
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical exfoliation of 1-dimensional antiferromagnetic nanoribbons from a non-van der Waals material†
Mulan
Yang
 a,
Guangming
Cheng
b,
Nitish
Mathur
a,
Ratnadwip
Singha
a,
Fang
Yuan
a,
Nan
Yao
b and
Leslie M.
Schoop
*a
a,
Guangming
Cheng
b,
Nitish
Mathur
a,
Ratnadwip
Singha
a,
Fang
Yuan
a,
Nan
Yao
b and
Leslie M.
Schoop
*a
aDepartment of Chemistry, Princeton University, Princeton, NJ 08544, USA. E-mail: lschoop@princeton.edu
bPrinceton Materials Institute, Princeton, NJ 08544, USA
First published on 18th January 2024
Abstract
As the demand for increasingly varied types of 1-dimensional (1D) materials grows, there is a greater need for new methods to synthesize these types of materials in a simple and scalable way. Chemical exfoliation is commonly used to make 2-dimensional (2D) materials, often in a way that is both straightforward and suitable for making larger quantities, yet this method has thus far been underutilized for synthesizing 1D materials. In the few instances when chemical exfoliation has been used to make 1D materials, the starting compound has been a van der Waals material, thus excluding any structures without these weak bonds inherently present. We demonstrate here that ionically bonded crystals can also be chemically exfoliated to 1D structures by choosing KFeS2 as an example. Using chemical exfoliation, antiferromagnetic 1D nanoribbons can be yielded in a single step. The nanoribbons are crystalline and closely resemble the parent compound both in structure and in intrinsic antiferromagnetism. The facile chemical exfoliation of an ionically bonded crystal shown in this work opens up opportunities for the synthesis of both magnetic and non-magnetic 1D nanomaterials from a greater variety of starting structures.
New conceptsChemical exfoliation is a scalable and facile synthesis technique for making nanomaterials, as evidenced by the many 2-dimensional materials that have been made in this way. However, despite the utility of this method, chemical exfoliation has rarely been applied to the synthesis of 1-dimensional (1D) materials. A few studies have shown that nanowires can be exfoliated from quasi-1D van der Waals materials, but we seek to broaden the applicability of chemical exfoliation to many other 1D materials by removing this limitation. Using KFeS2 as a case study, we show that quasi-1D crystal structures containing ionically bonded chains can be exfoliated to make nanoribbons of the same structure. Using transmission electron microscopy, we also determined the mechanism by which the nanoribbons form. The resultant nanomaterials also maintain the intrinsic antiferromagnetism of the parent compound, confirming that liquid exfoliation does not eliminate magnetism from the system. We hope this example emboldens researchers to use chemical exfoliation when trying to synthesize all manner of 1D materials; a van der Waals bonded structure is no longer a prerequisite. |
Introduction
The development of new nanomaterials has given rise to a variety of possible applications and theoretical studies, generally due to their high surface area-to-volume ratio and possible changes in their properties due to quantum confinement effects.1,2 These materials are categorized by their dimensionality, i.e., 2-dimensional (2D) materials are nanoscale in one dimension, 1-dimensional (1D) materials in two, and 0-dimensional (0D) materials in all three.3 Materials such as nanowires and nanoribbons are categorized as 1D and can be used in sensing,4 catalysis,5 and filtration.6 Magnetic nanowires have further uses as nanobarcodes,7 detectors of biopolymers,8 spintronic devices,9,10 data storage,11 and as model materials for unconventional physics, such as magnetic frustration12 and curvature-induced changes in magnetic field textures.13Given this widespread interest, scalable methods that are applicable to a broad range of 1D structures are needed. Currently, one of the most common syntheses involves using metal nanoparticles, either in the liquid or solid phase, to catalyze the bottom-up growth of nanowires.14,15 Potential issues arise if a metal nanoparticle used as a catalyst is incorporated into the nanowire.16 Doping the materials as they are grown can also be difficult due to issues such as the different rates of incorporation of different elements.14 There are top-down approaches, including lithography, that can circumvent these challenges by first growing the material as a bulk crystal. However, this limits the materials to thermodynamically stable phases that often need to be lattice matched with the substrate to avoid defects or dislocations from strain.14 An alternate synthetic route is chemical exfoliation, which has the advantage of being tunable to a wide range of materials, high yielding, and structurally consistent in its products.1 In addition, this solution processing method can be used to make inks of nanomaterials with magnetic and electronic properties of interest for applications like thin-film transistors and flexible electronics.17–19
Even though chemical exfoliation methods have been developed for synthesizing a wide variety of 2D materials, these methods have yet to be commonly applied to 1D materials.20 In the rare instances where liquid exfoliation is used, the starting materials are vdW compounds and have clear cleavage planes at these weak bonds. The first use of liquid exfoliation to make nanowires (as opposed to the simple dispersal of bulk material into solution)21 was in 2018 when Tian et al. made KP15 nanowires by sonication in ethanol. Although the bulk structure of KP15 incorporates both vdW and ionic bonding between the chains, the authors indicated that the cleavage into nanowires occurred at the vdW gaps between the planes.22 The next usage of chemical exfoliation was by Liu et al. in 2020, who used a similar sonication method to make semiconducting nanowires from the vdW material Ta2Pd3Se8.23 Qu et al. extended this method in 2021 to the magnetic vdW material CrSbSe3.20 They were able to synthesize high-quality nanowires that notably also retained the magnetic properties of the bulk material.
