
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePVA–FeCl3 composites as substrate and packaging materials for the controlled degradation of non-degradable metals in transient electronics†
Neeru
Mittal‡
 a,
Tae-Min
Jang‡
b,
Suk-Won
Hwang
*bcd and
Markus
Niederberger
*a
a,
Tae-Min
Jang‡
b,
Suk-Won
Hwang
*bcd and
Markus
Niederberger
*a
aLaboratory for Multifunctional Materials, Department of Materials, ETH Zürich, Vladimir-Prelog-Weg 5, 8093 Zurich, Switzerland. E-mail: markus.niederberger@mat.ethz.ch
bKU-KIST Graduate School of Converging Science & Technology, Korea University, Seoul, 02841, South Korea. E-mail: dupong76@korea.ac.kr
cDepartment of Integrative Energy Engineering, Korea University, Seoul, 02841, South Korea
dCenter for Biomaterials, Biomedical Research Institute, Korea Institute of Science and Technology (KIST), Seoul, 02792, South Korea
First published on 11th April 2023
Abstract
Progress in transient electronics depends largely on the availability of components and materials that can decompose in aqueous solutions. However, some of the most important electrically conductive materials, such as copper or aluminum, do not fall into this category. Here, we report a concept for solving this problem based on the preparation of a new water-soluble polymer composite as a packaging material that, when dissolved, releases a chemical etchant that decomposes these two metals. We investigate the synthesis, chemical properties, and solubility kinetics of a polyvinyl alcohol–iron chloride (PVA–FeCl3) composite, its degradation properties, and the associated dissolution mechanisms of metallic Al and Cu films and traces. The results show that Cu films dissolve in a rapid and uniform fashion and produce copper(I) chloride as the end product, while Al films exhibit inconsistent dissolution behavior. Moreover, the timescale for complete dissolution of Cu and Al can be adjusted by simply varying the amount of FeCl3 in the composite. The distinct advantages of this triggered transience mode include low cost, simplicity, precise control of the dissolution process by varying the polymer composition, and a universal degradation mechanism that can be extended to numerous transient electronic devices.
10th Anniversary StatementAs a doctoral student (M. N.), the predecessor journal, Journal of Materials Chemistry, was an important companion to my research, and my first article in that journal not only made me incredibly proud, but gave me the feeling of belonging to the scientific community. This did not change after the Journal of Materials Chemistry was split into three sister journals 10 years ago: the Journal of Materials Chemistry A remains one of the leading journals in the field of materials chemistry, and even now as a laboratory head I am delighted when a paper of ours is accepted for publication there. Although I sometimes miss the days when you could go to the library every week to read the thin issues of the few relevant scientific journals, I am pleased that the Journal of Materials Chemistry A has maintained its commitment to the highest quality even as the number of articles has increased significantly. |
1. Introduction
We, as a human society, are addicted to electronic devices, which are continuously being equipped with new features to expand their potential applications. The most recent addition is the “transience” nature, wherein devices are built with an expiry date.1 These devices operate for a well-defined time and then harmlessly disintegrate into their surroundings in a controlled fashion when exposed to external stimuli.2 Transient electronics have broad potential applications, including biomedical devices, environmental monitoring, military, and homeland security, to name a few.3The design and development of physically transient materials and components are crucial for further progressing the field of transient electronics and their applications. So far, many types of transient materials have been explored, including biodegradable and bioresorbable metals,4 metal oxides and nitrides,5 stimuli-responsive polymers,6 and nanomaterials.7 For most of these materials, transiency has been tailored for aqueous conditions involving chemical dissolution or physical disintegration due to the dissolution of the underlying substrate. For such reaction environments, the choice of materials for the manufacture of transient electronics is highly limited. Many materials such as copper (Cu), nickel (Ni), gold (Au) and aluminum (Al), which are indispensable in conventional electronics due to their excellent electrical conductivity, are excluded because of their insolubility in water. Clearly, a gap exists here, and hence a strategy that can assist in degrading these non-transient metallic components can significantly widen the scope of transient electronics, especially for transient batteries.3,8 Ensuring complete degradation of these non-transient metals can greatly benefit the development of novel transient batteries (both primary and secondary) and taking a step further, can also drive the ultimate vision of combining transiency with recycling to achieve circular economy.9–11
There are not many, but some interesting concepts on how to make non-transient metals suitable for transient electronics. Gao et al. developed moisture-triggered transient electronics wherein the hydrolysis process of the substrate generates corrosive organic acids that can digest various inorganic electronic materials and components.12 Chen et al. investigated the dissolution chemistry and kinetics of Au in potassium ferrocyanide solution using light as the trigger source.13 Rogers et al. developed a microfluidic system using thermally expandable polymers and utilized etching chemistry to enable on-demand and complete degradation of the microfluidic system.14 Yu et al. adopted the same concept of using chemical etchants for the on-demand degradation of transient electronics, but instead of heat, they applied electrical current as the trigger.15 All these approaches designed to degrade non-transient electronics are novel. But still, they either involve complicated fabrication processes, require specialized materials, are device-specific, or do not allow precise control over the transience behavior of the device.
