
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpot the difference in reactivity: a comprehensive review of site-selective multicomponent conjugation exploiting multi-azide compounds
Hiroki
Tanimoto
 * and
Takenori
Tomohiro
* and
Takenori
Tomohiro
Faculty of Pharmaceutical Sciences, University of Toyama, 2630 Sugitani, Toyama 930-0194, Japan. E-mail: tanimoto@pha.u-toyama.ac.jp
First published on 20th September 2024
Abstract
Going beyond the conventional approach of pairwise conjugation between two molecules, the integration of multiple components onto a central scaffold molecule is essential for the development of high-performance molecular materials with multifunctionality. This approach also facilitates the creation of functionalized molecular probes applicable in diverse fields ranging from pharmaceuticals to polymeric materials. Among the various click functional groups, the azido group stands out as a representative click functional group due to its steric compactness, high reactivity, handling stability, and easy accessibility in the context of multi-azide scaffolds. However, the azido groups in multi-azide scaffolds have not been well exploited for site-specific use in molecular conjugation. In fact, multi-azide compounds have been well used to conjugate to the same multiple fragments. To circumvent problems of promiscuous and random coupling of multiple different fragments to multiple azido positions, it is imperative to distinguish specific azido positions and use them orthogonally for molecular conjugation. This review outlines methods and strategies to exploit specific azide positions for molecular conjugation in the presence of multiple azido groups. Illustrative examples covering di-, tri- and tetraazide click scaffolds are included.
 Hiroki Tanimoto | Hiroki Tanimoto received his BS degree in 2004 and his PhD degree in 2009 from Keio University in Japan (Professor Noritaka Chida). After one year as a postdoctoral fellow at Vanderbilt University in the United States (Professor Gary A. Sulikowski), he joined the faculty of Nara Institute of Science and Technology (NAIST) in Japan in 2010 to start his academic career as an assistant professor. In 2020, he was appointed as an associate professor at the University of Toyama. His main research focuses on new synthetic methods including click strategies, synthesis of alkaloids, heterocycles, and functional materials toward chemical biology. |
 Takenori Tomohiro | Takenori Tomohiro received both BSc (1986) and PhD degrees (1992) from the University of Tsukuba. He had worked at the National Chemical Laboratory for Industry (1986–2002). During this period, he worked as a postdoctoral fellow at the University of Oxford for two years from 1992 and as a visiting professor at the Louis Pasteur University in Strasbourg (2002). He joined the faculty at the Toyama Medical and Pharmaceutical University (Currently University of Toyama) in 2002 as an associate professor and is now full professor. His research interests are in the areas of chemical biology related to biomolecular interaction and medicinal chemistry. |
1. Introduction
Coupling reactions to combine molecules into a single molecule play a key role in the functionalization of compounds for a wide range of functional materials. In particular, click chemistry, which facilitates the formation of robust chemical bonds between two or more components, has been operated under mild conditions.1–5 This property allows click conjugation reactions to be performed under aqueous, open-air, and even live-cell conditions with simple experimental manipulations, which has gained worldwide research applications. Today, click chemistry is well-established in a variety of research and development fields, including medicine, pharmaceuticals, polymers, applied materials chemistry, and chemical biology.6–20 Conjugation between the pair of azido and ethynyl (alkyne) functional groups is a representative method of click reactions. Over time, numerous functional group pairs have been developed to achieve faster, more bioorthogonal, and site-selective click conjugation (Fig. 1(a)).3,14,17,19,20 | ||
| Fig. 1 (a) Representative click conjugation functional groups. (b) Strategy for the synthesis of multifunctional materials using multi-click modular hub molecules. (c) Multi-azide hub scaffolds for multicomponent conjugation to functional materials. | ||
After the development of click chemistry, a multi-click modular hub strategy has emerged to integrate multiple compounds onto a single scaffold compound beyond the conventional one-to-one click conjugation (Fig. 1(b)).21–24 Click chemistry proves advantageous in this context due to the well-developed and established click reactions and functional groups that facilitate coupling between specific functional groups. Thus, the incorporation of multiple and appropriate click functional groups onto a hub molecule becomes highly efficient for the precise synthesis of multicomponent-integrated functional materials. These include versatile element-block polymers,25–28 dendrimers,29,30 and high-performance chemical probes with multi-responsive, multi-color, and multi-point recognition or cross-linking sites.31–34
Despite its utility, the introduction of multiple click functionalities onto a hub molecule is a complex undertaking that is far from straightforward. The multi-step process requires the sequential introduction of each click group prior to conjugation, resulting in lengthy synthetic procedures. In addition, the stepwise introduction of different click groups is challenging, especially when dealing with non-bench stable click functional groups.
On the contrary, among the various click functional groups, the azido group has held a prominent position since the early stages of click chemistry research.1,2 As a result, various methods of combining molecules have emerged. Advantageous features of the azido group include: (1) easy introduction into single and multiple positions of substrate molecules using inorganic azide or sulfonyl azide reagents via SN2, SNAr, cross-coupling, or diazo transfer reactions; (2) sufficient stability for easy handling; (3) compact size, which facilitates access to hindered target positions for click reactions; (4) various click conjugation methods have been developed, such as Staudinger–Bertozzi ligation,35–40 CuAAC (copper-catalyzed azide–alkyne cycloaddition),41–44 and SPAAC (strain-promoted azide–alkyne cycloaddition).45–48 These robust reactivities have also found applications in main-group element chemistry.49 In addition, the azido group is efficient for macrocyclization through intramolecular reactions.50 Moreover, the clickability of organic azides has recently been exploited for the direct detection of endogenous biomarkers related to oxidative stress-induced diseases and their radioisotope therapy in the causative pathogenic tissues.51–55
For these valuable points, especially on synthetic merit, multi-component conjugation with the molecules possessing multiple azido groups, multi-azide compounds have been focused as multi-click hub compounds (Fig. 1(c)).24 At first sight, multi-azide compounds appear to undergo unselective and promiscuous conjugation with azido groups at different sites. However, understanding the characteristics and differences of each functional group allows its versatile use even in the presence of multiple identical groups. In the case of hydroxy and amino groups, regioselective protection and functionalization have been successfully achieved through the strategic use of the steric environment and the substituent difference between alkyl and aryl positions.56–62 As with these functional groups, targeting a specific position of the azido group in the presence of multiple azido groups also becomes feasible by identifying differences in its properties and surrounding environment. Recent advances have led to the development of methods for linking multiple molecules into easily synthesized multi-azide compounds with precise control over linkage positions. As a result, multi-azide molecules now serve as molecular hubs capable of integrating various functional molecules in a targeted manner by orthogonally utilizing the multiple azido groups. The development of this technique holds great promise for the rapid and easy synthesis of multifunctional molecular probes and polymeric materials. In this context, we provide a comprehensive review of the strategy for the site-selective coupling reaction of azido groups in multi-azide compounds, facilitating multicomponent conjugation.
2. A brief review of the basic properties of organic azides, including common handling and recautions
Before discussing specific cases, a brief overview of organic azides is provided to elucidate their basic properties. The azido (N3) group is a 1,3-dipolar functional group consisting of three nitrogen atoms linked in a linear (but slightly bent) fashion (Fig. 2(a)).63–70 Currently, the azido group is widely recognized as a click-functional group in numerous applications. It is also incorporated into the structure of certain pharmaceutical molecules such as zidovudine (azidothymidine, AZT, Retrovir) and azidocillin. Conversely, only one example has been documented as a natural product.71,72 | ||
| Fig. 2 A brief review of organic azides: (a) resonance structures of organic azides and the examples of bioactive or natural azides. (b) Reaction properties of the organic azides. (c) General preparation methods. | ||
While the internal (N1) and terminal (N3) nitrogen atoms are negatively charged, the N1–N2 bond is longer than the N2–N3 bond. As the negative charge disappears through reaction at N1, the remaining diazonium moiety (N2–N3) exhibits high reactivity due to its strong electrophilicity. Consequently, the organic azide reaction is commonly conceptualized as involving the diazonium moiety of N2–N3 and N1 of the amide anion. In principle, N1 functions as a nucleophile, N3 as an electrophile, and N1–N3 as a 1,3-dipolar entity (Fig. 2(b)). Since the nucleophilicity of organic azides tends to be low in intermolecular reactions, the electrophilic and 1,3-dipolar properties of organic azides are used for coupling reactions including click conjugation. The electrophilicity is involved in Staudinger–Bertozzi ligation35–40 and sulfo-click reactions, while the 1,3-dipolar reactivity is associated with SPAAC45–48 and enolate/enamine-mediated triazole synthesis (see Section 4.2). In addition, in terms of nucleophilicity, the metal coordination step at the N1 position is a critical component in CuAAC.41–44
Typical methods for the synthesis of organic azides are shown in Fig. 2(c).73 Alkyl azides are commonly prepared by SN2 reactions of the corresponding halides or sulfonates with commercial sodium azide. In the case of tert-alkyl azides, SN1 reactions using Lewis acids are the method of choice. Diazo transfer reactions are accessible to primary, secondary, and tertiary alkyl azides. Aryl azides are typically synthesized by cross-coupling reactions (often using copper catalysts) with aryl halides. Diazo transfer reactions of anilines with sulfonyl azides or diazotization of anilines followed by SNAr reactions with sodium azide are also commonly used. In addition, recent advances have added decarboxylative azidation, electrochemical azidation, radical hydroazidation from olefins, and C–H azidation to the repertoire of azide synthesis methods.67,70
Despite their usefulness, it is important to recognize that organic azides, especially multi-azide compounds, pose potential hazards and explosive risks. Therefore, they should be handled with care in a hood equipped with a shield (usually made of polycarbonate) in order to reduce the risk of incidents arising from sudden detonation. Azide salts for the azidation reaction, such as the commonly used sodium azide, should be handled with a plastic spatula, especially in the case of heavy metal azide salts, and care should be taken to avoid scratching. In particular, sulfonyl azides have been reported to exhibit strong detonation properties.
Because SN2 azidation reactions, in general, are often performed in polar solvents, residual halogenated solvents, such as dichloromethane, used immediately before the azidation process may form explosive species due to azidation in polar solvents. For example, dichloromethane can form diazidomethane through azidation.63,74–76 To prevent unexpected detonation, it is advisable to thoroughly remove residual halogenated solvents before performing azidation reactions, especially in large-scale reactions.
In addition, special attention should be paid to compounds with (C + O)/N < 3 (C, N, O: number of individual atoms in a molecule). This metric also applies to organic azides, including multi-azide compounds, but molecules must be designed and prepared with careful consideration of the structure, stability, and reactivity of the azido groups, regardless of the ratio values.77 Despite the above precautions, researchers may still encounter unforeseen reactions. In the case of Tanimoto and co-workers’ approach to diazide 3 from 1 or 4, the synthetic steps to the azidoacetohydrazide structure unexpectedly yielded amine 7 in high yields (Scheme 1).78 This nitrogen–nitrogen bond cleavage may have occurred by cyclization of the desired 3 to the cyclic tetrazinone 5, followed by elimination of 6. Similarly, synthetic efforts on other α-azidoacetohydrazide substrates were unsuccessful or resulted in very low product yields, whereas the reaction of the hydroxamate derivative afforded the azide product in high yield without N–O bond cleavage. Therefore, for the safety of all researchers, it is important to report side reactions or unexpected observations along with successful research results.
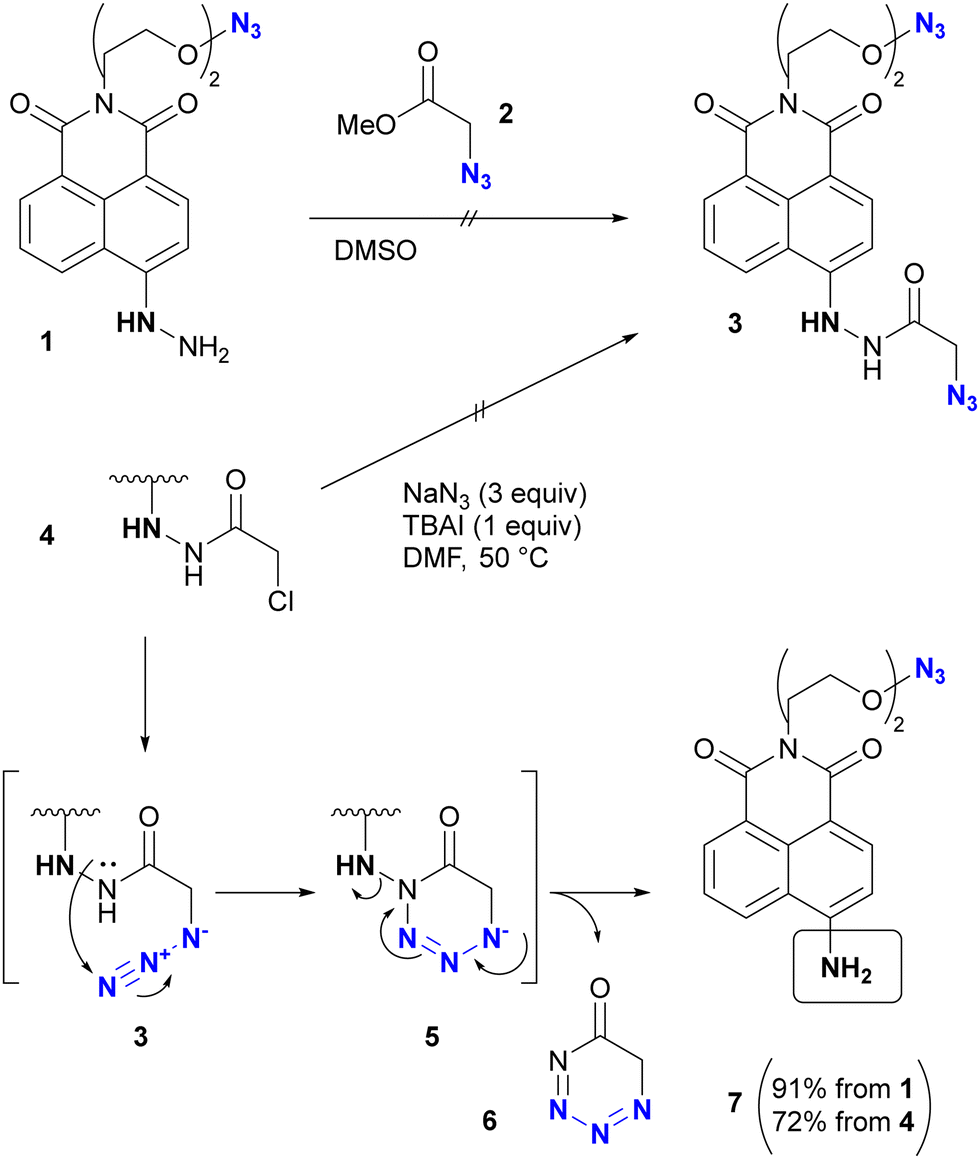 | ||
| Scheme 1 Nitrogen–nitrogen bond cleavage occurred in the azidation reaction. | ||
3. Multiple conjugation of the same components to multi-azide compounds
Azidation of substrates, especially via the SN2 reaction, provides precise access to multi-azide compounds through global azidation. Examples and their applications for multifunctionalization are shown in Scheme 2. In Ikeda's synthesis of ionic liquids,79 four azido groups were successfully introduced at the neopentyl positions in one step from pentaerythrityl tetrabromide 8.80,81 The resulting tetraazide 9 underwent a quadruple CuAAC reaction followed by methylation and anion exchange to yield the desired tetrahedral ionic liquids 10. Conversely, direct SN2 introduction of four imidazole compounds to 8 proved unsuccessful due to steric hindrance, demonstrating the accessibility of the azidation by the small structure and strong nucleophilicity of the azide ion.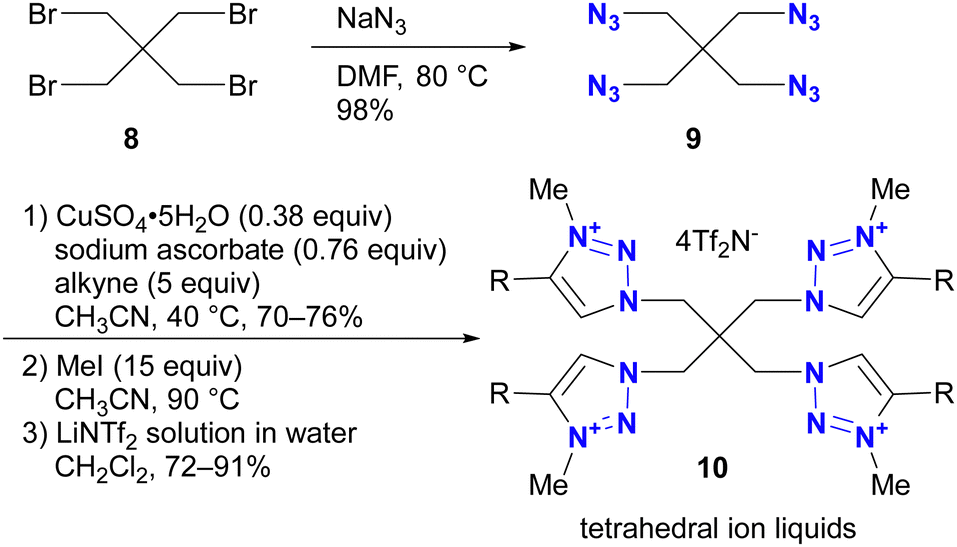 | ||
| Scheme 2 Synthesis of tetrahedral tetra-cationic ionic liquids by quadruple CuAAC. | ||
Because of their ready availability, multi-azide compounds have found utility in polymer functionalization (Fig. 3).6–13 For example, in addition to chain-growth polymerization of diazides with diynes,7 main-chain compounds composed of azide monomers acquire special properties through global click conjugation at the side chains. These include photoreaction and thermal reaction, resulting in the formation of crosslinked network polymers10 and porous materials.13
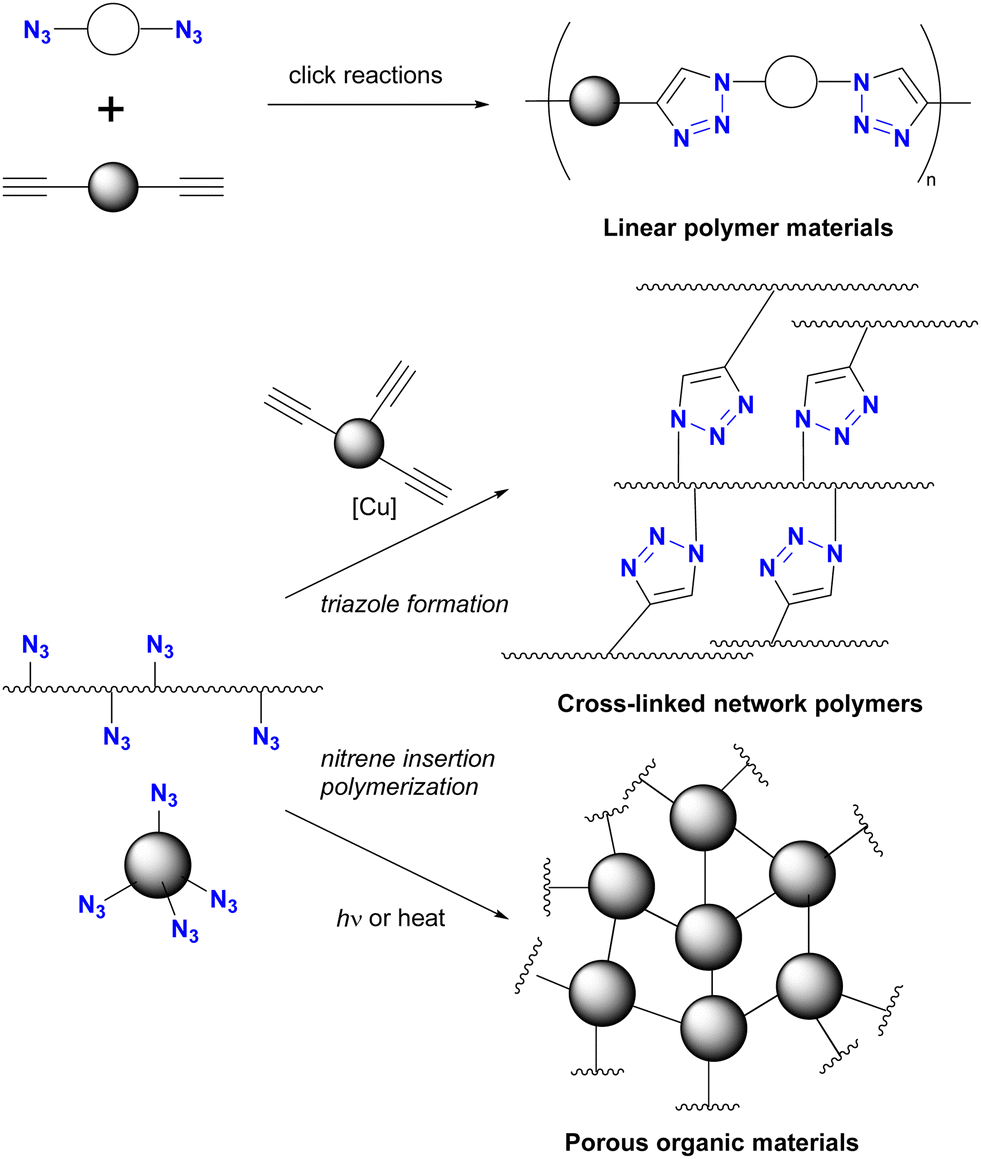 | ||
| Fig. 3 The general approach to crosslinked polymers by multiple CuAAC. | ||
As shown in Fig. 3, multi-azide molecules offer excellent accessibility for global functionalization via click conjugation reactions. However, due to the identical functional group, all azido positions are often coupled to the same conjugating component. In essence, multiple positions are endowed with only one function. To enable the attachment of different functional components to multi-azide platforms for the synthesis of multifunctional materials, it becomes essential to conjugate each azido position with different components. This raises the issue of site-selectivity in conjugation, as illustrated in Fig. 1(c). Random conjugation must be avoided as it has a significant impact on synthetic reproducibility, production efficiency and product quality. Therefore, achieving azide-site selective conjugation reactions is challenging but highly rewarding.
An example by Kirsch and colleagues is shown in Scheme 3.82 They investigated the conjugation of multiple components to each azido group using a sequential approach.83–85 Starting with the geminal diazide 11, CuAAC with two equivalents of alkyne followed by the retro–aldol reaction afforded the bis-triazole product 12. By reducing the amount of alkyne to 1.1 equivalents, a thermal [3+2] reaction with tosylacetylene 13 successfully afforded the mono-triazole product 14, which retained an azido group. The CuAAC reaction with methyl propiolate 15 afforded the product 16 with two different triazole rings. Following the IBX-SO3K-mediated decarboxylative azidation, the third triazole formation reaction by CuAAC afforded the tris-triazole 19. Despite the successful conjugation of three different components onto one molecule, this work also highlights the challenges associated with site-selective azide coupling reactions in the presence of multiple azido groups.
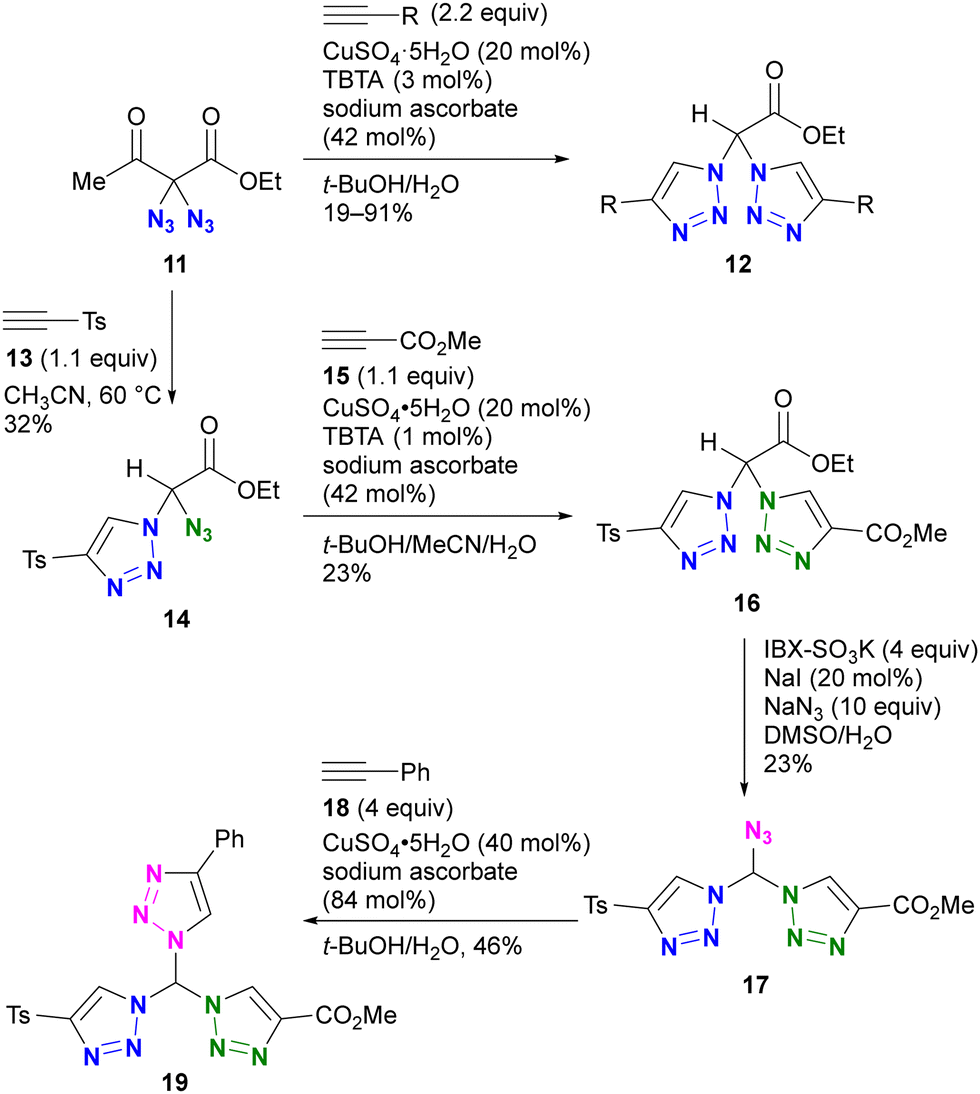 | ||
| Scheme 3 Stepwise selective azide conjugation to give tris(triazolyl)methane with three different substituents. IBX: 2-Iodoxybenzoic acid. TBTA: tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine. | ||
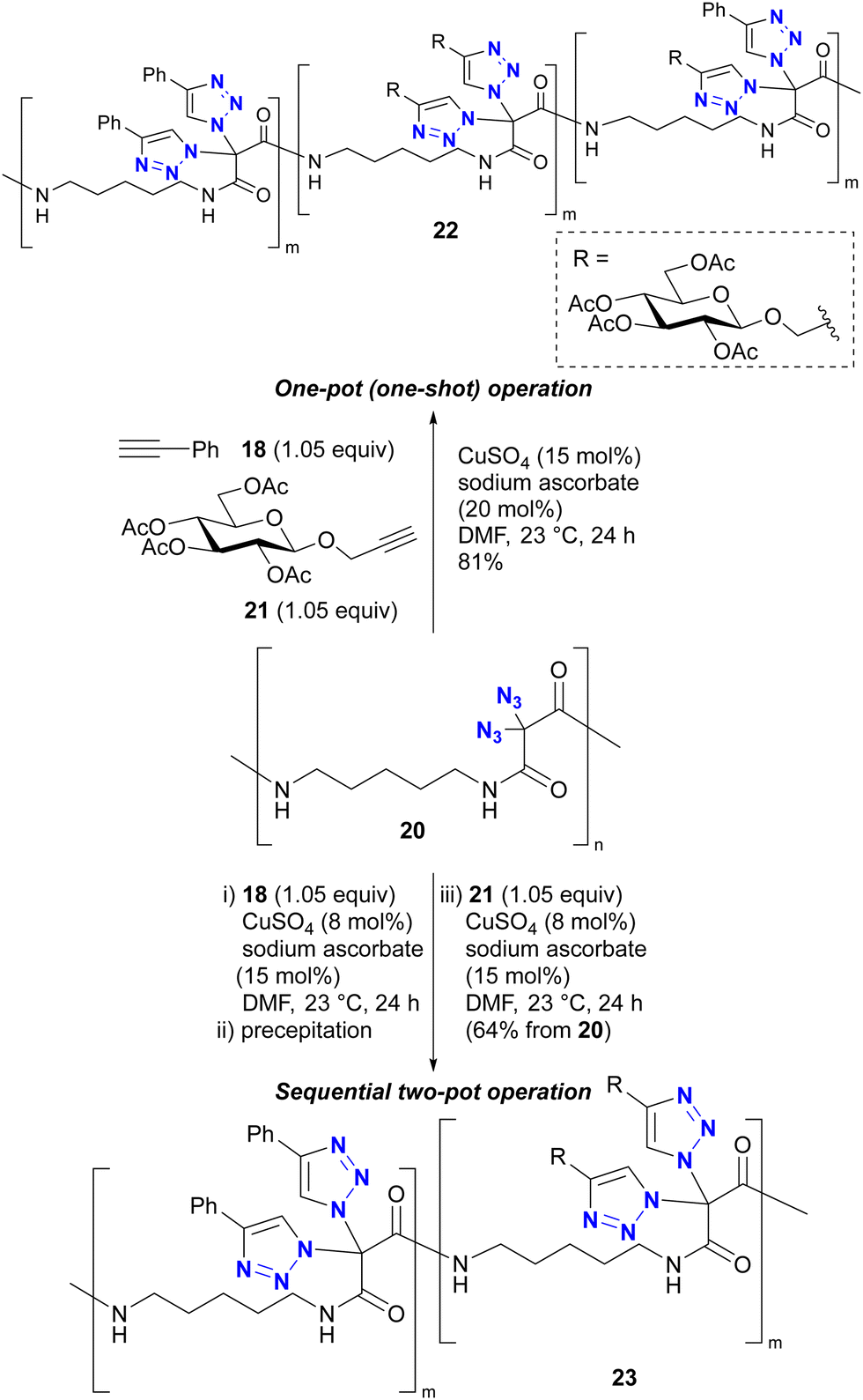
In addition, Kirsch's group also explored the polymer chemistry of their geminal diazide (Scheme 4).86 Using polymer 20 with a geminal diazide moiety in the main chain, one-pot CuAAC in the presence of two alkynes 18 and 21 produced the click-functionalized polymer 22, but in a randomly conjugated manner. Conversely, a sequential two-pot operation (CuAAC with 18 follwed by 21 as independent reactions) successfully yielded the site-selectively click-functionalized polymer 23 with the same alkynes attached to the diazidomethylene moiety. The reason for the azide-site selectivity observed in the two-pot operation is not described in this paper. However, Kirsch et al. also reported the strong acceleration of the second CuAAC of geminal diazides in Scheme 3.82 The copper catalyst on the triazole ring soon after the first CuAAC would be very close to the neighbouring azido group on the same carbon atom position. Thus, this proximity or possible coordinating effect (see also Section 4.4) of the azido group and the resulting triazole ring as metal ligand could accelerate the second CuAAC at the same carbon position to give the polymer with the double CuAAC region and the unreacted diazide region at the stage of the first CuAAC with 18. This case illustrates that multiple azido groups can be distinguished for site-selective conjugation through an appropriate strategy or molecular design that takes into account the characteristics of organic azide structures.
 | ||
| Scheme 4 CuAAC functionalization of the geminal diazide polymers and click site control. | ||
The following chapter provides a summary of how chemists distinguish multiple azido groups to achieve azido site-selective reactions, such as click conjugation for multicomponent integration.
4. Azide-site selective reaction strategies for diazides (N3 × 2)
We begin with a review of reported synthetic examples that distinguish two azido groups for inter- or intramolecular azide-site selective multicomponent conjugation reactions. Each section in this chapter is organized based on the structures of the organic azides, the reactions used, or the overall strategy.4.1. Distinction through enclosure with sterically bulky groups
Distinguishing reactive positions based on steric bulkiness is one of the most intuitive methods and is often used in azide click coupling reactions. Due to the different accessibility of the reactive species to the azido groups, the azido groups surrounded by bulky substituents persist while the less hindered azido groups readily undergo the reaction (Fig. 4). | ||
| Fig. 4 Differentiation of reactive azido sites by steric bulkiness. | ||
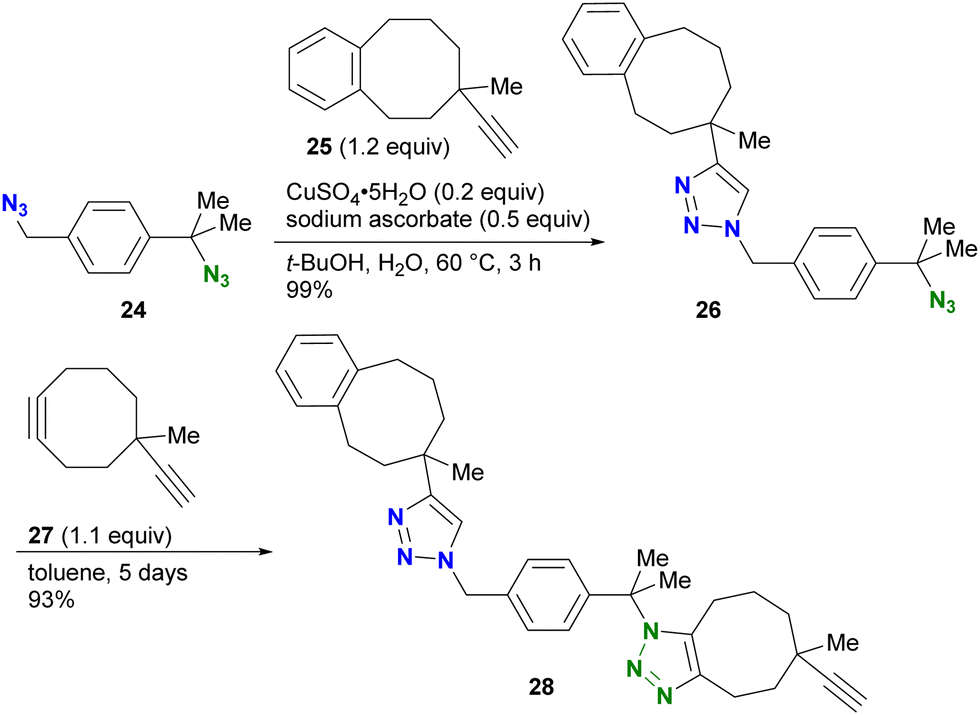
For example, in the study by Koert and colleagues, CuAAC of the diazide compound 24 with primary and tertiary alkyl azido groups with the sterically bulky alkyne 25 selectively occurred only at the primary alkyl azido position, yielding 26 (Scheme 5).87 Subsequent SPAAC conjugation to the remaining sterically hindered azido position led to the formation of the doubly clicked product 28. However, the use of steric hindrance for distinguishing the reactivity is a double-edged sword, especially when the researchers want to react the sterically hindered functions. Indeed, SPAAC of the tert-alkyl azido group in 26 with 27 required five days to complete the reaction.
 | ||
| Scheme 5 CuAAC example demonstrating azide-site click conjugation of the diazide by steric bulkiness. | ||
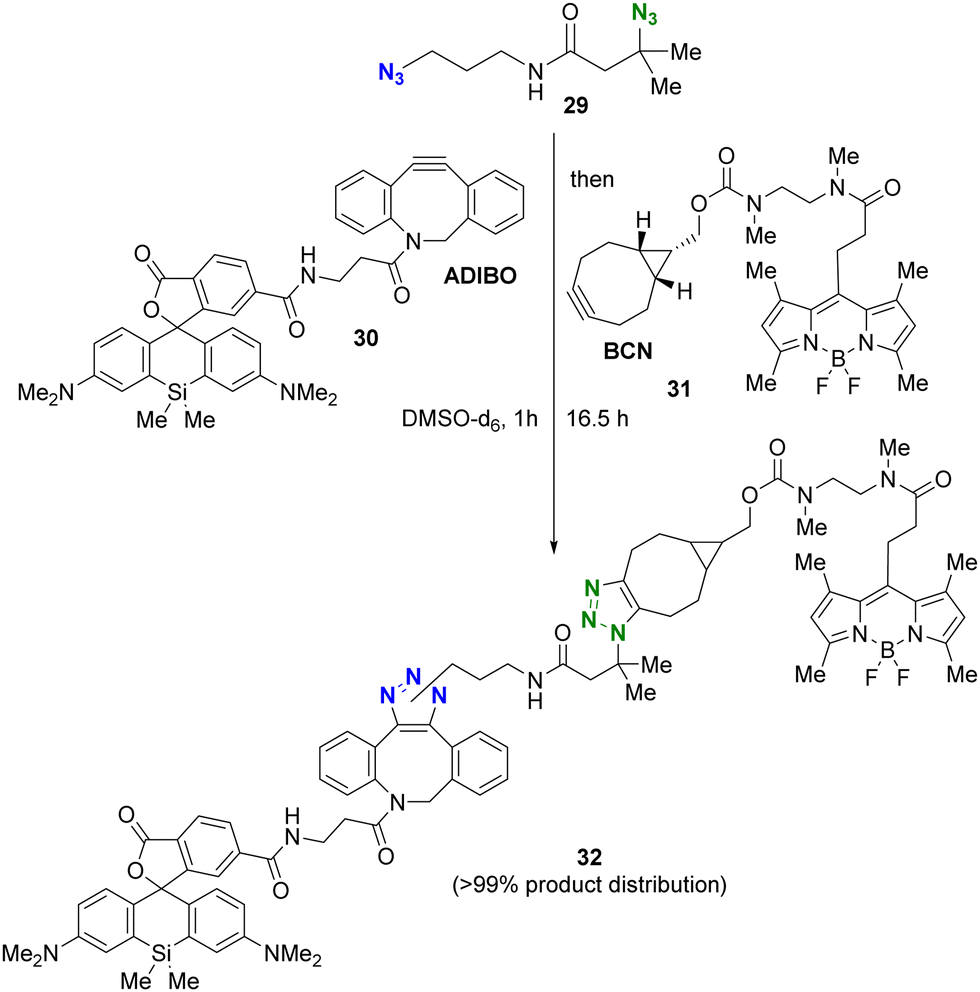
The use of more reactive cycloalkynes offers the potential to enhance the rapid SPAAC reaction with bulky alkyl azides. In a chemoselectivity study conducted by the Bickelhaupt and Mikula group (Scheme 6),88,89 the primary alkyl azido moiety of diazide 29 was first subjected to CuAAC with the ADIBO (AzaDIBenzocycloOctyne) conjugated fragment 30. Upon completion of the first click reaction, compound 31 containing BCN (BiCycloNonyne), a more reactive cycloalkyne compared to dibenzocyclooctyne-type alkynes, was introduced to undergo SPAAC with the persistent tert-alkyl azido moiety in a one-pot fashion. After 17 hours, the final product of the double-labeled conjugate 32 with sila-rhodamine and BODIPY was obtained with a distribution of more than 99% among the click products obtained.
 | ||
| Scheme 6 Sequential double SPAAC conjugation with the diazide through the difference in steric hindrance. | ||
The azido site-selective click reaction using steric repulsion is also demonstrated for vinyl azides (Scheme 7). The research group of Hosoya and Yoshida developed the vinyl azide of acrylamide (α-azidoacrylamide) as a versatile modular synthetic platform.90 First, para-ethynyltoluene 33 was attached to the less hindered vinyl azido moiety in diazide 34 by CuAAC, yielding 35 in excellent yield. The second CuAAC was then performed using ethynylbenzene 18 at the tert-alkyl azide position, yielding 36. In addition, using the acrylamide skeleton, the aza-Michael addition of amine 37 afforded the ethynyl chain conjugate 38 as part of the triple click product.