The library of accessible 1D materials would expand greatly if we could chemically exfoliate 1D nanostructures from materials with bonds stronger than those in vdW compounds. Non-vdW compounds, including Bi2O2Se,24 Na3Ni2BiO6,25 NaCrS2,26 and FeF3,27 have previously been chemically exfoliated to 2D sheets and platelets. By utilizing a wet ball-milling technique, even the covalently-bonded crystal α-germanium could be delaminiated into nanolayers.28 Certain materials, such as α-Fe2O329 and FeTiO3,30 retain their magnetic ordering even after undergoing liquid exfoliation. In addition, the non-vdW crystal RbNbO3 has been chemically exfoliated to make HxRb1−xO3 “nanobelts,” which suggests that extending this technique to 1D is possible.31 Another study showed that Li2Mo6Se6, which is a quasi-1D compound, can be dispersed in anhydrous methanol to produce nanowires of MoSe, although the process is hampered by the air and moisture sensitivity of the crystals.32 Given this precedent, we investigated the possibility of applying similar techniques under ambient conditions to an ionically bonded, quasi-1D chain compound and chose to study KFeS2, which has chains made of edge-sharing FeS4 tetrahedra that are ionically bonded by K+ (Fig. 1). Previous work has shown that KFeS2 nanowires can be synthesized using a bottom-up method, which requires the nanowires to be anchored to an Fe foam.33 An alternative bottom-up synthesis in solution results in “whiskers” of KFeS2.34
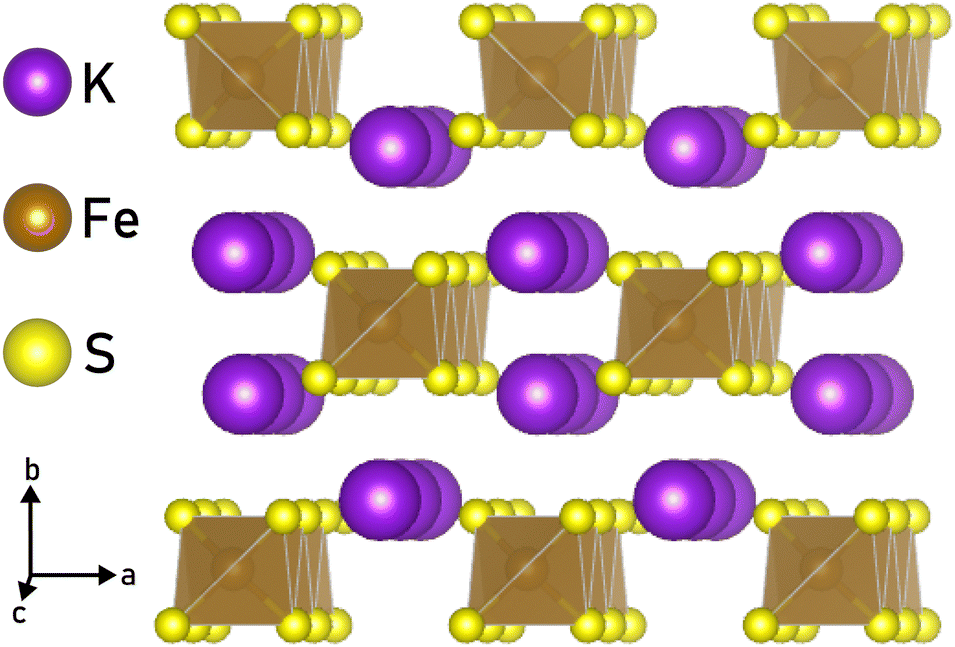 | ||
| Fig. 1 Structure of KFeS2. Notice the quasi-1D chain motif, with ionic bonds to an alkali ion. | ||
In addition to determining the feasibility of using liquid exfoliation to make these compounds, we also sought to determine whether the magnetic properties could be preserved. Magnetic studies on bulk KFeS2 show that it is antiferromagnetic (AFM) within the chains, with a Néel temperature (TN) of 250 ± 5 K; there are ferromagnetic (FM) interactions between the chains, but due to the significantly larger spacing between inter-chain Fe ions (6.6 Å) versus intra-chain Fe ions (2.7 Å), these interactions are less significant.35–39 Because the main magnetic interactions take place within the chain, the exfoliation process should not disrupt them greatly.
In this work, we implement a simple chemical exfoliation method to synthesize free-standing, crystalline KFeS2 nanoribbons and nanosheets. Note that we use “nanoribbon” here instead of “nanowire” to describe these 1D materials to clarify that their cross-section is flatter compared to most nanowires, following the terminology of Kong et al.40 However, unlike the nanowires and nanoribbons they grew, the crystal growth direction of these nanoribbons is the same as that of the previously referenced KFeS2 nanowires.33 The nanoribbons may be more easily related to the structures commonly formed from transition metal dichalcogenides as well.41 We show the variety of morphologies that can be formed and offer mechanistic reasoning for this behavior elucidated from electron diffraction imaging of individual samples. We also show that post-exfoliation, these materials retain their AFM character, albeit with a decrease in the Néel temperature. A magnetic hysteresis occurs in the nanostructures that is not present in the bulk material, potentially due to sulfur vacancies or other factors that cause uncompensated magnetic moments.42,43
Results and discussion
To investigate the mechanism by which ionically bonded crystals can be chemically exfoliated to 1D nanomaterials, we chose to study KFeS2 as a model. This material was chosen for its structure, as seen in Fig. 1, which is suitable for exfoliation to 1D as it consists of FeS4 chains that run along the c-axis with K+ ions in between to hold the chains together. This is reminiscent of materials in the AxMS2 family, where A = Li, Na, K and M = Mo, W, Nb, Ta, Ti, that have been chemically exfoliated to ultra-thin 2D nanosheets.44,45 These A ions tend to react readily with water, which can facilitate exfoliation through the intercalation of water molecules to separate the layers.31,46 In MAX phases, which are similarly layered and non-vdW, the M–A bond is known to be more reactive than the M–S bond, which allows the M–A bond to be broken preferentially during exfoliation.46,47 Assuming these bonds behave similarly in their 1D counterparts, we propose that during exfoliation the crystals will cleave K–S bonds before Fe–S bonds, leaving the chains intact. Therefore, the two possible cleavage planes are between the chains, along the ac or bc planes, which are equivalent to the (010) and (100) planes respectively. Because the K–S bonds required to break are nearly identical in length (3.34 Å and 3.31 Å, respectively), the crystals should cleave at both planes with perhaps a slight preference for cleaving at the ac plane.Nanomaterials were successfully synthesized