
Colloidal TiO2 nanocrystals with engineered defectivity and optical properties†
Julia J.
Chang
a,
Bin
Yuan
b,
Sandro
Mignuzzi
c,
Riccardo
Sapienza
c,
Francesco
Mezzadri
 d and
Ludovico
Cademartiri
*d
d and
Ludovico
Cademartiri
*d
aDepartment of Materials Science & Engineering, Iowa State University of Science and Technology, 2220 Hoover Hall, Ames, IA 50011, USA
bDepartment of Chemical & Biological Engineering, Iowa State University of Science and Technology, Gilman Hall, Ames, IA 50011, USA
cThe Blackett Laboratory, Department of Physics, Imperial College London, London, SW7 2AZ, UK
dDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parma, 43124, Italy. E-mail: ludovico.cademartiri@unipr.it
First published on 5th July 2024
Abstract
Partially reduced forms of titanium dioxide (sometimes called “black” titania) have attracted widespread interest as promising photocatalysts of oxidation due to their absorption in the visible region. The main approaches to produce it rely on postprocessing at high temperatures (up to 800 °C) and high pressures (up to 40 bar) or on highly reactive precursors (e.g., TiH2), and yield powders with poorly controlled sizes, shapes, defect concentrations and distributions. We describe an approach for the one-step synthesis of TiO2 colloidal nanocrystals at atmospheric pressure and temperatures as low as 280 °C. The temperature of the reaction allows the density of oxygen vacancies to be controlled by nearly two orders of magnitude independently of their size, shape, or colloidal stability. This synthetic pathway appears to produce vacancies that are homogeneously distributed in the nanocrystals, rather than being concentrated in an amorphous shell. As a result, the defects are protected from oxidation and result in stable optical properties in oxidizing environments.
New conceptsThe application of nanomaterials in catalysis, energy harvesting, and storage is increasingly dependent on their defects. This leads to a conundrum: we need to push materials further from equilibrium, but the further we push them, the harder it is to stabilize their steady state, and the harder it is to understand the structure/property relationships. It is therefore essential to develop systematic, reproducible ways to “synthesize” defects in relevant materials, in a way that controls their structures, spatial distribution, concentration, etc. In this paper, we show none of the classic approaches to the synthesis of oxide nanocrystals – the decomposition of alkoxide precursors in the presence of a Lewis-acid “crystallization catalyst” – can avoid the limitations of classic nanocrystal growth mechanisms (i.e., monomer addition) in the stabilization of point defects. By producing initially amorphous seed particles which are then crystallized through the “catalytic” influence of the Lewis acid, this low-temperature, catalysed “devitrification” is much more likely than near-epitaxial growth processes to trap point defects as diffusion in the solid state is much slower than on surfaces. On the basis of this reasoning, we feel the findings of this paper might very well translate to a host of other compositions. |
Introduction
This paper describes a direct, green(er) route to the synthesis of “black” TiO2 in the form of colloidal nanocrystals by the condensation/decomposition of titanium(IV) isopropoxide (TTIP) in the presence of titanium(IV) chloride (TiCl4) and trioctylphosphine oxide (TOPO). This reaction was previously shown to form nanocrystals with uniform sizes, shape, and colloidal stability in organic solvents.1 The off-white color of the product has been seldomly reported2–4 but not, to our knowledge, investigated.As we show here, the yellowish color does not result from a byproduct of the reaction or from interactions between capping ligands and the TiO2 nanoparticle surface,5 but rather from oxygen vacancies inside the material. Furthermore, decreasing the temperature of the reaction increases the concentration of oxygen vacancies and their absorption of visible light by >70 times.
“Black” TiO2 – titanium dioxide that has undergone partial reduction leading to broad absorption of visible light – has been the subject of significant interest since 2011,6 due to its enhanced photocatalysis in the visible region.7,8 Its absorption of visible light is thought to originate from a variety of oxygen-deficient defects leading to energy levels within the bandgap.8
These defect structures are usually produced by (i) energy-intensive (e.g., high power H2 plasma processing9,10), unsafe (e.g., high pressure H2 treatment11,12), or poorly scalable (e.g., anodization13,14) post-processing steps that partially reduce15–17 a crystalline TiO2 structure into a metastable,18 highly defective, partially amorphous state, or (ii) the partial oxidation of highly unstable low-valence titanium species (e.g., TiH2,19 TiCl320). These synthetic approaches are not green, sustainable, and are often inaccessible to academic laboratories due to safety concerns or regulations. Furthermore, the instability of these forms of TiO2 makes them highly susceptible to oxidation towards the crystalline, Ti4+ ground state.
