
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn effective descriptor for the screening of electrolyte additives toward the stabilization of Zn metal anodes†
Lin
Hong
 a,
Jingzhuo
Guan
a,
Yiwei
Tan
a,
Yao
Chen
a,
Yu-Si
Liu
b,
Wei
Huang
*a,
Chunyang
Yu
a,
Yongfeng
Zhou
a,
Jie-Sheng
Chen
a and
Kai-Xue
Wang
*a
a,
Jingzhuo
Guan
a,
Yiwei
Tan
a,
Yao
Chen
a,
Yu-Si
Liu
b,
Wei
Huang
*a,
Chunyang
Yu
a,
Yongfeng
Zhou
a,
Jie-Sheng
Chen
a and
Kai-Xue
Wang
*a
aSchool of Chemistry and Chemical Engineering, State Key Laboratory of Metal Matrix Composites, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China. E-mail: hw66@sjtu.edu.cn; k.wang@sjtu.edu.cn
bCollege of Smart Energy, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, P. R. China
First published on 2nd April 2024
Abstract
The introduction of additives into aqueous electrolyte has great potential in suppressing dendrite growth and improving the stability of Zn anodes. However, no relevant theory or descriptor could effectively support the screening and design of suitable additives. Herein, desolvation activation energy was proposed as a critical descriptor for the screening of electrolyte additives. The effectiveness of this descriptor was proved by anionic polysaccharide additives with a low desolvation barrier. Anionic polysaccharides could spontaneously form a protective layer on the Zn anode due to the good Zn affinity, promoting the desolvation behavior through hydrogen bonding and elevating the energy barrier of H2O dissociation. Consequently, the interfacial polarization was effectively decreased and the hydrogen evolution was inhibited, contributing to the enhanced stability of the Zn anode. As a proof of concept, pectin, one type of anionic polysaccharide was selected as the electrolyte additive for the systematic electrochemical performance studies. Significantly improved cycling stability with high reversibility, suppressed dendrite growth and low overpotential was achieved in the symmetric Zn cells.
Broader contextRechargeable aqueous Zn-ion batteries (AZIBs) are regarded as one of the most promising substitutes for LIBs owing to the distinctive merits of Zn metal anodes, including low cost, high safety, low redox potential (−0.762 V vs. SHE), and high volumetric capacity (5855 mA h cm−3). However, the poor reversibility and low cycling stability of rechargeable ZIBs caused by the uncontrolled dendrite growth, the obvious hydrogen evolution reaction (HER), and the serious Zn corrosion of the Zn metal anodes, hinder the practical application of AZIBs. The introduction of additives into aqueous electrolyte has great potential in stabilizing Zn anodes. However, no relevant theory or descriptor has yet been defined to guide the screening and design of suitable additives. In this work, we proposed desolvation activation energy as a critical descriptor for the screening of electrolyte additives. The effectiveness of this descriptor was proved using anionic polysaccharide additives with a low desolvation barrier. As a proof of concept, symmetric cells with a pectin additive achieved improved cycling stability, high reversibility, suppressed dendrite growth and low overpotential. |
1. Introduction
Rechargeable aqueous zinc-ion batteries (ZIBs) with the features of intrinsic safety, high abundance, high volumetric capacity (5855 mA h cm−3), and low redox potential (−0.762 V vs. SHE) of the Zn metal anode are considered as one of the most promising next-generation energy storage devices for large-scale energy storage applications.1–3 However, the poor reversibility and low cycling stability of rechargeable ZIBs associated with the uncontrolled dendrite growth, the obvious hydrogen evolution reaction (HER), and the serious Zn corrosion of the Zn metal anodes, hinder the practical application of these batteries.4–7 During the Zn deposition in mildly acidic electrolyte, the HER would inevitably occur on the Zn surface due to the low redox potential of Zn2+/Zn. The continuous HER would increase the pH value near the Zn anode, leading to the corrosion of the Zn metal and the formation of byproduct, Zn4(OH)6SO4·xH2O on the Zn surface.8–10 The irregular formation of Zn4(OH)6SO4·xH2O would increase the roughness of the Zn anode, thereby altering the distribution of the interfacial electric field and consequently triggering uncontrolled dendrite growth.Various strategies, such as the construction of artificial solid/electrolyte interphases,11–14 the design of zincophilic coatings,8,15–18 the modification of current collectors,19–21 the addition of electrolyte additives,5,22,23 and the regulation of oriented Zn growth,24–26 have been proposed to address the reversibility and stability issues of Zn anodes. Among these strategies, electrolyte additive regulation has proven to be a simple and efficient approach to stabilizing Zn electrodes. For instance, dimethyl sulfoxide (DMSO) as an electrolyte additive was added into aqueous ZnCl2 electrolyte.27 H2O molecules in the Zn2+ solvation sheath could be replaced due to the preferential solvation of DMSO with Zn2+ and meanwhile, a SEI layer composed of [Zn12(SO4)3Cl3(OH)15]·5H2O, ZnSO3, and ZnS was generated, consequently alleviating the H2O decomposition and suppressing the dendrite growth. 1,4-Dioxane (DX) as an electrolyte additive was also demonstrated to be able to modulate the solvation structure of Zn2+ in aqueous ZnSO4 electrolyte,28 promoting the preferential growth of the Zn (002) facets. In addition, other organic solvents, such as acetonitrile (AN),29 γ-butyrolactone (GBL),30N-methyl pyrrolidone (NMP),31N,N-dimethylformamide (DMF),32 and sulfolane (SL),33 were also reported to be capable of decreasing the number of solvating H2O molecules in the Zn2+ solvation shell. However, the symmetric cells with these organic solvents usually exhibit increased polarization voltages, which in turn would trigger the hydrogen evolution and side reactions. Currently, the intrinsic factors responsible for the high interfacial polarization caused by electrolyte additives still remain unclear. Therefore, a descriptor for the screening of effective electrolyte additives to lower the polarization and suppress the HER is highly desired.
In this work, we revealed that the high polarization of organic solvent additives is associated with the difficult desolvation behavior of Zn2+, and proposed the desolvation activation energy as a critical descriptor for the electrolyte additive screen. Anionic polysaccharides with a low desolvation energy barrier, including carrageenan, hyaluronic acid, and pectin,34–36 were thus screened as additives to reduce the interfacial polarization and suppress the hydrogen evolution. Due to the good Zn affinity, anionic polysaccharides would form an adsorption layer on the Zn anode surface and effectively guide the uniform Zn deposition. Moreover, the as-formed adsorption layer could promote the desolvation process of Zn2+ through the strong hydrogen bonding and elevate the energy barrier of H2O dissociation, leading to the lowered interfacial polarization and suppressed hydrogen evolution. Taking pectin, a typical anionic polysaccharide as a good example, high plating/stripping reversibility and remarkably high cycling stability were achieved for ZIBs. Moreover, a high reversible capacity of 175.1 mA h g−1 was still maintained for the Zn‖MnO2 full cells charged and discharged at 2C for over 500 cycles. The proposed descriptor based on the desolvation activation energy provides effective guidance for the screening of suitable additives for Zn-ion batteries.
