
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAutomation and machine learning augmented by large language models in a catalysis study
Yuming
Su
 ac,
Xue
Wang
a,
Yuanxiang
Ye
b,
Yibo
Xie
b,
Yujing
Xu
a,
Yibin
Jiang
*c and
Cheng
Wang
*ac
ac,
Xue
Wang
a,
Yuanxiang
Ye
b,
Yibo
Xie
b,
Yujing
Xu
a,
Yibin
Jiang
*c and
Cheng
Wang
*ac
aiChem, State Key Laboratory of Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, P. R. China. E-mail: 20520200156127@stu.xmu.edu.cn; 20520221152116@stu.xmu.edu.cn; yujingxu@xmu.edu.cn
bInstitute of Artificial Intelligence, Xiamen University, Xiamen 361005, P. R. China. E-mail: 36920221153140@stu.xmu.edu.cn; wangduphunsukh@stu.xmu.edu.cn
cInnovation Laboratory for Sciences and Technologies of Energy Materials of Fujian Province (IKKEM), Xiamen 361005, P. R. China. E-mail: yibin_jiang@outlook.com; wangchengxmu@xmu.edu.cn
First published on 26th June 2024
Abstract
Recent advancements in artificial intelligence and automation are transforming catalyst discovery and design from traditional trial-and-error manual mode into intelligent, high-throughput digital methodologies. This transformation is driven by four key components, including high-throughput information extraction, automated robotic experimentation, real-time feedback for iterative optimization, and interpretable machine learning for generating new knowledge. These innovations have given rise to the development of self-driving labs and significantly accelerated materials research. Over the past two years, the emergence of large language models (LLMs) has added a new dimension to this field, providing unprecedented flexibility in information integration, decision-making, and interacting with human researchers. This review explores how LLMs are reshaping catalyst design, heralding a revolutionary change in the fields.
1 Introduction
The field of catalyst design and discovery is undergoing a profound transformation, facilitated by the convergence of artificial intelligence (AI)1–3 and automation systems,4–6 as well as utilization of large data. This shift is propelled by advancements in four crucial areas: high-throughput information extraction,7–16 automated robotic systems for chemical experimentation,4–6,17–19 real-time active machine learning (ML) with on-line data processing and feedback for iterative optimization,4,20–35 and interpretable machine learning for generating knowledge,36–39 each playing a pivotal role in evolving traditional methodologies. Central to this modern era are self-driving labs40 that are further integrated with theoretical simulations and extensive databases, revolutionizing how catalysts are created and optimized.Recently, large language models (LLMs) such as GPT-x, ERNIE Bot, Claude-x, and Lamma-x,41 have begun to dramatically enhance these four technological pillars. By processing natural language, automating code generation and data analysis, optimizing design of experiment (DoE) algorithms, and facilitating human–computer interaction,16,42–47 LLMs are setting new standards for efficiency and innovation in catalysis research (Fig. 1). These capabilities allow for the extraction and utilization of data from diverse and unstructured sources such as scattered texts, videos, and images, previously inaccessible to more traditional ML technologies that relied on well-organized datasets.
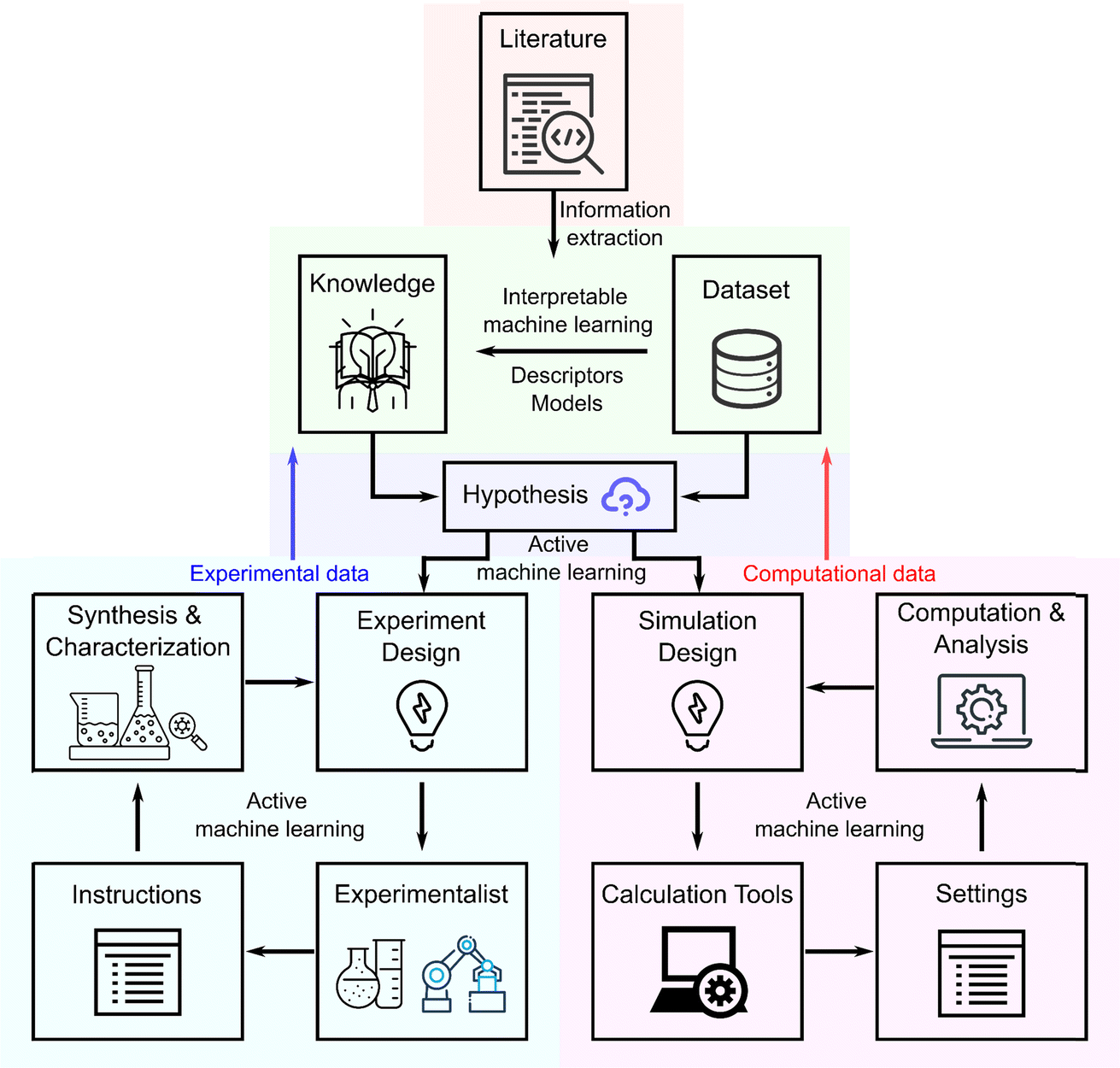 | ||
| Fig. 1 The workflow of catalyst design and discovery with information extraction, automated chemical experimentation, active machine learning, and interpretable machine learning. | ||
Moreover, automated and intelligent robotic systems, which have seen significant adoption over the last decade, spanning from flow systems19,48,49 to desktops50,51 and humanoid mobile robots,4,5 now seamlessly integrate with advanced LLMs. This synergy is reshaping decision-making strategies within the field, transitioning from traditional methods like Bayesian optimization4 and active learning32 to more sophisticated, LLM-enhanced approaches,45,47 towards more talented self-driving labs for closed-loop discovery. This is only the beginning of a shifting paradigm to on-demand catalyst development and in silico performance scanning for catalyst design and optimization.
Despite these technological advances, the role of the human researcher remains indispensable. The interpretability of ML methods is crucial for harnessing human intellectual engagement and deriving scientific insights that can inform new design principles for high-performance catalysts.36–39 Artificial neural networks (ANNs)52 used to be regarded as black-box models that are hard to explain, but recent innovations such as SHapley Additive exPlanations (SHAP)53 for graph neural networks (GNNs) and attention mechanisms in transformer models are enhancing the transparency of artificial neural networks, which were previously considered opaque. In addition, LLMs have also showcased their capabilities in extracting data mapping and articulating them in a clear plain language format.
Given the rapid pace of these advancements, it is timely to review the revolutionary shift in AI applications for catalysis research and development. This review will delve into how the integration of LLMs is redefining the four foundational ML technologies in catalysis, providing a historical perspective and discussing recent implementations that foreshadow the future of AI-assisted catalyst design.
2 High-throughput chemical information extraction
Traditionally, data extraction required manual efforts, which has successfully underpinned the establishment of chemical databases like Reaxys54 and SciFinder.55 With the increasing demand to autonomously gather and standardize chemical information effectively, the development of automated data extraction methods has split into two primary directions: the extraction of chemical information from figures including optical chemical structure recognition (OCSR),7–10 and text information extraction. Both avenues benefit significantly from enhancements provided by pre-trained LLMs.15,162.1 Information extraction from figures
A considerable amount of chemical information resides in figures, rendering Optical Chemical Structure Recognition (OCSR) essential for converting these complex visual data into accessible and interpretable formats. The primary task of OCSR is to transform visual representations of chemical structures into formats ready for computer processing. We now list and briefly discuss these different computer-ready formats.SMARTS (SMILES Arbitrary Target Specification): an extension of SMILES, SMARTS allows for defining substructural patterns within molecules, enhancing search and analysis capabilities.
InChI (International Chemical Identifier): provides a structured and layered representation of chemical data, facilitating interoperability across different data systems.
SELFIES (Self-referencing Embedded Strings): designed to ensure the validity of molecules represented, enhancing data integrity.
These string representations, integral to systematic chemical naming, have become increasingly valuable with the advent of language models. The seamless integration of these formats into LLMs enhances their utility, making them more than just systematic nomenclature but a dynamic part of molecular data processing. Furthermore, the development of multi-modal large models allows for directly translating structural drawings to the string representations without prior conversion, marking a significant advancement in the field.56
2.1.4.1 Segmentation challenges. The initial and crucial step in rule-based OCSR is the segmentation of chemical structures from potentially complex images. This task is challenging and critical as it sets the foundation for all subsequent analyses. Early rule-based models such as optical recognition of chemical structures (OROCS), chemical literature data extraction (CLiDE),65–67 the optical structure recognition application (OSRA) and Imago68,69 faced significant challenges in accurately segmenting chemical structures. These systems often struggled with noisy data and the presence of fragmented characters or text lines adjacent to the chemical structures.
In 2014, Simone Marinai et al.70 made an improvement by introducing a Markov logic-based probabilistic logic inference engine (Fig. 2). This development improved the ability to clean up noisy extractions, although challenges with fragmented elements persisted. More recently, in 2021, Yifei Wang et al.59 advanced the field further by employing a Single Shot MultiBox Detector (SSD) neural network combined with a Non-Maximum Area Suppression (NMAS) algorithm. This combination was specifically designed to enhance object identification within a single frame, significantly improving segmentation accuracy to 89.5% on a dataset of 2100 handwritten cyclic compound samples.
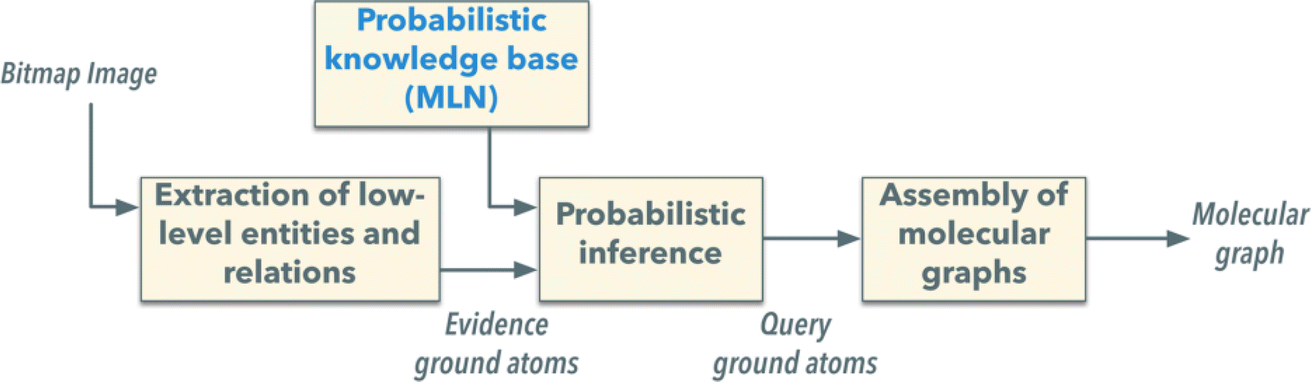 | ||
| Fig. 2 Scheme of the Markov logic OCSR with low-level image information extraction and probabilistic logic inference. Reproduced with permission from ref. 70 Copyright 2014, American Chemical Society. | ||
2.1.4.2 Inherent limitations. Despite these advancements, rule-based systems are often limited by two major factors:
(1) Insufficient understanding of embedded rules: the complexity of the embedded rules can lead to misinterpretations and errors in data extraction.
(2) Susceptibility to noise: the intricate rules are prone to interference from noisy data, which can degrade the quality of the output.
2.1.5.1 Innovative developments in machine learning for OCSR.
2.1.5.1.1 MSE-DUDL. Introduced in 2019 by Kyle Marshall et al.,71 MSE-DUDL combines a convolutional neural network (CNN) known for its prowess in visual pattern recognition, and a long short-term memory (LSTM) network equipped with an “attention” mechanism. This attention mechanism allows the model to focus selectively on different parts of the molecular structure, facilitating accurate SMILES prediction. While the method achieved an accuracy of 83% on a specialized test set, it faced limitations in recognizing certain complex chemical structures and stereochemical details, and struggled with images presented in inverted formats.
2.1.5.1.2 DECIMER. Developed by Christoph Steinbeck et al. in 2020,72 DECIMER employs an autoencoder architecture that includes a CNN encoder for converting images into vectors and a gated recurrent unit (GRU)-based decoder for translating these vectors into SMILES strings. Initially trained with data images created by the Chemical Development Kit (CDK), DECIMER has shown success in extracting structural representations from millions of examples. Enhancements such as DECIMER segmentation were introduced in 2021 (ref. 73) to improve chemical element detection in documents, and by 2023,74 DECIMER.ai further automated the segmentation, classification, and translation of chemical structures from printed literature into the SMILES format (Fig. 3).
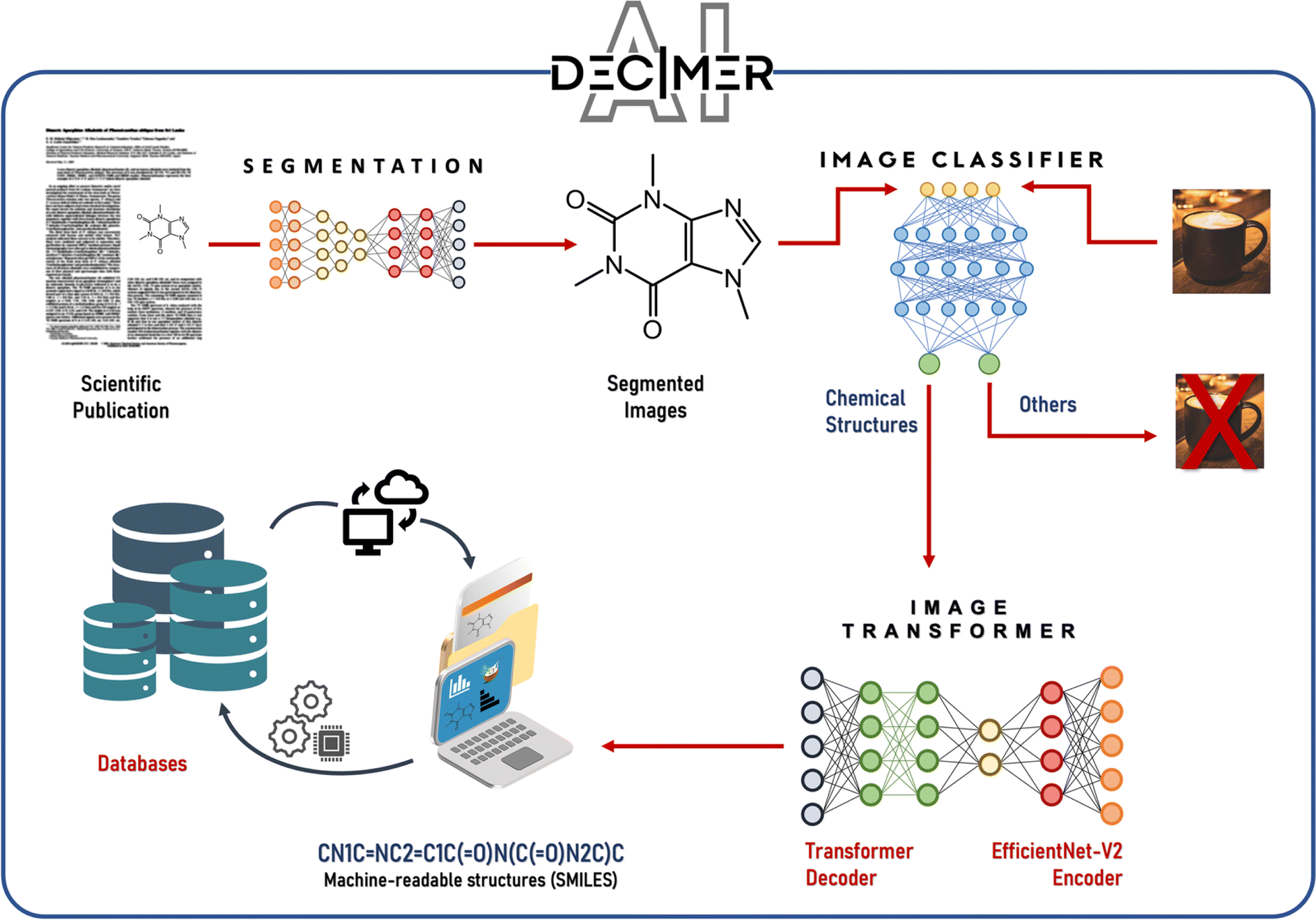 | ||
| Fig. 3 Overview of the integrated DECIMER workflow including image segmentation, classification, and translation to obtain SMILES. Reproduced with permission from ref. 74 under CC BY license. | ||
2.1.5.1.3 MolMiner. In 2022, Jianfeng Pei et al. developed MolMiner,75 a deep learning-based OCSR system that directly recognizes atoms and chemical bonds in images, circumventing traditional vectorization methods. It demonstrates superior accuracy and speed by extracting chemical structures from PDFs and outputting them in standardized formats, showcasing its efficacy over other OCSR systems like MolVec, OSRA, and Imago.
2.1.5.1.4 MolScribe. Representing the cutting edge, MolScribe is an image-to-graph generation model76 that merges neural network capabilities with rule-based methods. It predicts atoms and bonds along with their geometric layouts to construct 2D molecular graphs, applying symbolic chemistry constraints to recognize complex chemical patterns, including chirality and abbreviations. Enhanced by data augmentation strategies, MolScribe effectively handles domain shifts and various drawing styles found in chemical literature. Its robustness has been confirmed through testing, showing an accuracy of 76–93% on public benchmarks.
The accuracy and reliability of OCSR continue to improve as newer models are developed and refined. The use of multiple models for cross-validation purposes enhances robustness, offering better performance than what could be achieved by a single model. This progress is vital as it addresses the significant challenge of extracting organic reaction data on a large scale, a task that is increasingly crucial due to the exponential growth of available chemical data.
2.1.6.1 Advancements in multimodal large models. Recent advancements in AI have introduced multimodal large models, such as GPT-4, Gemini, and Claude, which have demonstrated promising capabilities in summarizing information from diverse sources. These models can be adept at extracting and synthesizing comprehensive experimental data from the scientific literature on catalysis.
2.1.6.2 Capabilities of multimodal large models in chemical data analysis.
2.1.6.2.1 Graphical data analysis. Many of these advanced models are now capable of interpreting trends and patterns directly from graphical representations, although the variability in data presentation styles continues to challenge the accuracy and reliability of the extractions.
2.1.6.2.2 Recognition of hand-drawn structures. Multimodal LLMs have shown an ability to recognize even simple hand-drawn chemical structures, which opens up possibilities for more intuitive interfaces between researchers and computational systems.
2.1.6.2.3 Integration with OSRA. Efforts are ongoing to integrate systems like the Optical Structure Recognition Application (OSRA) with multimodal LLMs to enhance the extraction of chemical structures from the literature. For instance, DP Technology's introduction of the Uni-Finder module represents a step forward (still at a testing stage on May 5th 2024). This module is designed for the comprehensive reading of scientific documents, including journal papers and patents, which facilitates a deeper understanding and utilization of published research.
The continuous improvement of multimodal LLMs is expected to revolutionize how scientific results are communicated and utilized. As these models become more sophisticated, they will enable the scientific community to integrate vast amounts of data in unprecedented ways. This integration is anticipated to lead to the development of new tools that could dramatically enhance the efficiency and creativity of catalyst design processes. The ability to compile and analyze the extensive data generated globally by researchers represents a transformative shift towards data-driven science, promising significant advancements in how we discover and develop new materials.
2.2 Text information extraction with language models
Before the advent of large language models (LLMs), there was significant effort in natural language processing (NLP) dedicated to extracting chemical information from texts. This process involved several traditional NLP tasks such as named entity recognition, relation extraction, and the construction of knowledge graphs.77,78 In named entity recognition, entities (which could be single words or phrases) are identified and categorized within the text, facilitating the detection of reagents, products, catalysts, and other chemical entities. Relation extraction focuses on identifying the connections between these entities, while knowledge graphs organize these entities and their relationships into structured representations. This foundation has enabled the creation of catalysis datasets related to topics like hydrogen production,12 CO2 reduction,13,14 and single-atom heterogeneous catalysis.792.2.1.1 ChemDataExtractor. The ChemDataExtractor tool,80,81 developed as early as 2016, utilizes word tokenization, clustering, and traditional machine-learning models to extract chemical knowledge from the literature. This tool can identify compounds and their properties, setting a precedent for the integration of more sophisticated models. In 2021, Regina Barzilay et al. developed the ChemRxnExtractor,82 a two-stage deep learning architecture based on transformer models. This system uses product extraction and reaction role labelling to structure chemical data. The transformer architecture's attention mechanism allows the model to concentrate on relevant parts of the data for different tasks, and its adaptive pre-training on large-scale unlabelled text has significantly improved its ability to identify and organize chemical information from textual sources (Fig. 4). It achieved notable F1 scores of 76.2% for product extraction and 78.7% for reaction role labelling on a specialized dataset.