Here, we report a new design concept for substrates or packaging materials of transient electronics to generate on-demand an active chain reaction by water using ferric chloride (FeCl3) incorporated in water-soluble polyvinyl alcohol (PVA). Upon triggering with water, the released ferric (Fe3+) and chloride (Cl−) ions readily react with non-soluble metals (e.g., Cu and Al) to cause complete dissolution in a given time. Compared to other encapsulation strategies, this approach has a few distinct advantages: (a) stable operation of devices until triggered by water; (b) the ability to dissolve a wide range of non-transient metals such as Cu, Al, Ni, Mn, Sn, etc., thus expanding the list of materials suitable for constructing transient electronics; (c) tunability of the degradation behavior by varying the composition of the polymer composite; and (d) facile fabrication procedure and low cost. Systematic investigations provide details of dissolution behaviors with underlying chemistries through the interaction between thin conductive metals and PVA–FeCl3 composites. Circuit demonstration offers the potential for complex operation via time-dynamic dissolution in sophisticated electronic systems.
2. Results and discussion
2.1 Physico-chemical properties of PVA composite films
Polyvinyl alcohol (PVA) is a non-toxic, biocompatible, and water-soluble synthetic polymer with excellent physical and chemical properties.16 Its superb film-forming ability, programmable water-solubility, and good dielectric properties make it a suitable substrate and packaging material for transient devices. PVA is commercially produced by the controlled alkaline hydrolysis of polyvinyl acetate, also called saponification reaction, where the ester group of polyvinyl acetate is partially replaced with the hydroxyl group.17 The length of the reaction time used for saponification determines the degree of hydrolysis of PVA, which is defined below:where x and y are the molar fractions of the hydroxyl and the acetate groups, respectively.18 The chemical structures of fully hydrolyzed and partially hydrolyzed PVA with repeating units are shown in Fig. S1.† The physico-chemical and mechanical properties of a polymer depend significantly on the degree of crystallinity, hydrolysis level, and molecular weight. For PVA, it is well documented that there is a strong inter- and intra-chain hydrogen bonding between the polar hydroxyl groups that determine its crystallinity, and thus its solubility in an aqueous solution.18 Clearly, one can tune the dissolution behavior of PVA in water by modifying the extent of hydrogen bonding between PVA chains. This can simply be done by exploring PVA with different degrees of hydrolysis and molecular weights and accordingly, the transience behavior of PVA films with molecular weights in the range of 9000–124
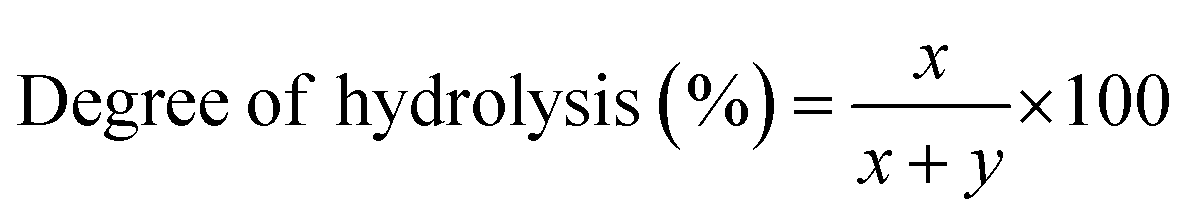
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 and degrees of hydrolysis from 80% to >99% was firstly investigated.
000 g mol−1 and degrees of hydrolysis from 80% to >99% was firstly investigated.
As shown in Fig. S2,† the PVA film with 80% degree of hydrolysis dissolved more readily in water than those with degrees of hydrolysis above or close to 98%, because the increasing number of acetate groups in 80% hydrolyzed PVA disrupts its intra/inter-chain hydrogen bonding and facilitates its rapid solubility. In contrast, a very high degree of hydrolysis makes PVA more crystalline and supports stronger inter- and intra-chain hydrogen bonding, which then significantly decreases the water solubility, as indicated by the transience times of more than 60 and 600 min for 98–99% and >99% hydrolyzed PVA films, respectively. Throughout this paper, transiency is defined as the moment when the film has lost its structural integrity and degraded to an unrecognizable level. Besides the degree of hydrolysis, higher molecular weights also decrease the water-solubility of PVA films due to increased crystallinity. It is important to note that once the PVA film is submerged in water, its solubility is influenced not only by inter/intra-chain hydrogen bonding but also by the newly formed hydrogen bonds between the water molecules and PVA chains. To achieve complete dissolution of the PVA films, the hydrogen bonding between PVA chains and water molecules must dominate over the PVA–PVA chains interactions.19 Therefore, it is essential to include both kinds of hydrogen bonding when interpreting the transiency of different PVA films in water.
After obtaining fully soluble PVA films, the next step included inspecting and finding a suitable chemical trigger that can be used to degrade non-soluble metals, especially Cu and Al. As reported thus far in transient electronics, especially for transient batteries, Cu metal film has been shown to undergo disintegration into smaller chunks due to the stresses exerted on the film from the swelling of the underlying water-soluble substrates such as sodium alginate.20 On the other hand, thin Al film exhibited complete dissolution in a particular pH-controlled environment generated by the reaction between different constituent materials of the transient device.8 However, both these approaches are device- and materials-specific, and extending them to other transient devices using entirely different raw materials is difficult. To solve this problem and to make use of these essential yet non-transient metals, we applied the basic principles of redox chemistry and carefully incorporated a strong oxidizing agent in PVA films that can readily oxidize Cu and Al metals. However, the selection of an appropriate oxidizing agent requires the following: (a) it should be water-soluble and safe to handle; (b) it must be capable of forming stable composites with PVA; and (c) it must allow tunable degradation kinetics for both metals.