2. Results and discussion
Fig. 1a compares the voltage profiles of symmetric cells with different organic solvents additives, including 1,4-dioxane (DX), N-methyl pyrrolidone (NMP), N,N-dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) at 1.0 mA cm−2 with 0.5 mA h cm−2. As expected, the symmetric cells with organic solvents exhibit much higher polarization overpotential than that with pure ZnSO4. Specifically, the overpotential reaches 67, 72, 76, and 93 mV for the symmetric cells with the additives of DMSO, DMF, NMP, and DX, respectively. This high polarization phenomenon is probably related to the interfacial kinetics of the Zn anodes. In aqueous electrolyte, the Zn2+ ion exists in a hydrated form of [Zn(H2O)6]2+.37,38 Due to the strong interaction between the Zn2+ core and surrounding H2O sheath, a large energy barrier is encountered in the removal of the H2O solvent sheath before Zn deposition. Thus, the desolvation process of the hydrated [Zn(H2O)6]2+ is the rate-determining step in the interfacial kinetics process, which could be evaluated by the activation energy (Ea).39 To explore the effect of organic solvent additives on the desolvation behavior of the hydrated Zn2+ ion, the Ea value was calculated based on electrochemical impedance spectroscopy (EIS) analysis with the Arrhenius equation eqn (1):40,41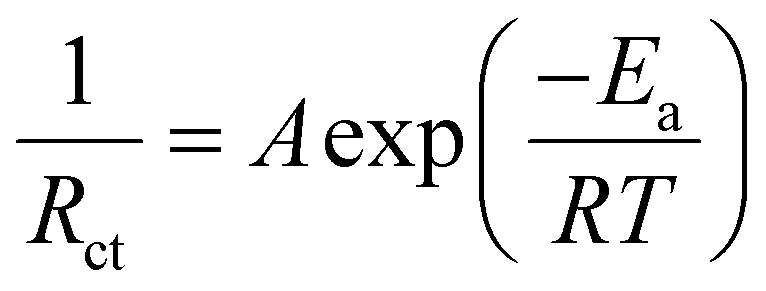 | (1) |
 | ||
| Fig. 1 Investigation of desolvation behavior and interfacial adsorption. (a) Voltage profiles of the Zn symmetric cells with different organic solvents. (b) Comparison of overpotentials and desolvation activation energies in the symmetric cells. (c) Fitting Arrhenius curves of the symmetric cells with different anionic polysaccharide additives obtained from the Nyquist plots at different temperatures. (d) ESP of the carrageenan, hyaluronic acid and pectin molecules. Adsorption configuration and charge density difference of H2O adsorbed on (e) and (f) Zn metal and (g) and (h) pectin. (i) Comparison of adsorption energies of H2O and different sites of pectin molecules on the (001) plane of Zn. (j) LUMO and HOMO energy levels with the corresponding isosurfaces of H2O and pectin molecules. | ||
To demonstrate the validity of this descriptor, anionic polysaccharides, a type of natural macromolecular material with abundant anion groups, including carrageenan, hyaluronic acid, and pectin were screened as the electrolyte additives. These polysaccharides show Ea values ranging from 17.5 to 20.2 kJ mol−1 (Fig. S3, ESI† and Fig. 1c), significantly lower than that of pure ZnSO4. Fig. 1d displays the electrostatic potential (ESP) distribution over the structural unit of carrageenan, hyaluronic acid, and pectin. The negative electrons primarily accumulate on the O atom of oxygen-containing groups, such as the –OH, –COOH and –COOCH3 in pectin, the –OSO3− and –OH in carrageenan, and the –OH, –COOH and –NHCOCH3 in hyaluronic acid. These electron-abundant groups serve as zincophilic sites and Zn2+ migration sites to homogenize the Zn2+ distribution over the surface of Zn and induce uniform Zn deposition. To demonstrate the feasibility of using anionic polysaccharides for the Zn stabilization, the electrochemical performances of symmetric cells with different additives were investigated. As depicted in Fig. S4 (ESI†), at 1.0 mA cm−2 with 0.5 mA h cm−2, the addition of carrageenan, hyaluronic acid, and pectin into 2.0 M ZnSO4 extends the lifespan of the Zn anode from 48 h to 2700 h, 2000 h, and 3700 h, respectively. More importantly, the symmetric cells with anionic polysaccharides exhibit significantly reduced overpotentials compared with those with organic solvents. Given the lowest Ea (17.5 kJ mol−1) and the longest lifespan of Zn anode achieved, pectin was selected as the representative for further systematic characterization and mechanism illustration.
To further reveal the desolvation mechanism of the hydrated Zn2+ ion with the pectin additive, DFT calculations were carried out to analyze the interaction between pectin and H2O molecules. It was found that pectin adsorbed on the Zn anode has a strong adsorption energy (−0.45 eV) towards H2O through hydrogen bonds between –COOCH3 and H2O (Fig. 1g). The formation of strong hydrogen bonds contributes to the effective separation of coordinated H2O from hydrated Zn2+, reducing the energy barrier for desolvation and decreasing the interfacial polarization. Consequently, the low interfacial polarization and the removal of H2O in advance could effectively suppress the hydrogen evolution. The delocalized electron in the charge density difference map also demonstrates the strong interaction between coordinated H2O and pectin (Fig. 1h). In contrast, the bare Zn surface exhibits a rather weak binding interaction (−0.37 eV) with H2O (Fig. 1e and f), resulting in a slow desolvation process and high polarization. In addition, the O–H bond strength of coordinated H2O is weaker than that of free H2O.42 Both the high voltage polarization and strength-decreased O–H bond would more easily trigger the hydrogen evolution and Zn corrosion. The released H2O during the desolvation process upon the Zn surface would also promote the HER.
Fig. 1i shows the optimized structures of different configurations of pectin adsorbed on the Zn(001) surface. Compared with H2O molecules, pectin displays larger binding energies towards the Zn surface, demonstrating the good Zn affinity of pectin. The preferential adsorption of pectin would allow the formation of a protective layer over the Zn surface, prohibiting the direct contact of Zn with H2O and thus mitigating the parasitic reactions. Among the different adsorption configurations, the parallel adsorption of pectin on the Zn surface via the –COOCH3 group exhibits the highest adsorption energy of −1.73 eV, indicating that pectin molecules prefer to adsorb on the Zn(001) facet in a parallel state (Fig. S5, ESI†). The charge density difference map also indicates the electron transfer from pectin to metallic Zn (Fig. S6, ESI†), further showing its strong chemisorption interactions. The interaction between Zn metal and pectin was also elucidated by the molecular orbital energy level. As shown in Fig. 1j, the highest occupied molecular orbital (HOMO) energy level of pectin is higher than that of H2O (−7.11 eV vs. −7.92 eV), demonstrating that pectin can more easily lose electrons when absorbed on the Zn surface. This result is in agreement with the charge density difference map. Therefore, the pectin molecules with abundant zincophilic sites would be spontaneously adsorbed on the Zn surface to form an artificial solid–electrolyte interphase (SEI) film, consequently regulating the homogeneous Zn deposition.