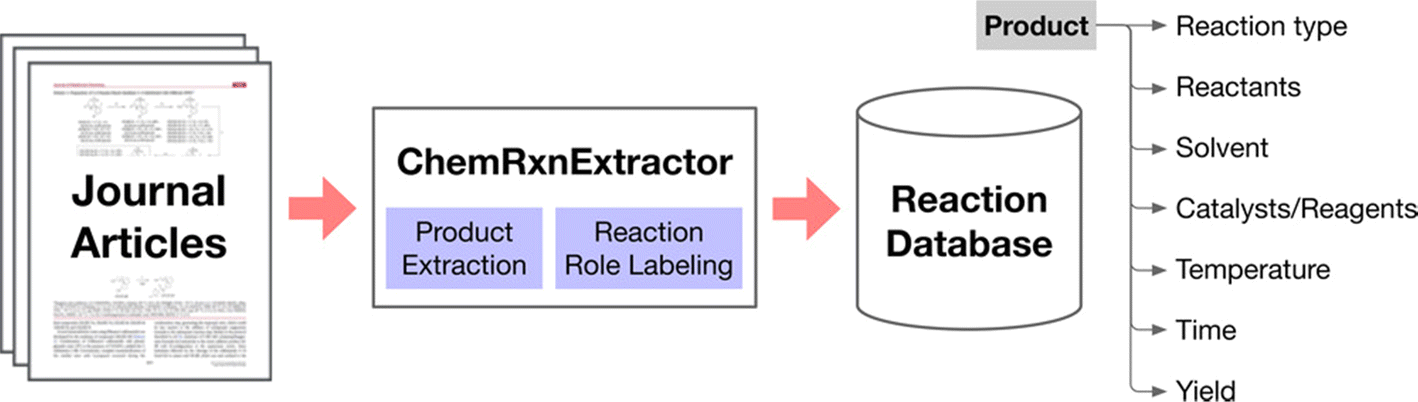 | ||
| Fig. 4 Scheme of the automated chemical reaction extraction from scientific literature. Reproduced with permission from ref. 82 Copyright 2019, American Chemical Society. | ||
2.2.1.2 SciBERT. Introduced in 2019, SciBERT11 leverages the BERT (Bidirectional Encoder Representations from Transformers) architecture, which is specifically trained on scientific texts to enhance performance in tasks like entity recognition and relation extraction.12–14 Following the surge in LLM advancements in 2022, models such as SciBERT, GPT-3, GPT-3.5, and GPT-4 have become integral to text-based data extraction in catalysis.12–16 These models have effectively turned the extraction of text-based data from scientific papers into a nearly solved challenge.
2.2.1.3 LLMs. Omar M. Yaghi et al.16 utilized OpenAI's GPT-3.5 to extract and format synthesis information of metal–organic frameworks (MOFs) from the literature. They addressed the hallucination issue in LLMs through careful prompt engineering and context provision (Fig. 5). The process involved segmenting the text, creating numeric vectors to represent each segment, comparing vectors to the ones of predefined synthesis descriptions, and choosing the segments with high similarity. GPT-3.5 then classified these segments as ‘synthesis’ or ‘non-synthesis’ using in-context learning (ICL), before formatting the synthesis information into tables. This approach, which also led to the development of a chemistry chatbot, demonstrates a promising framework for using LLMs for extracting and organizing scientific information.
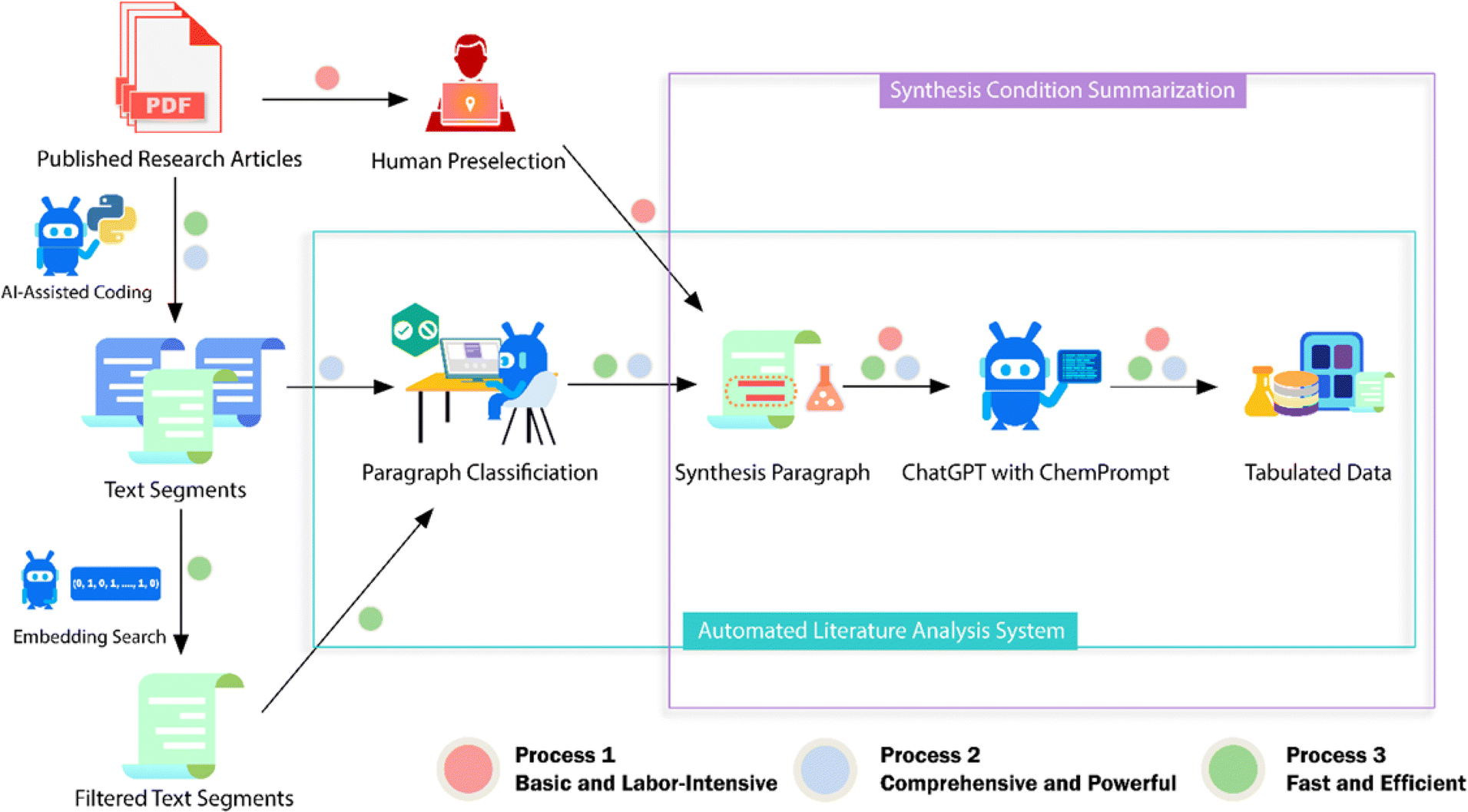 | ||
| Fig. 5 Scheme of the ChatGPT chemistry assistant workflow to extract synthesis information of MOFs from the literature. Reproduced with permission from ref. 16 Copyright 2023, American Chemical Society. | ||
2.3 Summary
In the domain of chemical information extraction, advancements have been marked by the development and deployment of diverse methods and tools. These technologies are succinctly summarized in Table 1 and are broadly categorized into three primary types based on the underlying technology: rule-based OCSR, machine learning-based (ML-based) OCSR, and language model-based (LM-based) systems.| Method | Type | Extracted content | Supported modality | Open source | Reference |
|---|---|---|---|---|---|
| CLiDE | Rule-based | Molecular structures and charge | Text & image | Yes | 66 |
| OSRA | Rule-based | Molecular structures | Text & image | Yes | 68 |
| Imago | Rule-based | Depicted molecules with up and down stereo bonds and pseudostems | Text & image | Yes | 69 |
| MSE-DUDL | ML-based | Structures of natural products and peptide sequences | Image | No | 71 |
| DECIMER | ML-based | Chemical classes, species, organism parts, and spectral data | Image | Yes | 72 |
| MolMiner | ML-based | Molecule structures | Image | No | 75 |
| ChemDataExtractor | LM-based | Identifiers, spectroscopic attributes, and chemical property attributes (e.g., melting point, oxidation/reduction potentials, photoluminescence lifetime, and quantum yield) | Text | Yes | 80 and 81 |
| SciBERT | LM-based | Identifiers of chemicals | Text | Yes | 11 |
| ChemRxnExtractor | LM-based | Reactants, catalysts, and solvents for reactions | Text | Yes | 82 |
| GPT-3.5 | LM-based | MOF synthesis | Text | No | 16 |
| GPT-4 | LM-based | Text & image | No |
The rule-based OCSR systems, once dominant, are now increasingly complemented or surpassed by neural network-based methods due to their flexibility and growing accuracy. These machine learning-based systems are not only more adaptable but also continue to improve as they learn from more data. The incorporation of rule-based techniques as a supplementary approach provides a layered methodological depth that enhances the overall robustness and generalizability of these technologies.
Language model-based systems, particularly those utilizing advanced LLMs, represent the frontier of chemical information extraction. Although their full potential is yet to be realized, the rapid evolution into multimodal models suggests that transformative developments could emerge shortly. These models are particularly promising for handling the vast and complex data typical in catalysis research.
The transition to open-source methods has also played a critical role in this field. Beginning with systems like OSRA in the 1990s, the move towards open-source has not only facilitated wider access to advanced tools but has also spurred innovation and customization, enhancing the collective capability of the research community.
This evolving landscape of chemical information extraction methods underscores the importance of continual adaptation and development to harness the ever-increasing volumes of data in catalysis and other fields of chemistry.
3 Automated and intelligent chemical robotic system
Automation technologies have profoundly transformed modern manufacturing, yet their integration into chemical research remains limited. This is primarily due to the challenges in meeting the diverse and flexible synthesis and characterization requirements of various chemical systems. Effective machine learning applications in this context demand a densely populated dataset within the search space to develop reliable models and derive meaningful insights. Consequently, the experimental systems employed must be both high-throughput and dependable.Over the past few decades, significant advancements in automation have led to reductions in costs and enhancements in the efficiency, accuracy, and reproducibility of experiments.83–86 The origins of chemical automation date back to the 1960s and 1970s with the development of automated devices like automated peptide synthesizers,87 DNA synthesizers,88 and organic synthesis modules.89 This was followed by the emergence of high-throughput automated synthesis systems in the era of combinatorial chemistry.90–96 More recently, the introduction of humanoid chemical robots4–6 and autonomous flow-based synthesis platforms17–19 has marked a new era of innovation in intelligent chemical synthesis.
A notable feature of this latest advancement is the interactive “ask and tell” process, such as active learning, where models are continuously trained on current observations and actively request additional data. This interactive approach can significantly accelerate discovery efficiency compared to traditional screening strategies.97 Therefore, experimental processes must be designed to be not only high-throughput but also sufficiently flexible to allow frequent access and modifications. This is also the stage where LLMs can contribute, integrating crucial domain knowledge to enhance exploration and decision-making processes.
In this section, we will discuss how advancements in hardware design, coupled with LLMs, enhance operational flexibility. Later, we will explore the promising potential of LLM-driven active learning in the subsequent section.
3.1 Automated and intelligent chemical experiment platform
To address diverse research tasks, various hardware design principles and methods were employed in building automation systems. This review will cover two categories of the systems:(1) Humanoid robotic systems: this approach relies on the usage of multi-axis arms that provide a high degree of operation flexibility, mimicking the behavior of human operators.
(2) Automated flow chemical systems: these systems are designed on the foundation of fluid dynamics and transport pipelines to achieve precise chemical operations, which can be seamlessly interfaced with analytical instruments.
3.2 Humanoid robotic system
In a laboratory environment, a robotic arm coupled with automated guided vehicles (AGVs) and advanced computer vision systems5 can robustly complete tasks such as sample preparation and handling, control of instruments, and integration of data recording, analysis, and experiment design. Key to this scheme is the flexibility introduced by AGVs and robotic arms as compared to that of their predecessors.The AGV-based autonomous mobile robot system launched by Andrew I. Cooper et al.4 is a remarkable advance in chemical automation. The team found improved photocatalysts for producing hydrogen from water after autonomous running for 8 days, completing 688 experiments in a design space of 10 variables. The robot (Fig. 6) can handle sample vials among eight workstations distributed around the lab, including a solid reagent dispensing system, a mixed liquid dispensing system and capping module, an ultrasound module, a photolysis module, a gas chromatography (GC) analysis module, and three separate sample storage modules to achieve a variety of experimental tasks.
 | ||
| Fig. 6 Autonomous mobile robot and experimental stations. The mobile robotic chemist (a), the roadmap of the whole laboratory (b) and several workstations (c–e) are shown. Reproduced with permission from ref. 4 Copyright 2020, Springer Nature. | ||
Despite the great advances, the mobile robotic chemist from Cooper's group is purely driven by Bayesian algorithms and does not capture existing chemical knowledge or include theoretical or physical models. Later, a comprehensive artificial intelligence chemistry laboratory (Fig. 7) was developed by Jun Jiang's team.5 This AI-Chemist consists of three modules, including a machine-reading module, a mobile robot module, and a computational module. The AI-Chemist system responds to scientific questions posed by researchers by tapping into vast amounts of literature. It digitizes and standardizes experimental protocols, enriching its knowledge base. The platform manages tasks, monitors the mobile robots, customizes experiment workflows, and stores the data for future use. The research team used the platform to find the best combinations of several Martian meteorite rocks to synthesize efficient water oxidation catalysts for future use in Martian exploration.98
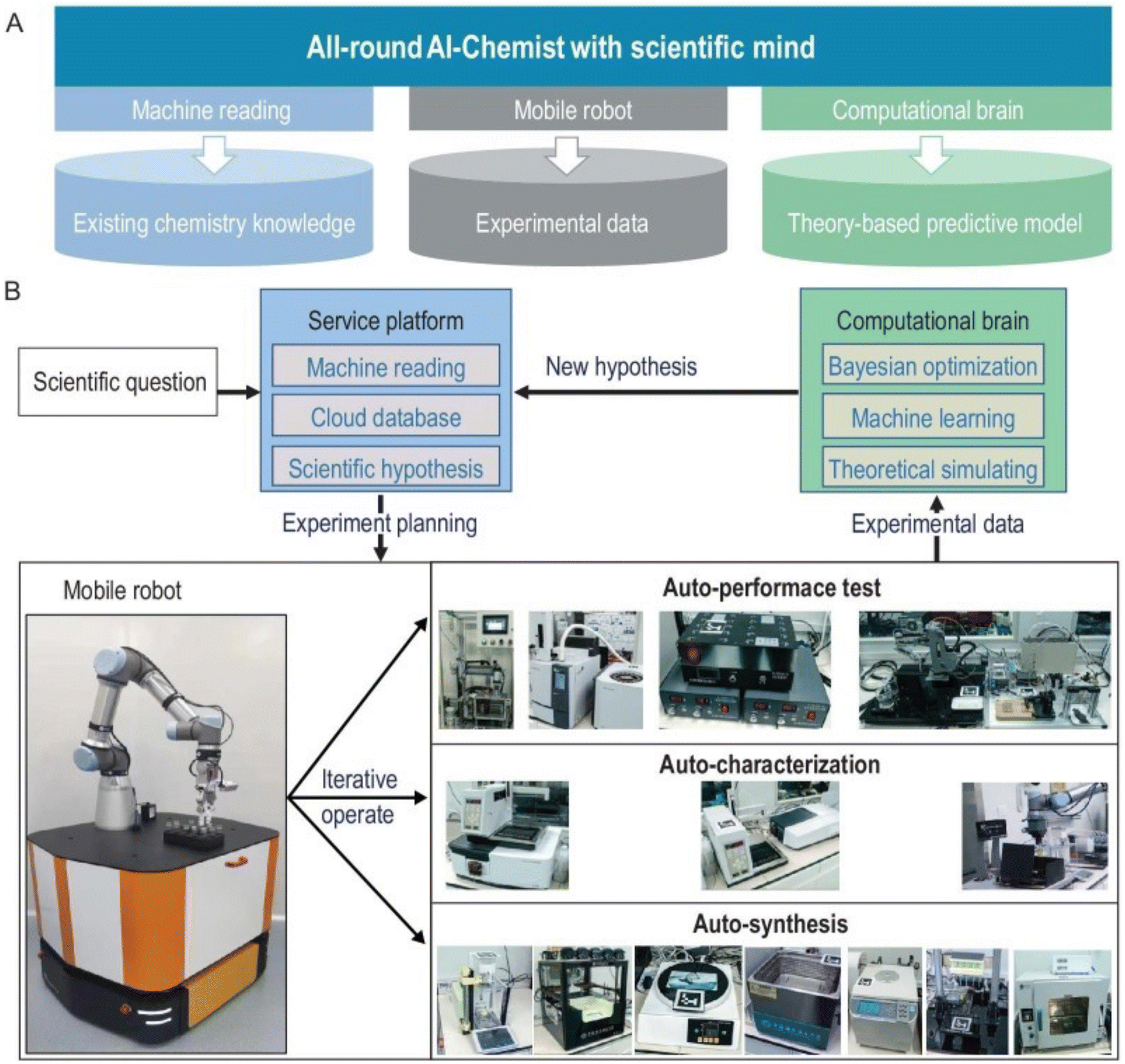 | ||
| Fig. 7 Design of the all-round AI-Chemist with a scientific mind. It includes three modules for chemistry knowledge, autonomous experimentation, and theoretical computation and machine learning (A). The workflow of the AI-Chemist to study various systems are shown in (B). Reproduced with permission from ref. 5 Copyright 2022, China Science Publishing & Media Ltd. | ||
The recent A-lab, developed by Gerbrand Ceder et al.,6 represents a significant advancement in the field of solid material synthesis. Despite some controversy on the actual phases of the fabricated materials, the hallmark of the A-lab is its high degree of automation, which encompasses the entire synthesis and characterization process, including critical steps such as powder dosing, sample heating, and X-ray diffraction (XRD) for product characterization.
One critical issue with the robotic arm system in laboratory settings is its moderate capacity to parallelize experimental tasks. While robotic arms bring automation and precision to the table, they still mimic human researchers to conduct multiple operations one by one. This constraint is particularly evident in high-throughput settings where speed and efficiency are paramount. To address this, integrating robotic systems with other automated solutions might be necessary.
3.3 Automated flow chemical system
Automated chemical synthesis systems based on flow pipelines are widely applied in many fields such as chemical pharmaceuticals99,100 and organic synthesis.17–19,101–104 The reactors used in the flow system can be categorized into two distinct types: batch reactors connected by pipelines and continuous flow reactors. The major advantage of the flow system comes from low-cost modularity, where the reaction module, product separation module, and detection module can all be connected to the same pipeline in sequence or parallel.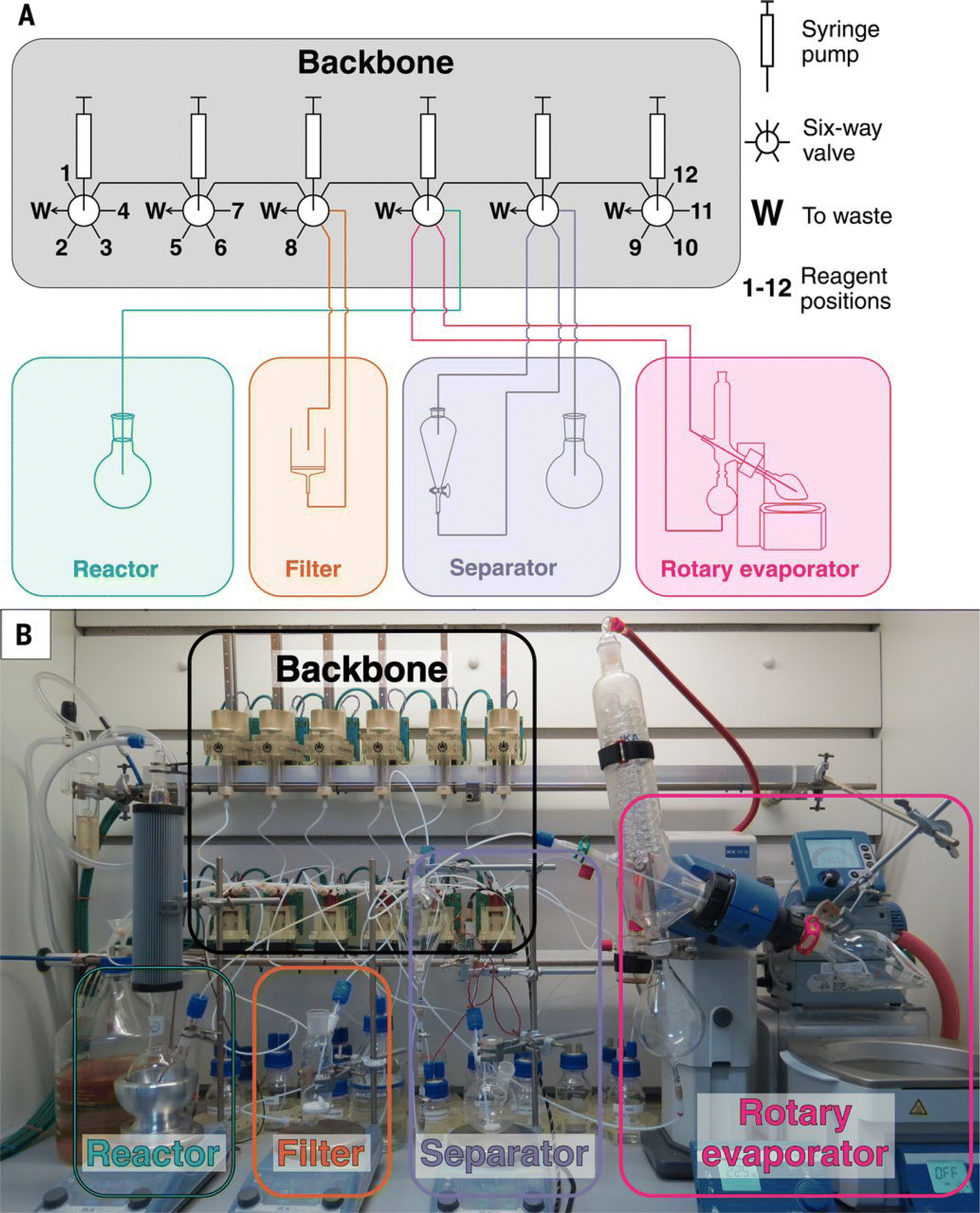 | ||
| Fig. 8 Physical implementation of the synthesis platform Chemputer. The scheme (A) and the actual set-up (B) of the Chemputer are shown respectively. Reproduced with permission from ref. 17 Copyright 2019, AAAS. | ||
One drawback of many flow systems is the lack of flexibility for different experiment tasks. One solution is to use general modules and their combination to support wider experiments. Alternatively, the modules can be reaction-specific as long as they can be designed and fabricated efficiently. Leroy Cronin et al.49 showcased a portable, suitcase-sized chemical synthesis platform with automated on-demand 3D printing of groups of reactors for different reactions. Researchers demonstrated the broad applicability of this system by synthesizing five organic small molecules, four oligopeptides, and four oligonucleotides, achieving good yields and purity.
The implementation of batch reactors with increased throughput has accelerated the search for catalysts in more complex systems that involve multiphase reactions. Cheng Wang et al.106 developed a fast screening platform with a coherent implementation of automated flow cell assembly and GC characterization. It was used for parallel synthesis, electrochemical characterization, and catalytic performance evaluation of electrocatalysts for the reduction of CO2 to C2+ products, which led to the discovery of a Mg–Cu bimetallic catalyst with competitive CO2 to C2+ performance and good stability compared to the top catalysts from other literature reports (Fig. 9).
