated for C59H111N12O16+: 1243.83, found 1243.60; [M + 2H]2+ calculated: 622.42, found 622.50; [M + 3H]3+ calculated: 415.28, found 415.30. This intermediate was used in subsequent steps without further purification.
:DCM (4.5 mL:0.5 mL) and left to stir for 16 h at r. t before removing the solvent in vacuo. The residue was resuspended in toluene (10 mL) and the solvent was removed in vacuo. The residue was further purified via HPLC and lyophilised to yield 3a (95.4 mg, 88.8 μmol, 20.7%) as a white powder. 13C NMR (600 MHz, D2O) δ 176.93, 174.79, 174.77, 173.80 (2C), 173.55, 172.09, 169.89, 169.75, 169.72, 56.90, 56.31 (2C), 55.06, 53.08, 51.78 (2C), 50.52, 50.39, 48.54, 48.39, 48.29, 48.01, 47.85, 47.82, 47.66, 39.27, 39.15, 39.08, 38.46, 30.42, 30.40, 30.06, 27.88, 27.85, 27.81, 27.61, 27.59, 27.20, 25.40 (3C), 23.08, 22.99 (2C), 21.00, 19.21. HRMS (H-ESI II) m/z: [M + H]+ calculated for C47H87N12O16+: 1075.63575, found 1075.63498; [M + 2H]2+ calculated: 538.321514, found 538.320867; [M + 3H]3+ calculated: 359.216768, found 359.216712.
:DCM (9:1) which was stirred at r. t for 16 h and the solvent was removed in vacuo. The residue was resuspended in toluene (10 mL) and the solvent was removed in vacuo. The residue was purified via solid phase extraction (100% ACN for 4 min, 100% H2O for 2.4 min, load, 100% ACN for 2 min, 20–80% ACN:H2O in 5% steps at 3 min each step, 95% ACN:H2O for 3 min, 10 mL min−1), and the collected fractions were combined, and the solvent was removed in vacuo. A fraction of the semi-pure product was further purified via HPLC and lyophilised to yield D2 (3) as a white-brown solid (3.0 mg, 2.27 μmol). 1H NMR (600 MHz, D2O) δ 8.42 (s, 0H), 8.42 (s, 0H), 4.01 (t, J = 6.7 Hz, 1H), 3.82 (qd, J = 16.8, 7.8 Hz, 5H), 3.64 (d, J = 10.3 Hz, 5H), 3.61 (d, J = 6.8 Hz, 5H), 3.48–3.41 (m, 14H), 3.18 (q, J = 8.7, 6.8 Hz, 10H), 3.11 (s, 7H), 3.01 (d, J = 15.5 Hz, 3H), 2.91 (d, J = 15.3 Hz, 3H), 2.80 (t, J = 7.1 Hz, 5H), 2.50 (t, J = 7.1 Hz, 5H), 2.15–2.10 (m, 4H), 1.90 (q, J = 7.0 Hz, 3H), 1.66–1.41 (m, 21H), 1.31 (t, J = 7.8 Hz, 8H). 13C NMR (600 MHz, D2O) δ 172.39, 172.34 (3C), 172.10 (2C), 171.88, 171.84, 170.6 (2C), 170.3, 70.22 (3C), 70.18, 70.11, 70.05, 69.97, 67.28, 58.92 (2C), 56.29, 55.93 (3C), 53.16, 51.55 51.22, 50.3 (2C), 47.54 (2C), 47.24 (3C), 38.99, 38.90 (2C), 39.77, 38.71, 36.30, 30.49, 30.43, 29.28 (3C), 29.09 (3C), 29.02, 28.13, 28.09, 23.97 (2C), 23.90, 23.21, 20.82 (2C). LRMS (ESI) m/z: [M + 2H]2+ calculated for C58H109N13O212+: 661.89, found 662.0; [M + 3H]3+ calculated: 441.60, found 441.80; [M + 4H]4+ calculated: 331.45, found 331.60.