Lastly, the current methods for producing “black” TiO2 are based on hydrolytic sol–gel approaches that yield irregular, polydisperse powders6,21,22 or commercially available TiO2 materials.23–25 The lack of control over the size of the particles prevents the increase of surface areas, while the lack of colloidal stability hinders their deposition and self-assembly in the liquid phase, and facilitates agglomeration that can significantly reduce the accessible surface area.26 The thermal treatments necessary to crystallize the atomic structure can easily cause the accumulation of black carbon from the incomplete combustion of the organic binders. The effect of carbon residues has not been conclusively investigated in “black” TiO2 but recent results in ZrO2 indicate that they can significantly alter interfacial properties such as grain growth kinetics.27
The approach we present here uses atmospheric pressure and moderate temperatures (<300 °C). It produces relatively uniform particles (polydispersity = 25%) with a well-defined octahedral shape, high colloidal stability (that enable thin-film deposition and self-assembly), and controlled concentration of homogeneously distributed oxygen vacancies that are highly resistant to oxidation. As such, these particles represent a highly controlled model system for the study of defect states in nanostructured TiO2.
Results and discussion
Our synthesis was based on the decomposition/condensation of titanium(IV) isopropoxide (TTIP) at high temperatures in the presence of titanium(IV) chloride (TiCl4, which acts as a crystallization catalyst1) and trioctylphosphine oxide (TOPO, which acts as a ligand and a solvent). Specifically, TiCl4 and TTIP were injected into degassed TOPO at 120 °C, and the reaction mixture was heated to 280 °C, 300 °C, 320 °C or 340 °C and kept at that temperature for 2 h. The particles were purified thrice by precipitation (using acetone as a nonsolvent) and redispersed in hexane.XRD characterization of the purified product (Fig. 1a) showed broadened diffraction peaks at angles that match bulk anatase (PDF#00-021-1272). The relative intensities closely matched the bulk diffractogram, indicating the absence of preferred orientation. Importantly, the diffractograms are inconsistent with the presence of any significant amounts of either brookite or rutile phases.
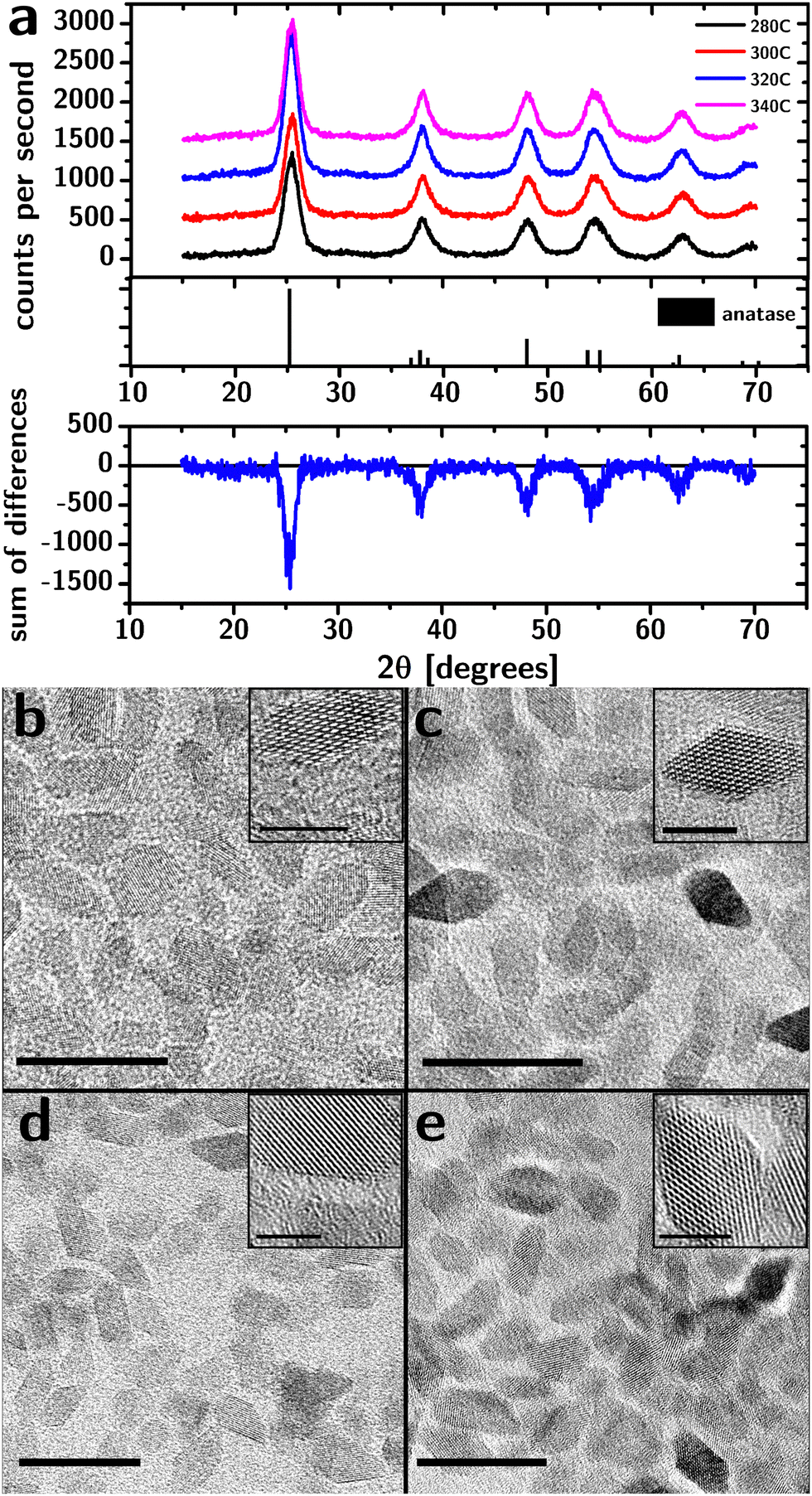 | ||