by sonicating bulk KFeS2 (Fig. 2a) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 solution of isopropyl alcohol (IPA) and water for 90 min. Two rounds of centrifugation were performed to isolate the exfoliated materials from any remaining bulk material and the resultant solids were dried under vacuum (details under Materials and Methods). Approximately 0.5–1 mg of nanomaterials was synthesized from each exfoliation of approximately 35 mg of bulk KFeS2. Both bulk and exfoliated KFeS2 are slightly water sensitive and so the materials were stored under argon when not in use to prevent oxidation. Post-exfoliation, we consistently observed both nanoribbon and nanosheet morphologies. To determine their crystal structures and properties, we used several characterization methods including diffraction, microscopy, and magnetic measurements. The samples were sufficiently air-stable for characterization to be done under atmospheric conditions. To establish the applicability of similar exfoliation methods to other quasi-1D chain compounds, we performed preliminary experiments on bulk KSbS2 (Fig. S1, ESI†) and showed that nanostructures could be exfoliated from this material as well (Fig. S2, ESI†), although further optimization to obtain smaller particles is necessary in this case. The exfoliation method and possible cleavage mechanism are described in the ESI.†
:2 solution of isopropyl alcohol (IPA) and water for 90 min. Two rounds of centrifugation were performed to isolate the exfoliated materials from any remaining bulk material and the resultant solids were dried under vacuum (details under Materials and Methods). Approximately 0.5–1 mg of nanomaterials was synthesized from each exfoliation of approximately 35 mg of bulk KFeS2. Both bulk and exfoliated KFeS2 are slightly water sensitive and so the materials were stored under argon when not in use to prevent oxidation. Post-exfoliation, we consistently observed both nanoribbon and nanosheet morphologies. To determine their crystal structures and properties, we used several characterization methods including diffraction, microscopy, and magnetic measurements. The samples were sufficiently air-stable for characterization to be done under atmospheric conditions. To establish the applicability of similar exfoliation methods to other quasi-1D chain compounds, we performed preliminary experiments on bulk KSbS2 (Fig. S1, ESI†) and showed that nanostructures could be exfoliated from this material as well (Fig. S2, ESI†), although further optimization to obtain smaller particles is necessary in this case. The exfoliation method and possible cleavage mechanism are described in the ESI.†
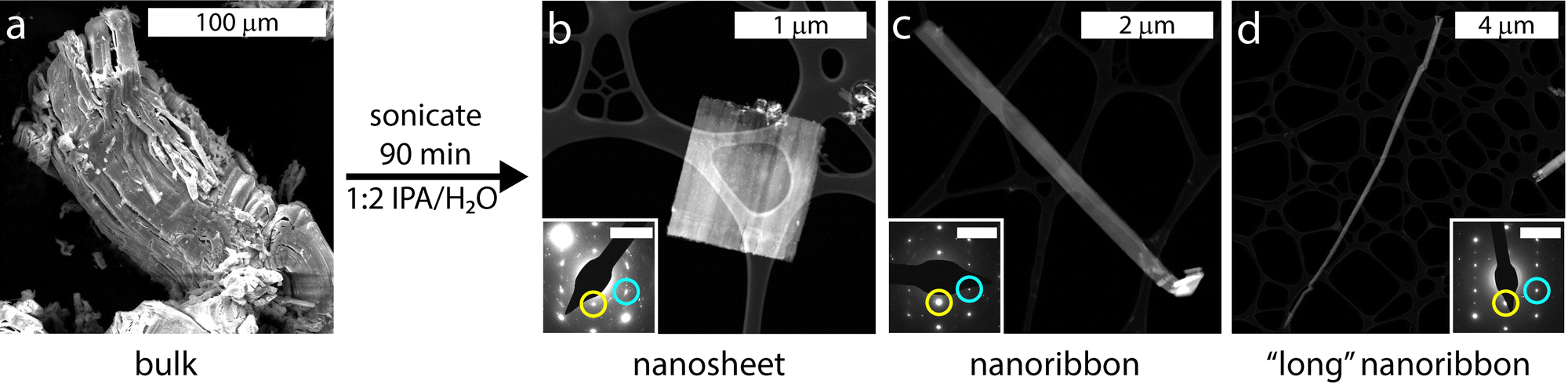 | ||
| Fig. 2 When (a) bulk KFeS2 is sonicated in 1:2 IPA/H2O for 90 min, it exfoliates into a mixture of (b) nanosheets and (c) nanoribbons, as well as occasional (d) particularly long nanoribbons. Although the morphologies are different, they all share the same crystal structure, as confirmed by the (b)–(d) insets) selected area electron diffraction patterns indexed to the [010] zone axis. The (002) diffraction spot is circled in cyan and the (200) is circled in yellow. The scale bar represents 5 nm−1. Note that the patterns have been rotated to the same direction for ease of comparison. | ||
The rest of this work focuses solely on the results from exfoliating KFeS2. An ensemble of nanomaterials was collected by centrifuging the exfoliation mixture and removing the supernatant. Powder X-ray diffraction (PXRD) data (Fig. 3) on such an ensemble confirmed that the exfoliation process did not affect the structure. Rietveld refinement of the PXRD shows that it matches well with the parent structure (Rwp = 5.9%). Although there is no clear change in peak shape, which can be an indication of the nanoscale nature of a material, high-angle annular dark-field scanning transmission electron microscope (HAADF-STEM) images of the PXRD sample (Fig. S3a, ESI†) showed successful exfoliation.
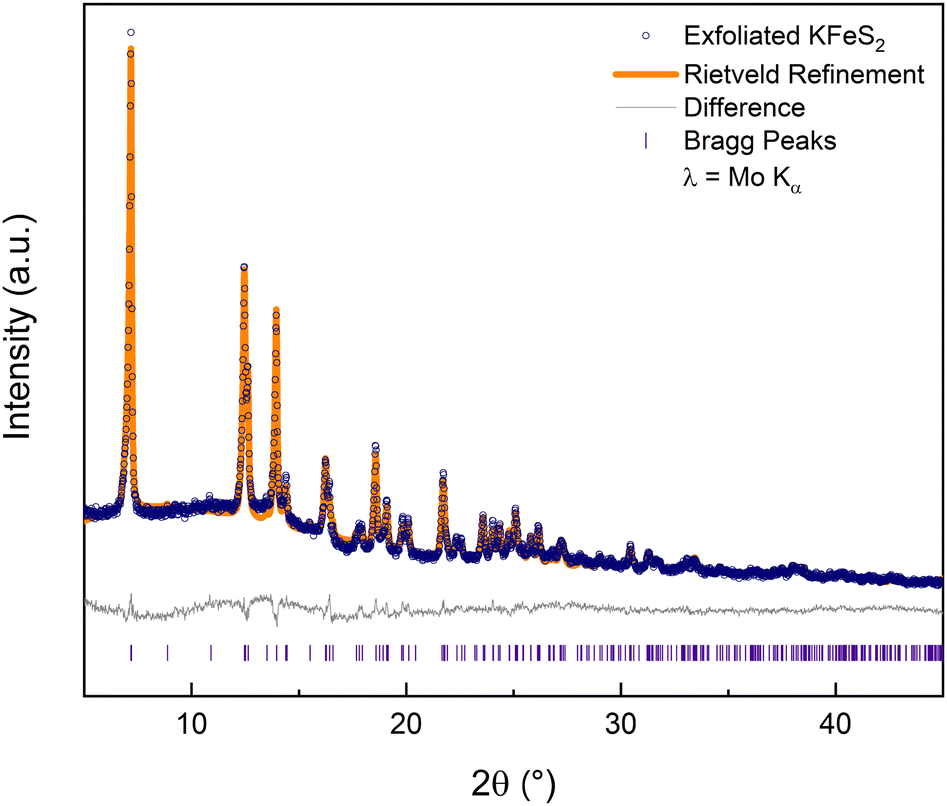 | ||
| Fig. 3 PXRD data of the exfoliated KFeS2 agrees well with the Rietveld refinement data. | ||
Transmission electron microscopy (TEM) and HAADF-STEM were also used to visualize the different morphologies of the KFeS2 nanomaterials. A statistical analysis was performed on 237 individual samples to determine the distribution of nanostructures (Fig. S4, ESI†). These categories were defined by their aspect ratios: greater than 5 (1D nanoribbons) and less than 5 (2D nanosheets) (Fig. 2b and c). The aspect ratios ranged from approximately 1 to 88, so the nanoribbon group includes extra “long” nanoribbons (Fig. 2d). Over half (59%) of the exfoliated materials were nanoribbons and the rest were nanosheets. Energy-dispersive X-ray spectroscopy (EDX) shows a near 1:1:2 stoichiometry of K:Fe:S regardless of the morphology (Fig. S5, ESI†) and no impurities were found.