:H2O (3:7) and combined before further purification using HPLC. Fractions containing the product were lyophilised to yield D2-Ph-NCS (3b) as a white solid (32.5 mg, 21.47 μmol, 24.7%). 1H NMR (700 MHz, D2O) δ 7.36 (q, J = 8.7 Hz, 4H), 4.19 (dd, J = 8.7, 5.7 Hz, 1H), 3.76 (t, J = 6.2 Hz, 4H), 3.74–3.56 (m, 31H), 3.23–3.12 (m, 11H), 2.79 (td, J = 7.1, 2.7 Hz, 5H), 2.58–2.50 (m, 4H), 2.49 (t, J = 7.1 Hz, 5H), 2.13 (s, 3H), 1.72–1.61 (m, 5H), 1.61 (dd, J = 9.6, 5.7 Hz, 5H), 1.51 (ddt, J = 14.3, 10.6, 7.2 Hz, 12H), 1.34 (s, 3H), 1.29 (tp, J = 14.9, 7.9, 6.6 Hz, 8H). Acquired using water suppression. 13C NMR (700 MHz, D2O) δ 179.82, 174.84, 174.04, 173.93, 173.85, 173.63, 134.81, 126.91, 126.37, 69.77, 69.73, 69.66, 66.73, 55.33, 54.24, 51.59, 47.97, 47.93, 47.78, 44.32, 39.26, 39.20, 35.77, 30.84, 30.56, 28.05, 28.01, 27.98, 27.91, 27.77, 27.73, 25.57, 25.53, 23.14, 22.99, 22.63, 19.66, 19.34. HRMS (H-ESI II) m/z: [M + H]+ calculated for C66H112N15O21S2+: 1514.75931, found 1514.75500; [M + 2H]2+ calculated: 757.883295, found 757.881144; [M + 3H]3+ calculated: 505.591289, found 505.593039. A larger-scale sample of D2-Ph-NCS provided by AusPep as a custom synthesis (90% pure by RP-HPLC) was used for selected studies (Fig. 5–10; S12–S15, S26–S30 and Tables S1–S7†), with all remaining work using compounds synthesized in-house (Fig. 2–4, S1–S11 and S16–S25†).
Data availability
All relevant data are presented in the main text and ESI† (general information, NMR spectra, LC-MS data, preparation of D2-mAb, 1-mAb, 2-mAb, radiolabelling, biology).Author contributions
The study was conceptualized by RC, MPW, and JLW. All authors contributed to the design of the study methods and the analysis of results. Experimental data was generated by JLW, ZHH, SG, NLF, JH, DTA, KM and WT. Clinical grade HuJ591 was provided by MPW and AI. The first draft of the manuscript was written by RC and JLW, with all authors contributing to manuscript review, editing, and data presentation, and approving the final submission.Conflicts of interest
The authors declare the following competing financial interest(s). Intellectual property related to this research includes Rachel Codd and James L. Wood as listed inventors. Michael P. Wheatcroft and Alesia Ivashkevich are employees of Telix Pharmaceuticals.Acknowledgements
Acknowledgement is made to The University of Sydney (Industry Engagement Seed Fund, Commercialisation CDIP Fund) and Telix Pharmaceuticals for funding, and to the Australian Government with the support of an Australian Government Research Training Program (RTP) Scholarship (J. L. W). Prof. T. Pukala (University of Adelaide) is acknowledged for initial MALDI measurements of D2-mAb. This research was facilitated by access to Sydney Mass Spectrometry, a core research facility at the University of Sydney. We thank Dr B. Mohanty for assistance with NMR spectroscopy carried out at Sydney Analytical, a core research facility at the University of Sydney. We thank the National Imaging Facility (NIF) at the University of Queensland and the National Collaborative Research Infrastructure Strategy (NCRIS) for use of the imaging facilities.Notes and references
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS PubMed.
- L. Bodei, K. Herrmann, H. Schöder, A. M. Scott and J. S. Lewis, Nat. Rev. Clin. Oncol., 2022, 19, 534–550 CrossRef CAS PubMed.
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Nat. Rev. Drug Discovery, 2020, 19, 589–608 CrossRef CAS PubMed.
- K. A. Morgan, S. E. Rudd, A. Noor and P. S. Donnelly, Chem. Rev., 2023, 123, 12004–12035 CrossRef CAS PubMed.
- R. J. Hicks, P. Jackson, G. Kong, R. E. Ware, M. S. Hofman, D. A. Pattison, T. A. Akhurst, E. Drummond, P. Roselt, J. Callahan, R. Price, C. M. Jeffery, E. Hong, W. Noonan, A. Herschtal, L. J. Hicks, A. Hedt, M. Harris, B. M. Paterson and P. S. Donnelly, J. Nucl. Med., 2019, 60, 777–785 CrossRef CAS PubMed.
- C. Cullinane, C. M. Jeffery, P. D. Roselt, E. M. van Dam, S. Jackson, K. Kuan, P. Jackson, D. Binns, J. van Zuylekom, M. J. Harris, R. J. Hicks and P. S. Donnelly, J. Nucl. Med., 2020, 61, 1800–1805 CrossRef CAS PubMed.