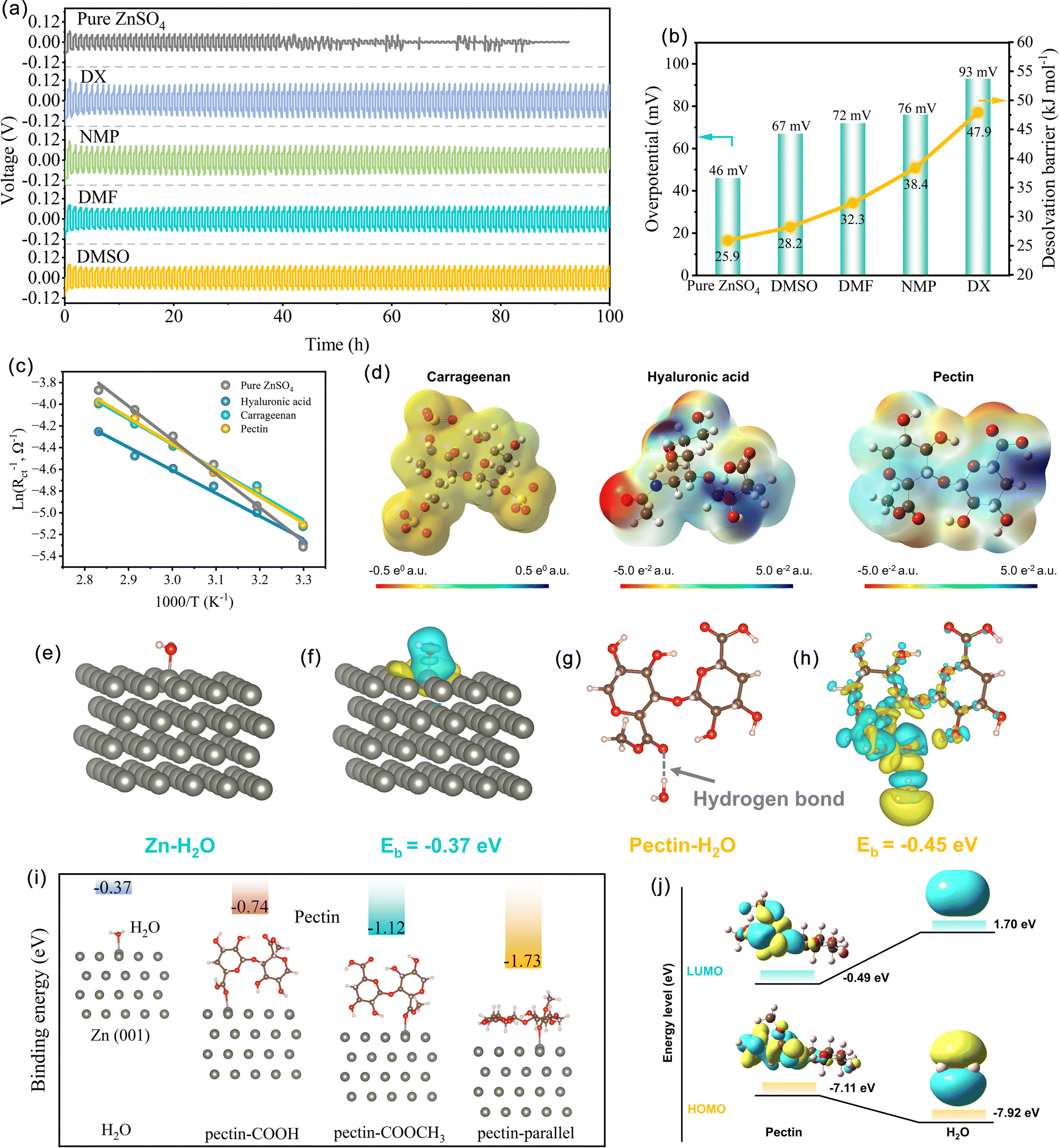
On the basis of the above analyses, the pectin layer adsorbed on the Zn surface is believed to have positive effects on Zn deposition. The surface morphology evolution of Zn anodes at various Zn loading capacities ranging from 0.5 to 5.0 mA h cm−2 was characterized using a confocal laser scanning microscope (CLSM) (Fig. 2a–d). Without pectin, the unevenly distributed Zn grains emerge at the initial plating stage (0.5 mA h cm−2), and continuous growth of these grains occurs with the loading capacity. Eventually, Zn protrusions with an altitude intercept over 49.8 μm generated at the capacity of 5.0 mA h cm−2. The formation of Zn protrusions is attributed to the uneven distribution of Zn2+ over the electrode surface due to the poor Zn affinity of bare Zn. Upon cycling, the continuous growth of protrusions would induce the formation of dendrites, which would then puncture the separator and consequently lead to the short-circuit failure of the batteries. Obviously, the introduction of zincophilic pectin could alleviate the issue of dendrite growth (Fig. 2c and d). Guided by the pectin layer covering on Zn, the Zn2+ ions prefer to interact with the zincophilic sites of pectin, inducing the formation of evenly distributed Zn nuclei at the initial nucleation stage. With the increase of loading capacity, these Zn nuclei would guide uniform Zn plating. Even at a high capacity of 5.0 mA h cm−2, the electrode shows a relatively flat and dense plating layer with an altitude intercept of less than 32.5 μm. COMSOL calculations were further conducted to simulate the order parameter distribution (ξ) and surface evolution on the Zn anode (Fig. 2e and f). The solid–liquid interface can be distinguished by different ξ values. With pure ZnSO4, the uneven nucleation sites are formed on the Zn surface and gradually grow into Zn dendrites as the plating time increases from 1 to 50 s. In contrast, with the addition of pectin, the uniform growth of Zn metal is achieved during the whole deposition process. These simulation results are consistent with CLSM observation.
 | ||
| Fig. 2 Zn nucleation and deposition process. (a) and (c) CLSM optical images and (b) and (d) corresponding height images of Zn deposition with different capacities from 0.5 to 5.0 mA h cm−2 in the pure ZnSO4 and the electrolyte with pectin additive. COMSOL simulation of the morphology evolution on Zn anode using (e) pure ZnSO4 and (f) ZnSO4–pectin after Zn electrodeposition for 1, 10, 30 and 50 s, respectively. | ||
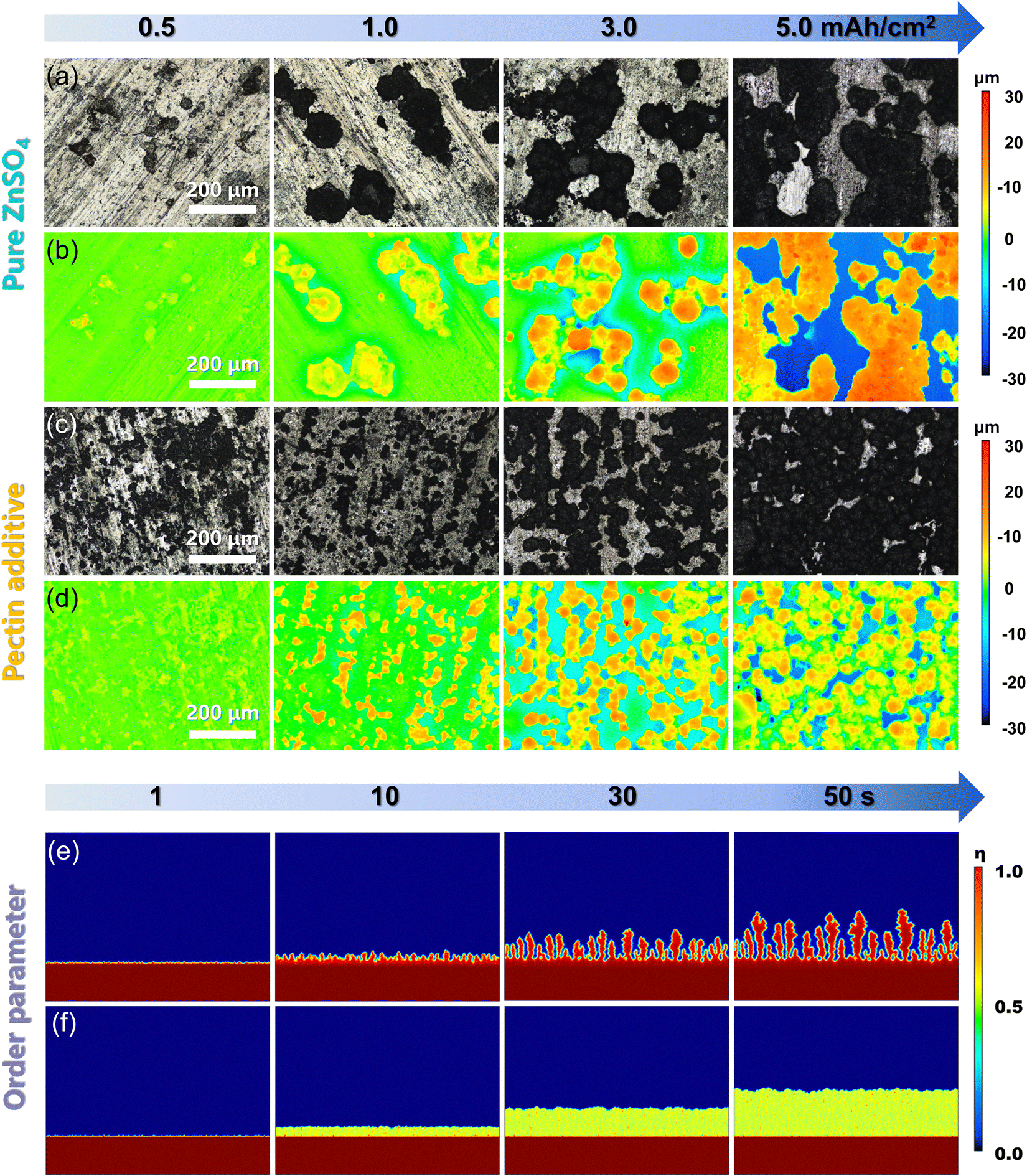
In order to investigate the suppression effect of pectin on the HER, the H2O dissociation energy barrier was calculated based on the adsorption models of the H2O molecule, activated H2O, OH* and H* intermediates on Zn(001) and Zn(001)–pectin, respectively (Fig. 3a and b). The activation barrier of H2O dissociation in ZnSO4–pectin increases from 0.67 eV (pure ZnSO4) to 3.85 eV, indicating that the HER activity on the Zn electrode is significantly inhibited in the ZnSO4–pectin electrolyte (Fig. 3c and d). The increased H2O splitting energy barrier is attributed to the shielding effect of pectin. The pectin molecule covers the Zn surface to form a protective layer, effectively avoiding the cleavage of H–OH bonds. To demonstrate the suppression of HER activity, H2 evolution polarization experiments were performed in 2.0 M ZnSO4 electrolyte with or without pectin additive (Fig. 3e). The introduction of pectin results in a much lower H2 evolution current density within a wide voltage range (−0.9 to −1.4 V vs. Ag/AgCl) compared with that in pure ZnSO4 electrolyte. In particular, the pure ZnSO4 electrolyte exhibits a H2 evolution current density of 37.73 mA cm−2 at −1.3 V, while that of the electrolyte with pectin additive decreases to 15.91 mA cm−2, indicating the difficulty in the occurrence of HER after the addition of pectin. The Tafel slopes increase from 83.2 mV dec−1 in pure ZnSO4 to 174.6 mV dec−1 in ZnSO4–pectin electrolyte (Fig. 3f), indicating the sluggish rate of HER with pectin. The suppressed HER activity is associated with the elevated H2O dissociation energy barrier. Meanwhile, the low interfacial polarization induced by the promoted desolvation also alleviates the HER. In addition, the formation of intermolecular hydrogen bonding between pectin and H2O would break the original hydrogen bond network of H2O molecules, reducing the free H2O activity. These synergistic effects contribute to the suppression of hydrogen evolution.