Selected area electron diffraction (SAED) images (Fig. (2b–d insets) confirm the structure to be that of the parent compound; the measured d-spacings are within 3% of the known structure (Table S3, ESI†) and the pattern matches SAED images from KFeS2 nanowires grown by Li et al.33 SAEDs were taken on samples of each morphology to confirm that they all share the same structure. High resolution (HR)-TEM further corroborates this result, as shown in Fig. 4a. The pattern of atoms matches that of KFeS2 as seen from the b-axis (Fig. 4b inset) and the fast Fourier transform (FFT) (Fig. 4c) indexes to the same peaks as the SAED. The measured atomic spacings are also within 3% error of the known values.
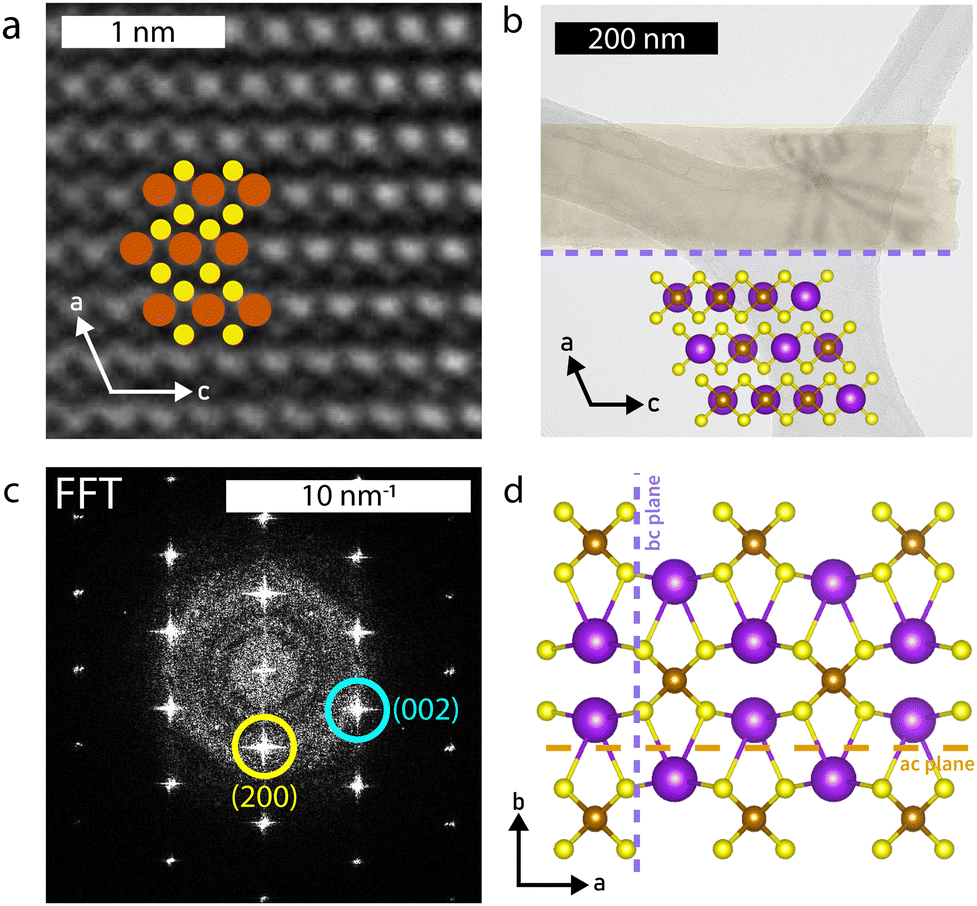 | ||
| Fig. 4 (a) HR-TEM image showing arrangement of atoms within the nanoribbon. (b) TEM image of a nanoribbon, showing cleavage along both ac and bc planes, as well as the atomic structure viewed along the b-axis. (c) The FFT of (a) indexed along the [010] zone axis. (d) Cleavage planes in KFeS2. Cleavage along the ac plane (orange, dashed line) exfoliates the bulk crystal to a 2D structure and cleavage along the bc plane (purple, dotted line) further reduces the crystal to a 1D structure. | ||
The SAED patterns additionally give insight into the mechanism of formation of different morphologies of KFeS2 (Fig. 4d). Most of the patterns show the ac plane of the material. This suggests that one of the easiest cleavage planes is the ac plane, which agrees with the prediction based on bond distances as discussed previously. By cleaving here, nanosheets of the material are generated. However, as mentioned above, it should be similarly easy to cleave along the bc plane. As suspected, cleavage occurs at the bc plane in addition to the ac plane, resulting in nanoribbons. In a few SAEDs (Fig. S3c, ESI†), the bc plane is visible, which supports this being the other cleavage plane. By measuring the distance between diffraction spots, we see that the Fe–Fe distance within the chain is within 3% of that of the parent structure. However, the inter-chain Fe–Fe distance has decreased significantly (Table S4, ESI†), which may be due to a loss of K+ (∼6%–20%) during the exfoliation process that causes the chains to collapse towards each other. Although this structural change is not seen in the XRD data, the SAED confirms its occurrence in this particular sample, as the SAED pattern shown in Fig. S3c (ESI†) was taken directly from the sample used for XRD. The effect may be too minimal to be seen on XRD, but detectable by more sensitive techniques, as seen in the magnetic data.
Atomic force microscopy was performed on individual nanoribbons to determine the average thickness of these samples (Fig. 5 and Fig. S6, ESI†). The samples ranged from 20–30 nm in height and were fairly uniform along the entire sample length.