- I. Captain, G. J.-P. Deblonde, P. B. Rupert, D. D. An, M.-C. Illy, E. Rostan, C. Y. Ralston, R. K. Strong and R. J. Abergel, Inorg. Chem., 2016, 55, 11930–11936 CrossRef CAS PubMed.
- K. P. Carter, G. J.-P. Deblonde, T. D. Lohrey, T. A. Bailey, D. D. An, K. M. Shield, W. W. Lukens and R. J. Abergel, Commun. Chem., 2020, 3, 1–7 CrossRef.
- J. Xu, F. Cai, Z. Luo, W. Fan, J. Dai, J. Cui, S. Li, C. Geng, Q. Zheng, Z. Wang and X. Tang, Eur. J. Nucl. Med. Mol. Imaging, 2022, 49, 2618–2633 CrossRef CAS PubMed.
- L. Wharton, C. Zhang, H. Yang, J. Zeisler, V. Radchenko, C. Rodríguez-Rodríguez, M. Osooly, B. O. Patrick, K.-S. Lin, F. Bénard, P. Schaffer and C. Orvig, Bioconjugate Chem., 2022, 33, 505–522 CrossRef CAS PubMed.
- B. A. Vaughn, C. S. Loveless, S. J. Cingoranelli, D. Schlyer, S. E. Lapi and E. Boros, Mol. Pharmaceutics, 2021, 18, 4511–4519 CrossRef CAS PubMed.
- Z. Baranyai, G. Tircsó and F. Rösch, Eur. J. Inorg. Chem., 2020, 36–56 CrossRef CAS.
- R. C. Mease, C. M. Kang, V. Kumar, S. R. Banerjee, I. Minn, M. Brummet, K. L. Gabrielson, Y. Feng, A. Park, A. P. Kiess, G. Sgouros, G. Vaidyanathan, M. R. Zalutsky and M. G. Pomper, J. Nucl. Med., 2022, 63, 259–267 CrossRef CAS PubMed.
- J. R. Dilworth and S. I. Pascu, Chem. Soc. Rev., 2018, 47, 2554–2571 RSC.
- N. B. Bhatt, D. N. Pandya and T. J. Wadas, Molecules, 2018, 23, 638 CrossRef PubMed.
- M. Chomet, G. A. M. S. van Dongen and D. J. Vugts, Bioconjugate Chem., 2021, 32, 1315–1330 CrossRef CAS PubMed.
- V. Radchenko, S. Busse and F. Roesch, Nucl. Med. Biol., 2014, 41, 721–727 CrossRef CAS PubMed.
- P. M. Smith-Jones, B. Stolz, C. Bruns, R. Albert, H. W. Reist, R. Fridrich and H. R. Macke, J. Nucl. Med., 1994, 35, 317–325 CAS.
- C. J. Mathias, M. R. Lewis, D. E. Reichert, R. Laforest, T. L. Sharp, J. S. Lewis, Z.-F. Yang, D. J. Waters, P. W. Snyder, P. S. Low, M. J. Welch and M. A. Green, Nucl. Med. Biol., 2003, 30, 725–731 CrossRef CAS PubMed.
- J. P. Holland, in Handbook of Radiopharmaceuticals: Methodology and Applications, ed. M. R. Kilbourn and P. J. Scott, John Wiley & Sons Ltd, Hoboken, NJ, 2nd edn, 2021, ch. 11, pp. 343–374 Search PubMed.
- U. Pandey, N. Gamre, Y. Kumar, P. Shetty, H. Dev Sarma and A. Dash, J. Radioanal. Nucl. Chem., 2016, 307, 187–194 CrossRef CAS.
- C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 3003–3016 CrossRef CAS.
- R. Codd, Coord. Chem. Rev., 2008, 252, 1387–1408 CrossRef CAS.
- L. Bíró, P. Buglyó and E. Farkas, Curr. Med. Chem., 2021, 28, 7209–7237 CrossRef.
- M. Patra, A. Bauman, C. Mari, C. A. Fischer, O. Blacque, D. Haussinger, G. Gasser and T. L. Mindt, Chem. Commun., 2014, 50, 11523–11525 RSC.
- S. E. Rudd, P. Roselt, C. Cullinane, R. J. Hicks and P. S. Donnelly, Chem. Commun., 2016, 52, 11889–11892 RSC.
- T. Richardson-Sanchez, W. Tieu, M. P. Gotsbacher, T. J. Telfer and R. Codd, Org. Biomol. Chem., 2017, 15, 5719–5730 RSC.