| Fig. 1 Structural characterization of “black” TiO2 nanocrystals. (a) XRD spectra of TiO2 nanocrystal powders synthesized at different temperatures. Below is the differences in intensity between the spectra showing the absence of a residual signal between the peaks, indicating the absence of an amorphous phase. (b)–(e) TEM images of “black” TiO2 nanocrystals synthesized at 280 °C (b), 300 °C (c), 320 °C (d), and 340 °C (e). Scale bars are 20 nm (5 nm in the insets). | ||
The XRD characterization does not support the presence of an amorphous fraction. After subtraction of the background signal (Fig. S1, ESI†), the diffraction intensity between the main peaks and 2θ < 30° was analyzed for evidence of amorphous material. While the diffraction intensity at the peaks varied by up to 45% between samples (due to differences in the amount of material on the holder), this difference in intensity vanished between the peaks demonstrating the absence of a broad, amorphous background.
TEM characterization (Fig. 1b–e) shows the particles to be approximately octahedral at all reaction temperatures. The fringes associated with the crystalline lattice reached the edges of the particles indicating the lack of an amorphous shell, consistent with XRD data.
The side length of the octahedral particles (Table 1) estimated from TEM images suggests a small increase in size with reaction temperature and a relatively constant polydispersity (∼25%). The distribution of the side lengths was lognormal (i.e., the Kolmogorov–Smirnov test28 failed to reject the distribution).
| 280 °C | 300 °C | 320 °C | 340 °C | |
|---|---|---|---|---|
| a TEM [nm] | 6.20 ± 0.18 | 6.49 ± 0.14 | 6.52 ± 0.13 | 6.40 ± 0.23 |
| p XRD [nm] | 4.64 | 4.94 | 5.03 | 5.14 |
| p TEM [nm] | 4.82 ± 0.14 | 5.05 ± 0.11 | 5.08 ± 0.10 | 4.98 ± 0.18 |
| Band gap [eV] | 3.498 ± 0.002 | 3.468 ± 0.002 | 3.503 ± 0.002 | 3.477 ± 0.002 |
The approximately normal distribution of particle sizes, the small crystal size, the relatively well-defined crystal shape, and the isolated diffraction peaks allow for a quantitative application of Scherrer's analysis. After subtraction of the background, the diffraction peaks could be fitted with Gaussian peaks (the Kolmogorov–Smirnov test28 failed to reject the normal distribution). The “true crystallite size” p (cube root of the crystallite volume;29Table 1) was obtained by using the appropriate Scherrer constant (0.8613) and making the appropriate conversions to account for the octahedral shape of the particles (cf. ESI†).
The true crystallite size increases slightly with an increase in reaction temperature and is within the uncertainty of the true crystallite size obtained from TEM data (for all temperatures >280 °C), consistent with a lack of polycrystallinity or an amorphous phase (cf. Fig. S2, ESI†).