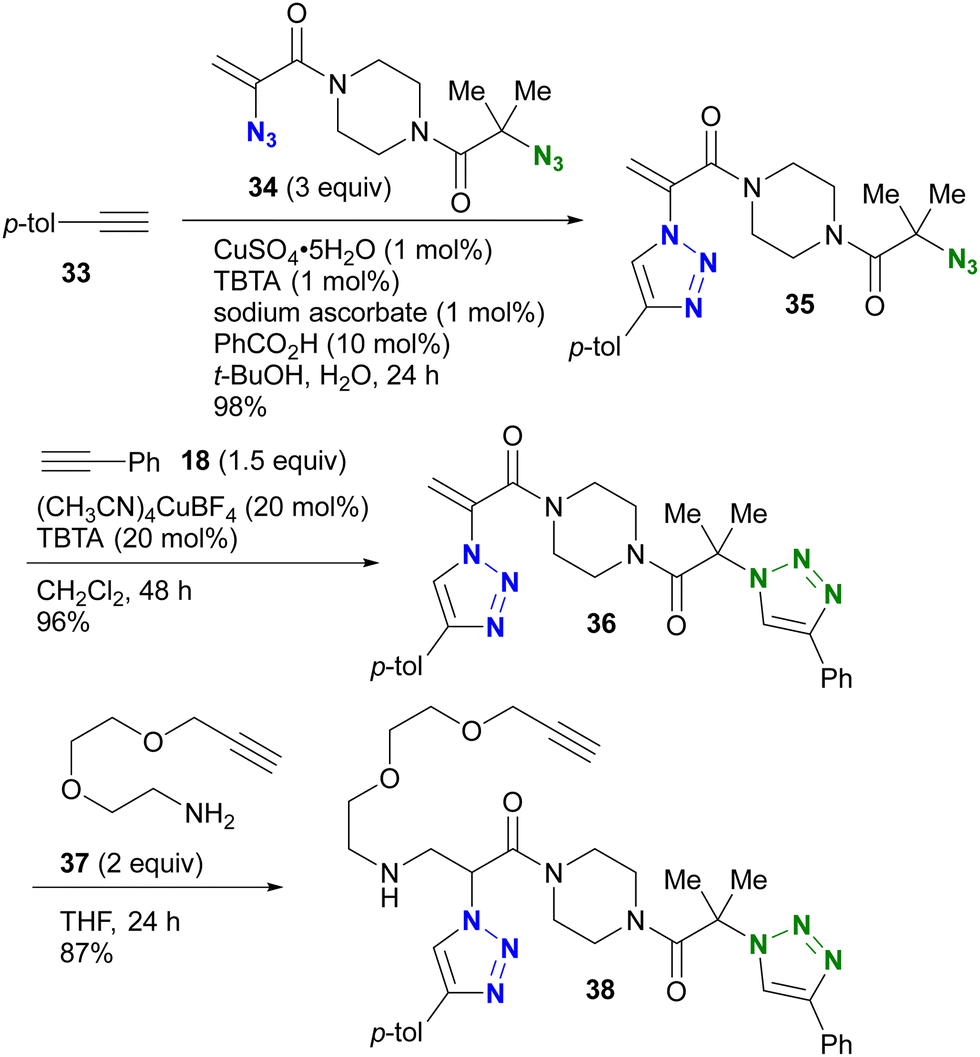 | ||
| Scheme 7 Sequential triple click conjugation of the vinyl azide-containing diazido platform via steric azido site-selective CuAAC reactions and Michael addition. | ||
As mentioned above, steric bulkiness is commonly used to attenuate azide reactivity. However, for Csp2 azides, such as aryl and alkenyl azides, the opposite reactivity occurs in certain cases (Fig. 5).91 The research group of Hosoya and Yoshida reported the distinct and rapid click reactivity of 1-azido-2,6-diisopropylbenzene structures compared to typical aryl azides, despite their apparent bulkiness. While the azido groups in aryl azides are typically conjugated to aromatic rings to maintain planarity, thereby reducing the nucleophilicity of the azido groups (as discussed later in Section 4.2), the steric bulkiness induced by the 2,6-disubstitution disrupts the planarity. As a result, the azido groups are no longer conjugated to the aryl groups. In addition, the steric repulsion induces a bent conformation in the azido group, making it more reactive. Cosequently, these aryl azides can be distinguished from other aryl azides. The research group also reported that apparently sterically hindered aryl azides exhibit accelerated reaction rates in the presence of para-electron donating groups. Similar reactivity patterns have been observed in sterically hindered alkenyl azides.92,93
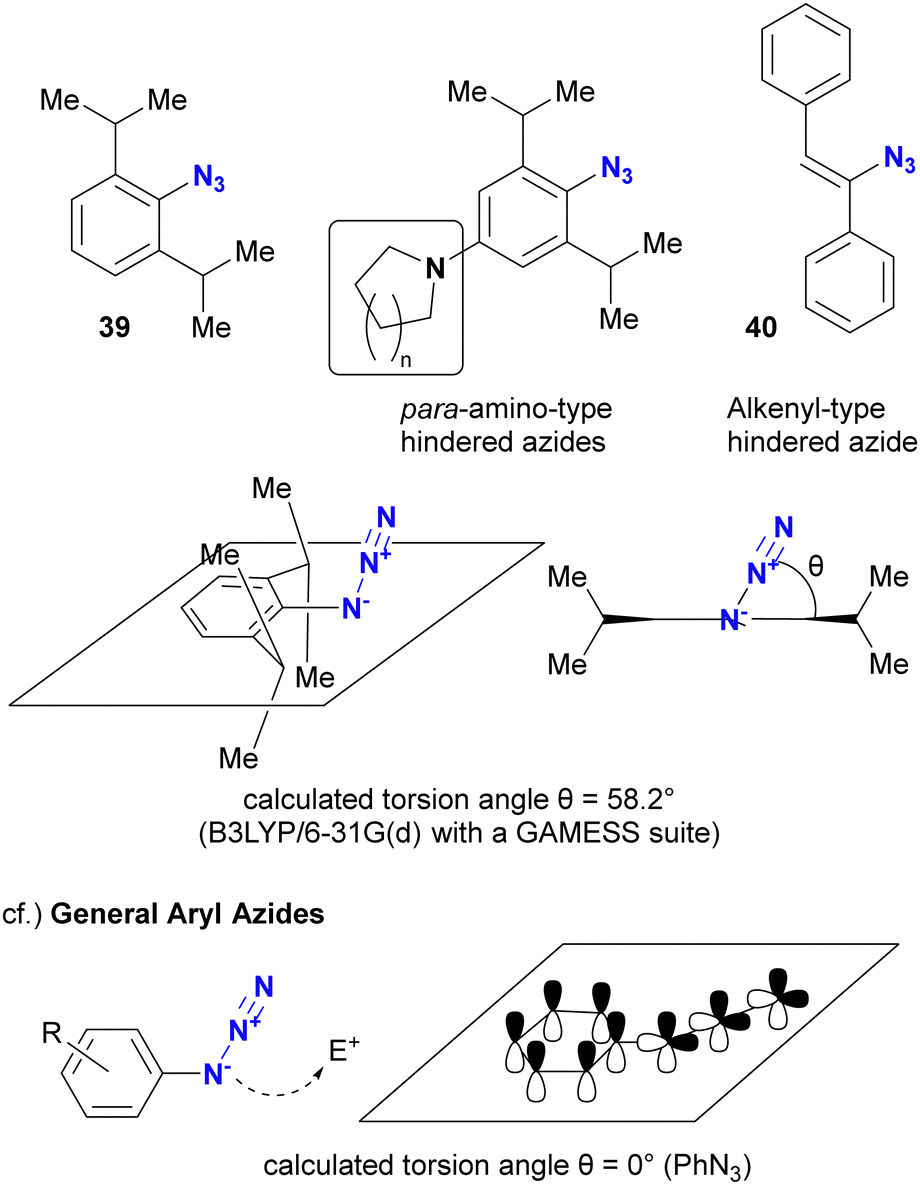 | ||
| Fig. 5 Reactivity of sterically hindered aryl and alkenyl azides. | ||
The difference in reactivity between bulky aryl azides and typical aryl azides is evident in nucleophilic addition reactions. For example, Tanimoto and co-workers reported the propargyl cation-mediated rapid formation of triazole skeletons, such as 42, and its application in three-component coupling reactions and the synthesis of pharmaceutical candidates (Scheme 8(a)).94,95 Although this reaction is also discussed in Section 4.4 as an azide-site selective reaction, this section will focus exclusively on the case of bulky aryl azides. In this reaction, the nucleophilicity of the organic azide attacking the propargyl cation of the strong electrophile is critical. Although nucleophilic alkyl azides, including the bulky 1-azidoadamantane 44, gave triazole products in high yields, the non-nucleophilic azidobenzene 45 and sulfonyl azide 46 did not give the desired results. However, 2,6-diisopropyl-azidobenzene 39, despite its apparently sterically shielded azido group, easily gave the coupled product in 85% yield. While the azide-selective reaction is not explored in this context, this result highlights the significant difference between bulky and general aryl azides.
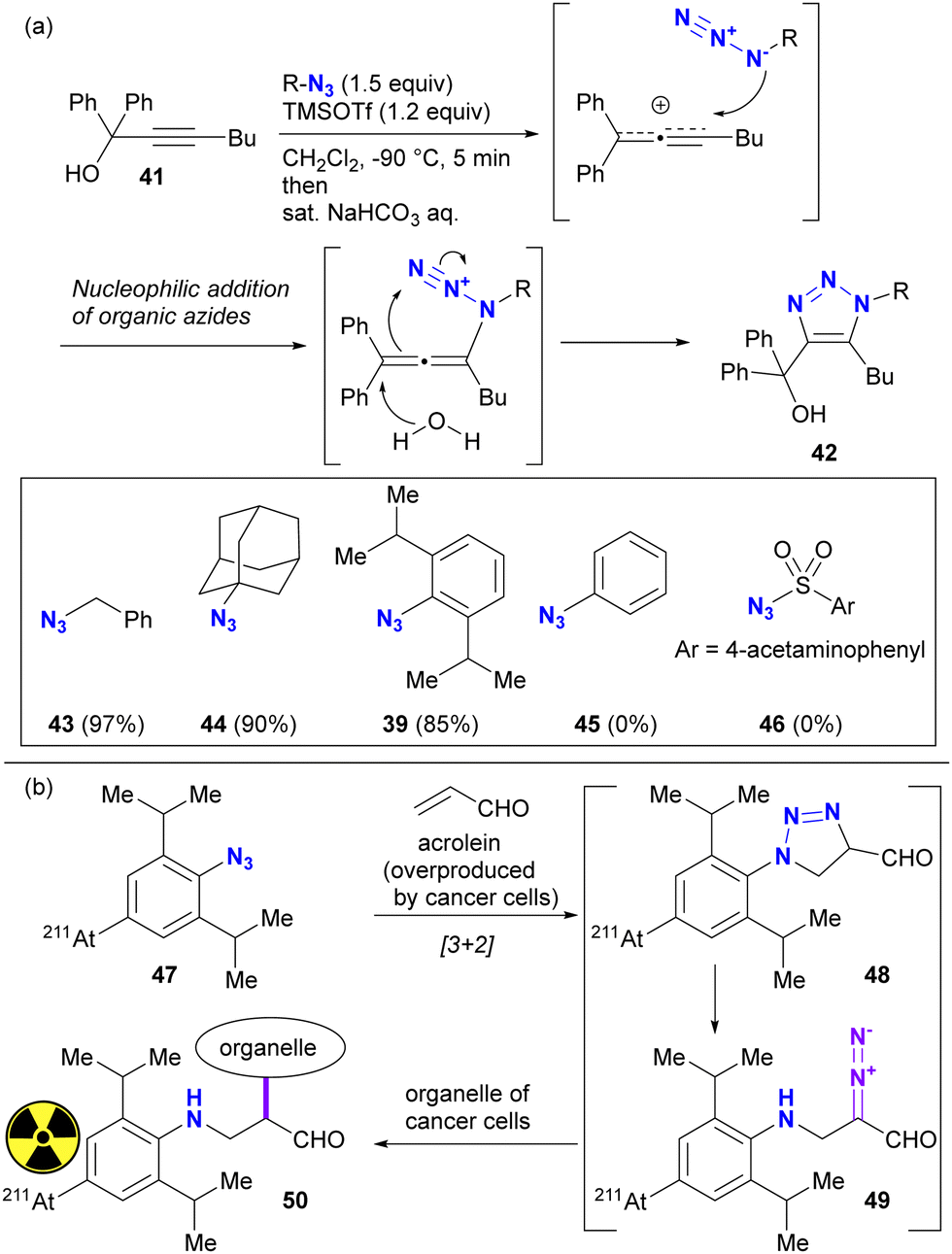 | ||
| Scheme 8 Examples of the reactivities of 1-azido-2,6-diisopropylbenzenes; (a) propargyl cation-mediated triazole synthesis and the reaction scope of organic azides, including bulky aryl azide; (b) radioactive element-containing bulky aryl azide for labeling the cancer cells through [3+2] reaction with acrolein. | ||
The specific reactivities of the bulky aryl azides have also been utilizied in the potential therapeutic purposes (Scheme 8(b)). Acrolein has been known to be overexpressed by most cancer cells. Tanaka and Pradipta's group developed the acrolein-targeting labeling strategies using 2,6-diisopropylazidobenzenes,51–55 and recently reported cancer cell-anchoring with the radioactive astatine-containing azides.54 This sequence consists of [3+2] cycloaddition with acrolein. The resulting triazoline 48 was converted to a diazo compound 49 by spontaneous ring opening. The diazo moiety in this intermediate was incorporated into the labeling of the cancer cell organelle near the acrolein.
Taking advantage of these properties, the group of Hosoya and Yoshida demonstrated the azide-site selective reaction using diazides composed of sterically bulky aryl azide and non-bulky aryl azide (Scheme 9).91–93 The SPAAC of DIBO (DIBenzocycloOctyne 51) with diazide 52 gave SPAAC products 53a and 53b, with the predominant product being 53a, which was obtained by cycloaddition at the bulky group-surrounded azide moiety due to the bent azido structure and deconjugation from the aryl group (Scheme 9(a)). Conversely, triazole formation with acetylacetone46 from diketone (this reaction is discussed in the next Section 4.2) occurred at the less sterically hindered azido site in 52, yielding 55, which exhibited the opposite selectivity to SPAAC (Scheme 9(b)). This result clearly illustrates that the reactivity of sterically hindered aryl azides differs significantly from that of general aryl azides. Furthermore, the introduction of an electron-donating group at the para-position of the bulky azide enhances its click reactivity. The amino group of the electron-donating group serves as an effective scaffold for tethering molecules, allowing the versatile design of reactive multi-azide platform compounds. Indeed, diazides 56 with the cyclic amine piperazine linker, recently selected as the PROTAC linker,96–98 demonstrated successive and azide-site selective integration via SPAAC, followed by CuAAC with propargyl alcohol 57, yielding 58 in excellent yields. In addition, the same research group reported a case involving sterically hindered vinyl azides. In the competitive reaction of DIBO 51 with bulky 40 and non-bulky 59, the bulky azide-selective SPAAC proceeded, yielding 60a as the main product (Scheme 9(c)). This strategy is also presented in the final chapter on multicomponent azide-selective conjugation.
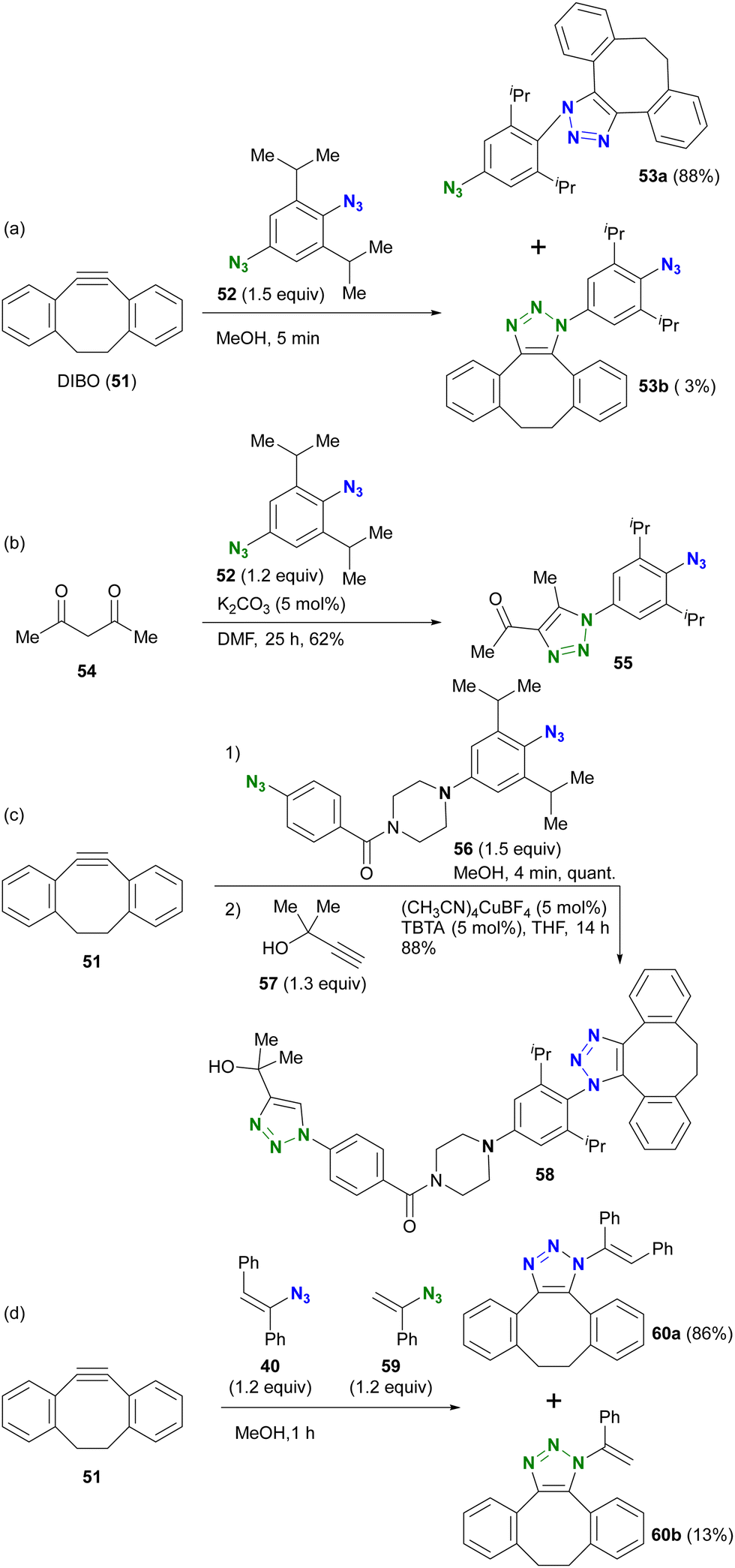 | ||
| Scheme 9 Azide-site selective conjugation reactions using sterically bulky aryl azides. | ||
As discussed in this section, steric hindrance usually imparts an inert reactivity to the alkyl azido groups, resulting in azide-site selective conjugation of the less hindered azido position. However, this factor leads to different properties in the case of aryl azides. Therefore, the exploitation of azido nucleophilicity is expected to play a key role in future efforts related to the multi-azide compounds as well as general synthetic organic chemistry to achieve complex functionalization.
4.2. Distinction between alkyl and aryl azides based on different properties
In addition to steric hindrance, one of the most noticeable differences between azide compounds is whether they are alkyl or aryl in nature. Alkyl azides typically exhibit both nucleophilic reactivity at the N1 position and electrophilic reactivity at the N3 position. In contrast, aryl azides function predominantly as electrophiles and exhibit low nucleophilicity (see Scheme 8 and Fig. 6). Some of their properties have already been outlined in the previous section (Section 4.1). This variance is due to the conjugation of aryl and azido groups. In addition to reducing the N1 nucleophilicity and increasing the electrophilicity, this extended conjugation can also affect the 1,3-dipolar properties of the azido group in terms of HOMO/LUMO levels. As a result, aryl and alkenyl azides exhibit a different nature and reactivity compared to alkyl azides. This section presents azide-type selective reactions between alkyl and aryl azides.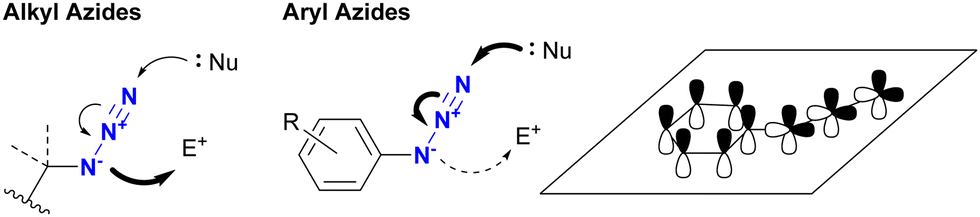 | ||
| Fig. 6 The difference in reactivity between alkyl and aryl azides. | ||
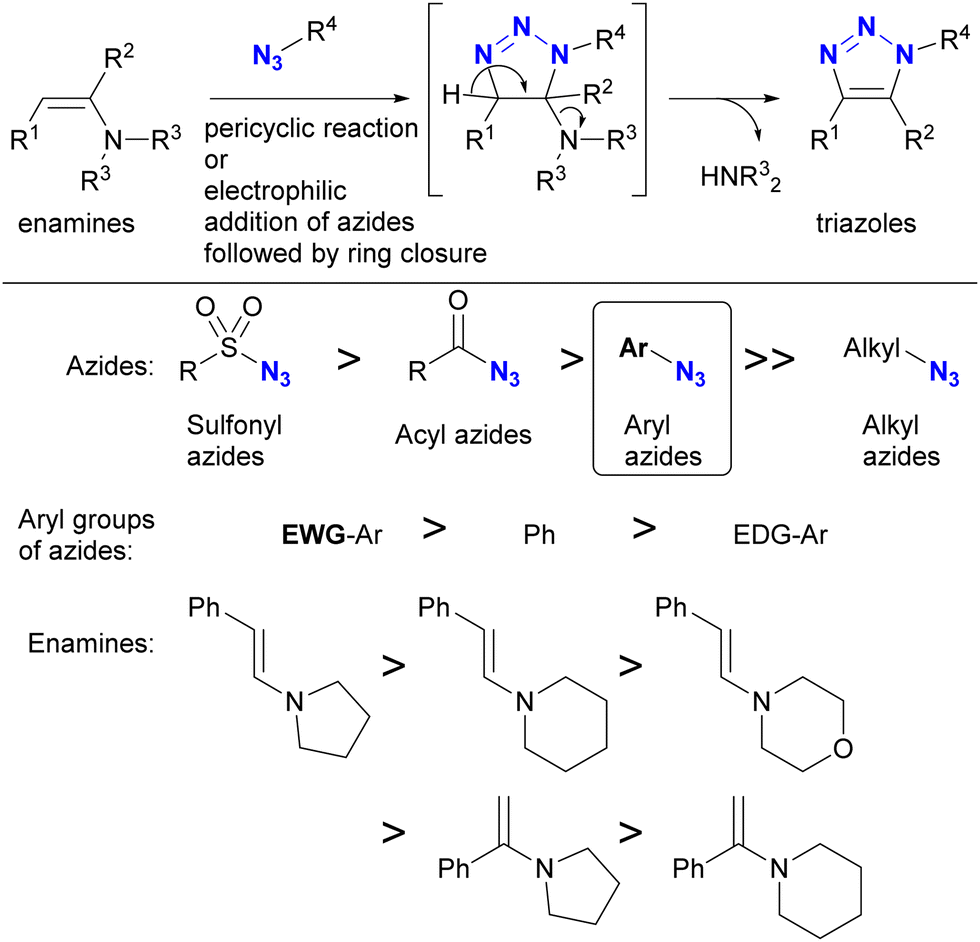 | ||
| Scheme 10 Enamine-azide cycloaddition for the synthesis of triazole rings and the reactivity orders of enamines and azides. | ||
As with enamines, the reaction proceeds with enol and enolate, and most reactions are not performed with ketones or aldehydes as starting materials (Scheme 11). Enolates or enamines are formed in the flask with a base or amine catalyst.104–106 For example, from the pioneering work of the Ramachary group,104 aldehydes 61a–d with activated α-methylenes were converted to the corresponding triazoles 62a–d using aryl azides and the amine base catalyst (Scheme 11(a)). The azide species were critical for successful results, and aryl azides provided the desired products in high yields. On the other hand, the benzyl azide 43 of the alkyl azide provided the triazole 62g in low yields, even when a stronger alkoxide base was used. In the case of the aldehyde enolate, as mentioned above, the tosyl azide of the sulfonyl azide did not afford the N-substituted triazole 62h. Due to the accessibility of enolization, β-ketoesters or 1,3-diketones are often chosen as conjugation scaffolds.105 Indeed, acetylacetone 54 and methyl acetoacetate 64 converted 4-nitrophenyl azide 63 to the triazoles 65a and 65b of the coupled products in good yields. As with aryl azides, vinyl azides 66 can be used as substrates.106
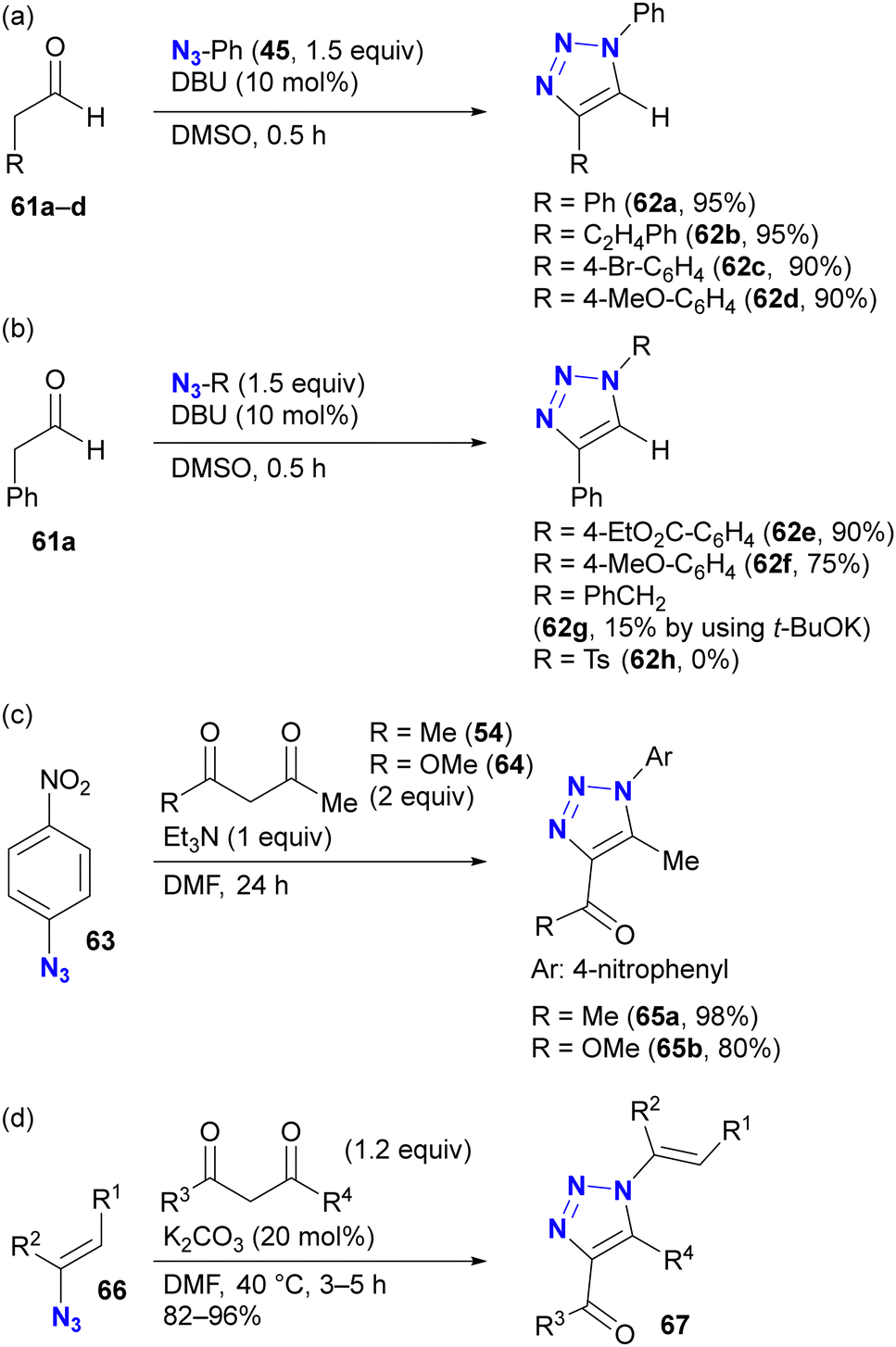 | ||
| Scheme 11 Enolate-mediated triazolization reactions with organic azides. | ||
The enol/enamine-type triazolization reactions described herein successfully facilitated azide-selective triazole synthesis (Scheme 12). Using Chiba's reaction conditions with acetylacetone 54,106 Hosoya et al. demonstrated that enolate-mediated triazolization exhibited aryl azide selectivity in the presence of alkyl, aryl, and bulky aryl azides, resulting in the preparation of 68b from phenyl azide 45 in 84% yield.107 The sterically unhindered aryl azide-site selective reactions in the aryl diazides have already been presented in Scheme 9.
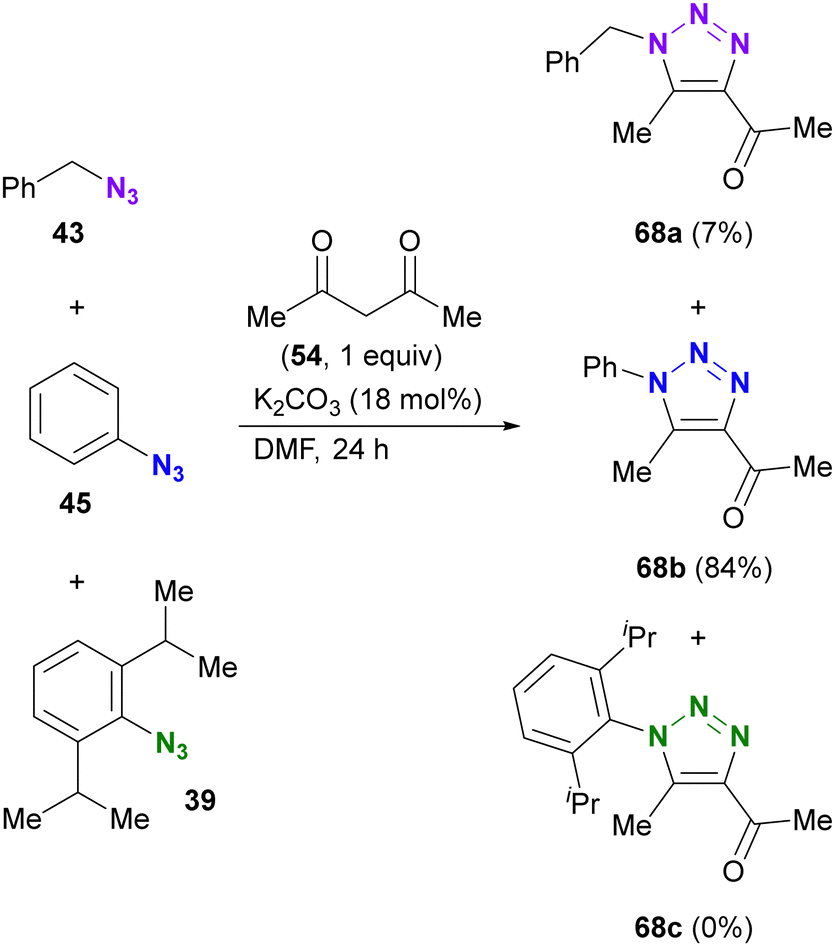 | ||
| Scheme 12 Aryl azide-selective enolate-mediated triazolization reaction in the presence of alkyl, aryl, and bulky aryl azides. | ||
 | ||
| Fig. 7 Staudinger reactions of electron-poor aryl azides to stable iminophosphoranes and the aryl azide scaffolds. | ||
Taking advantage of the acceleration effect in this reaction, Yang, Rangström, and co-workers have developed chemistry using the 4-azido-2,3,5,6-tetrafluorobenzoyl group as a fast Staudinger reaction azide scaffold to construct robust aza-ylide structures.114,115 Separately, Yi and Xi's group has also explored o-fluorinated aryl azides for labeling or sensing applications in chemical biology.116–120 In their research, azide-site selective one-pot double conjugation was demonstrated (Scheme 13).121 Using diazide 69, which consists of an aryl azide of tetrafluorobenzamide and an alkyl azide, the first Staudinger reaction selectively formed the iminophosphorane at the aryl azide moiety with NBD (NitroBenzoxaDiazole)-conjugated phosphine 70 due to the enhanced electrophilicity of the fluoroaryl groups. Subsequently, the second Staudinger reaction of the traceless type with azo-benzene-linked phosphine 71 provided the ligated amido bond. Overall, the three-component conjugated product 72 was obtained in 36% yield (60% per conjugation step) with excellent azide site selectivity. A comparison of reaction times (overnight for aryl azide to 96 h for alkyl azide) illustrates the improved reactivity of the aryl azido group. It should also be noted that aryl iminophosphoranes containing the electron-difficient arenes are stable and do not undergo further Staudinger–Bertozzi ligation, whereas the alkyl azides form the amide through the ligation reaction.
 | ||
| Scheme 13 Azide-site-selective sequential double Staudinger reaction conjugation of aryl- and alkyl-azido-containing diazide. | ||
In addition to electron density, steric factors also play an important role in the reactivity of aryl azides. Hosoya and Yoshida's group developed a chemistry involving 1-azido-2,6-dichlorobenzenes (Scheme 14).122 Due to their electron-withdrawing nature and steric hindrance, the Staudinger reaction rapidly yields water- and air-stable iminophosphoranes. Taking advantage of this property, diazide 73, consisting of alkyl azide and 4-alkoxycarbonyl-2,6-dichloroaryl azide, was introduced into the solution of triphenylphosphine and DIBO 51, yielding the azide-site-selectively clicked product 74 in 93% yield. This orthogonality is due to the rapid Staudinger reaction of the o-dichloroaryl azides, which exceeds the SPAAC of 73. The researchers also demonstrated that Staudinger–Bertozzi ligation to the amide bond does not occur with the iminophosphoranes of the 2,6-dichloroarylazido group. The robustness of this aza-ylide has found applications in the chemical modification of proteins in living cells by their group122 and also in the synthesis of hydrolysis-resistant amphiphilic molecules by the group of Yamashina and Toyota.123
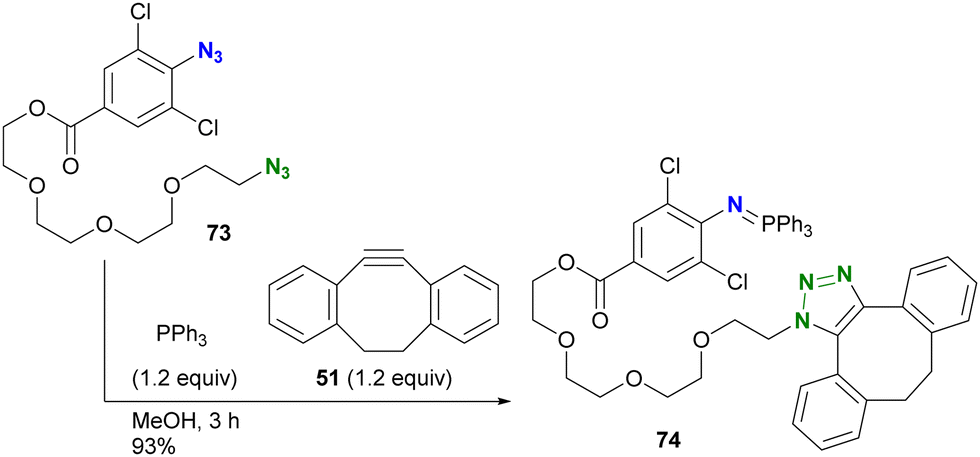 | ||
| Scheme 14 Three-component coupling reaction of diazide by o-dichloroaryl azide-selective Staudinger reaction forming the stable aza-ylide and by SPAAC of alkyl azide. | ||
As shown in Schemes 13 and 14, 2,3,5,6-tetrafluoroaryl and 2,6-dichloroaryl azides can provide azaylides much more rapidly than alkyl azides. Recently, Yoshida and co-workers demonstrated the azide-site selectivity between these two aryl azides in the Staudinger reaction (Scheme 15).124 Notably, tetrafluoroaryl azides showed the best reactivity among 2,6-dichloroaryl and alkyl azides, and excellent selectivity was achieved with o-alkoxycarbonyl-substituted aryl phosphines. This selectivity was also exploited as a triple click platform compound 76 of the ethynylated diazide. Successive conjugation to each click group was achieved to obtain 79 in good yields.