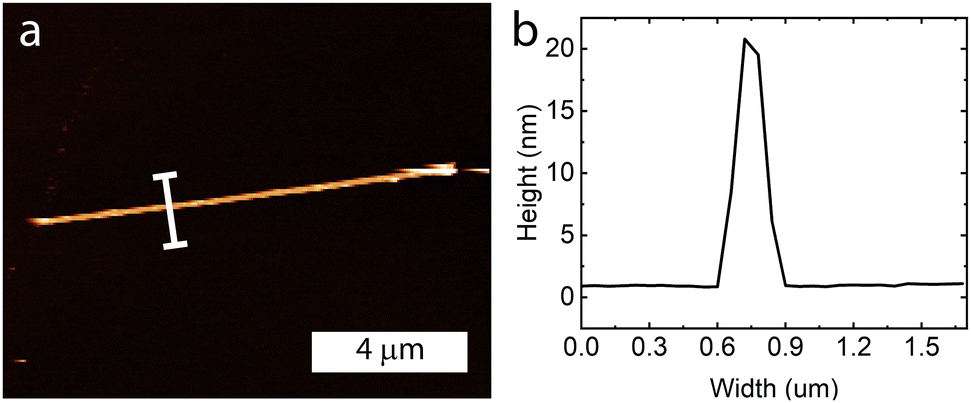 | ||
| Fig. 5 (a) Atomic force microscopy image of a nanoribbon, with (b) the height profile showing it is approximately 20 nm in height. | ||
To investigate if the magnetic properties of KFeS2 were retained post-exfoliation, magnetic susceptibility measurements were performed at 500 Oe on bulk (Fig. 6a) and an ensemble of exfoliated KFeS2 (Fig. 6b). The bulk data is comparable to literature results from powder samples; there is no obvious transition at TN, but there is a broad, higher temperature (T = 353 K) transition that was also previously observed and had been attributed to short range correlations occurring above TN.37,38 Literature measurements agree that this broad peak is present, but show it occurring at a range of temperatures: 565 K in a single crystal sample38 and closer to 700 K in a powder sample.35 The susceptibility of the ensemble of exfoliated nanostructures shows a similar broad peak, which could be attributed to either of two causes. This peak may be due to the same short range interactions causing the broad peak seen in the bulk data, but the AFM transition in quasi-1D compounds is also known to be broader than ones seen in 3D compounds.48–50 In either case, the dramatic decrease in the temperature at which this peak occurs indicates that there is a decrease in the TN. Sulfur vacancies may be limiting the amount of AFM interactions between the Fe ions by disrupting the superexchange mechanism, thus reducing the AFM character.42 Any decrease in distance between the chains could also encourage more inter-chain FM interactions to occur, which may disrupt the intra-chain AFM interactions as well. The bifurcation of the field-cooled (FC) data may be due to superparamagnetism of sparse iron impurities.51,52
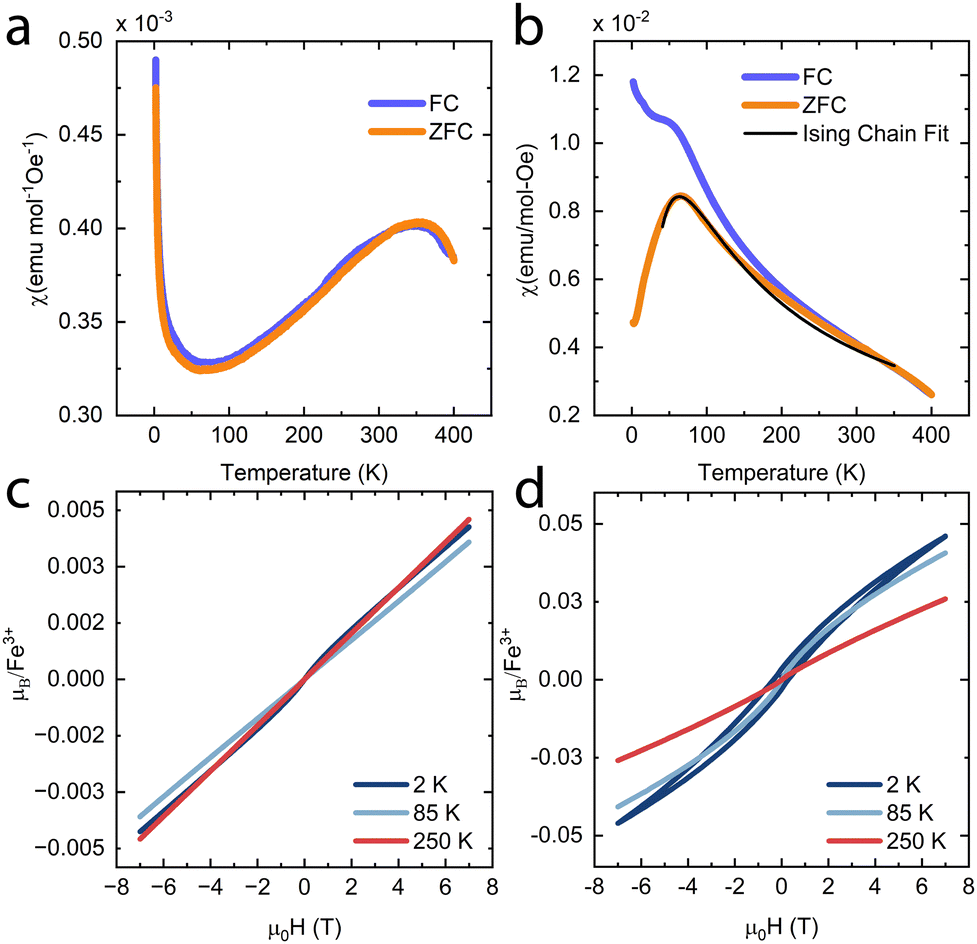 | ||
| Fig. 6 Magnetic susceptibility data at 500 Oe of (a) bulk and (b) exfoliated KFeS2. The exfoliated KFeS2 zero-field-cooled data (ZFC) has been fit as an Ising chain, shown in black. Field-dependent magnetization data of (c) bulk and (d) exfoliated KFeS2 is also shown; note the hysteresis at T = 2 K that occurs only in the exfoliated sample. Additional data shown was collected at possible temperatures of interest: T = 85 K (broad peak) and TN = 250 K. | ||
The magnetic susceptibility data of quasi-1D AFM chain compounds is usually fit using either the Heisenberg model (in the form of the Bonner–Fisher equation)48 or the Ising model. We also tried to fit the data to the Curie–Weiss law (Fig. S7, ESI†), but the high-temperature data is non-linear; one cause of this may be a small peak at T = 350 K from residual bulk material. The best fit for the zero-field-cooled (ZFC) data of the exfoliated materials was to eqn (1) for a spin-5/2 Ising model. This equation was adapted from Keene et al., who found it to model the behavior of Fe(II) more accurately than does the Bonner–Fisher equation because it better accounted for iron-specific qualities such as strong spin–orbit coupling.53 This choice is also supported by Sweeney and Coffman, who showed that KFeS2 appeared to fit better to an Ising model than to the Heisenberg model.54
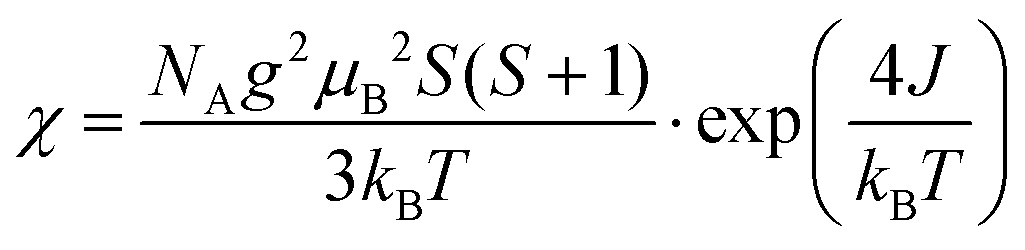 | (1) |
In eqn (1), NA is Avogadro's number, S is the spin state, μB is the Bohr magneton, and kB is the Boltzmann constant. We solve for g, the g-factor of the magnetic ion, and J, the intra-chain exchange constant. It was determined that J/kB = −16 K and g = 1.2 in the nanoribbon/nanosheet ensemble. The published g-factor of bulk KFeS2 is 2.0,55 so this value is significantly lower than expected. There are multiple possibilities for why this value differs so greatly from the previously measured value: Fe being difficult to fit, as Keene et al. had stated,53 and possible variation in the Fe oxidation state due to slight loss of K or S during the exfoliation process, which would differ from the model which assumes a spin-5/2 state. Although this calculated g-factor is low, we will continue with this model as it gave the best fit and discuss the resultant values in a general sense.