While the structural characterization does not indicate a significant influence of reaction temperature, the optical properties tell a different story. The yellowish color of the particles (both as a dispersion and as a powder) becomes increasingly intense as the reaction temperature decreases (Fig. 2a). The absorbance spectra, normalized to their band edge absorption (cf.Fig. 2b), show a significant increase in the absorption of visible light as the reaction temperature is decreased. The absorption occurs as a broad band that monotonically decreases with wavelength.
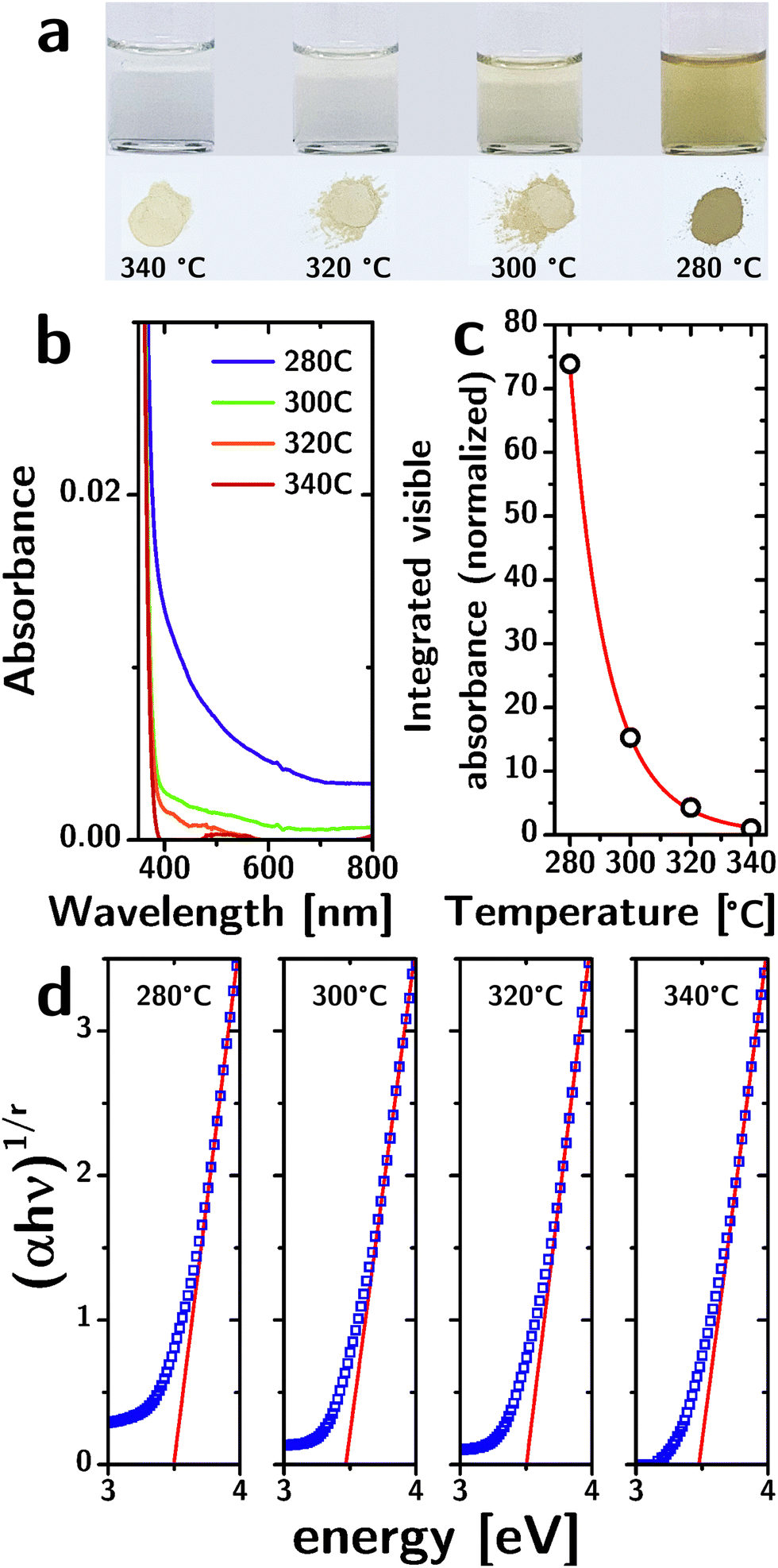 | ||
| Fig. 2 Optical properties of “black” TiO2 nanocrystals. (a) Photos of 10 mg mL−1 colloidal TiO2 nanoparticle dispersions produced at different reaction temperatures and the corresponding dry powders. (b) Optical absorption spectrum of dispersions synthesized at different temperatures. (c) Comparison of the experimentally determined visible absorbance (black empty circles) with an Arrhenius model (red line). (d) Tauc plot of the nanoparticle dispersions synthesized at different reaction temperatures. | ||
The integrated absorbance in the visible region (from 400 nm to 800 nm, normalized to the absorbance for T = 340 °C) has a nearly perfect (R2 = 0.99994) exponential dependence on the reaction temperature T of the type: A = a·exp(−b/T) (cf.Fig. 2c). Since the absorbance of dilute absorbers is linearly dependent on their concentration (according to Lambert–Beer's law), the exponential dependence strongly suggests that the absorbers are removed by a thermally activated process during particle growth, consistent with their origin as defects. The b parameter in the exponential fit allowed us to estimate the activation energy associated with this process at 0.91 × 10−19 J. Importantly, the single exponentiality of the data (within the admittedly narrow range of temperature samples) is a strong indicator that the defectivity responsible for these optical features is of a single kind.