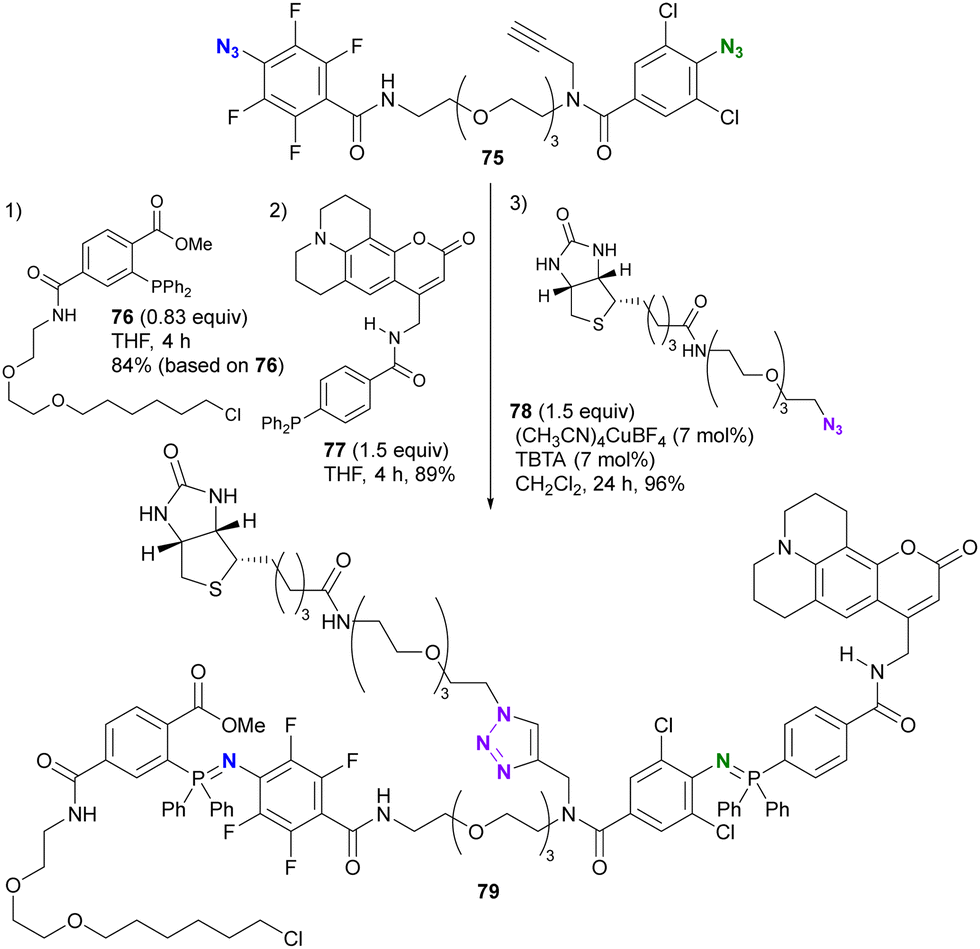 | ||
| Scheme 15 Selective Staudinger reaction distinguishing the tetrafluoroaryl and o-dichloroaryl azide moieties in the alkynyl diazide platform compound. | ||
As highlighted in the introduction to this section, the stability of iminophosphoranes also depends on the choice of phosphines. When chemists want to reduce azido groups on aromatic rings, they often face the problem of forming stable and robust aza-ylides. The group of Yoshida and Hosoya investigated suitable phosphines for the Staudinger reduction of electron-withdrawing aryl azides (Scheme 16(a)).125 The results show a complex trend influenced by various parameters, including electron donating/withdrawing properties, cone angles of the phosphines, and steric bulkiness, which affect the stability of the products (aza-ylides or phosphazides) and reduction reactivity. After extensive research, they identified tributylphosphine or its salt as a suitable and readily available reagent for Staudinger reduction without the formation of stable aza-ylides. The successful choice of phosphine also allowed the selective reduction of the aryl azide group in the presence of the alkyl azido moiety (Scheme 16(b)). Although this is a reduction reaction, understanding the reactivity of phosphines with organic azides and the properties of the reaction products could be expected to improve the efficiency of site-specific reactions in multi-azide scaffolds and their versatile conjugation to multi-component systems.
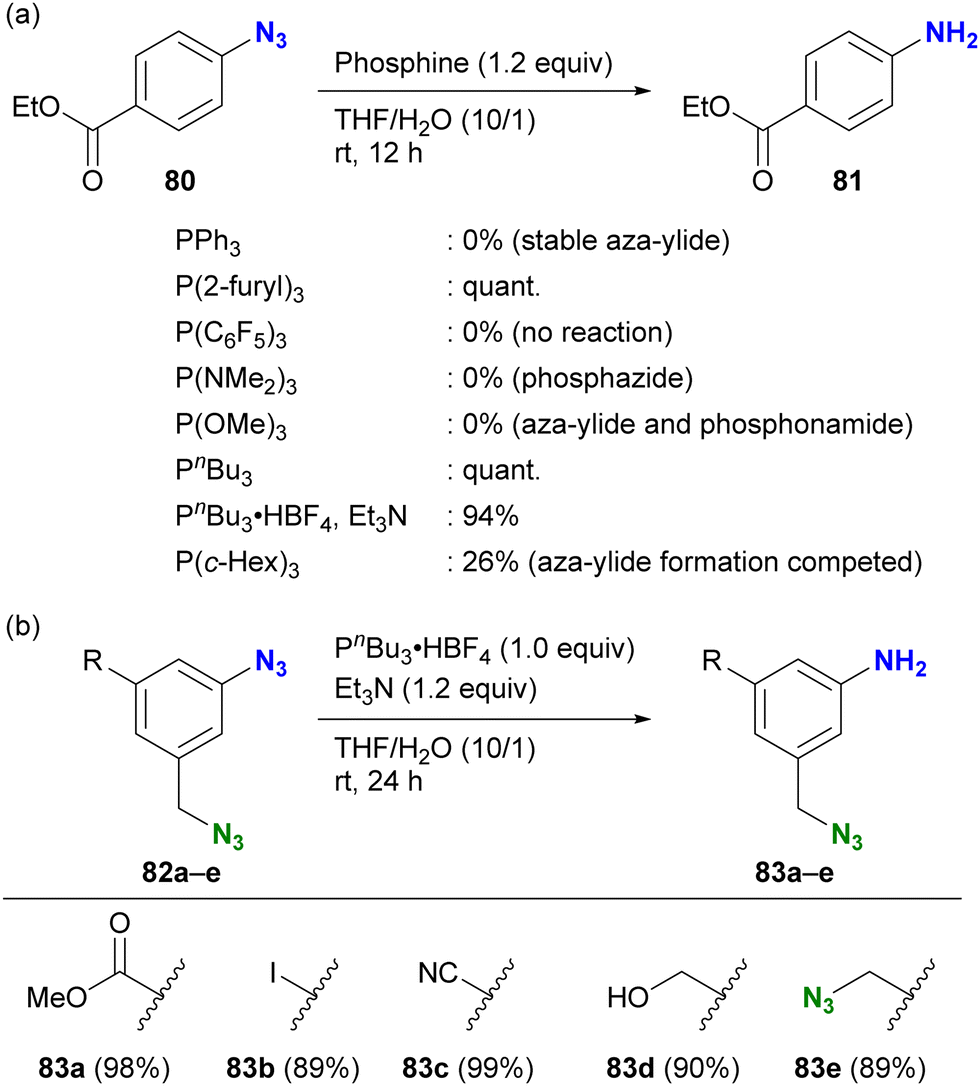 | ||
| Scheme 16 Aryl azide-selective Staudinger reduction (a) scope of phosphines without formation of aza-ylides (b) application to the di- and triazide substrates. | ||
The incorporation of sterically bulky and electron-withdrawing aryl azide moieties can increase the selectivity of azide sites in SPAAC reactions. As shown in Table 1 from the research of Bickelhaupt and van Delft,126 SPAAC reactivities are influenced by the combination of organic azides and cycloalkyne azidophiles. Reaction rate trends with organic azides also vary among different cycloalkyne species. In particular, the steric hindrance of the azido moiety affects DIBAC (DIBenzo-aza-cyclooctyne, 91). In Table 1, the electron-poor aryl azide pyridinium salt 89 showed the most significant rate constant with BCN 90 and the lowest value with DIBAC 91, resulting in excellent cycloalkyne selectivity.
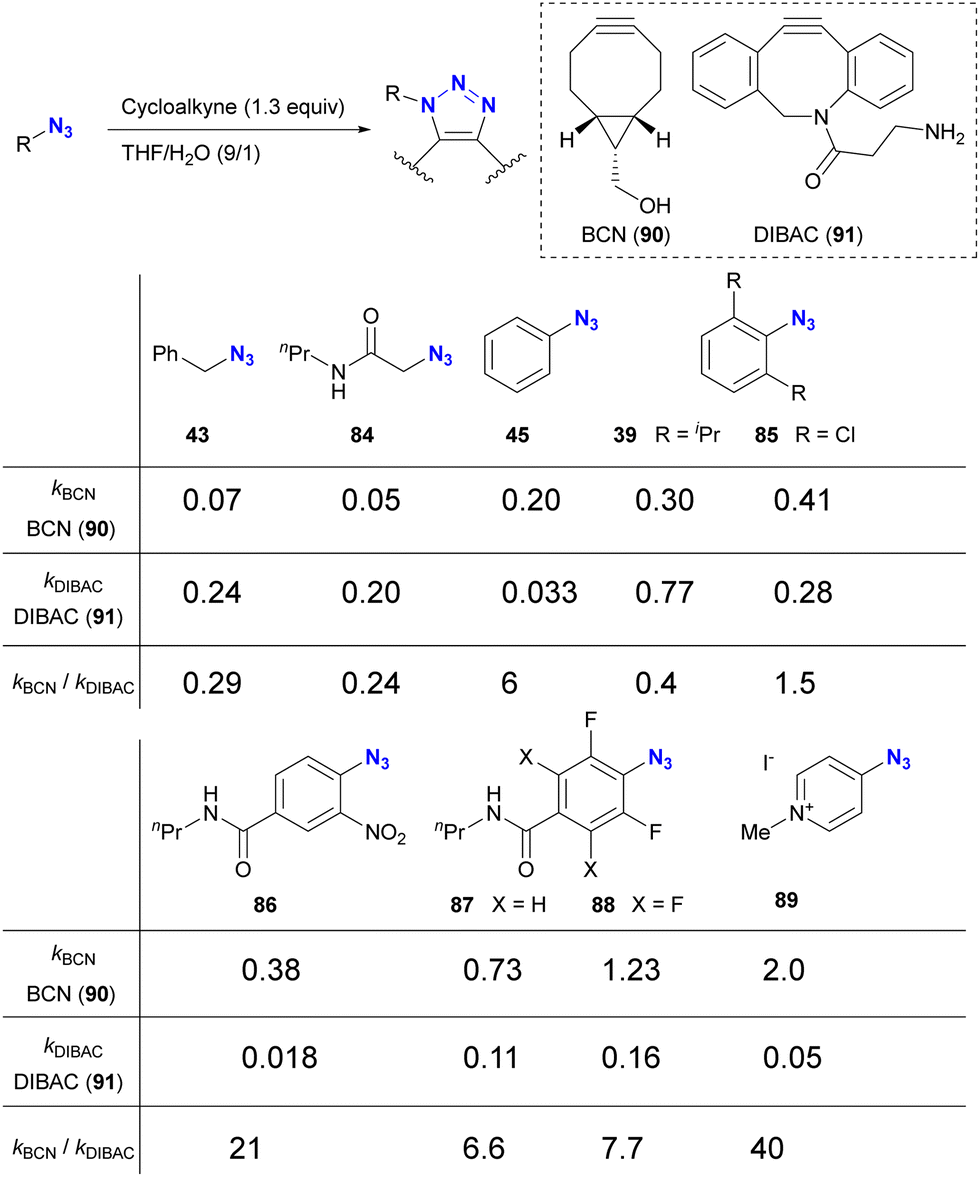
|
However, taking into account stability, availability, and multifunctionality, Bickelhaupt, van Delft, and their colleagues found that 4-azido-2-nitrobenzamide 86 was the most efficient organic azide structure for maximizing the reactivity difference between BCN 90 and DIBAC 91 in SPAAC reactions.126 Building on this discovery, the diazide 92, composed of 4-azido-2-nitrobenzamide and a primary alkyl azide, successfully underwent a double SPAAC reaction to yield the site-selective click product 93 in 83% yield (Scheme 17). As introduced here, both electron-withdrawing and steric factors play a crucial role in achieving selective conjugation reactions and also allow the incorporation of additional functions into the click scaffolds.
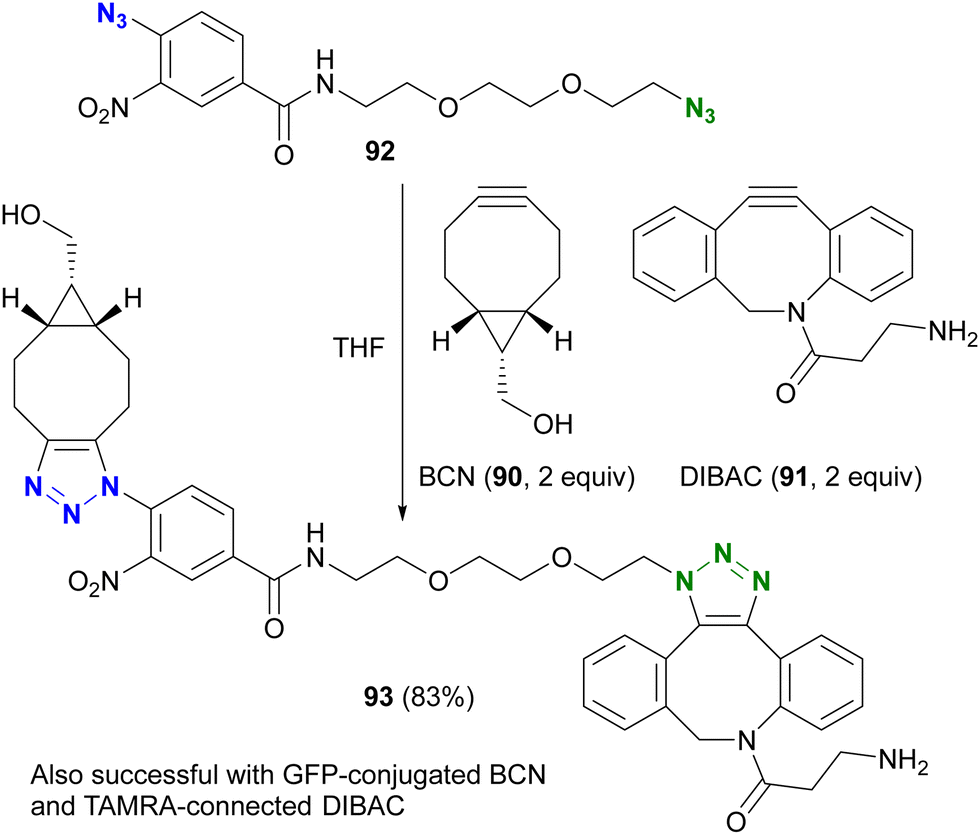 | ||
| Scheme 17 Azide-type selective double SPAAC conjugation with diazide 92 of alkyl and electron-poor aryl azido moieties. GFP: green fluorescent protein. TAMRA: carboxytetramethylrhodamine. | ||
Direct photoactivation of aryl azides under UV light irradiation (approximately 250–400 nm) results in the elimination of dinitrogen from the azido groups, leading to the formation of singlet nitrenes (Scheme 18(a)). These generated nitrenes then undergo further rearrangement to form benzazirine and seven-membered ring ketenimine. Each of these species can trap appropriate functional groups, particularly nucleophilic species, allowing crosslinking of target compounds such as proteins. Recent advances in photocatalysis now allow the use of less harmful visible light.129
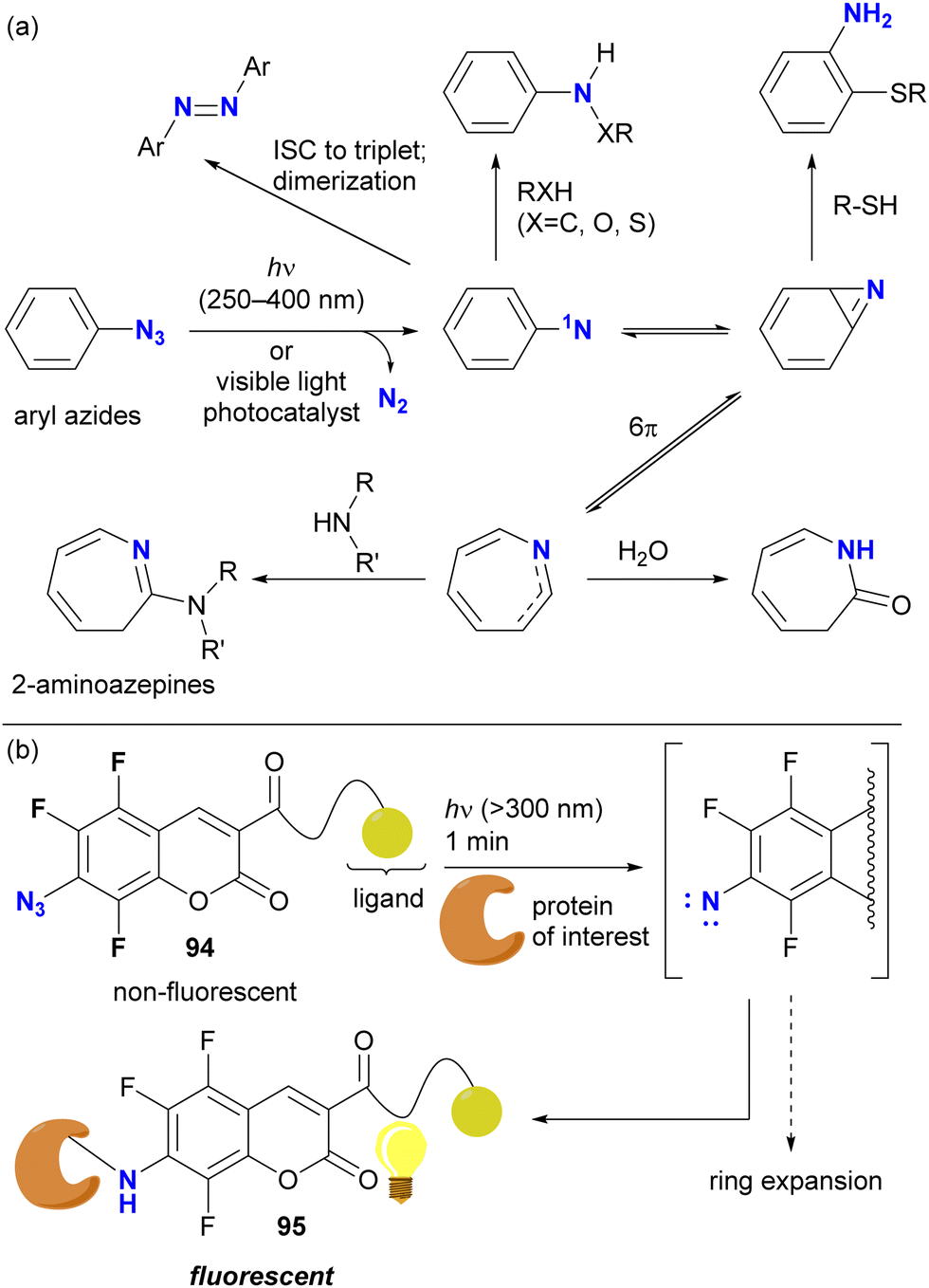 | ||
| Scheme 18 Photochemical generation of aryl nitrenes followed by the ring expansion and amine capture reactions. (a) General reaction mechanisms. (b) Photolabeling of fluorinated azidocoumarins suppressing aromatic ring expansion. ISC: intersystem crossing. | ||
With the aforementioned properties, aryl azides have been established as scaffolds for photoaffinity labeling through the generation of aryl nitrenes. In most cases, however, the contribution of the nitrene itself to the photolabeling is insufficient due to the short lifetime and the associated aromatic ring expansion, as illustrated. Because of the different nature of nitrenes and (cyclic) ketimines, the specificity of labeling varies. To suppress or slow down the nitrene rearrangement, the aryl moiety should be highly electron-deficient, such as ortho-difluorinated azidoarenes.127 Recently, trifluoro-7-azidocoumarin 94, a turn-on photolabeling probe, has been reported (Scheme 18(b)).132 Photo-crosslinking with a protein, achieved by nitrene generation while retaining the benzene ring, resulted in the N-protein-substituted 7-aminocoumarin 95, which showed fluorescence emission. Although azide-site selectivity was not explicitly mentioned, the labeling conditions for 94 (>300 nm, such as UV-A light irradiation in 1 min) are expected to facilitate the development of further complex functionalization via azide-selective reactions.
In contrast to aryl azides, alkyl azides remain intact due to the slower formation of nitrene under the photoirradiation conditions. The unique properties of the organic azide photoreactions allow the selective use of aryl azides in the presence of alkyl azides, this feature enhances the design of complex molecular probes and contributes to the efficiency of functional molecular probe synthesis. The group of Hosoya and Suzuki pioneered the photoaffinity labeling of aryl azides in the presence of alkyl azides (Scheme 19).133 Photoirradiation of triazide 96, containing two alkyl azides and one aryl azide, selectively decomposed the aryl azido moiety at 254 nm. The capture of the intermediate with diethylamine resulted in the formation of the seven-membered ring azepine 97, along with the preservation of the two unreacted alkyl azides.
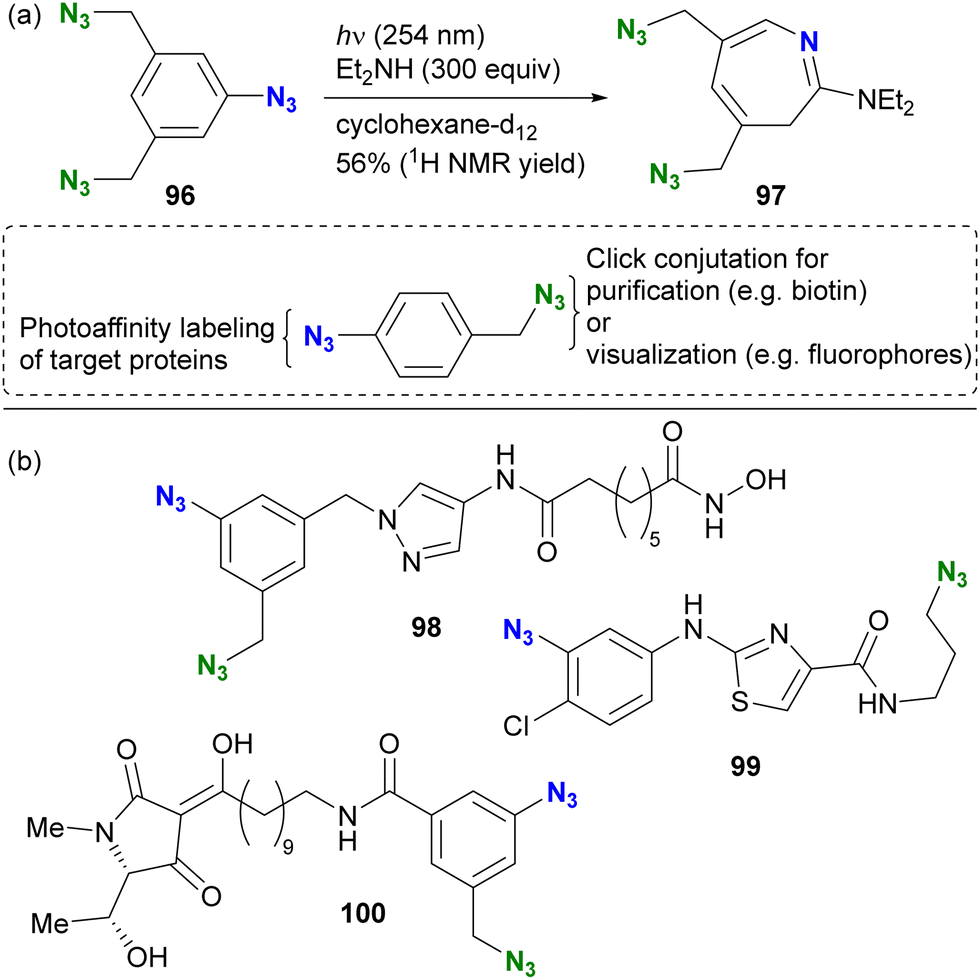 | ||
| Scheme 19 (a) Azide-site selectivity in the triazide consisting of aryl and alkyl azides. (b) Diazide photoaffinity probe molecules with bioactive ligand moieties. | ||
This azide-site selectivity allows post-labeling click reactions of alkyl azides, which is useful for purification or visualization. Several examples of reported diazido chemical probes with aryl and alkyl azido groups are shown in Scheme 19(b). For example, pyrazole 98 showed potent inhibition of histone deacetylase (HDAC8) at 17 nM without affecting neuroprotective properties.134 Thiazole 99 showed significant anti-influenza activity with an EC50 of 9.1 μM.135 Photoaffinity labeling experiments were performed with influenza neuroproteins at 350 nm for 5 minutes, followed by CuAAC to the remaining alkyl azido moiety with a biotin-tethered alkyne. Subsequent trypsin digestion and LC–MS/MS analysis revealed the binding pockets and labeled positions. In addition, Schobart et al. reported the total synthesis of the natural product penicillenol C1, and its diazido analog 100 was also prepared for a photoaffinity labeling assay.136 As the recent revisiting of the photoreaction of aryl azides on skeletal editing strategy,130,131 this photochemical approach is expected to be further developed as well as in multifunctional click chemistry.
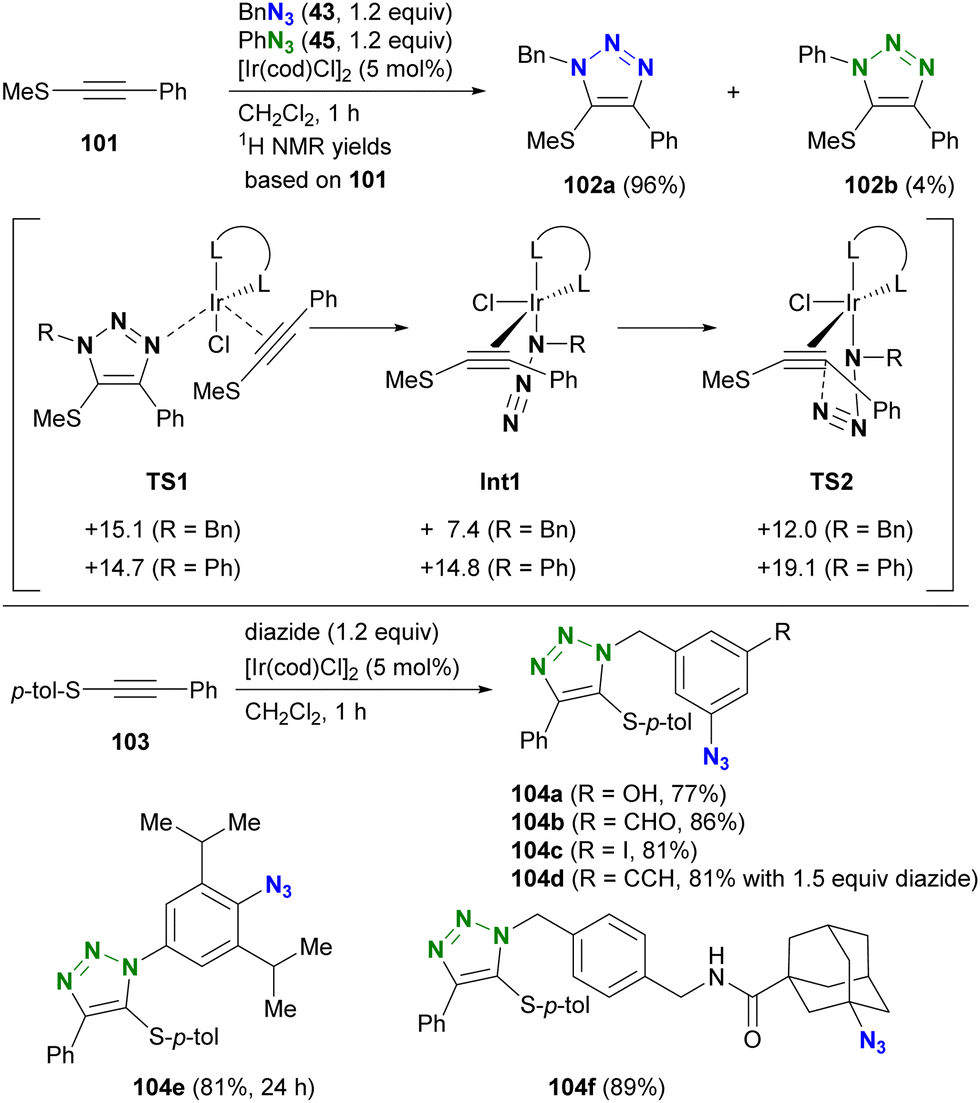 | ||
| Scheme 20 Iridium-catalyzed azido-type-selective triazolization reactions with thioalkynes. | ||
The selectivity of this process is elucidated by density functional theory (DFT) calculations at the M06/LanL2DZ (Ir), M06/6-31G(d) (N, Cl, S, and C atoms of the alkynes and alkenes in the cod ligands), and M06/6-31G (others) levels. Although the initial coordination states (TS1) have similar energy levels, there are differences in the azide-coordinated iridium catalysts (Int1 and TS2), which have lower energy levels with alkyl azides compared to those with aryl azides. Consequently, the iridium-catalyzed reaction of thioalkynes 103 with diazides containing both aryl and alkyl azido moieties selectively yields alkyl azide site-cyclized products 104a–d. The higher energies observed in reactions with aryl azides are attributed to resonance between the aryl and azido groups. Migration to structure TS2, characterized by a highly twisted azido group detached from conjugation with the aryl group, accounts for the increased energies of Int1 and TS2 in aryl azide reactions. In addition, steric repulsion has a significant effect on this reaction. In particular, the less sterically hindered azido moieties lead to the formation of mono-triazole products 104e and 104f. This strategy will be further discussed in the final chapter (Chapter 5) on azide-site selective multicomponent conjugation.
4.3. Distinction by the use of sulfonyl azides
Despite the absence of the C–N3 bond, sulfonyl azides are known to be highly reactive organoazido compounds (Scheme 21(a)). The unique reactivity of sulfonyl azides is due to the highly electrophilic azido moiety introduced by the sulfonyl group. As a result, sulfonyl azides have been widely used in diazo transfer reactions.138–144 These compounds also facilitate molecular coupling. While CuAAC reactions proceed similarly to other organic azides, the strong electron-withdrawing nature of sulfonyl azides allows ring opening of triazole structures, leading to the formation of α-diazoimines (Scheme 21(b)). In the presence of metal catalysts such as copper and rhodium, subsequent transformations involving C–H insertion and ketimine formation become feasible.145–148 Sulfonyl azides also show reactivity with acetylide nucleophiles.149–152 In contrast to CuAAC, which yields 1,4-disubstituted triazoles, this reaction yields 1,5-disubstituted heterocycles (Scheme 21(c)). Conversely, reactions of sulfonyl azides with enamines yield a variety of products, including amidines, N-“unsubstituted” triazoles, and N-sulfonyl enamides, depending on the substrate.103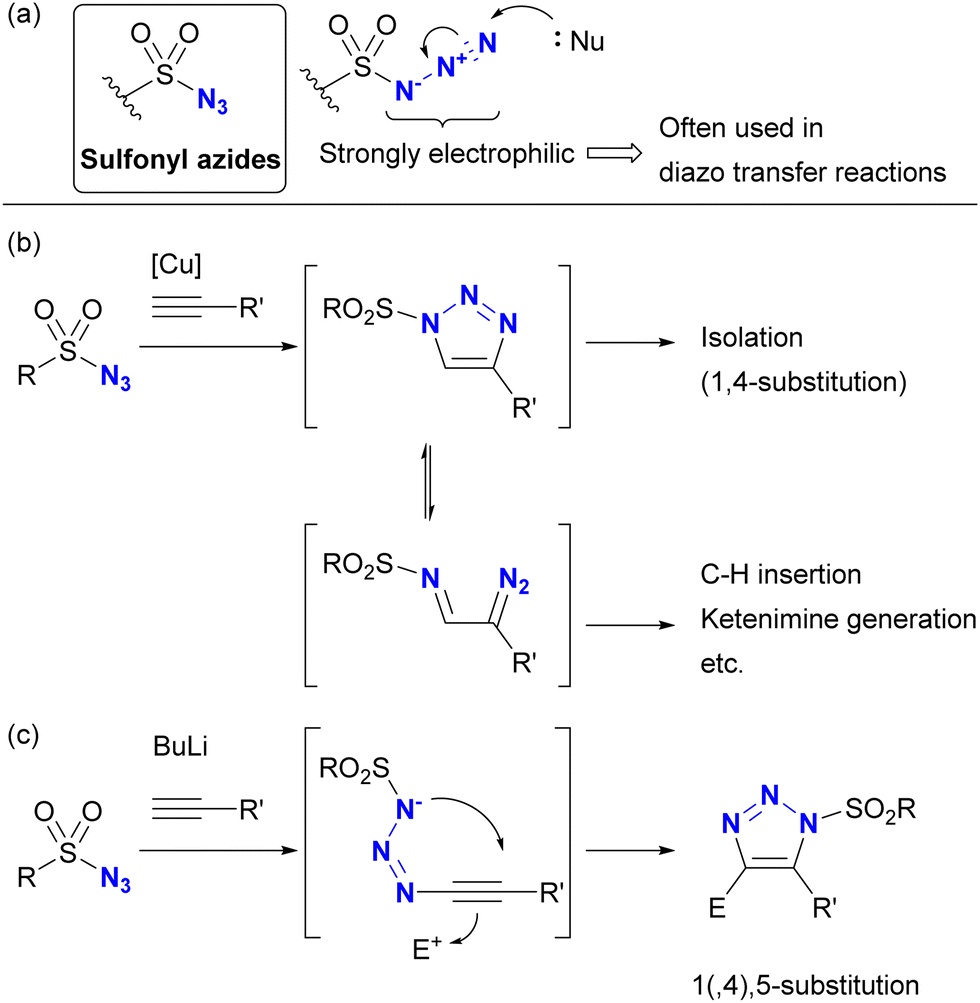 | ||
| Scheme 21 (a) Sulfonyl azides and the triazolization reactions by (b) CuAAC and (c) anion-mediated cycloaddition. | ||
Due to their strong electrophilicity, Staudinger reactions with sulfonyl azides proceed smoothly even in the presence of other azido moieties. Demonstrating the utility of this property, Gothelf's group reported the azide-site selective Staudinger reaction of diazides with sulfonyl and alkyl azido moieties (Scheme 22).153 The solid-supported oligonucleotide 105, linked to the terminal nucleic acid by a phosphite linkage, underwent an azide-site selective Staudinger reaction with diazide 106 containing sulfonyl and alkyl azido moieties, yielding 107. Subsequent solid-supported DNA synthesis followed by the removal of CPG (controlled pore glass) resulted in the formation of 108, which retained a click-functional azido group.
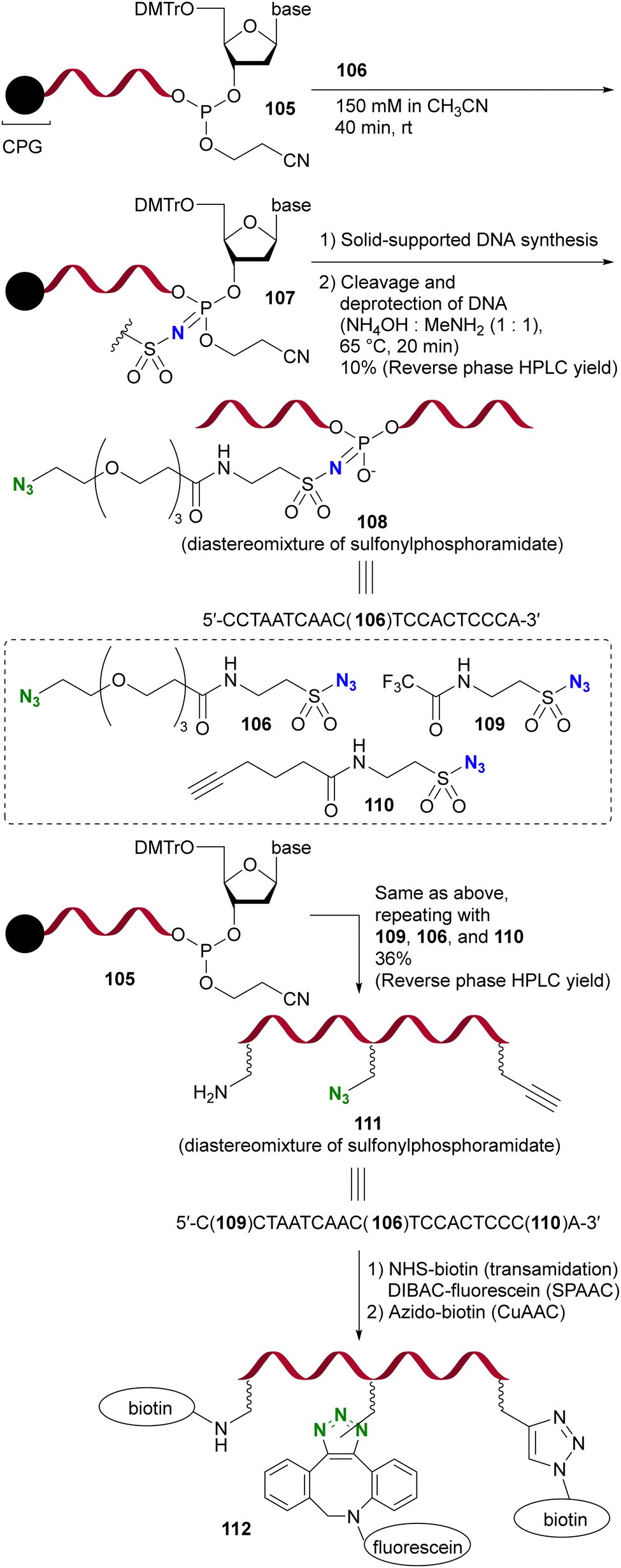 | ||
| Scheme 22 Sulfonyl azide-selective Staudinger reaction in the synthesis of the modified DNA oligonucleotides. CPG: controlled pore glass. DMTr: 4,4′-dimethoxytrityl. | ||
Furthermore, this azido site-selective Staudinger reaction was used for the sequential introduction of multiple chemical handle groups, resulting in the synthesis of 111 by the iterative performance of the Staudinger reaction and DNA synthesis. With the introduced amino, azido, and ethynyl groups, the functionalized single-stranded DNA oligomer 112 was further obtained by additional conjugation, including click reactions.