- W. Tieu, T. Lifa, A. Katsifis and R. Codd, Inorg. Chem., 2017, 56, 3719–3728 CrossRef CAS.
- C. J. M. Brown, M. P. Gotsbacher and R. Codd, Aust. J. Chem., 2020, 73, 969–978 CAS.
- J. P. Holland, Inorg. Chem., 2020, 59, 2070–2082 CrossRef CAS PubMed.
- A. K. Salih, S. J. Raheem, M. Dominguez Garcia, W. K. Ahiahonu and E. W. Price, Inorg. Chem., 2022, 61, 20964–20976 CrossRef CAS PubMed.
- R. Raavé, G. Sandker, P. Adumeau, C. B. Jacobsen, F. Mangin, M. Meyer, M. Moreau, C. Bernhard, L. Da Costa, A. Dubois, V. Goncalves, M. Gustafsson, M. Rijpkema, O. Boerman, J.-C. Chambron, S. Heskamp and F. Denat, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1966–1977 CrossRef.
- H. Damerow, X. Cheng, V. von Kiedrowski, R. Schirrmacher, B. Wängler, G. Fricker and C. Wängler, Pharmaceutics, 2022, 14, 2214 CrossRef PubMed.
- E. Khozeimeh Sarbisheh, K. L. Summers, A. K. Salih, J. J. H. Cotelesage, A. Zimmerling, I. J. Pickering, G. N. George and E. W. Price, Inorg. Chem., 2023, 62, 2637–2651 CrossRef PubMed.
- Y. Toporivska and E. Gumienna-Kontecka, J. Inorg. Biochem., 2019, 198, 110753 CrossRef CAS PubMed.
- B. Borgias, A. D. Hugi and K. N. Raymond, Inorg. Chem., 1989, 28, 3538–3545 CrossRef CAS.
- B. J. Hernlem, L. M. Vane and G. D. Sayles, Inorg. Chim. Acta, 1996, 244, 179–184 CrossRef CAS.
- W. P. Cacheris, S. K. Nickle and A. D. Sherry, Inorg. Chem., 1987, 26, 958–960 CrossRef CAS.
- V. Kubíček, J. Havlíčková, J. Kotek, G. Tircsó, P. Hermann, E. Tóth and I. Lukeš, Inorg. Chem., 2010, 49, 10960–10969 CrossRef.
- C. G. Pippin, T. J. McMurry, M. W. Brechbiel, M. McDonald, R. Lambrecht, D. Milenic, M. Roselli, D. Colcher and O. A. Gansow, Inorg. Chim. Acta, 1995, 239, 43–51 CrossRef CAS.
- M. T. Ma, L. K. Meszaros, B. M. Paterson, D. J. Berry, M. S. Cooper, Y. M. Ma, R. C. Hider and P. J. Blower, Dalton Trans., 2015, 44, 4884–4900 RSC.
- D. N. Pandya, N. Bhatt, H. Yuan, C. S. Day, B. M. Ehrmann, M. Wright, U. Bierbach and T. J. Wadas, Chem. Sci., 2017, 8, 2309–2314 RSC.
- D. N. Pandya, K. E. Henry, C. S. Day, S. A. Graves, V. L. Nagle, T. R. Dilling, A. Sinha, B. M. Ehrmann, N. B. Bhatt, Y. Menda, J. S. Lewis and T. J. Wadas, Inorg. Chem., 2020, 59, 17473–17487 CrossRef CAS.
- S. Aime, M. Botta and G. Ermondi, Inorg. Chem., 1992, 31, 4291–4299 CrossRef CAS.
- S. Aime, M. Botta, M. Fasano, M. P. M. Marques, C. F. G. C. Geraldes, D. Pubanz and A. E. Merbach, Inorg. Chem., 1997, 36, 2059–2068 CrossRef CAS.
- J. Blahut, P. Hermann, Z. Tošner and C. Platas-Iglesias, Phys. Chem. Chem. Phys., 2017, 19, 26662–26671 RSC.
- M. Briand, M. L. Aulsebrook, T. L. Mindt and G. Gasser, Dalton Trans., 2017, 46, 16387–16389 RSC.
- A. Guillou, D. F. Earley, S. Klingler, E. Nisli, L. J. Nüesch, R. Fay and J. P. Holland, Bioconjugate Chem., 2021, 32, 1263–1275 CrossRef CAS PubMed.
- D. Novak, T. Tomašič, M. Krošelj, U. Javornik, J. Plavec, M. Anderluh and P. K. Peitl, ChemMedChem, 2021, 16, 155–163 CrossRef CAS PubMed.