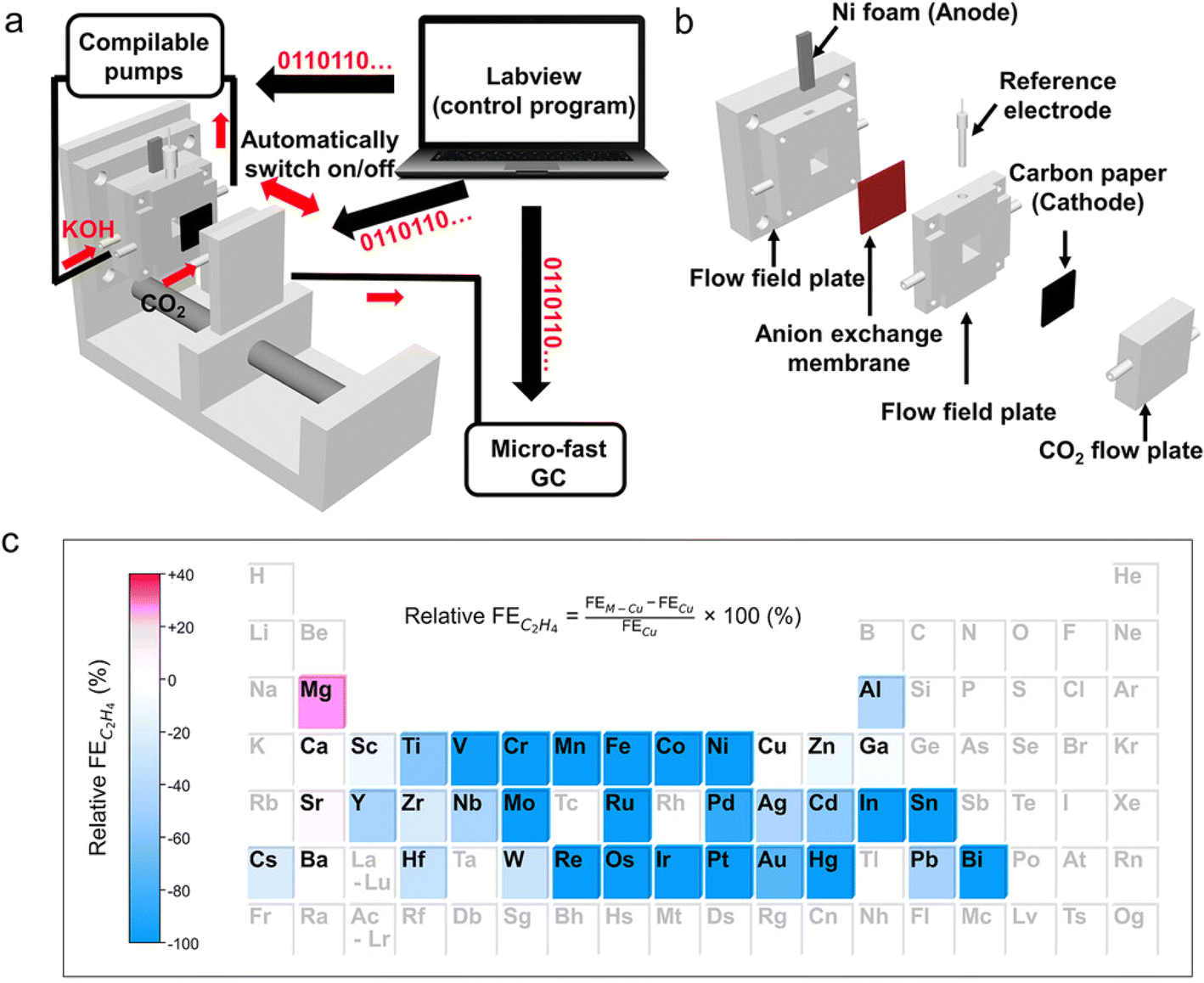 | ||
| Fig. 9 Fast screening platform for screening bimetallic catalysts for the CO2RR. (a) Schematic illustration of the fast screening platform for the CO2RR. (b) Exploded view of a 3D-printed flow cell. (c) Heat map of the relative FE of C2H4 over Cu-based bimetallic catalysts. Elements in black font represent tested metal salt additives and elements in grey font represent the untested ones. Reproduced with permission from ref. 106 Copyright 2022, Wiley. | ||
Timothy F. Jamison et al.115 developed a flexible, manually reconfigurable benchtop flow chemistry platform (Fig. 10), including various reactor modules for heating/cooling, photochemical reaction, and packed bed reaction. In addition, the platform integrates liquid–liquid separation technology and is equipped with inline analysis tools such as high performance liquid chromatography (HPLC), Fourier transform infrared spectroscopy (FTIR), Raman spectroscopy, and mass spectrometry.
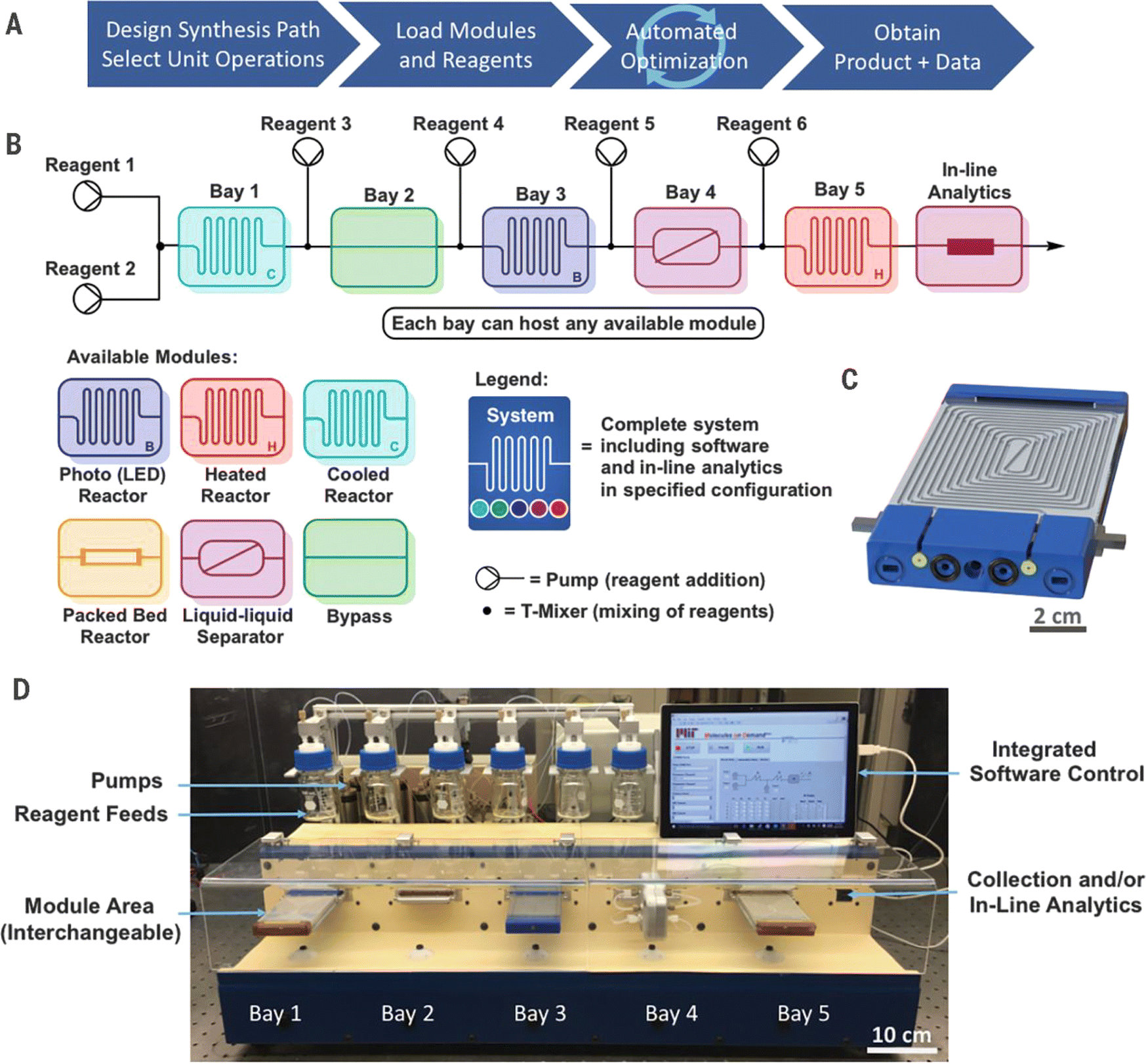 | ||
| Fig. 10 Plug-and-play, reconfigurable, continuous-flow chemical synthesis system. The workflow (A), the design of the flow system (B) and its actual setup (C) with interchangeable modules (D) are shown in the figure. Reproduced with permission from ref. 115 Copyright 2018, AAAS. | ||
One issue of the continuous flow system is its high cost in paralleling and adaptation. To partly address this issue, Kerry Gilmore et al.116 reported a “radial synthesizer” based on a series of continuous flow modules arranged radially around a central switching station, which allows selective access to individual reactors and avoids equipment redundancies and reconfiguration among different reactions. Storing stable intermediates inside fluidic pathways enables simultaneous optimization of subsequent steps during route development. Online monitoring via infrared (IR) and 1H/19F NMR spectroscopy enables fast post-reaction analysis and feedback. The performance of this system has been demonstrated in transition metal-catalyzed C–C and C–N cross-coupling, olefination, reductive amination, nucleophilic aromatic substitution reactions, light-driven oxidation-reduction catalysis, and continuous multi-step reactions. In addition, flow selection valve technology can be used to create different process combinations, as demonstrated by Nathan Collins et al.117 in an advanced automated continuous flow synthesizer called AutoSyn, which can access 3800 unique process combinations and up to seven consecutive reaction steps for efficiently preparing a variety of pharmaceutical small molecule compounds with a scale from milligrams to grams within hours.
To make the fluidic system even more adaptive, Klavs F. Jensen et al.123 combined the robotic arm and the flow system (Fig. 11): the robotic arm is responsible for assembling modular process units, including reactors and separators, into a continuous flow path. After the synthesis, the robotic arm can disconnect the reagent lines and move the processing module to the appropriate storage location. Pneumatic grippers are used to ensure tight connections between process chambers. In 2023, the same group introduced a prototype that further incorporates machine learning with robotics to autonomously design, synthesize, and analyze dye-like molecules with minimal human intervention.124 This system successfully synthesized and characterized 303 new dyes, advancing the efficiency of chemical discovery.
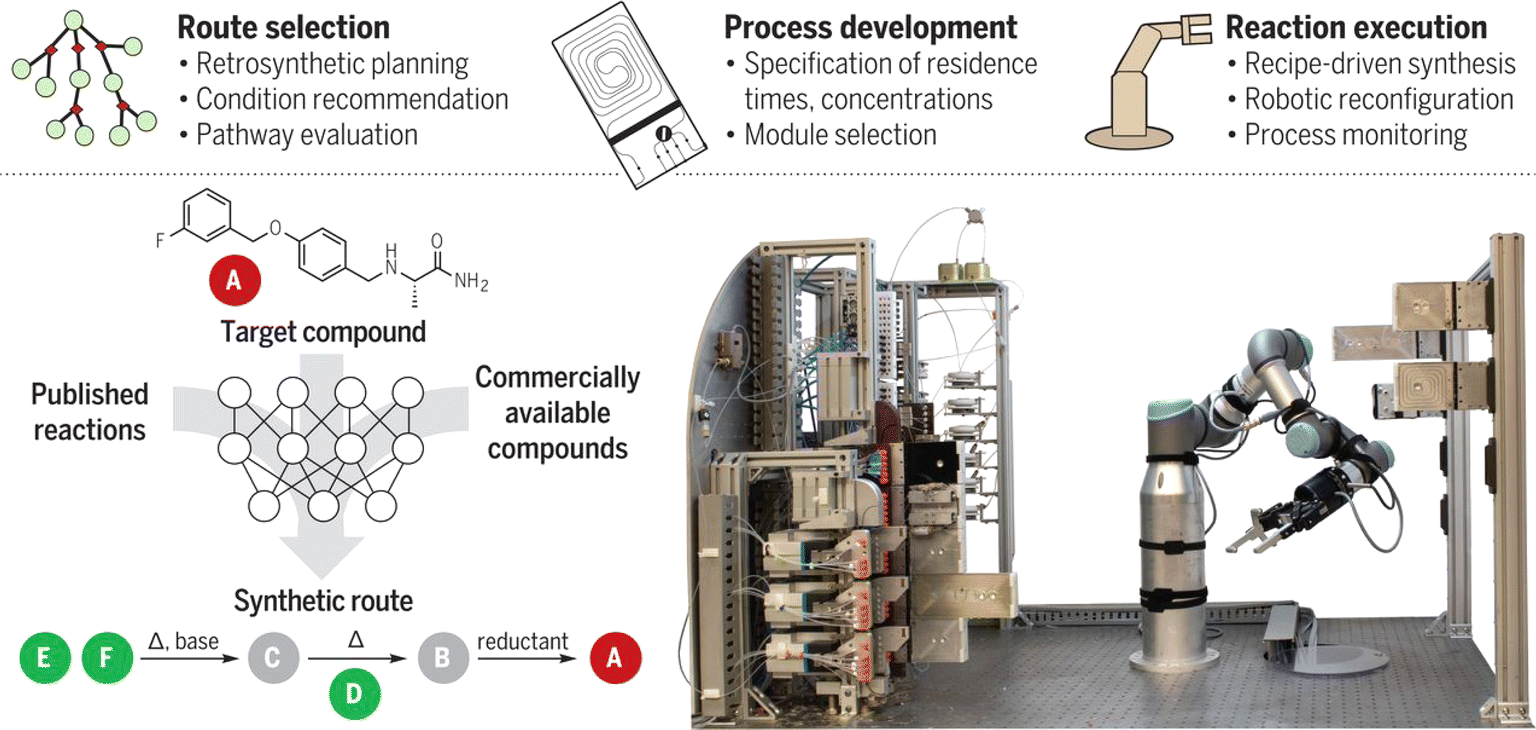 | ||
| Fig. 11 A robotically reconfigurable flow chemistry platform. Reproduced with permission from ref. 123 Copyright 2019, AAAS. | ||
Flow chemistry systems, while revolutionizing chemical synthesis and processing, present several limitations in automation. The setup and maintenance of these systems are complex and resource-intensive. Establishing precise control over flow rates, temperature, and pressure requires specialized equipment and expertise. This complexity also extends to scalability issues; while flow systems excel in scaling up certain types of reactions, they may be less adaptable for reactions requiring long residence times or intricate synthesis steps. Additionally, the rigidity in altering reaction conditions can limit their flexibility, making them less suitable for laboratories that frequently switch between diverse chemical processes. Material compatibility is another concern, as the construction materials of the flow reactors must withstand a wide range of chemicals and conditions, limiting their use with highly reactive or corrosive substances. Furthermore, while adept at handling large-scale production, flow chemistry systems can be less efficient for small-scale synthesis, often leading to inefficiencies and wastage when dealing with minute quantities.
3.4 Large language models and robots
The introduction of LLMs to robotic systems defines a new frontier in automation.First, LLMs have facilitated the development of robotics, including log information extraction, assisted robot design,125 and task generation and planning.42,43,126,127 As pointed out by Francesco Stella et al.,125 LLMs can be the creator for designing the automating system, be the mentor and copilot for domain scientists who do not have the necessary educational background to implement automation in their research, and be an assistant to debugging, troubleshooting, and method selection during the technology implementation phase to accelerate the process.
Second, LLMs, especially the multimodal ones, can help develop next-generation robots with increased flexibility. Vemprala and others from the Microsoft team126 proposed a strategy that combines prompt engineering and a high-level feature library to enable ChatGPT to handle various robotic tasks and scenarios. An open-source tool called PromptCraft was introduced, which includes a collaboration platform and a ChatGPT-integrated sample robot simulator. However, the LLM-controlled robotic movement is not robust enough for direct use in chemistry experiments where safety and reliability are of primary concern.
Third, LLMs also offer solutions to program robots. Kourosh Darvish et al. introduced the CLAIRIFY method,42 which combines automatic iterative prompting with program verification to ensure the syntactic accuracy of task plans and their alignment with environmental constraints. The system's objective is to produce a syntactically correct task plan suitable for robotic action as a prompt for LLMs to generate a program. However, the generated plan needs to be verified to detect any compilation error and pass the error messages as subsequent input prompts for iterative interaction with the LLMs. The capability of this method was demonstrated by translating natural language to an abstract and concise high-level chemical description language (χDL), which was originally developed and used in the control of Chemputers.18
Compared to high-level descriptive codes, generating low-level operational codes to interface directly with the robotic system can be more complicated. Genki N. Kanda et al.43 demonstrated that GPT-4 can generate low-level operational Python scripts for automated robots like Opentrons-2 (OT-2) from natural language instructions. They designed a pipeline based on GPT-4 to automatically translate natural language experimental descriptions into Python scripts compatible with OT-2. Leveraging OpenAI, this approach iteratively queries the model, extracts, and validates scripts using a simulator of OT-2, and provides feedback on any errors for correction. This shift towards natural language instruction simplifies the automation process, making it accessible to a broader range of researchers and promoting the automation of biological experiments.
3.5 Summary
Automated and intelligent chemical robotic systems are promising to significantly enhance the efficiency, accuracy, and reproducibility of experiments. Table 2 summarizes various types of automated and intelligent chemical robotic systems, detailing their specific functions, supported operations, characterization techniques, and chemical spaces they explored. These systems range from humanoid robotic systems to batch reactors and continuous flow reactors, each with unique capabilities and applications to study different chemical systems.| Type | Description | Synthesis operations | Characterization | Target compounds | Reference |
|---|---|---|---|---|---|
| Humanoid | Mobile robotic chemist | Solid dispensing, liquid dispensing, capping/uncapping, heating, and sonication | Gas chromatography | Catalysts for photolysis of water to produce hydrogen | 4 |
| An all-round AI-Chemist | Solid dispensing, liquid dispensing, magnetic stirring, sonication, drying, centrifugation, and liquid extraction | UV-vis, fluorescence, and Raman spectroscopy, and gas chromatography | Materials for electrocatalysts, photocatalysts, and luminescence | 5 | |
| A-lab, an autonomous laboratory | Powder dosing and sample heating | X-ray diffraction (XRD) | Primarily oxides and phosphates identified through extensive ab initio phase-stability data | 6 | |
| Flow: Batch reactors | Modular robotic synthesis system | Mixing, filtration, liquid–liquid separation, evaporation, and chromatographic separation | — | Organic molecules | 17 |
| A portable suitcase-sized chemical synthesis platform | Liquid transfer, temperature control, evaporation, filtration, and separation | — | Organic molecules | 49 | |
| Fast screening platform for the CO2RR | Liquid handling, electric cell preparation, and electrolysis | Micro-fast gas chromatography | Electrocatalysts for the CO2RR | 106 | |
| Flow: continuous flow reactors | Benchtop flow chemistry platform | Liquid handling, heating, cooling, photoreaction, extraction and purification | High-performance liquid chromatography (HPLC), IR spectroscopy, Raman spectroscopy, and mass spectrometry | Reconfigurable system for automated optimization of diverse chemical reactions | 115 |
| Radial synthesizer system | Liquid transfer, mixing, and dilution | IR spectrometry and nuclear magnetic resonance | Cross-coupling, olefination, reductive amination, nucleophilic aromatic substitution reactions, light-driven redox catalysis, and continuous multi-step reactions | 116 | |
| An automated multistep chemical synthesizer | Heating, liquid–liquid separation, gas–liquid separation, and heterogeneous catalysis | Liquid chromatography-mass spectrometry (LC-MS) | Pharmaceutical small molecules | 117 | |
| Humanoid robotic system with flow reactors | A robotic platform for flow synthesis of organic compounds | Liquid handling, separation, and temperature adjustment | High-performance liquid chromatography and nuclear magnetic resonance | Organic molecules | 123 |
We expect a much enhanced automation level in chemistry research. However, current automation in chemistry still faces challenges, particularly in the trade-offs between the flexibility and throughput of automated systems. For instance, although capable of vast amounts of operations compared to flow systems, humanoid robotic systems are usually slower in operational speed to ensure accuracy and safety. On the other hand, flow chemistry systems can handle hundreds or thousands of experiments per day, but are more task-specific with limited flexibility. New developments in these strategies are required to enhance flexibility, throughput, and robustness at the same time.
Another challenge lies in the control part of the robotic systems. Although digital twins are very common for humanoid robotics and in industry, the development of digital twins for the whole automated chemistry system is still at its initial stage despite a few efforts.4,18,128 Ensuring the integrity and safety of experimental procedures remains paramount in automation labs. Therefore, greater attention must be directed toward enhancing the capability to simulate experimental procedures and detect any potential physical or chemical issues during the development of various robotic systems. Furthermore, despite the rapid advancements in novel algorithms, such as reinforcement learning, the control of robots in chemistry labs often relies on hardcoded programming. This limitation restricts their ability to perform complex tasks and adds challenges to the maintenance, transferability, and future development of the systems. LLMs appear promising in introducing flexibility to control systems. However, the reliability of LLM-generated code must be verified either by human experts or through digital twins. It is foreseeable that digital twins and LLMs will soon be more cohesively integrated into the control of chemical robotic systems.
4 Design and discovery of catalysts with active machine learning
In the discovery of catalysts, the search space is often vast and grows exponentially with the number of parameters. This inherent complexity makes the traditional trial-and-error approach for catalyst screening both labor and computationally intensive and time-consuming. The emergence of ML and LLMs has provided opportunities to address this problem. By utilizing ML and LLMs to guide experimental design with experimental or theoretical feedback, the search for catalysts can be significantly accelerated.4.1 Design of catalysts guided by machine learning
The implementation of ML in experimental design can lead to more efficient and cost-effective research. Olsson129 defines active machine learning as a supervised machine learning technique in which the learner (i.e., the machine learning model) determines the sampling point from which it learns. Bayesian optimization (BO)130 and active learning (AL) are two important branches of active machine learning that are applied in catalyst design.BO is an optimization strategy that balances the exploration of uncertain regions and the exploitation of known regions with superior objective values. It is generally used to optimize a black-box function and consists of three key components:
(1) Surrogate model: this is a predictive model designed to approximate the underlying function. A wide range of machine learning models can be employed for this purpose, such as the Gaussian process,131 ensembles of artificial neural networks,132 and Bayesian neural networks.133–136
(2) Acquisition function: an acquisition function is a scoring function used to rank sampling points within the input space based on the surrogate model's predictions. Examples of such functions include expected improvement (EI),137,138 probability of improvement (PI),139 and upper confidence boundary (UCB).140 The acquisition function is instrumental in selecting the most promising candidates for further evaluation.
(3) Bayesian inference:141 this is a foundational technique in Bayesian optimization, utilized for training the surrogate model. It uses Bayes' theorem to update the probability of a hypothesis or event based on observed evidence.
On the other hand, AL is a family of machine learning techniques that aims to minimize the number of labelled data points while obtaining a high-performance model. It can usually be achieved through an adaptive sampling strategy, which prioritizes the labelling of data points with the highest uncertainty and information gain for the model.