Ferric chloride (FeCl3) is one of the most commonly used oxidizing agents and has been used to etch metals such as Cu, Al, Ni, and Sn in industrial photochemical machining.21 It is water-soluble and forms stable composites with PVA for applications in electrochromic, display materials, sensors, etc.22 In addition, FeCl3 is widely used in wastewater treatment to remove phosphorus to prevent eutrophication,23 thus suitable for eco-friendly, disposable, and water-soluble electronics.
PVA–FeCl3 composite films were synthesized by mixing an appropriate amount of FeCl3 as an additive to the aqueous polymer solution, as illustrated in Scheme S1.† The inorganic additive was found to significantly impact the dissolution behavior of PVA-based films (see Fig. 1). Based on the previous experiments, it is clear that PVA with >99% degree of hydrolysis and higher molecular weight failed to form water-soluble films; therefore, only PVA with lower molecular weights and degrees of hydrolysis were considered for these sets of experiments. Preliminary experiments using 98–99% hydrolyzed PVA with 10 and 15 wt% FeCl3 indicated the non-soluble nature of the composite films. Both films kept their original shape with noticeable swelling and failed to dissolve in distilled water even after 4 days, as shown in Fig. S3.† This might happen due to the increase in the degree of crystallinity of PVA films with the addition of a transition metal salt as additive. On the contrary, composite films prepared from PVA with 80% degree of hydrolysis and a much lower molecular weight fully dissolved in water within a few minutes. Surprisingly, their dissolution times were faster than that of pristine PVA films, as shown in Fig. 1.
 | ||
| Fig. 1 Digital photographs showing the time-sequential dissolution in distilled water of (a) PVA and (b) PVA–15% FeCl3 films with Mw of 9000–10000 g mol−1 and 80% degree of hydrolysis. | ||
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) experiments were performed to understand the chemical modifications induced by the added FeCl3 within the PVA polymer matrix. The ATR-FTIR spectra of pure and FeCl3-containing PVA films with varying additive concentrations are shown in Fig. 2a and b. The broad bands observed from 3500–3200 cm−1 correspond to O–H stretching from the inter/intra-chain hydrogen bonds. The vibration band between 3000–2800 cm−1 refers to C–H stretching from alkyl groups, and peaks between 1750–1690 cm−1 are due to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds from acetate groups that are left from the partial hydrolysis of polyvinyl acetate.24 The bands at 1430 cm−1, 1370 cm−1, and 1088 cm−1 correspond to O–H and C–H bending, C–H in-plane bending, and acetyl C–O stretching, respectively. For PVA–FeCl3 composite films, the characteristic bands mainly occur in lower wavenumbers from 650–1850 cm−1 and show considerable frequency shifts (Fig. 2b). The intensity of the CO stretching vibration at 1727 cm−1 decreases with increasing additive concentration, followed by the occurrence of a new peak at around 1710 cm−1. The frequencies of CC stretching at 1638 cm−1, acetyl C–O at 1088 cm−1, and skeletal vibration of PVA at 844 cm−1 shifts toward lower wavenumbers while the stretching frequency of acetate C–O–C at 1240 cm−1 has moved towards higher wavenumber.25
O and C–O bonds from acetate groups that are left from the partial hydrolysis of polyvinyl acetate.24 The bands at 1430 cm−1, 1370 cm−1, and 1088 cm−1 correspond to O–H and C–H bending, C–H in-plane bending, and acetyl C–O stretching, respectively. For PVA–FeCl3 composite films, the characteristic bands mainly occur in lower wavenumbers from 650–1850 cm−1 and show considerable frequency shifts (Fig. 2b). The intensity of the CO stretching vibration at 1727 cm−1 decreases with increasing additive concentration, followed by the occurrence of a new peak at around 1710 cm−1. The frequencies of CC stretching at 1638 cm−1, acetyl C–O at 1088 cm−1, and skeletal vibration of PVA at 844 cm−1 shifts toward lower wavenumbers while the stretching frequency of acetate C–O–C at 1240 cm−1 has moved towards higher wavenumber.25
 | ||
| Fig. 2 ATR-FTIR spectra of (a) PVA–FeCl3 films with additive concentrations of 0, 1, 15, and 30 wt% and (b) enlarged spectra in the wavenumber region from 650 to 1850 cm−1. (c) XRD patterns and (d) transience behavior of PVA–FeCl3 films with different additive concentrations. | ||
All the observed changes in the ATR-FTIR spectra of PVA-based composite films are understood based on the chemical interactions between PVA and the additive, FeCl3. Many studies have shown that the interactions between polymer and additive in composite films occur via an acceptor–donor mechanism.26 FeCl3 has a high spin d5 electron configuration, and this half-filled d-subshell is expected to have a stabilizing effect. Therefore, when this potent Lewis acid, FeCl3, is added to a polymer, it is expected to alter the properties of the polymer through charge-transfer mechanisms such as interaction with the electron donating –OH/CO groups of PVA to form stable complexes.26 These interactions are reflected in the fluctuated intensities and wavenumbers shift of different peaks in the obtained ATR-FTIR spectra. For example, the decreasing intensity of the CO band at 1727 cm−1 points toward the attachment of Fe3+ to the CO group, facilitating complex formation. In other words, FeCl3 strongly interacts with the carbonyl group of PVA (no change in –OH peak is observed), whose total amount in the PVA matrix depends on its degree of hydrolysis. PVA used here has a degree of hydrolysis of 80% and hence contains a lot of acetate groups (–OCOCH3) that destroy inter/intra-chain hydrogen bonding, lower crystallinity, and facilitate high water solubility. Further interaction of these carbonyl groups with Fe3+ leads to an even more reduced crystallinity and destruction of hydrogen bonds, resulting in an accelerated dissolution of PVA composite films in an aqueous solution. This interaction is confirmed by the affected frequencies of the acetyl CO group at 1638 cm−1, the acetyl C–O group at 1088 cm−1, and the acetate C–O–C group at 1240 cm−1. The possible chemical interactions between various chemical groups of PVA and FeCl3 is presented in Scheme S2.†
The decrease in the degree of crystallinity of the composite films with increasing additive concentration is also observed in the X-ray diffraction patterns, shown in Fig. 2c. The XRD pattern of pristine PVA film shows a broad diffraction peak centered at 2θ = 19.4°, corresponding to the (101) plane of PVA.27 The presence of a broad halo and the absence of any sharp peaks indicate the semicrystalline nature of PVA. With the increased concentration of FeCl3, the diffraction peak at ∼19.4° becomes broader and shows a significant decrease in intensity. The disappearance of the (101) peak in PVA–FeCl3 composite films indicates that the distribution of FeCl3 as filler in the polymer matrix is random, distorting the hydrogen bonding within the polymer chains by forming charge-transfer complexes.22 Thus, both ATR-FTIR and XRD results imply that PVA–FeCl3 composite films have even weaker inter/intra-chain hydrogen bonds than pristine PVA films and are expected to exhibit faster water-transience behavior. To confirm this, the transiency of composite films was examined in distilled water, and as expected, the composite films displayed a rapid dissolution in aqueous environment. As shown in Fig. 2d, the polymer film containing the highest amount of FeCl3 dissolved in distilled water within 150 s compared to the pure PVA films that took more than double the time (350 s) to dissolve.