The increased absorbance in the visible region is not accompanied by a decrease in the bandgap energy. We identified the exponent r in Tauc's relation and the range of energies to consider by a second-derivative analysis of (αhν)1/r as a function of hν for different values of r and found that the second derivative reaches zero across the band-edge for r = 1/2, consistent with a direct bandgap (Fig. S3, ESI†).
The Tauc plots (Fig. 2d) indicate a bandgap of ∼3.5 eV with no significant dependence on reaction temperature (cf.Table 1). The errors listed for the bandgap energies were obtained from the linear fits, but are certainly underestimated (the exact linear fit depends on the range of energies chosen). This bandgap is larger than the reported bandgap for bulk anatase (3.2 eV).30 The larger bandgap could be a result of quantum confinement given that the particle radii (rp ∼ 2.3 nm) are comparable to the Bohr radius aH of anatase (1.5 nm).31
The particles are luminescent in the visible region (cf.Fig. 3a). The emission spectrum (excitation at 440 nm) peaks at 575 nm but is rather broad (∼320 meV) and exhibits a pronounced tail toward higher wavelengths. The luminescence lifetime collected from dried samples can be fitted with two exponentials (cf.Fig. 3c) with lifetimes of ∼1.5 and ∼5.5 ns that are largely independent of the reaction temperature (cf. Fig. S4, ESI†). The broad luminescence spectrum and the wide Stokes shift (∼660 meV) are consistent with emission from defect states.
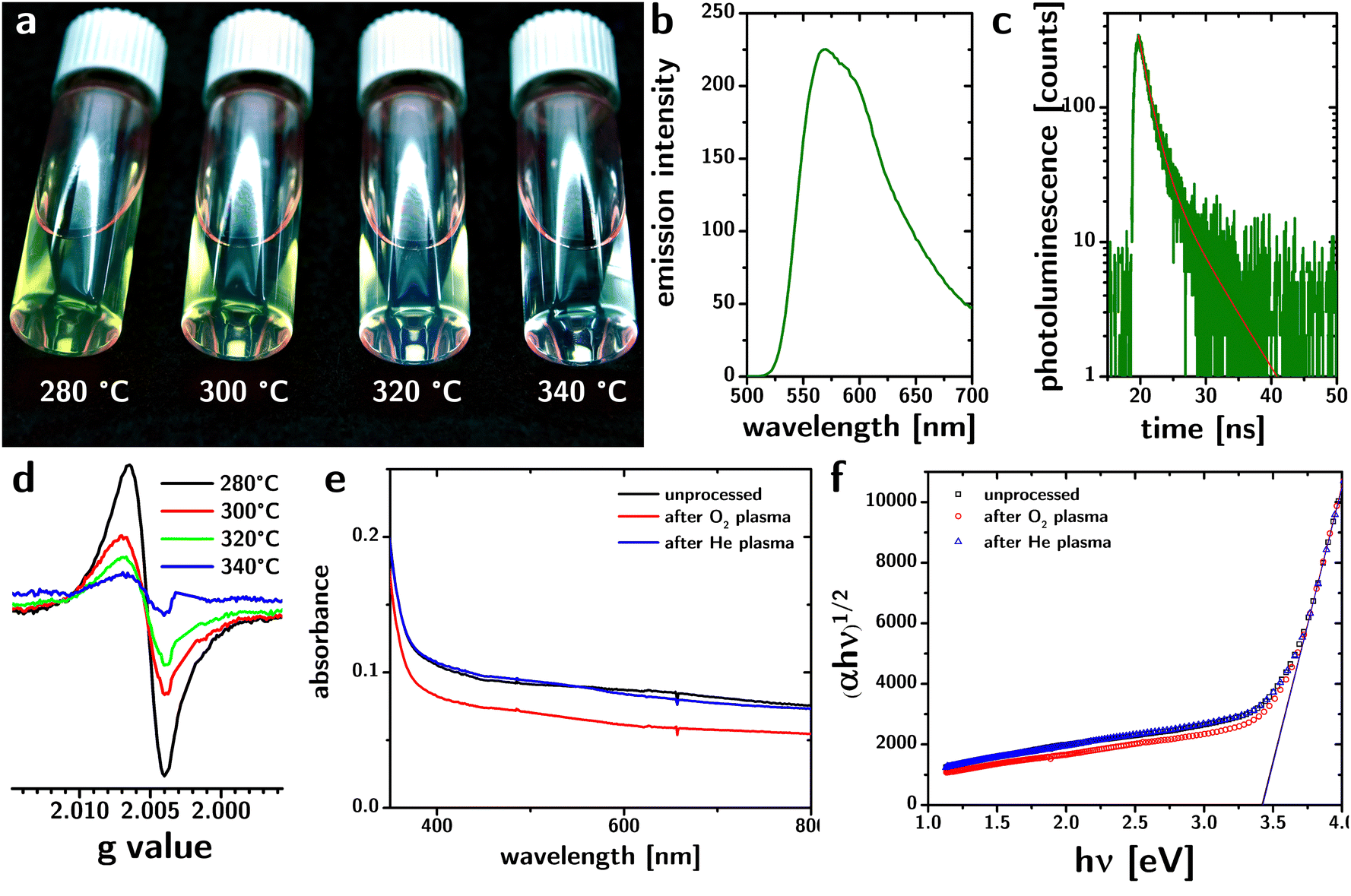 | ||
| Fig. 3 (a) Photograph of yellow–green luminescence from “black” TiO2 nanoparticle dispersions synthesized at different reaction temperatures. (b) and (c) Photoluminescence spectrum and lifetime from “black” TiO2 nanoparticle dispersions synthesized at 280 °C. The red line in panel (c) indicates the bi-exponential fit. (d) EPR spectrum from “black” TiO2 nanoparticles synthesized at different reaction temperatures. The signal at g = 2.007 is consistent with oxygen vacancies in anatase. (e) and (f) Optical absorption spectrum and Tauc plots of thin films of “black” TiO2 nanoparticles before (black) and after O2 plasma treatment (red) and He plasma treatment (blue). | ||