Both BO4,20–28 and AL29–34 have been applied in the design of and search for catalysts. BO can efficiently explore the vast parameter space of catalyst design and select experiments that are likely to yield the desired products. By iteratively updating the ML model and selecting new experiments based on the retrained model, BO can guide the search for optimal catalysts. AL, in the meantime, can assist in selecting the most informative data points for labelling, reducing labelling costs while improving model performance. It has been applied in many fields including materials design,142,143 retrosynthesis,144,145 and drug discovery.146,147 Besides the original purpose of AL, its application in catalyst design also demonstrated its capability for global optimization, presenting a remarkable analogy to the BO framework. The applications of BO and AL in the field of catalysis will be discussed respectively below.
4.2 Bayesian optimization
BO effectively balances exploration and exploitation to identify the best candidates within the design space. The method can significantly reduce the number of experiments required to find the optimal reaction parameters or formulations. For example, in 2020, Yusuke Yamauchi and coworkers20 employed BO to efficiently discover ternary PtPdAu alloy catalysts. They exhibited excellent catalytic activity in electrochemical methanol oxidation (Fig. 12). Remarkably, through only 47 experiments, which is less than 1% of the potential composition space, the authors successfully discovered the optimal composition with a high catalytic performance. More interestingly, the sampling scheme using current density as the performance metric yielded a precursor composition with minimal Au content, which would have been challenging for chemists to predict. Thus, the implementation of BO can not only accelerate the search for catalysts but also offer new insights into the design of catalysts.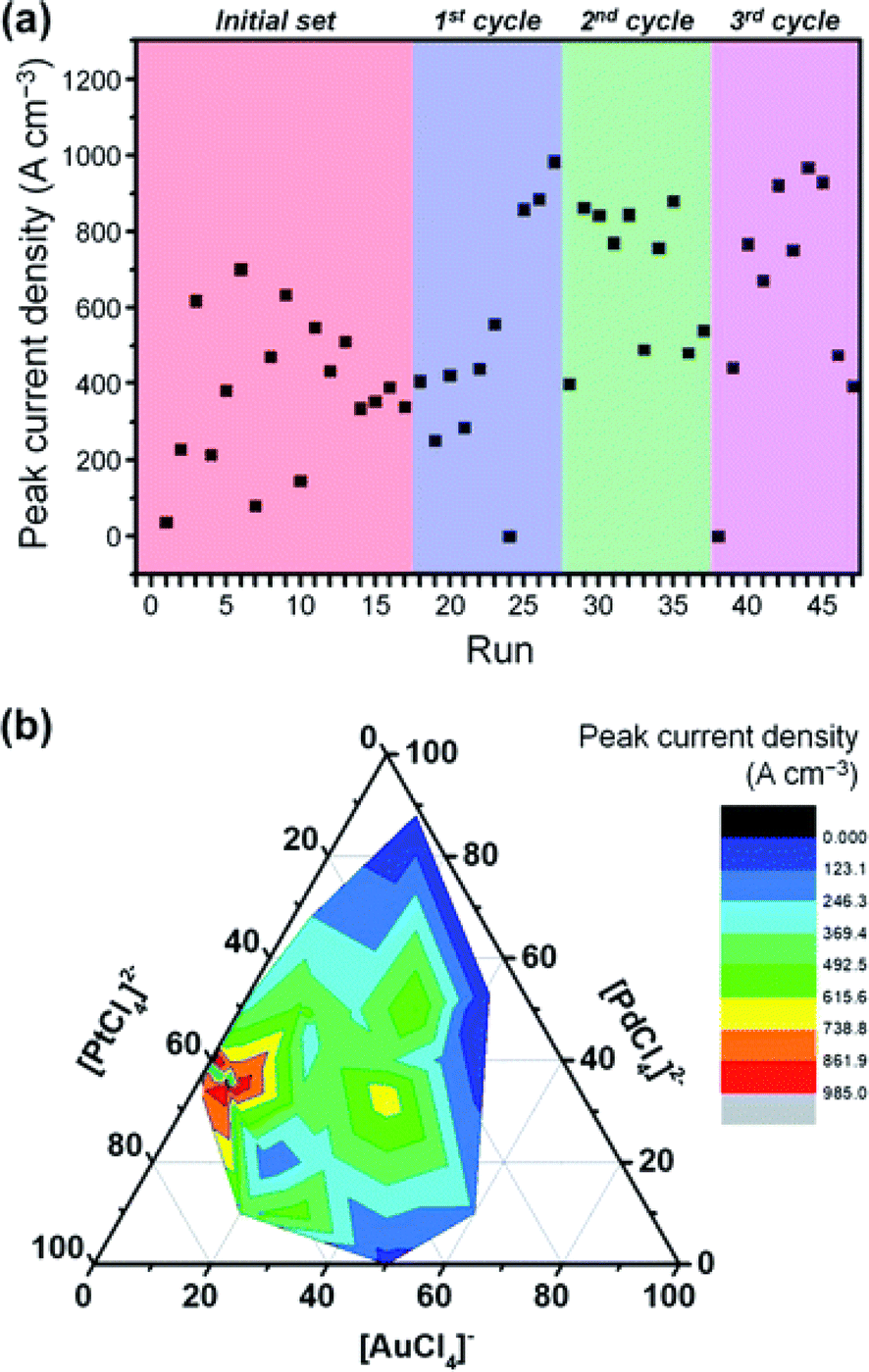 | ||
| Fig. 12 Bayesian optimization of the methanol electro-oxidation process. (a) Peak current density of methanol electro-oxidation as a function of the number of BO rounds. (b) A contour plot showing the peak current density and a ternary plot depicting the chemical composition in the electrolyte solution. Reproduced with permission from ref. 20 Copyright 2020, Royal Chemical Society. | ||
In 2020, Bayesian experiments for autonomous researchers (BEAR)21 combined BO with high-throughput automated experiment systems to achieve self-driven material discovery—a cycle of the design of experiments, automated experiment feedback, and retraining of machine learning models to design new experiments. As discussed before, Andrew I. Cooper et al.4 developed an AI chemist to improve the catalytic performance for hydrogen production with BO (Fig. 13). It successfully discovered a mixture of photocatalysts that exhibited six times higher activity than the original formulation. Compared to manual operations, the experimental time cost is reduced by approximately 60 times.
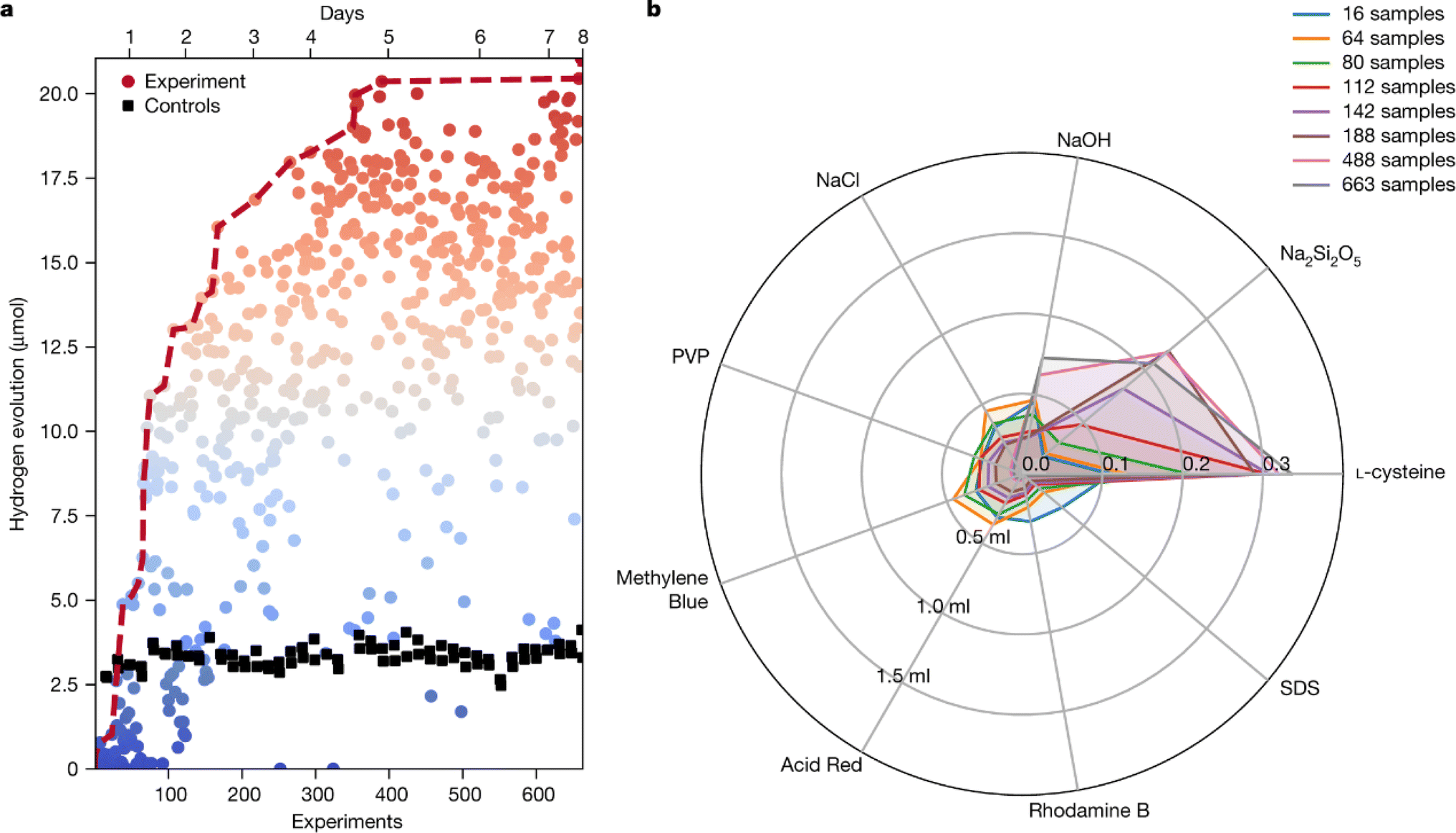 | ||
| Fig. 13 (a) Maximum rate of hydrogen evolution from photolyzed water reaching 21.05 μmol h−1 after 688 experiments during an 8-day autonomous search. (b) Radar plot illustrating the sampling in the search space during experimentation. Reproduced with permission from ref. 4 Copyright 2020, Springer Nature. | ||
In 2021, Jan Rossmeisl et al.22 developed a computational framework that combines density functional theory (DFT) calculations, ML-driven kinetic modelling, and BO to explore a wide range of composition space to search for multi-component high entropy alloys for the oxygen reduction reaction (ORR). To accelerate catalyst discovery, the authors integrated kinetic modelling with BO, where a Gaussian-process-based surrogate model provided suggestions for alloy compositions. The proposed compositions were evaluated using the kinetic model, and the surrogate model was updated based on the ORR activity predicted by the kinetic model. BO effectively identified optimal compositions through 150 iterations, including Ag18Pd82, Ir≈50Pt≈50, and Ir≈10Pd≈60Ru≈30 (Fig. 14). These compositions closely matched the optimal compositions found through grid search in the same chemical space. Experimental confirmation of the three optimized compositions by high-throughput thin-film synthesis and ORR testing in the Ag–Pd, Ir–Pt, and Pd–Ru binary alloy spaces, reveals the best-performing compositions of Ag14Pd86, Ir35Pt65, and Pd65Ru35. The experimental results reasonably matched the results of BO, and BO can accelerate the discovery of optimal catalysts by up to 20 times.
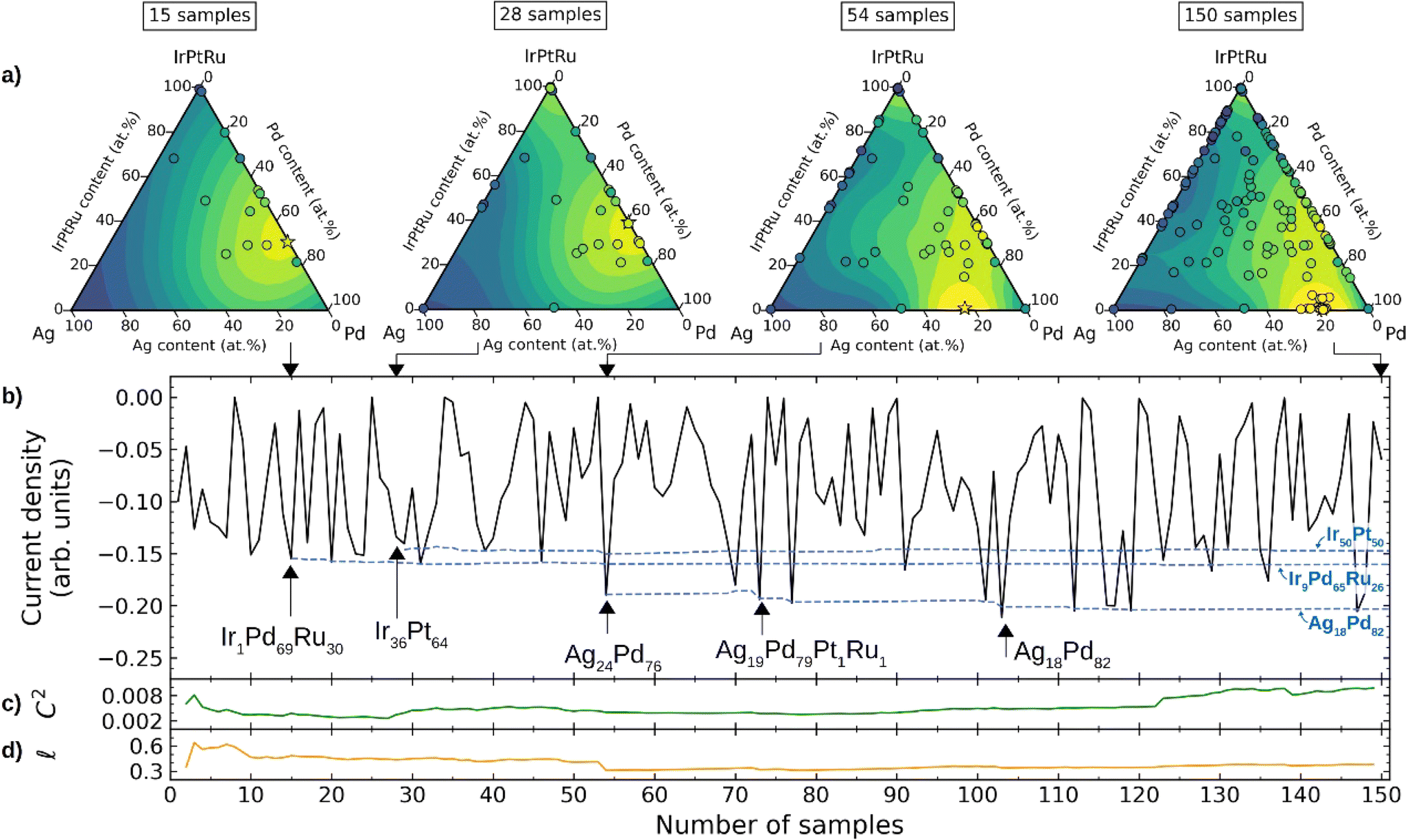 | ||
| Fig. 14 BO for composition optimization of an Ag–Ir–Pd–Pt–Ru system for the ORR. (a) Pseudo-ternary plots (Ir, Pt, and Ru collected at one concentration) showing pseudo-functions after sampling 15, 28, 54, and 150 compositions. Yellow indicates regions with higher absolute values of the simulated current densities, and blue indicates regions corresponding to lower values. (b) Current densities sampled during BO (black solid line) and the emergence of the three most active locally optimal compositions (blue dashed line). (c) and (d) Variation of the GP-squared exponential kernel function with respect to the constant term (c) and the length scale (d) hyper-parameters. Reproduced with permission from ref. 22 Copyright 2021, Wiley. | ||
4.3 Active learning
Active learning is a strategy that explores the design space to establish a precise and reliable mapping from it to an output space (e.g. various properties of compounds) and optimizes toward high-performance solutions. Active learning can be used to reduce the number of expensive DFT simulations for the design and screening of catalysts in a large space.Yousung Jung et al.30 proposed an active learning method in the discovery of catalysts for the CO2RR driven by uncertainty and prediction error. It utilizes cost-effective non-ab initio input features, i.e., LMTO d-band width and electronegativity, as chemisorption descriptors to predict adsorption energies on alloy surfaces. Screening of large-scale materials is carried out by combining these descriptors with two machine learning models: an ensemble of artificial neural networks (ANNs) and kernel ridge regression (KRR). The catalytic performance of a set of 263 alloy systems was studied by predicting *CO binding energy using the models. During the active learning process, an ensemble consisting of five neural networks with the same architecture but varied initial weights was trained on an initial dataset. The ensemble was used to predict the *CO binding energy on the rest of the dataset to find candidates with the highest prediction variance, which will be included in the next training process. As an alternative machine learning model, the performance of KRR148,149 was also elaborated. It involves the training of a KRR model on the initial dataset with *CO binding energy as the output. Then, an additional KRR model was trained on the prediction error from the previously trained model as an error predictor.148,150 Later, the KRR error predictor was used to estimate the error rate for the rest of the dataset, which helps select candidates for the next round of training. Both models (ensemble of ANNs and the KRR model) were used to predict the adsorption energy of CO on (100) crystalline surfaces. The best model gives an RMSE of only 0.05 eV without the d-band center as a descriptor. The authors discovered Cu3Y@Cu* to be a highly active and cost-effective catalyst for the CO2RR.
Besides the original purpose of using active learning to establish an accurate and reliable model, it can also be utilized for global optimization. In 2018, Zachary W. Ulissi et al.31 proposed a cyclic workflow with ideas from agent-based model optimization and active learning for screening electrocatalysts for the CO2RR and HER. This workflow, illustrated in Fig. 15, involves machine learning screening, DFT validation, and machine learning retraining. To start, the researchers obtained a search space of intermetallic crystals and their corresponding surfaces from the Materials Project.151 They then selected a series of materials as optimal candidates for catalysis using a machine-learning model. DFT calculations for the selected candidates were performed, providing more accurate predictions of the catalytic properties. The DFT results were then used to retrain the machine learning model, creating an iterative process for continuously improving the catalyst database. In their study, the authors considered a total of 31 elements, composed of 50% d-block elements and 33% p-block elements. The search space consists of 1499 intermetallics for potential catalysis applications. 131 possible surfaces from 54 alloys and 258 possible surfaces from 102 alloys were identified as valid candidates for the CO2RR and HER, respectively. The number of candidate alloy catalysts can be further reduced to 10 and 14 for the CO2RR and HER. This comprehensive screening approach allowed for the identification of theoretically promising catalysts for the CO2RR and HER.
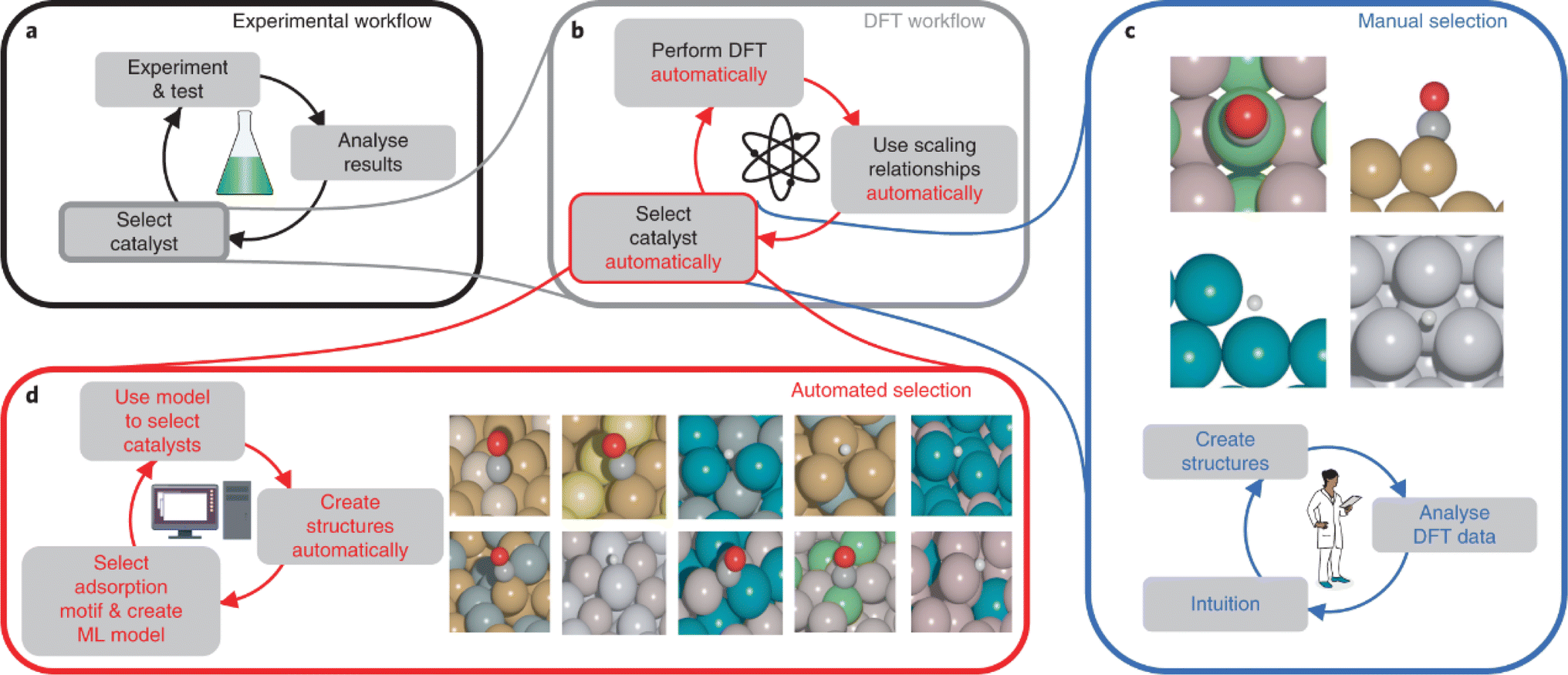 | ||
| Fig. 15 Workflow for automating theoretical materials discovery. (a) and (b) The experimental workflow for catalyst discovery is accelerated by the ab initio DFT workflow. (c) Scientists relied on their expertise and experimental results to screen data for DFT calculations traditionally. (d) This work uses ML to select DFT data automatically and systematically. Reproduced with permission from ref. 31 Copyright 2018, Springer Nature. | ||
In 2020, Edward H. Sargent et al.32 developed a machine learning-accelerated, high-throughput DFT framework for rapid screening of CO2RR electrocatalysts (Fig. 16) similar to the one from Zachary W. Ulissi's group31 described above. The researchers studied a dataset of 244 different copper-containing intermetallics, forming a search space of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 229 surfaces and 228969 adsorption sites. DFT simulations were performed on a subset of these sites to calculate the CO adsorption energies. These data were then used to train machine learning models to predict the CO adsorption energy on the adsorption sites. The researchers encoded each adsorption site as a numeric array and used a combination of random forest and boosted trees to enhance prediction performance. The framework combined the machine learning predicted CO adsorption energy with the volcano scaling relationship to identify sites with the highest catalytic activity. These optimal points were then simulated using DFT to provide additional training data for the machine-learning model. Thus, an active learning workflow was established, cycling between DFT simulations, machine learning regression, and machine learning prioritization, to continuously query and construct a DFT database. This workflow performed over 300 regressions, which guided DFT calculations for CO binding energies at approximately 4000 different adsorption sites to identify Cu–Al as the most promising material for the CO2RR in the search space. Furthermore, the authors synthesized de-alloyed nanoporous Cu–Al catalysts for validation, which achieved over 80% Faraday efficiency (compared to ∼66% for pure Cu) at a current density of 400 mA cm−2 (1.5 V vs. NHE). It showed a 2.8-fold improvement in cathodic power conversion efficiency (PCE) at 400 mA cm−2 compared to previous state-of-the-art results. This work demonstrated an effective method for high-throughput catalyst screening, combining machine learning and DFT calculations.