Previous studies of single crystal KFeS2 have used a spin-1/2 Heisenberg model to calculate J/kB to be −441 K,36,38 but a spin-5/2 Heisenberg model on a powder sample resulted in a very different J/kB of −66 K.35 Overall, the absolute value is always higher for the bulk than what is reported here for the nanostructures, indicating a greater intra-chain Fe–Fe exchange interaction in the bulk than in the exfoliated material. This agrees with the lower AFM character of the nanomaterials. Using the Green function method from Oguchi,56 we estimate that the ratio of inter- to intra-chain exchange is slightly less than 1. This value is higher than that reported for the bulk, which is on the order of 0.1,35,36 perhaps due to the increase in inter-chain interactions from the chains getting closer together.
Field-dependent magnetization measurements on bulk KFeS2 (Fig. 6c) show AFM ordering with a minor FM impurity, most likely some form of Fe such as KFeO2.39 When measuring the nanostructure ensemble (Fig. 6d), however, a small hysteresis opens up, which has multiple possible causes. Because the chains interact FM with each other, it is possible that the weakened AFM interactions allow the FM interactions between the chains to become more prominent. Although not seen in the overall PXRD of the exfoliated samples, the chains may also be closer to each other in some nanostructures, as seen in the occasional SAED of a nanoribbon, which would also increase the FM interactions. Alternatively, the hysteresis may be caused by uncompensated magnetic moments which taken together appear FM. These may result from sulfur vacancies weakening the AFM interactions.42 Finally, another possible explanation is the development of spin-glass-like behavior. In the closely related material KFeO2, competition between the major AFM state and a small FM component can lead to spin-glass behavior presenting as hysteresis in the magnetization measurements. This behavior may be the result of geometric frustration or uncompensated spins on the surface of the material.43 The development from an AFM state to a spin-glass-like one may be exacerbated by the exfoliation to nanoscale dimensions, as this transition has been observed in nanostructures such as LaFeO3 nanoparticles57 and hollow γ-Fe2O3 nanoparticles.58 Overall, however, the AFM behavior is retained. The small measured magnetic moments also support this. The higher moment of the nanostructures may be attributed to a greater Fe impurity, but the magnetic moment is still significantly lower than what would be expected in a FM material. In general, the analysis of the magnetic properties shows that, although differing from the bulk, magnetic order is retained in the exfoliated nanostructures.
Conclusions
We have shown here that in the synthesis of 1D nanostructures, liquid exfoliation is not limited solely to layered vdW materials, but can instead be extended to synthesize 1D materials from a variety of chain-containing crystals, including those that are magnetic and ionically bonded. The quasi-1D compound KFeS2 was exfoliated into nanoribbons averaging 25 nm in height. The exfoliation was facile and resulted in a majority of nanoribbons, thus making them simple to mass-produce. Magnetic measurements showed that the exfoliated materials retain AFM behavior with the TN lower compared to that of the bulk. A small magnetic hysteresis opens up in the exfoliated material, which may be due to sulfur vacancies slightly disrupting the AFM state and causing some magnetic moments to become uncompensated. Having shown the efficacy of this technique for making KFeS2 nanoribbons, as well as preliminary work on making KSbS2 nanostructures, we encourage other researchers to employ this method in the synthesis and study of 1D nanostructures from any chain-containing, quasi-1D compounds.Materials and methods
Synthesizing KFeS2
Bulk KFeS2 was synthesized using a 1:0.8:2 molar ratio of potassium (cubes in mineral oil, Oakwood Chemical), iron (≥ 99%, reduced, powder (fine), Sigma Aldrich), and sulfur (powder, sublimed, −100 mesh, 99.5%, Alfa Aesar). The reagents were loaded into an alumina crucible under an argon atmosphere and sealed inside a quartz tube under vacuum. This was heated to 700 °C at 90 °C h−1 and held at this temperature for 5 days. It was then cooled at 25 °C h−1 to 300 °C before cooling to room temperature.
Exfoliating KFeS2
Nanoribbons and nanosheets were exfoliated from bulk KFeS2 in a 1:2 IPA:H2O solution, similar to the method used by Qu et al.20 The powder was added at 1.2 mg mL−1 and the mixture was then sonicated for 90 min on low power using a sonicator (Branson CPX1800). To minimize possible oxidation of KFeS2 in water, the mixture was then centrifuged (Sigma 3-30KS) at 14000 rpm for 45 min to collect all of the material at the bottom of the tube. Most of the solvent was removed and was replaced with 21 mL of IPA. The nanostructures were redispersed by sonicating for 5 min. To remove any remaining bulk KFeS2, the mixture was centrifuged at 2000 rpm for 10 min and the supernatant from this step was centrifuged at 3000 rpm for a further 10 min. Finally, to collect the exfoliated materials, the liquid was centrifuged at 13000 rpm for 30 min. The nanostructures were dried under vacuum to ensure no solvent remained.