 | ||
| Fig. 3 Inhibition of the side reactions by pectin. DFT calculated adsorption models for H2O and activated H2O, OH−, and H+ on the surface of (a) Zn metal, and (b) pectin molecule. The corresponding dissociation energy barriers of H2O in (c) ZnSO4–H2O and (d) ZnSO4–H2O–pectin. (e) Hydrogen evolution polarization curves of Zn anodes in pure ZnSO4 and pectin electrolyte. (f) The corresponding Tafel slope curves. (g) Corrosion curves of Zn anodes in pure ZnSO4 and pectin electrolyte. (h) XRD patterns of Zn anodes in pure ZnSO4 and pectin electrolyte after 50 cycles. | ||
Zn corrosion would reduce the utilization rate of active Zn and compromise the reversibility of the Zn anode. The suppressed Zn corrosion by the pectin additive was evidenced by the linear polarization curves (Fig. 3g). The corrosion potential of ZnSO4–pectin increases from −0.97 V (vs. Ag/AgCl) of pure ZnSO4 to −0.95 V, while the corrosion current density decreases from 0.773 mA cm−2 of pure ZnSO4 to 0.159 mA cm−2. Both the increase of corrosion potential and decrease of corrosion current density indicate the significant role of pectin as an effective inhibitor for parasitic Zn corrosion. To confirm the chemical component of the byproduct induced by the Zn corrosion, the cycled Zn anodes were characterized by XRD. After 50 cycles, several new peaks located at 8.3°, 16.8°, and 25.5° are distinctly observed for the Zn anode cycled with pure ZnSO4 electrolyte, indexed to Zn4(OH)6SO4·5H2O (JCPDS no. 39-0688) (Fig. 3h). In the case of ZnSO4–pectin electrolyte, however, no peaks associated with Zn4(OH)6SO4·5H2O are detected after cycling, suggesting that the formation of Zn4(OH)6SO4·5H2O can be well suppressed by the pectin additive.
The electrochemical performances of Zn anodes in 2.0 M ZnSO4 electrolyte with different amounts of pectin were investigated by symmetric cells. Fig. S7 (ESI†) shows the voltage profiles of symmetric cells at 1.0 mA cm−2 with a capacity of 0.5 mA h cm−2. Notably, the symmetric cells with pectin-0.10 wt% electrolyte operate stably for more than 3700 h, approximately 70 times longer than that of the cells with pure ZnSO4 electrolyte. In addition, the electrolytes with pectin additive exhibit a wider electrochemical window than that of pectin-free electrolyte (Fig. S8, ESI†). Meanwhile, the pectin-0.10 wt% electrolyte enables the fastest plating/stripping kinetics among the tested electrolytes (Fig. S9, ESI†). In this work, an optimized electrolyte with 0.10 wt% pectin was selected for further evaluation.
The stability and reversibility of Zn/Zn2+ conversion chemistry were investigated using the Cu‖Zn batteries with pure ZnSO4 and ZnSO4–pectin electrolytes at 2.0 mA cm−2 with a capacity of 1.0 mA h cm−2. The coulombic efficiency (CE) of Cu‖Zn batteries, the ratio of the stripping capacity from Cu substrate to the plating capacity on Cu, could directly reflect the reversibility of Zn plating/stripping. As depicted in Fig. 4a, the Cu‖Zn batteries with pure ZnSO4 electrolyte could operate stably for less than 90 h, and then drastic fluctuation is observed around 85 cycles. The fluctuation could be further confirmed by the corresponding voltage curves (Fig. S10, ESI†). At the 87th cycle, the stripping capacity is only 0.44 mA h cm−2, leading to a low CE of only 43.50%. The loss of stripping capacity might be caused by the formation of “dead Zn” as well as the occurrence of side reactions. While at the 89th cycle, the stripping capacity reaches as high as 1.60 mA h cm−2, corresponding to a high CE value of 159.67%. The extra stripping capacity is a result of the incomplete stripping from Cu substrate into electrolyte, demonstrating the inferior reversibility of Zn/Zn2+ chemistry. In contrast, the Cu‖Zn batteries with the ZnSO4–pectin electrolyte show highly stable and reversible plating/stripping over 2300 cycles with an average CE of 99.4%. The corresponding plating/stripping curves of the 200th, 600th, 1000th, 1600th, and 2200th cycles are well overlapped (Fig. 4b), further demonstrating the high reversibility of Zn plating/stripping. The greatly improved reversibility is attributed to the formation of a pectin layer over the electrode surface, effectively guiding the homogeneous Zn deposition and inhibiting the parasitic reactions.