In addition to the previously mentioned reactions, sulfonyl azides are known to be involved in another specific click conjugation reaction, commonly referred to as the “sulfo-click” reaction.154–157 In this reaction, the click partner for the sulfonyl azide is a thiocarboxylic acid (or thioamide in some cases).158,159 The reaction mechanism is shown in Scheme 23. Analogous to the Staudinger–Bertozzi ligation, the sulfo-click reaction begins with the nucleophilic addition of the thiolate of thiocarboxylates to the azido groups in sulfonyl azides, followed by intramolecular transamidation, resulting in N-sulfonyl amides (N-acyl sulfonamides) of carboxylic acid bioisosteres as click products. This reaction is accompanied by the formation of dinitrogen gas and elemental sulfur. Reaction with thioamides instead of thiocarboxylic acids yields N-sulfonyl amidines.158,159 Notably, electron-poor aryl azides, such as the 4-azido tetrafluorobenzoyl group, have also been reported to undergo sulfo-click reactions.160
 | ||
| Scheme 23 Reaction mechanism of the sulfo-click reaction. | ||
The azide-site selective sulfo-click reaction was investigated by the group of Clavé and Smietana (Scheme 24).161,162 Using the thymidine derivative 113 with an alkyl azide at the 3′ position and a sulfonyl azide attached at the 5′ position through the amide bond, coupling with the thiocarboxylate derivative of uridine 114 occurred via the sulfo-click reaction, resulting in 115 attached via N-sulfonylamide. The remaining alkyl azido moiety in 115 was then combined with the alkyne 116via a one-pot CuAAC to give the three-component coupled product 117 in 86% yield.
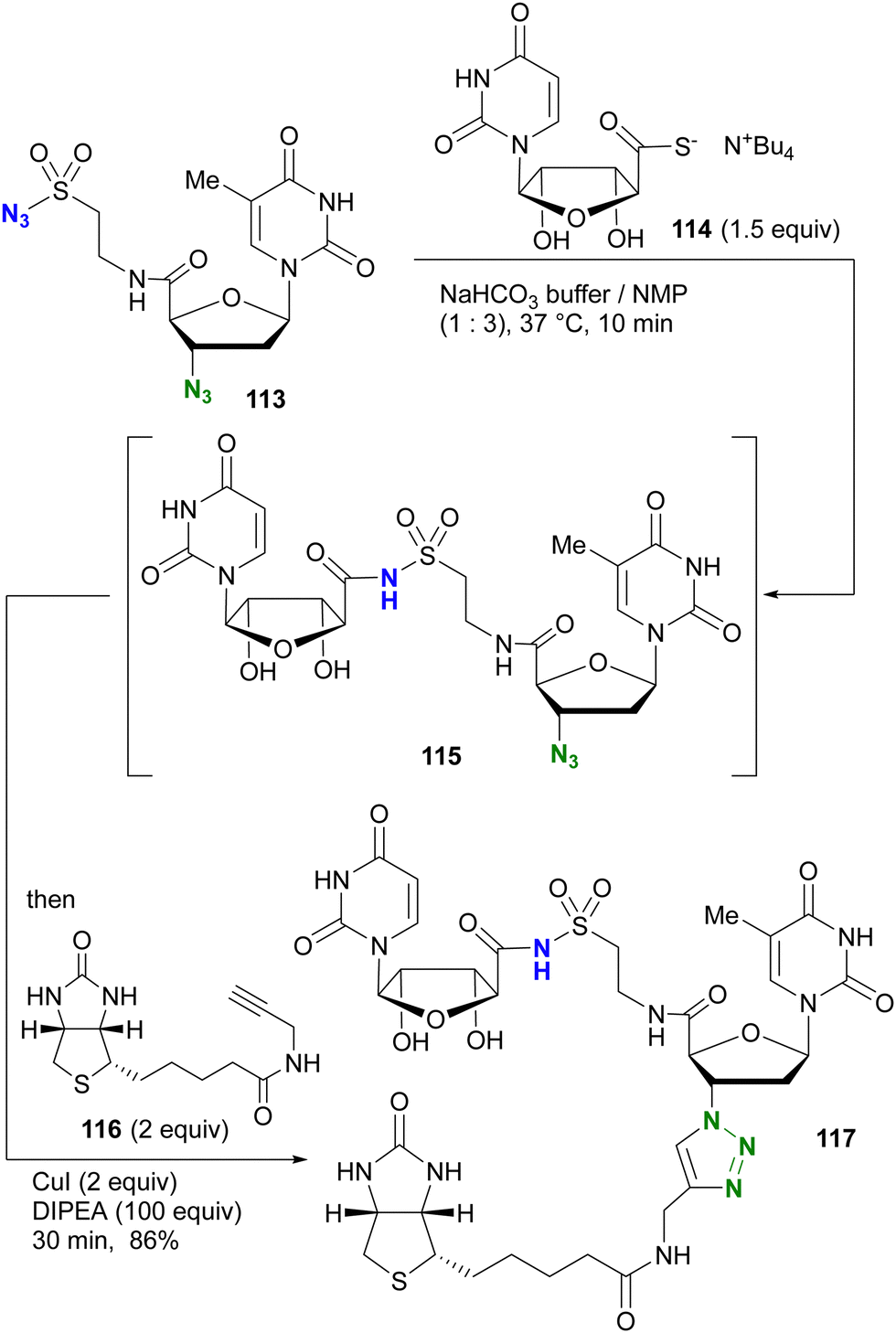 | ||
| Scheme 24 One-pot orthogonal dual conjugation of a diazido nucleoside 113 by sulfo-click and CuAAC reactions. | ||
4.4. Distinction by coordination and hydrogen bonding
The azido group is a 1,3-dipolar species. Therefore, coordination, such as protonation or metal complexation of the azido group, is typically not the primary focus. Nevertheless, a limited number of examples have used the coordination-to-azide strategy, particularly at the N1 position of the azido group, to modify azido reactivities (Fig. 8). As described, alkyl azides, unlike aryl azides, lack additional conjugation extensions that alter the azido properties. As a result, it is difficult to distinguish between their reactivities other than steric hindrance. The coordination-to-azide strategy is proving effective in alkyl azide cases, as it circumvents the need for steric hindrance, which significantly inhibits reactivity. This section presents reported cases of metal catalyst coordination and intramolecular hydrogen bonding aimed at distinguishing between two alkyl azido groups.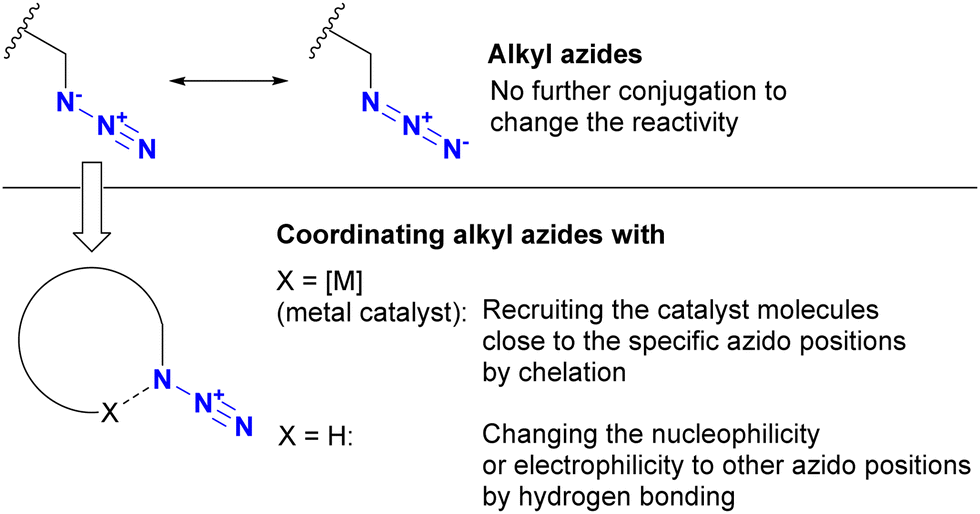 | ||
| Fig. 8 Coordination-to-azide strategy to alter the reactivity of the azido groups. | ||
While general CuAAC reactions typically employ copper(I) catalysts, Zhu and co-workers made the remarkable discovery that copper(II) reagents can also facilitate CuAAC reactions without the need for additional reducing reagents. In addition, they found that chelating azides, such as 2-picolyl azides (Scheme 25), further accelerate these reactions.163,164 A detailed mechanistic study proposed the involvement of key complexes, including the mixed-valence dicopper 120, which consists of a copper acetylide and an azide-chelating copper center.165 In this state, the azido and ethynyl groups are positioned in close proximity, facilitating the CuAAC reaction.
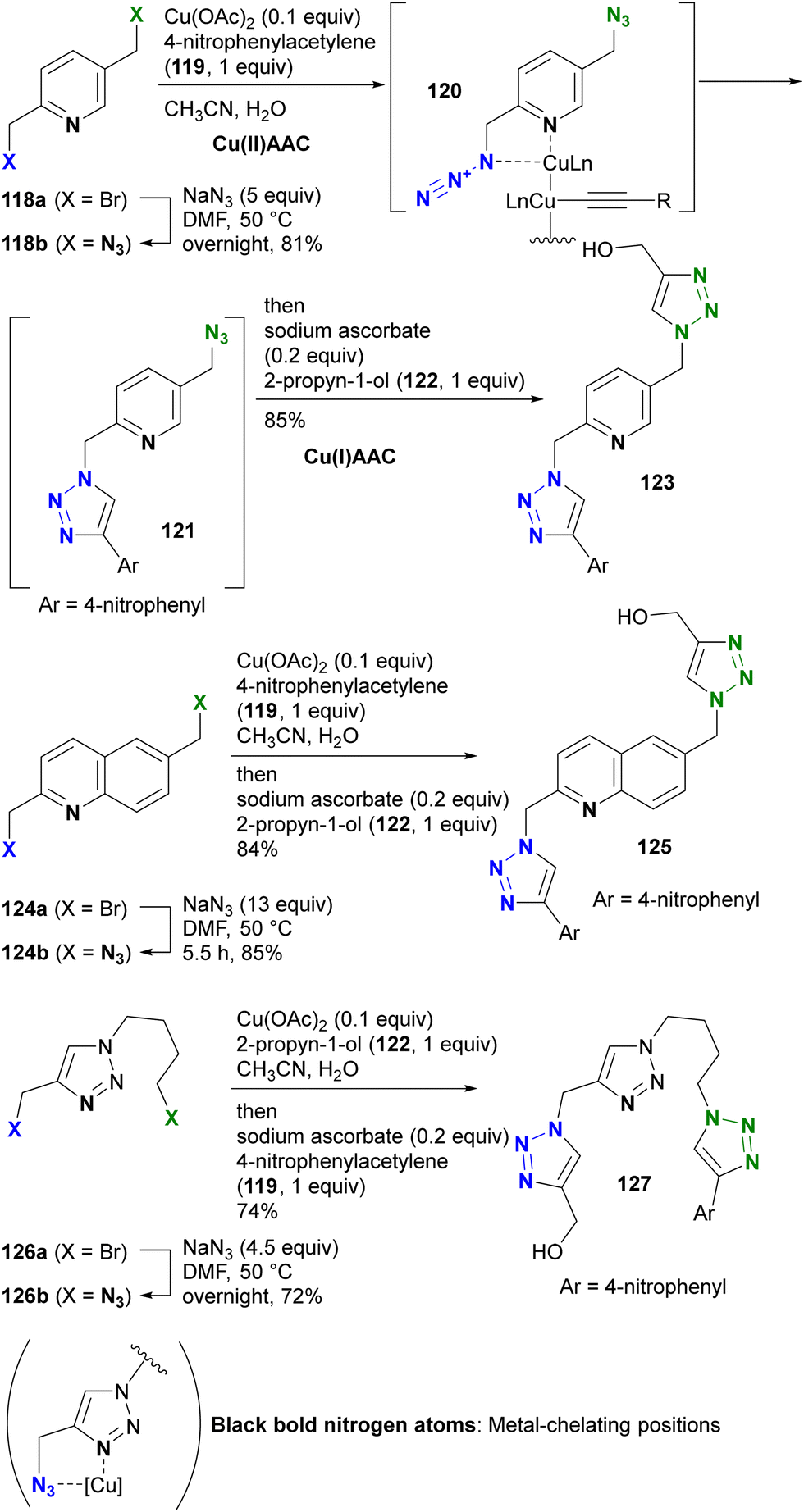 | ||
| Scheme 25 Metal-coordination-induced azide-site-selective CuAAC reaction of alkyl diazides possessing heterocycles. | ||
Zhu et al. extended this chelation strategy to achieve azide-site selective conjugation of alkyl diazides containing a heterocycle as a chelating scaffold, prepared by double SN2 azidation of corresponding dibromides with a heterocycle as a chelating scaffold.166 Using 2,5-di(azidomethyl)pyridine 118b, they achieved a single CuAAC conjugation with copper(II) acetate in a chelating azide-selective manner, resulting in the formation of 121. Subsequent one-pot reduction of the copper(II) catalyst in the mixture to copper(I) by the addition of sodium ascorbate initiated rapid Cu(I)AAC on the remaining non-chelating azido moiety of 121, yielding the bistriazole 123 of the three-component conjugate.
The selective Cu(II)AAC reaction can also be extended to diazides with the quinoline core skeleton 124b and even the triazole 126b, resulting in the formation of azide-site selective coupling products 125 and 127 after the one-pot Cu(II) reduction and Cu(I)AAC reaction. In addition, this chelation strategy not only preserves catalyst activity but also effectively limits the use of toxic copper catalysts. Consequently, this click strategy has found application in chemical biology for azide linker structures containing copper ligand moieties, such as 128a–d, to increase reaction rates and reduce copper toxicity (Fig. 9).167–173
 | ||
| Fig. 9 Azide linkers with metal-azide chelating structures. | ||
More recently, this chelation-based click acceleration for site-selective CuAAC has been reported in a multi-azide compound (Scheme 26).174 siRNA, antisense oligonucleotides, or mRNA for pharmaceutical applications, such as SARS-CoV-2 mRNA vaccines, require improvement of stability, specific targeting, or cellular uptake by chemical modification. In particular, modification of the 3′-end of oligonucleotides with a triple-N-acetylgalactosamine (GalNAc) moiety is known to be important for siRNA. To simplify functionalization, Carell and co-workers used site-selective and sequential tandem CuAAC with tetraazide 133, which consists of three PEG azides and a picolyl azide. Similar to the histidine side chain moiety in peptides,173 CuAAC of oligonucleotides is also enhanced by complexation of copper ions with the base moieties. Nevertheless, CuAAC of the 3′-end alkynylated oligonucleotide 132 with 133 was successfully achieved at the picolyl azide moiety close to the metal coordination site. In one pot, subsequent triple click conjugation at the three PEG-azide moieties with alkynyl GalNAc 134 efficiently afforded the site-selectively clicked product 135 in 70–80% yield, which has the same sequence and chemical modification pattern as the double-stranded siRNA therapeutic inclisiran.
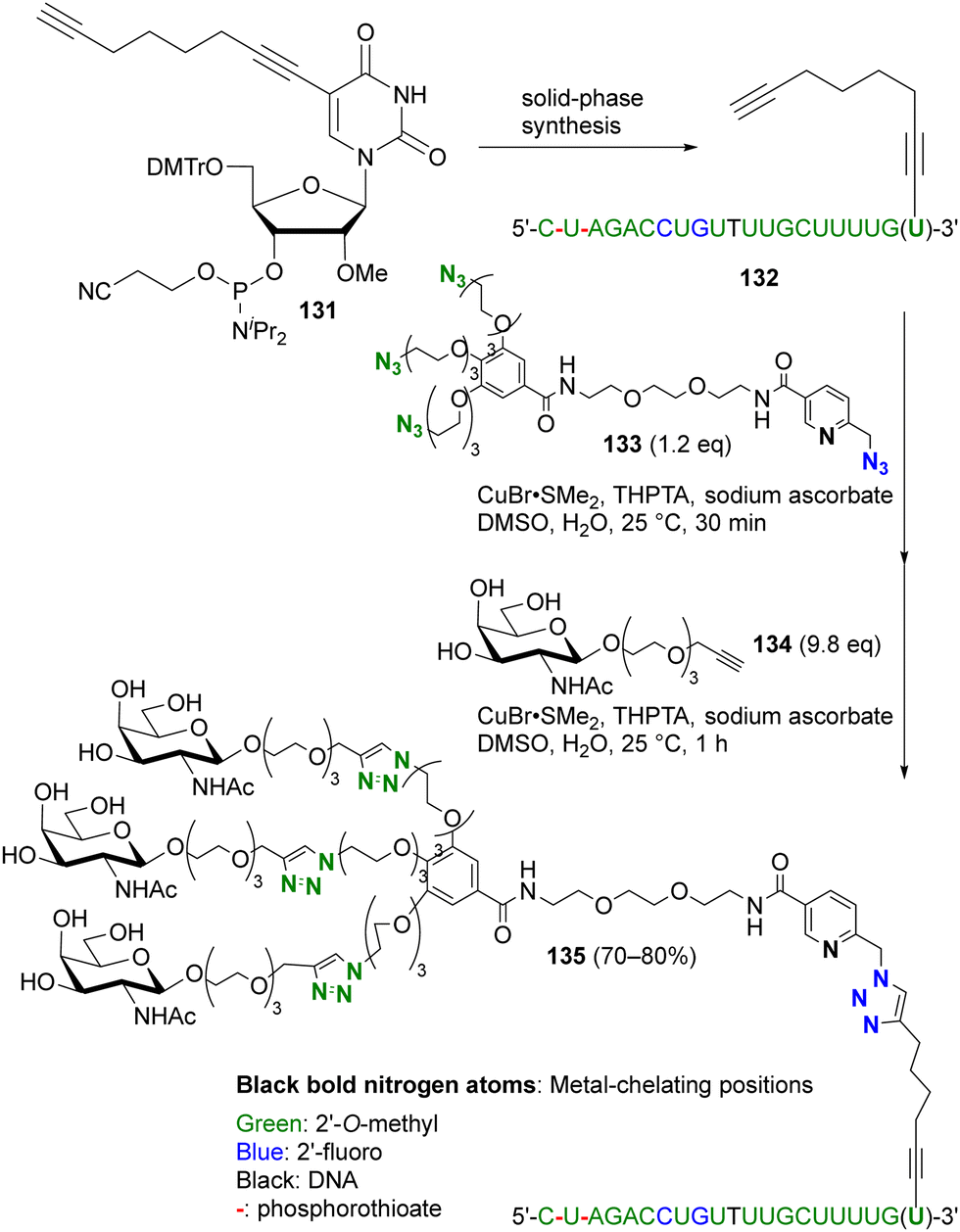 | ||
| Scheme 26 Metal-coordination-induced azide-site-selective sequential CuAAC reaction of tetraazide with oligonucleotide. THPTA: tris(3-hydroxypropyltriazolylmethyl)amine. | ||
In addition to metal coordination, hydrogen bonding is a prevalent interaction in nature. In particular, intramolecular hydrogen bonding influences the bioactivity and programmed hierarchical structures of molecules. Tanimoto and co-workers exploited the molecular design of organic azides with intramolecular hydrogen bonding for azide-site selective conjugation (Fig. 10).175 Using DFT calculations (Gaussian09 program suite with the B3LYP-D3/6-311G** method), they estimated the intramolecular interaction between the azido and amido N–H positions in α-azido-secondary amides (α-AzSA). The antiperiplanar positioning of the N3–C bond to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is primarily attributed to dipolar repulsion. However, the electron density of the N1 atom in α-AzSA is estimated to increase as a result of this intramolecular interaction. In the case of other organic azides, no such increase is observed. In α-AzSA, this interaction accelerates electrophilic reactions such as the Staudinger reaction/ligation, facilitated by anion stabilization in the adduct intermediates. Conversely, nucleophilic reactions of organic azides are suppressed due to the reduced nucleophilicity of the adjacent protic N–H group. Due to the absence of intramolecular interactions, β-azido substrates do not exhibit specific reactivity differences from general alkyl azides.
O is primarily attributed to dipolar repulsion. However, the electron density of the N1 atom in α-AzSA is estimated to increase as a result of this intramolecular interaction. In the case of other organic azides, no such increase is observed. In α-AzSA, this interaction accelerates electrophilic reactions such as the Staudinger reaction/ligation, facilitated by anion stabilization in the adduct intermediates. Conversely, nucleophilic reactions of organic azides are suppressed due to the reduced nucleophilicity of the adjacent protic N–H group. Due to the absence of intramolecular interactions, β-azido substrates do not exhibit specific reactivity differences from general alkyl azides.
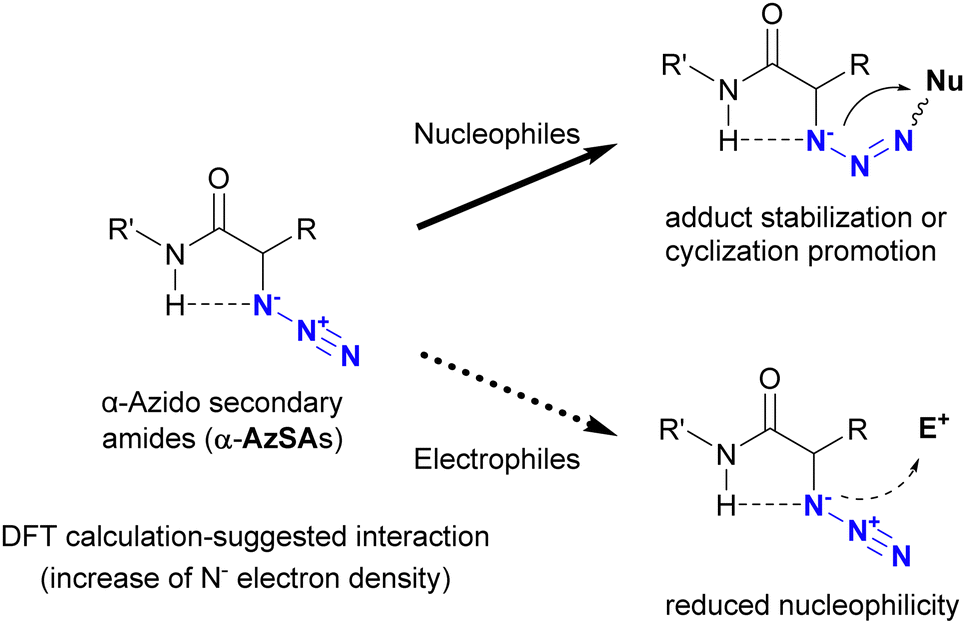 | ||
| Fig. 10 Characteristic reactivity of α-azido secondary amides induced by the intramolecular interaction between azido and amido N–H groups. | ||
This hydrogen bonding strategy has been demonstrated in azide site-selective conjugation reactions.175 Treatment of the primary alkyl diazide 136, composed of α-AzSA and benzylic azide, with phosphine 137 afforded the azobenzene conjugated product 138 (Scheme 27(a)). The facilitating effect of α-AzSA on the traceless Staudinger ligation in the electrophilic reaction, shown in Fig. 10, ensured excellent site selectivity. The remaining benzylic azido moiety in 138 was then conjugated to biotin via the second traceless Staudinger ligation, yielding 140. Due to significant differences in reactivity, the double traceless Staudinger ligation was performed as a one-pot reaction, resulting in a higher overall yield. Furthermore, the azide-site selective reaction demonstrated the inhibitory influence of α-AzSA (Scheme 27(b)). Consistent with the steric hindrance effect (Section 4.1), the SPAAC of diazide 141 with DIBO 51 occurred selectively at the less sterically hindered α-AzSA position, yielding 142. Conversely, the propargyl cation-mediated triazole synthesis of the nucleophilic reaction (Scheme 8)94,95 with propargyl alcohol 143 occurred exclusively at the sterically hindered adamantyl azide position, yielding mono-triazole 140 while retaining the α-AzSA moiety of the primary alkyl azide, contrary to the general reactivity trend of steric hindrance. This strategy will also be discussed in the final chapter on multicomponent conjugation.
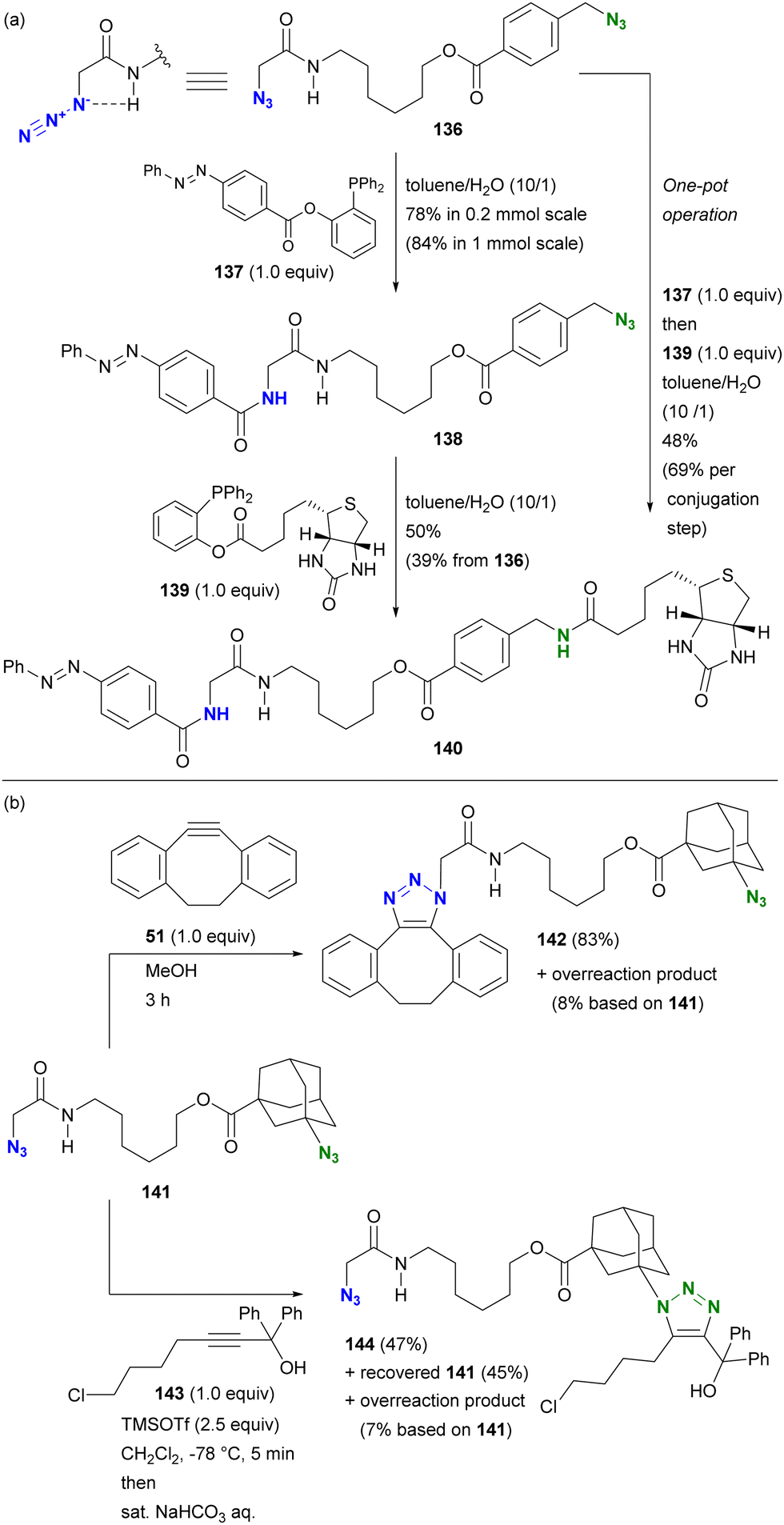 | ||
| Scheme 27 Azide-site selective conjugation of diazides through (a) promoting and (b) suppressing properties of the α-AzSA structures. | ||
After reading the above, readers may be curious about selectivity in cases involving SPAAC and CuAAC, which are common click reactions. Tanimoto et al. also investigated the comparison of SPAAC reactions, but no selectivity for azides was observed between α-AzSA 145 and alkyl azide 146 (Scheme 28).175 This lack of selectivity may be due to the pericyclic nature of the SPAAC reaction with DIBO 51 where, unlike stepwise reactions, there are no intermediates affected by intramolecular hydrogen bonding interactions (Fig. 10).
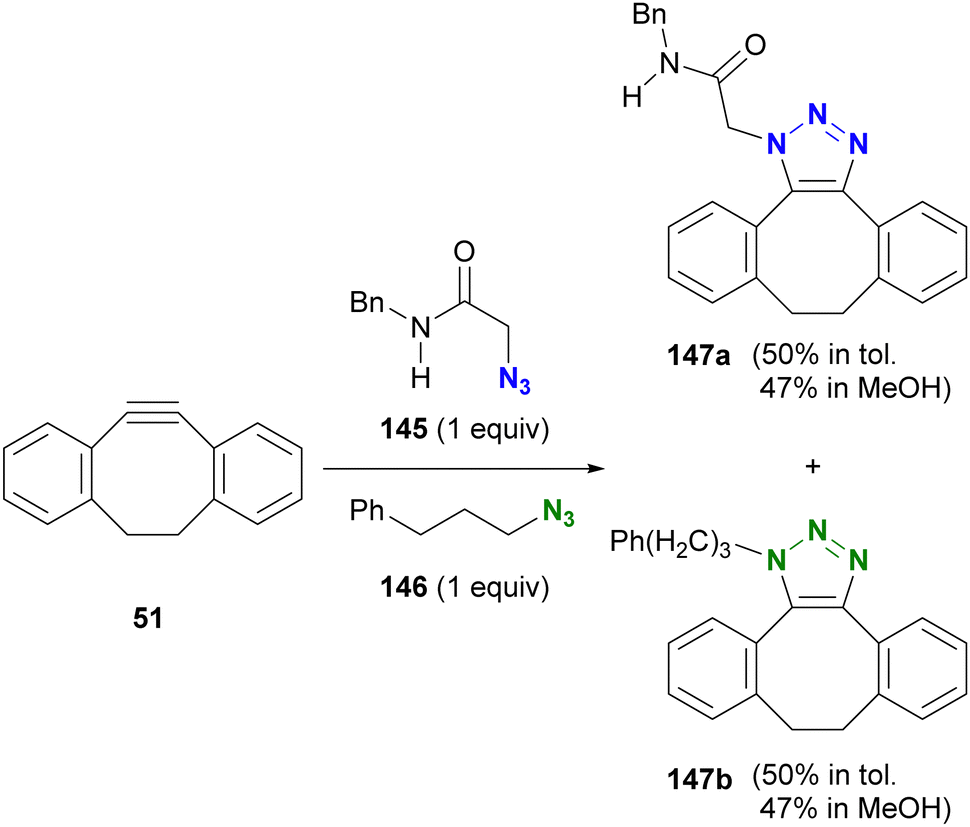 | ||
| Scheme 28 Competitive SPAAC of DIBO 51 with α-AzSA 145 and alkyl azide 146. | ||
On the other hand, Raines reported the acceleration of SPAAC of α-azido and α-diazo secondary amides using ABC (2-azabenzo-benzocyclooctyne, 2-ABC, 148), which can be synthesized from the commercial precursor of the antihistamine loratadine (Claritin) (Table 2).176 Compared to DIBO 51, ABC 148 increased the rate of SPAAC with the azido and diazo substrates 145 and 149. In the case of α-AzSA, NBO (natural bond orbital) analysis (calculated at the M06-2X/6-311++G** level) suggests intramolecular hydrogen bonding, as shown in Table 2, and the intermolecular nN of the ABC → πCO interaction, stabilizing the transition state of cycloaddition. Although a rate comparison between organic azides lacking intramolecular hydrogen bonding is not provided, the SPAAC reaction of ABC 148 with 151, which lacks one N–H group compared to 145, resulted in the formation of the cycloadduct isomer. In addition to the azido compounds, the SPAAC of the diazo-secondary amide with DIBO 51 was significantly accelerated, although no intramolecular hydrogen bond was formed. In contrast, the SPAAC of ABC 148 with the diazo ester 150, which has no intra- or intermolecular hydrogen bonding via N–H, was much slower than that of the diazoamide 149. These results suggest that intermolecular hydrogen bonding plays a crucial role in regioselectivity and acceleration of reaction rates in SPAAC with ABC 148. However, in this case, the intramolecular hydrogen bonding between the azido and amido N–H groups in 145 would be expected to serve only for conformational fixation.
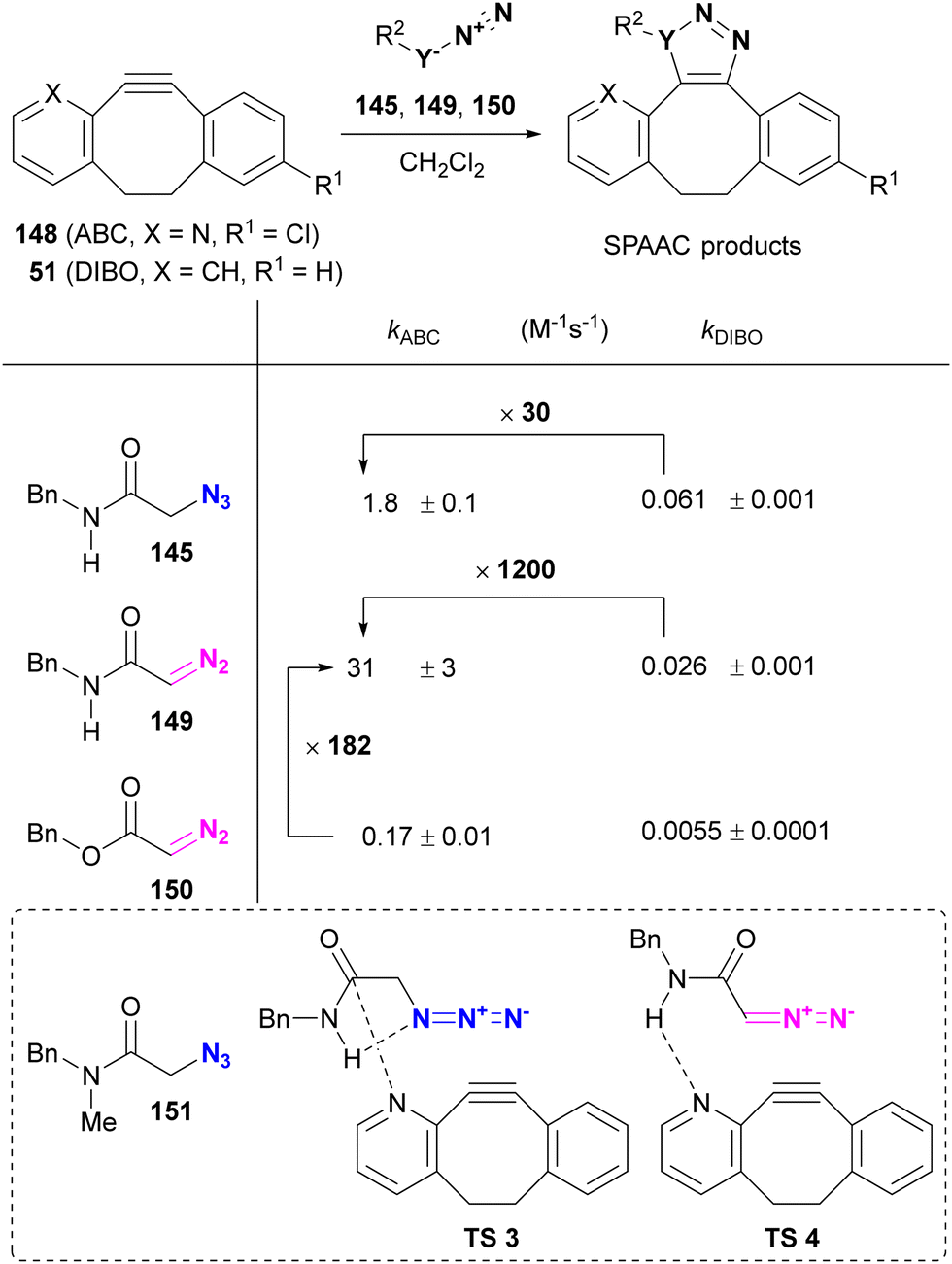
|
The case of CuAAC is reported in the study of Sun and Wang et al. (Scheme 29).177 In their study of the copper-NHC (N-heterocyclic carbene)-catalyzed intermolecular azide-silyl alkyne cycloaddition reaction, substrate selectivity was investigated, although only ratios are reported. When an unprotected terminal alkyne was used, no selectivity was observed between α-AzSA 145 and benzyl azide 43. Conversely, the use of the silylated alkyne increased the selectivity, yielding the α-AzSA-derived triazole 153a as the primary product, while no product derived from γ-AzSA 156, such as 153d, was observed. However, intramolecular hydrogen bonding is not proposed to be involved in this reaction. Instead, the complexation of copper with the alkyne and azido group facilitates desilylation by the adjacent amido group via complex 157. This sequence accelerates the formation of the azide-coordinated copper acetylide, allowing the coupling reaction with α-AzSA to proceed.