229 surfaces and 228969 adsorption sites. DFT simulations were performed on a subset of these sites to calculate the CO adsorption energies. These data were then used to train machine learning models to predict the CO adsorption energy on the adsorption sites. The researchers encoded each adsorption site as a numeric array and used a combination of random forest and boosted trees to enhance prediction performance. The framework combined the machine learning predicted CO adsorption energy with the volcano scaling relationship to identify sites with the highest catalytic activity. These optimal points were then simulated using DFT to provide additional training data for the machine-learning model. Thus, an active learning workflow was established, cycling between DFT simulations, machine learning regression, and machine learning prioritization, to continuously query and construct a DFT database. This workflow performed over 300 regressions, which guided DFT calculations for CO binding energies at approximately 4000 different adsorption sites to identify Cu–Al as the most promising material for the CO2RR in the search space. Furthermore, the authors synthesized de-alloyed nanoporous Cu–Al catalysts for validation, which achieved over 80% Faraday efficiency (compared to ∼66% for pure Cu) at a current density of 400 mA cm−2 (1.5 V vs. NHE). It showed a 2.8-fold improvement in cathodic power conversion efficiency (PCE) at 400 mA cm−2 compared to previous state-of-the-art results. This work demonstrated an effective method for high-throughput catalyst screening, combining machine learning and DFT calculations.
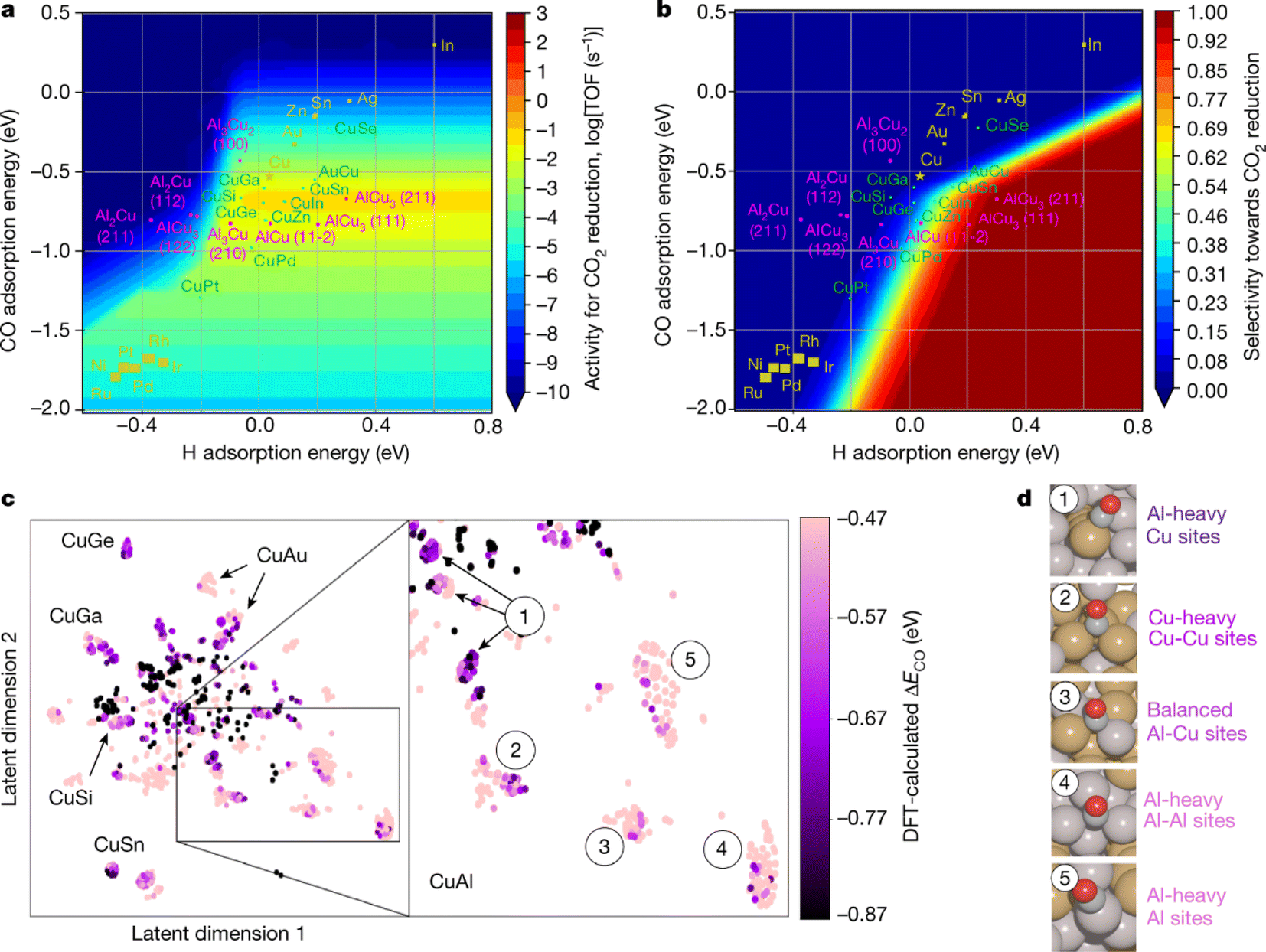 | ||
| Fig. 16 Screening of CO2RR electrocatalysts using an active learning algorithm based on the DFT framework. (a) A two-dimensional activity volcano plot of the CO2RR. (b) A two-dimensional selectivity volcano plot of the CO2RR. (c) DFT calculations were performed on approximately 4000 adsorption sites of Cu-containing alloys identified by t-SNE. On the right, the Cu–Al clusters are labeled numerically. (d) Representative coordination sites for each cluster are labeled in the t-SNE. Reproduced with permission from ref. 32 Copyright 2020, Springer Nature. | ||
While BO and AL are initially different approaches, they tend to converge on the catalyst optimization task. BO usually uses a probabilistic model with the goal of optimization, while AL can adopt more diverse models with the goal of efficiently constructing a machine learning model. When AL also used a probabilistic model and assessed uncertainty in making the decision about which point to explore next, it is equivalent to exploration-oriented BO, but the ultimate goal of AL is to improve the model most efficiently, which is beyond the uncertainty strategy.
When all the obtainable information about the system comes from the previous experimental/calculation results, BO and AL are mathematically sound methods to most efficiently explore the space. However, when domain knowledge is available, it is possible to come up with a more efficient strategy by combining the testing information with domain knowledge. The addition of domain knowledge into the process can be achieved by using LLMs.
4.4 Design and synthesis of catalysts guided by large language models
The diverse and interdisciplinary knowledge spanning chemistry, materials science, computer science, and data science, which are needed for the data-driven design and discovery of catalysts, can present a formidable challenge for researchers. LLMs152,153 offer a promising solution to overcome the knowledge gaps from multiple fields efficiently. LLMs have been used by chemists for tasks such as catalytic reaction prediction,45 property prediction,154–157 and synthesis condition design.156,158In BO and AL, a machine learning model (or a surrogate model) is necessary to approximate a mapping. Traditional machine learning models can take continuous, discrete, or categorical variables as the input. In contrast, LLMs, with their inherent capabilities to process natural language descriptions and generate new content accordingly, can be potentially used as a surrogate model, which can support a versatile input format. To incorporate the training data into the models, in-context learning (ICL), a technique that includes training data as examples in the prompt for LLMs, can be used. Alternatively, fine-tuning the models using the existing dataset represents another viable approach.
Andrew D. White's group45 demonstrated the usage of LLMs as the surrogate model in Bayesian optimization. The aim is to use a generative pre-trained transformer (GPT) as a surrogate model to predict the properties of the product according to the experimental procedure. Both fine-tuning and ICL were used for model training. To introduce prediction uncertainty when querying the LLMs, they designed two prompting strategies, a (1) multiple-choice option template and (2) top k completions template for regression. With the multiple-choice template, the LLM will treat the regression problem as a multi-option question to give a predicted value in one of the five ranges. Furthermore, the probability of selecting each option can be accessed. In the top k completion template, the question will be queried k times to the LLM to generate k answers. Both strategies generated a discrete probability distribution of the output, which can be used in Bayesian optimization. The authors used a series of models from OpenAI (text-curie-001, text-davinci-003, GPT-4, etc.) with in-context learning or fine-tuning to predict the C2 yield for oxidative coupling of methane based on synthesis procedures. Gaussian process regression was used as a baseline with text embedding to convert the synthesis description to a numeric input. Among the LLMs, GPT-4 is the best model in either ICL or fine-tuning. When GPT-4 and the top-k completion strategies were used, the ICL model showed comparable performance (mean absolute error, which is abbreviated as MAE, of 1.854) to the Gaussian process regression (MAE of 1.893). When the fine-tuning was implemented, the MAE of the model was further decreased to 1.325. Later, the authors implemented Bayesian optimization using the Gaussian process or LLMs with ICL as the surrogate model. The ICL model reached 99% quantile after 15 samples, after which the performance did not improve significantly and failed to find the maximum value in the sample pool. Although the GPR model also failed to find the maximum in the sample pool, it was a little closer to the maximum and showed a higher efficiency in the optimization. Due to the token size limitation and the complexity of the C2 data, the authors only selected the five most relevant examples during ICL, which can be the reason why the ICL model did not perform as well as the GPR model in Bayesian optimization. However, as a proof-of-concept, it is enough to demonstrate that LLMs have the potential to guide researchers in decision-making.
The in-context learning ability of the LLMs is promising for building an interactive workflow where an AI agent iteratively assists and instructs human experts to increase search efficiency through experimental feedback. Recently, Omar M. Yaghi and his coworkers have built such a workflow and demonstrated its capability in the synthesis of MOFs with prompt engineering and in-context learning.47 This innovative workflow involves three components: ChemScope, ChemNavigator, and ChemExecutor (Fig. 17). With the usage of ChemScope, the human researchers offer GPT-4 the project goals and necessary information like the literature of reticular chemistry and availability of lab resources to generate a project blueprint. Here, GPT-4 reads the general concepts of reticular chemistry and constructs a scheme of the project with multiple stages, where each stage contains well-defined objectives and indicators for their completion. Then, ChemNavigator and ChemExecutor were used coherently to go through the stages and achieve the objectives defined by ChemScope. ChemNavigator was used to define tasks to complete the objectives of the current stage. It takes the project scheme from ChemScope, previous trial-and-error summaries, human feedback, and current situations to update the summaries and generate three tasks accordingly. With the updated summary and tasks from ChemNavigator, the ChemExecutor outputs step-by-step instructions to complete the task. Additionally, ChemExecutor also defines a template to record the experimental feedback from the human researchers, which will be used later in the next iteration. At this point, the human researchers will perform the experiments and fill up the template. The interaction among ChemExecutor, ChemExecutor, and human researchers was iterated several times until the completion of the project. The recording of experimental feedback and consistent updating of the summary enabled GPT-4 to learn from experiment outcomes and optimize protocols to complete the complex tasks. Using this human–computer interactive workflow, the researchers successfully discovered and characterized a series of isomorphic MOF-521s. This work highlights the advantages of the large language model in interacting with human experts in natural language without coding skills, making it easy to use for all chemists. Additionally, the in-context learning facilitated by GPT-4 can continuously optimize experimental protocols to complete complicated research tasks. When such a workflow is integrated with automated robotic systems, it paves the way for a new paradigm of self-driving labs, where the design and discovery of catalysts go beyond a purely data-driven approach.
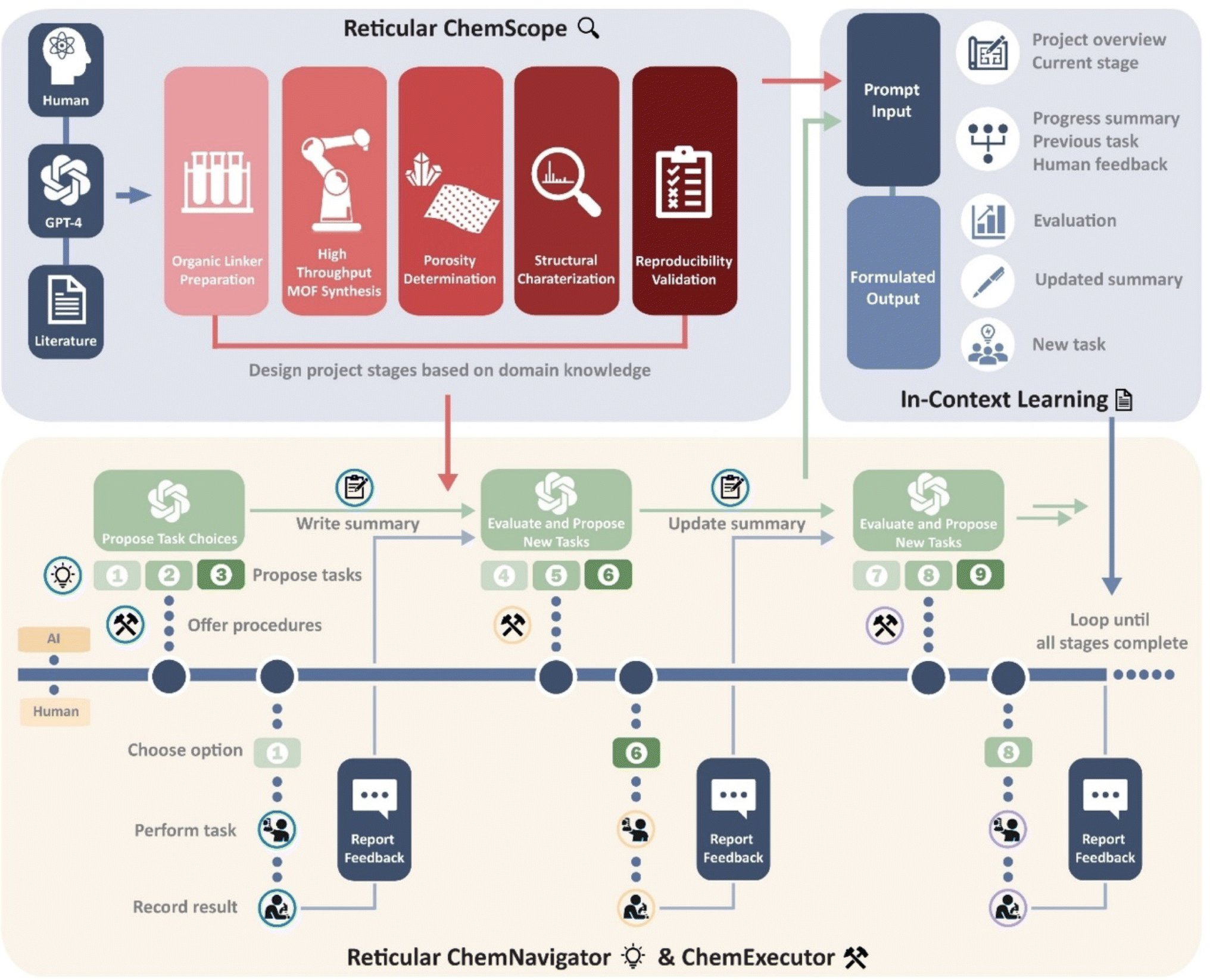 | ||
| Fig. 17 Framework diagram for GPT-4-directed MOF synthesis. The workflow consists of three phases: Reticular ChemScope, Reticular ChemNavigator, and Reticular Executor. The ICL capability of GPT-4 is achieved by combining pre-designed prompt systems with continuous human feedback. Reproduced with permission from ref. 47 Copyright 2021, Wiley. | ||
Despite the potential applications of LLMs in the design of and search for catalysts, there are still some problems to be addressed. The major problem is the well-known hallucinations in the context generated by LLMs. Although researchers have tried to mitigate this issue through methods such as prompt engineering, in-context learning, and fine-tuning, further improvements are needed to enable the accuracy and reliability of these models. Secondly, LLMs with direct domain expertise are still lacking. Thus, when dealing with domain-specific scientific problems, the models need to be fine-tuned; otherwise they can show low accuracy and misunderstanding. While LLMs hold promise in chemical research, further research and improvements are necessary to overcome the existing limitations and bring the application of artificial intelligence in the research of catalysts into a new era.
4.5 Summary
Traditional trial-and-error methods require a significant cost of time in screening and testing candidate catalysts, together with inference through expert knowledge and occasionally serendipity. Active machine learning can lower the knowledge barrier and greatly accelerate the discovery process by utilizing experimental data to build surrogate models, avoiding brute-force or uniform search of the entire chemical space. The implementation of active machine learning in the optimization of catalyst search is summarized in Table 3.| Type | Surrogate models | Variables (input) | Target (output) | Research systems | Reference |
|---|---|---|---|---|---|
| Bayesian optimization | Random forest and Gaussian process | Ratio of a metal precursor (continuous) | Current density | Electrocatalytic oxidation of methanol | 20 |
| Gaussian process | Reagent concentration for catalyst synthesis (continuous) | Hydrogen evolution rate | Photocatalytic hydrogen generation | 4 | |
| Gaussian process | Alloy compositions (continuous) | Current density | Electrocatalytic O2 reduction | 22 | |
| Large language models from open AI | Experimental procedure as text | C2 yield | Oxidative coupling of methane | 45 | |
| Active learning | Artificial neural networks and kernel ridge regression | Electronegativity and d-band width of alloys (continuous) | *CO binding energy (*CO refers to adsorbed CO on a solid surface) | Electrocatalytic CO2 reduction | 30 |
| Extra tree regressor, random forest, Gaussian process,etc. | Fingerprints of the surface and sites of intermetallics (discrete) | Adsorption energies of CO and H | Electrocatalytic CO2 reduction and H2 evolution | 31 | |
| Random forest and boosted tree | Fingerprints of adsorption sites from copper-containing metals (discrete) | CO adsorption energy | Electrocatalytic CO2 reduction | 32 | |
| GPT-4 | Synthesis procedure as text input | Success or failure of the synthesis | MOF synthesis | 47 |
Several challenges persist in implementing active machine learning, particularly related to surrogate models. These models excel in interpolation rather than extrapolation, making them prone to overfitting and necessitating training data of a specific scale. Many efforts are made to improve the surrogate models for higher generality (e.g., Phoenics135) and extend variables from simple continuous variables to discrete or categorical variables (e.g., Gryffin136). Additionally, a crucial challenge lies in selecting relevant catalysis features compatible with surrogate models. Incorporating irrelevant descriptors can impede the effectiveness of active learning algorithms, reducing their performance to that of uniform random search. The difficulty in feature selection confines certain closed-loop searches to mere recipe optimization, treating the process as a black box and adjusting only continuous variables such as reagent ratios or concentrations (Table 3). However, catalytic reaction activity and selectivity are closely linked to explicit factors such as intermediate adsorption energy, d-band center, electronegativity, and steric hindrance, which inherently serve as valid features. These features can be assessed through ab initio theoretical calculations or in situ characterization. While the advent of automated laboratories has alleviated concerns regarding insufficient data acquisition, it remains a costly endeavor, especially considering the challenges in automating certain characterization techniques. Consequently, strategies for evaluating and selecting an appropriate subset from these explicit features require further refinement. The subsequent section will delve into the detailed elaboration of chemical descriptors employed in machine learning algorithms.
5 Interpretable machine learning for catalysis
In catalysis research, the pursuit of knowledge extends beyond mere data collection; true understanding stems from interpretable models that can elucidate observations in ways that are comprehensible to human scientists.39,159 In this section, we explore the potential role of large language models (LLMs) in identifying suitable descriptors for catalysis systems and enhancing model-agnostic methods for interpretability. These aspects are crucial for advancing catalyst design and facilitating iterative research and development processes.36–385.1 Descriptors for traditional machine learning
Understanding catalysis data begins with the identification of the correct descriptors of catalytic systems. Descriptors are crucial in interpretable machine learning because they must not only capture relevant information but also minimize redundancy. The range of available descriptors provides substantial flexibility in modelling various aspects of catalytic processes.P, molar refractivity (MR), electronegativity, and atomic charges. Steric effects are captured by descriptors such as the Tolman cone angle, Sterimol values, torsion angles, bite angle, buried volume, dispersion descriptor, and solvent accessible surface area (Fig. 18).174,177–179 Tools like PaDEL-Descriptor software180 and SPOC descriptors181 package them into comprehensive descriptor suites for broader applications in research.
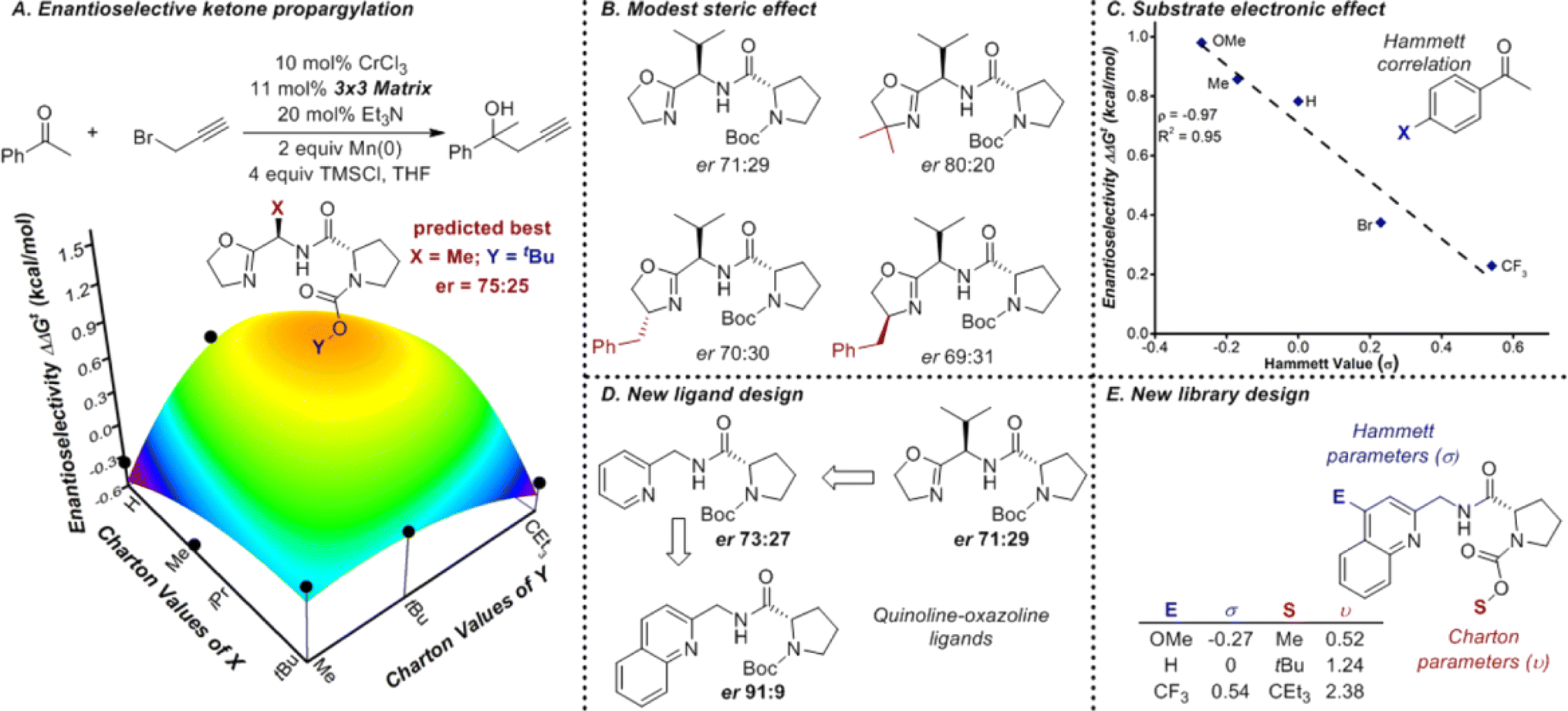 | ||
| Fig. 18 The design of a new ligand library for enantioselective ketone propargylation (A). It was shown the steric effect from the oxazoline group had limited influence on the reaction (B) while the electronic effect from the substract is more dominant (C). Thus, a new ligand library with a quinoline group and varied steric effects is design for further screening, as shown in (D and E). Reproduced with permission from ref. 177 Copyright 2016, American Chemical Society. | ||
5.2 Descriptor selection and machine learning
Descriptor selection is a crucial step in the machine learning process, involving the elimination of irrelevant and redundant descriptors. This task is particularly challenging in catalysis research, where data sets are often limited. An overly large set of descriptors can lead to spurious correlations that do not reflect underlying chemical phenomena. Traditional machine learning techniques vary in how they select descriptors:Artificial Neural Networks (ANNs): these models automatically extract and continuously refine descriptors through the iterative updating of network weights.