Electron paramagnetic resonance (EPR) has been previously used to detect oxygen vacancies in anatase.32 EPR spectra from our samples (cf.Fig. 3d) show signals at room temperature with a g value equal to 2.007, which is consistent with oxygen vacancies in the anatase lattice.32 The increased signal with decreased reaction temperature is consistent with an increase in defect concentration.
The picture provided by all data indicates that the absorption of visible light by the particles is caused by oxygen vacancies. Differently from prior approaches to produce “black” TiO2, our structural and optical characterization suggests that the defects are distributed throughout the particles, rather than being concentrated in an amorphous shell.
To test this conclusion, we exposed the particles to O2 plasma. Oxidizing conditions have been shown to reduce (and, in some cases, eliminate) the absorption of visible light by “black” TiO2,18 consistent with the localization of the defects at the surface of the material. We hypothesized that a homogeneous distribution would render the defects in our material less susceptible to oxidizing environments.
Therefore, we deposited the particles as thin films by spin coating and characterized the UV and visible absorption of light by the films, before and after treatment with O2 plasma.33,34 As a control, we used a non-oxidizing plasma environment (He plasma35). The results are shown in Fig. 3e. While the He plasma treatment does not affect the absorption of the films in the visible region, the O2 plasma reduces absorbance in the visible region by 20–30%. 48 hour O2 plasma treatment could remove all the organic parts of capping ligands,36 and thus the persistence of visible absorption after O2 plasma processing proves that the organic-TiO2 interaction is not the dominant source of the optical properties of the particles.