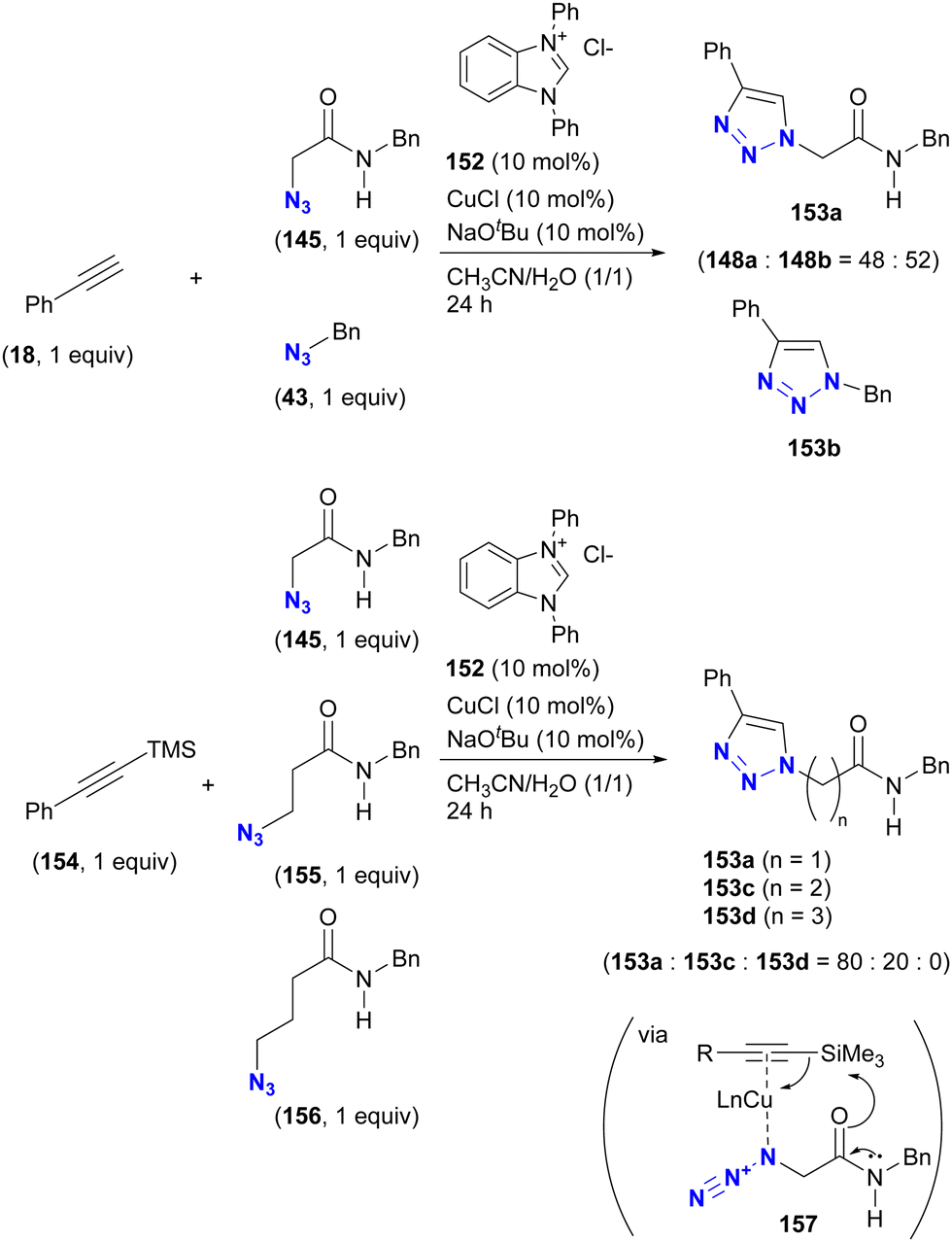 | ||
| Scheme 29 Copper-NHC-catalyzed CuAAC reactions of silylated and unsilylated alkynes with alkyl azides and the substrate selectivity. | ||
In the cases of hydrogen bonding illustrated here, it is important to note that the interaction itself may not directly promote the conjugation reaction. Nevertheless, these interactions play a supporting role by facilitating the progression of reaction intermediates or transition states, thereby enabling azide-site selective reactions. Further exploration of this interaction has the potential to improve the selectivity and speed of organic azide reactions, including click reactions.
4.5. Distinctive functionalization through azido group protection
One of the most intuitive and commonly used strategies to achieve regioselective or site-selective reactions is protection.178 By protecting the functional groups to preserve their reactivity for subsequent functionalization, those in unprotected positions can be selectively modified. For example, the ethynyl group associated with click chemistry can be protected by masking the terminal C–H position, typically with a silyl group such as trimethylsilyl or dimethylhydroxymethyl (Fig. 11). Since the masked ethynyl groups remain inert to CuAAC, multi-component click reactions with various alkyne-containing compounds have been successfully demonstrated in many cases.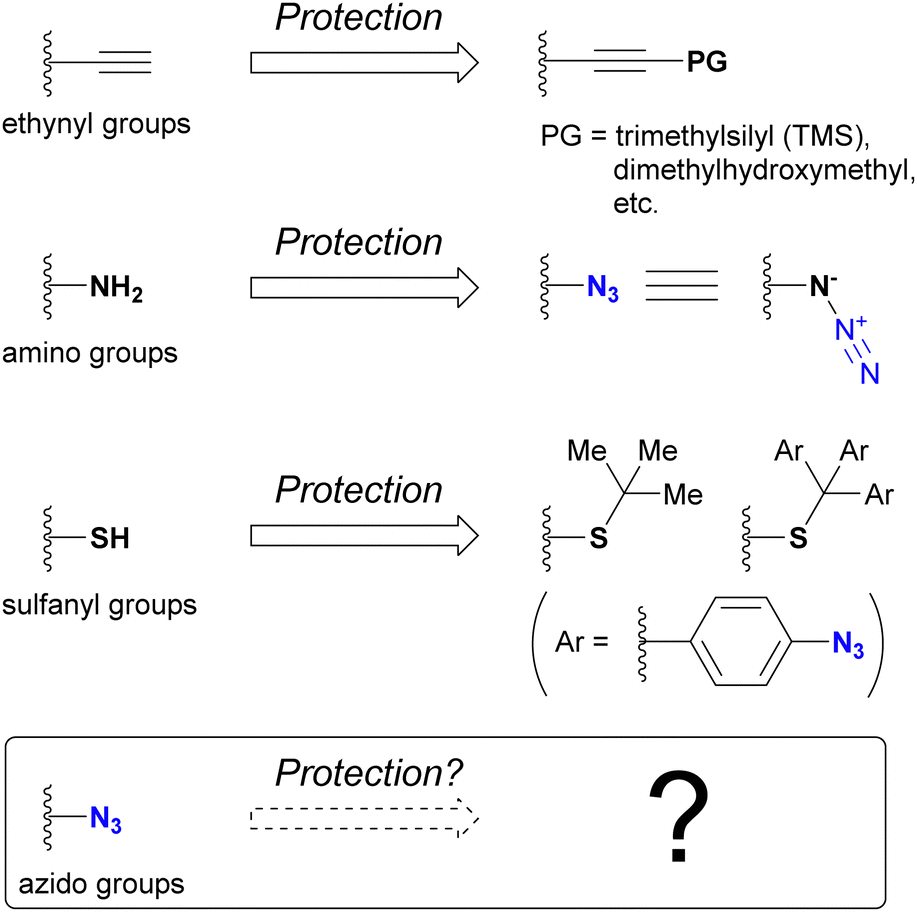 | ||
| Fig. 11 Protection of functional groups. | ||
Conversely, the azido group itself is recognized as an amino-protecting group. Due to its ease of introduction and ability to generate amines by reduction, azide has found utility as a protecting group in natural product synthesis. For sulfhydryl groups (thiols), the tert-butyl group has been commonly used as a protecting group. Recently, as the same concept of 4-azidobenzyl-type protective groups for hydroxy and amino groups,179–181 the tris(4-azidophenyl)methyl group, has been developed.182 Removal of the azidobenzyl-type protecting groups is accomplished by reduction of the azido group, which initiates elimination of the protected groups. As shown here, the azido group itself usually contributes as a protective group, and the deprotection steps are performed by decomposing the azido groups. On the contrary, which group can protect azido groups? Approaches to answering this question are presented in this section.
Azido groups are susceptible to reduction conditions such as the Staudinger reaction with phosphines and reduction by thiols (Fig. 12). In particular, aryl azides are susceptible to nucleophilic reduction due to the stabilizing properties of triazenes in nucleophilic adducts. In addition, organometallic reagents, including Grignard reagents, transition metal catalysts, and DIBAL-H, can damage the azido groups of aryl azides.183–193 In contrast, alkyl azides remain unaffected under Grignard reactions and DIBAL-H reduction conditions and react more slowly with nucleophiles than aryl azides. Consequently, stable alkyl azides have found utility in protecting amino groups in natural product synthesis.63,194–199 From a click chemistry perspective, protection of specific azido positions could alter the reactivity order of conjugated azido sites. Therefore, protective groups for organic azides, especially aryl azides, are of significant value.
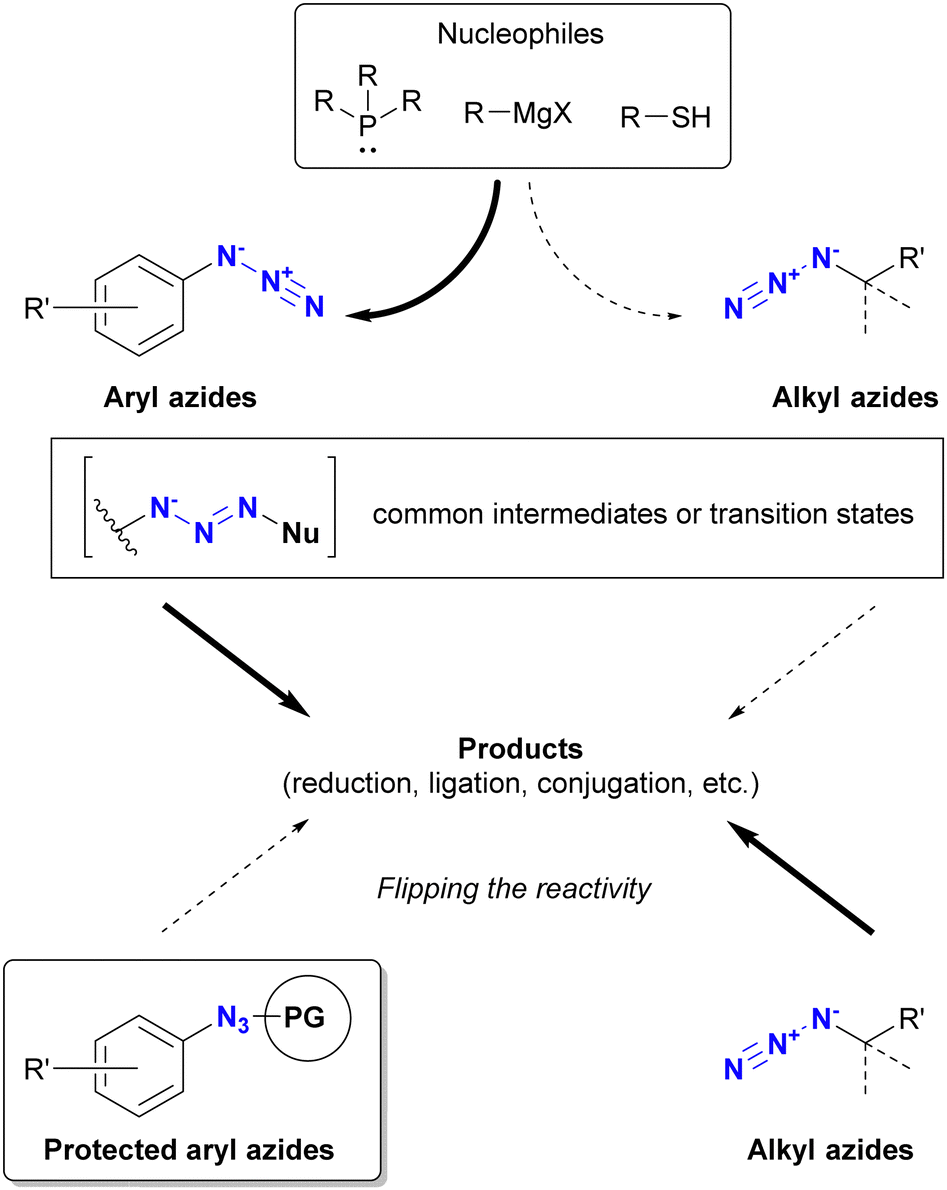 | ||
| Fig. 12 Reactivity of aryl and alkyl azides toward nucleophiles and azide protection strategies. | ||
The conventional approach to azido protection involves tautomerization, leading to the formation of cyclic structures (Scheme 30).200 2-Azido azaheterocycles, such as 2-azidopyridine, are often observed as a tautomeric mixture. In other words, the cyclization of 2-azido azaheterocycles results in the tetrazole structures, which are no longer considered an azido group. Chapyshev and co-workers have reported this strategy for azido site-selective reactions.201,202 Treatment of the 2,4,6-triazido-3,5-dibromopyridine 158 with triethyl phosphite at room temperature yields 159, the mono aza-ylide at the 4-position, since this position is the most electron-withdrawing, as discussed earlier (Section 4.2). Subsequently, the environmental situation of the remaining two azido groups appears to be identical. Nevertheless, cyclization with the nitrogen in the pyridine ring occurs, yielding the monoazido tetrazolopyridine. At this point, only one azido group remains, and an additional Staudinger reaction under reflux conditions selectively produces the monoazido product 160a and the tetrazolo[1,5,a]pyridine 160b as a tautomeric mixture in 100% yield. Subsequent hydrolysis yields the diphosphoramidated azidopyridine 161. This case demonstrates excellent azido site selectivity.
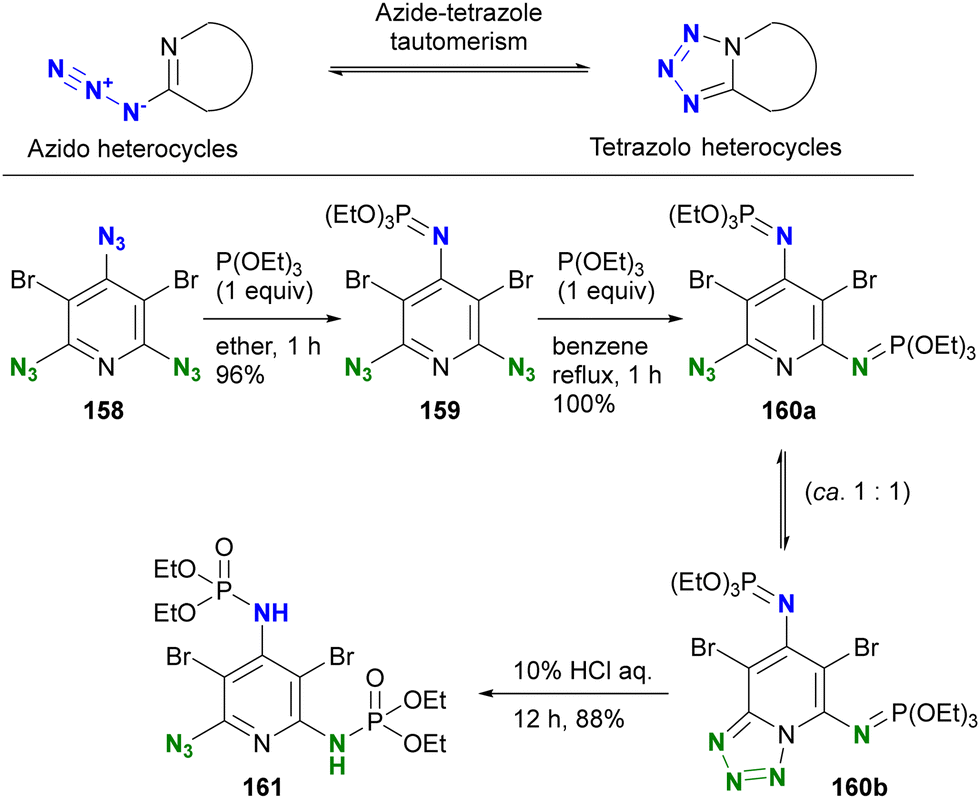 | ||
| Scheme 30 Regioselective azido group protection through tetrazole ring formation. | ||
The phenomenon of tautomerism in azido heterocycles certainly extends beyond 2-azidopyridines. Although not used for azido group protection, Lovely and colleagues recently documented in the supplementary report203 of the total synthesis204 of calcaridine A 166 and its epimer 167 that the oxidation products with oxaziridine 163 were not the 2-azidodihydroimidazol-4-ones 164a and 165a, but rather their tautomers, the tetrazole-fused lactams 164b and 165b (Scheme 31).
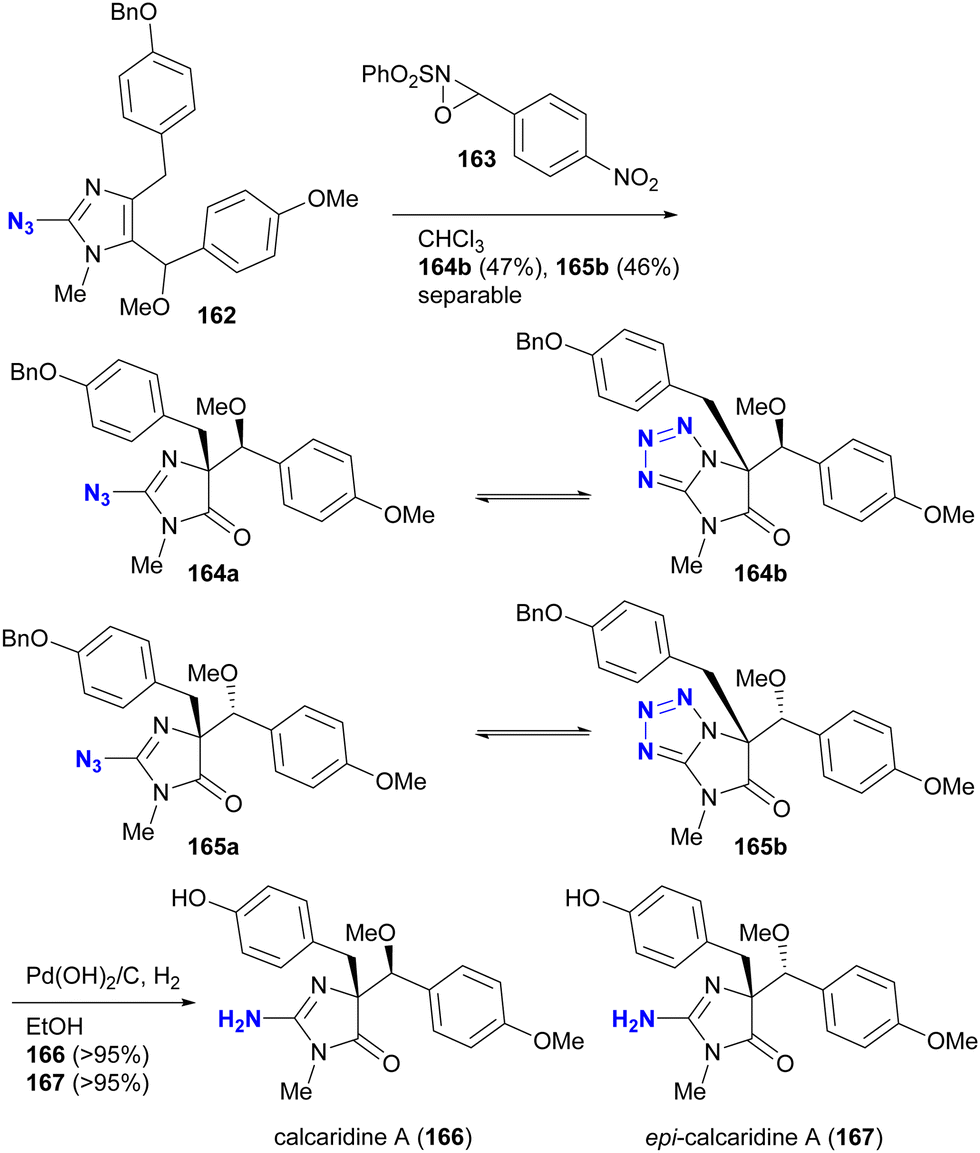 | ||
| Scheme 31 Tautomerism of the azido imidazolines in the total synthesis of calcaridine A 166 and its epimer 167. | ||
However, protection of the azido groups by tetrazole formation essentially limits the substrates to azido heterocycles. Recently, the group of Yoshida and Hosoya reported a more versatile strategy using a transient azido protection method (Scheme 32).205–207 Treatment of aryl azides (benzyl azide in one case) with Amphos (also described as APhos) resulted in the formation of stable phosphazides 168. The key is to suppress aza-ylide formation by cyclization of the phosphazide intermediate, facilitated by the bulky tert-butyl groups. In these non-azido forms, SPAAC reactions no longer occurred. In addition, treatment of these phosphazides 168 with elemental sulfur readily reverted to the starting material azides.
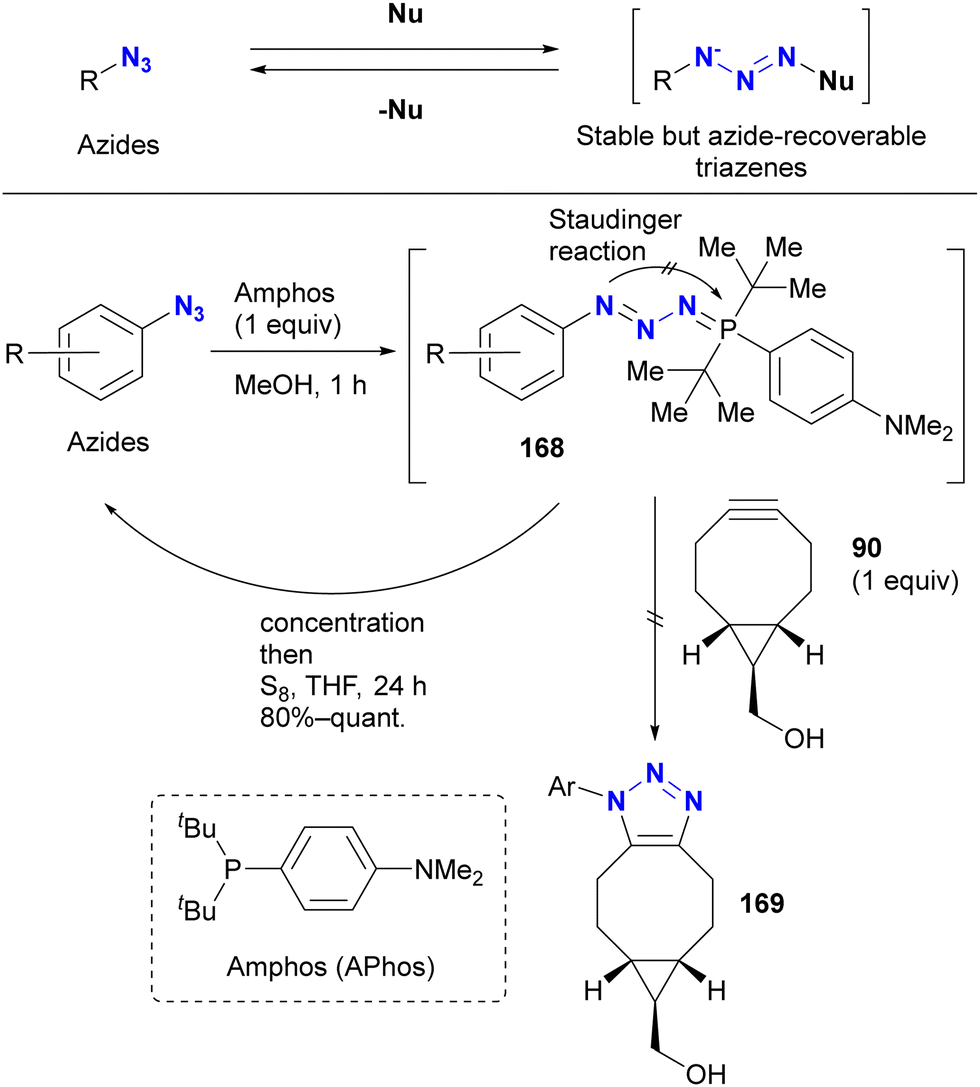 | ||
| Scheme 32 Transient protection of azido groups with Amphos (Aphos). | ||
Yoshida and Hosoya's group also demonstrated their own phosphine-mediated transient azide protection in an azide-site selective reaction (Scheme 33).206 While the alkyl aryl diazide 170 is efficient as a diazido probe, as shown in Scheme 19, conversion of the aldehyde moiety with Grignard reagents is challenging because they can damage both the formyl group and the aryl azido group. However, treatment of 170 with Amphos selectively masked the aryl azido group as the phosphazide 171.207 With only one azido group present at this point, a one-pot SPAAC reaction was then performed at the alkyl azido position, yielding 172. Grignard reagent was then added to the mixture to convert the formyl group, followed by regeneration of the azido group with elemental sulfur to give 173. In this sequence, the aryl azido group in 173 was protected from both SPAAC and Grignard reactions by Amphos. This newly developed protection strategy is expected to open new horizons in organic azide chemistry, potentially finding applications in complex molecular transformations and selective functionalization (a modified method by another group is presented in the next chapter).
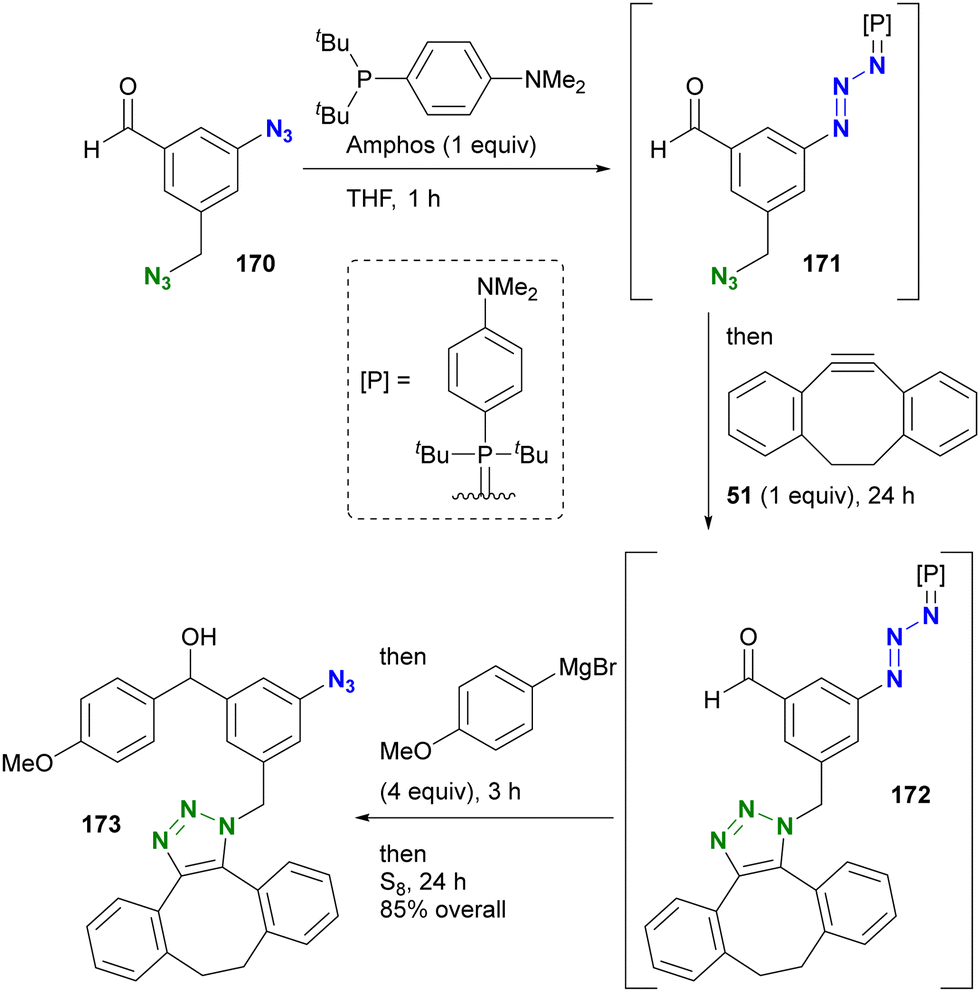 | ||
| Scheme 33 Site-selective one-pot conversion of diazido benzaldehyde 170 through transient protection of aryl azido moiety. | ||
4.6. Distinguishing through site-selective conversion from azido to different click-functional groups
Beyond their direct application, converting specific azido positions to other click functional groups offers a promising strategy for site-selective azide utilization, capitalizing on the ease of azido group introduction. One approach to achieve this is through transient protection via phosphazide formation, as discussed in Section 4.5. Further examples of this strategy are discussed in this section.As we have discussed, various methods of azide site selectivity have been developed to distinguish between aryl and alkyl azides. However, in most cases, with the exception of the coordination strategy in Section 4.4, the distinction of reactivity between alkyl azides is based on steric hindrance and is still difficult. On the other hand, α-azido carbonyl structures are known to undergo β-elimination upon basic treatment, resulting in the formation of unstable α-imino carbonyl intermediates, which have been applied to various organic syntheses.208–210 Tanimoto et al. focused on this feature and demonstrated a site-selective azido group transformation as shown in Fig. 13.211,212 The α-imino-carbonyl intermediates generated by the base treatment of α-azido-carbonyl materials were subjected to in situ condensation with sulfonyl hydrazides or hydroxylamines to produce the corresponding sulfonyl hydrazones or oximes. Under the same conditions, the sulfonyl hydrazones were further converted to diazo compounds. Unlike the azido group in the carbonyl α-position, common alkyl and aryl azides tolerated basic conditions due to the absence of the acidic hydrogen atom. Consequently, the diazo group was selectively generated at the stable carbonyl α-position.
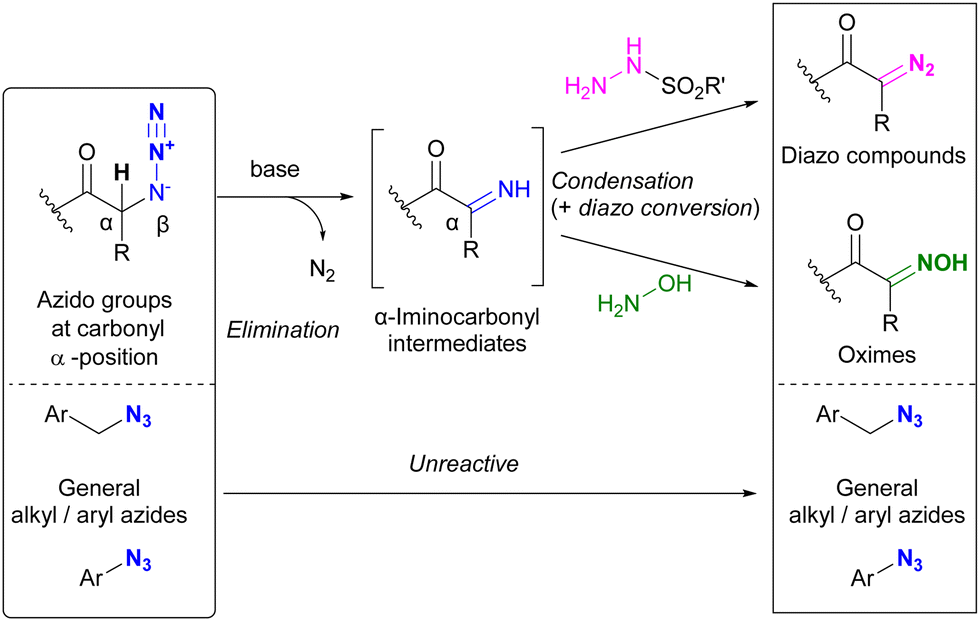 | ||
| Fig. 13 Carbonyl α-position selective conversion of azido groups to diazo and oxime groups by β-elimination. | ||
Under the established conditions, azide-site selective conversions to diazo211 and oxime212 groups were investigated (Scheme 34). The azido groups at the α-position of tert-amides and ketones were converted, even in the presence of non-functionalized alkyl-azido or aryl-azido positions. In the presence of α-azido ketones and amides, the α-keto position with the more acidic C–H group was selectively converted. The resulting product, diazo azido compounds, is readily accessible for site-selective conjugation. Indeed, 174a enabled azido site-selective conjugation by Staudinger–Bertozzi ligation, yielding 177, and diazo site-selective conjugation by [3+2] cycloaddition with acrylate 178, as developed by Raines and co-workers,211–217 yielding 179. The use of the oxime moiety in site-selective conjugation and the stepwise diazo conversion of the multiple azido positions are presented in the final chapter.
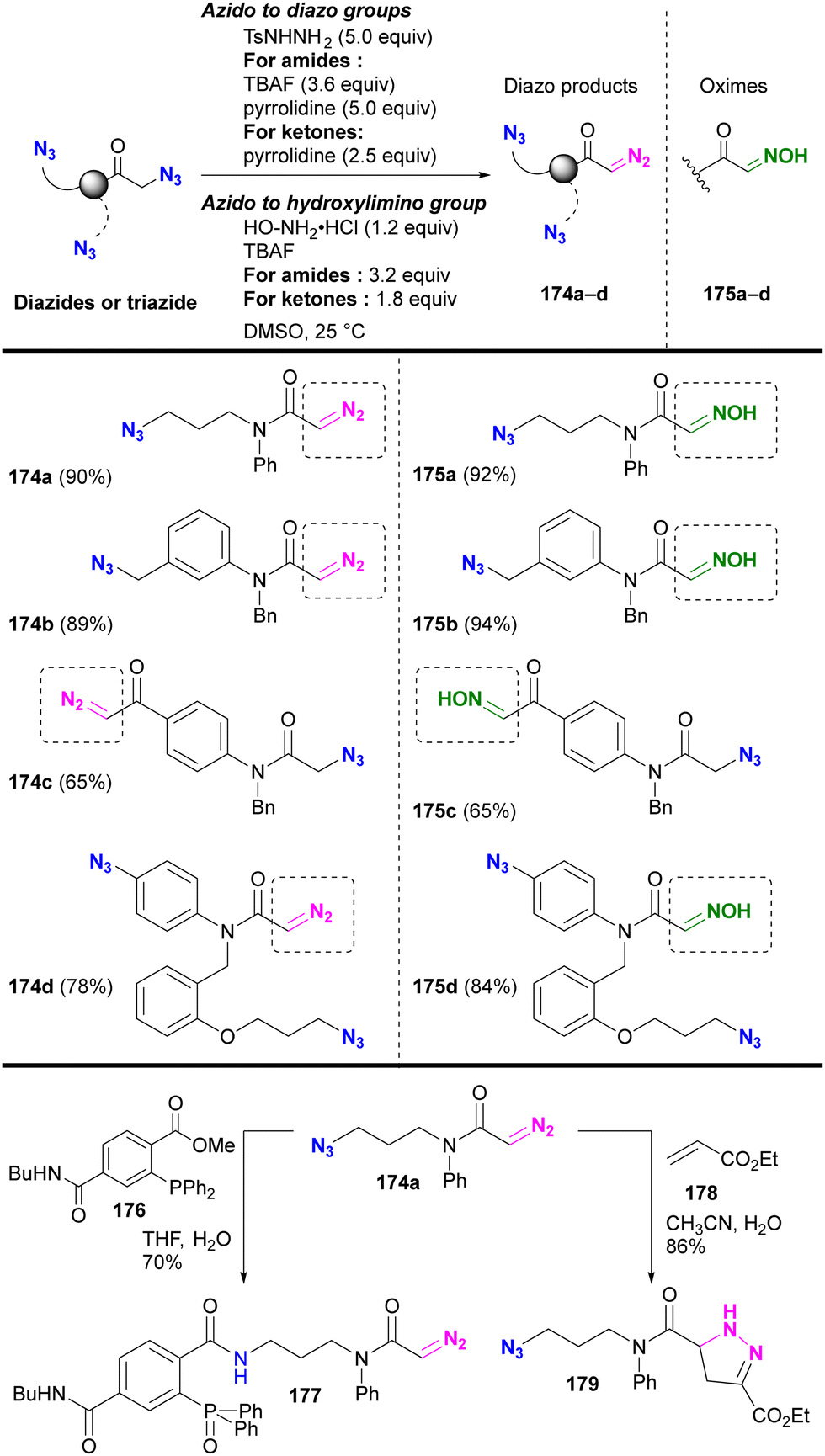 | ||
| Scheme 34 Azide-site-selective conversion to diazo and oxime groups. | ||
In Section 4.5, we introduced azide protection strategies and discussed site selectivity. A recent advancement in this methodology involves an improved deprotection step that utilizes a deprotective conversion from the azido to the diazo group (Scheme 35).218 Treatment of α-azido carbonyl compounds of alkyl azides, such as 145, with Amphos, similar to the approach used for aryl azides, yielded the stable phosphazide intermediate 180 (Schemes 32 and 33). This intermediate 180 was then converted to the α-diazo compound 149 by addition of silica gel under aqueous conditions, presumably via the decomposition of the phosphazide by protonation of the phosphine oxide imine moiety of the phosphazide followed by deprotonation of the carbonyl α-hydrogen atom.
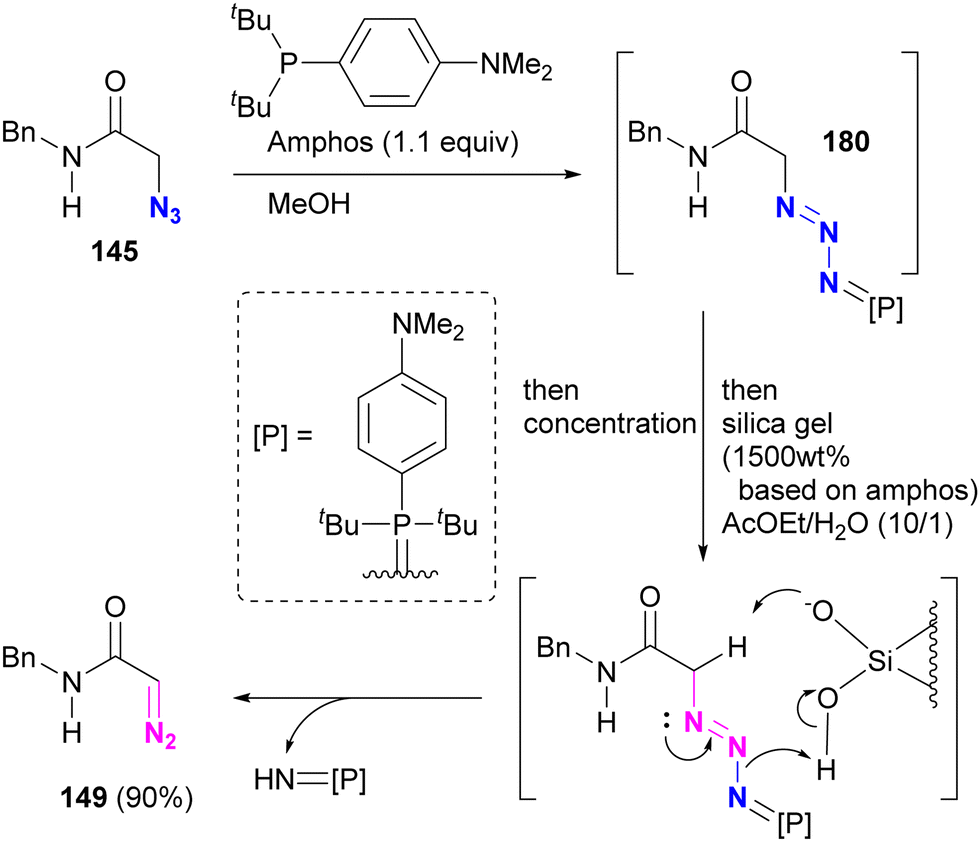 | ||
| Scheme 35 Amphos-mediated conversion of azido to diazo group via transient azide protection. | ||
In this sequence, the alkyl azido group was first protected as a phosphazide and then converted to an alkyl diazo group. This advantageous transformation was further demonstrated by competitive reactions involving two azido molecules and diazide compounds (Scheme 36(a)). A one-pot competition between tertiary amide 181 and alkyl azide 146, facilitated by 1.1 equivalents of Amphos to protect 181, followed by SPAAC of the unmasked azide 146 with 51 and subsequent deprotective conversion of the masked azido group to a diazo group, successfully afforded diazoamide 182 in 89% yield. Similarly, SPAAC triazole 147b was obtained from alkyl azide 146 in a high yield of 90%.