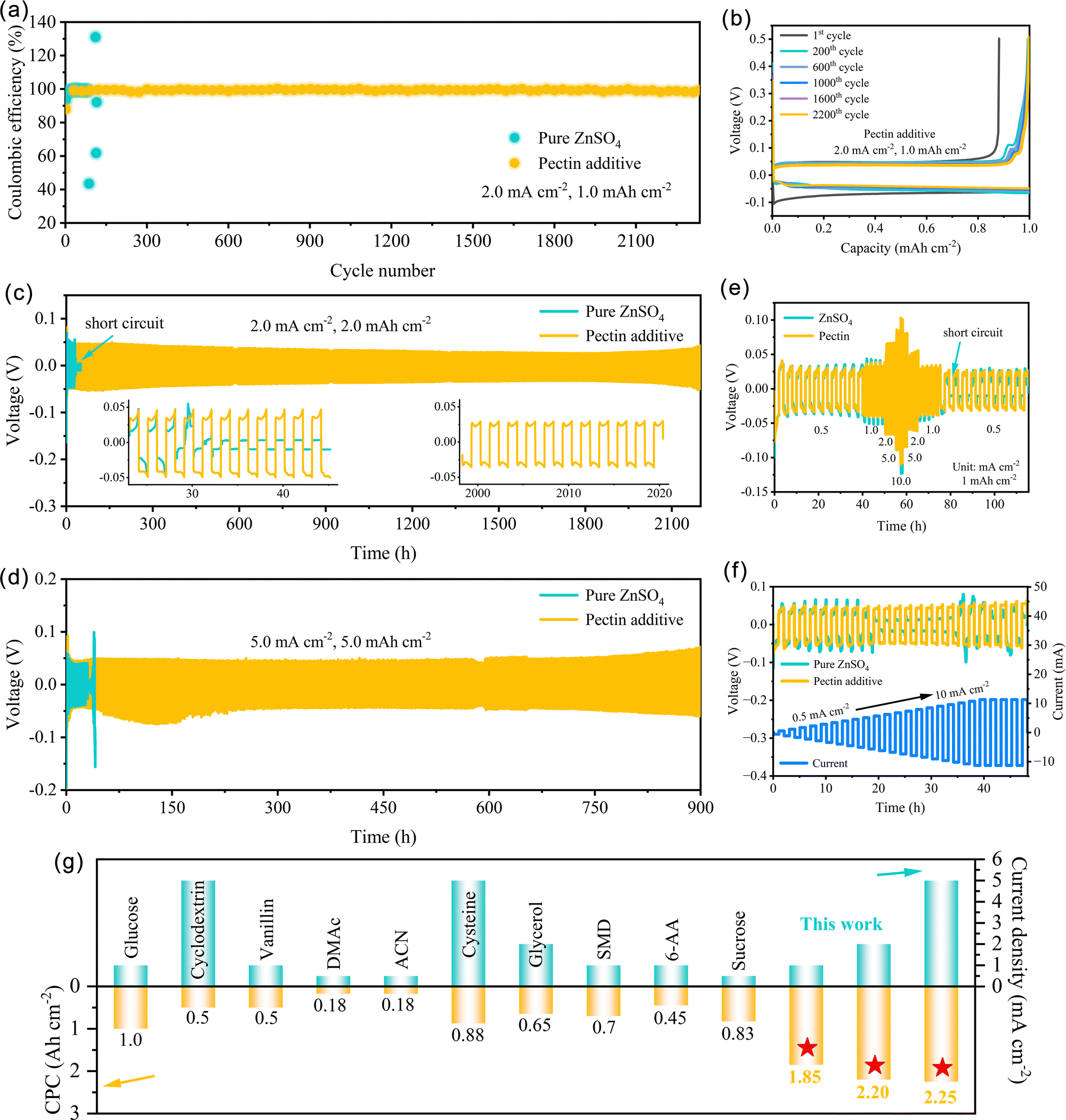 | ||
| Fig. 4 Effect of pectin additive on the electrochemical performance of the Zn anode. (a) Coulombic efficiency of Zn plating/stripping in Cu‖Zn cells with pure ZnSO4 and pectin electrolyte at 2.0 mA cm−2 for 1.0 mA h cm−2. (b) Voltage profiles of the Cu‖Zn cell with pectin additive at the 1st, 200th, 600th, 1000th, 1600th, and 2200th cycles. Cycling performance of the Zn symmetric cells at (c) 2.0 mA cm−2 for 2.0 mA h cm−2, and (d) 5.0 mA cm−2 for 5.0 mA h cm−2. (e) Rate performance of Zn symmetric cells. (f) Potential evolution of Zn symmetric cells at step-increased current densities. (g) The CPC comparison of the Zn symmetric cells between this work and other reports. | ||
The cycling stability of Zn anodes was further evaluated using the symmetric cells. Cycled at 1.0 mA cm−2 for 1.0 mA h cm−2, the symmetric cell with pectin additive exhibits an ultralong lifespan of 2500 h and stable overpotential (Fig. S11, ESI†). On the contrary, the symmetric cell with pure ZnSO4 electrolyte encounters a sudden voltage drop after only 20 cycles, attributed to a short-circuit. Further increasing the current density to 2.0 mA cm−2 and capacity to 2.0 mA h cm−2, the Zn anode in the ZnSO4–pectin electrolyte can still operate for more than 2200 h, approximately 1100 cycles with a stable and low overpotential of around 35 mV (Fig. 4c). Stable and flat voltage hysteresis was observed in the corresponding voltage curves of the symmetric cell with the ZnSO4–pectin electrolyte (insets of Fig. 4c), demonstrating the stable and reversible plating/stripping behavior of the batteries. Even under deep cycling conditions (5.0 mA cm−2, 5.0 mA h cm−2, DOD ≈ 28.5%), the symmetric cell with the ZnSO4–pectin electrolyte maintains stable cycling for more than 900 h with stable voltage hysteresis (Fig. 4d), showing an almost thirtyfold enhancement over that of the symmetric cell without pectin. Notably, the overpotential at high current density (5.0 mA cm−2) is only approximately 45 mV, indicating the low polarization and superb interfacial kinetics of the Zn anode endowed by the pectin. The remarkable cycling life and cumulative plated capacity (CPC) achieved in this work outperform most of the Zn anodes reported in the literature (Fig. 4g and Table S2, ESI†).38,43–51
The interfacial kinetics of the Zn anode was evaluated by the rate performance of symmetric cells (Fig. 4e). With the aid of pectin, the symmetric cells exhibit lower voltage hysteresis and more stable voltage plateau than those without additive, indicating fast interfacial kinetics and low polarization enabled by the pectin adsorbed layer. Conversely, short-circuit failure is observed for the cells without pectin when the current density increases to 10.0 mA cm−2 and recovers to 0.5 mA cm−2. The short circuit is attributed to the random dendrite growth triggered by the high current density. The rate capability was further evaluated with a step-increased current density (0.5 mA cm−2) and a fixed plating/stripping time of 1 h (Fig. 4f). No distinct increase in the overpotential is observed for the cell with pectin additive even with the current densities ranging from 0.5 to 10.0 mA cm−2. This result is probably associated with promoting the desolvation behavior and interfacial Zn2+ transport kinetics due to the formation of a pectin adsorbed layer on the Zn anode surface.