Characterization
Structures were visualized using the software VESTA.59 For PXRD, the dried nanomaterials were collected into a 0.5 mm capillary. The powder diffractometer (STOE STADI P) used Mo Kα 1 radiation and a Dectris Mythen 2R 1K detector. Rietveld refinements were performed using the TOPAS software. SEM images were collected using a Quanta 200 FEG Environmental-SEM in high-vacuum mode. TEM, SAED, EDX, HAADF-STEM, and HR-TEM images were collected on a Talos F200X Scanning/Transmission Electron Microscope with a SuperX EDX detector. Electron diffraction images were simulated using the software CrystalMaker, which also showed the theoretical distances between each diffraction spot. The distances between diffraction spots were measured using the software ImageJ. For atomic force microscopy, samples were drop-cast on silicon wafers and allowed to dry in air. The measurements were performed using a Bruker Dimension Icon3 Atomic Force Microscope in tapping mode and the data was processed using the Gwyddion software. Magnetic measurements were collected in a straw holder on a Quantum Design MPMS 3 using SQUID-VSM.Author contributions
M. Y. developed the methodology, collected and analyzed the data, and wrote the original and final draft. G. C. and R. S. helped collect and interpret data. N. M. helped validate and interpret the results. F. Y. and L. M. S. conceptualized the idea. N. Y. and L. M. S. supervised the project. All authors reviewed and edited the draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the DOD's office of naval research (ONR) (award number N00014-21-1-2733), the Princeton Center for Complex Materials, a National Science Foundation (NSF)-MRSEC program (DMR-2011750), the Princeton Catalysis Initiative (PCI), and the Gordon and Betty Moore Foundation’S EPIQS initiative (grant number GBMF9064). The authors acknowledge the use of the Imaging and Analysis Center (IAC) operated by the Princeton Materials Institute at Princeton University, which is supported in part by the Princeton Center for Complex Materials (PCCM), a National Science Foundation (NSF) Materials Research Science and Engineering Center (MRSEC; DMR-2011750).References
- J. Bae, M. Kim, H. Kang, T. Kim, H. Choi, B. Kim, H. W. Do and W. Shim, Adv. Mater., 2021, 33, 2006043 CrossRef CAS PubMed.
- A. V. Kolobov and J. Tominaga, Two-Dimensional Transition-Metal Dichalcogenides, Springer, Cham, 2016 Search PubMed.
- B. Mekuye and B. Abera, Nano Select, 2023, 4, 486–501 CrossRef CAS.
- T. Yasui, T. Yanagida, S. Ito, Y. Konakade, D. Takeshita, T. Naganawa, K. Nagashima, T. Shimada, N. Kaji, Y. Nakamura, I. A. Thiodorus, Y. He, S. Rahong, M. Kanai, H. Yukawa, T. Ochiya, T. Kawai and Y. Baba, Sci. Adv., 2017, 3, e1701133 CrossRef PubMed.
- T. Wasiak and D. Janas, J. Alloys Compd., 2022, 892, 162158 CrossRef CAS.
- E. Horváth, J. Gabathuler, G. Bourdiec, E. Vidal-Revel, M. Benthem Muñiz, M. Gaal, D. Grandjean, F. Breider, L. Rossi, A. Sienkiewicz and L. Forró, npj Clean Water, 2022, 5, 1–11 CrossRef.
- M. R. Zamani Kouhpanji and B. Stadler, Part. Part. Syst. Charact., 2021, 38, 2000227 CrossRef.
- M. R. Zamani Kouhpanji and B. J. H. Stadler, AIP Adv., 2020, 10, 125231 CrossRef CAS.
- A. Mourachkine, O. V. Yazyev, C. Ducati and J.-P. Ansermet, Nano Lett., 2008, 8, 3683–3687 CrossRef CAS PubMed.
- S. Aryal, D. Paudyal and R. Pati, Nano Lett., 2021, 21, 1856–1862 CrossRef CAS PubMed.
- G. Hrkac, J. Dean and D. A. Allwood, Philos. Trans. R. Soc., A, 2011, 369, 3214–3228 CrossRef CAS PubMed.
- A. May, M. Hunt, A. Van Den Berg, A. Hejazi and S. Ladak, Commun. Phys., 2019, 2, 1–9 CrossRef.
- C. Donnelly, A. Hierro-Rodríguez, C. Abert, K. Witte, L. Skoric, D. Sanz-Hernández, S. Finizio, F. Meng, S. McVitie, J. Raabe, D. Suess, R. Cowburn and A. Fernández-Pacheco, Nat. Nanotechnol., 2022, 17, 136–142 CrossRef CAS PubMed.
- P. C. McIntyre and A. Fontcuberta i Morral, Mater. Today Nano, 2020, 9, 100058 CrossRef.
- N. P. Dasgupta, J. Sun, C. Liu, S. Brittman, S. C. Andrews, J. Lim, H. Gao, R. Yan and P. Yang, Adv. Mater., 2014, 26, 2137–2184 CrossRef CAS PubMed.
- J. E. Allen, E. R. Hemesath, D. E. Perea, J. L. Lensch-Falk, Z. Y. Li, F. Yin, M. H. Gass, P. Wang, A. L. Bleloch, R. E. Palmer and L. J. Lauhon, Nat. Nanotechnol., 2008, 3, 168–173 CrossRef CAS PubMed.
- F. Torrisi and J. N. Coleman, Nat. Nanotechnol., 2014, 9, 738–739 CrossRef CAS PubMed.
- A. G. Kelly, T. Hallam, C. Backes, A. Harvey, A. S. Esmaeily, I. Godwin, J. Coelho, V. Nicolosi, J. Lauth, A. Kulkarni, S. Kinge, L. D. A. Siebbeles, G. S. Duesberg and J. N. Coleman, Science, 2017, 356, 69–73 CrossRef CAS PubMed.
- X. Song, R. Singha, G. Cheng, Y.-W. Yeh, F. Kamm, J. F. Khoury, B. L. Hoff, J. W. Stiles, F. Pielnhofer, P. E. Batson, N. Yao and L. M. Schoop, Sci. Adv., 2023, 9, eadd6167 CrossRef CAS PubMed.
- Y. Qu, M. Q. Arguilla, Q. Zhang, X. He and M. Dincă, J. Am. Chem. Soc., 2021, 143, 19551–19558 CrossRef CAS PubMed.
- P. Topolovsek, C. Gadermaier, D. Vengust, M. Strojnik, J. Strle and D. Mihailovic, Nano Lett., 2015, 15, 813–818 CrossRef CAS PubMed.
- N. Tian, D. Liu, B. Zhou, Y. Yang, G. Zhang, D. Zhang and Y. Zhang, Mater. Lett., 2018, 228, 89–91 CrossRef CAS.
- X. Liu, S. Liu, L. Y. Antipina, Y. Zhu, J. Ning, J. Liu, C. Yue, A. Joshy, Y. Zhu, J. Sun, A. M. Sanchez, P. B. Sorokin, Z. Mao, Q. Xiong and J. Wei, Nano Res., 2020, 13, 1627–1635 CrossRef CAS.
- W. Chen, R. Zhang, Y. Sun, J. Wang, Y. Fan and B. Liu, Adv. Powder Mater., 2023, 2, 100080 CrossRef.
- B. L. Hoff, G. Cheng, G. Villalpando, F. Yuan, N. Yao and L. M. Schoop, J. Phys. Mater., 2022, 5, 044004 CrossRef.
- X. Song, G. Cheng, D. Weber, F. Pielnhofer, S. Lei, S. Klemenz, Y.-W. Yeh, K. A. Filsinger, C. B. Arnold, N. Yao and L. M. Schoop, J. Am. Chem. Soc., 2019, 141, 15634–15640 CrossRef CAS PubMed.