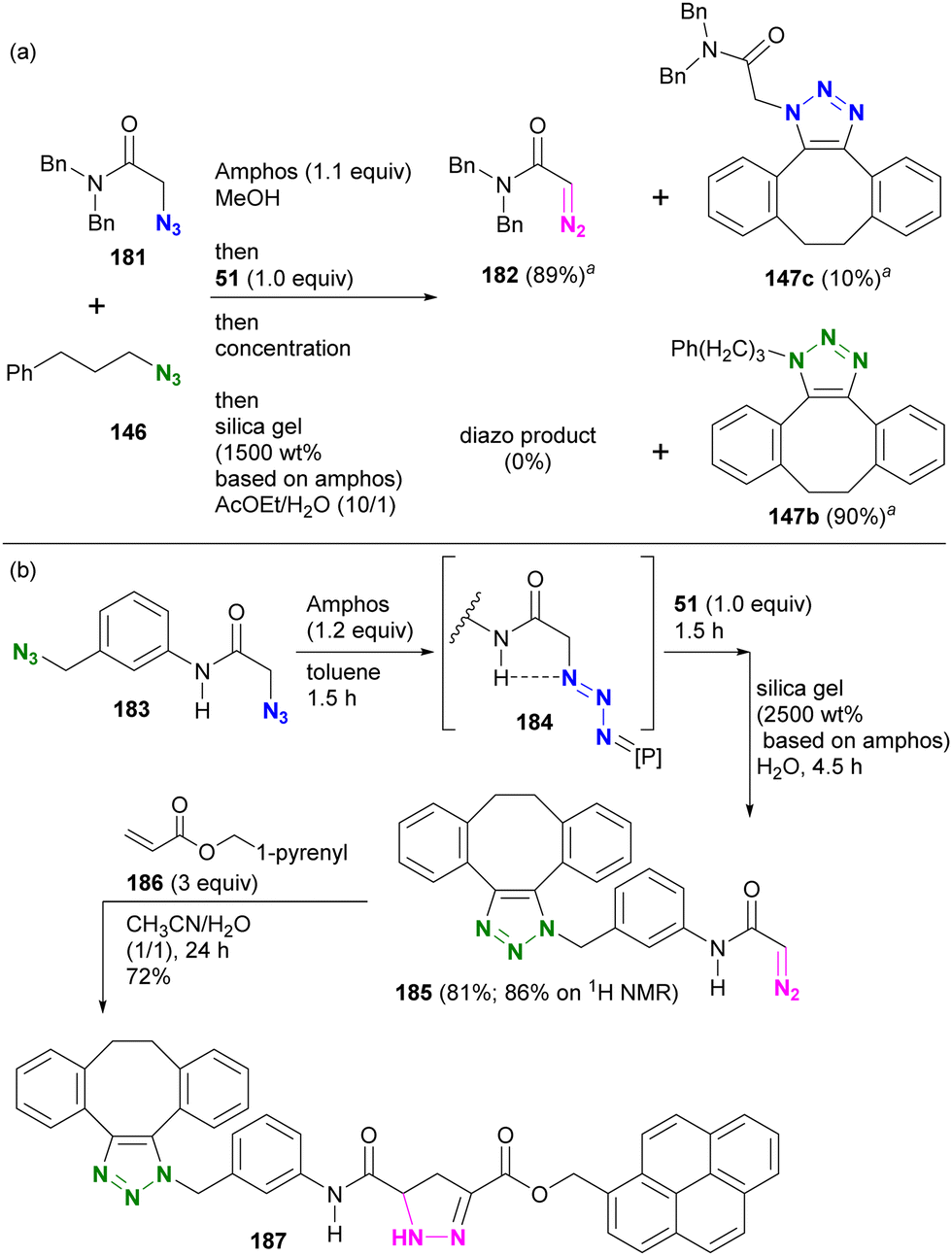 | ||
| Scheme 36 (a) Inter- and (b) Intramolecular azide site-selective utilization through sequential protection, SPAAC of unmasked azide, and the deprotective conversion to diazo group.a 1H NMR yields. | ||
In the case of diazide 183, which has an azido group at the amido-α and benzylic positions, although the benzylic position was reported to be protectable,205 the azido group at the amido-α position was selectively masked to give the phosphazide intermediate 184 (Scheme 36(b)). The one-pot operation was then continued to facilitate SPAAC of the unprotected benzylic azido group with 51. Subsequently, the addition of silica gel under wet conditions proceeded with the deprotective conversion of the phosphazide to the diazo group to afford 185 in 81% overall yield. The newly generated diazo moiety in 185 was then coupled to acrylate 186 by a diazo-acrylate cycloaddition reaction to give 187.213–217
Although site selectivity has not been investigated for the conversion of functional groups from azido to diazo, other methods based on phosphine chemistry have been developed previously. Following the pioneering work of Feigelson by nitrosation of iminophosphoranes prepared by the Staudinger reaction of azides (Scheme 37(a)),219 the Raines group introduced direct conversion of azide to diazo using ester-tethering phosphines 192a or water-soluble 192b (Scheme 37(b)).220,221 More recently, Cao, Li, and colleagues reported a similar conversion method using secondary phosphine oxides 195 that can switch between diazo conversion to 194 and azido reduction to 196 (Scheme 37(c)).222
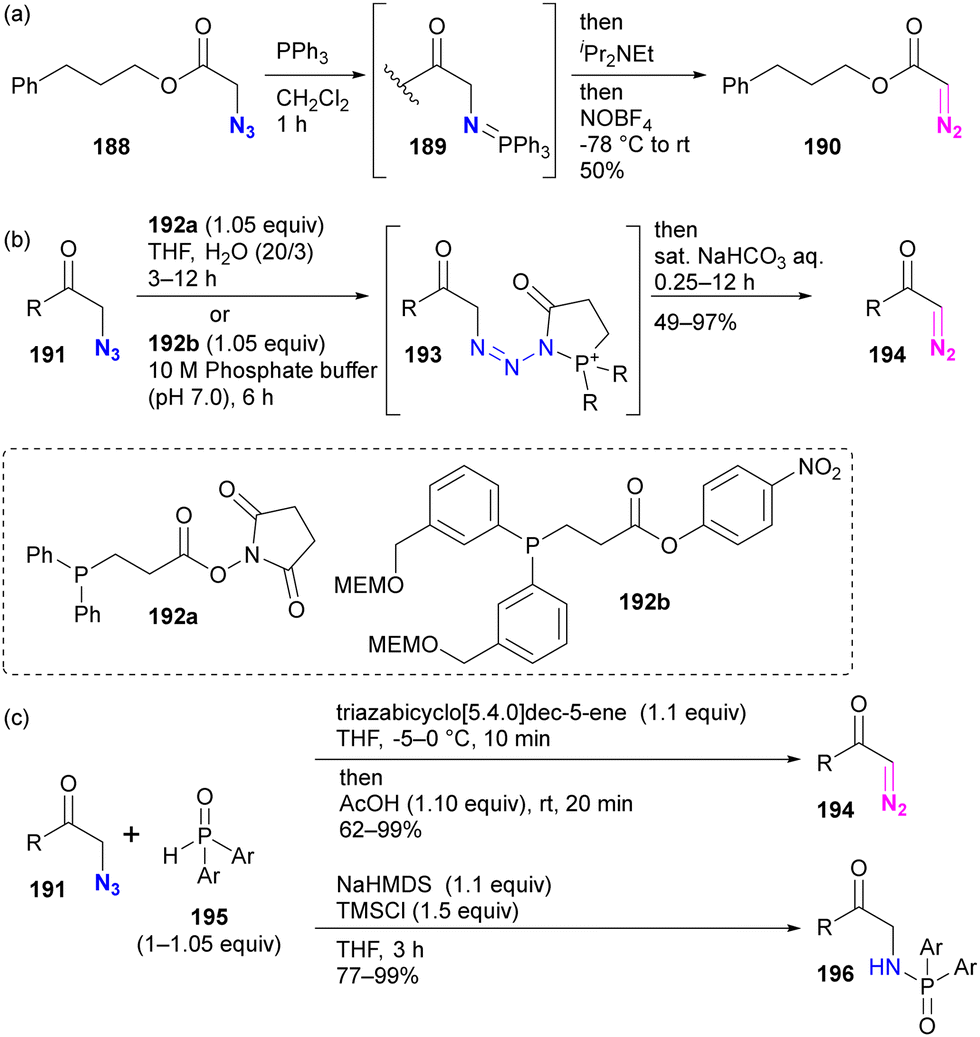 | ||
| Scheme 37 Other methods converting azido to diazo groups. | ||
With these recent advances in functional group conversion methods, the azido group is now also recognized as a protected form of the unstable diazo group. Consequently, further development of azide-converting strategies, including site selectivity and conversion to various functional groups, would be valuable not only in the field of click chemistry, but also in the broader field of synthetic organic chemistry.
4.7. Distinguishing the azido positions of allylic azides in equilibrium
Molecules occasionally exhibit distinct structures due to the presence of equilibrium states. In such cases, functional groups can occupy different environments. Allylic azides are an example of organic azides that exhibit this phenomenon. Specifically, the allylic azido group in 197a migrates to the alternate allylic position to form 197b and vice versa (Fig. 14). This rearrangement reaction is known as the Winstein rearrangement and occurs even at room temperature.223–225 The Winstein rearrangement is primarily a [3,3]-sigmatropic rearrangement, but recent studies by Topczewski and co-workers propose that it involves an ionic pathway through 197c.226 In this case, although the molecule has only one azido group, the azido group can exist in two or more states, including E/Z isomers and stereoisomers. The [3,3]-sigmatropic rearrangement also occurs in propargyl azides with a C–C triple bond. However, this rearrangement is irreversible due to the subsequent formation of allenyl azides, which promptly cyclize to triazoles (Banert cascade).227–231 Similar cycloaddition has been demonstrated in Scheme 8(a).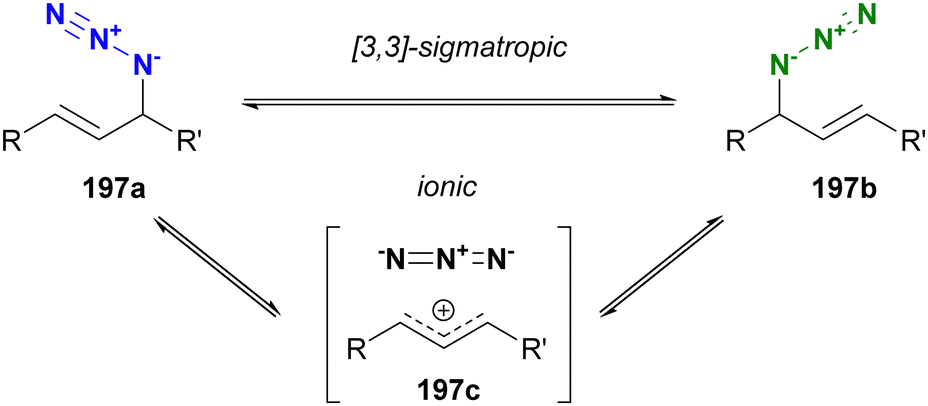 | ||
| Fig. 14 Winstein rearrangement of allyl azides. | ||
A simple strategy for distinguishing between the two (or more) azides in equilibrium is to capture the azido groups at specific positions through an intramolecular reaction. The key is to incorporate a reactive functional group into allylic azide compounds (Fig. 15). The design of these molecules should ensure that the reactive group is positioned far away from the azido group in 198a, but close enough to react after the Winstein rearrangement in 198b. This approach allows for azide-site selective reactions even under equilibrium conditions.
 | ||
| Fig. 15 Strategy for differentiation of reactive positions in allyl azides by intramolecular trapping. | ||
Aubé and co-workers applied their Schmidt reaction strategy in conjunction with the Winstein rearrangement for the formal synthesis of pinnaic acid 205, a cytosolic phospholipase A2 (cPLA2) inhibitor (Scheme 38).232 The bicyclic cyclobutanones 201a and 201b, which have an allylic azide side chain, existed in a 76:24 equilibrium. Although 201b was a minor component, treatment with titanium chloride under reflux conditions facilitated the intramolecular Schmidt reaction of 201b to the seven-membered ring intermediate 202, resulting in the formation of 203b as the major product. Since the formation of the nine-membered ring is required, the Schmidt reaction of 201a did not proceed. Subsequent hydroboration-oxidation of 203b using in situ prepared disiamylborane afforded Kibayashi's synthetic intermediate 204, a key precursor of pinnaic acid 205.233
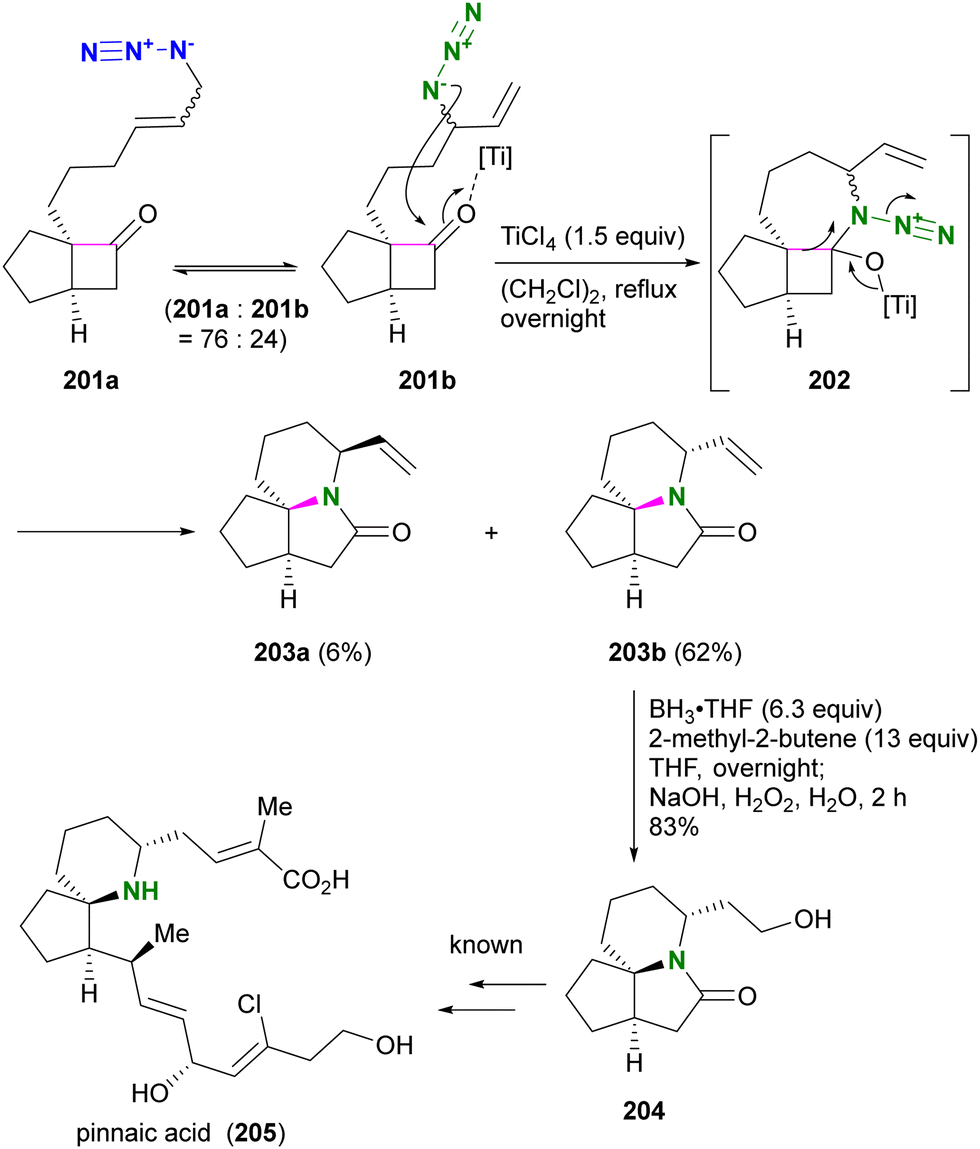 | ||
| Scheme 38 Formal synthesis of pinnaic acid 205 by the Winstein rearrangement-mediated intramolecular Schmidt reaction. | ||
Recently, Liu and co-workers reported the combination cascade reaction of Winstein rearrangement and intermolecular Schmidt reaction (Scheme 39).234 In general, the Schmidt reaction is inactive in intermolecular cases in the absence of strong electrophiles, such as activated sulfonium ions,235 due to the low nucleophilicity of organic azides. Therefore, the formation of oxocarbenium ions from ketones using azido alcohols is advantageous both in the intramolecular reaction process and in the activation of the carbonyl groups.236 Liu's group demonstrated the intermolecular Schmidt reaction using alcohols 207a,b consisting of an allyl azide structure. Upon formation of oxocarbenium ion intermediates208 derived from ketones 206 and azido alcohols 207, the primary alkylazido groups (208a), which are thermodynamically favored but far from the carbonyl group, are unreactive, similar to Scheme 38. With the rearranged secondary alkylazido groups (208b) close to the carbonyl group, intramolecular Schmidt reactions proceeded to afford the corresponding lactams 209 with branched hydroxyalkenyl substituents. The acyclic ketone moiety was less reactive than the cyclic ketone structure and the site-selective reaction was achieved, as shown in the case of the diketone substrate to give 209fa and 209fb.
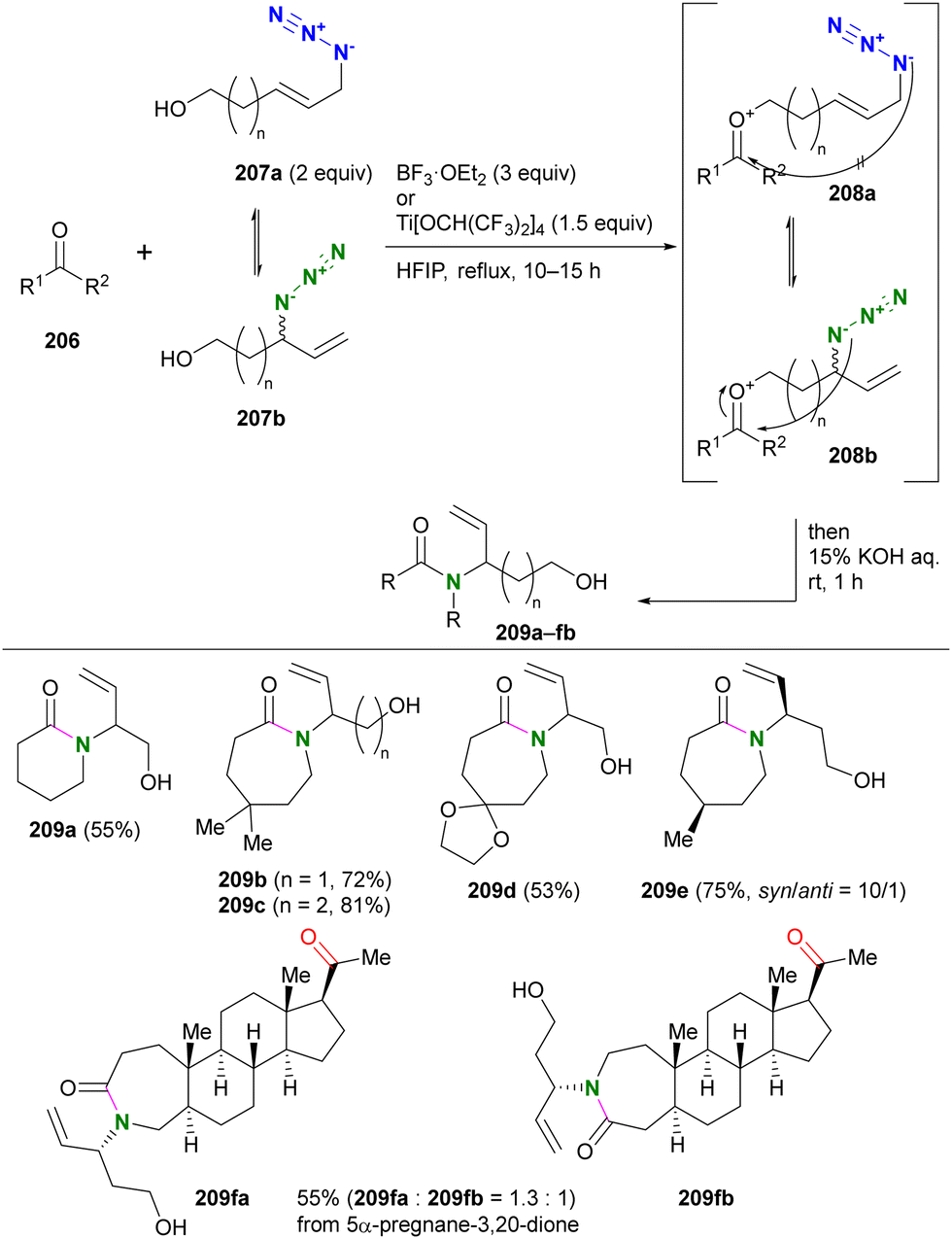 | ||
| Scheme 39 Winstein rearrangement-associated intermolecular Schmidt reaction. HFIP: 1,1,1,3,3,3-hexafluoro-2-propanol. | ||
Aubé and co-workers also used the Winstein rearrangement equilibrium for the azide–alkyne cycloaddition reaction (Scheme 40).237 In the presence of the primary allylic alkyl azide 210a and the secondary alkyl azide 210b in equilibrium, intramolecular cycloaddition with the alkynyl group took place under heating conditions. This resulted in the formation of the 6/6/5 tricyclic triazole 211 from the minor component secondary alkyl azide 210b. In the case of the acyclic enyne compound 212a, the CuAAC reaction at room temperature yielded only the dimeric bistriazole 213. Intramolecular CuAAC to produce 214 from the (Z)-olefin formed by the Winstein equilibrium did not occur. However, under heating conditions, the equilibrium shifted, facilitating the intramolecular azide–alkyne cycloaddition of the secondary alkyl azide 213b, resulting in the formation of 215. This result highlights the ability of reaction conditions to selectively differentiate the two equilibrium azides.
 | ||
| Scheme 40 Winstein rearrangement-associated intramolecular azide–alkyne [3+2] cycloaddition. | ||
A similar strategy was also exemplified in the synthesis of iminosugars by Murphy and co-workers (Scheme 41).238 The allyl azide 216a with the vinyl moiety was subjected to heating to facilitate the Winstein rearrangement and azide-olefin [3+2] cycloaddition. As shown in Scheme 41, the triazoline formation by the [3+2] reaction occurred exclusively from the rearranged internal azide 216b. In a one-pot reaction, the triazole ring-opening reactions of 217 were carried out with the addition of acetic acid or thiol as a nucleophile to give the corresponding iminosugars 218 and 219.
 | ||
| Scheme 41 Synthesis of iminosugars by intramolecular azide–alkene cycloaddition triggered by the Winstein rearrangement. | ||
In contrast to intramolecular trapping, it is difficult to distinguish the azide position by intermolecular reactions. This difficulty arises from the 1,3-dipolar nature of the azido group, where only weak interactions or coordination can be expected. To address this issue, Sharpless and Fokin's group demonstrated a case of copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) based on the peripheral environment of the allylic azido group in the early stages of their click chemistry (Scheme 42).239 CuAAC of primary and secondary alkyl azides 220a and 220b at equilibrium gave the corresponding triazole products 221a and 221b in a ratio similar to that of the starting materials. Conversely, triazole 223 from primary alkyl azide 222a was the only product obtained from primary and tertiary alkyl azides 222a and 222b. Even in the case of secondary and tertiary alkyl azides 224a and 224b, CuAAC product 225 was derived exclusively from secondary alkyl azide 220a without any other isomers. This demonstrates the significant influence of steric hindrance in achieving selective reactions at the less hindered position. However, the incorporation of a bulky group is not always feasible due to molecular design constraints. The challenge of distinguishing between multiple azido groups is discussed further in the following section on enantioselective reactions.
 | ||
| Scheme 42 Azide-site selective intermolecular CuAAC reactions under the Winstein rearrangement equilibrium conditions. | ||
On the other hand, converting the olefinic positions in the allyl azides is also an efficient way to differentiate. In this scenario, the azido groups remain intact. Here we show recent examples of the construction of vicinal diamine structures by cascade sigmatropic rearrangement (Scheme 43). Following the report of the combination with Claisen rearrangement by Craig and co-workers,240 Batey's group provided an example of distinguishing Winstein equilibrium states without capturing the azido group (Scheme 43(a)).241 The allylic azido groups in the starting trichloroacetimidates 226a and 226b were in equilibrium. Upon heating the starting materials under microwave irradiation conditions, the Overman rearrangement of the [3,3]-sigmatropic rearrangement is possible only from 226b, yielding the doubly [3,3]-rearranged vicinal diamine precursor materials 227. This cascade (domino) [3,3] rearrangement was also demonstrated for the cyclic compounds 228a and 228b, resulting in the formation of 229.
 | ||
| Scheme 43 [3,3]-Sigmatropic rearrangement cascade differentiating the azido position under equilibrium by Winstein rearrangement. | ||
Even if the Winstein rearrangement is set as the last step in the double rearrangement cascade, a conformational change can solve the equilibrium problem. In the synthetic study of jogyamycin 234, which has a densely substituted and aminated cyclopentane skeleton, Schomaker and co-workers chose the [3,3]-sigmatropic rearrangement cascade consisting of an Ichikawa rearrangement followed by a Winstein rearrangement as the key step (Scheme 43(b)).242 Both rearrangements occurred at ambient temperature, but the Winstein rearrangement as the second step involved the reverse reaction, preserving 232a with the two adjacent tetrasubstituted carbon centers. The group found that the conversion of the resulting isocyanates in 232a,b to urea resulted in the exclusive formation of 233, the doubly rearranged compound. In contrast to 232a and 232b, the reverse Winstein rearrangement of 233 occurred only when the temperature was raised to 100 °C in 1,4-dioxane. On the other hand, the Cope-Winstein rearrangement cascade forming a seven-membered ring by Meyer et al. produced single isomers.243 This difference demonstrates the conformation-dependent equilibrium of the Winstein rearrangement.
As introduced here, the development of the Winstein rearrangement has been prominent recently and has also been applied to asymmetric reactions (see next chapter). Although the equilibrium ratios are strongly dependent on the substrates and the substituents, site-selective use of the azido group positions is not impossible. In addition, recent mechanistic studies have suggested that the heteroatom functional group may influence the Winstein equilibrium ratio through a weak electrostatic interaction or negative hyperconjugation (rather than hydrogen bonding).244 These detailed studies would also help to further improve the accuracy of azide-site selective reactions.
4.8. Differentiation by enantioselective desymmetrization
As mentioned above, it is difficult to distinguish the azido positions in the absence of steric hindrance. In particular, achieving enantioselective reactions of organic azides is a formidable challenge. Due to the 1,3-dipolar nature of the azido group, positive intermolecular interactions with external components are not expected. Furthermore, in CuAAC reactions, the reaction of organic azides with copper acetylide complexes is typically faster than the copper acetylide formation step, which is generally considered to be the rate-determining step.245,246 This makes the desymmetrization of diazido compounds difficult. As a result, chemoselective reactions, particularly in asymmetric click reactions, are typically explored using diyne compounds to selectively couple an organic monoazido compound to one side of the alkynes.247,248 Despite this background, chemists are also addressing the challenging problem of achieving enantioselective or desymmetrization reactions in multi-azide systems. We review these efforts in this section.In the early stages of click chemistry, Meng, Fokin, and Finn reported the first enantioselective CuAAC coupling of diazides and the kinetic resolution of racemic monoazido compounds (Scheme 44).249 The scheme illustrates the desymmetrization of the geminal diazide 235. Using the PyBOX type ligand 236, phenylacetylene 18 was converted into mono- and over-clicked products. Despite the use of an excess of geminal diazide 235 (two equivalents to the alkynes), the major product was the over-clicked compound 237b, with only moderate optical purity observed for the mono-clicked product 237a. This landmark achievement also underscores the challenges of distinguishing between multiple azido positions in enantioselective reactions.
 | ||
| Scheme 44 Enantioselective desymmetrization of geminal diazides by CuAAC. | ||
Fifteen years later, Zhou's group developed a highly enantioselective CuAAC desymmetrization of alkyl diazides (Scheme 45).250 Using the PyBOX-type ligand 240 under low-temperature conditions, the CuAAC reaction of the prochiral diazido alcohol 238 proceeded enantioselectively, yielding 241a with up to 95%ee. In addition, iodoalkynes 239b were suitable substrates for this reaction, yielding 5-iodo-1,4-disubstituted triazoles 241b with enantiomeric excesses ranging from 74% to 96%. Notably, the formation of achiral bistriazole compounds by further click reactions is suppressed due to the mismatch with the chiral copper complex in mono-CuAAC products. Conversely, in the mirror-image mono-CuAAC pathway, the first CuAAC is slow while the second CuAAC is fast for the formation of achiral bistriazoles, effectively consuming the unnecessary mono-triazole enantiomer. These factors contribute to the high enantioselectivity observed in diazide desymmetrization.
 | ||
| Scheme 45 Enantioselective desymmetrization of alkyl diazides by CuAAC. | ||
In the context of kinetic resolution of racemic azides, Topczewski et al. reported the CuAAC-catalyzed kinetic resolution of cyclic alkyl azides (Scheme 46).251 Using the PyBOX-type ligand 244, the azido group at the benzylic position in racemic 243 was successfully enantioselectively coupled to alkynes 242, yielding chiral triazoles 245 with up to 94%ee. A notable feature of this work is the recycling of the unreactive chiral antipode starting material 243 by epimerization to the racemic form, which can then be reintroduced for subsequent kinetic resolution.
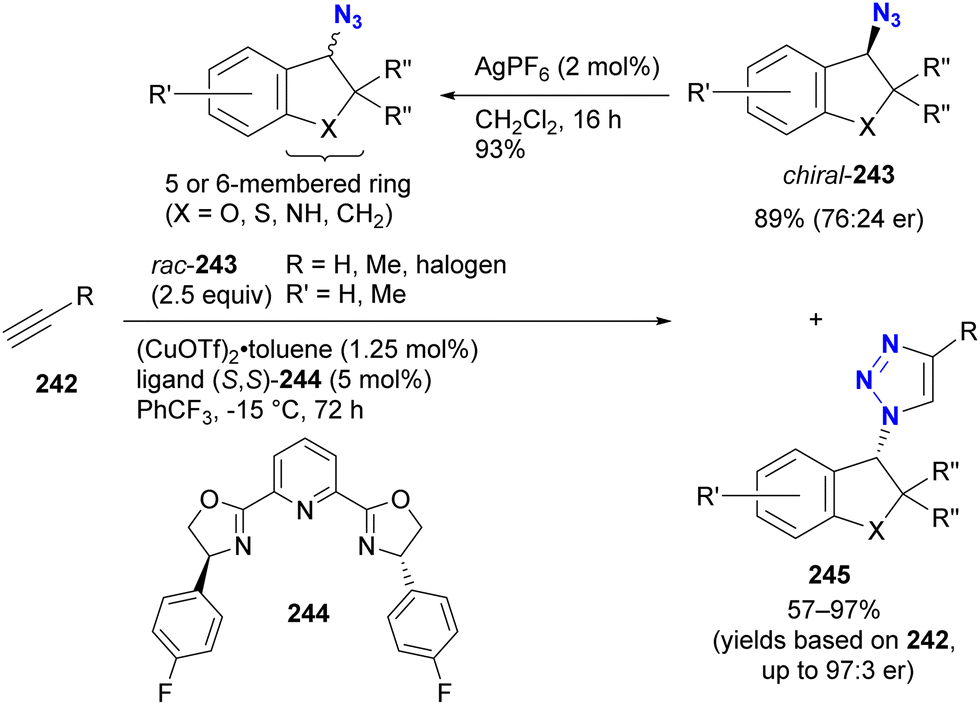 | ||
| Scheme 46 Kinetic resolution of racemic benzylic azides by CuAAC reaction and recycling of unreactive azides by racemization. | ||
Recently, Wang and Zhou's research group developed a kinetic resolution method for racemic bulky tert-alkyl azides, as shown in Scheme 47.252 This advance builds upon their azide desymmetrization chemistry shown in Scheme 45. The copper catalyst, in conjunction with the chiral sulfonyl PyBOX ligand 248, effectively converted the sterically hindered racemic azides 246 to the corresponding chiral triazoles 248. Remarkably, the starting azide was recovered as a chiral material by the enantioselective CuAAC reaction. Mechanistic insights gained from DFT calculations and control experiments indicate that the sulfonyl group decreases the Lewis basicity of the ligand while increasing the electrophilicity of the copper center. This improved coordination facilitates the interaction of the α-carbonyl moiety or the azide with the dicopper complex, thereby enabling enantiomeric recognition.
 | ||
| Scheme 47 Kinetic resolution of racemic tert-alkyl azides by the CuAAC reaction and application to the synthesis of the chiral kjellmanianone aza analog 252. | ||
Furthermore, they demonstrated the application of their kinetic resolution method using racemic tert-alkyl azide 250 to selectively obtain the unreactive enantiomer (R)-250. Following the Staudinger reaction of (R)-250, they successfully synthesized the optically active compound 247, an aza analog of the anti-Gram-positive bacterial kjellmanianone 248.
An intriguing case of simultaneous (the authors termed it “coetaneous”) kinetic resolution of both racemic alkyne 254 and azide 255 by CuAAC was reported by Buckley and Fossey (Scheme 48).253 Using the PyBOX ligand 256, two diastereomeric CuAAC products, including their enantiomers, were obtained. Among them, the major product (R,S)-257 was obtained with good enantiopurity (90%ee), while the minor diastereomer (R,R)-257 was nearly racemic.
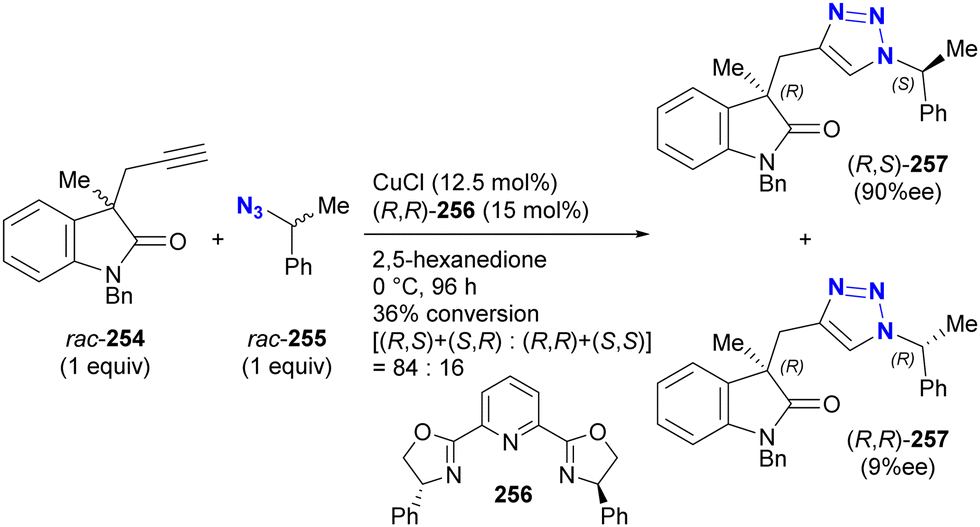 | ||
| Scheme 48 Coetaneous CuAAC kinetic resolution of racemic alkyne and azide. | ||
In Section 4.7, we introduced that the Winstein rearrangement is also associated with racemization. For example, the Winstein rearrangement product of the chiral compound 258 is ent-258 (Scheme 49). Therefore, the dynamic kinetic resolution of organic azides by the rearrangement equilibrium is promising. This significant advance has been documented by Topczewski and co-workers.254 Under elevated temperature conditions to facilitate rearrangement, CuAAC dynamic kinetic resolution of allyl azides using the PyBOX ligand 260 was successfully achieved with good isolated yields and enantiomeric excess values. This methodology proved effective for both cyclic (five- to seven-membered ring, containing either an ethereal oxygen atom or dimethylmethylene) and acyclic materials, as well as for alkynes linked to complex compounds such as bioactive natural products.