To reveal the failure mechanism of Zn anodes, the morphology evolution of Zn anodes was in situ investigated using optical microscopy. With the pure ZnSO4 electrolyte, the generation of initial Zn nuclei is heterogeneous. Plenty of protrusions were observed after deposition for 10 min and these protrusions gradually evolve into Zn dendrites at the edges of the Zn electrode with the plating time (Fig. 5a), accounting for the premature failure of the Zn anode. In comparison, the addition of pectin into electrolyte enables a smooth plating layer without visible protrusion generated during the whole process (Fig. 5b), benefitting from the uniform Zn2+ flux enabled by the zincophilic groups of pectin. The morphology evolution of Zn anodes before and after cycling was characterized by CLSM and SEM (Fig. 5c–h). After 100 cycles, island-like Zn dendrites with a maximum altitude intercept of 22.9 μm are observed on the Zn anode cycled in pure ZnSO4 (Fig. 5d and g). The growth of sharp Zn dendrites would easily pierce the separator of the battery, which is responsible for the short cycling lifespan and the fast capacity decay of the battery. In contrast, with pectin, the cycled Zn anode shows a smooth surface with negligible altitude intercept (Fig. 5e and h), contributing to the long-term reversible cycling process. A similar morphology evolution tendency was observed in the SEM images (Fig. 5i–l). Obviously, the Zn anode cycled in the ZnSO4–pectin electrolyte exhibits a planar and dendrite-free surface, ascribed to the regulation of the zincophilic pectin layer adsorbed on the Zn anode. The effective suppression of Zn dendrite significantly enhances the reversibility of the Zn anode and thereby extends the cycle lifespan of the battery. Overall, the elimination of dendrite, the promotion of desolvation, together with the suppression of the HER and Zn corrosion synergistically contribute to the excellent electrochemical performance.
 | ||
| Fig. 5 Morphology evolution of the Zn deposition and cycling process. (a) and (b) In situ optical microscopy observation of Zn plating in pure ZnSO4 and pectin electrolyte at 5.0 mA cm−2. Scale bars: 200 μm. (c)–(e) CLSM optical images, (f)–(h) corresponding 3D height images and (i)–(l) SEM images of Zn anodes in (d), (g) and (j) pure ZnSO4 and (e), (h), (k) and (l) pectin electrolyte before cycling and after 100 cycles at 1.0 mA cm−2 with the capacity of 0.5 mA h cm−2. The wavy lines in (c)–(e) show the corresponding height profiles for the horizontal line segments. | ||
The advantage of the ZnSO4–pectin electrolyte was further evaluated with Zn‖MnO2 full cells. The MnO2 cathode with nanorod morphology was prepared using the hydrothermal method. As shown in Fig. S12 (ESI†), the cyclic voltammetry (CV) curves display two pairs of characteristic redox peaks, attributed to Zn2+ or H+ insertion/extraction reactions.52 Notably, the oxidation peaks of the cells with pectin shift to the direction of lower voltage, indicating that the promotion of Zn2+ desolvation could also decrease the polarization voltage of the cathode reaction. In addition, the addition of pectin could lower the charge transfer resistance (Rct) of the full cells (Fig. 6a). Specifically, the Rct of the cell with pectin electrolyte is only 108.4 Ω, much lower than that of the cells without pectin (349.9 Ω), attributing to the accelerated desolvation behavior and the fast interfacial ion diffusion kinetics. The fast kinetics would reduce the polarization, accelerate the electrode reaction rate, and increase the reversibility. Self-discharge behavior is an important indicator to reveal the stability of full cells (Fig. 6b and c). After a rest of 24 h, the Zn‖MnO2 cells with the ZnSO4–pectin electrolyte maintains 92.76% of its charge capacity, much better than those with the pure ZnSO4 electrolyte (86.59%). Meanwhile, the voltage of the cells with the ZnSO4–pectin electrolyte only drop to 1.53 V after 24 h rest, higher than that of the cells with the pure ZnSO4 electrolyte. The long-term cycling performance was evaluated at a high rate of 2C (Fig. 6d). The cells with the pure ZnSO4 electrolyte show irreversible capacity decay with a remaining capacity of only 42.2 mA h g−1 after 500 cycles. In contrast, a high specific capacity of 175.1 mA h g−1 with a high average CE of nearly 99% is retained for the cells with the ZnSO4–pectin electrolyte after 500 cycles. The capacity fading mechanism can be further revealed by the corresponding charge/discharge curves (Fig. 6e). Without the addition of pectin, the voltage plateaus of the full cells gradually disappear after 200 cycles, while the charge/discharge curves of the full cells with pectin are nearly overlapped from the 5th to 500th cycle, indicating the high reversibility of the full cells with the pectin additive.
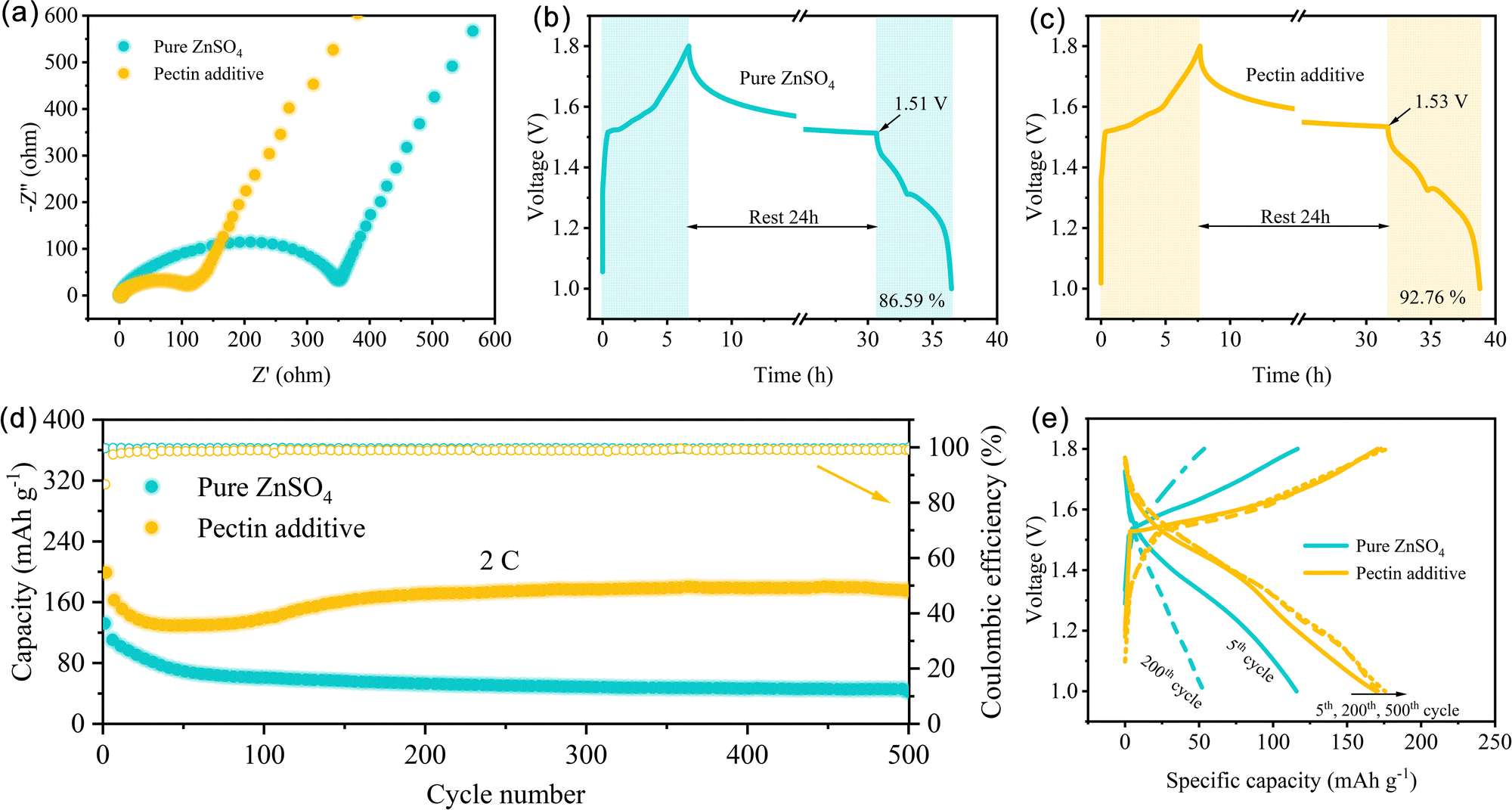 | ||