- M. W. Giese, R. H. Woodman, G. T. Hermanson and P. D. Davis, in Chemical Linkers in Antibody–Drug Conjugates (ADCs), ed. F. L. van Delft and J. M. Lambert, The Royal Society of Chemistry, 2022, vol. 81, ch. 9, pp. 286–376 Search PubMed.
- H. D. King, D. Yurgaitis, D. Willner, R. A. Firestone, M. B. Yang, S. J. Lasch, K. E. Hellström and P. A. Trail, Bioconjugate Chem., 1999, 10, 279–288 CrossRef CAS PubMed.
- G. Liao, Z. Zhou, S. Burgula, J. Liao, C. Yuan, Q. Wu and Z. Guo, Bioconjugate Chem., 2015, 26, 466–476 CrossRef CAS PubMed.
- C. Abate-Shen and F. Nunes de Almeida, Cancer Res., 2022, 82, 1689–1691 CrossRef CAS PubMed.
- D. M. Nanus, M. I. Milowsky, L. Kostakoglu, P. M. Smith-Jones, S. Vallabahajosula, S. J. Goldsmith and N. H. Bander, J. Urol., 2003, 170, S84–S88 CrossRef PubMed.
- N. T. Viola-Villegas, K. K. Sevak, S. D. Carlin, M. G. Doran, W. H. Evans, D. W. Bartlett, A. M. Wu and J. S. Lewis, Mol. Pharmaceutics, 2014, 11, 3965–3973 CrossRef CAS PubMed.
- S. Vallabhajosula, P. M. Smith-Jones, V. Navarro, S. J. Goldsmith and N. H. Bander, Prostate, 2004, 58, 145–155 CrossRef CAS PubMed.
- R. I. J. Merkx, D. Lobeek, M. Konijnenberg, L. D. Jiménez-Franco, A. Kluge, E. Oosterwijk, P. F. A. Mulders and M. Rijpkema, Eur. J. Nucl. Med. Mol. Imaging, 2019, 48, 3277–3285 CrossRef PubMed.
- R. I. J. Merkx, M. Rijpkema, G. M. Franssen, A. Kip, B. Smeets, A. Morgenstern, F. Bruchertseifer, E. Yan, M. P. Wheatcroft, E. Oosterwijk, P. F. A. Mulders and S. Heskamp, Pharmaceuticals, 2022, 15, 570 CrossRef CAS PubMed.
- S. Vallabhajosula, A. Nikolopoulou, Y. S. Jhanwar, G. Kaur, S. T. Tagawa, D. M. Nanus, N. H. Bander and S. J. Goldsmith, Curr. Radiopharm., 2016, 9, 44–53 CrossRef CAS PubMed.
- J. M. White, F. E. Escorcia and N. T. Viola, Theranostics, 2021, 11, 6293–6314 CrossRef CAS PubMed.
- G. L. DeNardo, S. J. Kennel, J. A. Siegel and S. J. Denardo, Clin. Lymphoma, 2004, 5, S5–S10 CrossRef CAS PubMed.
- M. Chomet, M. Schreurs, M. J. Bolijn, M. Verlaan, W. Beaino, K. Brown, A. J. Poot, A. D. Windhorst, H. Gill, J. Marik, S. Williams, J. Cowell, G. Gasser, T. L. Mindt, G. A. M. S. van Dongen and D. J. Vugts, Eur. J. Nucl. Med. Mol. Imaging, 2021, 48, 694–707 CrossRef CAS PubMed.
- U. Eberlein, M. Cremonesi and M. Lassmann, J. Nucl. Med., 2017, 58(Suppl 2), 97S–103S CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc02851a |
| ‡ Abbreviations: mCRPC, metastatic castration-resistant prostate cancer; DFOB, desferrioxamine B; DOTA, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid; HBED-CC, 3,3′-(((ethane-1,2-diylbis((carboxymethyl)azanediyl))bis(methylene)) bis(4-hydroxy-3,1-phenylene))dipropionic acid; NET, neuroendocrine tumours; PSMA, prostate-specific membrane antigen. |
| § Examples of other activating groups are NHS-, maleimide-, or ethyl squaramide groups installed from reactions of D2 with bis-N-succinimidyl glutarate, N-succinimidyl-3-maleimidopropionate, or diethyl squarate, respectively. |
| ¶ All studies involving animals were conducted in accordance with the guidelines of the Animal Ethics Committee of The University of Queensland, and the Australian Code for the Care and Use of Animals for Scientific Purposes (AEC approval number: 2022/AE000135). |
| This journal is © The Royal Society of Chemistry 2024 |