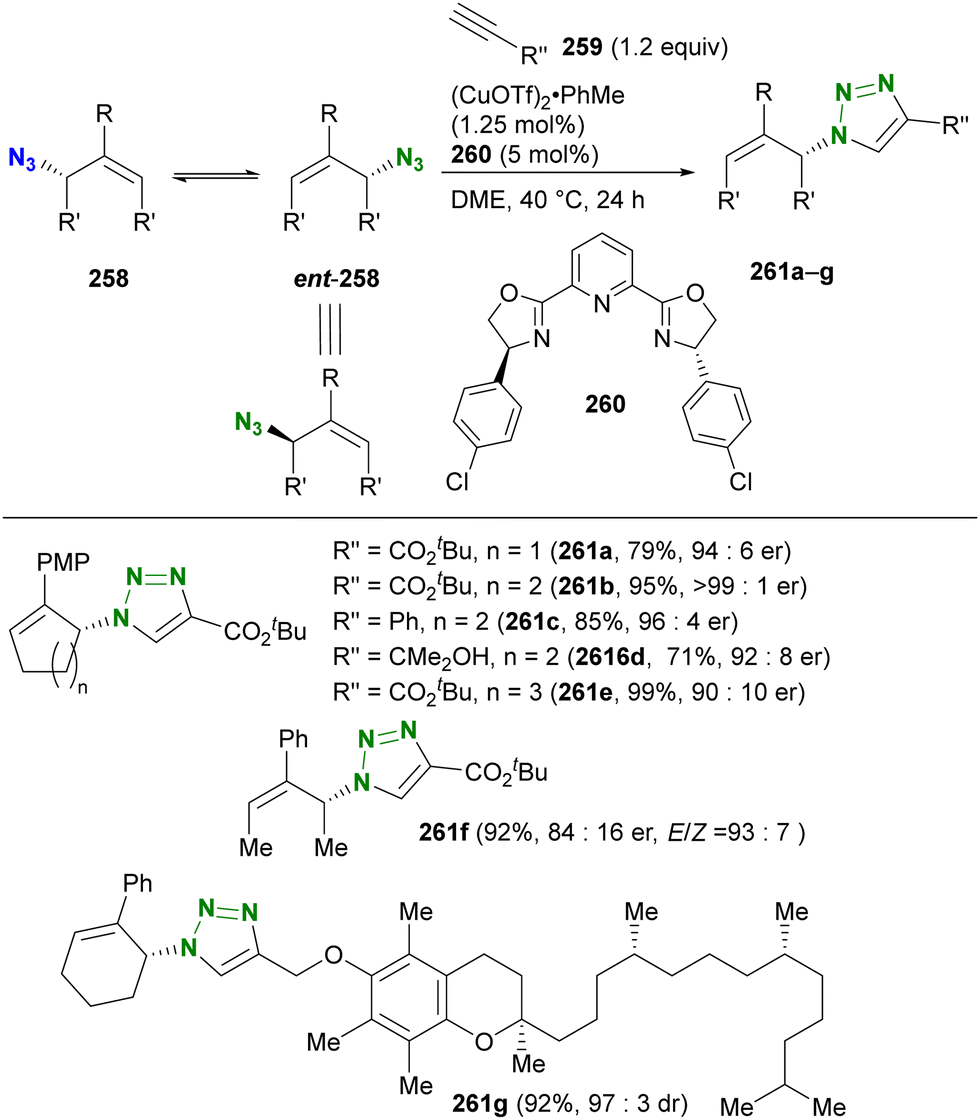 | ||
| Scheme 49 Enantioselective CuAAC by differentiation of allyl azide structures under Winstein rearrangement equilibrium. | ||
In addition, Topczewski et al. demonstrated the dynamic kinetic resolution of allyl azides by nickel-catalyzed azide–alkyne cycloaddition (NiAAC) using the chiral aminophosphine ligand 264via Winstein rearrangement (Scheme 50).255 While the substrates are similar to those of CuAAC, the nickel-catalyzed reaction exhibited acceptance of internal alkynes, in contrast to CuAAC. The NiAAC products 265a–f were obtained in moderate to excellent yields, accompanied by good enantioselectivity and regioselectivity with respect to the 4,5-position substituents.
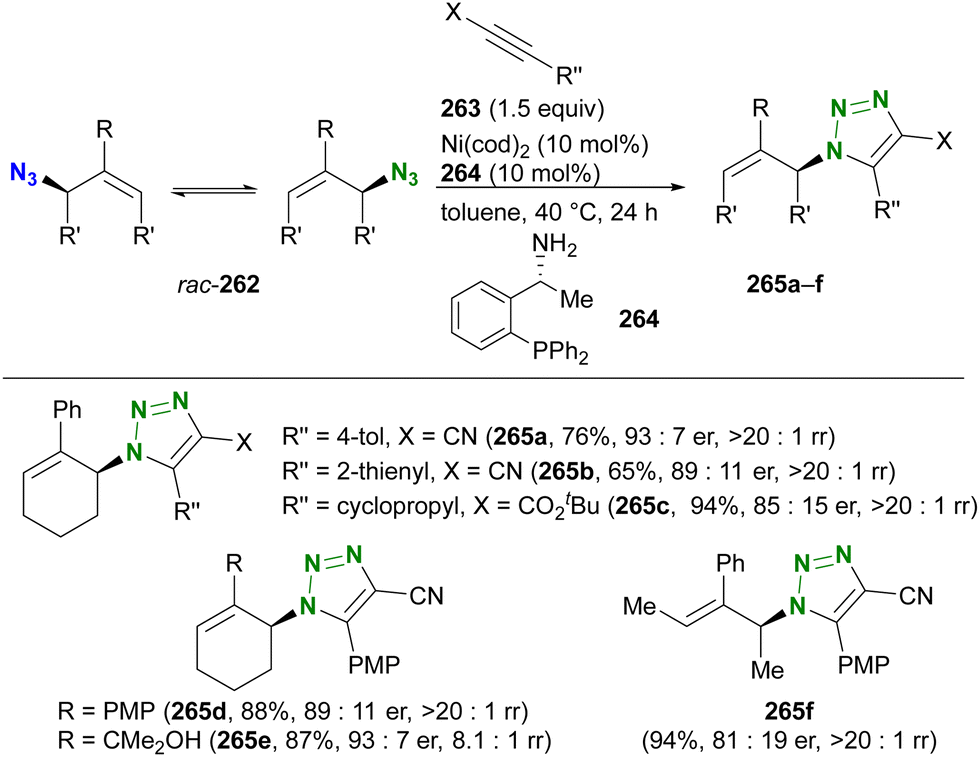 | ||
| Scheme 50 Enantioselective NiAAC by differentiation of allyl azide structures under the Winstein rearrangement equilibrium. | ||
Without converting azido groups, the functionalization of the olefin moiety provides a means of differentiating the azido position in equilibrium, although it does not constitute a click conjugation. Here we show an example of enantioselective reactions (Scheme 51).256 Topczewski's group also reported the dynamic kinetic resolution of allyl azides by asymmetric dihydroxylation of the alkene moiety via Winstein rearrangement.
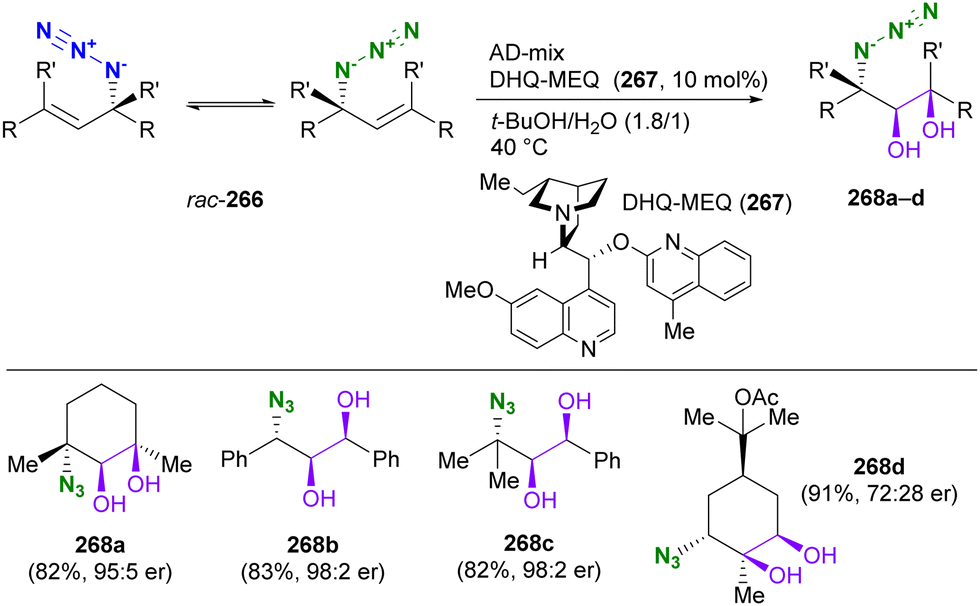 | ||
| Scheme 51 Enantioselective dihydroxylation of the olefins under the Winstein rearrangement equilibrium. | ||
In this section, we have reviewed recent significant advances in the catalytic asymmetric reactions of racemic organic azides, including dynamic kinetic resolution. With the exception of nickel-catalyzed reactions, PYBOX-type ligands have proven to be efficient for copper-catalyzed asymmetric click reactions. Future developments in this research area would be promising for applications in the chiral organization of supramolecules and the potential of selective click labeling in a bioorthogonal environment.
5. Azide-site selective multicomponent integration on multi-azide scaffolds (N3 × 2, 3, and 4)
In the previous chapter, we introduced a variety of methods and strategies for site-selective use of the multiple azido groups. In this chapter, we present their application to azide-site selective multicomponent conjugation to multi-azide scaffolds containing more than two types of azido groups.Using a hydrogen bonding strategy (Section 4.4), Tanimoto and co-workers investigated the azide-site selective conjugation of diazide 136 with the bis-primary alkyl azide compound (Scheme 52(a)).175 In Scheme 27(a), the double Staudinger–Bertozzi ligation was demonstrated with initial utilization of the α-AzSA position followed by the benzylic azido group. Conversely, Scheme 52(a) illustrates the conjugation in reverse order. Using the carbocation-mediated triazole construction procedure with propargyl alcohol 143 (Schemes 8(a) and 27(b)), the reaction at the α-AzSA position of diazide 136 was suppressed, allowing selective progress at the benzylic azido position to introduce the HaloTag ligand-type moiety. During this step, the addition of trimethylsilyl azide instead of aqueous quenching again yielded a new diazide, 269, composed of primary and tertiary alkyl azides. Subsequently, α-AzSA of the less hindered primary alkyl azide in 269 was selectively conjugated with biotin via traceless Staudinger–Bertozzi ligation with phosphine 139, yielding 270 (Section 4.1). Finally, the remaining bulky tert-alkyl azido moiety in 270 was coupled to alkyne 271 with the fluorescent coumarin moiety via CuAAC, yielding compound 272.
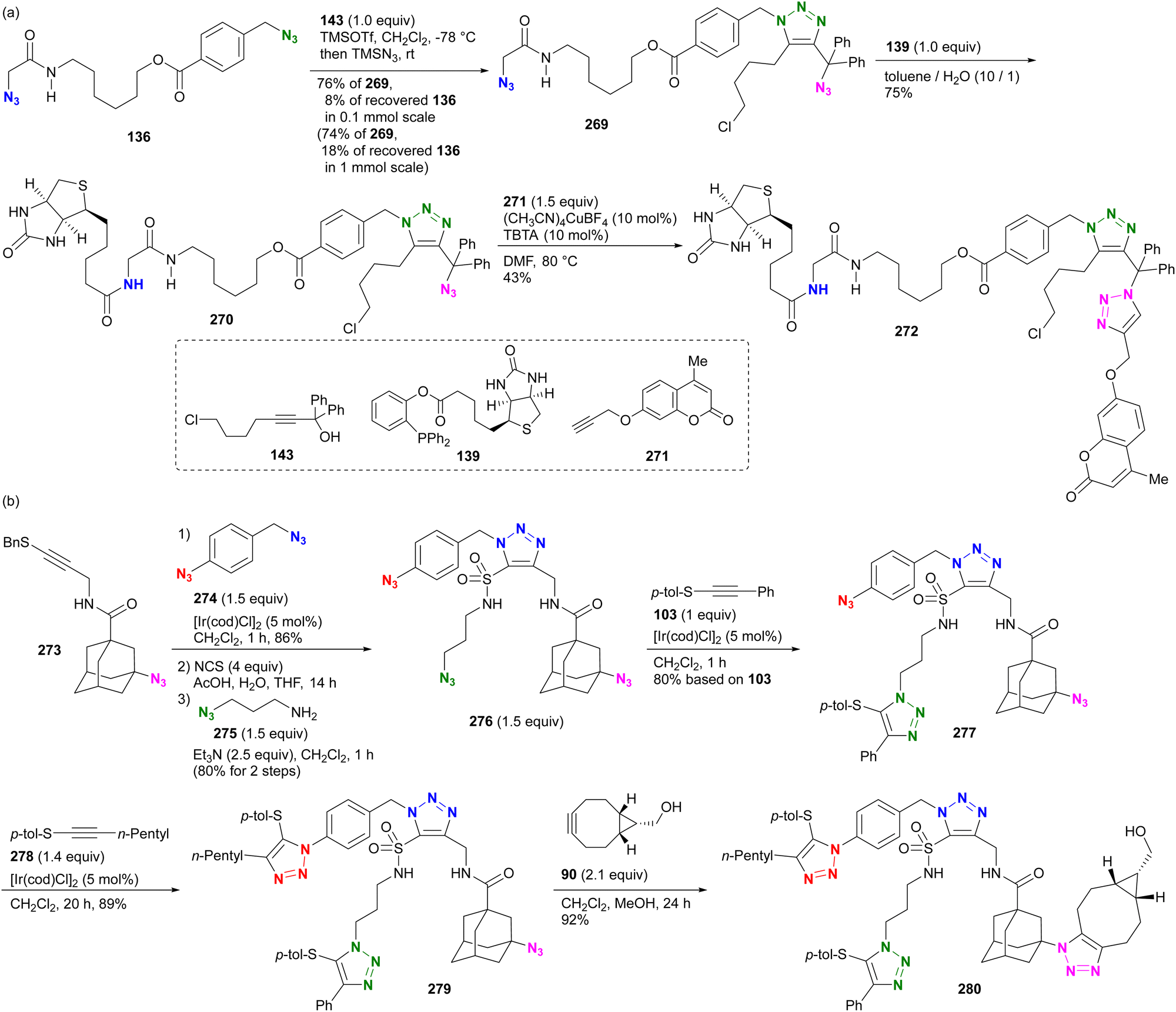 | ||
| Scheme 52 Differentiation of azido sites in in the (a) di- and (b) triazide platform compounds by successive introduction of azido groups. | ||
The iridium-catalyzed alkyl azide-selective triazole synthesis with thioalkynes (Scheme 20), which allows the prior use of alkyl azides in the presence of aryl azides, has also been demonstrated in azide-site selective conjugation onto a denser multi-azide platform by Hosoya and co-workers (Scheme 52(b)).137 The thioalkyne 273 containing the bulky adamantyl azide was selectively coupled to the alkyl aryl diazide 274 using an iridium catalyst. The benzyl sulfide moiety was then converted to a sulfonyl chloride by NCS oxidation and combined with the azido alkylamine 275 to synthesize the triazide 276 consisting of aryl, primary alkyl, and tertiary alkyl azido moieties. The primary alkyl azido moiety in triazide 276 was then first coupled to thioalkyne 103via the second iridium-catalyzed reaction. Next, the third iridium-catalyzed reaction coupled thioalkyne 278 with the remaining aryl azido moiety in diazide 277 to form 279. In the final step, the sequential multi-component integration was completed by conjugation to the remaining bulky adamantyl azide moiety in 279 with BCN 90, resulting in the formation of 280.
Highly azide-site-selective multicomponent conjugations in the presence of three or more azides have been achieved (Scheme 53). Using the enhanced clickability strategy of bulky aryl azides (Fig. 5), Hosoya and colleagues demonstrated azide-site selective conjugation in the presence of three azido groups (Scheme 53(a)).107 Conjugation to triazide 281, composed of primary alkyl, aryl, and 2,6-diisopropyl-flanked aryl azides, began with enolate-mediated aryl azide-selective triazolization with β-ketoamide 282 containing the HaloTag ligand. Subsequently, the alkyl azido moiety was selectively combined with BODIPY-alkyne 283via ruthenium-catalyzed azide–alkyne cycloaddition (RuAAC), resulting in the formation of 1,5-disubstituted triazoles. Finally, the 2,6-diisopropyl-aryl-azido moiety was coupled with biotin-tethered DIBO 284, yielding 285 as a four-component coupled product.
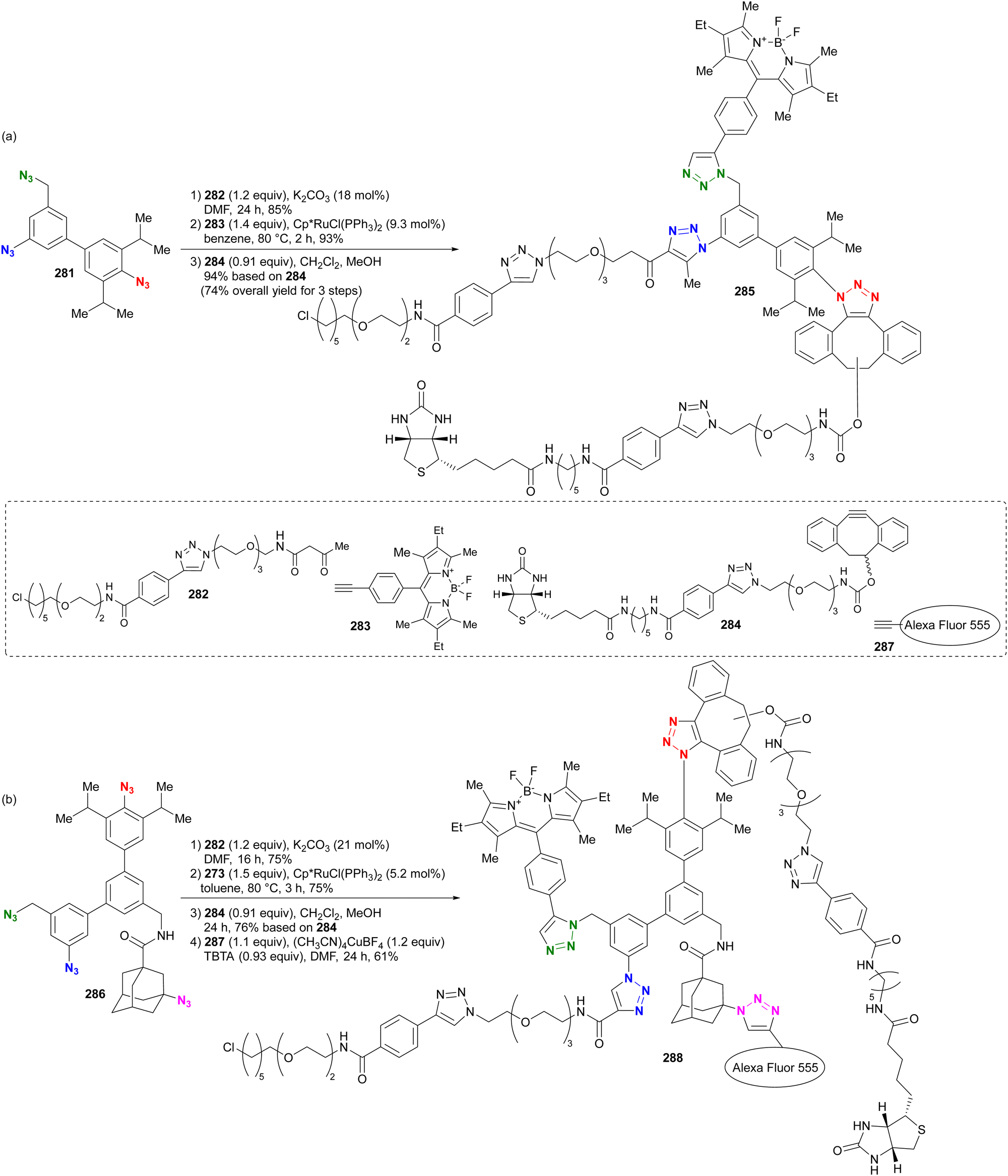 | ||
| Scheme 53 Sequential conjugation of the multi-components to the azido groups in the (a) tri- and (b) tetraazide platform compounds. | ||
Subsequently, the research group of Yoshida and Hosoya reported an advanced multicomponent conjugation by differentiating the four azide groups (Scheme 53(b)).257 The tetraazide platform molecule 286, comprising primary alkyl, aryl, 2,6-diisopropyl aryl, and tertiary alkyl azides, was subjected to sequential conjugation with four different components (1. Enolate-mediated triazolization of aryl azide with β-ketoamide 282; 2. RuAAC of primary alkyl azide with alkyne 283; 3. SPAAC of 2,6-diisopropyl aryl azide with DIBO 284; 4. CuAAC of tertiary alkyl azide with AlexaFluor-conjugated alkyne 287) to yield 288. Despite the presence of three or four potential click sites, each coupling step resulted in the desired position-conjugated products with good to excellent yields and selectivity.
In addition to azide-differentiating sequential conjugation, azide-site selective functional conversion of multi-azide compounds is used in multicomponent coupling reactions. The conversion of carbonyl-α-azides to diazo or oxime groups, as studied by Tanimoto and co-workers (Fig. 13 and Scheme 34), is an example of such reactions in multicomponent coupling reactions.211,212 The starting material, tri(alkylazide) compound 290, containing α-azido ketone, α-azido tert-amide, and general primary alkyl azide groups, was synthesized from 289 by global azidation of the three positions (Scheme 54(a)).211 First, the azido group at the α-position of ketocarbonyl moiety in 290 was converted to the diazo group to afford 291. Following a diazo site-selective [3+2] cycloaddition with 1,4-naphthoquinone 292, the azido group at the aminocarbonyl α-position was converted to the diazo group, yielding 293, while retaining the general alkyl azide moiety. Subsequent diazo site-selective [3+2] coupling with acrylate 178,213–216 followed by SPAAC on the remaining alkyl azido moiety with DIBO 51 resulted in the formation of 294.
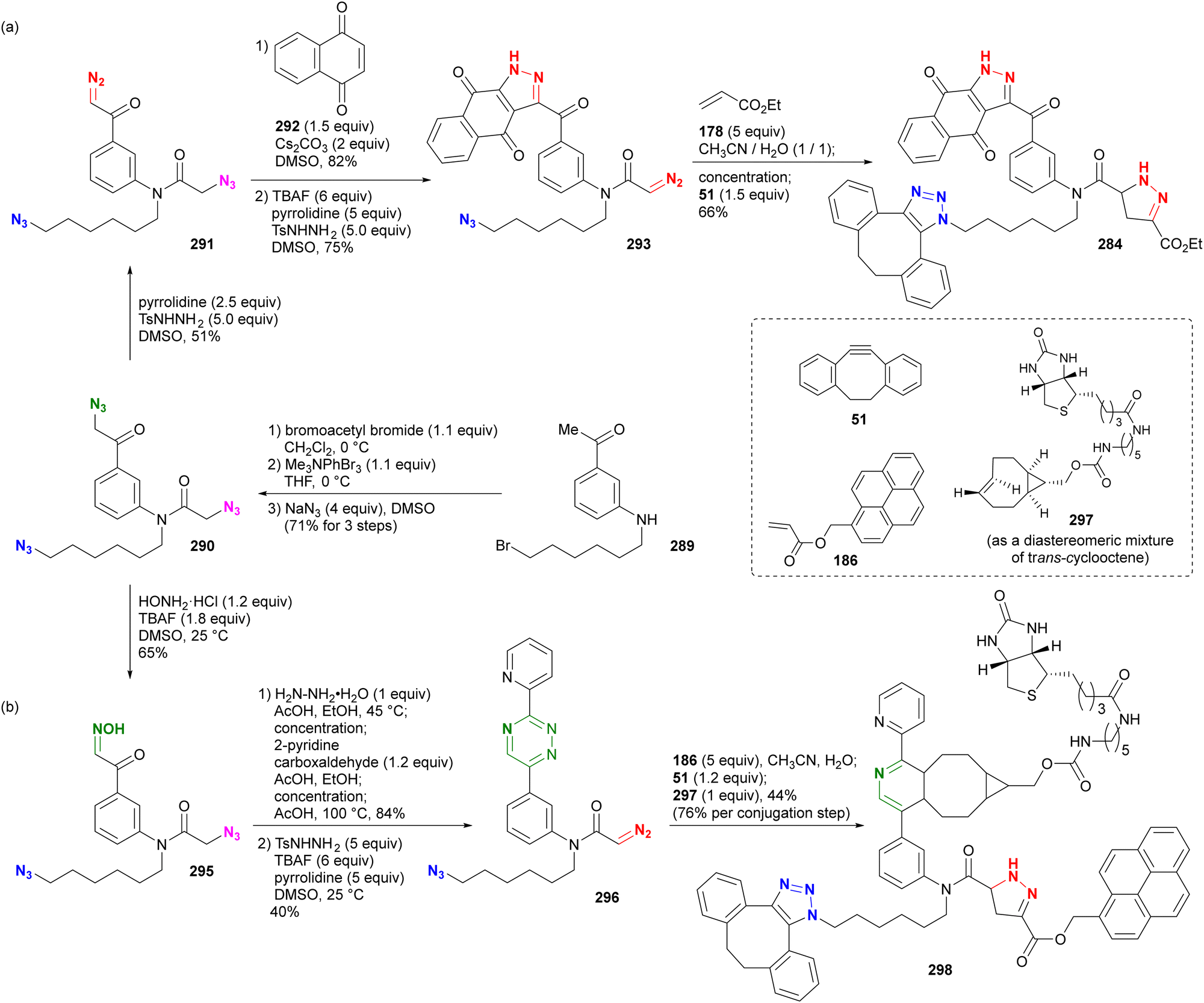 | ||
| Scheme 54 Multicomponent conjugation to the tris(alkylazido) compound. (a) Successive conjugation by diazo conversion. (b) One-pot conjugation by conversion of the triazide compound to the triple click scaffold. | ||
Furthermore, the same research group extended the synthesis of triazide 290 to the triple-click scaffold compound 296 with three different click functional groups (Scheme 54(b)).212 First, the azide was converted to an oxime at the ketone moiety (α-hydroxyiminoketone), yielding compound 295. Using Vrabel's method,258 the α-hydroxyimino ketone moiety was further converted in a single reaction to the 1,2,4-triazine skeleton of the selectively conjugated group with trans-cyclooctenes.259,260 After a second conversion of the azide on the amido moiety to the diazo group, the triple-click scaffold 296 consisting of triazine, diazo, and azido click groups was formed. Subsequently, by sequentially adding (1-pyrenyl)methyl acrylate 186 to the diazo group, DIBO 51 to the azido groups, and biotin-linked trans-cyclooctene 298 to the triazine group, the site-selective multicomponent integrated material 298 was successfully obtained from 296 in one pot.
6. Conclusions and outlook
In summary, this review provides a comprehensive overview of strategies to selectively exploit specific positions of azido groups, even in the presence of multiple other azido groups. Organic azides, known for their clickability, have proven to be efficient compounds in synthetic organic chemistry and especially in chemical biology over the last two decades. As a result, multi-azide molecules with multiple azido groups are attractive scaffolds for multicomponent conjugation to functional materials.To further enhance the multifunctionality of multi-azide-derived materials, the independent use of each azido group in a multi-azide compound can facilitate the integration of diverse components onto a single scaffold. Even when the same functional group is present, site-selective use among the multiple azido groups has been achieved by focusing on different properties related to the situation or environment of the azido groups, such as alkyl, aryl, sterically hindered, and carbonyl group adjacent positions as well as those in rearrangement equilibrium. Based on these findings, di-, tri-, and tetraazide compounds have proven to be versatile platforms for site-selective conjugation with various functional components.
The advancement of azide chemistry to distinguish molecular coupling or conjugation reactions facilitates more sophisticated applications of click chemistry beyond simple couplings. In addition, as suggested in a recent perspective report,261 there is a growing industrial need to establish efficient synthesis in azide chemistry, such as through continuous flow reactions or automated processes for safety.262–264 From this perspective, future efforts on new accurate azide site differentiation methods that take into account potential hazards could also further develop the practical application of the multi-azide strategies presented in this review, and chemoinformatics such as parameterization analysis, productivity optimization, and data analysis may also contribute to this issue. These advances are expected to have a significant impact on precise and accurate multi-component conjugation for the discovery of novel functional materials in materials science and the development of chemical biology through the use of efficient chemical probes in the life sciences.
Author contributions
HT conceptualized this review topic and wrote the first draft of the manuscript. All authors contributed to the review and editing of the original manuscript.Data availability
This Featured Article is a review of published research contributions, including those from the authors’ group. All schemes and figures have been prepared and reconstructed by the authors based on the peer-reviewed literature cited in the reference section. No primary or new research results, software, or code are included.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by grants from JSPS KAKENHI (C, JP22K06523) and the Takeda Science Foundation.Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021, DOI:10.1002/1521-3773(20010601)40:11%3C2004::AID-ANIE2004%3E3.0.CO;2-5.
- C. W. Tornøe, C. Christensen and M. Meldal, Chem. Rev., 2008, 108, 2952–3015, DOI:10.1021/cr0783479.
- N. J. Agard, J. M. Baskin, J. A. Prescher, A. Lo and C. R. Bertozzi, ACS Chem. Biol., 2006, 1, 644–648, DOI:10.1021/cb6003228.
- L. Zhu, C. J. Brassard, X. Zhang, P. M. Guha and R. J. Clark, Chem. Rec., 2016, 16, 1501–1517, DOI:10.1002/tcr.201600002.
- E. Kim and H. Koo, Chem. Sci., 2019, 10, 7835–7851, 10.1039/C9SC03368H.
- A. Qin, J. W. Y. Lamb and B. Z. Tang, Chem. Soc. Rev., 2010, 39, 2522–2544, 10.1039/B909064A.
- A. Qin, Y. Liu and B. Z. Tang, Macromol. Chem. Phys., 2015, 216, 818–828, DOI:10.1002/macp.201400571.
- Y. Shi, X. Cao and H. Gao, Nanoscale, 2016, 8, 4864–4881, 10.1039/C5NR09122E.
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, Polym. Chem., 2018, 9, 2853–2867, 10.1039/C7PY02047C.
- D. Huang, Y. Liu, A. Qin and B. Z. Tang, in Click Polymerization, ed. A. Qin, B. Z. Tang, The Royal Society of Chemistry, Croydon, 2018 Search PubMed.
- M. Schock and S. Bräse, Molecules, 2020, 25, 1009, DOI:10.3390/molecules25041009.
- K. Li, D. Fong, E. Meichsner and A. Adronov, Chem. – Eur. J., 2021, 27, 5057–5073, DOI:10.1002/chem.202003386.
- For a recent application of multiazides to porous materials, see J. Brückel, Y. Matt, L. Schmidt, C. Mattern, U. Schepers, S. Leopold, M. Calkovsky, D. Gerthsen and S. Bräse, ChemNanoMat, 2024, 10, e202300222, DOI:10.1002/cnma.202300222.
- Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley & Sons, West Sussex, 2009 Search PubMed.
- M. F. Debets, C. W. J. van der Doelen, F. P. J. T. Rutjes and F. L. van Delft, ChemBioChem, 2010, 11, 1168–1184, DOI:10.1002/cbic.201000064.
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676, DOI:10.1021/ar200148z.
- D. M. Patterson, L. A. Nazarova and J. A. Prescher, ACS Chem. Biol., 2014, 9, 592–605, DOI:10.1021/cb400828a.
- A. Lossouarn, P.-Y. Renard and C. Sabot, Bioconjugate Chem., 2021, 32, 63–72, DOI:10.1021/acs.bioconjchem.0c00568.
- K. Porte, M. Riomet, C. Figliola, D. Audisio and F. Taran, Chem. Rev., 2021, 121, 6718–6743, DOI:10.1021/acs.chemrev.0c00806.
- T. Luu, K. Gristwood, J. C. Knight and M. Jörg, Bioconjugate Chem., 2024, 35, 715–731, DOI:10.1021/acs.bioconjchem.4c00084.
- D. M. Beal and L. H. Jones, Angew. Chem., Int. Ed., 2012, 51, 6320–6326, DOI:10.1002/anie.201200002.
- D. Sato, Z. Wu, H. Fujita, J. S. Lindsey, D. Sato, Z. Wu, H. Fujita and J. S. Lindsey, Organics, 2021, 2, 161–273, DOI:10.3390/org2030013.
- D. Goyard, A. M.-S. Ortiz, D. Boturyn and O. Renaudet, Chem. Soc. Rev., 2022, 51, 8756–8783, 10.1039/D2CS00640E.
- S. Yoshida, Org. Biomol. Chem., 2020, 18, 1550–1562, 10.1039/C9OB02698C.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 633–643, DOI:10.1246/bcsj.20150081.
- M. Gon, K. Tanaka and Y. Chujo, Polym. J., 2018, 50, 109–126, DOI:10.1038/pj.2017.56.
- New Polymeric Materials Based on Element-Blocks, ed. Y. Chujo, Springer, Singapore, 2019 Search PubMed.
- M. Gon, S. Ito, K. Tanaka and Y. Chujo, Bull. Chem. Soc. Jpn., 2021, 94, 2290–2301, DOI:10.1246/bcsj.20210235.
- A.-M. Caminade, R. Laurent, B. Delavaux-Nicota and J. P. Majoral, New J. Chem., 2012, 36, 217–226, 10.1039/C1NJ20458K.
- F. Najafi, M. Salami-Kalajahi and H. Roghani-Mamaqani, J. Mol. Liq., 2022, 347, 118396, DOI:10.1016/j.molliq.2021.118396.
- S.-S. Ge, B. Chen, Y.-Y. Wu, Q.-S. Long, Y.-L. Zhao, P.-Y. Wang and S. Yang, RSC Adv., 2018, 8, 29428–29454, 10.1039/C8RA03538E.
- R. Miyajima, K. Sakai, Y. Otani, T. Wadatsu, Y. Sakata, Y. Nishikawa, M. Tanaka, Y. Yamashita, M. Hayashi, K. Kondo and T. Hayashi, ACS Chem. Biol., 2020, 15, 2364–2373, DOI:10.1021/acschembio.0c00335.
- D. Korovesis, H. A. Beard, C. Mérillat and S. H. L. Verhelst, ChemBioChem, 2021, 22, 2206–2218, DOI:10.1002/cbic.202000874.
- S.-S. Xue, Y. Li, W. Pan, N. Li and B. Tang, Chem. Commun., 2023, 59, 3040–3049, 10.1039/D2CC07008A.
- E. Saxon and C. R. Bertozzi, Science, 2000, 287, 2007–2010, DOI:10.1126/science.287.5460.2007.
- E. Saxon, S. J. Luchansky, H. C. Hang, C. Yu, S. C. Lee and C. R. Bertozzi, J. Am. Chem. Soc., 2002, 124, 14893–14902, DOI:10.1021/ja027748x.
- For recent reviews, see: Z.-P. A. Wang, C.-L. Tiana and J.-S. Zheng, RSC Adv., 2015, 5, 107192, 10.1039/C5RA21496C.
- S. Liu and K. J. Edgar, Biomacromolecules, 2015, 16, 2556–2571, DOI:10.1021/acs.biomac.5b00855.
- C. Bednarek, I. Wehl, N. Jung, U. Schepers and S. Bräse, Chem. Rev., 2020, 120, 4301–4354, DOI:10.1021/acs.chemrev.9b00665.
- T. K. Heiss, R. S. Dorn and J. A. Prescher, Chem. Rev., 2021, 121, 6802–6849, DOI:10.1021/acs.chemrev.1c00014.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599, DOI:10.1002/1521-3773(20020715)41:14%3C2596::AID-ANIE2596%3E3.0.CO;2-4.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057–3064, DOI:10.1021/jo011148j.
- For recent reviews, see: M. Meldal and F. Diness, Trends Chem., 2020, 2, 569–584, DOI:10.1016/j.trechm.2020.03.007.
- S. Neumann, M. Biewend, S. Rana and W. H. Binder, Macromol. Rapid Commun., 2020, 41, 1900359, DOI:10.1002/marc.201900359.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047, DOI:10.1021/ja044996f.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 9422–9425, DOI:10.1002/anie.201003761.
- For recent reviews, see: J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16, DOI:10.1007/s41061-016-0010-x.
- E. G. Chupakhin and M. Y. Krasavin, Chem. Heterocycl. Compd., 2018, 54, 483–501, DOI:10.1007/s10593-018-2295-x.
- L. Zhu and R. Kinjo, Chem. Soc. Rev., 2023, 52, 5563–5606, 10.1039/D3CS00290J.
- M. K. Jaiswal and V. K. Tiwari, Chem. Rec., 2023, 23, e202300167, DOI:10.1002/tcr.202300167.
- For review, see: A. R. Pradipta and K. Tanaka, Chem. Commun., 2021, 57, 9798–9806, 10.1039/D1CC03590H.
- For the research papers published after the review, see A. R. Pradipta, P. Ahmadi, K. Terashima, K. Muguruma, M. Fujii, T. Ichino, S. Maeda and K. Tanaka, Chem. Sci., 2021, 12, 5438–5449, 10.1039/D0SC06083F.
- A. R. Pradipta, H. Michiba, A. Kubo, M. Fujii, T. Tanei, K. Morimoto, K. Shimazu and K. Tanaka, Bull. Chem. Soc. Jpn., 2022, 95, 421–426, DOI:10.1246/bcsj.20210387.
- Y. Ode, A. R. Pradipta, P. Ahmadi, A. Ishiwata, A. Nakamura, Y. Egawa, Y. Kusakari, K. Muguruma, Y. Wang, X. Yin, N. Sato, H. Haba and K. Tanaka, Chem. Sci., 2023, 14, 8054–8060, 10.1039/D3SC02513F.
- Y. Ode, A. R. Pradipta, A. Ishiwata, A. Nambu, K. Ohnuki, H. Mizuma, H. Habac and K. Tanaka, Chem. Commun., 2024, 60, 3291–3294, 10.1039/D4CC00048J.
- For selected recent reviews, see: V. Dimakos and M. S. Taylor, Chem. Rev., 2018, 118, 11457–11517, DOI:10.1021/acs.chemrev.8b00442.
- Y. Ueda, Chem. Pharm. Bull., 2021, 69, 931–944, DOI:10.1248/cpb.c21-00425.
- For a selected recent example, see: H. Tang, Y.-B. Tian, H. Cui, R.-Z. Li, X. Zhang and D. Niu, Nat. Commun., 2020, 11, 5681, DOI:10.1038/s41467-020-19348-x.
- For selected recent papers, see: V. Perron, S. Abbott, N. Moreau, D. Lee, C. Penney and B. Zacharie, Synthesis, 2009, 283–289, DOI:10.1055/s-0028-1083290.
- I. B. Seiple, S. Su, I. S. Young, A. Nakamura, J. Yamaguchi, L. Jørgensen, R. A. Rodriguez, D. P. O’Malley, T. Gaich, M. Köck and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 14710–14726, DOI:10.1021/ja2047232.
- H. Chen, R. Huang, Z. Li, W. Zhu, J. Chen, Y. Zhan and B. Jiang, Org. Biomol. Chem., 2017, 15, 7339–7345, 10.1039/C7OB01866E.
- A. Fryszkowska, C. An, O. Alvizo, G. Banerjee, K. A. Canada, Y. Cao, D. DeMong, P. N. Devine, D. Duan, D. M. Elgart, I. Farasat, D. R. Gauthier, E. N. Guidry, X. Jia, J. Kong, N. Kruse, K. W. Lexa, A. A. Makarov, B. F. Mann, E. M. Milczek, V. Mitchell, J. Nazor, C. Neri, R. K. Orr, P. Orth, E. M. Phillips, J. N. Riggins, W. A. Schafer, S. M. Silverman, C. A. Strulson, N. Subramanian, R. Voladri, H. Yang, J. Yang, X. Yi, X. Zhang and W. Zhong, Science, 2022, 376, 1321–1327, DOI:10.1126/science.abn2009.