| Fig. 6 The electrochemical performance of Zn‖MnO2 full cells. (a) Nyquist plots. (b) and (c) Self-discharge tests after a rest of 24 h. (d) Long-term cycling performance at 2C and (e) the corresponding charge–discharge curves. | ||
3. Conclusions
In summary, desolvation activation energy was proposed as a critical descriptor for the screening of electrolyte additives. Anionic polysaccharides with low desolvation activation energy were demonstrated to be effective electrolyte additives to address the issues of high polarization and hydrogen evolution of Zn anodes. Taken as a good example, pectin polysaccharide, a typical anionic polysaccharide was systematically investigated as the electrolyte additive. According to the DFT calculations, the pectin molecules are inclined to adsorb in parallel on the Zn anode surface to form a protective layer. With the hydrogen bonding interaction between pectin and H2O, the desolvation activation energy is decreased and the removal of H2O from hydrated Zn2+ is accelerated, significantly lowering the interfacial voltage polarization. The introduction of pectin greatly elevates the energy barrier of H2O splitting, effectively suppressing the HER. In addition, the oxygen-containing groups of pectin molecules serve as zincophilic sites to induce the uniform Zn deposition. Consequently, the asymmetric cell with pectin achieves highly reversible plating/stripping behavior with a CE of 99.4% over 2300 cycles. The symmetric cell steadily operates for more than 900 h under deep cycling conditions (5.0 mA cm−2, 5.0 mA h cm−2). A high capacity of approximately 175.1 mA h g−1 was maintained for the Zn‖MnO2 full cells with pectin additive after 500 cycles at 2C. The descriptor of desolvation activation energy proposed in this work will provide effective guidance for the design of low-polarization electrolyte additives.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (22133005, and 22208209) and the Program for Basic Research of Shanghai Science and Technology Commission (19JC1412600). The computations in this paper were run on the π 2.0 cluster supported by the Center for High Performance Computing at Shanghai Jiao Tong University.References
- F. Wang, J. Zhang, H. Lu, H. Zhu, Z. Chen, L. Wang, J. Yu, C. You, W. Li, J. Song, Z. Weng, C. Yang and Q.-H. Yang, Nat. Commun., 2023, 14, 4211 CrossRef CAS.
- H. Lu, J. Hu, X. Wei, K. Zhang, X. Xiao, J. Zhao, Q. Hu, J. Yu, G. Zhou and B. Xu, Nat. Commun., 2023, 14, 4435 CrossRef CAS PubMed.
- P. Xiao, Y. Wu, K. Liu, X. Feng, J. Liang, Y. Zhao, C. Wang, X. Xu, T. Zhai and H. Li, Angew. Chem., Int. Ed., 2023, 62, e202309765 CrossRef CAS PubMed.
- Y. Zhong, X. Xie, Z. Zeng, B. Lu, G. Chen and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202310577 CrossRef CAS PubMed.
- K. Zhou, Z. Li, X. Qiu, Z. Yu and Y. Wang, Angew. Chem., Int. Ed., 2023, 62, e202309594 CrossRef CAS PubMed.
- J. Li, Z. Guo, J. Wu, Z. Zheng, Z. Yu, F. She, L. Lai, H. Li, Y. Chen and L. Wei, Adv. Energy Mater., 2023, 13, 2301743 CrossRef CAS.
- Y. Li, Y. Wang, Y. Xu, W. Tian, J. Wang, L. Cheng, H. Yue, R. Ji, Q. Zhu, H. Yuan and H. Wang, Small, 2022, 18, 2202214 CrossRef CAS.
- L. Hong, L. Y. Wang, Y. Wang, X. Wu, W. Huang, Y. Zhou, K. X. Wang and J. S. Chen, Adv. Sci., 2022, 9, 2104866 CrossRef CAS PubMed.
- L. Hong, X. Wu, L.-Y. Wang, M. Zhong, P. Zhang, L. Jiang, W. Huang, Y. Wang, K.-X. Wang and J.-S. Chen, ACS Nano, 2022, 16, 6906–6915 CrossRef CAS PubMed.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, 2007406 CrossRef CAS PubMed.
- L. Hong, X. Wu, C. Ma, W. Huang, Y. Zhou, K.-X. Wang and J.-S. Chen, J. Mater. Chem. A, 2021, 9, 16814–16823 RSC.
- L. Hong, X. Wu, Y. S. Liu, C. Yu, Y. Liu, K. Sun, C. Shen, W. Huang, Y. Zhou, J. S. Chen and K. X. Wang, Adv. Funct. Mater., 2023, 33, 2300952 CrossRef CAS.
- W. Dong, C. Liu, X. Ji, H. Yao, J. Li, H. Du and S. Cheng, Small Methods, 2023, 2300799 CrossRef PubMed.
- J. Yu, C. Chen, F. Shi, R. Li, F. Chen, J. Tang, K. C. Chan and Z.-L. Xu, Energy Storage Mater., 2023, 63, 102966 CrossRef.
- S. Li, J. Fu, G. Miao, S. Wang, W. Zhao, Z. Wu, Y. Zhang and X. Yang, Adv. Mater., 2021, 33, 2008424 CrossRef CAS PubMed.
- D. Han, S. Wu, S. Zhang, Y. Deng, C. Cui, L. Zhang, Y. Long, H. Li, Y. Tao, Z. Weng, Q. H. Yang and F. Kang, Small, 2020, 16, 2001736 CrossRef CAS.
- W. Liu, Q. Zhao, H. Yu, H. Wang, S. Huang, L. Zhou, W. Wei, Q. Zhang, X. Ji, Y. Chen and L. Chen, Adv. Funct. Mater., 2023, 33, 2302661 CrossRef CAS.
- R. Zhao, X. Dong, P. Liang, H. Li, T. Zhang, W. Zhou, B. Wang, Z. Yang, X. Wang, L. Wang, Z. Sun, F. Bu, Z. Zhao, W. Li, D. Zhao and D. Chao, Adv. Mater., 2023, 35, 2209288 CrossRef CAS PubMed.
- O. Blumen, G. Bergman, K. Schwatrzman, S. Harpaz, S. H. Akella, M. S. Chae, N. Bruchiel-Spanier, N. Shpigel and D. Sharon, J. Mater. Chem. A, 2023, 11, 19970–19980 RSC.