These experiments clearly show that our substrates support the degradation of non-transient metals efficiently. However, they do not provide information on the long-term stability, i.e., whether there are adverse effects on the conductivity of the metals during storage under ambient conditions, e.g., due to accidental release of FeCl3. These issues, which are important for the reliable operation of transient devices until their programmed decomposition, need to be investigated in future studies.
2.2 Water-triggered degradation of non-transient metals
The transience behavior and possible degradation mechanism of thin Cu film on PVA–FeCl3 substrates are presented first. Fig. 3a and b shows the time-sequential images of Cu dissolution using 100 μm thick PVA–15% FeCl3 as packaging material (forming top layer of the electronic) in the presence of water as a trigger. At a macroscopic level, the surface of Cu film looks rough after one minute of dissolution, followed by the development of a few micropores and then complete dissolution within two minutes. The dissolution process appears uniform, similar to that reported for metals like Mg, Mg alloys, W, and Mo.28 Ferric ions are one of the most reactive oxidants and looking at the standard electrode potentials of Fe3+/Fe2+ (0.771 V), O2/H2O (1.229 V), and Cu2+/Cu (0.34 V), it is easy to conclude that thermodynamically, metallic copper can be oxidized to Cu2+ in the presence of Fe3+.29 Also, molecular oxygen can accelerate the dissolution process as it acts as an oxidizing agent in the aqueous solution.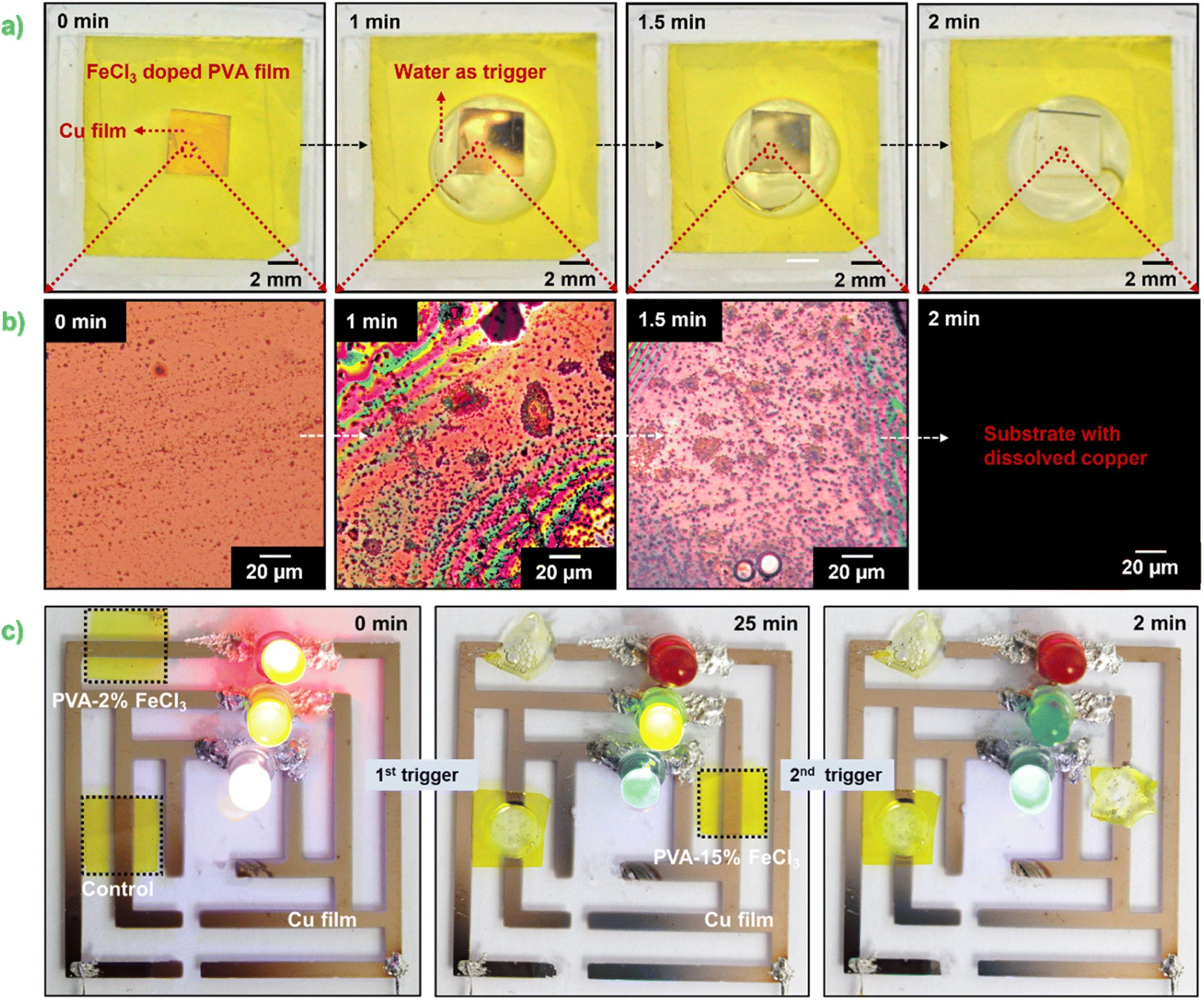 | ||
| Fig. 3 Water-triggered dissolution behavior of a 100 nm thick Cu film encapsulated with PVA–15% FeCl3 film. (a) Digital photographs and (b) optical microscopy images showing the structural changes occurring in the Cu film during dissolution. (c) Digital photographs showing the multistep degradation of Cu traces attached to LEDs by triggering PVA–FeCl3 substrates with different additive concentrations with distilled water. The composite film marked as “control” is PVA–15% FeCl3. | ||
The chemical reactions resulting in the dissolution of metallic Cu films in the presence of an acidic FeCl3 solution can be described as follows:29