5.3 Incorporating chemical knowledge through LLMs
All the above descriptor selection processes have neglected the physical meanings of descriptors, which can lead to models that lack interpretability or generalizability. Large language models (LLMs) have the potential to revolutionize this process by embedding chemical knowledge into the selection process. They can track the physical significance of descriptors and, when data alone are insufficient, use embedded chemistry knowledge to guide the selection process. Recent advancements have demonstrated the utility of augmenting traditional models with LLMs to leverage the linguistic implications of descriptors, providing a novel perspective on model training and interpretability.209One such pre-trained model, Uni-Mol, incorporates 3D information in its self-training reconstruction process and has outperformed state-of-the-art models in molecular property prediction. It demonstrates strong performance in tasks that require spatial information, such as predicting protein-ligand binding poses and generating molecular conformations.211 Similarly, Payel Das et al. showed that a motif-based transformer applied to 3D heterogeneous molecular graphs (MOLFORMER) excels by utilizing attention mechanisms to capture spatial relationships within molecules.157 Another innovative approach, the Chemical Space Explorer (ChemSpacE), uses pre-trained deep generative models for exploring chemical space in an interpretable and interactive manner.212 The ChemSpacE model has exhibited impressive capabilities in molecule optimization and manipulation tasks across both single-property and multi-property scenarios. This process not only enhances the interpretability of deep generative models by navigating through their latent spaces but also facilitates human-in-the-loop exploration of chemical spaces and molecule design.
Despite these advancements, caution is necessary when considering the information used during pre-training. Unlike natural languages, which are imbued with rich contextual and cultural knowledge, pure chemical structures typically contain limited information, often constrained to basic chemical rules such as the octet rule. Pre-training models solely on 2D chemical structures or 1D SMILES strings without incorporating additional chemical knowledge may lead to models that lack substantial chemical understanding.
Pre-trained models, with their capacity for insightful interpretations and enhancements in molecular predictions, hold significant promise for transforming areas in catalyst design, molecular property prediction, and reaction optimization.
Beyond the RAG agent, there are several examples of using language model architectures to train on chemistry data using SMILES or other molecular representations. These molecular pre-training models, discussed in the previous section, are developed from scratch purely with chemistry data and, as such, do not inherit the broader knowledge typically embedded in LLMs. Notable examples include SELFormer, which utilizes a transformer-based chemical language model to learn high-quality molecular representations called SELFIES.213 Born and Manica proposed the Regression Transformer (RT), a method that abstracts regression as a conditional sequence modelling problem.214 Alán Aspuru-Guzik and his team investigated the ability of simple language models to learn complex molecular distributions, demonstrating their powerful generative capabilities through the prediction of distributions of the highest scoring penalized logP molecules in ZINC15.215 Francesca Grisoni provided a comprehensive overview of the current state, challenges, and future prospects of chemical language models in drug discovery.216
Recent initiatives have leveraged the capabilities of pre-trained language models like GPT-3, fine-tuning them with chemically curated data. In 2023, Berend Smit et al. published an influential paper titled “Is GPT-3 all you need for low-data discovery in chemistry”15 first on preprint. The title was apparently inspired by the seminal Google paper on transformers. The paper was later published in Nature Machine Intelligence with a modified title “Leveraging large language models for predictive chemistry”.156 They experimented with fine-tuning GPT-3 using chemistry data written in a sentence and used it as a general machine learning model for classification and regression. The chemicals are represented by either SMILES or IUPAC names in natural language, which makes no difference in the prediction performance. The fine-tuned GPT-3 model achieved superior performance over traditional models in predicting material properties and reaction yields, especially in data-scarce scenarios. Its ability to accept the IUPAC names of chemicals as inputs facilitates non-specialist use. The authors explored the model's potential in generating molecules based on specific requirements and tested its in-context learning capabilities, which also showed promising results.
It is interesting to discuss what aspect of the LLM's ability is used in the task of learning chemistry data. Most likely, the LLM's abilities to learn new patterns and apply basic chemical logic are critical in these tasks. It is not clear if the LLM's general knowledge about specific molecule or functional groups is used or not. It is important to recognize that these models may not fully “understand” the underlying chemistry and should be used with caution due to their potential for producing misleading results or hallucinations. Despite these limitations, this work introduces a novel paradigm in machine learning that utilizes language models to foster advancements in low-data learning within the field of chemistry.
5.4 Interpreting machine learning results
For data-driven research in catalysis to be fully beneficial, it's crucial for models to be understandable so that human scientists can actively participate and apply their findings. LLMs introduce both new challenges and opportunities for achieving this goal.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O and output yield of the reaction is 40%, please derive the output from the input”. If we only asked the LLM to give the answer, we have no way to know how the model actually works. We need to add some prompt to ask the LLM to explain how the answer is arrived at. It is yet to be tested what prompt can accurately achieve the purpose and eliminate any hallucination. The ideal prompt may also be model specific and fulfill two critical criteria:
O)O and output yield of the reaction is 40%, please derive the output from the input”. If we only asked the LLM to give the answer, we have no way to know how the model actually works. We need to add some prompt to ask the LLM to explain how the answer is arrived at. It is yet to be tested what prompt can accurately achieve the purpose and eliminate any hallucination. The ideal prompt may also be model specific and fulfill two critical criteria:
1. Interpretability: ideally, prompts should be phrased in natural language to ensure they are easily understood by human users.
2. Accuracy: prompts must accurately map input features to outputs, providing a clear and logical explanation of the data.
There are a variety of auto-prompting methods based on gradient descent to search for a prompt that can map the input feature to the output values.222–224 However, as a result of gradient descent, it is not guaranteed that these searched prompts are generally interpretable. Additionally, gradient-descent-based methods are usually computationally expensive. To address these two problems, Jianfeng Gao et al.44 introduced an interpretable auto-prompting method (iPrompt) using LLMs to directly generate and modify the prompts. There are three steps to search for ideal prompts in this method:
(1) Prompt proposal: in this stage, a prefix of data points is fed to the LLMs, requiring them to complete the prompts that map the input features to the output values. It generates a series of candidate prompts that will be evaluated further.
(2) Reranking: the performance of the candidate prompts from (1) is evaluated, and those that maximize the accuracy will be maintained.
(3) Iterate with exploration: the top candidate prompts from (2) will be truncated randomly. Then the truncated prompts will be fed to LLMs to regenerate new prompts while maintaining accuracy.
This iterative process continues until no further improvements are observed. The direct generation and modification of prompts by LLMs in steps 1 and 3 enhance interpretability, while accuracy is optimized in step 2. However, despite their impressive capabilities, LLMs may still lack depth in mathematical rigor, theoretical simulation, or specialized domain knowledge required for some catalysis applications. Incorporating AI agents equipped with a comprehensive toolkit could potentially address these limitations, enhancing both the interpretability and accuracy of machine learning models in catalysis.
5.5 Summary
Interpretable machine learning models are becoming indispensable in chemical research for exploring complex chemical processes and catalytic mechanisms. These models allow chemists to extract diverse chemical information from data and elucidate structure–activity relationships with precision and efficiency. The shift towards models that prioritize excellent interpretability and continuity, such as those employing physicochemical and theory-based descriptors, marks a significant advance over traditional Boolean fingerprints, which often lack intuitive insights and demonstrate poor extrapolative capabilities. The use of LLMs in the learning process to bring in domain knowledge and consider chemical meaning of different descriptors in the learning process is still limited.Recent developments in graph-based and latent space descriptors of pre-trained models are attracting increasing attention, despite sometimes not providing direct insights. These descriptors are valued for their potential to bridge sophisticated computational models with practical chemical understanding, a connection that is strengthening due to ongoing algorithmic improvements.
Model-agnostic methods like SHAP, LIME, and PDP provide robust frameworks for interpreting machine learning models. However, the methods need a significant update to meet the new challenge due to the involvement of LLMs.
As we look to the future, the enhancement of interpretable models and the expansion of model-agnostic methods are set to increase AI's utility beyond mere speed and accuracy. By integrating tailored, interpretable descriptors across different systems, this approach not only deepens chemical insights but also empowers the use of machine learning to quantitatively analyze structure–activity relationships, thus broadening AI's impact on scientific discovery.
6 Conclusions and perspectives
The design and discovery of optimal catalysts is a complex endeavor due to the inherent complexity of catalytic processes and the vast search space. Traditional trial-and-error approaches are laborious, time-consuming, and often fail to provide sufficient insights. However, the recent advancements in high-throughput information extraction, automated chemical experimentation, active machine learning, and interpretable machine learning have revolutionized this field.Automated extraction of unstructured chemical data, facilitated by optical character recognition and large language models (LLMs), lays the groundwork for robust data-driven approaches. Automated robotic platforms streamline experimentation, enabling real-time decision-making and facilitating closed-loop optimizations. Active learning algorithms optimize experiment selection based on accumulated data to minimize trial numbers. Interpretable machine learning models disclose underlying structure–property relationships, providing critical insights for rational catalyst design.
Despite these advances, challenges persist. Information extraction needs to evolve to handle diverse unstructured data formats reliably. Current technologies like image segmentation tools225,226 are still advancing towards fully autonomous capabilities for extracting and analyzing raw chemical data from figures. Moreover, the integration of text and figure data demands enhanced anaphora resolution and inference capabilities to support detailed analyses. Future developments in multimodal AI, capable of processing text, images, video, and voice, will be crucial in this aspect.
LLMs have demonstrated potential in comprehending complex data and have been applied successfully in projects like the one-pot synthesis conditions of MOFs. Yet, the full scope of their capabilities, especially in formatting conditions for multi-step synthesis procedures, remains underexplored. The cost and operational speed of robotic systems also limits their widespread adoption in chemical laboratories, necessitating innovations in specialized post-synthesis processing and auto-sampling for diverse catalytic systems.
The variability in control interfaces across different laboratory equipment poses another challenge, limiting hardware transferability among research communities. Standardizing control languages or systems could enhance collaborative efforts. Although natural language is commonly used to instruct experiments, its ambiguity necessitates sophisticated mapping to specific robotic operations, a task where LLMs could play a transformative role if their reliability is proven in more complex scenarios.
Furthermore, the high-dimensional nature of catalysis design and the chemical consumption in high-throughput processes suggest that automated platforms should be capable of managing varied reaction scales, from small-scale synthesis and characterization to larger-scale production.
As machine learning approaches become more integrated into catalyst design, it is anticipated that they will address increasingly complex design problems. Incorporating scientific hypotheses into the discovery process requires an iterative approach, where hypotheses are generated and modified, and data are queried for validation. AI agents,227e.g., ChemCrow228 equipped with tools for automated experimentation, information retrieval, and machine learning, show promise in bridging these capabilities to create a self-evolving, intelligent system.
Although human feedback should ideally not exist in the process, it can be used for safety checks or as alternative solutions if any of the functions (e.g., automated experimentation) are missing in the toolset, as demonstrated by Omar M. Yaghi et al.47 In the iteration, the AI agents should be instructed to generate or modify hypotheses together with their validation procedures within the toolset. Later the toolset can be utilized to give feedback to the AI agents for further improvement of the hypotheses via LLMs directly or Bayesian inference.
In conclusion, the last decade's advances have shifted the paradigm from traditional methods to a more efficient, systematic approach to experimental design in catalyst research. The integration of LLMs and AI agents promises to further enhance the capability, flexibility, and efficiency of these systems, paving the way for a future where intelligent systems can autonomously explore vast chemical spaces and contribute to scientific discovery in unprecedented ways.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
All the authors wrote the review together.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We acknowledge the financial support from the National Key R&D Program of China (2021YFA1502500), the National Natural Science Foundation of China (22125502, 22071207, and 22121001), and the Fundamental Research Funds for the Central Universities (No. 20720220011).Notes and references
- J. Gasteiger, Chemistry in times of artificial intelligence, Chemphyschem, 2020, 21, 2233–2242 CrossRef CAS PubMed.
- T. Mueller, A. G. Kusne and R. Ramprasad, Machine learning in materials science: Recent progress and emerging applications, Rev. Comput. Chem., 2016, 29, 186–273 CAS.
- P. Raccuglia, K. C. Elbert, P. D. Adler, C. Falk, M. B. Wenny, A. Mollo, M. Zeller, S. A. Friedler, J. Schrier and A. J. Norquist, Machine-learning-assisted materials discovery using failed experiments, Nature, 2016, 533, 73–76 CrossRef CAS PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, A mobile robotic chemist, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- Q. Zhu, F. Zhang, Y. Huang, H. Xiao, L. Zhao, X. Zhang, T. Song, X. Tang, X. Li, G. He, B. Chong, J. Zhou, Y. Zhang, B. Zhang, J. Cao, M. Luo, S. Wang, G. Ye, W. Zhang, X. Chen, S. Cong, D. Zhou, H. Li, J. Li, G. Zou, W. Shang, J. Jiang and Y. Luo, An all-round AI-Chemist with a scientific mind, Natl. Sci. Rev., 2022, 9, nwac190 CrossRef CAS PubMed.
- N. J. Szymanski, B. Rendy, Y. Fei, R. E. Kumar, T. He, D. Milsted, M. J. McDermott, M. Gallant, E. D. Cubuk, A. Merchant, H. Kim, A. Jain, C. J. Bartel, K. Persson, Y. Zeng and G. Ceder, An autonomous laboratory for the accelerated synthesis of novel materials, Nature, 2023, 624, 86–91 CrossRef CAS PubMed.
- K. Rajan, H. O. Brinkhaus, A. Zielesny and C. Steinbeck, A review of optical chemical structure recognition tools, J. Cheminf., 2020, 12, 60 CAS.
- K. T. Mukaddem, E. J. Beard, B. Yildirim and J. M. Cole, ImageDataExtractor: A Tool To Extract and Quantify Data from Microscopy Images, J. Chem. Inf. Model., 2020, 60, 2492–2509 CrossRef CAS PubMed.
- E. J. Beard and J. M. Cole, ChemSchematicResolver: a toolkit to decode 2D chemical diagrams with labels and R-groups into annotated chemical named entities, J. Chem. Inf. Model., 2020, 60, 2059–2072 CrossRef CAS PubMed.
- F. Musazade, N. Jamalova and J. Hasanov, Review of techniques and models used in optical chemical structure recognition in images and scanned documents, J. Cheminf., 2022, 14, 61 Search PubMed.
- I. Beltagy, K. Lo and A. Cohan, SciBERT: A Pretrained Language Model for Scientific Text, arXiv, 2019, preprint, arXiv:1903.10676, DOI:10.48550/arXiv.1903.10676.
- A. Kumar, S. Ganesh, D. Gupta and H. Kodamana, A text mining framework for screening catalysts and critical process parameters from scientific literature - A study on Hydrogen production from alcohol, Chem. Eng. Res. Des., 2022, 184, 90–102 CrossRef CAS.
- L. Wang, Y. Gao, X. Chen, W. Cui, Y. Zhou, X. Luo, S. Xu, Y. Du and B. Wang, A corpus of CO2 electrocatalytic reduction process extracted from the scientific literature, Sci. Data, 2023, 10, 175 CrossRef CAS PubMed.
- Y. Gao, L. Wang, X. Chen, Y. Du and B. Wang, Revisiting Electrocatalyst Design by a Knowledge Graph of Cu-Based Catalysts for CO2 Reduction, ACS Catal., 2023, 13, 8525–8534 CrossRef CAS.
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Is GPT-3 all you need for low-data discovery in chemistry?, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-fw8n4.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, ChatGPT Chemistry Assistant for Text Mining and the Prediction of MOF Synthesis, J. Am. Chem. Soc., 2023, 145, 18048–18062 CrossRef CAS PubMed.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Organic synthesis in a modular robotic system driven by a chemical programming language, Science, 2019, 363, eaav2211 CrossRef CAS PubMed.
- S. H. M. Mehr, M. Craven, A. I. Leonov, G. Keenan and L. Cronin, A universal system for digitization and automatic execution of the chemical synthesis literature, Science, 2020, 370, 101–108 CrossRef CAS PubMed.
- S. Rohrbach, M. Šiaučiulis, G. Chisholm, P.-A. Pirvan, M. Saleeb, S. H. M. Mehr, E. Trushina, A. I. Leonov, G. Keenan, A. Khan, A. Hammer and L. Cronin, Digitization and validation of a chemical synthesis literature database in the ChemPU, Science, 2022, 377, 172–180 CrossRef CAS PubMed.
- A. S. Nugraha, G. Lambard, J. Na, M. S. A. Hossain, T. Asahi, W. Chaikittisilp and Y. Yamauchi, Mesoporous trimetallic PtPdAu alloy films toward enhanced electrocatalytic activity in methanol oxidation: unexpected chemical compositions discovered by Bayesian optimization, J. Mater. Chem. A, 2020, 8, 13532–13540 RSC.
- A. E. Gongora, B. Xu, W. Perry, C. Okoye, P. Riley, K. G. Reyes, E. F. Morgan and K. A. Brown, A Bayesian experimental autonomous researcher for mechanical design, Sci. Adv., 2020, 6, eaaz1708 CrossRef PubMed.
- J. K. Pedersen, C. M. Clausen, O. A. Krysiak, B. Xiao, T. A. A. Batchelor, T. Löffler, V. A. Mints, L. Banko, M. Arenz, A. Savan, W. Schuhmann, A. Ludwig and J. Rossmeisl, Bayesian Optimization of High-Entropy Alloy Compositions for Electrocatalytic Oxygen Reduction, Angew. Chem., Int. Ed., 2021, 60, 24144–24152 CrossRef CAS PubMed.
- G. O. Kayode, A. F. Hill and M. M. Montemore, Bayesian optimization of single-atom alloys and other bimetallics: efficient screening for alkane transformations, CO2 reduction, and hydrogen evolution, J. Mater. Chem. A, 2023, 11, 19128–19137 RSC.
- X. Wang, Y. Huang, X. Xie, Y. Liu, Z. Huo, M. Lin, H. Xin and R. Tong, Bayesian-optimization-assisted discovery of stereoselective aluminum complexes for ring-opening polymerization of racemic lactide, Nat. Commun., 2023, 14, 3647 CrossRef CAS PubMed.
- Y. Okazaki, Y. Fujita, H. Murata, N. Masuyama, Y. Nojima, H. Ikeno, S. Yagi and I. Yamada, Composition-Designed Multielement Perovskite Oxides for Oxygen Evolution Catalysis, Chem. Mater., 2022, 34, 10973–10981 CrossRef CAS.
- Y. Zhang, T. C. Peck, G. K. Reddy, D. Banerjee, H. Jia, C. A. Roberts and C. Ling, Descriptor-Free Design of Multicomponent Catalysts, ACS Catal., 2022, 12, 10562–10571 CrossRef CAS.
- V. A. Mints, J. K. Pedersen, A. Bagger, J. Quinson, A. S. Anker, K. M. Ø. Jensen, J. Rossmeisl and M. Arenz, Exploring the Composition Space of High-Entropy Alloy Nanoparticles for the Electrocatalytic H2/CO Oxidation with Bayesian Optimization, ACS Catal., 2022, 12, 11263–11271 CrossRef CAS.
- Q. Liang, A. E. Gongora, Z. Ren, A. Tiihonen, Z. Liu, S. Sun, J. R. Deneault, D. Bash, F. Mekki-Berrada, S. A. Khan, K. Hippalgaonkar, B. Maruyama, K. A. Brown, J. Fisher Iii and T. Buonassisi, Benchmarking the performance of Bayesian optimization across multiple experimental materials science domains, npj Comput. Mater., 2021, 7, 188 CrossRef.
- Y. Ureel, M. R. Dobbelaere, O. Akin, R. J. Varghese, C. G. Pernalete, J. W. Thybaut and K. M. Van Geem, Active learning-based exploration of the catalytic pyrolysis of plastic waste, Fuel, 2022, 328, 125340 CrossRef CAS.
- J. Noh, S. Back, J. Kim and Y. Jung, Active learning with non-ab initio input features toward efficient CO2 reduction catalysts, Chem. Sci., 2018, 9, 5152–5159 RSC.
- K. Tran and Z. W. Ulissi, Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution, Nat. Catal., 2018, 1, 696–703 CrossRef CAS.
- M. Zhong, K. Tran, Y. Min, C. Wang, Z. Wang, C.-T. Dinh, P. De Luna, Z. Yu, A. S. Rasouli, P. Brodersen, S. Sun, O. Voznyy, C.-S. Tan, M. Askerka, F. Che, M. Liu, A. Seifitokaldani, Y. Pang, S.-C. Lo, A. Ip, Z. Ulissi and E. H. Sargent, Accelerated discovery of CO2 electrocatalysts using active machine learning, Nature, 2020, 581, 178–183 CrossRef CAS PubMed.
- M. Kim, Y. Kim, M. Y. Ha, E. Shin, S. J. Kwak, M. Park, I.-D. Kim, W.-B. Jung, W. B. Lee, Y. Kim and H.-T. Jung, Exploring Optimal Water Splitting Bifunctional Alloy Catalyst by Pareto Active Learning, Adv. Mater., 2023, 35, 2211497 CrossRef CAS PubMed.
- M. Kim, M. Y. Ha, W.-B. Jung, J. Yoon, E. Shin, I.-d. Kim, W. B. Lee, Y. Kim and H.-t. Jung, Searching for an Optimal Multi-Metallic Alloy Catalyst by Active Learning Combined with Experiments, Adv. Mater., 2022, 34, 2108900 CrossRef CAS PubMed.