- T. Chen, H. Kaur, M. McCrystall, R. Tian, A. Roy, R. Smith, D. V. Horvath, J. Maughan, B. Konkena, M. Venkatesan, K. Synnatschke, T. Carey, J. Liu, J. Pepper, R. Zhang, C. Backes, V. Nicolosi, H. Xia and J. N. Coleman, FlatChem, 2022, 33, 100360 CrossRef CAS.
- C. Gibaja, D. Rodríguez-San-Miguel, W. S. Paz, I. Torres, E. Salagre, P. Segovia, E. G. Michel, M. Assebban, P. Ares, D. Hernández-Maldonado, Q. Ramasse, G. Abellán, J. Gómez-Herrero, M. Varela, J. J. Palacios and F. Zamora, Adv. Mater., 2021, 33, 2006826 CrossRef CAS PubMed.
- A. Puthirath Balan, S. Radhakrishnan, C. F. Woellner, S. K. Sinha, L. Deng, C. D. L. Reyes, B. M. Rao, M. Paulose, R. Neupane, A. Apte, V. Kochat, R. Vajtai, A. R. Harutyunyan, C.-W. Chu, G. Costin, D. S. Galvao, A. A. Martí, P. A. van Aken, O. K. Varghese, C. S. Tiwary, A. M. M. Ramaswamy Iyer and P. M. Ajayan, Nat. Nanotechnol., 2018, 13, 602–609 CrossRef CAS PubMed.
- A. P. Balan, A. B. Puthirath, S. Roy, G. Costin, E. F. Oliveira, M. A. S. R. Saadi, V. Sreepal, R. Friedrich, P. Serles, A. Biswas, S. A. Iyengar, N. Chakingal, S. Bhattacharyya, S. K. Saju, S. C. Pardo, L. M. Sassi, T. Filleter, A. Krasheninnikov, D. S. Galvao, R. Vajtai, R. R. Nair and P. M. Ajayan, Mater. Today, 2022, 58, 164–200 CrossRef CAS.
- F. Cheng, X. Jiang, Z. Zhang, R. Ma, T. Sasaki, F. Pan and X. Jing, Chem. Commun., 2019, 55, 2417–2420 RSC.
- L. Venkataraman, Y. S. Hong and P. Kim, Phys. Rev. Lett., 2006, 96, 076601 CrossRef PubMed.
- L. Li, T. Gao, Y. Ge, Q. Zhang, J. Wang, Z. Ma, W. Guo, S. Yu and Y. Fan, J. Mater. Chem. A, 2021, 9, 27727–27735 RSC.
- D. Liang, Y. Chen, S. Zhu, Y. Gao, T. Sun, K. Ri and X. Xie, Sustainable Environ. Res., 2021, 31, 25 CrossRef CAS.
- Z. Tomkowicz, A. Szytula and H. Bak-ptasiewicz, Phys. Status Solidi A, 1980, 57, K25–K30 CrossRef CAS.
- S. K. Tiwary and S. Vasudevan, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 7812–7814 CrossRef CAS.
- W. Bronger, A. Kyas and P. Müller, J. Solid State Chem., 1987, 70, 262–270 CrossRef CAS.
- S. K. Tiwary and S. Vasudevan, Solid State Commun., 1997, 101, 449–452 CrossRef CAS.
- D. C. Johnston, S. C. Mraw and A. J. Jacobson, Solid State Commun., 1982, 44, 255–258 CrossRef CAS.
- D. Kong, J. C. Randel, H. Peng, J. J. Cha, S. Meister, K. Lai, Y. Chen, Z.-X. Shen, H. C. Manoharan and Y. Cui, Nano Lett., 2010, 10, 329–333 CrossRef CAS PubMed.
- T. Chowdhury, E. C. Sadler and T. J. Kempa, Chem. Rev., 2020, 120, 12563–12591 CrossRef CAS PubMed.
- F. Wang, N. Mathur, A. N. Janes, H. Sheng, P. He, X. Zheng, P. Yu, A. J. DeRuiter, J. R. Schmidt, J. He and S. Jin, Sci. Adv., 2021, 7, eabj4086 CrossRef CAS PubMed.
- Q. Li, X. Qian, C. Cheng, B. Kang, S. Cao, J. Zhang and Z. Feng, Phys. B: Condens. Matter, 2021, 608, 412878 CrossRef CAS.
- C. Guo, H. Li, W. Zhao, J. Pan, T. Lin, J. Xu, M. Chen and F. Huang, J. Mater. Chem. C, 2017, 5, 5977–5983 RSC.
- A. Anto Jeffery, C. Nethravathi and M. Rajamathi, J. Phys. Chem. C, 2014, 118, 1386–1396 CrossRef CAS.
- J. Peng, Z.-j Chen, B. Ding and H.-M. Cheng, Research, 2023, 6, 0040 CrossRef CAS PubMed.
- E. Lee and D.-J. Kim, J. Electrochem. Soc., 2019, 167, 037515 CrossRef.
- J. C. Bonner and M. E. Fisher, Phys. Rev., 1964, 135, A640–A658 CrossRef.
- S. Eggert, I. Affleck and M. Takahashi, Phys. Rev. Lett., 1994, 73, 332–335 CrossRef CAS PubMed.
- S. J. Sebastian, K. Somesh, M. Nandi, N. Ahmed, P. Bag, M. Baenitz, B. Koo, J. Sichelschmidt, A. A. Tsirlin, Y. Furukawa and R. Nath, Phys. Rev. B, 2021, 103, 064413 CrossRef CAS.
- N. Paunović, Z. V. Popović and Z. D. Dohčević-Mitrović, J. Phys.: Condens. Matter, 2012, 24, 456001 CrossRef PubMed.
- A. Ghosh, S. Paul and S. Raj, Solid State Commun., 2015, 208, 1–6 CrossRef CAS.
- T. D. Keene, I. Zimmermann, A. Neels, O. Sereda, J. Hauser, M. Bonin, M. B. Hursthouse, D. J. Price and S. Decurtins, Dalton Trans., 2010, 39, 4937 RSC.
- W. V. Sweeney and R. E. Coffman, Biochim. Biophys. Acta, Gen. Subj., 1972, 286, 26–35 CrossRef CAS PubMed.
- R. S. De Biasi and C. A. Taft, J. Mater. Sci., 1978, 13, 2274–2275 CrossRef CAS.
- T. Oguchi, Phys. Rev., 1964, 133, A1098–A1099 CrossRef.
- D. Alshalawi, J. M. Alonso, A. R. Landa-Cánovas and P. de la Presa, Nanomaterials, 2023, 13, 1657 CrossRef CAS PubMed.
- H. Khurshid, P. Lampen-Kelley, Ò. Iglesias, J. Alonso, M.-H. Phan, C.-J. Sun, M.-L. Saboungi and H. Srikanth, Sci. Rep., 2015, 5, 15054 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00408b |
| This journal is © The Royal Society of Chemistry 2024 |