The loss of absorbance is instead consistent with the loss of defects on the surface of the particles. Considering an octahedral shape, the sizes reported in Table 1, and the length of a Ti–O bond (∼2 Å), the fraction of atoms located at the surface is 18%. Therefore, the loss of absorbance caused by plasma treatment is consistent with the oxidation of the oxygen vacancies on the surface of the particles. An analysis of the absorption spectrum of the films using the Tauc plot (cf.Fig. 3f) shows that the plasma processing does not change the bandgap of the material.
Conclusion
In summary, the combined characterization by XRD, TEM, UV-vis spectroscopy, EPR, and photoluminescence, as well the characterization of colloids synthesized at various temperatures and surface oxidized by plasma, is only internally consistent with the presence of point defects rather than extended defects (see XRD and TEM data), which indicates that these point defects are of one kind (absorption intensity vs. reaction temperature), that they are most likely O vacancies (EPR) and that their distribution in the material is uniform (plasma oxidation of the surfaces and consequent reduction of absorption).Fundamentally, we have shown in this paper that the decomposition and crystallization of alkoxide precursors of Ti allows for the control of point defects inside the structure in a way that is quantitatively predictable on thermodynamic grounds. The control of defectivity in nanostructures is usually strategized by starting from a thermodynamic ground state and then pushing the system out of equilibrium. This is the approach most commonly used in the synthesis of defective TiO2.15–17 This is inherently a costly process since pushing systems out of equilibrium requires the application of energy. The further out of equilibrium one goes, the more difficult it is to maintain reproducibility as the system becomes sensitive to its history. Also, the influx of energy tends to produce graded structures (e.g., the forced reduction by H2 leads to surface reduction and reconstruction).
It is often thought that starting instead from a metastable state rather than a ground state would not allow such precise control. We here show that the devitrification approach to the creation of nanocrystals allows instead for a “controlled descent” into a tuned but still defective end state using temperature and time as synthetic variables.
The growth mechanism by devitrification of an amorphous template bypasses the difficulties in the introduction of point defects in colloidal nanocrystals by forcing their diffusion in bulk rather than on surfaces. This work further clarifies decades of inconsistent observations on the optical properties of TiO2 nanocrystals.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work described in this paper has been supported by the Member-Specific-Research-Intel program of Semiconductor Research Corporation under Award No. 2015-IN-2582.References
- T. J. Trentler, T. E. Denler, J. F. Bertone, A. Agrawal and V. L. Colvin, J. Am. Chem. Soc., 1999, 121, 1613–1614 CrossRef CAS.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS.
- B. Wu, C. Guo, N. Zheng, Z. Xie and G. D. Stucky, J. Am. Chem. Soc., 2008, 130, 17563–17567 CrossRef CAS.
- T. Xia, J. W. Otto, T. Dutta, J. Murowchick, A. N. Caruso, Z. Peng and X. Chen, J. Mater. Res., 2013, 28, 326–332 CrossRef CAS.
- D. Q. Vo, E.-J. Kim and S. Kim, J. Colloid Interface Sci., 2009, 337, 75–80 CrossRef CAS PubMed.
- X. Chen, L. Liu, Y. Y. Peter and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS.
- X. Chen, L. Liu and F. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- X. Liu, G. Zhu, X. Wang, X. Yuan, T. Lin and F. Huang, Adv. Energy Mater., 2016, 6, 1600452 CrossRef.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin and X. Xie, Adv. Funct. Mater., 2013, 23, 5444–5450 CrossRef CAS.
- Y. Yan, B. Hao, D. Wang, G. Chen, E. Markweg, A. Albrecht and P. Schaaf, J. Mater. Chem. A, 2013, 1, 14507–14513 RSC.
- N. Liu, C. Schneider, D. Freitag, M. Hartmann, U. Venkatesan, J. Müller, E. Spiecker and P. Schmuki, Nano Lett., 2014, 14, 3309–3313 CrossRef CAS.
- C. Sun, Y. Jia, X.-H. Yang, H.-G. Yang, X. Yao, G. Q. Lu, A. Selloni and S. C. Smith, J. Phys. Chem. C, 2011, 115, 25590–25594 CrossRef CAS.
- J. Dong, J. Han, Y. Liu, A. Nakajima, S. Matsushita, S. Wei and W. Gao, ACS Appl. Mater. Interfaces, 2014, 6, 1385–1388 CrossRef CAS.