- V. Tshitoyan, J. Dagdelen, L. Weston, A. Dunn, Z. Rong, O. Kononova, K. A. Persson, G. Ceder and A. Jain, Unsupervised word embeddings capture latent knowledge from materials science literature, Nature, 2019, 571, 95–98 CrossRef CAS PubMed.
- M. Erdem Günay and R. Yıldırım, Recent advances in knowledge discovery for heterogeneous catalysis using machine learning, Catal. Rev., 2021, 63, 120–164 CrossRef.
- T. Toyao, Z. Maeno, S. Takakusagi, T. Kamachi, I. Takigawa and K.-i. Shimizu, Machine Learning for Catalysis Informatics: Recent Applications and Prospects, ACS Catal., 2019, 10, 2260–2297 CrossRef.
- T. Mou, H. S. Pillai, S. Wang, M. Wan, X. Han, N. M. Schweitzer, F. Che and H. Xin, Bridging the complexity gap in computational heterogeneous catalysis with machine learning, Nat. Catal., 2023, 6, 122–136 CrossRef.
- A. J. Medford, M. R. Kunz, S. M. Ewing, T. Borders and R. Fushimi, Extracting Knowledge from Data through Catalysis Informatics, ACS Catal., 2018, 8, 7403–7429 CrossRef CAS.
- M. Abolhasani and E. Kumacheva, The rise of self-driving labs in chemical and materials sciences, Nat. Synth., 2023, 2, 483–492 CrossRef.
- W. X. Zhao, K. Zhou, J. Li, T. Tang, X. Wang, Y. Hou, Y. Min, B. Zhang, J. Zhang, Z. Dong, Y. Du, C. Yang, Y. Chen, Z. Chen, J. Jiang, R. Ren, Y. Li, X. Tang, Z. Liu, P. Liu, J.-Y. Nie and J.-R. Wen, A Survey of Large Language Models, arXiv, 2023, preprint, arXiv:2303.18223, DOI:10.48550/arXiv.2303.18223.
- N. Yoshikawa, M. Skreta, K. Darvish, S. Arellano-Rubach, Z. Ji, L. Bjørn Kristensen, A. Z. Li, Y. Zhao, H. Xu, A. Kuramshin, A. Aspuru-Guzik, F. Shkurti and A. Garg, Large language models for chemistry robotics, Auton. Robots, 2023, 47, 1056–1086 CrossRef.
- T. Inagaki, A. Kato, K. Takahashi, H. Ozaki and G. N. Kanda, LLMs can generate robotic scripts from goal-oriented instructions in biological laboratory automation, arXiv, 2023, preprint, arXiv:2304.10267, DOI:10.48550/arXiv.2304.10267.
- C. Singh, J. X. Morris, J. Aneja, A. M. Rush and J. Gao, Explaining Patterns in Data with Language Models via Interpretable Autoprompting, arXiv, 2022, preprint, arXiv:2210.01848, DOI:10.48550/arXiv.2210.01848.
- M. Caldas Ramos, S. S. Michtavy, M. D. Porosoff and A. D. White, Bayesian Optimization of Catalysts With In-context Learning, arXiv, 2023, preprint, arXiv:2304.05341, DOI:10.48550/arXiv.2304.05341.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Autonomous chemical research with large language models, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- Z. Zheng, Z. Rong, N. Rampal, C. Borgs, J. T. Chayes and O. M. Yaghi, A GPT-4 Reticular Chemist for Guiding MOF Discovery, Angew. Chem., Int. Ed., 2023, 62, e202311983 CrossRef CAS PubMed.
- D. S. Salley, G. A. Keenan, D.-L. Long, N. L. Bell and L. Cronin, A modular programmable inorganic cluster discovery robot for the discovery and synthesis of polyoxometalates, ACS Cent. Sci., 2020, 6, 1587–1593 CrossRef CAS PubMed.
- J. S. Manzano, W. Hou, S. S. Zalesskiy, P. Frei, H. Wang, P. J. Kitson and L. Cronin, An autonomous portable platform for universal chemical synthesis, Nat. Chem., 2022, 14, 1311–1318 CrossRef CAS PubMed.
- A. R. Frisbee, M. H. Nantz, G. W. Kramer and P. L. Fuchs, Laboratory automation. 1: Syntheses via vinyl sulfones. 14. Robotic orchestration of organic reactions: Yield optimization via an automated system with operator-specified reaction sequences, J. Am. Chem. Soc., 1984, 106, 7143–7145 CrossRef CAS.
- B. P. MacLeod, F. G. L. Parlane, T. D. Morrissey, F. Hse, L. M. Roch, K. E. Dettelbach, R. Moreira, L. P. E. Yunker, M. B. Rooney, J. R. Deeth, V. Lai, G. J. Ng, H. Situ, R. H. Zhang, M. S. Elliott, T. H. Haley, D. J. Dvorak, A. Aspuru-Guzik, J. E. Hein and C. P. Berlinguette, Self-driving laboratory for accelerated discovery of thin-film materials, Sci. Adv., 2020, 6, eaaz8867 CrossRef CAS PubMed.
- F. Emmert-Streib, Z. Yang, H. Feng, S. Tripathi and M. Dehmer, An Introductory Review of Deep Learning for Prediction Models With Big Data, Front. Artif. Intell., 2020, 3 DOI:10.3389/frai.2020.00004.
- S. Lundberg and S.-I. Lee, A Unified Approach to Interpreting Model Predictions, arXiv, 2017, preprint, arXiv:1705.07874, DOI:10.48550/arXiv.1705.07874.
- Reaxys, https://www.reaxys.com, accessed May 12, 2021.
- SciFinder, https://scifinder.cas.org, accessed May 12, 2021.
- X. Zeng, H. Xiang, L. Yu, J. Wang, K. Li, R. Nussinov and F. Cheng, Accurate prediction of molecular properties and drug targets using a self-supervised image representation learning framework, Nat. Mach. Intell., 2022, 4, 1004–1016 CrossRef.
- R. Rozas and H. Fernandez, Automatic processing of graphics for image databases in science, J. Chem. Inf. Comput. Sci., 1990, 30, 7–12 CrossRef CAS.
- S. S. Bukhari, Z. Iftikhar and A. Dengel, Chemical structure recognition (CSR) system: automatic analysis of 2D chemical structures in document images presented in part at the 2019 International Conference on Document Analysis and Recognition (ICDAR), 2019 Search PubMed.
- Y. Wang, T. Zhang and X. Yu, A component-detection-based approach for interpreting off-line handwritten chemical cyclic compound structures presented in part at the 2021 IEEE International Conference on Engineering, Technology & Education (TALE), 2021 Search PubMed.
- J. R. McDaniel and J. R. Balmuth, Kekule: OCR-optical chemical (structure) recognition, J. Chem. Inf. Comput. Sci., 1992, 32, 373–378 CrossRef CAS.
- N. M. Sadawi, A. P. Sexton and V. Sorge, Chemical structure recognition: a rule-based approach presented in part at the Document Recognition and Retrieval XIX, 2012 Search PubMed.
- A. Fujiyoshi, K. Nakagawa and M. Suzuki, Robust method of segmentation and recognition of chemical structure images in cheminfty presented in part at the Pre-proceedings of the 9th IAPR international workshop on graphics recognition, GREC, 2011 Search PubMed.
- C. Hong, X. Du and L. Zhang, Research on chemical expression images recognition presented in part at the 2015 Joint International Mechanical, Electronic and Information Technology Conference (JIMET-15), 2015 Search PubMed.
- J. Park, G. R. Rosania, K. A. Shedden, M. Nguyen, N. Lyu and K. Saitou, Automated extraction of chemical structure information from digital raster images, Chem. Cent. J., 2009, 3, 4 CrossRef PubMed.
- R. Casey, S. Boyer, P. Healey, A. Miller, B. Oudot and K. Zilles, Optical recognition of chemical graphics presented in part at the Proceedings of 2nd International Conference on Document Analysis and Recognition (ICDAR'93), 1993 Search PubMed.
- P. Ibison, M. Jacquot, F. Kam, A. Neville, R. W. Simpson, C. Tonnelier, T. Venczel and A. P. Johnson, Chemical literature data extraction: the CLiDE Project, J. Chem. Inf. Comput. Sci., 1993, 33, 338–344 CrossRef CAS.
- A. T. Valko and A. P. Johnson, CLiDE Pro: the latest generation of CLiDE, a tool for optical chemical structure recognition, J. Chem. Inf. Model., 2009, 49, 780–787 CrossRef CAS PubMed.
- I. V. Filippov and M. C. Nicklaus, Optical structure recognition software to recover chemical information: OSRA, an open source solution, J. Chem. Inf. Model., 2009, 49, 740–743 CrossRef CAS PubMed.
- V. Smolov, F. Zentsev and M. Rybalkin, Imago: Open-Source Toolkit for 2D Chemical Structure Image Recognition presented in part at the TREC, 2011 Search PubMed.
- P. Frasconi, F. Gabbrielli, M. Lippi and S. Marinai, Markov logic networks for optical chemical structure recognition, J. Chem. Inf. Model., 2014, 54, 2380–2390 CrossRef CAS PubMed.
- J. Staker, K. Marshall, R. Abel and C. M. McQuaw, Molecular Structure Extraction from Documents Using Deep Learning, J. Chem. Inf. Model., 2019, 59, 1017–1029 CrossRef CAS PubMed.
- K. Rajan, A. Zielesny and C. Steinbeck, DECIMER: towards deep learning for chemical image recognition, J. Cheminf., 2020, 12, 65 CAS.
- K. Rajan, H. O. Brinkhaus, M. Sorokina, A. Zielesny and C. Steinbeck, DECIMER-Segmentation: Automated extraction of chemical structure depictions from scientific literature, J. Cheminf., 2021, 13, 20 CAS.
- K. Rajan, H. O. Brinkhaus, M. I. Agea, A. Zielesny and C. Steinbeck, DECIMER. ai-An open platform for automated optical chemical structure identification, segmentation and recognition in scientific publications, Nat. Commun., 2023, 14, 5045 CrossRef CAS PubMed.
- Y. Xu, J. Xiao, C.-H. Chou, J. Zhang, J. Zhu, Q. Hu, H. Li, N. Han, B. Liu, S. Zhang, J. Han, Z. Zhang, S. Zhang, W. Zhang, L. Lai and J. Pei, MolMiner: You only look once for chemical structure recognition, J. Chem. Inf. Model., 2022, 62, 5321–5328 CrossRef CAS PubMed.
- Y. Qian, J. Guo, Z. Tu, Z. Li, C. W. Coley and R. Barzilay, MolScribe: Robust Molecular Structure Recognition with Image-to-Graph Generation, J. Chem. Inf. Model., 2023, 63, 1925–1934 CrossRef CAS PubMed.
- W. Hemati and A. Mehler, LSTMVoter: chemical named entity recognition using a conglomerate of sequence labeling tools, J. Cheminf., 2019, 11, 3 Search PubMed.
- D. Mahendran, C. Tang and B. T. McInnes, Graph Convolutional Networks for Chemical Relation Extraction presented in part at the Companion Proceedings of the Web Conference 2022, Virtual Event, Lyon, France, 2022 Search PubMed.
- M. Suvarna, A. C. Vaucher, S. Mitchell, T. Laino and J. Pérez-Ramírez, Language models and protocol standardization guidelines for accelerating synthesis planning in heterogeneous catalysis, Nat. Commun., 2023, 14, 7964 CrossRef CAS PubMed.
- M. C. Swain and J. M. Cole, ChemDataExtractor: A Toolkit for Automated Extraction of Chemical Information from the Scientific Literature, J. Chem. Inf. Model., 2016, 56, 1894–1904 CrossRef CAS PubMed.
- J. Mavračić, C. J. Court, T. Isazawa, S. R. Elliott and J. M. Cole, ChemDataExtractor 2.0: Autopopulated Ontologies for Materials Science, J. Chem. Inf. Model., 2021, 61, 4280–4289 CrossRef PubMed.
- J. Guo, A. S. Ibanez-Lopez, H. Gao, V. Quach, C. W. Coley, K. F. Jensen and R. Barzilay, Automated chemical reaction extraction from scientific literature, J. Chem. Inf. Model., 2021, 62, 2035–2045 CrossRef PubMed.
- R. B. Merrifield and J. M. Stewart, Automated peptide synthesis, Nature, 1965, 207, 522–523 CrossRef CAS PubMed.
- C. Porte, W. Debreuille, F. Draskovic and A. Delacroix, Automation and optimization by simplex methods of 6-chlorohexanol synthesis, Process Control Qual., 1996, 4, 111–122 Search PubMed.
- R. W. Wagner, F. Li, H. Du and J. S. Lindsey, Investigation of cocatalysis conditions using an automated microscale multireactor workstation: Synthesis of m eso-tetramesitylporphyrin, Org. Process Res. Dev., 1999, 3, 28–37 CrossRef CAS.
- J. S. Lindsey, A retrospective on the automation of laboratory synthetic chemistry, Chemom. Intell. Lab. Syst., 1992, 17, 15–45 CrossRef CAS.
- R. B. Merrifield, J. M. Stewart and N. Jernberg, Instrument for automated synthesis of peptides, Anal. Chem., 1966, 38, 1905–1914 CrossRef CAS PubMed.
- G. Alvarado-Urbina, G. M. Sathe, W.-C. Liu, M. F. Gillen, P. D. Duck, R. Bender and K. K. Ogilvie, Automated synthesis of gene fragments, Science, 1981, 214, 270–274 CrossRef CAS PubMed.
- M. Legrand and A. Foucard, Automation on the laboratory bench, J. Chem. Educ., 1978, 55, 767 CrossRef CAS.
- J. Bajorath, Integration of virtual and high-throughput screening, Nat. Rev. Drug Discovery, 2002, 1, 882–894 CrossRef CAS PubMed.
- R. L. Martin, C. M. Simon, B. Smit and M. Haranczyk, In silico design of porous polymer networks: high-throughput screening for methane storage materials, J. Am. Chem. Soc., 2014, 136, 5006–5022 CrossRef CAS PubMed.
- R. Macarron, M. N. Banks, D. Bojanic, D. J. Burns, D. A. Cirovic, T. Garyantes, D. V. S. Green, R. P. Hertzberg, W. P. Janzen, J. W. Paslay, U. Schopfer and G. Sitta Sittampalam, Impact of high-throughput screening in biomedical research, Nat. Rev. Drug Discovery, 2011, 10, 188–195 CrossRef CAS PubMed.
- S. M. Senkan, High-throughput screening of solid-state catalyst libraries, Nature, 1998, 394, 350–353 CrossRef CAS.
- X. D. Xiang, X. Sun, G. Briceno, Y. Lou, K.-A. Wang, H. Chang, W. G. Wallace-Freedman, S.-W. Chen and P. G. Schultz, A combinatorial approach to materials discovery, Science, 1995, 268, 1738–1740 CrossRef CAS PubMed.
- W. F. Maier, K. Stoewe and S. Sieg, Combinatorial and high-throughput materials science, Angew. Chem., Int. Ed., 2007, 46, 6016–6067 CrossRef CAS PubMed.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, Accelerating electrolyte discovery for energy storage with high-throughput screening, J. Phys. Chem. Lett., 2015, 6, 283–291 CrossRef CAS PubMed.
- J. S. Smith, B. Nebgen, N. Lubbers, O. Isayev and A. E. Roitberg, Less is more: Sampling chemical space with active learning, J. Chem. Phys., 2018, 148 DOI:10.1063/1.5023802.
- Q. Zhu, Y. Huang, D. Zhou, L. Zhao, L. Guo, R. Yang, Z. Sun, M. Luo, F. Zhang, H. Xiao, X. Tang, X. Zhang, T. Song, X. Li, B. Chong, J. Zhou, Y. Zhang, B. Zhang, J. Cao, G. Zhang, S. Wang, G. Ye, W. Zhang, H. Zhao, S. Cong, H. Li, L.-L. Ling, Z. Zhang, W. Shang, J. Jiang and Y. Luo, Automated synthesis of oxygen-producing catalysts from Martian meteorites by a robotic AI chemist, Nat. Synth., 2024, 3, 319–328 CrossRef.
- A. Adamo, R. L. Beingessner, M. Behnam, J. Chen, T. F. Jamison, K. F. Jensen, J.-C. M. Monbaliu, A. S. Myerson, E. M. Revalor, D. R. Snead, T. Stelzer, N. Weeranoppanant, S. Y. Wong and P. Zhang, On-demand continuous-flow production of pharmaceuticals in a compact, reconfigurable system, Science, 2016, 352, 61–67 CrossRef CAS PubMed.
- M. Baumann and I. R. Baxendale, The synthesis of active pharmaceutical ingredients (APIs) using continuous flow chemistry, Beilstein J. Org. Chem., 2015, 11, 1194–1219 CrossRef CAS PubMed.
- J. Wegner, S. Ceylan and A. Kirschning, Flow chemistry–a key enabling technology for (multistep) organic synthesis, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- S. Kobayashi, Flow “fine” synthesis: high yielding and selective organic synthesis by flow methods, Chem.–Asian J., 2016, 11, 425–436 CrossRef CAS PubMed.
- M. E. Briggs, A. G. Slater, N. Lunt, S. Jiang, M. A. Little, R. L. Greenaway, T. Hasell, C. Battilocchio, S. V. Ley and A. I. Cooper, Dynamic flow synthesis of porous organic cages, Chem. Commun., 2015, 51, 17390–17393 RSC.
- G. Jas and A. Kirschning, Continuous flow techniques in organic synthesis, Chem. - Eur. J., 2003, 9, 5708–5723 CrossRef CAS PubMed.
- D. Angelone, A. J. S. Hammer, S. Rohrbach, S. Krambeck, J. M. Granda, J. Wolf, S. Zalesskiy, G. Chisholm and L. Cronin, Convergence of multiple synthetic paradigms in a universally programmable chemical synthesis machine, Nat. Chem., 2021, 13, 63–69 CrossRef CAS PubMed.
- M. Xie, Y. Shen, W. Ma, D. Wei, B. Zhang, Z. Wang, Y. Wang, Q. Zhang, S. Xie, C. Wang and Y. Wang, Fast Screening for Copper-Based Bimetallic Electrocatalysts: Efficient Electrocatalytic Reduction of CO2 to C2+ Products on Magnesium-Modified Copper, Angew. Chem., Int. Ed., 2022, 61, e202213423 CrossRef CAS PubMed.
- M. B. Plutschack, B. u. Pieber, K. Gilmore and P. H. Seeberger, The hitchhiker's guide to flow chemistry, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS PubMed.
- A. R. Bogdan and Y. Wang, A high-throughput synthesis of 1, 2, 4-oxadiazole and 1, 2, 4-triazole libraries in a continuous flow reactor, RSC Adv., 2015, 5, 79264–79269 RSC.
- S. E. Lohse, J. R. Eller, S. T. Sivapalan, M. R. Plews and C. J. Murphy, A simple millifluidic benchtop reactor system for the high-throughput synthesis and functionalization of gold nanoparticles with different sizes and shapes, ACS Nano, 2013, 7, 4135–4150 CrossRef CAS PubMed.
- E. J. Roberts, S. E. Habas, L. Wang, D. A. Ruddy, E. A. White, F. G. Baddour, M. B. Griffin, J. A. Schaidle, N. Malmstadt and R. L. Brutchey, High-throughput continuous flow synthesis of nickel nanoparticles for the catalytic hydrodeoxygenation of guaiacol, ACS Sustain. Chem. Eng., 2017, 5, 632–639 CrossRef CAS.
- M. Ago, S. Huan, M. Borghei, J. Raula, E. I. Kauppinen and O. J. Rojas, High-throughput synthesis of lignin particles (∼30 nm to ∼2 μm) via aerosol flow reactor: Size fractionation and utilization in pickering emulsions, ACS Appl. Mater. Interfaces, 2016, 8, 23302–23310 CrossRef CAS PubMed.
- E. M. Chan, C. Xu, A. W. Mao, G. Han, J. S. Owen, B. E. Cohen and D. J. Milliron, Reproducible, high-throughput synthesis of colloidal nanocrystals for optimization in multidimensional parameter space, Nano Lett., 2010, 10, 1874–1885 CrossRef CAS PubMed.
- H. Long, T.-S. Chen, J. Song, S. Zhu and H.-C. Xu, Electrochemical aromatic C--H hydroxylation in continuous flow, Nat. Commun., 2022, 13, 3945 CrossRef CAS PubMed.
- B. Winterson, T. Rennigholtz and T. Wirth, Flow electrochemistry: a safe tool for fluorine chemistry, Chem. Sci., 2021, 12, 9053–9059 RSC.
- A.-C. Bédard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Reconfigurable system for automated optimization of diverse chemical reactions, Science, 2018, 361, 1220–1225 CrossRef PubMed.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Automated radial synthesis of organic molecules, Nature, 2020, 579, 379–384 CrossRef CAS PubMed.
- N. Collins, D. Stout, J.-P. Lim, J. P. Malerich, J. D. White, P. B. Madrid, M. Latendresse, D. Krieger, J. Szeto and V.-A. Vu, Fully automated chemical synthesis: toward the universal synthesizer, Org. Process Res. Dev., 2020, 24, 2064–2077 CrossRef CAS.
- C. D. Scott, R. Labes, M. Depardieu, C. Battilocchio, M. G. Davidson, S. V. Ley, C. C. Wilson and K. Robertson, Integrated plug flow synthesis and crystallisation of pyrazinamide, React. Chem. Eng., 2018, 3, 631–634 RSC.
- G. Niu, L. Zhang, A. Ruditskiy, L. Wang and Y. Xia, A droplet-reactor system capable of automation for the continuous and scalable production of noble-metal nanocrystals, Nano Lett., 2018, 18, 3879–3884 CrossRef CAS PubMed.
- E. W. Yeap, D. Z. Ng, D. Lai, D. J. Ertl, S. Sharpe and S. A. Khan, Continuous flow droplet-based crystallization platform for producing spherical drug microparticles, Org. Process Res. Dev., 2018, 23, 93–101 CrossRef.
- B. Rimez, J. Septavaux and B. Scheid, The coupling of in-flow reaction with continuous flow seedless tubular crystallization, React. Chem. Eng., 2019, 4, 516–522 RSC.
- O. Okafor, K. Robertson, R. Goodridge and V. Sans, Continuous-flow crystallisation in 3D-printed compact devices, React. Chem. Eng., 2019, 4, 1682–1688 RSC.
- C. W. Coley, D. A. Thomas III, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A robotic platform for flow synthesis of organic compounds informed by AI planning, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- B. A. Koscher, R. B. Canty, M. A. McDonald, K. P. Greenman, C. J. McGill, C. L. Bilodeau, W. Jin, H. Wu, F. H. Vermeire, B. Jin, T. Hart, T. Kulesza, S.-C. Li, T. S. Jaakkola, R. Barzilay, R. Gómez-Bombarelli, W. H. Green and K. F. Jensen, Autonomous, multiproperty-driven molecular discovery: From predictions to measurements and back, Science, 2023, 382, eadi1407 CrossRef CAS PubMed.
- F. Stella, C. Della Santina and J. Hughes, How can LLMs transform the robotic design process?, Nat. Mach. Intell., 2023, 1–4 Search PubMed.