| FeCl3 + 3H2O = Fe(OH)3 + 3H+ + 3Cl− |
| 2Fe3+ + Cu = Cu2+ + 2Fe2+ |
| O2 + 4H+ + 2Cu = 2Cu2+ + 2H2O |
The acidic nature of the FeCl3 solution resulting from the dissolution of PVA-based composite films is confirmed by the pH measurements shown in Fig. S4a.† As the dissolution progresses, the concentration of Cu2+ in the solution increases, and its subsequent reaction with metallic Cu also starts occurring, as shown below:
| Cu2+ + Cu = 2Cu+ |
Cu+ is gradually consumed in other reactions, such as oxidation–reduction reactions with Fe3+ and H+ to form Cu2+ and complex ions like CuCln(n−1) with the chloride anions.30 These complex ions get further oxidized, releasing Cl− back into the reaction mixture. The chloride anions then repeat the whole process of complexation and oxidation, and thus appear to have an accelerating effect on the dissolution process. The presence of these complex ions is confirmed by grazing incidence angle XRD studies conducted on the Cu film before and after the dissolution process. The grazing incidence angle XRD patterns in Fig. S4b† show the characteristic peak at 2θ = 47.5°, corresponding to the (220) crystal plane of copper chloride (CuCl).31 Hence, FeCl3 functions as both an oxidant and a complexing agent during the dissolution process of the metallic Cu film.
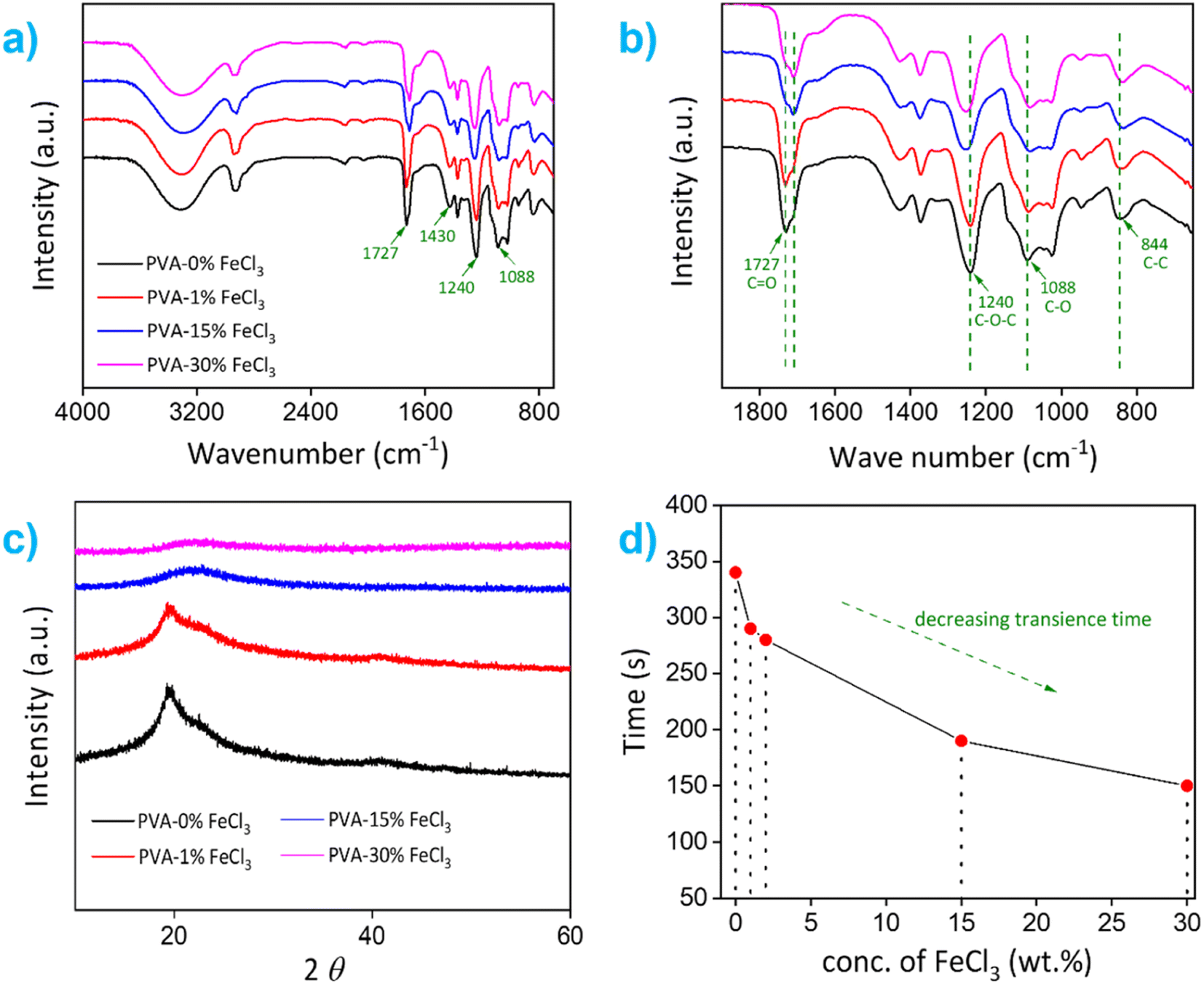
Next, the reaction kinetics of the Cu dissolution process were studied using composite films integrated with an in-house-built electronic device that contained three different colored light-emitting diodes (LEDs) connected in parallel with thin Cu traces (see Fig. S5†). In the electronic circuit, a small area of the Cu trace connected to red and blue LEDs was covered/encapsulated with polymer films containing different amounts of FeCl3, 2 and 15 wt%, respectively. These films were triggered with water in a stepwise fashion to release Fe3+ and Cl−, which initiated the dissolution process of Cu traces. As shown in Fig. 3c, the Cu trace covered with PVA–2% FeCl3 film completely dissolved within ∼25 min after the first trigger, causing the red LED to switch off. The