- L. Zheng, H. Cheng, F. Liang, S. Shu, C. K. Tsang, H. Li, S.-T. Lee and Y. Y. Li, J. Phys. Chem. C, 2012, 116, 5509–5515 CrossRef CAS.
- L. Mascaretti, R. Matarrese, A. Ravanelli, M. Isacchi, P. Mazzolini, C. S. Casari, V. Russo, I. Nova, G. Terraneo and C. Ducati, Int. J. Hydrogen Energy, 2017, 42, 26639–26651 CrossRef CAS.
- L. Mascaretti, S. Ferrulli, P. Mazzolini, C. S. Casari, V. Russo, R. Matarrese, I. Nova, G. Terraneo, N. Liu and P. Schmuki, Sol. Energy Mater. Sol. Cells, 2017, 169, 19–27 CrossRef CAS.
- N. Liu, H.-G. Steinrück, A. Osvet, Y. Yang and P. Schmuki, Appl. Phys. Lett., 2017, 110, 072102 CrossRef.
- M. I. Nandasiri, V. Shutthanandan, S. Manandhar, A. M. Schwarz, L. Oxenford, J. V. Kennedy, S. Thevuthasan and M. A. Henderson, J. Phys. Chem. Lett., 2015, 6, 4627–4632 CrossRef CAS.
- L. R. Grabstanowicz, S. Gao, T. Li, R. M. Rickard, T. Rajh, D.-J. Liu and T. Xu, Inorg. Chem., 2013, 52, 3884–3890 CrossRef CAS.
- Z. Zhao, H. Tan, H. Zhao, Y. Lv, L.-J. Zhou, Y. Song and Z. Sun, Chem. Commun., 2014, 50, 2755–2757 RSC.
- S.-T. Myung, M. Kikuchi, C. S. Yoon, H. Yashiro, S.-J. Kim, Y.-K. Sun and B. Scrosati, Energy Environ. Sci., 2013, 6, 2609–2614 RSC.
- Z. Wang, C. Yang, T. Lin, H. Yin, P. Chen, D. Wan, F. Xu, F. Huang, J. Lin and X. Xie, Energy Environ. Sci., 2013, 6, 3007–3014 RSC.
- C. Barzan, E. Groppo, S. Bordiga and A. Zecchina, ACS Catal., 2014, 4, 986–989 CrossRef CAS.
- X. Jiang, Y. Zhang, J. Jiang, Y. Rong, Y. Wang, Y. Wu and C. Pan, J. Phys. Chem. C, 2012, 116, 22619–22624 CrossRef CAS.
- H. Lu, B. Zhao, R. Pan, J. Yao, J. Qiu, L. Luo and Y. Liu, RSC Adv., 2014, 4, 1128–1132 RSC.
- J. P. Kelly and O. A. Graeve, in Sintering: mechanisms of convention nanodensification and field assisted processes, ed. R. Castro and K. van Benthem, Springer Science & Business Media, 2012, vol. 35 Search PubMed.
- P. Mohapatra, S. Shaw, D. Mendivelso-Perez, J. M. Bobbitt, T. F. Silva, F. Naab, B. Yuan, X. Tian, E. A. Smith and L. Cademartiri, Nat. Commun., 2017, 8, 2038 CrossRef.
- F. J. Massey Jr, J. Am. Stat. Assoc., 1951, 46, 68–78 CrossRef.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS.
- H. Tang, K. Prasad, R. Sanjines, P. Schmid and F. Levy, J. Appl. Phys., 1994, 75, 2042–2047 CrossRef CAS.
- X. Yu, B. Kim and Y. K. Kim, ACS Catal., 2013, 3, 2479–2486 CrossRef CAS.
- S. Shaw, J. L. Colaux, J. L. Hay, F. C. Peiris and L. Cademartiri, Adv. Mater., 2016, 28, 8900–8905 CrossRef CAS PubMed.
- S. Shaw, T. F. Silva, P. Mohapatra, D. Mendivelso-Perez, X. Tian, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Phys. Chem. Chem. Phys., 2019, 21, 1614–1622 RSC.
- S. Shaw, X. Tian, T. F. Silva, J. M. Bobbitt, F. Naab, C. L. Rodrigues, E. A. Smith and L. Cademartiri, Chem. Mater., 2018, 30, 5961–5967 CrossRef CAS.
- S. Shaw, J. L. Colaux, J. L. Hay, F. C. Peiris and L. Cademartiri, Adv. Mater., 2016, 28, 8900–8905 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00143e |
| This journal is © The Royal Society of Chemistry 2024 |