- Y. Mu, Z. Li, B.-k Wu, H. Huang, F. Wu, Y. Chu, L. Zou, M. Yang, J. He, L. Ye, M. Han, T. Zhao and L. Zeng, Nat. Commun., 2023, 14, 4205 CrossRef CAS PubMed.
- J.-H. Wang, L.-F. Chen, W.-X. Dong, K. Zhang, Y.-F. Qu, J.-W. Qian and S.-H. Yu, ACS Nano, 2023, 17, 19087–19097 CrossRef CAS.
- D. Wang, D. Lv, H. Peng, C. Wang, H. Liu, J. Yang and Y. Qian, Angew. Chem., Int. Ed., 2023, 62, e202310290 CrossRef CAS PubMed.
- X. Wang, K. Feng, B. Sang, G. Li, Z. Zhang, G. Zhou, B. Xi, X. An and S. Xiong, Adv. Energy Mater., 2023, 13, 2301670 CrossRef CAS.
- X. Song, L. Bai, C. Wang, D. Wang, K. Xu, J. Dong, Y. Li, Q. Shen and J. Yang, ACS Nano, 2023, 17, 15113–15124 CrossRef CAS.
- S. D. Pu, C. Gong, Y. T. Tang, Z. Ning, J. Liu, S. Zhang, Y. Yuan, D. Melvin, S. Yang, L. Pi, J. J. Marie, B. Hu, M. Jenkins, Z. Li, B. Liu, S. C. E. Tsang, T. J. Marrow, R. C. Reed, X. Gao, P. G. Bruce and A. W. Robertson, Adv. Mater., 2022, 34, 2202552 CrossRef CAS.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. Quilty, G. Renderos, X. Liu, Y. Deng, L. Wang, D. Bock, C. Jaye, D. Zhang, E. Takeuchi, K. Takeuchi, A. Marschilok and L. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS.
- T. Wei, Y. Ren, Y. Wang, L. e Mo, Z. Li, H. Zhang, L. Hu and G. Cao, ACS Nano, 2023, 17, 3765–3775 CrossRef CAS.
- Z. Hou, H. Tan, Y. Gao, M. Li, Z. Lu and B. Zhang, J. Mater. Chem. A, 2020, 8, 19367–19374 RSC.
- P. Xiao, Y. Wu, J. Fu, J. Liang, Y. Zhao, Y. Ma, T. Zhai and H. Li, ACS Energy Lett., 2022, 8, 31–39 CrossRef.
- D. Wang, D. Lv, H. Liu, S. Zhang, C. Wang, C. Wang, J. Yang and Y. Qian, Angew. Chem., Int. Ed., 2022, 61, e202212839 CrossRef CAS PubMed.
- L. Zhou, F. Wang, F. Yang, X. Liu, Y. Yu, D. Zheng and X. Lu, Angew. Chem., Int. Ed., 2022, 61, e202208051 CrossRef CAS PubMed.
- M. Wang, J. Ma, Y. Meng, J. Sun, Y. Yuan, M. Chuai, N. Chen, Y. Xu, X. Zheng, Z. Li and W. Chen, Angew. Chem., Int. Ed., 2022, 62, e202214966 CrossRef PubMed.
- P. Perumal, S. Selvasekarapandian, K. P. Abhilash, P. Sivaraj, R. Hemalatha and P. C. Selvin, Vacuum, 2019, 159, 277–281 CrossRef CAS.
- M. Muthukrishnan, C. Shanthi, S. Selvasekarapandian, R. Manjuladevi, P. Perumal and P. Chrishtopher Selvin, Ionics, 2018, 25, 203–214 CrossRef.
- S. Eswaragomathy, S. Selvanayagam, S. Selvasekarapandian, N. Muniraj Vignesh, S. Aafrin Hazaana and R. Meera Naachiyar, Ionics, 2023, 29, 2329–2340 CrossRef CAS.
- Q. Ma, R. Gao, Y. Liu, H. Dou, Y. Zheng, T. Or, L. Yang, Q. Li, Q. Cu, R. Feng, Z. Zhang, Y. Nie, B. Ren, D. Luo, X. Wang, A. Yu and Z. Chen, Adv. Mater., 2022, 34, 2207344 CrossRef CAS PubMed.
- K. Zhao, G. Fan, J. Liu, F. Liu, J. Li, X. Zhou, Y. Ni, M. Yu, Y.-M. Zhang, H. Su, Q. Liu and F. Cheng, J. Am. Chem. Soc., 2022, 144, 11129–11137 CrossRef CAS PubMed.
- H. Yu, Y. Chen, W. Wei, X. Ji and L. Chen, ACS Nano, 2022, 16, 9736–9747 CrossRef CAS PubMed.
- Q. Yang, L. Li, T. Hussain, D. Wang, L. Hui, Y. Guo, G. Liang, X. Li, Z. Chen, Z. Huang, Y. Li, Y. Xue, Z. Zuo, J. Qiu, Y. Li and C. Zhi, Angew. Chem., Int. Ed., 2021, 61, e202112304 CrossRef PubMed.
- S. Jiao, J. Fu, M. Wu, T. Hua and H. Hu, ACS Nano, 2021, 16, 1013–1024 CrossRef PubMed.
- Q. Zhang, Y. Ma, Y. Lu, X. Zhou, L. Lin, L. Li, Z. Yan, Q. Zhao, K. Zhang and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 23357–23364 CrossRef CAS PubMed.
- P. Jiang, Q. Du, M. Shi, W. Yang and X. Liang, Small Methods, 2023, 2300823 CrossRef PubMed.
- Z. Zhang, Z. He, N. Wang, F. Wang, C. Du, J. Ruan, Q. Li, D. Sun, F. Fang and F. Wang, Adv. Funct. Mater., 2023, 33, 2214648 CrossRef CAS.
- S. Yang, K. Xue, J. Zhang, Y. Guo, G. Wu, C. Li, C. Xia, Y. Zhang, Y. Chen and L. Zhou, Energy Storage Mater., 2023, 62, 102929 CrossRef.
- F. Wu, Y. Chen, Y. Chen, R. Yin, Y. Feng, D. Zheng, X. Xu, W. Shi, W. Liu and X. Cao, Small, 2022, 18, 2202363 CrossRef CAS PubMed.
- S.-H. Huh, Y. J. Choi, S. H. Kim, J.-S. Bae, S.-H. Lee and S.-H. Yu, J. Mater. Chem. A, 2023, 11, 19384–19395 RSC.
- S. Zheng, Y. Wang, B. Luo, L. Sun, G. Duan, J. Huang and Z. Ye, Chem. Eng. J., 2023, 473, 145313 CrossRef CAS.
- L. Zhou, R. Yang, S. Xu, X. Lei, Y. Zheng, J. Wen, F. Zhang and Y. Tang, Angew. Chem., Int. Ed., 2023, 62, e202307880 CrossRef CAS.
- K. Zhao, F. Liu, G. Fan, J. Liu, M. Yu, Z. Yan, N. Zhang and F. Cheng, ACS Appl. Mater. Interfaces, 2021, 13, 47650–47658 CrossRef CAS.
- P. Sun, L. Ma, W. Zhou, M. Qiu, Z. Wang, D. Chao and W. Mai, Angew. Chem., Int. Ed., 2021, 60, 18247–18255 CrossRef CAS PubMed.
- N. Zhang, S. Huang, Z. Yuan, J. Zhu, Z. Zhao and Z. Niu, Angew. Chem., Int. Ed., 2020, 60, 2861–2865 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00199k |
| This journal is © The Royal Society of Chemistry 2024 |