- Organic Azides: Synthesis and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, West Sussex, 2010 Search PubMed.
- For selected reviews published after the perspective book ref. 58, see: H. Tanimoto and K. Kakiuchi, Nat. Prod. Commun., 2013, 8, 1021–1034, DOI:10.1177/1934578X1300800730.
- D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440–11453, 10.1039/C4CC03016H.
- X.-R. Song, Y.-F. Qiu, X.-Y. Liu and Y.-M. Liang, Org. Biomol. Chem., 2016, 14, 11317–11331, 10.1039/C6OB01965J.
- K. Wu, Y. Liang and N. Jiao, Molecules, 2016, 21, 352, DOI:10.3390/molecules21030352.
- G. Huang and G. Yan, Adv. Synth. Catal., 2017, 359, 1600–1619, DOI:10.1002/adsc.201700103.
- R. Sala, C. Loro, F. Foschi and G. Broggini, Catalysts, 2020, 10, 1173, DOI:10.3390/catal10101173.
- H. Lindner, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 22347–22353, DOI:10.1021/jacs.3c09122.
- M. B. Hossain, D. van der Helm, R. Sanduja and M. Alam, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 1199–1202, DOI:10.1107/S0108270185007132.
- A. G. N. Avila, B. Deschênes-Simard, J. E. Arnold, M. Morency, D. Chartrand, T. Maris, G. Berger, G. M. Day, S. Hanessian and J. D. Wuest, J. Org. Chem., 2022, 87, 6680–6694, DOI:10.1021/acs.joc.2c00369.
- E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297–368, DOI:10.1021/cr00084a001.
- A. Hassner, M. Stern, H. E. Gottlieb and F. Frolow, J. Org. Chem., 1990, 55, 2304–2306, DOI:10.1021/jo00295a014.
- R. E. Conrow and W. D. Dean, Org. Process Res. Dev., 2008, 12, 1285–1286, DOI:10.1021/op8000977.
- For perspective, see: D. S. Treitler and S. Leung, J. Org. Chem., 2022, 87, 11293–11295, DOI:10.1021/acs.joc.2c01402.
- R. R. Farias, A. J. S. Mascarenhas, T. J. Santos and M. M. Victor, Tetrahedron Lett., 2020, 61, 152574, DOI:10.1016/j.tetlet.2020.152574.
- H. Tanimoto, R. Adachi, A. Otsuki and T. Tomohiro, Organics, 2022, 3, 520–533, DOI:10.3390/org3040035.
- T. Ikeda and Y. Matsushita, Chem. Lett., 2020, 49, 14–16, DOI:10.1246/cl.190744.
- R. Butera, A. Shrinidhi, K. Kurpiewska, J. Kalinowska-Tłuścik and A. Dömling, Chem. Commun., 2020, 56, 10662–10665, 10.1039/D0CC04522E.
- W. Hayes, H. M. I. Osborn, S. D. Osborne, R. A. Rastall and B. Romagnoli, Tetrahedron, 2003, 59, 7983–7996, DOI:10.1016/j.tet.2003.08.011.
- H. Erhardt, F. Mohr and S. F. Kirsch, Chem. Commun., 2016, 52, 545–548, 10.1039/C5CC08163G.
- Kirsch group has studied the chemistry of geminal diazides (sometimes triazides), and the following reviews and accounts summarize various reactions controlling their characteristics. See: K. Bensberg and S. F. Kirsch, Synthesis, 2022, 4447–4460, DOI:10.1055/s-0042-1751355.
- I. E. Celik and S. F. Kirsch, Eur. J. Org. Chem., 2021, 53–63, DOI:10.1002/ejoc.202001020.
- A. P. Häring and S. F. Kirsch, Molecules, 2015, 20, 20042–20062, DOI:10.3390/molecules201119675.
- P. Biallas, J. Heider and S. F. Kirsch, Polym. Chem., 2019, 10, 60–64, 10.1039/C8PY01087K.
- N. Münster, P. Nikodemiak and U. Koert, Org. Lett., 2016, 18, 4296–4299, DOI:10.1021/acs.orglett.6b02048.
- D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chem. – Eur. J., 2019, 25, 754–758, DOI:10.1002/chem.201805215.
- D. Svatunek, N. Houszka, T. A. Hamlin, F. M. Bickelhaupt and H. Mikula, Chem. – Eur. J., 2022, 28, e202200414, DOI:10.1002/chem.202200414.
- H. Takemura, S. Goto, T. Hosoya and S. Yoshida, Chem. Commun., 2020, 56, 15541–15544, 10.1039/D0CC07212E.
- S. Yoshida, A. Shiraishi, K. Kanno, T. Matsushita, K. Johmoto, H. Uekusa and T. Hosoya, Sci. Rep., 2011, 1, 82, DOI:10.1038/srep00082.
- S. Yoshida, J. Tanaka, Y. Nishiyama, Y. Hazama, T. Matsushita and T. Hosoya, Chem. Commun., 2018, 54, 13499–13502, 10.1039/C8CC05791E.
- S. Yoshida, S. Goto, Y. Nishiyama, Y. Hazama, M. Kondo, T. Matsushita and T. Hosoya, Chem. Lett., 2019, 48, 1038–1041, DOI:10.1246/cl.190400.
- H. Zhang, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Org. Lett., 2013, 15, 5222–5225, DOI:10.1021/ol402387w.
- H. Zhang, H. Tanimoto, T. Morimoto, Y. Nishiyama and K. Kakiuchi, Tetrahedron, 2014, 70, 9828–9835, DOI:10.1016/j.tet.2014.10.076.
- S. He, G. Dong, J. Cheng, Y. Wu and C. Sheng, Med. Res. Rev., 2022, 42, 1280–1342, DOI:10.1002/med.21877.
- R. I. Troup, C. Fallan and M. G. J. Baud, Explor. Target Anti-Tumor Ther., 2020, 1, 273–312, DOI:10.37349/etat.2020.00018.
- For recent research on the piperazine as a PROTAC linker, see: J. Desantis, A. Mammoli, M. Eleuteri, A. Coletti, F. Croci, A. Macchiarulo and L. Goracci, RSC Adv., 2022, 12, 21968–21977, 10.1039/D2RA03761K.
- For reviews, see: V. A. Bakulev, T. Beryozkina, J. Thomas and W. Dehaen, Eur. J. Org. Chem., 2018, 262–294, DOI:10.1002/ejoc.201701031.
- J. John, J. Thomas and W. Dehaen, Chem. Commun., 2015, 51, 10797–10806, 10.1039/C5CC02319J.
- For the mechanistic study, see: F. V. Gaspar, M. F. M. F. Azevedo, L. S. A. Carneiro, S. B. Ribeiro, P. M. Esteves and C. D. Buarque, Tetrahedron, 2022, 120, 132856, DOI:10.1016/j.tet.2022.132856.
- For the report of sulfonyl azides, see: G. Cheng, X. Zeng, J. Shen, X. Wang and X. Cui, Angew. Chem., Int. Ed., 2013, 52, 13265–13268, DOI:10.1002/anie.201307499.
- For a recent application example on the DNA-encoded library synthesis, see: K. Pan, Y. Yao, Y. Zhang, Y. Gu, Y. Wang, P. Ma, W. Hou, G. Yang, S. Zhang and H. Xu, Bioconjugate Chem., 2023, 34, 1459–1466, DOI:10.1021/acs.bioconjchem.3c00235.
- D. B. Ramachary, A. B. Shashank and S. Karthik, Angew. Chem., Int. Ed., 2014, 53, 10420–10424, DOI:10.1002/anie.201406721.
- S. Zeghada, G. Bentabed-Ababsa, A. Derdour, S. Abdelmounim, L. R. Domingo, J. A. Sáez, T. Roisnel, E. Nassar and F. Mongin, Org. Biomol. Chem., 2011, 9, 4295–4305, 10.1039/C1OB05176H.
- E. Pei, J. Ng, Y.-F. Wang, B. W.-Q. Hui, G. Lapointe and S. Chiba, Tetrahedron, 2011, 67, 7728–7737, DOI:10.1016/j.tet.2011.08.006.
- S. Yoshida, K. Kanno, I. Kii, Y. Misawa, M. Hagiwara and T. Hosoya, Chem. Commun., 2018, 54, 3705–3708, 10.1039/C8CC01195H.
- For selected recent examples, see: M. Sundhoro, J. Park, B. Wu and M. Yan, Macromolecules, 2018, 51, 4532–4540, DOI:10.1021/acs.macromol.8b00618.
- Y. Li, N. Busatto and P. J. Roth, Macromolecules, 2021, 54, 3101–3111, DOI:10.1021/acs.macromol.0c02833.
- K. Wang and J. A. Gladysz, Macromolecules, 2022, 55, 8883–8891, DOI:10.1021/acs.macromol.2c01552.
- L.-Y. Hsia, H.-N. Chen, C.-H. Chiang, M.-Y. Hung, H.-K. Wei, C.-W. Luo, M.-Y. Kuo, S.-Y. Luo and C.-C. Chu, J. Org. Chem., 2020, 85, 9361–9366, DOI:10.1021/acs.joc.0c00901.
- T. Umeno, K. Usui and S. Karasawa, Asian J. Org. Chem., 2021, 10, 1123–1130, DOI:10.1002/ajoc.202100015.
- H.-Y. Lin, C.-F. Chen, C.-H. Chen, J.-L. Yeh, T.-T. Huang, Y.-C. Chu and C.-C. Chu, ACS Appl. Polym. Mater., 2022, 4, 7518–7527, DOI:10.1021/acsapm.2c01206.
- S. Xie, S. A. Lopez, O. Ramström, M. Yan and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 2958–2966, DOI:10.1021/ja511457g.
- M. Sundhoro, S. Jeon, J. Park, O. Ramström and M. Yan, Angew. Chem., Int. Ed., 2017, 56, 12117–12121, DOI:10.1002/anie.201705346.
- C. Wei, R. Wang, L. Wei, L. Cheng, Z. Li, Z. Xi and L. Yi, Chem. – Asian J., 2014, 9, 3586–3592, DOI:10.1002/asia.201402808.
- J. Zhang, Y. Gao, X. Kang, Z. Zhu, Z. Wang, Z. Xi and L. Yi, Org. Biomol. Chem., 2017, 15, 4212–4217, 10.1039/C7OB00830A.
- Y. Xie, L. Cheng, Y. Gao, X. Cai, X. Yang, L. Yi and Z. Xi, Chem. – Asian J., 2018, 13, 1791–1796, DOI:10.1002/asia.201800503.
- D. Ma, X. Kang, Y. Gao, J. Zhu, L. Yi and Z. Xi, Tetrahedron, 2019, 75, 888–893, DOI:10.1016/j.tet.2019.01.002.
- For minireview, see: X. Kang, X. Cai, L. Yi and Z. Xi, Chem. – Asian J., 2020, 15, 1420–1429, DOI:10.1002/asia.202000005.
- L. Cheng, X. Kang, D. Wang, Y. Gao, L. Yi and Z. Xi, Org. Biomol. Chem., 2019, 17, 5675–5679, 10.1039/C9OB00528E.
- T. Meguro, N. Terashima, H. Ito, Y. Koike, I. Kii, S. Yoshida and T. Hosoya, Chem. Commun., 2018, 54, 7904–7907, 10.1039/C8CC00179K.
- M. Yamashina, H. Suzuki, N. Kishida, M. Yoshizawa and S. Toyota, Angew. Chem., Int. Ed., 2021, 60, 17915–17919, DOI:10.1002/anie.202105094.
- M. Hamada, G. Orimoto and S. Yoshida, Chem. Commun., 2024, 60, 7930–7933, 10.1039/D4CC02723J.
- T. Meguro, S. Yoshida and T. Hosoya, Chem. Lett., 2017, 46, 473–476, DOI:10.1246/cl.161159.
- J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaup and F. L. van Delft, Nat. Commun., 2014, 5, 5378, DOI:10.1038/ncomms6378.
- N. Gritsan and M. Platz, In Organic Azides: Synthesis and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, West Sussex, 2010, pp. 311–372 Search PubMed.
- S. Yoshida and T. Hosoya, in Cutting-Edge Organic Synthesis and Chemical Biology of Bioactive Molecules: The Shape of Organic Synthesis to Come, ed. Y. Kobayashi, Springer, 2019, pp. 335–355 Search PubMed.
- For a review of visible-light cross-linking using photocatalysts, see: Y. Zhang, J. Tan and Y. Chen, Chem. Commun., 2023, 59, 2413–2420, 10.1039/D2CC06987C.
- Recently, this photoreaction of aryl azides, generating aryl nitrenes, has been developed in aromatic ring skeleton editing. See: S. C. Patel and N. Z. Burns, J. Am. Chem. Soc., 2022, 144, 17797–17802, DOI:10.1021/jacs.2c08464.
- T. J. Pearson, R. Shimazumi, J. L. Driscoll, B. D. Dherange, D.-I. Park and M. D. Levin, Science, 2023, 381, 1474–1479, DOI:10.1126/science.adj5331.
- C. Bousch, B. Vreulz, K. Kansal, A. El-Husseini and S. Cecioni, Angew. Chem., Int. Ed., 2023, 62, e202314248, DOI:10.1002/anie.202314248.
- T. Hosoya, T. Hiramatsu, T. Ikemoto, M. Nakanishi, H. Aoyama, A. Hosoya, T. Iwata, K. Maruyama, M. Endo and M. Suzuki, Org. Biomol. Chem., 2004, 2, 637–641, 10.1039/B316221D.
- R. Neelarapu, D. L. Holzle, S. Velaparthi, H. Bai, M. Brunsteiner, S. Y. Blond and P. A. Petukhov, J. Med. Chem., 2011, 54, 4350–4364, DOI:10.1021/jm2001025.
- P.-H. Chiu, W. Huang, H.-T. Hsu, W.-F. Huang, Y.-T. Wu, T.-J. R. Cheng and J.-M. Fang, Eur. J. Med. Chem. Rep., 2022, 6, 100091, DOI:10.1016/j.ejmcr.2022.100091.
- K. Kempf, A. Raja, F. Sasse and R. Schobert, J. Org. Chem., 2013, 78, 2455–2461, DOI:10.1021/jo3026737.
- K. Sugiyama, Y. Sakata, T. Niwa, S. Yoshida and T. Hosoya, Chem. Commun., 2022, 58, 6235–6238, 10.1039/D2CC01739C.
- M. Kitamura, S. Kato, M. Yano, N. Tashiro, Y. Shiratake, M. Sando and T. Okauchi, Org. Biomol. Chem., 2014, 12, 4397–4406, 10.1039/C4OB00515E.
- M. Kitamura, M. Yano, N. Tashiro, S. Miyagawa, M. Sando and T. Okauchi, Eur. J. Org. Chem., 2011, 458–462, DOI:10.1002/ejoc.201001509.
- H. Ye, R. Liu, D. Li, Y. Liu, H. Yuan, W. Guo, L. Zhou, X. Cao, H. Tian, J. Shen and P. G. Wang, Org. Lett., 2013, 15, 18–21, DOI:10.1021/ol3028708.
- N. Fischer, E. D. Goddard-Borger, R. Greiner, T. M. Klapötke, B. W. Skelton and J. Stiestorfer, J. Org. Chem., 2012, 77, 1760–1764, DOI:10.1021/jo202264r.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797–3800, DOI:10.1021/ol701581g.
- For a safer diazo-transfer strategy by in situ sulfonyl azide preparation in water, see: D. Dar’in, G. Kantina and M. Krasavin, Chem. Commun., 2019, 55, 5239–5242, 10.1039/C9CC02042J.
- P. A. Zhmurov, D. V. Dar’in, O. Y. Bakulina and M. Krasavin, Mendeleev Commun., 2020, 30, 311–312, DOI:10.1016/j.mencom.2020.05.016.
- For selected reviews, see: T. Miura and M. Murakami, in Rhodium Catalysis in Organic Synthesis: Methods and Reactions, ed. K. Tanaka, Wiley-VCH, Weinheim, 2019, pp. 449–470 Search PubMed.
- H. M. L. Davies and J. S. Alford, Chem. Soc. Rev., 2014, 43, 5151–5162, 10.1039/C4CS00072B.
- P. Anbarasan, D. Yadagiri and S. Rajasekar, Synthesis, 2014, 3004–3023, DOI:10.1055/s-0034-1379303.
- For a recent application of carbene insertion and SPAAC of sulfonyl azides, see: M. B. Williams, R. J. Wells and A. Boyer, Chem. Commun., 2022, 58, 12495–12498, 10.1039/D2CC03648G.
- M. E. Meza-Aviña, M. K. Patel and M. P. Croatt, Tetrahedron, 2013, 69, 7840–7846, DOI:10.1016/j.tet.2013.05.048.
- M. E. Meza-Aviña, M. K. Patel, C. B. Lee, T. J. Dietz and M. P. Croatt, Org. Lett., 2011, 13, 2984–2987, DOI:10.1021/ol200696q.
- J. Boyer, C. Mack, N. Goebel and J. L. Morgan, J. Org. Chem., 2003, 23, 1051–1053, DOI:10.1021/jo01101a604.
- For recent development of the triazolization of sulfonyl azides with aryl acetylides followed by further cycloaddition to generate benzosultams, see: S. Aggarwal, A. Vu, D. B. Eremin, R. Persaud and V. V. Fokin, Nat. Chem., 2023, 15, 764–772, DOI:10.1038/s41557-023-01188-z.
- A. Santorelli and K. V. Gothelf, Nucleic Acids Res., 2022, 50, 7235–7246, DOI:10.1093/nar/gkac566.
- N. Shangguan, S. Katukojvala, R. Greenberg and L. J. Williams, J. Am. Chem. Soc., 2003, 125, 7754–7755, DOI:10.1021/ja0294919.
- R. V. Kolakowski, N. Shangguan, R. R. Sauers and L. J. Williams, J. Am. Chem. Soc., 2006, 128, 5695–5702, DOI:10.1021/ja057533y.
- D. T. S. Rijkers, R. Merkx, C.-B. Yim, A. J. Brouwer and R. M. J. Liskamp, J. Pept. Sci., 2010, 16, 1–5, DOI:10.1002/psc.1197.
- N. K. Namelikonda and R. Manetsch, Chem. Commun., 2012, 48, 1526–1528, 10.1039/C1CC14724B.
- M. Aswad, J. Chiba, T. Tomohiro and Y. Hatanaka, Chem. Commun., 2013, 49, 10242–10244, 10.1039/C3CC46055J.
- For a review of thioamide reaction with diazo and azido compounds, see: V. Bakulev, Y. Shafran and W. Dehaen, Tetrahedron Lett., 2019, 60, 513–523, DOI:10.1016/j.tetlet.2019.01.032.
- For the sulfo-click-like ligation with electron-poor aryl azides, see: S. Xie, R. Fukumoto, O. Ramström and M. Yan, J. Org. Chem., 2015, 80, 4392–4397, DOI:10.1021/acs.joc.5b00240.
- G. Clavé, E. Dursun, J.-J. Vasseur and M. Smietana, Org. Lett., 2020, 22, 1914–1918, DOI:10.1021/acs.orglett.0c00265.
- G. Clavé, J.-J. Vasseur and M. Smietana, Curr. Protoc. Nucleic Acid Chem., 2020, 83, e120, DOI:10.1002/cpnc.120.
- W. S. Brotherton, H. A. Michaels, J. T. Simmons, R. J. Clark, N. S. Dalal and L. Zhu, Org. Lett., 2009, 11, 4954–4957, DOI:10.1021/ol9021113.
- G.-C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540–6548, DOI:10.1021/jo101305m.
- G.-C. Kuang, P. M. Guha, W. S. Brotherton, J. T. Simmons, L. A. Stankee, B. T. Nguyen, R. J. Clark and L. Zhu, J. Am. Chem. Soc., 2011, 133, 13984–14001, DOI:10.1021/ja203733q.
- Z. Yuan, G.-C. Kuang, R. J. Clark and L. Zhu, Org. Lett., 2012, 14, 2590–2593, DOI:10.1021/ol300899n.
- C. Uttamapinant, A. Tangpeerachaikul, S. Grecian, S. Clarke, U. Singh, P. Slade, K. R. Gee and A. Y. Ting, Angew. Chem., Int. Ed., 2012, 51, 5852–5856, DOI:10.1002/anie.201108181.
- C. Uttamapinant, M. I. Sanchez, D. S. Liu, J. Z. Yao, K. A. White, S. Grecian, S. Clark, K. R. Gee and A. Y. Ting, Nat. Protoc., 2013, 8, 1620–1634, DOI:10.1038/nprot.2013.096.
- V. Bevilacqua, M. King, M. Chaumontet, M. Nothisen, S. Gabillet, D. Buisson, C. Puente, A. Wagner and F. Taran, Angew. Chem., Int. Ed., 2014, 53, 5872–5876, DOI:10.1002/anie.201310671.
- A. Sallustrau, S. Bregant, C. Chollet, D. Audisio and F. Taran, Chem. Commun., 2017, 53, 7890–7893, 10.1039/C7CC03247A.
- Y. Su, L. Li, H. Wang, X. Wang and Z. Zhang, Chem. Commun., 2016, 52, 2185–2188, 10.1039/C5CC08466K.
- N. Inoue, A. Onoda and T. Hayashi, Bioconjugate Chem., 2019, 30, 2427–2434, DOI:10.1021/acs.bioconjchem.9b00515.
- For the incorporation of copper ligand peptides with an alkyne moiety, see: A. G. Aioub, L. Dahora, K. Gamble and M. G. Finn, Bioconjugate Chem., 2017, 28, 1693–1701, DOI:10.1021/acs.bioconjchem.7b00161.
- A. J. Tölke, J. F. Gaisbauer, Y. V. Gärtner, B. Steigenberger, A. Holovan, F. Streshnev, S. Schneider, M. Müller and T. Carell, Angew. Chem., Int. Ed., 2024, 63, e202405161, DOI:10.1002/anie.202405161.
- K. Maegawa, H. Tanimoto, S. Onishi, T. Tomohiro, T. Morimoto and K. Kakiuchi, Org. Chem. Front., 2021, 8, 5793–5803, 10.1039/D1QO01088C.
- J. M. Dones, N. S. Abularrage, N. Khanal, B. Gold and R. T. Raines, J. Am. Chem. Soc., 2021, 143, 9489–9497, DOI:10.1021/jacs.1c03133.
- Y. Xia, L.-Y. Chen, S. Lv, Z. Sun and B. Wang, J. Org. Chem., 2014, 79, 9818–9825, DOI:10.1021/jo5011262.
- Protective Groups in Organic Synthesis, ed. P. G. M. Wuts, John Wiley & Sons, Hoboken, NJ, 4th edn, 2014 Search PubMed.
- B. Loubinoux and P. Gerardin, Tetrahedron Lett., 1991, 32, 351–354, DOI:10.1016/S0040-4039(00)92626-1.
- K. Fukase, M. Hashida and S. Kusumoto, Tetrahedron Lett., 1991, 32, 3557–3558, DOI:10.1016/0040-4039(91)80832-Q.
- R. J. Griffin, E. Evers, R. Davison, A. E. Gibson, D. Layton and W. J. Irwin, J. Chem. Soc., Perkin Trans. 1, 1996, 1205–1211, 10.1039/P19960001205.
- X. Qiu, J. Brückel, C. Zippel, M. Nieger, F. Biedermann and S. Bräse, RSC Adv., 2023, 13, 2483–2486, 10.1039/D2RA05997E.
- P. A. S. Smith, C. D. Rowe and L. B. Bruner, J. Org. Chem., 1969, 34, 3430–3433, DOI:10.1021/jo01263a047.
- D. H. Sieh, D. J. Wilbur and C. J. Michejda, J. Am. Chem. Soc., 1980, 102, 3883–3887, DOI:10.1021/ja00531a033.
- B. M. Trost and W. H. Pearson, J. Am. Chem. Soc., 1981, 103, 2483–2485, DOI:10.1021/ja00399a089.
- B. M. Trost and W. H. Pearson, J. Am. Chem. Soc., 1983, 105, 1054–1056, DOI:10.1021/ja00342a069.
- K. Nishiyama and N. Tanaka, J. Chem. Soc., Chem. Commun., 1983, 1322–1323, 10.1039/C39830001322.
- R. H. Smith, Jr. and C. J. Michejda, Synthesis, 1983, 476–477, DOI:10.1055/s-1983-30389.
- G. W. Kabalka and G. Li, Tetrahedron Lett., 1997, 38, 5777–5778, DOI:10.1016/S0040-4039(97)01295-1.
- A. Nakhai, B. Stensland, P. H. Svensson and J. Bergman, Eur. J. Org. Chem., 2010, 6588–6599, DOI:10.1002/ejoc.201000328.
- A. A. Suleymanov, R. Scopelliti, F. F. Tirani and K. Severin, Org. Lett., 2018, 20, 3323–3326, DOI:10.1021/acs.orglett.8b01214.
- S. Graßl, J. Singer and P. Knochel, Angew. Chem., Int. Ed., 2020, 59, 335–338, DOI:10.1002/anie.201911704.
- S. Xu, H. Guo, Y. Liu, W. Chang, J. Feng, X. He and Z. Zhang, Org. Lett., 2022, 24, 5546–5551, DOI:10.1021/acs.orglett.2c02053.
- S. Yaragorla and R. Muthyala, Tetrahedron Lett., 2010, 51, 467–470, DOI:10.1016/j.tetlet.2009.10.120.
- I. B. Seiple, S. Su, I. S. Young, A. Nakamura, J. Yamaguchi, L. Jørgensen, R. A. Rodriguez, D. P. O’Malley, T. Gaich, M. Köck and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 14710–14726, DOI:10.1021/ja2047232.
- J. R. Frost, C. M. Pearson, T. M. Snaddon, R. A. Booth and S. V. Ley, Angew. Chem., Int. Ed., 2012, 51, 9366–9371, DOI:10.1002/anie.201204868.
- H. Miyaoka, Y. Abe, N. Sekiya, H. Mitome and E. Kawashima, Chem. Commun., 2012, 48, 901–903, 10.1039/C1CC16468F.
- H. Miyaoka, Y. Abe and E. Kawashima, Chem. Pharm. Bull., 2012, 60, 1224–1226, DOI:10.1248/cpb.c12-00485.
- S. Hanessian, R. R. Vakiti, S. Dorich, S. Banerjee, F. Lecomte and B. Deschênes-Simard, J. Org. Chem., 2012, 77, 9458–9472, DOI:10.1021/jo301638z.
- For a recent review on the chemistry of azide-tetrazole tautomerism of azido heterocycles, see: D. Moderhack, Heterocycles, 2023, 106, 3–65, DOI:10.3987/REV-22-988.
- S. V. Chapyshev, A. V. Chernyak and I. K. Yakushchenko, J. Heterocycl. Chem., 2016, 53, 970–974 CrossRef CAS.
- For review, see: S. V. Chapyshev, Molecules, 2015, 20, 19142–19171, DOI:10.1002/jhet.2339.
- P. B. Koswatta, R. Sivappa, H. V. R. Dias and C. J. Lovely, Org. Lett., 2023, 25, 6234, DOI:10.1021/acs.orglett.3c02502.
- P. B. Koswatta, R. Sivappa, H. V. R. Dias and C. J. Lovely, Org. Lett., 2008, 10, 5055–5058, DOI:10.1021/ol802018r.
- T. Meguro, S. Yoshida, K. Igawa, K. Tomooka and T. Hosoya, Org. Lett., 2018, 20, 4126–4130, DOI:10.1021/acs.orglett.8b01692.
- T. Aimi, T. Meguro, A. Kobayashi, T. Hosoya and S. Yoshida, Chem. Commun., 2021, 57, 6062–6065, 10.1039/D1CC01143J.
- R. Namioka, M. Suzuki and S. Yoshida, Front. Chem., 2023, 11, 1237878, DOI:10.3389/fchem.2023.1237878.
- T. Patonay and R. V. Hoffman, J. Org. Chem., 1995, 60, 2368–2377, DOI:10.1021/jo00113a015.
- J. Patel, G. Clavé, P.-Y. Renard and X. Franck, Angew. Chem., Int. Ed., 2008, 47, 4224–4227, DOI:10.1002/anie.200800860.
- For a review, see: T. Patonay, K. Kónya and É. Juhasz-Tóth, Chem. Soc. Rev., 2011, 40, 2797–2847, 10.1039/C0CS00101E.
- T. Yokoi, H. Tanimoto, T. Ueda, T. Morimoto and K. Kakiuchi, J. Org. Chem., 2018, 83, 12103–12121, DOI:10.1021/acs.joc.8b02074.
- T. Yokoi, T. Ueda, H. Tanimoto, T. Morimoto and K. Kakiuchi, Chem. Commun., 2019, 55, 1891–1894, 10.1039/C8CC09415B.
- N. A. McGrath and R. T. Raines, Chem. Sci., 2012, 3, 3237–3240, 10.1039/C2SC20806G.
- B. Gold, M. R. Aronoff and R. T. Raines, J. Org. Chem., 2016, 81, 5998–6006, DOI:10.1021/acs.joc.6b00948.
- M. R. Aronoff, G. Gold and R. T. Raines, Org. Lett., 2016, 18, 1538–1541, DOI:10.1021/acs.orglett.6b00278.
- M. R. Aronoff, G. Gold and R. T. Raines, Tetrahedron Lett., 2016, 57, 2347–2350, DOI:10.1016/j.tetlet.2016.04.020.
- For a review, see: K. A. Mix, M. R. Aronoff and R. T. Raines, ACS Chem. Biol., 2016, 11, 3233–3244, DOI:10.1021/acschembio.6b00810.
- H. Tanimoto, R. Adachi, K. Tanisawa and T. Tomohiro, Org. Lett., 2024, 26, 2409–2413, DOI:10.1021/acs.orglett.4c00566.
- G. B. Feigelson, Tetrahedron Lett., 1998, 39, 1129–1130, DOI:10.1016/S0040-4039(97)10783-3.
- E. L. Myers and R. T. Raines, Angew. Chem., Int. Ed., 2009, 48, 2359–2363, DOI:10.1002/anie.200804689.
- H.-H. Chou and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 14936–14939, DOI:10.1021/ja407822b.
- W. Luo, F. Xu, Z. Wang, J. Pang, Z. Wang, Z. Sun, A.-Y. Peng, X. Cao and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202310118, DOI:10.1002/anie.202310118.
- A. Gagneuz, S. Winstein and W. G. Young, J. Am. Chem. Soc., 1960, 82, 5956–5957, DOI:10.1021/ja01507a045.
- For reviews, see: A. S. Carlson and J. J. Topczewski, Org. Biomol. Chem., 2019, 17, 4406–4429, 10.1039/C8OB03178A.
- A. A. Ott and J. J. Topczewski, ARKIVOC, 2020, 2019(vi), 1–17, DOI:10.24820/ark.5550190.p010.819.
- A. A. Ott, M. H. Packard, M. A. Ortuño, A. Johnson, V. P. Suding, C. J. Cramer and J. J. Topczewski, J. Org. Chem., 2018, 83, 8214–8224, DOI:10.1021/acs.joc.8b00961.
- K. Banert, in Organic Azides: Synthesis and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, West Sussex, 2010, pp. 116–166 Search PubMed.
- K. Banert, M. Hagedorn, C. Hemeltjen, A. Ihle, K. Weigand and H. Priebe, ARKIVOC, 2017, 338–361, DOI:10.24820/ARK.5550190.P009.846.
- For the recent discussion on the reaction mechanism, see: S. Bhattacharyya and K. Hatua, R. Soc. Open Sci., 2018, 5, 171075, DOI:10.1098/rsos.171075.
- J. R. Alexander, M. H. Packard, A. M. Hildebrandt, A. A. Ott and J. J. Topczewski, J. Org. Chem., 2020, 85, 3174–3181, DOI:10.1021/acs.joc.9b03061.
- M. Villarreal-Parra, G. E. Di Gresia, G. R. Labadie and M. M. Vallejos, J. Org. Chem., 2023, 88, 9750–9759, DOI:10.1021/acs.joc.3c00371.
- R. Liu, O. Gutierrez, D. J. Tantillo and J. Aubé, J. Am. Chem. Soc., 2012, 134, 6528–6531, DOI:10.1021/ja300369c.
- Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2004, 6, 965–968, DOI:10.1021/ol0301431.
- R. Liu, J. Wang, H. Wu, X. Quan, S. Wang, J. Guo, Y. Wang and H. Li, Chem. Commun., 2024, 60, 4362–4365, 10.1039/D4CC00907J.
- B. Ardiansah, H. Tanimoto, T. Tomohiro, T. Morimoto and K. Kakiuchi, Chem. Commun., 2021, 57, 8738–8741, 10.1039/D1CC02770K.
- S. Grecian and J. Aubé, inOrganic Azides: Synthesis and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, West Sussex, 2010, pp. 191–237 Search PubMed.
- R. H. Vekariya, R. Liu and J. Aubé, Org. Lett., 2014, 16, 1844–1847, DOI:10.1021/ol500011f.
- L. Moynihan, R. Chadda, P. McArdle and P. V. Murphy, Org. Lett., 2015, 17, 6226–6229, DOI:10.1021/acs.orglett.5b03209.
- A. K. Feldman, B. Colasson, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 13444–13445, DOI:10.1021/ja050622q.
- D. Craig, J. W. Harvey, A. G. O’Brien and A. J. P. White, Org. Biomol. Chem., 2011, 9, 7057–7061, 10.1039/C1OB05972F.
- A. A. Tjeng, K. L. Handore and R. A. Batey, Org. Lett., 2020, 22, 3050–3055, DOI:10.1021/acs.orglett.0c00801.
- K. A. Nicastri, N. C. Gerstner and J. M. Schomaker, Org. Lett., 2023, 6, 8279–8283, DOI:10.1021/acs.orglett.3c03286.
- T. Abegg, T. Abegg, J. Cossy and C. Meyer, Org. Lett., 2022, 24, 4954–4959, DOI:10.1021/acs.orglett.2c01888.
- M. M. Vallejos and G. R. Labadie, RSC Adv., 2020, 10, 4404–4413, 10.1039/C9RA10093H.
- For representative papers, see B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457–460, DOI:10.1126/science.1229506.
- C. P. Seath, G. A. Burley and A. J. B. Watson, Angew. Chem., Int. Ed., 2017, 56, 3314–3318, DOI:10.1002/anie.201612288.
- For a review and a perspective paper on asymmetric CuAAC reactions, see: C.-Q. Qin, C. Zhao, G.-S. Chen and Y.-L. Liu, ACS Catal., 2023, 13, 6301–6311, DOI:10.1021/acscatal.3c00911.
- W. D. G. Brittain, B. R. Buckley and J. S. Fossey, ACS Catal., 2016, 6, 3629–3636, DOI:10.1021/acscatal.6b00996.
- J.-c Meng, V. V. Fokin and M. G. Finn, Tetrahedron Lett., 2005, 46, 4543–4546, DOI:10.1016/j.tetlet.2005.05.019.
- C. Wang, R.-Y. Zhu, K. Liao, F. Zhou and J. Zhou, Org. Lett., 2020, 22, 1270–1274, DOI:10.1021/acs.orglett.9b04522.
- J. R. Alexander, A. A. Ott, E.-C. Liu and J. J. Topczewski, Org. Lett., 2019, 21, 4355–4358, DOI:10.1021/acs.orglett.9b01556.
- Y. Gong, C. Wang, F. Zhou, K. Liao, X.-Y. Wang, Y. Sun, Y.-X. Zhang, Z. Tu, X. Wang and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202301470, DOI:10.1002/anie.202301470.
- W. D. G. Brittain, A. G. Dalling, Z. Sun, C. S. Le Duff, L. Male, B. R. Buckley and J. S. Fossey, Sci. Rep., 2019, 9, 15086, DOI:10.1038/s41598-019-50940-4.
- E.-C. Liu and J. J. Topczewski, J. Am. Chem. Soc., 2019, 141, 5135–5138, DOI:10.1021/jacs.9b01091.
- E.-C. Liu and J. J. Topczewski, J. Am. Chem. Soc., 2021, 143, 5308–5313, DOI:10.1021/jacs.1c01354.
- A. A. Ott, C. S. Goshey and J. J. Topczewski, J. Am. Chem. Soc., 2017, 139, 7737–7740, DOI:10.1021/jacs.7b04203.
- S. Yoshida, Y. Sakata, Y. Misawa, T. Morita, T. Kuribara, H. Ito, Y. Koike, I. Kii and T. Hosoya, Chem. Commun., 2021, 57, 899–902, 10.1039/D0CC07789E.
- S. J. Siegl, R. Dzijak, A. Vázquez, R. Pohl and M. Vrabel, Chem. Sci., 2017, 8, 3593–3598, 10.1039/C6SC05442K.
- D. N. Kamber, Y. Liang, R. J. Blizzard, F. Liu, R. A. Mehl, K. N. Houk and J. A. Prescher, J. Am. Chem. Soc., 2015, 137, 8388–8391, DOI:10.1021/jacs.5b05100.
- R. D. Row and J. A. Prescher, Acc. Chem. Res., 2018, 51, 1073–1081, DOI:10.1021/acs.accounts.7b00606.
- P. Bollini, M. Diwan, P. Gautam, R. L. Hartman, D. A. Hickman, M. Johnson, M. Kawase, M. Neurock, G. S. Patience, A. Stottlemyer, D. G. Vlachos and B. Wilhite, ACS Eng. Au, 2023, 3, 364–390, DOI:10.1021/acsengineeringau.3c00023.
- D. Ichinari, Y. Ashikari, K. Mandai, Y. Aizawa, J. Yoshida and A. Nagaki, Angew. Chem., Int. Ed., 2020, 59, 1567–1571, DOI:10.1002/anie.201912419.
- M. Kleoff, J. Schwan, L. Boeser, B. Hartmayer, M. Christmann, B. Sarkar and P. Heretsch, Org. Lett., 2020, 22, 902–907, DOI:10.1021/acs.orglett.9b04450.
- T. Jiang, G. Coin, S. Bordi, P. L. Nichols, J. W. Bode and B. M. Wanner, J. Org. Chem., 2024, 89, 7962–7969, DOI:10.1021/acs.joc.4c00603.
| This journal is © The Royal Society of Chemistry 2024 |