Cu trace covered with a composite film marked as "Control" also completely dissolved, however it did not electrically affect the operation of the other LEDs. After that, the second trigger dissolved the local Cu pattern overlaid with PVA–15% FeCl3 film in 2 minutes, which shut off the green LED, while the blue LED continued to operate due to the constant power supply through the bypass circuit. This controlled transiency of metallic Cu traces using PVA–FeCl3 films is achieved owing to the difference in the number of available reactive species (i.e., Fe3+ and Cl−) and variable pH values, both processes known to accelerate the Cu dissolution process. The PVA films with lower ferric chloride concentration released a smaller number of reactive ions and provided a comparatively high pH value (∼2.3, see Fig. S4a†), resulting in a slow Cu dissolution process while completely opposite happened in case of PVA–15% FeCl3 film. This carefully designed experiment demonstrates the programmable nature of our PVA-based composite films. It also confirms that the transience time of non-transient metals (e.g., Cu) is tunable by simply adjusting the additive concentration in the composite film.
The dissolution behavior of thin metallic Al films in the presence of PVA–FeCl3 as the encapsulating layer was also studied. Fig. 4 shows the time-sequential dissolution images of a thin layer of Al (80 nm) covered with 100 μm thick PVA–15% FeCl3 film when immersed in water. In sharp contrast to what is observed for Cu, the complete dissolution of the Al film (6 mm × 6 mm) occurred in a very slow mode over more than 2 hours. Furthermore, the optical microscopy images reveal a non-uniform dissolution process showing the occurrence of a lot of pits/cracks for Al in contrast to the uniform dissolution observed for Cu. According to the literature, chloride ion is one of the most common species that causes pitting corrosion in Al in different environments, such as seawater and the chemical industry.32 Although Al is highly resistant to various reactive agents due to a highly stable oxide layer, Cl− can easily break down this passivation layer and initiate the dissolution process.