- S. Vemprala, R. Bonatti, A. Bucker and A. Kapoor, Chatgpt for robotics: Design principles and model abilities, Microsoft Auton. Syst. Robot. Res, 2023, 2, 20 Search PubMed.
- L. Wang, Y. Ling, Z. Yuan, M. Shridhar, C. Bao, Y. Qin, B. Wang, H. Xu and X. Wang, GenSim: Generating Robotic Simulation Tasks via Large Language Models, arXiv, 2023, preprint, arXiv:2310.01361, DOI:10.48550/arXiv.2310.01361.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Organic synthesis in a modular robotic system driven by a chemical programming language, Science, 2019, 363, eaav2211 CrossRef CAS PubMed.
- F. Olsson, A literature survey of active machine learning in the context of natural language processing, Report 11003154 (ISSN), Swedish Institute of Computer Science, Kista, Sweden, 2009 Search PubMed.
- Y. Ureel, M. R. Dobbelaere, Y. Ouyang, K. De Ras, M. K. Sabbe, G. B. Marin and K. M. Van Geem, Active Machine Learning for Chemical Engineers: A Bright Future Lies Ahead, Engineering, 2023, 27, 23–30 CrossRef CAS.
- X. Wang, Y. Jin, S. Schmitt and M. Olhofer, Recent Advances in Bayesian Optimization, ACM Comput. Surv., 2023, 55, 287 Search PubMed.
- R. González Perea, E. Camacho Poyato, P. Montesinos and J. A. Rodríguez Díaz, Optimisation of water demand forecasting by artificial intelligence with short data sets, Biosyst. Eng., 2019, 177, 59–66 CrossRef.
- J. M. Hernández-Lobato and R. P. Adams, Probabilistic Backpropagation for Scalable Learning of Bayesian Neural Networks presented in part at the International Conference on Machine Learning, 2015 Search PubMed.
- A. Graves, Practical variational inference for neural networks presented in part at the Proceedings of the 24th International Conference on Neural Information Processing Systems, Granada, Spain, 2011 Search PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, Phoenics: A Bayesian Optimizer for Chemistry, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Gryffin: An algorithm for Bayesian optimization of categorical variables informed by expert knowledge, Applied Physics Reviews, 2021, 8 DOI:10.1063/5.0048164.
- J. Močkus, in Optimization Techniques IFIP Technical Conference: Novosibirsk, July 1–7, 1974, ed. G. I. Marchuk, Springer Berlin Heidelberg, Berlin, Heidelberg, 1975, pp. 400–404, DOI:10.1007/978-3-662-38527-2_55.
- A. Zilinskas, Optimization of one-dimensional multimodal functions, J. R. Stat. Soc., C: Appl. Stat., 1978, 27, 367–375 Search PubMed.
- H. J. Kushner, A New Method of Locating the Maximum Point of an Arbitrary Multipeak Curve in the Presence of Noise, J. Basic Eng., 1964, 86, 97–106 CrossRef.
- P. Auer, Using confidence bounds for exploitation-exploration trade-offs, J. Mach. Learn. Res., 2003, 3, 397–422 Search PubMed.
- J.-B. Masson, Counting biomolecules with Bayesian inference, Nat. Comput. Sci., 2022, 2, 74–75 CrossRef PubMed.
- L. Bassman Oftelie, P. Rajak, R. K. Kalia, A. Nakano, F. Sha, J. Sun, D. J. Singh, M. Aykol, P. Huck, K. Persson and P. Vashishta, Active learning for accelerated design of layered materials, npj Comput. Mater., 2018, 4, 74 CrossRef.
- K. M. Jablonka, G. M. Jothiappan, S. Wang, B. Smit and B. Yoo, Bias free multiobjective active learning for materials design and discovery, Nat. Commun., 2021, 12, 2312 CrossRef CAS PubMed.
- N. S. Eyke, W. H. Green and K. F. Jensen, Iterative experimental design based on active machine learning reduces the experimental burden associated with reaction screening, React. Chem. Eng., 2020, 5, 1963–1972 RSC.
- S. Viet Johansson, H. Gummesson Svensson, E. Bjerrum, A. Schliep, M. Haghir Chehreghani, C. Tyrchan and O. Engkvist, Using Active Learning to Develop Machine Learning Models for Reaction Yield Prediction, Mol. Inf., 2022, 41, e2200043 CrossRef PubMed.
- D. Reker, P. Schneider, G. Schneider and J. B. Brown, Active learning for computational chemogenomics, Future Med. Chem., 2017, 9, 381–402 CrossRef CAS PubMed.
- D. Reker and G. Schneider, Active-learning strategies in computer-assisted drug discovery, Drug Discovery Today, 2015, 20, 458–465 CrossRef PubMed.
- F. Douak, F. Melgani and N. Benoudjit, Kernel ridge regression with active learning for wind speed prediction, Appl. Energy, 2013, 103, 328–340 CrossRef.
- M. Sharma and M. Bilgic, Evidence-based uncertainty sampling for active learning, Data Min. Knowl. Discov., 2017, 31, 164–202 CrossRef.
- F. Douak, N. Benoudjit and F. Melgani, A two-stage regression approach for spectroscopic quantitative analysis, Chemom. Intell. Lab. Syst., 2011, 109, 34–41 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A materials genome approach to accelerating materials innovation, APL Mater., 2013, 1, 011002 CrossRef.
- T. Guo, K. Guo, B. Nan, Z. Liang, Z. Guo, N. Chawla, O. Wiest and X. Zhang, What can large language models do in chemistry? a comprehensive benchmark on eight tasks, arXiv, 2023, preprint, arXiv:2305.18365, DOI:10.48550/arXiv.2305.18365.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry and A. Askell, Language models are few-shot learners, Adv. Neural Inf. Process Syst., 2020, 33, 1877–1901 Search PubMed.
- A. E. Blanchard, J. Gounley, D. Bhowmik, M. Chandra Shekar, I. Lyngaas, S. Gao, J. Yin, A. Tsaris, F. Wang and J. Glaser, Language models for the prediction of SARS-CoV-2 inhibitors, Int. J. High Perform. Comput., 2022, 36, 587–602 CrossRef PubMed.
- C. Xu, Y. Wang and A. Barati Farimani, TransPolymer: a Transformer-based language model for polymer property predictions, npj Comput. Mater., 2023, 9, 64 CrossRef.
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Leveraging large language models for predictive chemistry, Nat. Mach. Intell., 2024, 6, 161–169 CrossRef.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Large-scale chemical language representations capture molecular structure and properties, Nat. Mach. Intell., 2022, 4, 1256–1264 CrossRef.
- Z. Yang, Y. Wang and L. Zhang, AI becomes a masterbrain scientist, bioRxiv, 2023, preprint, DOI:10.1101/2023.04.19.537579.
- J. Kim, D. Kang, S. Kim and H. W. Jang, Catalyze Materials Science with Machine Learning, ACS Mater. Lett., 2021, 3, 1151–1171 CrossRef CAS.
- S. Singh and R. B. Sunoj, Molecular Machine Learning for Chemical Catalysis: Prospects and Challenges, Acc. Chem. Res., 2023, 56, 402–412 CrossRef CAS PubMed.
- A. G. Maldonado and G. Rothenberg, Predictive modeling in homogeneous catalysis: a tutorial, Chem. Soc. Rev., 2010, 39, 1891–1902 RSC.
- B. Kim, S. Lee and J. Kim, Inverse design of porous materials using artificial neural networks, Sci. Adv., 2020, 6, eaax9324 CrossRef CAS PubMed.
- D. A. Carr, M. Lach-hab, S. Yang, I. I. Vaisman and E. Blaisten-Barojas, Machine learning approach for structure-based zeolite classification, Microporous Mesoporous Mater., 2009, 117, 339–349 CrossRef CAS.
- A. Jain and T. Bligaard, Atomic-position independent descriptor for machine learning of material properties, Phys. Rev. B, 2018, 98, 214112 CrossRef.
- K. Li and D. Xue, Estimation of Electronegativity Values of Elements in Different Valence States, J. Phys. Chem. A, 2006, 110, 11332–11337 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, MoleculeNet: a benchmark for molecular machine learning, Chem. Sci., 2018, 9, 513–530 RSC.
- P. Schlexer Lamoureux, K. T. Winther, J. A. Garrido Torres, V. Streibel, M. Zhao, M. Bajdich, F. Abild-Pedersen and T. Bligaard, Machine Learning for Computational Heterogeneous Catalysis, ChemCatChem, 2019, 11, 3581–3601 CrossRef CAS.
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, Reoptimization of MDL Keys for Use in Drug Discovery, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed.
- D. Butina, Unsupervised Data Base Clustering Based on Daylight's Fingerprint and Tanimoto Similarity: A Fast and Automated Way To Cluster Small and Large Data Sets, J. Chem. Inf. Comput. Sci., 1999, 39, 747–750 CrossRef CAS.
- X. He, Y. Su, J. Zhu, N. Fang, Y. Chen, H. Liu, D. Zhou and C. Wang, Uncovering the influence of the modifier redox potential on CO2 reduction through combined data-driven machine learning and hypothesis-driven experimentation, J. Mater. Chem. A, 2023, 11, 18106–18114 RSC.
- D. Rogers and M. Hahn, Extended-Connectivity Fingerprints, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- Y. Guo, X. He, Y. Su, Y. Dai, M. Xie, S. Yang, J. Chen, K. Wang, D. Zhou and C. Wang, Machine-Learning-Guided Discovery and Optimization of Additives in Preparing Cu Catalysts for CO2 Reduction, J. Am. Chem. Soc., 2021, 143, 5755–5762 CrossRef CAS PubMed.
- J. P. Janet and H. J. Kulik, Resolving Transition Metal Chemical Space: Feature Selection for Machine Learning and Structure–Property Relationships, J. Phys. Chem. A, 2017, 121, 8939–8954 CrossRef CAS PubMed.
- C. B. Santiago, J.-Y. Guo and M. S. Sigman, Predictive and mechanistic multivariate linear regression models for reaction development, Chem. Sci., 2018, 9, 2398–2412 RSC.
- X. Ma, Z. Li, L. E. K. Achenie and H. Xin, Machine-Learning-Augmented Chemisorption Model for CO2 Electroreduction Catalyst Screening, J. Phys. Chem. Lett., 2015, 6, 3528–3533 CrossRef CAS PubMed.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Machine Learning for Electrocatalyst and Photocatalyst Design and Discovery, Chem. Rev., 2022, 122, 13478–13515 CrossRef CAS PubMed.
- M. S. Sigman, K. C. Harper, E. N. Bess and A. Milo, The Development of Multidimensional Analysis Tools for Asymmetric Catalysis and Beyond, Acc. Chem. Res., 2016, 49, 1292–1301 CrossRef CAS PubMed.
- J. P. Reid and M. S. Sigman, Comparing quantitative prediction methods for the discovery of small-molecule chiral catalysts, Nat. Rev. Chem, 2018, 2, 290–305 CrossRef CAS.
- K. C. Harper and M. S. Sigman, Three-Dimensional Correlation of Steric and Electronic Free Energy Relationships Guides Asymmetric Propargylation, Science, 2011, 333, 1875–1878 CrossRef CAS PubMed.
- C. W. Yap, PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed.
- Q. Yang, Y. Liu, J. Cheng, Y. Li, S. Liu, Y. Duan, L. Zhang and S. Luo, An Ensemble Structure and Physicochemical (SPOC) Descriptor for Machine-Learning Prediction of Chemical Reaction and Molecular Properties, Chemphyschem, 2022, 23, e202200255 CrossRef CAS PubMed.
- E. L. Willighagen, H. M. G. W. Denissen, R. Wehrens and L. M. C. Buydens, On the Use of 1H and 13C 1D NMR Spectra as QSPR Descriptors, J. Chem. Inf. Model., 2006, 46, 487–494 CrossRef CAS PubMed.
- T. Jin, Q. Zhao, A. B. Schofield and B. M. Savoie, Machine learning models capable of chemical deduction for identifying reaction products, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-l6lzp.
- X. Wang, S. Jiang, W. Hu, S. Ye, T. Wang, F. Wu, L. Yang, X. Li, G. Zhang, X. Chen, J. Jiang and Y. Luo, Quantitatively Determining Surface–Adsorbate Properties from Vibrational Spectroscopy with Interpretable Machine Learning, J. Am. Chem. Soc., 2022, 144, 16069–16076 CrossRef CAS PubMed.
- D. Loffreda, F. Delbecq, F. Vigné and P. Sautet, Fast Prediction of Selectivity in Heterogeneous Catalysis from Extended Brønsted–Evans–Polanyi Relations: A Theoretical Insight, Angew. Chem., Int. Ed., 2009, 48, 8978–8980 CrossRef CAS PubMed.
- F. Wei and L. Zhuang, Unsupervised machine learning reveals eigen reactivity of metal surfaces, Sci. Bull., 2024, 69, 756–762 CrossRef CAS PubMed.
- S. Ringe, The importance of a charge transfer descriptor for screening potential CO(2) reduction electrocatalysts, Nat. Commun., 2023, 14, 2598 CrossRef CAS PubMed.
- W. T. Hong, R. E. Welsch and Y. Shao-Horn, Descriptors of Oxygen-Evolution Activity for Oxides: A Statistical Evaluation, J. Phys. Chem. C, 2015, 120, 78–86 CrossRef.
- C. Ren, S. Lu, Y. Wu, Y. Ouyang, Y. Zhang, Q. Li, C. Ling and J. Wang, A Universal Descriptor for Complicated Interfacial Effects on Electrochemical Reduction Reactions, J. Am. Chem. Soc., 2022, 144, 12874–12883 CrossRef CAS PubMed.
- M. Andersen, A. J. Medford, J. K. Nørskov and K. Reuter, Scaling-Relation-Based Analysis of Bifunctional Catalysis: The Case for Homogeneous Bimetallic Alloys, ACS Catal., 2017, 7, 3960–3967 CrossRef CAS.
- J. Liu, W. Luo, L. Wang, J. Zhang, X.-Z. Fu and J.-L. Luo, Toward Excellence of Electrocatalyst Design by Emerging Descriptor-Oriented Machine Learning, Adv. Funct. Mater., 2022, 32, 2110748 CrossRef CAS.
- X. Lin, Y. Wang, X. Chang, S. Zhen, Z. J. Zhao and J. Gong, High-Throughput Screening of Electrocatalysts for Nitrogen Reduction Reactions Accelerated by Interpretable Intrinsic Descriptor, Angew Chem. Int. Ed. Engl., 2023, 62, e202300122 CrossRef CAS PubMed.
- X. Wang, S. Ye, W. Hu, E. Sharman, R. Liu, Y. Liu, Y. Luo and J. Jiang, Electric Dipole Descriptor for Machine Learning Prediction of Catalyst Surface–Molecular Adsorbate Interactions, J. Am. Chem. Soc., 2020, 142, 7737–7743 CrossRef CAS PubMed.
- L. H. Mou, T. Han, P. E. S. Smith, E. Sharman and J. Jiang, Machine Learning Descriptors for Data-Driven Catalysis Study, Advanced Science, 2023, 10, e2301020 CrossRef PubMed.
- O. Isayev, C. Oses, C. Toher, E. Gossett, S. Curtarolo and A. Tropsha, Universal fragment descriptors for predicting properties of inorganic crystals, Nat. Commun., 2017, 8, 15679 CrossRef CAS PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, A graph-convolutional neural network model for the prediction of chemical reactivity, Chem. Sci., 2019, 10, 370–377 RSC.
- L. P. Hammett, Linear free energy relationships in rate and equilibrium phenomena, Trans. Faraday Soc., 1938, 34, 156–165 RSC.
- R. W. Taft Jr, Linear Free Energy Relationships from Rates of Esterification and Hydrolysis of Aliphatic and Ortho-substituted Benzoate Esters, J. Am. Chem. Soc., 1952, 74, 2729–2732 CrossRef.
- J. M. Crawford, C. Kingston, F. D. Toste and M. S. Sigman, Data Science Meets Physical Organic Chemistry, Acc. Chem. Res., 2021, 54, 3136–3148 CrossRef CAS PubMed.
- W. L. Williams, L. Zeng, T. Gensch, M. S. Sigman, A. G. Doyle and E. V. Anslyn, The Evolution of Data-Driven Modeling in Organic Chemistry, ACS Cent. Sci., 2021, 7, 1622–1637 CrossRef CAS PubMed.
- J. P. Reid and M. S. Sigman, Holistic prediction of enantioselectivity in asymmetric catalysis, Nature, 2019, 571, 343–348 CrossRef CAS PubMed.
- S. M. Lundberg, G. Erion, H. Chen, A. DeGrave, J. M. Prutkin, B. Nair, R. Katz, J. Himmelfarb, N. Bansal and S.-I. Lee, From local explanations to global understanding with explainable AI for trees, Nat. Mach. Intell., 2020, 2, 56–67 CrossRef PubMed.
- R. Ouyang, S. Curtarolo, E. Ahmetcik, M. Scheffler and L. M. Ghiringhelli, SISSO: A compressed-sensing method for identifying the best low-dimensional descriptor in an immensity of offered candidates, Phys. Rev. Mater., 2018, 2, 083802 CrossRef CAS.
- B. Weng, Z. Song, R. Zhu, Q. Yan, Q. Sun, C. G. Grice, Y. Yan and W. J. Yin, Simple descriptor derived from symbolic regression accelerating the discovery of new perovskite catalysts, Nat. Commun., 2020, 11, 3513 CrossRef CAS PubMed.
- C. J. Bartel, S. L. Millican, A. M. Deml, J. R. Rumptz, W. Tumas, A. W. Weimer, S. Lany, V. Stevanović, C. B. Musgrave and A. M. Holder, Physical descriptor for the Gibbs energy of inorganic crystalline solids and temperature-dependent materials chemistry, Nat. Commun., 2018, 9, 4168 CrossRef PubMed.
- W. Xu, M. Andersen and K. Reuter, Data-Driven Descriptor Engineering and Refined Scaling Relations for Predicting Transition Metal Oxide Reactivity, ACS Catal., 2020, 11, 734–742 CrossRef.
- Z.-K. Han, D. Sarker, R. Ouyang, A. Mazheika, Y. Gao and S. V. Levchenko, Single-atom alloy catalysts designed by first-principles calculations and artificial intelligence, Nat. Commun., 2021, 12, 1833 CrossRef CAS PubMed.
- M. E. Tipping and C. M. Bishop, Probabilistic principal component analysis, J. R. Stat. Soc., B: Stat. Methodol., 1999, 61, 611–622 CrossRef.
- C. Singh, A. Askari, R. Caruana and J. Gao, Augmenting interpretable models with large language models during training, Nat. Commun., 2023, 14, 7913 CrossRef CAS PubMed.
- J. Xia, Y. Zhu, Y. Du, Y. Liu and S. Z. Li, A Systematic Survey of Chemical Pre-trained Models, IJCAI, 2023 Search PubMed.
- G. Zhou, Z. Gao, Q. Ding, H. Zheng, H. Xu, Z. Wei, L. Zhang and G. Ke, Uni-Mol: A Universal 3D Molecular Representation Learning Framework, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- Y. Du, X. Liu, N. M. Shah, S. Liu, J. Zhang and B. Zhou, ChemSpacE: Interpretable and Interactive Chemical Space Exploration, ChemRxiv, 2022, preprint, DOI:10.26434/chemrxiv-2022-x49mh-v3.
- A. Yüksel, E. Ulusoy, A. Ünlü and T. Doğan, SELFormer: molecular representation learning via SELFIES language models, Mach. Learn.: Sci. Technol., 2023, 4, 025035 Search PubMed.
- J. Born and M. Manica, Regression Transformer enables concurrent sequence regression and generation for molecular language modelling, Nat. Mach. Intell., 2023, 5, 432–444 CrossRef.
- D. Flam-Shepherd, K. Zhu and A. Aspuru-Guzik, Language models can learn complex molecular distributions, Nat. Commun., 2022, 13, 3293 CrossRef CAS PubMed.
- F. Grisoni, Chemical language models for de novo drug design: Challenges and opportunities, Curr. Opin. Struct. Biol., 2023, 79, 102527 CrossRef CAS PubMed.
- A. G. Yohannes, C. Lee, P. Talebi, D. H. Mok, M. Karamad, S. Back and S. Siahrostami, Combined High-Throughput DFT and ML Screening of Transition Metal Nitrides for Electrochemical CO2 Reduction, ACS Catal., 2023, 13, 9007–9017 CrossRef CAS.
- A. Chakkingal, P. Janssens, J. Poissonnier, A. J. Barrios, M. Virginie, A. Y. Khodakov and J. W. Thybaut, Machine learning based interpretation of microkinetic data: a Fischer–Tropsch synthesis case study, React. Chem. Eng., 2022, 7, 101–110 RSC.
- M. T. Ribeiro, S. Singh and C. Guestrin, “Why Should I Trust You?”: Explaining the Predictions of Any Classifier presented in part at the Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, California, USA, 2016 Search PubMed.
- H. S. Pillai, Y. Li, S. H. Wang, N. Omidvar, Q. Mu, L. E. K. Achenie, F. Abild-Pedersen, J. Yang, G. Wu and H. Xin, Interpretable design of Ir-free trimetallic electrocatalysts for ammonia oxidation with graph neural networks, Nat. Commun., 2023, 14, 792 CrossRef CAS PubMed.
- K. Vellayappan, Y. Yue, K. H. Lim, K. Cao, J. Y. Tan, S. Cheng, T. Wang, T. Z. H. Gani, I. A. Karimi and S. Kawi, Impacts of catalyst and process parameters on Ni-catalyzed methane dry reforming via interpretable machine learning, Appl. Catal., B, 2023, 330 DOI:10.1016/j.apcatb.2023.122593.
- T. Shin, Y. Razeghi, R. L. Logan IV, E. Wallace and S. Singh, AutoPrompt: Eliciting Knowledge from Language Models with Automatically Generated Prompts, arXiv, 2020, preprint, arXiv:2010.15980, DOI:10.48550/arXiv.2010.15980.
- X. L. Li and P. Liang, Prefix-Tuning: Optimizing Continuous Prompts for Generation, arXiv, 2021, preprint, arXiv:2101.00190, DOI:10.48550/arXiv.2101.00190.
- K. Hambardzumyan, H. Khachatrian and J. May, WARP: Word-level Adversarial ReProgramming, Online, August, 2021 Search PubMed.
- A. Kirillov, E. Mintun, N. Ravi, H. Mao, C. Rolland, L. Gustafson, T. Xiao, S. Whitehead, A. C. Berg, W.-Y. Lo, P. Dollár and R. Girshick, Segment Anything, arXiv, 2023, preprint, arXiv:2304.02643, DOI:10.48550/arXiv.2304.02643.
- O. Aydin and M. Y. Yassikaya, Validity and Reliability Analysis of the PlotDigitizer Software Program for Data Extraction from Single-Case Graphs, Perspect. Behav. Sci., 2022, 45, 239–257 CrossRef PubMed.
- H. Yang, S. Yue and Y. He, Auto-GPT for Online Decision Making: Benchmarks and Additional Opinions, arXiv, 2023, preprint, arXiv:2306.02224, DOI:10.48550/arXiv.2306.02224.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Augmenting large language models with chemistry tools, Nat. Mach. Intell., 2024, 6(5), 525–535 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2024 |