 | ||
| Fig. 4 Water-triggered dissolution behavior of 80 nm thick Al film encapsulated with PVA–15% FeCl3 film. (a) Digital photographs and (b) optical microscopy images showing the structural changes occurring in the Al film during the course of dissolution. | ||
The Al dissolution process consists of four main steps described as follows. Initially, the outermost oxide layer present on the Al surface is covered with hydroxyl groups. These –OH groups will remain undissociated if the pH of the aqueous solution is the same as the isoelectric point of the oxide, which is reported to be 9.5.33 However, in the acidic environment produced by the dissolution of the PVA–15% FeCl3 packaging, the hydroxyl groups will get a positive charge that favors the adsorption of negatively charged Cl−via coulombic attraction. After adsorption, these Cl− penetrate through the oxide layer following several possible mechanisms, such as transport through oxygen vacancies,34 water channels,35 or localized film dissolution.36 When Cl− crosses the oxide layer and reaches the metal substrate, pitting corrosion gets initiated at the metal/oxide interface by chloride-assisted localized dissolution in the presence of water molecules as the electrolyte.37 The local dissolution of oxide film on the Al surface is called pitting. Once pitting has been initiated, it propagates on the Al surface and usually involves the formation of blisters produced by continuous localized reactions.38 These blisters subsequently rupture due to the accumulation of hydrogen gas in the localized environment. The formation and rupturing of blisters were indeed observed during the water-triggered dissolution of Al film using FeCl3 present in the composite film. The optical microscopy images showing one of those few ruptured blisters are shown in Fig. S6.† During this dissolution process, the reported degradation products are soluble aluminum complexes with chloride ions.39
Besides encapsulant, PVA–FeCl3 composite films were also utilized as substrates for transient electronics. In transient electronics, it is pretty common to deposit conductive materials, both organic and inorganic, directly on either water-soluble or biodegradable polymers to develop transient current collectors.40 In fact, novel techniques like 3D printing have been also employed to develop transient electronics containing water soluble substrates and carbon-based conductive materials. These devices showed well controlled and preprogrammed transience behavior owing to the tunability of the structural properties of 3D-printed substrate.40 So, we also investigated the feasibility of directly depositing Cu and Al metals on the composite films. Fig. S7† shows the free-standing PVA–15% FeCl3 film coated with 100 nm thick Cu film in different patterns. These Cu-coated polymer composite films completely dissolved within a few minutes after triggering with water. As already described, their transiency rate can be controlled by adjusting the additive concentration or thickness of the polymer substrate. Besides transiency, it is also vital for transient substrates to present stable electrical characteristics after metal deposition, as poor electrical conductivity may result in the loss of functionality of the whole transient device. To confirm this, the electrical resistance of metallic Al film deposited on top of the PVA–15% FeCl3 substrate was measured using a multimeter and presented in Fig. S8.† The Al film showed a low resistance value of 2.7 Ω and is considered suitable to work as a substrate for numerous electronic devices. The results presented here on water-triggered transience of various conductive metals with no transient properties, such as Al and Cu, lay the solid foundation for fabricating fully soluble transient electronics.
3. Conclusion
We developed a novel transient material that can initiate the degradation of non-dissolvable metals like Al and Cu after triggering with water. The transient PVA–FeCl3 composite decomposed these metals by chemical reactions, whose rate can be controlled by simply varying the polymer composition. The transience time scale for both metals can be tuned over a wide range from minutes to hours and if needed, even to days. The design concept presented offers innovative options for integrating electrically conductive materials into transient electronics, thus not only expanding their application potential but also opening up space for novel developments such as transient batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge ETH Zurich for financial support (ETH Research Grant ETH-45-18-1) and the Young Researcher Exchange Programme-National Research Foundation of Korea for sponsoring the 3 month research exchange in South Korea. This work was supported by Korea University grant, KU-KIST Graduate School of Converging Science and Technology Program, National Research Foundation of Korea (NRF) grant funded by the Korea government (the Ministry of Science, ICT, MSIT) (RS-2022-00165524), and the Ministry of the Science and ICT (MSIT), under the ICT Creative Consilience program (IITP-2023-2020-0-01819) supervised by the IITP (Institute for Information & communications Technology Planning & evaluation). The author would like to thank Dr WonBae Han for many helpful discussions.References
- S. W. Hwang, H. Tao, D. H. Kim, H. Cheng, J. K. Song, E. Rill, M. A. Brenckle, B. Panilaitis, S. M. Won, Y. S. Kim, Y. M. Song, K. J. Yu, A. A. Ameen, R. Li, Y. Su, M. Yang, D. L. Kaplan, M. R. Zakin, M. J. Slepian, Y. Huang, F. G. Omenetto and J. A. Rogers, Science, 2012, 337, 1640 CrossRef CAS PubMed.
- K. K. Fu, Z. Wang, J. Dai, M. Carter and L. Hu, Chem. Mater., 2016, 28, 3527–3539 CrossRef CAS.
- L. A. Wehner, N. Mittal, T. Liu and M. Niederberger, ACS Cent. Sci., 2021, 7, 231–244 CrossRef CAS PubMed.
- L. Yin, H. Cheng, S. Mao, R. Haasch, Y. Liu, X. Xie, S.-W. Hwang, H. Jain, S.-K. Kang, Y. Su, R. Li, Y. Huang and J. A. Rogers, Adv. Funct. Mater., 2014, 24, 645–658 CrossRef CAS.
- S. K. Kang, S. W. Hwang, H. Cheng, S. Yu, B. H. Kim, J. H. Kim, Y. Huang and J. A. Rogers, Adv. Funct. Mater., 2014, 24, 4427–4434 CrossRef CAS.
- H. L. Hernandez, S.-K. Kang, O. P. Lee, S.-W. Hwang, J. A. Kaitz, B. Inci, C. W. Park, S. Chung, N. R. Sottos, J. S. Moore, J. A. Rogers and S. R. White, Adv. Mater., 2014, 26, 7637–7642 CrossRef CAS PubMed.
- Y. K. Lee, K. J. Yu, E. Song, A. Barati Farimani, F. Vitale, Z. Xie, Y. Yoon, Y. Kim, A. Richardson, H. Luan, Y. Wu, X. Xie, T. H. Lucas, K. Crawford, Y. Mei, X. Feng, Y. Huang, B. Litt, N. R. Aluru, L. Yin and J. A. Rogers, ACS Nano, 2017, 11, 12562–12572 CrossRef CAS PubMed.
- N. Mittal, A. Ojanguren, N. Cavasin, E. Lizundia and M. Niederberger, Adv. Funct. Mater., 2021, 31, 2101827 CrossRef CAS.
- N. Mittal and M. Niederberger, Chimia, 2022, 76, 298–302 CrossRef CAS.
- N. Mittal, A. Ojanguren, M. Niederberger and E. Lizundia, Adv. Sci., 2021, 8, 2004814 CrossRef CAS PubMed.
- L. Teng, S. Ye, S. Handschuh-Wang, X. Zhou, T. Gan and X. Zhou, Adv. Funct. Mater., 2019, 29, 1808739 CrossRef.
- Y. Gao, Y. Zhang, X. Wang, K. Sim, J. Liu, J. Chen, X. Feng, H. Xu and C. Yu, Sci. Adv., 2017, 3, 1–8 Search PubMed.
- W. D. Chen, S. K. Kang, W. J. Stark, J. A. Rogers and R. N. Grass, Sens. Actuators, B, 2019, 282, 52–59 CrossRef CAS.
- C. H. Lee, J. W. Jeong, Y. Liu, Y. Zhang, Y. Shi, S. K. Kang, J. Kim, J. S. Kim, N. Y. Lee, B. H. Kim, K. I. Jang, L. Yin, M. K. Kim, A. Banks, U. Paik, Y. Huang and J. A. Rogers, Adv. Funct. Mater., 2015, 25, 1338–1343 CrossRef CAS.
- K. Sim, X. Wang, Y. Li, C. Linghu, Y. Gao, J. Song and C. Yu, J. Micromech. Microeng., 2017, 27, 065010 CrossRef.
- J. Pajak, M. Ziemski and B. Nowak, Chemik, 2010, 64, 523–530 CAS.
- M. I. Baker, S. P. Walsh, Z. Schwartz and B. D. Boyan, J. Biomed. Mater. Res., Part B, 2012, 100, 1451–1457 CrossRef PubMed.
- W. B. Chu, J. W. Yang, T. J. Liu, C. Tiu and J. Guo, Colloids Surf., A, 2007, 302, 1–10 CrossRef CAS.
- H. Acar, S. Çinar, M. Thunga, M. R. Kessler, N. Hashemi and R. Montazami, Adv. Funct. Mater., 2014, 24, 4135–4143 CrossRef CAS.
- K. Fu, Z. Liu, Y. Yao, Z. Wang, B. Zhao, W. Luo, J. Dai, S. D. Lacey, L. Zhou, F. Shen, M. Kim, L. Swafford, L. Sengupta and L. Hu, Nano Lett., 2015, 15, 4664–4671 CrossRef CAS PubMed.
- D. M. Allen and L. T. Ler, J. Environ. Monit., 1999, 1, 103–108 RSC.
- A. Tawansi, A. El-Khodary and M. M. Abdelnaby, Curr. Appl. Phys., 2005, 5, 572–578 CrossRef.
- K. Fytianos, E. Voudrias and N. Raikos, Environ. Pollut., 1998, 101, 123–130 CrossRef CAS.
- A. Strakšys, T. Kochane and S. Budriene, Chemija, 2013, 24, 160–169 Search PubMed.
- S. Wei, V. Pintus and M. Schreiner, J. Anal. Appl. Pyrolysis, 2012, 97, 158–163 CrossRef CAS.
- T. Çaykara, R. Inam, Z. Öztürk and O. Güven, Colloid Polym. Sci., 2004, 282, 1282–1285 CrossRef.
- A. G. El-Shamy, W. Attia and K. M. Abd El-Kader, J. Alloys Compd., 2014, 590, 309–312 CrossRef CAS.
- S. K. Kang, S. W. Hwang, S. Yu, J. H. Seo, E. A. Corbin, J. Shin, D. S. Wie, R. Bashir, Z. Ma and J. A. Rogers, Adv. Funct. Mater., 2015, 25, 1789–1797 CrossRef CAS.
- Z. Wang, J. Che and C. Ye, Hydrometallurgy, 2010, 105, 69–74 CrossRef CAS.
- O. Herreros, R. Quiroz, A. Restovic and J. Viñals, Hydrometallurgy, 2005, 77, 183–190 CrossRef CAS.
- Q. Li, S. W. Zhang, Y. Zhang and C. Chen, Nanotechnology, 2006, 17, 4981–4985 CrossRef CAS.
- P. M. Natishan and W. E. O'Grady, J. Electrochem. Soc., 2014, 161, C421–C432 CrossRef CAS.
- E. McCafferty and J. P. Wightman, J. Colloid Interface Sci., 1997, 194, 344–355 CrossRef CAS PubMed.
- J. O. M. Bockris and L. V. Minevski, J. Electroanal. Chem., 1993, 349, 375–414 CrossRef CAS.
- T. H. Nguyen and R. T. Foley, J. Electrochem. Soc., 1980, 127, 2563–2566 CrossRef CAS.
- D. Macdonald and G. Englehardt, ECS Trans., 2010, 28, 123–144 CrossRef CAS.
- E. McCafferty, Corros. Sci., 2003, 45, 1421–1438 CrossRef CAS.
- P. M. Natishan and E. McCafferty, J. Electrochem. Soc., 1989, 136, 53–58 CrossRef CAS.
- B. A. Abd-El-Naby, O. A. Abdullatef and W. A. El-Mahmody, Phys. Chem., 2017, 7, 1–7 CAS.
- J. Yoon, J. Han, B. Choi, Y. Lee, Y. Kim, J. Park, M. Lim, M. H. Kang, D. H. Kim, D. M. Kim, S. Kim and S. J. Choi, ACS Nano, 2018, 12, 6006–6012 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09507f |
| ‡ Neeru Mittal and Tae-Min Jang contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |