
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review on recent trends in selective hydrodeoxygenation of lignin derived molecules†
Jake G.
Tillou
,
Chigozie J.
Ezeorah
,
Joseph J.
Kuchta
III
,
Sachini C. D.
Dissanayake Mudiyanselage
,
James D.
Sitter
and
Aaron K.
Vannucci
 *
*
Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC 29208, USA. E-mail: vannucci@mailbox.sc.edu
First published on 18th August 2023
Abstract
A review of recent reports that focus on the selective hydrodeoxygenation of lignin biomass derived aromatic compounds is presented. Obtaining high value chemical feedstocks and fuels from lignin is recognized as an essential aspect of the economic feasibility of biorefineries. Lignin is both a non-edible part of biomass and a potential source of aromatic commodity chemicals. The selective catalytic conversion of lignin derived compounds to deoxygenated aromatic molecules represents the most direct route towards high value chemicals while also maximizing hydrogen use efficiency. This review aims to give an overview of reports within the last 3–4 years that have focused on the selective hydrodeoxygenation of lignin derived or lignin inspired oxygenated aromatic compounds. Many of these lignin derived dimers and monomers can be selectively obtained via reductive catalytic fractionation (RCF) of native lignin, thus a section of this review is dedicated to recent advancements of RCF. The observed trends with respect to catalyst composition and reaction conditions in these reports along with an outlook for selective catalytic hydrodeoxygenation is presented.
 Jake G. Tillou | Jake G. Tillou is a fourth-year graduate student in the in the Department of Chemistry and Biochemistry at the University of South Carolina. His research focuses on the selective hydrodeoxygenation of lignin model compounds using molecular catalysts. Jake obtained his B.S. from Newberry College. |
 Chigozie J. Ezeorah | Chigozie J. Ezeorah is currently a PhD student at the University of South Carolina, supervised by Prof. Aaron Vannucci and with a research focus on synthesis of single site catalysts and their application in hydrodeoxygenation of lignin model compounds. He holds lectureship position at the University of Nigeria Nsukka where he obtained BSc in Pure and Industrial Chemistry (2012) and MSc in Inorganic Chemistry (2018). He has also obtained MRes in Catalysis: Chemistry and Engineering from Imperial College London in 2019. |
 Joseph J. Kuchta III | Joseph J. Kuchta III earned his Bachelor's degree in Chemistry from Delaware Valley University in 2012, after which he gained valuable experience working in industry as a chemist, specializing in HPLC/UPLC. In 2020, he decided to transition back to academia and joined the research group of Professor Aaron Vannucci at the University of South Carolina. His current research is focused on small molecule activation using molecular catalysis with the aid of metal oxide supports and atomic layer deposition. |
 Sachini C. D. Dissanayake Mudiyanselage | Sachini C. D. Dissanayake Mudiyanselage obtained her B.S. from the University of Colombo, Sri Lanka in 2019. Her PhD research focuses on the photoelectrochemical reduction of naphthols for hydrogen atom transfer (HAT) reactions. |
 James D. Sitter | James D. Sitter recently defended his PhD dissertation and has accepted a position at the Naval Research Laboratories in Washington D.C. His PhD research focused on the synthetic and catalytic conversions of nitrogen-containing heterocycles. Previous to his graduate studies, James served in the U.S. Army and obtained his B.S. from the University of South Carolina in 2019. |
 Aaron K. Vannucci | Aaron K. Vannucci is an associate professor in the Department of Chemistry and Biochemistry at the University of South Carolina. His research focuses on sustainable transformations of small molecules including lignin conversion and photocatalytic bond forming reactions. Previous to his academic appointment he obtained his bachelor's degree from The College of Wooster in 2004 followed by his PhD in chemistry from The University of Arizona in 2009. He then spent time as a postdoctoral fellow at UNC Chapel Hill. He has been at the University of South Carolina since 2014. |
Sustainability spotlightEfficient biomass conversion remains an important goal for the development of renewable fuels and biorefineries focused on a more sustainable chemical industry. Specifically, lignin biomass, which historically was treated as waste, offers the possibility to produce high value aromatic molecules and high-octane rating fuels. The ability to selectively deoxygenate aromatics while avoiding ring hydrogenation offers the most direct and sustainable route towards high value chemical production from lignin biomass. Direct and selective deoxygenation of lignin derived molecules helps conserve H2 usage and limits the number of chemical steps necessary to achieve aromatic products. Biomass conversion addresses UN Sustainable Development Goals 7 (clean energy), 11 (sustainable cites), and 12 (responsible production). |
1. Introduction
Researchers and consumers alike are pushing towards the increased use of renewable energies. Biomass is the only renewable energy source that is also a potential feedstock of carbon for commodity chemical production. The selective conversion of biomass to fuels and commodity chemicals is thus an essential aspect towards the increased usage of renewable energy. While production of chemicals derived from biomass is already on an industrial scale; it is just a small fraction of production compared to fossil fuels. This leads to the need for continued research on the selective conversion of biomass.Naturally occurring biomass is composed of three substructures, lignin (Fig. 1a), cellulose (Fig. 1b), and hemicellulose (Fig. 1c). The cellulosic components are widely utilized in pulps for paper production and refineries for liquid fuels, such as ethanol.1 Lignin, however, is commonly considered a by-product of these processes and is burned on-site for a low-cost energy source.2 With lignin comprising nearly a third of biomass by weight, the selective conversion of lignin biomass represents an important research target.3 As shown in Fig. 1, lignin biomass is a phenolic polymer with a high oxygen content. Upgrading lignin into fuels and commodity chemicals therefore requires depolymerization followed by deoxygenation of the monomeric and dimeric units.4 The most direct route to the highest value products is selective deoxygenation while maintaining the aromaticity of the monomers and dimers. The resulting products of selective deoxygenation (i.e. benzene, toluene, styrene, etc.) are high value aromatic compounds that are essential towards making biorefineries economically feasible.4,5 Selective deoxygenation also helps conserve H2 usage and tends to prevent coking of catalysts during the conversion of lignin.6
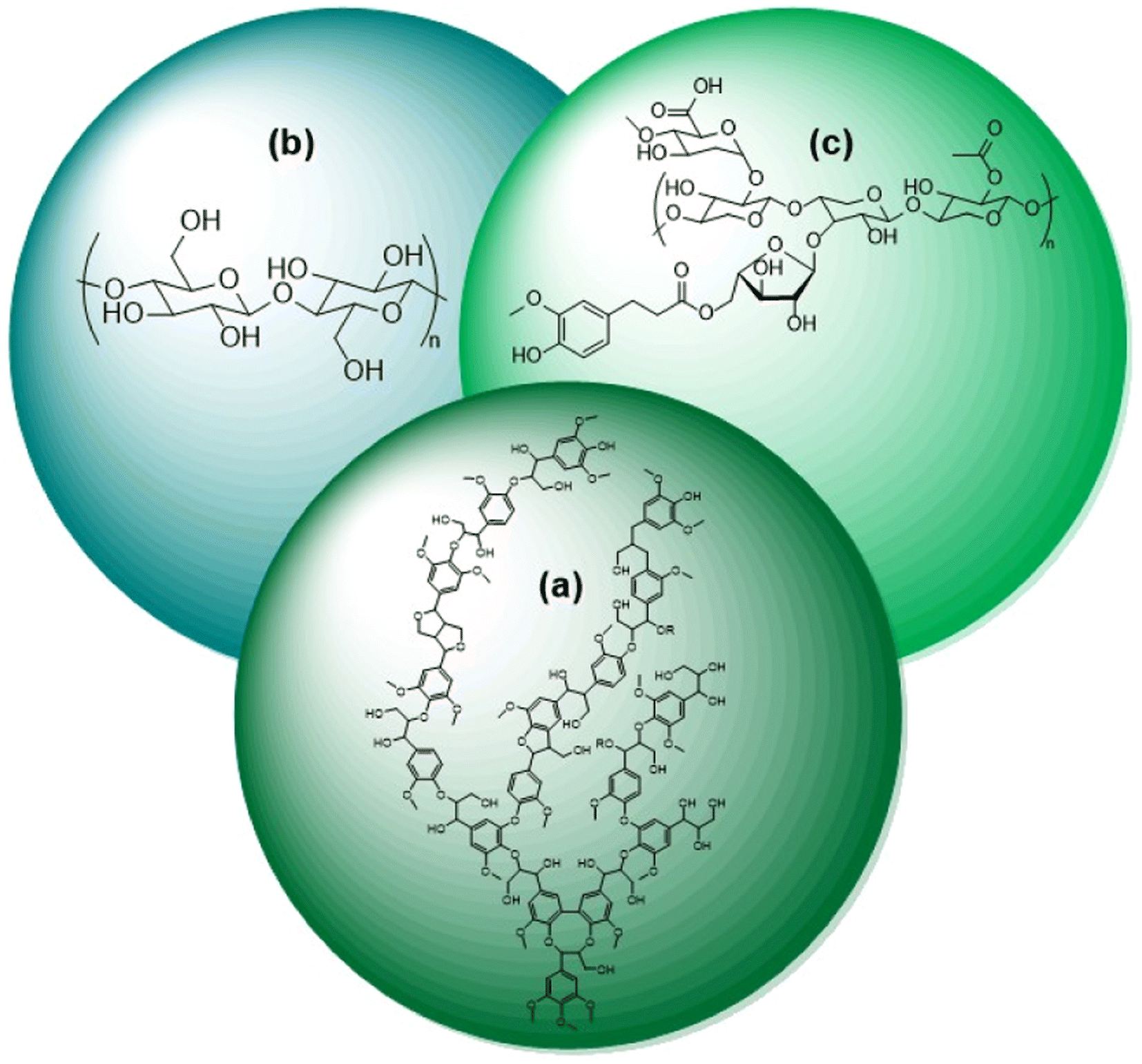 | ||
| Fig. 1 Substructures of biomass: (a) lignin, (b) cellulose, and (c) hemicellulose. | ||
Over the past couple of decades, extensive research has gone into the conversion of lignin biomass.7 Large advances have been achieved in the depolymerization of lignin to generate high yields of oxygenated monomeric and dimeric units, including reductive,8 oxidative,9 and “lignin-first” approaches.10 The lignin-first approach, which involves the reductive catalytic fractionation of lignin from the cellulosic components has been shown to result in some of the highest selectivity of monomeric and dimeric units.10 However, the resulting oxygenated small molecules (representative examples shown in Fig. 2) have low octane ratings and are common peroxide formers.11 The deoxygenation of these lignin-derived molecules is thus necessary to generate biofuels and a wider range of commodity chemicals. Reports have shown synthetic routes for converting lignin-derived compounds into specialty chemicals.12–14 However, selective hydrodeoxygenation (HDO), which is the removal of oxygen atoms with H2via C–O bond cleavage while maintaining the aromatic structure, represents the most direct route towards high value chemicals.15
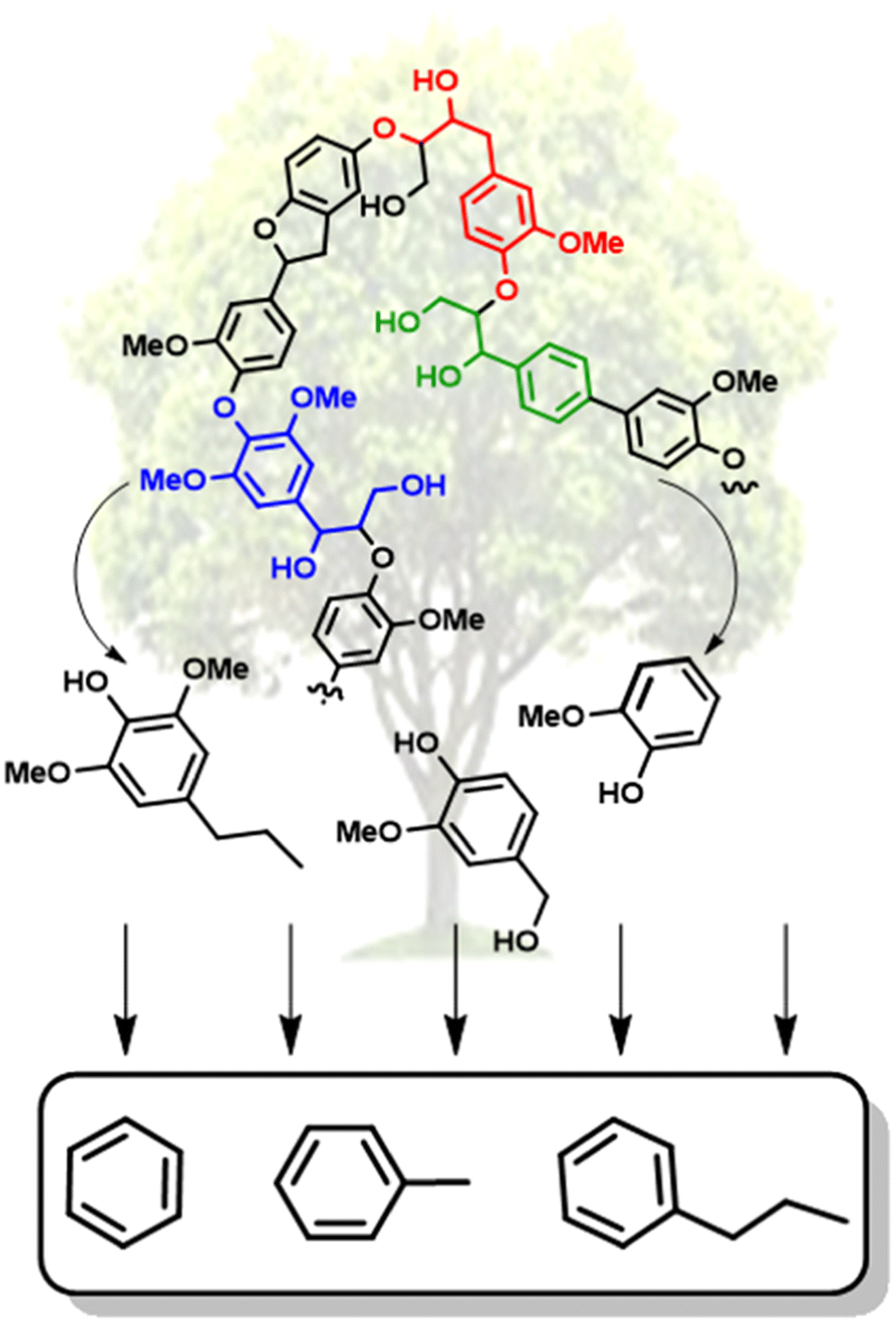 | ||
| Fig. 2 Illustrative conversion of lignin biomass to deoxygenated aromatic monomers. | ||
Hydrodeoxygenation of lignin-derived compounds has become a very active area of research in the biomass community. However, the breaking of C–O bonds during HDO reactions to remove water or small alcohols such as methanol, is often competitive with hydrogenation of the aromatic ring. Even with the goal of the selective removal of oxygen atoms,16–19 the majority of reports show that catalytic HDO of lignin-derived biomass results in both deoxygenation and ring hydrogenation. This distribution of products can be observed in recent reviews ranging from comprehensive overviews20,21 to focusing on specialized approaches22,23 or specific catalyst systems.24–30
The selective removal of C–O bonds and oxygenated functionality while maintaining aromaticity is difficult due to the high bond dissociation energy (BDE) of C–O bonds in aromatic compounds. The C–O BDE of phenol is 111 kcal mol−1, and the C–O BDE of the Csp2–O bond of methoxy groups of oxygenated aromatics is near 100 kcal mol−1.30 These high BDEs thus typically require high reaction temperatures >200 °C to achieve HDO reactions. However, high reaction temperatures may lead to catalyst deactivation through leaching, coking, or sintering.31 Changes in catalyst composition at high temperatures may also lead to a lack of product selectivity towards deoxygenation over ring hydrogenation.32 It has been reported, however, that reaction selectivity towards deoxygenation is more closely tied to the hydrogen pressure applied during the reaction.33 Higher H2 pressure (>30 atm) may lead to more observed deoxygenation of substrates, however, ring hydrogenation becomes unpreventable under these conditions.34 Conversely, ring hydrogenation can be suppressed with lower H2 pressure (1–10 atm).35 Product selectivity towards deoxygenation can also be affected by the choice of solvent. The solvent may block catalytic active sites or be incorporated into unwanted side products.17,36
Obviously, the composition of the catalyst – metal(s) used, support, catalyst size – will also influence product selectivity during HDO reactions. For example, transition metals in the nickel triad show high affinity for hydrogenation reactions, whereas more oxophilic metals may favor deoxygenation reactivity over ring hydrogenation.23 Bimetallic species have been shown to combine the ability of two different metals to achieve high catalyst activity towards selective HDO reactions.37 In addition, acidic supports have been shown to aid in the C–O bond activation process in the generation of bifunctional catalysts for HDO.38 Trends in metal nanoparticle size have also arisen in the literature as smaller particles have been reported to show greater selectivity towards C–O bond cleavage over ring hydrogenation.18,39 Molecular transition-metal-based catalysts have also shown excellent selectivity for deoxygenation over ring hydrogenation.17,40
This review will focus on recent studies being published within the last few years and on reports where selective C–O bond cleavage was targeted over ring hydrogenation. Selectivity in this report will be defined by the direct deoxygenation of substrates without observed ring hydrogenation. Reaction selectivity trends will be explored with respect to reaction conditions, such as solvent, temperature, hydrogen source/pressure, and with the catalyst utilized. A summary of the catalytic results of the manuscripts reviewed here can be found in Table S1† and consists mainly of reactions performed on model monomer and dimer substrates that can be derived from lignin biomass. This review is separated into metal catalysts commonly reported for HDO reactions and outlines the approaches and conditions where selective hydrodeoxygenation was favored over ring hydrogenation and high yields of aromatic products were achieved from lignin-derived or model lignin substrates.
2. Selective HDO of lignin derived compounds
2.1 Nickel catalysts
Nickel is an inexpensive Earth abundant metal that has been widely studied in monometallic and bimetallic systems for hydrodeoxygenation (HDO) activity. The high stability under hydrogenolysis and hydrodeoxygenation conditions has also made Ni a catalyst of recent interest. Nickel has been studied in bifunctional systems coupled with supports such as carbon, SiO2, or Al2O3.23,41–45 There also have been many recent reports utilizing heterogeneous nickel catalysts for the conversion of lignin or lignin model compounds to arene or cycloalkane monomers.46–50 Reports have also shown that nickel catalyst supports can enhance C–O bond activation by utilizing Lewis acid sites in the support while nickel activates H2.3,44,51 Product distributions for systems utilizing nickel catalysts consist mainly of over-hydrogenated products resulting in cycloalkanes. More recently, nickel catalysts have been studied for the hydrogenolysis of 4-O-5, α-O-4, and, most importantly, β-O-4 bonds in model lignin dimers (Fig. 3) under quite moderate conditions.3,42–44,51–55 Here we highlight a number of reported Nickel catalysts employed in the selective conversion of model lignin compounds.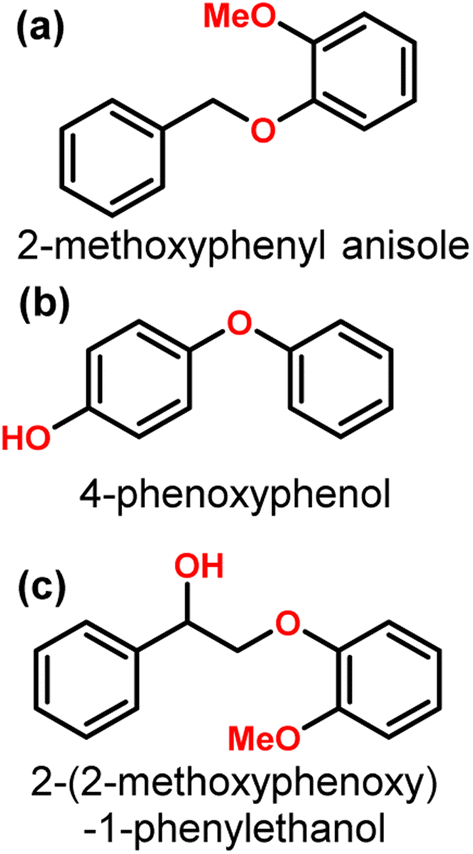 | ||
| Fig. 3 Structure of model compounds with common C–O–C linkages in lignin. | ||
A recent investigation into the cleavage of 4-O-5, α-O-4, and β-O-5 bonds was conducted by Guo et al. utilizing a Ni/CaO–H-ZSM-5 catalyst under low temperature reaction conditions.52 The H-ZSM-5 support was chosen due to the characteristic Lewis acidity stability. CaO was utilized as a basic site to facilitate hydrogen transfer and has been utilized as a lignin conversion catalyst on its own.56 Nickel and calcium oxide nanoparticles were loaded onto the zeolite support by deposition–precipitation (DP) and the final catalyst was characterized by XRD, TEM, BET, FT-IR, and ICP-AES. The average Ni particle size determined by TEM was 6.95 nm and a 45 wt% of Ni was determined by ICP-AES. This catalyst was tested against 2-methoxyphenyl anisole as an α-O-4 model compound (Fig. 3a), 4-phenoxyphenol as a 4-O-5 model compound (Fig. 3b), and 2-(2-methoxyphenoxy)-1-phenylethanol as β-O-4 model compound (Fig. 3c). Conversion of the β-O-4 model compound over Ni/CaO–H-ZSM-5(60) achieved up to 88.2% conversion where the product distribution favored 1-phenyl ethanol (42.7%) and guaiacol (49.6%) with ethylbenzene (7.4%) and 1-methoxy-2-phenethoxybenzene (0.3%). As shown in Fig. 4, increasing H2 pressure up to 2.5 MPa (25 atm) resulted in the increased production of hydrogenation products, following recent trends that higher pressures of H2 lead to more observed ring hydrogenation products.57 However, the α-O-4 model compound, 2-methoxyphenyl anisole, was readily converted to toluene (49.4%) and guaiacol (50.5%) and increasing H2 pressure did not result in further conversion of toluene and guaiacol to cycloalkanes. Furthermore, the lowest activity was observed for the hydrogenolysis of the 4-O-5 model compound due to the 4-O-5 bond having a higher bond energy than the α-O-4 and β-O-4 bonds. After 60 minutes at 140 °C only 40% of the 4-O-5 model compound was converted to phenol (66.1%), benzene (10.1%), cyclohexanol (20.6%), and cyclohexanone (3.2%). This reaction illustrates the need for elevated temperatures to help active the C–O bonds of compounds derived from lignin biomass.58
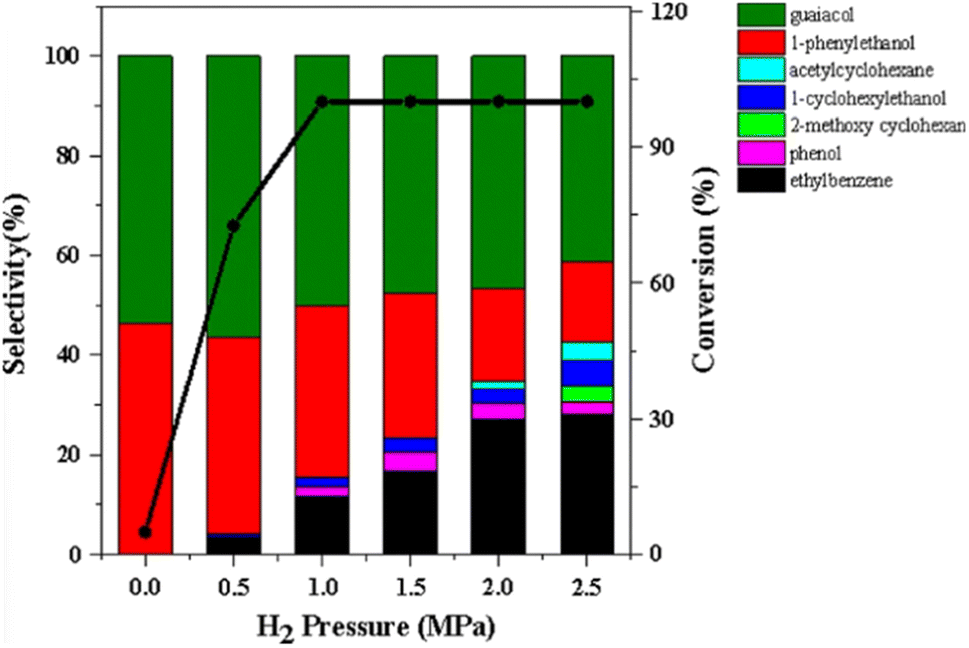 | ||
| Fig. 4 The product distributions for the conversion of 2-(2-methoxyphenoxy)-1-phenylethanol (β-O-4) over Ni/CaO–H-ZSM-5(60) under different H2 pressures. Reproduced from ref. 52 with permission from BMC Chem., copyright 2019. | ||
In an effort to reduce the need for high H2 pressures and reduce ring hydrogenation in the conversion of lignin dimers, Jiang et al. used isopropanol as a proton donor in catalytic transfer hydrogenation reactions performed by a series of Ni/Al2O3 catalysts.42 The Ni/Al2O3 catalysts were synthesized from the calcination of [Ni2Al(OH)6](NO3)·0.6H2O precursors followed by subsequent reduction under H2. Reduction temperatures were varied for each catalyst batch between 500–700 °C in 50 °C intervals. The catalysts were then passivated under 1 mol% O2/N2 mixture and stored under air for catalytic testing. The catalysts were analyzed by TEM, N2 adsorption/desorption, XRD, XPS, and ICP-AES. The average Ni particle size was found to increase with increasing reduction temperature from 5.5 nm for the Ni/Al2O3-500 catalyst to 10.2 nm for the Ni/Al2O3-700 catalyst. Ni loadings followed a similar trend as the particle size, where Ni/Al2O3-500 loading was 56.15 wt% and Ni/Al2O3-700 loading was 61.50 wt%. Catalytic screenings utilizing benzyl phenyl ether (BPE) as a model α-O-4 compound found that conversion of BPE using the series of Ni/Al2O3-T catalysts (T representing the temperature of reductions) resulted in product mixtures including toluene, phenol, cyclohexanone, and cyclohexanol. BPE conversion increased with increasing Ni particle size, however, selectivity towards direct deoxygenation decreased as ring hydrogenation products were observed with larger particle sizes. This trend of particle size and selectivity has been observed and hypothesized that smaller particle sizes have the correct geometry and a higher ratio of active sites to promote deoxygenation.39 For this study under review, catalysts with smaller particle sizes led to more selectivity towards the desired toluene and phenol products. However, these results were also obtained at lower reactions temperature so a direct correlation to particle size and catalysts selectivity could not be drawn from these experiments. Additional mechanistic studies led to the proposed mechanism for this conversion which is shown in Fig. 5. The nickel catalysts first activated isopropanol to generate reactive H species, which undergo subsequent C–O bond cleavage reactions. The authors show that higher reaction temperatures do lead to the increase of cyclohexanol formation, indicating that reaction temperature can have an adverse effect on obtaining selective deoxygenation.
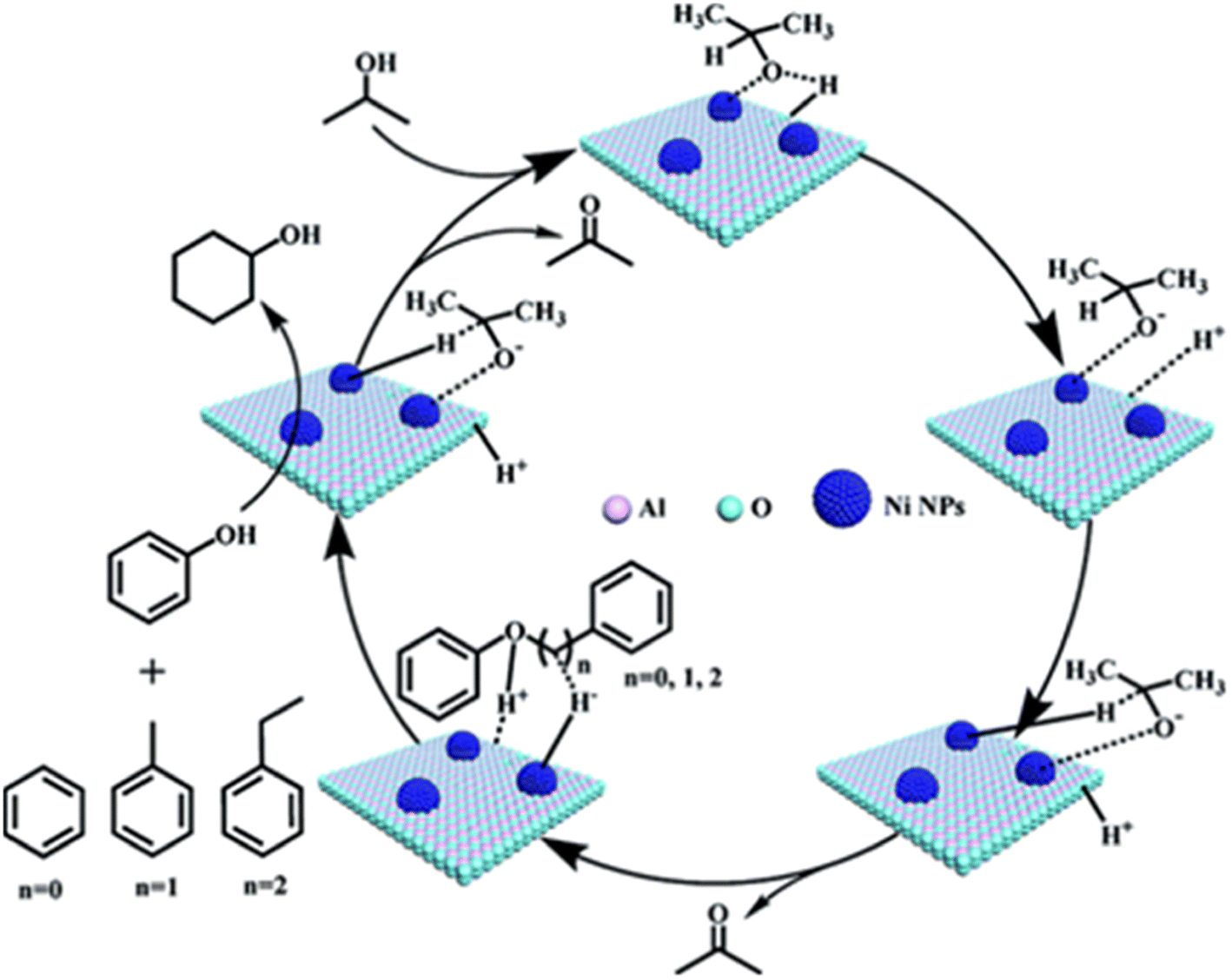 | ||
| Fig. 5 Proposed mechanisms for cleavage of C–O bonds in the α-O-4, β-O-4, and 4-O-5 compounds. Reproduced from ref. 42 with permission from RSC, copyright 2019. | ||
A Ni catalyst on an activated carbon support was utilized by Zhu et al. for the hydrolysis of BPE.54 Activated carbon (AC) was chosen as the support due to its known thermal and mechanical stability as well as uniform pore size distributions, efficient mass transfer of reactant molecules, and controllable textural properties. The Ni/AC catalyst was prepared by a wet-impregnation method using Ni(NO3)2·6H2O as the starting Ni source. Ni/AC was characterized by N2 adsorption–desorption analyses, BET analyses, XRD, SEM, and TEM. Metal loading of Ni on AC was 10 wt% and particle size was varied within each catalyst sample. Small particles around 3 nm were the most common, but Ni particles up to 20 nm were also detected. Hydrogenolysis using Ni/AC resulted in full conversion of BPE with a product mixture that included toluene, phenol and cyclohexanol. Fig. 6 shows the effect of temperature, H2 pressure, time, and catalyst loading on BPE conversion and arene product selectivity. The results show that increased temperature (beyond 145 °C) resulted in the consumption of phenol to cyclohexanol. This result once again shows that the elevated temperature is often needed to cleave stable C–O bonds, but also leads to increased, unwanted ring hydrogenation reactivity. Similarly, increasing H2 pressure above 2 MPa again resulted in consumption of phenol to cyclohexanol continuing the observed trend of greater H2 pressures leading to ring hydrogenation. The time studies by the authors, shown in the lower left of Fig. 6, also highlighted an interesting aspect towards selectivity. Conversion of the phenol product to the unwanted cyclohexanol did not appreciably occur until the BPE substrate was completely consumed. This result shows why batch reactions performed at full conversions may lead to additional reactivity. Performing reactions in flow reactors or under limited conversion may give more reliable insight into catalyst activity towards the chosen substrate.59
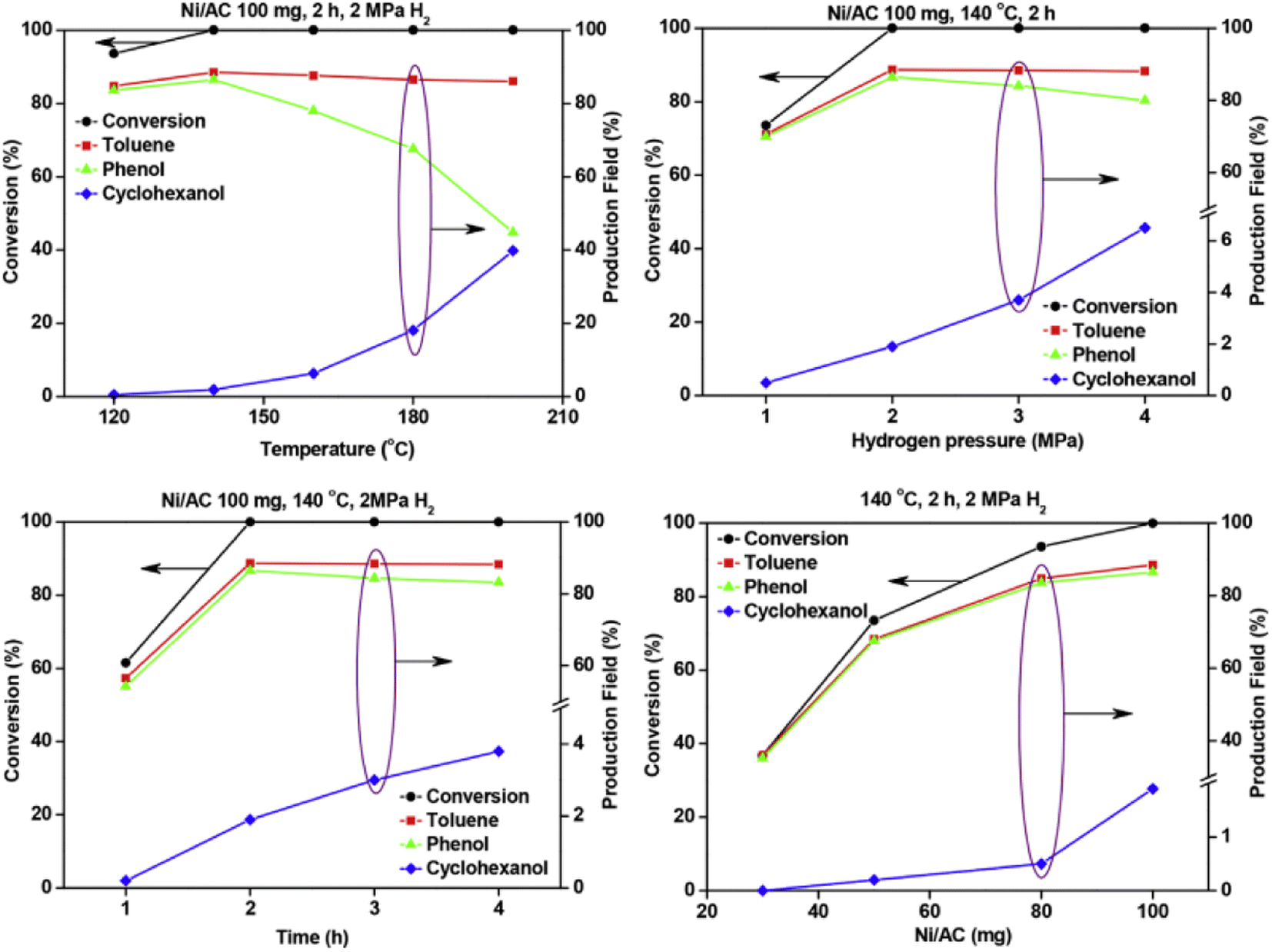 | ||
| Fig. 6 Effects of temperature (upper left), time (lower left), hydrogen pressure (upper right) and Ni loading (lower right) on the selective cleavage of BPE. Reproduced from ref. 54 with permission from J. Energy Inst., copyright 2019. | ||
Matsagar et al. explored the use of cosolvent systems and their effects on reagent solubility and catalyst stability. They utilized a carbon black (CB) supported Ni catalyst.53 The Ni/CB was prepared by an impregnation method using Ni(NO3)2·6H2O as the catalyst precursor. TEM imaging of Ni/CB shows the average Ni particle size was 8.1 nm and are homogenously distributed across the surface of the CB support with an average Ni loading of 6.8 wt% as determined by ICP-AES. Hydrogenolysis experiments were conducted using BPE as the lignin model compound. Cosolvents systems consisting of MeOH/H2O and EtOH/H2O resulted in the greatest BPE conversion of 83% and 91% respectively. The decreased conversion in the MeOH/H2O cosolvent system was attributed to deactivation of the catalyst by the formation of methoxy groups on the Ni surface. These results highlight how solvents can play a role in catalysts activity as discussed in the introduction. Manipulation of the volume fraction of EtOH in the EtOH/H2O cosolvent system also affected BPE conversion. As the volume fraction of EtOH increased from 0% to 30%, BPE conversion increased from 54% to 91%; however, further increasing the EtOH volume fraction to 100% decrease BPE conversion to 13%. The authors attributed this reaction trend to changing area ratios of metallic Ni, NiO, Ni(OH)2 on the catalyst surface. As seen in Fig. 7, finding the optimal volume fraction of EtOH/H2O proved to be important as a low volume fraction would result in decrease BPE solubility, and a high-volume fraction would result in a decreased ratio of catalytically active metallic Ni. Fig. 7 thus illustrates the important balance required of the solvent between lignin solubility and not interfering with catalyst active sites. Once again, the authors showed the trend of batch reactors, such that product mixtures consisted of toluene (46.7%) and phenol (45.5%) when the duration of the reaction was 1 hour in a batch reactor. Increasing the reaction duration led to the ring-hydrogenated products cyclohexanol and cyclohexanone from the over hydrogenation of phenol. Overall, compared to other similar noble metal catalysts such as Pd/C, RuC, and Rh/C, the reported Ni/CB catalyst displayed higher selectivity toward the activation of the aliphatic C–O bond in BPE without over hydrogenation.
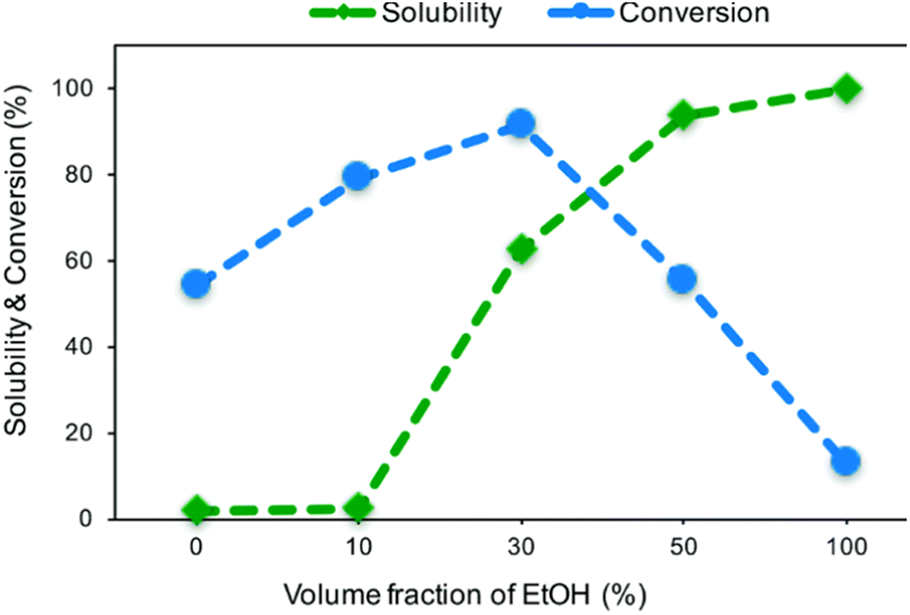 | ||
| Fig. 7 Solubility of BPE and its conversion in different volume fractions of H2O/EtOH co-solvent system. Reproduced from ref. 53 with permission from RSC, copyright 2019. | ||
As an alternative to carbon supports, Lin et al. doped Ni/H-ZSM-5 with CeO2.44 A series of Ni–xCeO2 catalysts were prepared by the precipitation method using Ni(NO3)2·6H2O as the catalyst precursor. TEM imaging showed the average Ni particle size to be 8.1 nm. The metal loading of Ni for each catalyst was 10 wt%. The loading of CeO2 varied from 5 to 15 wt%. Hydrogenolysis experiments were conducted using 2-(2-methoxyphenoxy)-1-phenylethan-1-ol, as a model compound for the β-O-4 linkage found in lignin. As seen in Fig. 8, Ni on ZSM-5 could activate the β-O-4 bond and form guaiacol and ethylbenzene, albeit at less than 50% selectivity. As CeO2 loading on the Ni-ZMS-5 catalyst increased, however, the selectivity towards guaiacol and ethylbenzene increased, indicating more β-O-4 bond cleavage. This result illustrates the importance of the acid/base properties of the support and the authors conclude that CeO2 helped regulate the hydrogenolysis process through the tuning of redox properties of the active nickel species. The authors furthermore examined the effect of catalyst structure on activity. As also shown in Fig. 8, when the nickel catalyst was synthesized with wet impregnation (Ni–10CeO2/H-ZSM-5i) instead of their standard precipitation method (Ni–10CeO2/H-ZSM-5) the conversion and selectivity of the 2-(2-methoxyphenoxy)-1-phenylethan-1-ol substrate decreased. This was attributed to larger Ni particles and lower nickel dispersion on the support, which continues the commonly observed trend that smaller catalyst particles with tighter particle distributions lead to more selective product formation.
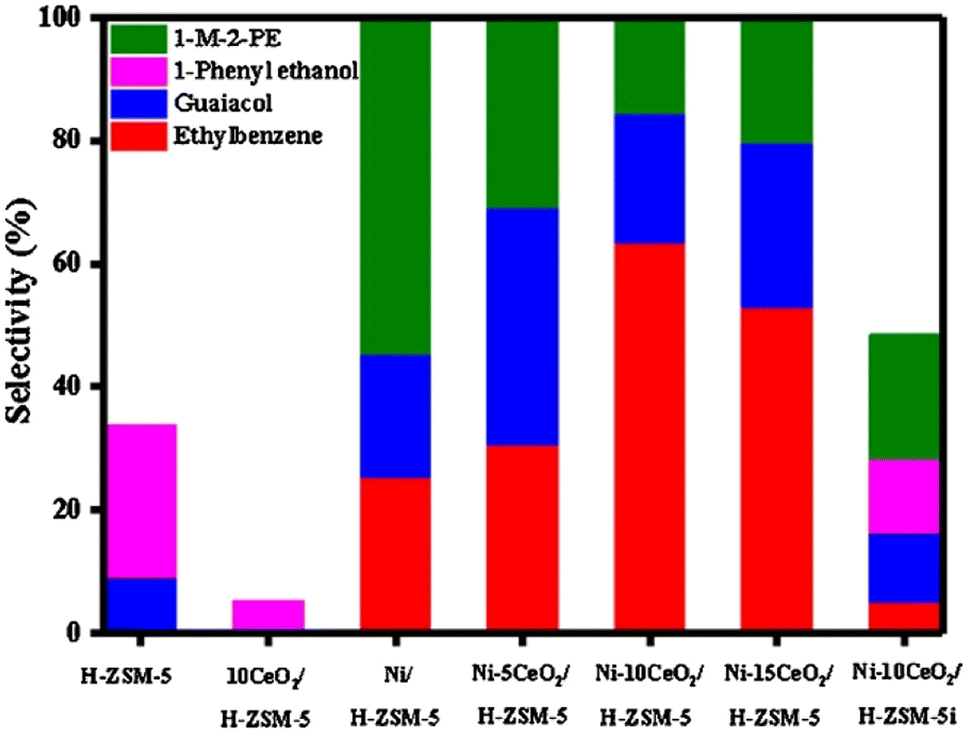 | ||
| Fig. 8 β-O-4 catalytic hydrogenolysis product distribution histogram. Reproduced from ref. 44 with permission from Elsevier, copyright 2020. | ||
A further illustration on how Ni particle size affected product selectivity was shown by Chen et al. when they investigated Ni3S2-doped carbon nanosheets (Ni3S2-CSs) for hydrogenolysis.55 Nickel sulfide was chosen due to its low cost and carbon nanosheets were chosen as the support due to previously being used with sodium lignosulfonate produced in papermaking. Ni3S2-CSs was synthesized first by dissolving boric acid, sodium lignosulfonate, and nickel nitrate hexahydrate in DI water. The solution was kept at 80 °C until evaporated to yield a dark yellow composite. This composite was annealed at the desired temperature for 2 h and then dried for another 12 h at 80 °C. TEM revealed the thickness of the sheets to be approximately 30 nm. Nickel particle size increased with increasing metal loading due to agglomeration. Hydrogenolysis experiments were conducted with 2-phenoxy-1-phenylethanol as a β-O-4 model compound. It was noted that increasing Ni content had the drawback of decreased deoxygenation selectivity as increased amounts of cyclohexanol were observed in product mixtures stemming from larger Ni particle sized catalysts. In addition, at 800 °C calcination temperatures, Ni particle sizes grew so large that conversion of the 2-phenoxy-1-phenylethanol began to drop below smaller Ni particle catalysts under identical conditions. This drop in conversion shows an even more concerning trend, that particle size not only affects product selectivity, but can also inhibit overall catalyst activity.
With the trend of larger catalyst particles resulting in lower product selectivity, preventing catalyst sintering during reaction becomes of greater importance for selective deoxygenation reactions. Thus, a nitrogen-doped carbon support with nickel catalyst was utilized by Tan et al. for the hydrogenolysis of several β-O-4 model compounds.60 Ni embedded in or encapsulated by N-doped carbon has shown remarkable stability and catalytic activity in hydrogenation reactions and thus, was chosen for the hydrogenolysis of model lignin β-O-4 compounds. The Ni@NC-800 catalysts were encapsulated by graphitic NC shells. Ni nanoparticle sizes Ni@NC-800 ranged between 10-20 nm at 10.17 wt%. Hydrogenolysis experiments were conducted with 2-phenoxy-1-phenylethanol using isopropanol as the hydrogen source. Acid leaching was also conducted to investigate the structure–activity relationship which resulted in Ni@NC-800-H retaining catalytic activity with improved selectivity toward arene products. The improved catalyst activity and selectivity toward arene products over Ni@NC-800-H compared to Ni/NC-800 can be observed in Fig. 9. In both cases, the catalysts exhibited excellent recyclability, indicating the high stability of the catalyst structure with the nickel particles being encapsulated by graphitic NC shells. This approach offers a route to prevent catalyst sintering and control particle size towards higher catalyst selectivity. Furthermore, the Ni-NC-800-H catalyst exhibited greater activity (higher conversions shown in Fig. 9), which illustrates the previously observed impact the acidity of the support can have on hydrodeoxygenation catalysis.61 The authors also examined and expanded the scope of methoxy-rich substrates and showed their catalyst was capable of reacting with C–O bonds with a wide range of bond dissociation energies, while maintaining catalyst structure stability over a range of temperatures.
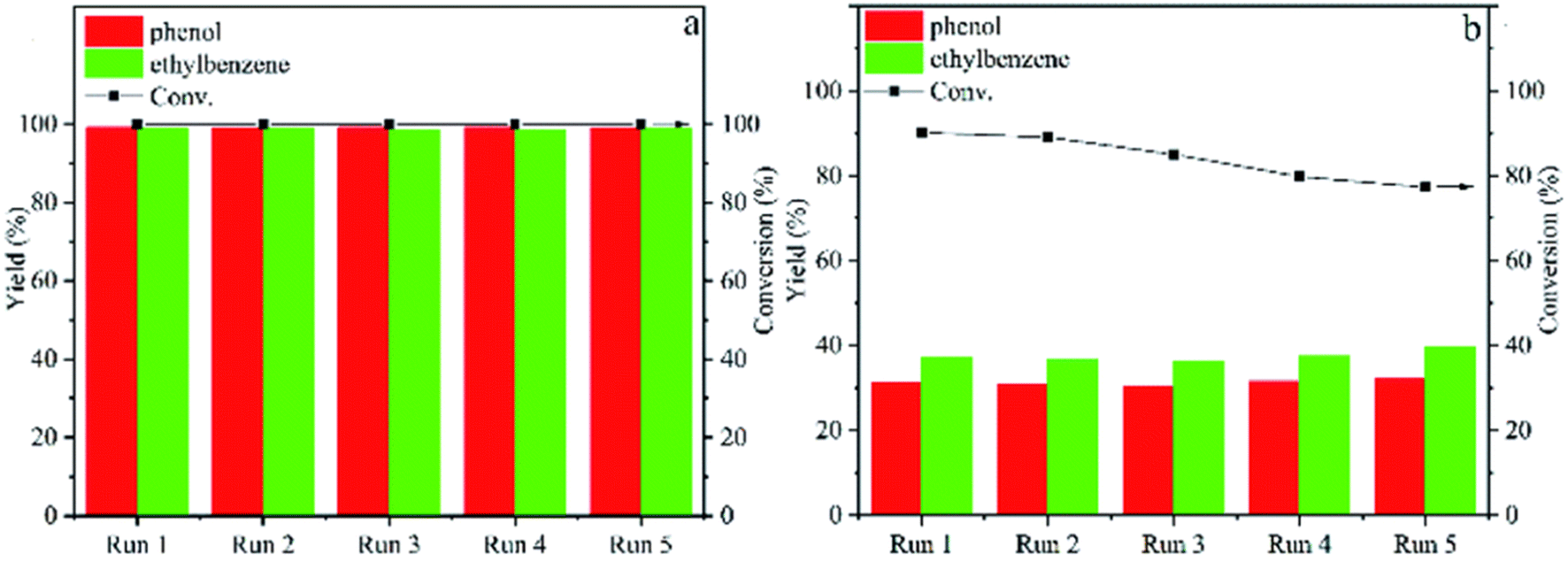 | ||
| Fig. 9 Catalytic cycling of Ni@NC-800-H (a) and Ni/NC-800 (b) for transfer hydrogenolysis of 2-phenoxy-1-phenylethanol at 180 °C with 4 h reaction time. Reproduced from ref. 60 with permission from RSC, copyright 2022. | ||
Lastly, several heterogeneous supported nickel compounds have been recently utilized to convert model lignin dimers and monomers to selectively produce ring-hydrogenated monomeric units and gaseous products. Xu et al. demonstrated a La–Ni/CMK-3 catalyst capable of converting phenol (80.9% conversion) to cyclohexanol (76.64%) at 240 °C in 4 h without an external hydrogen source.62 Similarly, Chen et al. utilized several NixLay/CNT catalysts for the conversion of BPE to methylcyclohexane, cylohexanol, and cyclohexanone.63 Chen et al. utilized a carbon nanotube supported Ni catalyst capable of converting diphenyl ether to cyclohexane (88%) and cyclohexanol (82%) using isopropanol as the hydrogen source.64 This Ni/CNT catalyst was also shown to convert modified diphenyl ethers, benzyloxybenzene, phenethoxybenzene and 2-(2-methoxyphenoxy)-1-phenylethan-1-ol into ring-hydrogenated monomeric units. Lin et al. utilized a heterogeneous HZ5-NS zeolite supported Ni catalyst for the conversion of 4-ethylguaiacol to cyclohexane and ethyl-cyclohexane products.65
2.2 Ruthenium catalysts
Many previous reports have shown that the use of heterogeneous ruthenium catalysts has resulted in ring hydrogenation during deoxygenation of lignin derived compounds.23,45,66–70 Ruthenium catalysts have garnered interest due to their innate oxophilicity allowing for a more direct interaction with the C–O bonds in lignin and lignin model compounds.71 Heterogeneous ruthenium catalyst have been recently studied on various supports such as carbon, Nb2O5, modified zeolites, and metal oxide doped silicas.72,73 Ruthenium catalyst have also been studied as both metal Ru nanoparticles and as RuO2 species for activity towards HDO.71 Ruthenium catalysts have shown HDO activity under a wide variety of conditions such as high temperatures, high pressures, and even in electrocatalytically driven HDO systems.70,73 Due to their use in fuel production, ruthenium catalysts tend to result in product mixtures with predominantly ring hydrogenated products when HDO reactions are conducted. More recently, reactivity and selectivity of ruthenium catalysts towards arene products has improved with modification to the catalyst support. Here we highlight ruthenium catalysts that demonstrated high selectivity during the conversion of lignin model compounds.Hossain, et al. sought to investigate the effects of Ru and RuO2 on HDO by utilizing carbon supported catalysts of different Ru/RuO2 (reduced to oxidized) ratios.71 The oxophilicity of metallic ruthenium was thought to allow for a more direct cleavage of C–O bonds while the Lewis acid sites of RuO2 had been previously shown to facilitate the HDO of furanic compounds. The catalysts were synthesized from commercial Ru/C by various pretreatment methods to tailor the composition. The catalysts were characterized by XRD, H2-TPR, EDS, and SEM. The Ru nanoparticle size for the Ru/C-red catalyst was 4.4 nm while the Ru/C–Ox catalyst has Ru nanoparticles between 19–25 nm in size. Ru/C–Ox also contained RuO2 ranging between 6–10 nm in size. H-TPR of these catalysts found that Ru/C-red had 4.09 wt% Ru with 0.69 wt% in RuO2 while Ru/C–Ox had 18.71 wt% Ru with 6.35 wt% in RuO2. HDO reactions were conducted using 2-phenyl ethyl phenyl ether as a model compound. HDO experiments using 2-phenethyl phenyl ether found that commercial Ru/C was capable of cleaving Cβ–O at 280 °C in the absence of H2 due to the presence of Cα–OH moieties on the surface of the support. While a hydrogen atom source would still be needed for prolonged catalytic activity from this catalyst, this catalyst is a great example of how the choice of support is important for promoting the desired catalytic activity. Fig. 10 shows the different proposed pathways that could result in the formation of the observed product mixture. It was hypothesized that Ru could activate Cα–OH to release hydrogen and form a “hydrogen pool” for hydrogenation to cleave the Cβ–O bond. Solvent and catalyst effects were also observed for this student. Protic ethanol was also found to promote HDO over deoxygenation when compared to catalysis conducted with 1,4-dioxane as the solvent. Furthermore, smaller particle catalysts were once again shown to be more active as Ru/C–Ox (the highest RuO2 content) had the lowest hydrogenolysis activity. This was attributed to the large crystallite size of RuO2 present in Ru/C–Ox.
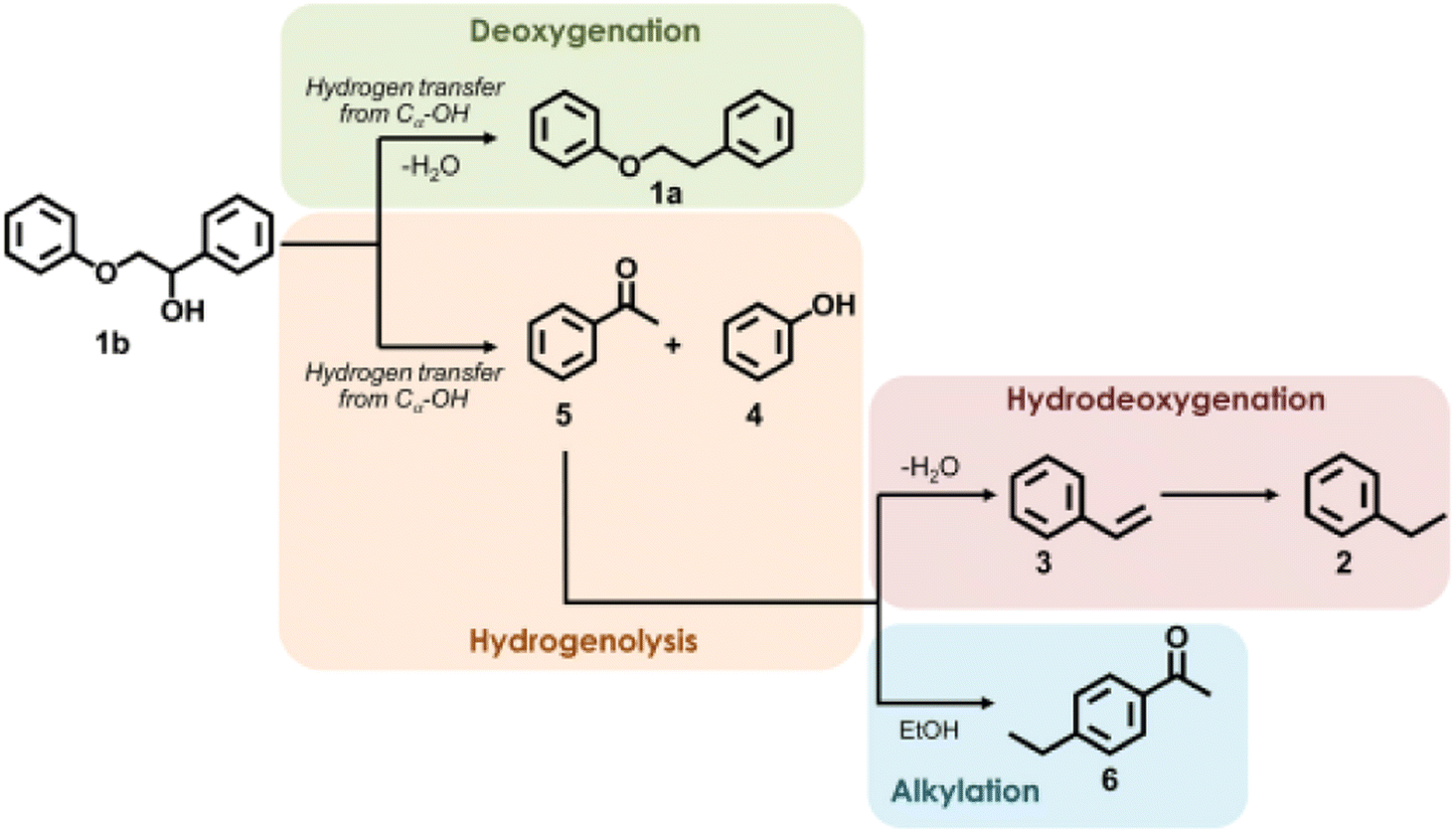 | ||
| Fig. 10 Proposed reaction pathway of hydrogenolysis of 2-phenoxy-1-phenylethanol over Ru/C. Reproduced from ref. 71 with permission from Elsevier, copyright 2019. | ||
A well-studied and effective support for heterogeneous ruthenium catalysts is Nb2O5. Xin et al. sought to investigate the effect of different morphologies of Nb2O5 on ruthenium reactivity.74 The Nb2O5 morphologies chosen for this study included flowers (F-Nb2O5), hollowed (H-Nb2O5), mesoporous (M-Nb2O5), and layered (L3-Nb2O5). All of the Ru/Nb2O5 catalysts were synthesized by the incipient wetness method. TEM images showed that the average particle size of F-Nb2O5 and H-Nb2O5 were 7.6 nm and 1.9 nm respectively. The Ru nanoparticles on M-Nb2O5 and L3-Nb2O5 could not be seen by TEM imaging. EDS-mapping revealed that Ru nanoparticles were present and homogenously dispersed on both M-Nb2O5 and L3-Nb2O5, but too small to be seen in TEM. The weight loading of Ru was 2.0 wt%, 1.6 wt%, 1.7 wt%, and 1.9 wt% for Ru/F-Nb2O5, Ru/H-Nb2O5, Ru/M-Nb2O5, and Ru/L3-Nb2O5 respectively. HDO experiments carried out with 4-methylphenol revealed Ru/L3-Nb2O5 to be the most active and most selective catalyst for deoxygenated aromatics due to small Ru particle size and the high Nb2O5 surface area. Excluding Ru particle size by synthesizing four additional catalysts of the same particle size resulted in a reactivity trend that correlated decreasing Nb2O5 surface area with decreasing reactivity. The authors indicated that increased surface area allowed for more unsaturated Nb![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites. Increasing the hydrothermal time of the layered Nb2O5 resulted in a less crystalline support with greater surface area for NbO sites. In addition, recyclability experiments using Ru/L1-Nb2O5 over three reaction cycles revealed no apparent catalyst deactivation or change in reaction selectivity (Fig. 11). Therefore, the authors show that not only does the metal catalyst support help promote reactivity, but also aids in metal catalyst stability. Post reaction characterization showed no change in the layered structure of the support and no obvious aggregation of Ru nanoparticles, analogous to the carbon nitride nickel catalysts discussed above. Catalysis utilizing Ru on this support (Ru/L1-Nb2O5) generally had higher conversion of 4-methylphenol that of Ru/L3-Nb2O3. The desired arene selectivity (deoxygenation over ring hydrogenation) was also shown to increase for reactions carried out in aqueous solutions, indicating the protic nature of water may play a role in the catalytic mechanism.
O sites. Increasing the hydrothermal time of the layered Nb2O5 resulted in a less crystalline support with greater surface area for NbO sites. In addition, recyclability experiments using Ru/L1-Nb2O5 over three reaction cycles revealed no apparent catalyst deactivation or change in reaction selectivity (Fig. 11). Therefore, the authors show that not only does the metal catalyst support help promote reactivity, but also aids in metal catalyst stability. Post reaction characterization showed no change in the layered structure of the support and no obvious aggregation of Ru nanoparticles, analogous to the carbon nitride nickel catalysts discussed above. Catalysis utilizing Ru on this support (Ru/L1-Nb2O5) generally had higher conversion of 4-methylphenol that of Ru/L3-Nb2O3. The desired arene selectivity (deoxygenation over ring hydrogenation) was also shown to increase for reactions carried out in aqueous solutions, indicating the protic nature of water may play a role in the catalytic mechanism.
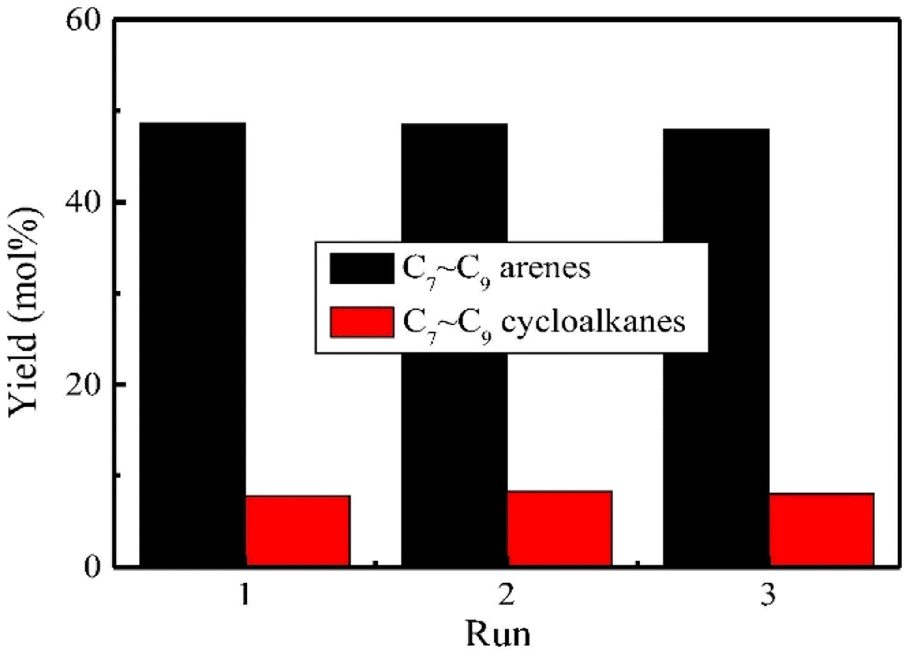 | ||
| Fig. 11 Products distribution in three successive runs during the HDO of enzymatic lignin over Ru/L1-Nb2O5 catalyst. Reproduced from ref. 74 with permission from Elsevier, copyright 2019. | ||
The use of alternative hydrogen sources with Ru catalysts was studied by Cao et al.75 Hydrogen gas that is typically used for hydrogenolysis is mainly sourced from fossil raw materials. Simple aliphatic alcohols, such as ethanol and methanol which can be sourced from biomass, are effective solvents for hydrogenolysis and could provide the hydrogen necessary for these reactions. The alcohols may also limit the amount of active hydrogen atoms at the catalyst surface, which has been proposed to limit unwanted ring hydrogenation.33 Different hydrogen sources were compared using a ruthenium on activated carbon (Ru/AC) hydrogenolysis catalyst. Ru/AC was prepared by incipient-wetness impregnation method and was characterized by N2 adsorption–desorption, XRD, CO pulse, and TEM. The Ru average particle size on Ru/AC was 1.8 nm with a narrow size distribution. Hydrogenolysis experiments were conducted using BPE. Hydrogenolysis using BPE and 1.0 MPa H2 resulted in 88.8% conversion and product mixture comprised of predominantly toluene and phenol. When hydrogen pressure was decreased to 0.1 MPa, conversion of BPE was decreased to 22.7% with arene selectivity increased to 90.8%. Increasing the pressure of hydrogen to 2.0 MPa resulted in improved conversion of BPE, but full ring hydrogenation in the product mixture. This is a clear indication that higher H2 pressures likely lead to an over saturation of activated hydrogen atoms on the catalyst surface, leading to unwanted ring hydrogenated products. Switching from gaseous hydrogen to an H-donor solvent (isopropanol) resulted in 98.8% conversion of BPE with arene selectivity above 95%. Isopropanol was found to be the best H-donor as isopropanol is capable of being both a H-bond donor and an H-bond acceptor. Isopropanol interacts with the catalyst surface as shown in Fig. 12 and helps mediate the hydrogen atom concentration on the surface of the catalyst compared to H2, which leads to more of the desired C–O bond activation and less observed ring hydrogenation.
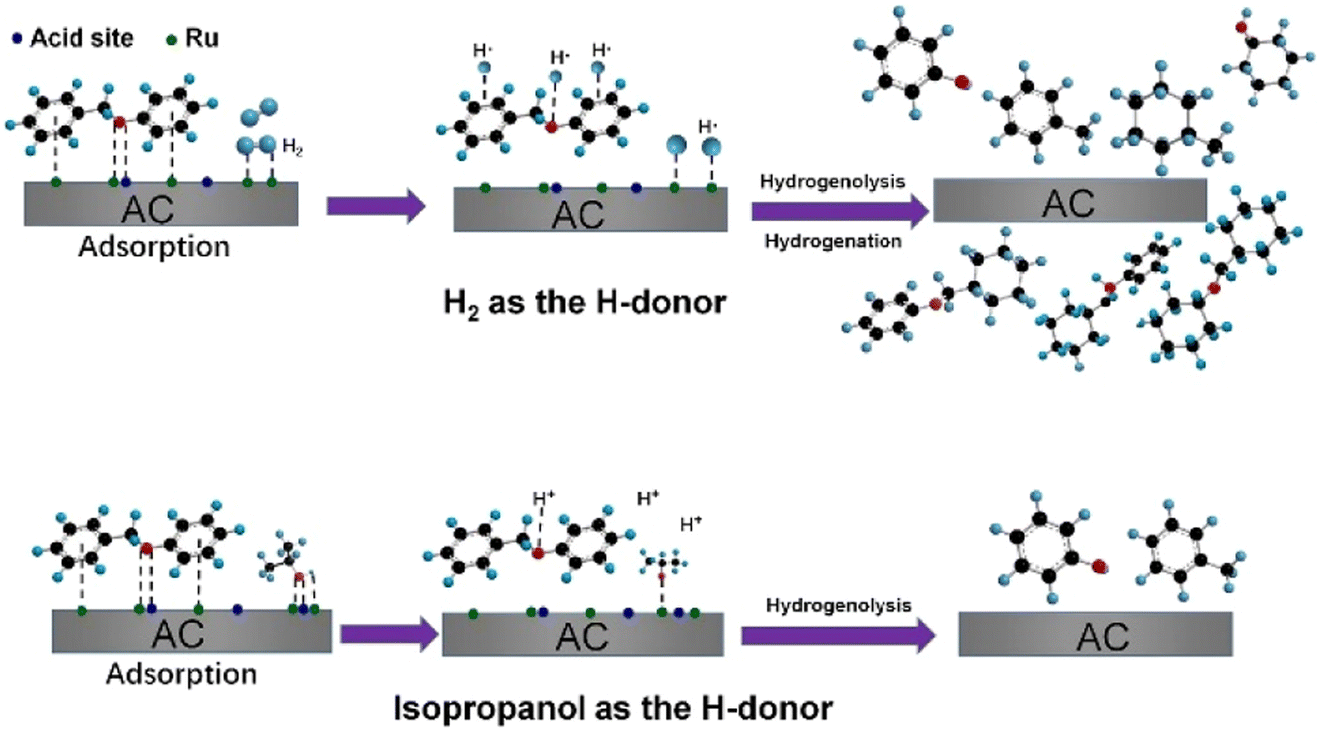 | ||
| Fig. 12 Proposed pathway for catalytic hydrogenolysis of BPE over Ru/AC. Reproduced from ref. 75 with permission from Elsevier, copyright 2021. | ||
A study conducted by Shu et al. sought to investigate the effect of catalyst preparation method on metal particle size and dispersion on the support.72 A polyol reduction method for catalyst synthesis was chosen as it has been used to regulate the morphology of metal nanomaterials including silver nanowires and gold nanorods. Ru/SiO2–ZrO2 catalysts were produced using the polyol reduction method using ethylene glycol and the impregnation method. The Ru loading for the catalysts studies was around 5 wt%. The dispersion of Ru increased across the support surface for the polyol synthesized catalyst (13.3%) compared to that of the impregnation synthesized catalyst (9.6%). The impregnated catalyst also had a larger Ru particle size (6.1 nm) than that of the polyol catalyst (4.1 nm). NH3-TPD revealed that the impregnated catalyst had weak acid sites indicated by two peaks below 240 °C while the polyol catalyst had both weak acid sites and strong acid sites indicated by a peak around 360 °C. The integrated area of the polyol catalyst was also significantly greater than that of the impregnated catalyst indicating that the polyol catalyst possessed many more acid sites. HDO experiments were conducted with guaiacol, phenolic compounds and pyrolysis lignin-oil as substrates. HDO experiments conducted at 240 °C using guaiacol as the substrate catalyzed by both polyol and impregnated Ru/SiO2–ZrO2 catalysts found that both catalysts produced only ring-hydrogenated products, but only the polyol catalyst achieved a product mixture composed of ∼98% deoxygenated products. Deconvoluting these results showed that at elevated temperatures both C–O bond activation and ring hydrogenation were thermodynamically capable and observed. However, the more acidic support and smaller particle size of the polyol synthesized Ru catalysts were much more efficient at performing deoxygenation reactions. While no selectivity towards deoxygenation over ring hydrogenation was observed in this study, the general reactivity trends (smaller particle size, acid/base support effects) were still observed. Further studies at lower reaction temperature may be able to tune the polyol synthesized catalysts toward exclusion deoxygenation selectivity.
Hu et al. studied the effect of mordenite zeolite supports on hydrodeoxygenation using Ru nanoparticle catalysts.76 Layered mordenite zeolites have microporous and mesoporous features and they have high specific surface area which can result in a well dispersed nanoparticle catalyst. Catalysts were synthesized using the deposition–precipitation method. The catalyst with the smallest nanoparticle size (1.68 nm) contained 2.98 wt% Ru and was labeled as Ru3%. XPS data revealed the presence of RuO2, suggesting that not all of the Ru3+ species were reduced to metallic Ru through H2 reduction. While conversion remained high amongst all substrates (diphenyl ether, anisole, and oxybis(methylene)dibenzene), the product mixtures consisted mainly of deoxygenated ring-hydrogenated products. Unfortunately, the authors only reported reactions performed at one H2 pressure (30 atm). As reaction trends reported previously34 and in this review show, lowered H2 pressures may have resulted in less ring hydrogenation products. These results, which use small catalyst particles but still obtain exclusively ring hydrogenated products, indicate that H2 pressure may be a larger factor than catalyst size when targeting selective deoxygenation.
Cao et al. also utilized a Ru catalyst supported on a zeolite.77 The bifunctional Ru/HZSM-5 catalyst was prepared by the incipient wetness impregnation method and studied for the deoxygenation of DPE. Ruthenium (5 wt%) was mostly distributed to the micropores (diameter of 0.6 nm) of the HZSM-5 support. Addition of Ru to the support resulted in a decrease in the support acidity. Strong acid sites disappeared completely upon the addition of Ru. HDO reactions with 5% Ru/HZSM-5 and DPE as the model compound resulted in the complete conversion of DPE to cyclohexane. Variations in the solvent resulted in lower activity and selectivity. Product mixtures included benzene, cyclohexane, cyclohexanol, phenol, and other minor products. When non-polar n-hexane was used as the solvent, full conversion of DPE was observed with complete selectivity toward cyclohexane. Subsequent reactions showed that catalytic activity was retained up to three reaction cycles. Reaction cycles 3 and 4 showed lowered activity and product selectivity. While these zeolites supported catalysts displayed selectivity toward specific products, they did not display selectivity toward the higher value, deoxygenated arene products.
2.3 Palladium catalysts
Hydrodeoxygenation utilizing palladium catalysts has received interest due to the current use of palladium catalysts in the petroleum industry for desulfurization processes.78 Early studies utilizing palladium catalysts resulted in over hydrogenation to cycloalkane products.23,45 More recently, studies have focused on carbon-supported palladium catalysts for selective HDO of lignin model compounds.79–81 These catalysts have been studied at temperatures up to 500 °C and pressures up to 30 bar.79,81 In addition to heterogeneous catalysts, molecular and hybrid palladium catalysts have been studied for selective HDO of benzylic substrates under more moderate conditions compared to the typical heterogeneous catalyst.40,78 These catalysts tend to be more selective than their heterogeneous counterparts, but are less stable at high temperatures and are more difficult to recycle.32,40,78 Here we highlight both heterogeneous and homogeneous palladium catalysts that demonstrated high selectivity during the conversion of lignin model compounds.A study conducted by Alijani et al. sought to investigate the effect of different carbon support properties on the HDO activity of a palladium heterogeneous catalyst.79 Carbon supports were chosen due to their high surface area, thermal stability in reductive environments, and their ability to facilitate nanoparticle deposition on neutral surfaces. This study focused on four carbon supports (Norit, KB, G60, and HHT). These supports vary in physical–chemical properties such as surface area, pore size and type of surface functionalities. Catalysts were synthesized via the sol immobilization method with sodium tetrachloropalladate(II) as the metal source and polyvinyl alcohol (PVA) as the capping agent. Metal loadings of each catalyst were 1 wt% determined by ICP analysis. BET analysis revealed the surface areas of Pd/Norit, Pd/KB, Pd/G60, and Pd/HHT to be 2000 m2 g−1, 1600 m2 g−1, 800 m2 g−1, and 40 m2 g−1, respectively. The pore diameters were found to be 5.5 nm, 2.0 nm, 18.7 nm and 25.4 nm, respectively. Finally, total pore volume was found to be 0.80 cm3 g−1, 1.59 cm3 g−1, 0.75 cm3 g−1, and 1.50 cm3 g−1, respectively. XPS was used to evaluate Pd oxidation state and the presence of oxygen functionalities on the support surface. TEM imaging revealed all catalyst samples to have homogenous dispersal of nanoparticles. Pd/Norit and Pd/KB had mean particle sizes around 2.5 and 2.7 nm, respectively. Pd/G60 and Pd/HHT showed higher mean particle sizes of 3.5 and 3.9, respectively. HDO experiments were conducted under mild conditions at 50 °C and 5 bar H2 with vanillin as the lignin model compound and isopropanol as the solvent. As shown in Fig. 13, catalytic activity was correlated with increasing graphitization order (Fig. 13 top) and decreasing oxygen functionalization (Fig. 13 bottom). The authors posited that higher graphitization order allowed for a stronger interaction with the aromatic ring, resulting in enhanced catalytic activity. Increased oxygen functionalization disrupted this graphitization, resulting in decreased catalytic activity. Each of these catalysts were able to selectively convert vanillin to creosol under mild conditions. Full conversion to creosol was observed with each of the catalysts, however the support had an impact on selectivity at lower conversions. Vanillyl alcohol was the main product of Pd/Norit, Pd/HHT, and Pd/KB with selectivity between 73–82%. For Pd/G60, high amounts of vanillyl isopropyl ether and creosol were produced at 50% conversion (23% and 21%, respectively). In all cases, the mild reaction temperature made complete deoxygenation of the substrate thermodynamically unfeasible. Unfortunately, higher reaction temperatures were not reported. This study does show that solvent choice is important as incorporation of the isopropanol into the product mixture was observed with the vanillyl isopropyl ether product.
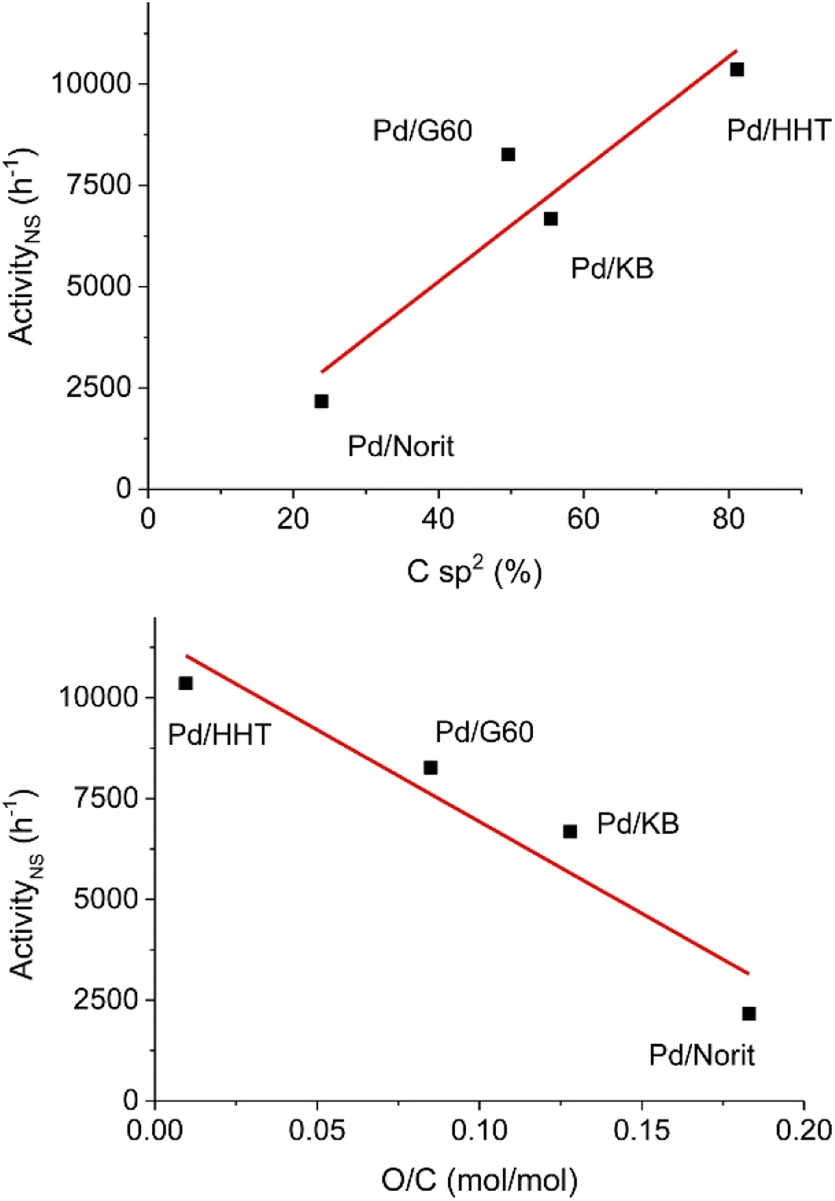 | ||
| Fig. 13 Correlation between catalytic activity and graphitization order (top) and correlation between catalytic activity and oxygen functionalization (bottom). Reproduced from ref. 78 with permission from ACS, copyright 2019. | ||
Zhang et al. conducted a study utilizing an N-doped carbon supported palladium catalyst for the cleavage of Caromatic–Cα and Cα–O bonds in veratryl alcohol.80 N-doped carbon was chosen as the support due to its stability during the preparation of palladium catalysts. N-doped carbon also minimizes over-oxidation of the metal nanoparticles. The Pd/CNx catalyst was synthesized by reduction of a solution of Pd(O2CCH3)2 by sodium borohydride in the presence of the carbon nitride support. TEM images showed an average Pd nanoparticle size of 2.8 nm and BET analysis showed an average pore size of 3.3 nm. The XPS data confirmed the Pd/CNx catalyst to contain 70.5% Pd(0). Catalytic experiments were conducted at 220 °C using veratryl alcohol as a model lignin compound and isopropanol as the solvent and hydrogen donor. Pd/CNx was shown to outperform a variety of previously reported Pd catalysts (Pd/C, Pd/C@TiO2, Fe–Pd/C@TiO2, and Pd/ZSM-5) for the conversion of veratryl alcohol to 1,2-dimethoxybenzene and 3,4-dimethoxytoluene. It should be noted that product distribution did not show a significantly favored product. The notable exception was an experiment conducted with the addition of 0.5 mL of a 1% formic acid solution which resulted in a product mixture comprised of 18.60% 1,2-dimethoxybenzene and 69.23% 3,4-dimethoxytoluene. This was explained by the formation of a formato-palladium intermediate, which blocked vacant coordination sites of Pd resulting in the increased selectivity toward 3,4-dimethoxytoluene. Time-on-stream performance was analyzed, and the results shown in Fig. 14. As shown, the initially formed products of the conversion of veratryl alcohol were the oxidation of the alcohol to form 3,4-dimethoxybenzaldehyde (product c) and 3,4-dimethoxybenzoic acid (product d). With further time on stream, the oxidized products were converted to 3,4-dimethoxytoluene (product a) and 1,2-dimethoxybenzene (product b). These products represent the deoxygenation of the benzylic alcohol moiety without ring hydrogenation. The Csp2-OMe bonds were also left unreacted. The authors posit that the isopropanol solution led to the initial oxidation of the benzylic alcohol, however, reactivity by oxidized PdOx on the surface was not considered.
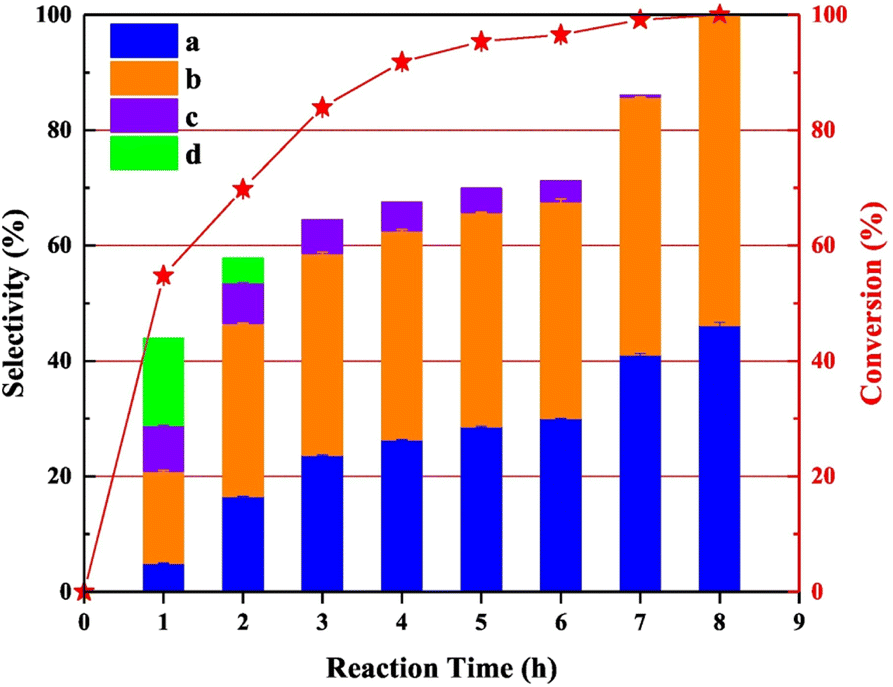 | ||
| Fig. 14 Time-onstream performance of Pd/CNx for the conversion of veratryl alcohol. Reproduced from ref. 79 with permission from Elsevier, copyright 2021. | ||
Another study utilizing an N-doped carbon supported palladium catalyst for the HDO of a lignin model compound was conducted by Liu et al.81 TEM imaging revealed uniform dispersal of the Pd particles across the N-doped carbon support. Palladium present on the surface of the support was shown by XPS to be comprised of mainly metallic Pd. Catalytic experiments were conducted between 300–500 °C using guaiacol as the model lignin compound in the gas phase without solvent. At 400 °C, the Pd/NC catalyst outperformed a similar Pd/C catalyst in regards to product selectivity. Both catalysts resulted in full conversion of the model compound, but the selectivity toward benzene was higher for that of Pd/NC (85%) compared to that of Pd/C (49%) with other products observed being 2-methoxycyclohexan-1-ol, cyclohexanone, and cyclohexene. Fig. 15 shows the time-on-stream (TOS) for Pd/C at 400 °C (white circles) and Pd/NC at various temperatures. Pd/C exhibits high selectivity toward benzene until around 15 min on stream when a stark decrease can be observed, and the reaction begins to favor the formation of phenol. For Pd/NC, at 300 °C and 400 °C, selectivity toward benzene, the completely deoxygenated product, remains high. At 500 °C, selectivity toward benzene drops quickly and phenol selectivity increases for Pd/NC after 5 min. The authors hypothesize that the Pd/C catalysts is unstable at 300 °C and sinters. Upon sintering the conversion of phenol (formed upon removal of methanol after Csp2-OMe bond activation) is no longer observed. In the case of the Pd/NC catalyst, the support helps stabilize the catalyst from sintering and the catalyst thus shows high selectivity to benzene up to 400 °C. At 500 °C sintering of the Pd/NC catalyst deactivates the catalyst to the activation of the phenol bond. However, the bottom of Fig. 15 also shows the lifetime of the catalyst over an extended period (6 h) at 300 °C. As shown, the benzene yield only decreases marginally with no observable increase in phenol production after 6 h indicating prolonged catalyst stability. This report highlights one of the best sets of results for the selective deoxygenation of lignin derived molecules. The catalyst can cleave high-energy Csp2-OMe and phenolic bonds while maintaining aromaticity. These results may be directly linked to the lack of solvent (gas phase reaction with guaiacol and H2/N2 feed only), stabilized catalyst through support interactions, and the use of a flow reactor instead of batch to remove aromatic products before further unwanted reactions can occur.
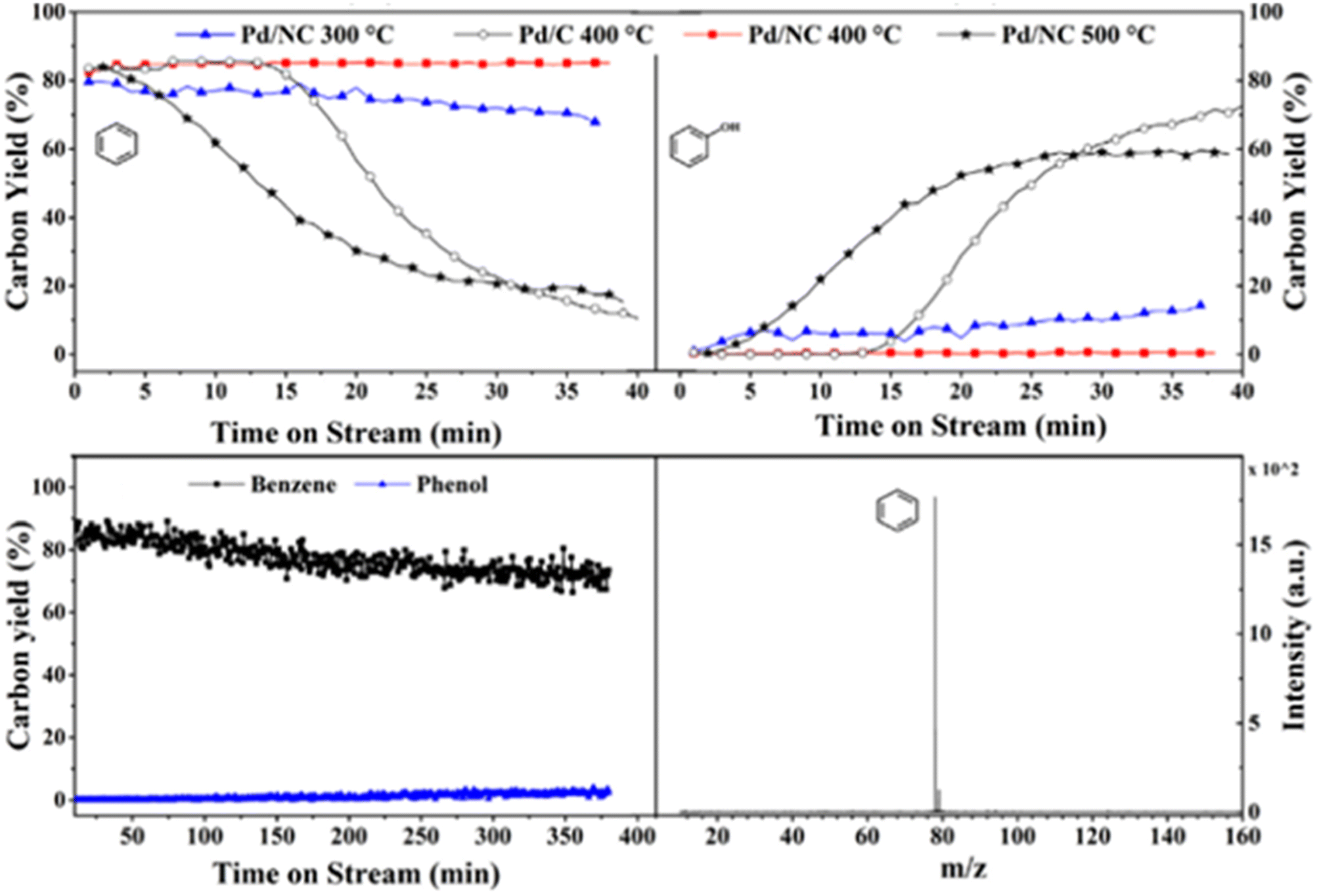 | ||
| Fig. 15 Time-onstream for Pd/C and Pd/NC at 400 °C and 500 °C (top). Catalyst stability for guaiacol HDO over Pd/NC (bottom). Reproduced from ref. 80 with permission from Elsevier, copyright 2021. | ||
Vannucci et al. took a different by approach by utilizing a molecular [Pd(tpy)Cl]Cl catalyst for the HDO of benzylic substrates.40 While molecular catalysts tend to have lower stability at elevated temperatures and are more difficult to recycle than their heterogeneous counterparts, they tend to exhibit a higher product selectivity. A 2,2’:6′,2′′-terpyridine (tpy) ligand was selected, as organometallic complexes utilizing this ligand with Ni and Pd have previously displayed the ability to activate C–O bonds. The catalyst was synthesized by refluxing a solution of 1.1 mmol tpy with 1.1 mmol K2PdCl4 in EtOH for 3 h. Catalytic experiments were conducted in H2 atmosphere using benzyl alcohol, benzophenone, and benzaldehyde as the lignin model compounds with methanol as the solvent. The [Pd(tpy)Cl]Cl catalyst was found to optimally perform at 100 °C and 20 bar H2 with full conversion of benzyl alcohol to toluene. Under identical conditions, the authors found that 2 wt% Pd/SBA-15 heterogeneous catalysts also performed full conversion of benzyl alcohol but with only 56% selectivity toward toluene with the second major product being methyl cyclohexane (23%). Reactions conducted with benzaldehyde and benzophenone resulted in 96% and >99% conversion, respectively. The major products of these reactions were toluene and diphenylmethane, respectively. Attempts to perform reactions at 200 °C resulted in catalyst decomposition. Thus, molecular catalysts with respect to temperature show similar trends to heterogeneous catalysts. Higher temperatures lead to either sintering or catalyst decomposition which leads to less reactivity and selectivity.
Taking this concept of molecular HDO catalysts a step further, DeLucia et al. immobilized a similar molecular catalyst onto a high surface area silica support to improve recyclability and stability.78 A300 silica was chosen as the catalyst support due to the ease of loading organometallic compounds with acid moieties such as carboxylic acids. The catalyst chosen [chloro(2,2’:6′,2′′-terpyridine-carboxylic acid)palladium(II)] was similar to the molecular catalyst previously mentioned, but with an additional carboxylic acid moiety in the 4-position of the central pyridine ring. The catalyst is able to bind to the surface of the silica through hydrogen bonding with the silica Si–OH bonds. The structure of the molecular catalyst and hybrid catalyst are shown in Fig. 16. ICP-MS analysis determined the metal loading of the hybrid catalyst was 2.1 wt% Pd. XRD analysis of the supported catalyst and the unmodified A300 silica revealed no observable peak corresponding to metallic palladium. Catalytic experiments were conducted using benzyl alcohol, benzaldehyde, benzophenone, vanillyl alcohol and vanillin as lignin model compounds. Experiments conducted with benzyl alcohol with different solvents concluded that dodecane was the best solvent for the selective HDO of benzyl alcohol to toluene at 100 °C and 20 bar H2. This was attributed to the nonpolar solvent stabilizing the catalyst binding to the oxide support and the noncoordinating nature of the solvent with Pd. Product monitoring over time showed the direct conversion of benzyl alcohol to toluene without side products being observed (Fig. 17). Product selectivity, however, was not affected by H2 pressure between 5 and 30 bar H2. This result shows that molecular catalysts may break the observed correlation between H2 pressure and product selectivity observed for heterogeneous catalysts. This could be due to molecular catalysts essentially being a “single-site” catalyst and not offering an extended hydrogenated surface for ring hydrogenation to occur. Reactions with benzaldehyde, benzophenone, vanillyl alcohol and vanillin resulted in 88% conversion for benzaldehyde and >99% conversion for the other substrates. The selectivities for these substrates were >99% toluene, 67% diphenylmethane, >99% creosol and >99% creosol, respectively. When reactions were performed under limited conversion (∼20%), the catalyst was recyclable with product selectivities being maintained and post-reaction XRD and XPS analysis supporting the lack of observable Pd decomposition products.
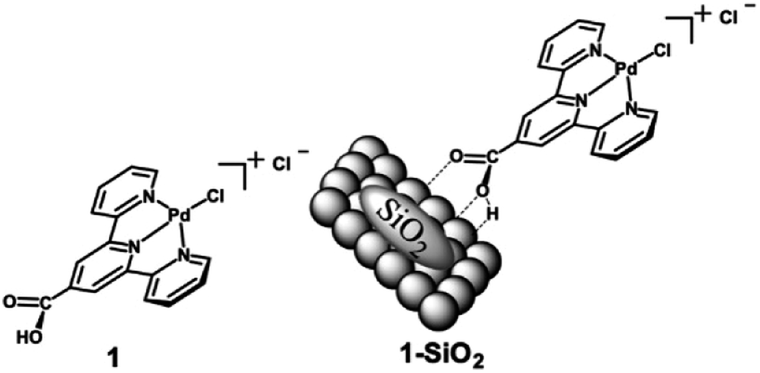 | ||
| Fig. 16 Structure of chloro(2,2’:6′,2′′-terpyridine-4'carboxylic acid)palladium(II) chloride (1) and a graphical representation of the molecular catalyst attached to a SiO2 support (1-SiO2). Figure taken from reference. Reproduced from ref. 17 with permission from ACS, copyright 2019. | ||
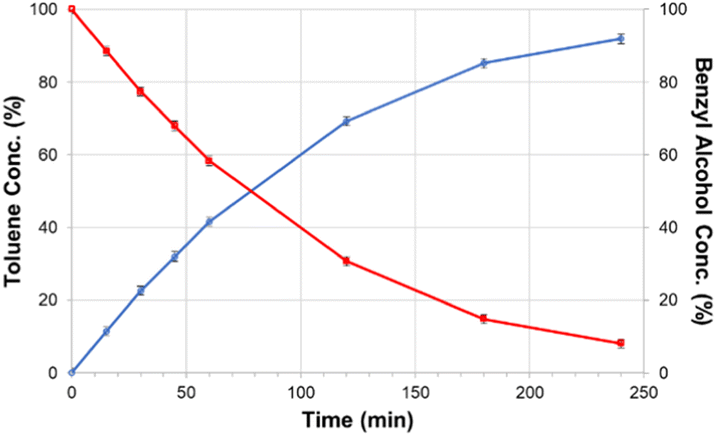 | ||
| Fig. 17 Product formation and substrate consumption curves for the selective catalytic hydrodeoxygenation of benzyl alcohol performed from a solid support immobilized molecular catalysts. Reproduced from ref. 17 with permission from ACS, copyright 2019. | ||
An additional study of a molecular catalyst by Tillou et al. sought to establish a series of experiments to determine the active catalytic species in under various HDO conditions.32 The catalyst was 2,6-bis(4-isopropyl-2-oxazolinyl)pyridine palladium(II) (catalyst 1) shown in Fig. 18. Catalytic experiments were conducted with benzyl alcohol under H2 with methanol as the solvent. At room temperature, 31% conversion of benzyl alcohol to toluene with complete selectivity was observed for a 4 h reaction. The conversion of benzyl alcohol increased with increasing temperature up to 98% at 200 °C. However, in all reactions conducted at ≥100 °C, black solid (catalyst 2) formation was observed. This solid particulate was isolated via filtration and catalytic experiments were conducted using 2 and benzyl alcohol. At temperatures <200 °C, no benzyl alcohol conversion was observed. At 200 °C, full conversion of benzyl alcohol to toluene was observed. From these two data sets, it was concluded that the catalytic activity observed from the molecular catalyst above 100 °C was most likely from 2. Catalyst 2 was characterized by XPS analysis and found that it was composed of Pd, C, O and N in addition to P and F from the counter anions. The binding energy of the Pd 3d was determined to be 335.25 eV corresponding to metallic Pd. XRD analysis showed that palladium forms a crystalline material with particle size between 3.5 and 5.5 nm. The recyclability of 2 was then investigated and found that by the 3rd cycle, 2 becomes inactive. Post reaction solutions of 1 and 2 were investigated for catalytically active leeched palladium species. It was found that post reaction solutions of 1 contained no catalytically active palladium species, but post reaction solutions of 2 appeared to be catalytically active. This coupled with the apparent deactivation of 2 led to the conclusion that palladium from 2 leeches into the reaction solution and remains catalytically active as 3. This study showed that the use of molecular catalysts for HDO reactions must be careful in assigning the active catalytic species, and that molecular catalysts can decompose under what would be considered mild conditions for many heterogeneous nanoparticle catalysts on supports. Furthermore, once molecular catalysts decompose, the reaction selectivity can change, analogous to changing particle size in heterogeneous catalysts.
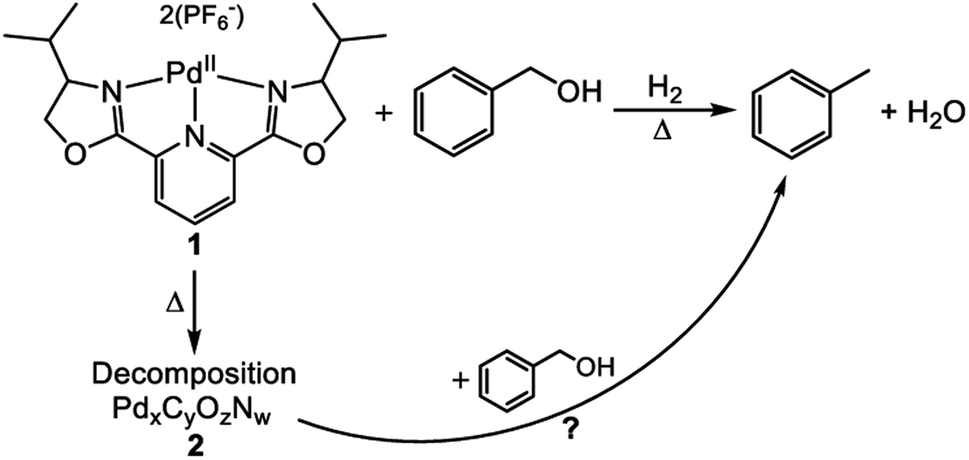 | ||
| Fig. 18 Reaction scheme showing the selective hydrodeoxygenation (DO) of benzyl alcohol by a molecular catalyst. Decomposition of the molecular catalyst could also lead to a catalytically active species. Reproduced from ref. 32 with permission from Elsevier, copyright 2021. | ||
2.4 Bimetallic catalysts
While traditional monometallic catalysts have remained a major focus for potential hydrodeoxygenation catalysts, bimetallic catalysts have received increasing attention due to synergistic effects.82–84 Traditionally, these bimetallic catalysts were used for the hydrogenation of LMCs with mixed results.85 Bimetallic catalysts utilized several different metals including Ni, Rh, Mo, Pt, Pd, and Co.85 These catalysts tend to be more active and more stable than their monometallic counterparts.82 Bimetallic catalysts have gained interest in the cleavage of the β-O-4, 4-O-5, and α-O-4 bonds. Bimetallic catalysts including Ni and another metal such as Pd or Ru have been especially effective for the selective cleavage of these bonds to aromatic monomers.82–84 These catalysts tend to be less selective toward arenes compared to those nickel bimetallic catalysts.82 Here we highlight several bimetallic catalysts that demonstrated high selectivity during the conversion of lignin biomass.Gao et al. studied a AuPd/CeO2 catalyst for the hydrogenolysis of aryl ether C–O bonds.82 The AuPd/CeO2 was synthesized by sol-immobilization method. Metal content was found to be 1.31 wt% Au and 0.72 wt% Pd by ICP-MS analysis. The average AuPd/CeO2 particle size was 5 nm. XPS data revealed the addition of Pd induced Au 4f peaks toward low binding energy while Pd 3d peaks shifted to high binding energy. The surface ratio of Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pd was estimated to be 1.2:1.0 by XPS. Catalytic experiments were conducted with benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4), and diphenyl ether (4-O-5) as lignin model dimers. Reactions were conducted in aqueous solution with formic acid as the hydrogen donor. AuPd/CeO2 was able to completely convert BPE to phenol and toluene with less than 10% ring hydrogenated products. The conversion and selectivity of the bimetallic catalysis was greatly improved from the conversion using either monometallic Au/CeO2 or Pd/CeO2. The conversion of 2-phenylethyl phenyl ether was 40.7% in 1 h and increased to 98.2% in 4 h. The product mixture was selective for ethylbenzene and phenol. Conversion of the DPE substrate was lower in comparison (24% after 24 h at 150 °C). Conversion, however, increased to 40.4% at 160 °C and finally 100% at 180 °C. The products of this reaction were predominantly phenol and benzene. Recyclability tests were conducted using BPE and DPE as shown in Fig. 19. For reactions with BPE, conversion seemingly did not decline across five cycles with product selectivity remaining consistent across each cycle. For DPE, conversion and product selectivity remained consistent across 5 cycles. The recycling tests show the catalyst was stable under mild conditions and that product selectivity could be maintained by keeping the substrate conversion under 50%. These results also highlight the benefit of bimetallic catalysts where one metal (Pd) is used to activate the hydrogen gas, and the second metal (Au) is utilized in the C–O bond cleavage.
:Pd was estimated to be 1.2:1.0 by XPS. Catalytic experiments were conducted with benzyl phenyl ether (α-O-4), 2-phenylethyl phenyl ether (β-O-4), and diphenyl ether (4-O-5) as lignin model dimers. Reactions were conducted in aqueous solution with formic acid as the hydrogen donor. AuPd/CeO2 was able to completely convert BPE to phenol and toluene with less than 10% ring hydrogenated products. The conversion and selectivity of the bimetallic catalysis was greatly improved from the conversion using either monometallic Au/CeO2 or Pd/CeO2. The conversion of 2-phenylethyl phenyl ether was 40.7% in 1 h and increased to 98.2% in 4 h. The product mixture was selective for ethylbenzene and phenol. Conversion of the DPE substrate was lower in comparison (24% after 24 h at 150 °C). Conversion, however, increased to 40.4% at 160 °C and finally 100% at 180 °C. The products of this reaction were predominantly phenol and benzene. Recyclability tests were conducted using BPE and DPE as shown in Fig. 19. For reactions with BPE, conversion seemingly did not decline across five cycles with product selectivity remaining consistent across each cycle. For DPE, conversion and product selectivity remained consistent across 5 cycles. The recycling tests show the catalyst was stable under mild conditions and that product selectivity could be maintained by keeping the substrate conversion under 50%. These results also highlight the benefit of bimetallic catalysts where one metal (Pd) is used to activate the hydrogen gas, and the second metal (Au) is utilized in the C–O bond cleavage.
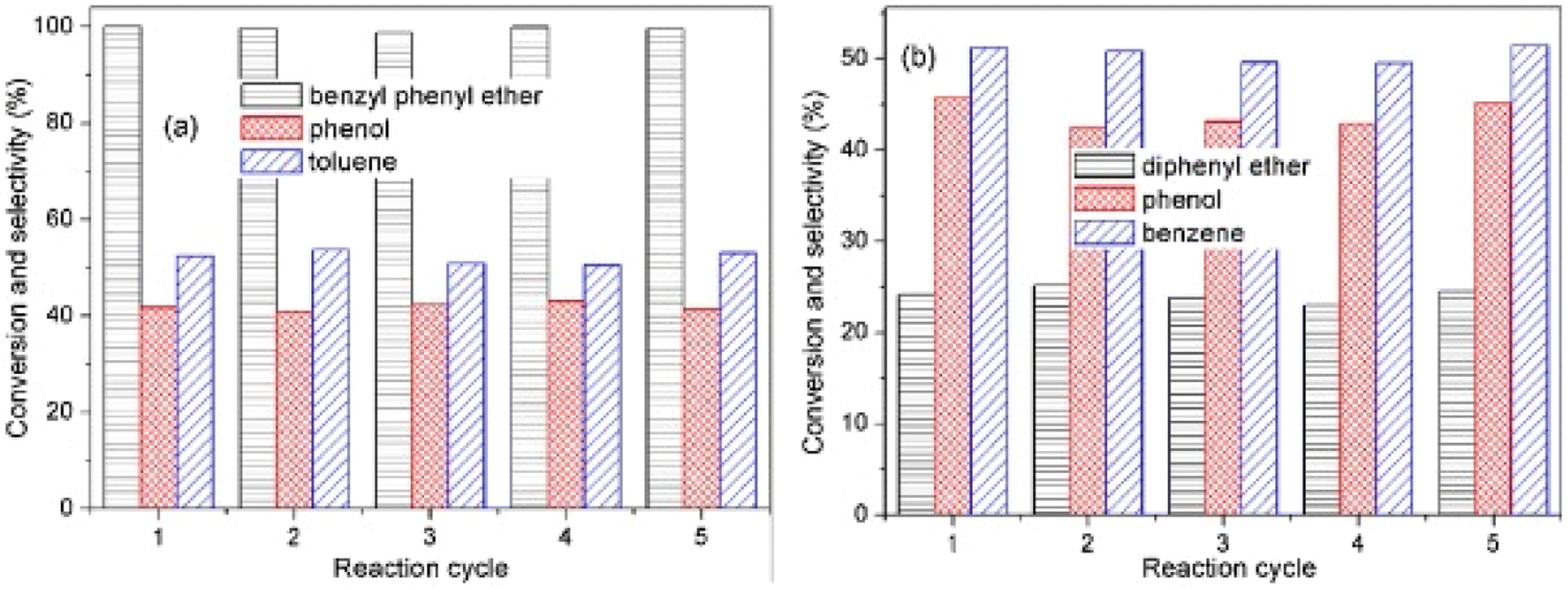 | ||
| Fig. 19 (a) Reusability tests for hydrogenolysis of benzyl phenyl ether using Au1Pd1/CeO2. (b) Reusability tests for hydrogenolysis of diphenyl ether with Au1Pd1/CeO2. Reproduced from ref. 81 with permission from Elsevier, copyright 2021. | ||
A study by Zhao et al. focused on the deoxygenation of DPE by a bimetallic Pd–Ni/HZSM-5 catalyst.83 Nickel was chosen as it has shown remarkable activity as a bimetallic catalyst toward the activation of the β-O-4 bond while Pd is efficient at generating active hydrogen atoms. HZSM-5 was chosen due to the presence of acid sites on the support. The average particle size was found by TEM to be 17 nm. The metal loading for Pd and Ni was 0.88 wt% and 9.23 wt%, respectively. The strength of the acid sites of the support were analyzed by NH3-TPD. The acid sites consisted of weak and moderate strength sites. The chosen substrate was diphenyl ether (DPE) as the model 4-O-5 compound. Reactions were conducted with n-hexane at 220 °C under 20 bar H2. After 2 h, DPE was fully hydrogenated to cyclohexane. This was likely due to the high H2 pressure as has been discussed throughout this review. Decreasing the hydrogen pressure to 1 bar H2 resulted in a reduction in the DPE conversion to 28.9%. Although the conversion saw a decline, the arene selectivity increased with a product mixture composed of 65% benzene, 15% phenol, and 18% cyclohexane. Although arene selectivity remained quite low, the efficiency for the conversion DPE to cyclohexane was higher than that of other Ni bimetallic catalysts. However, a recyclability study conducted on the catalyst showed that by the 4th cycle a large decline (84%) of substrate conversion was observed indicating a lack of catalyst stability under reaction conditions. This decrease in activity was explained through nanoparticle agglomeration and catalyst coking. Coking reactions can be common during ring hydrogenation86 and this highlights another reason to target C–O bond activation over ring hydrogenation reactions.
A nickel–ruthenium catalyst was studied by Zhu et al. for the hydrogenolysis of BPE to aromatic monomers.84 Activated carbon (AC) was chosen as the catalyst support due to its high surface area, porosity, electron conductivity, and relative chemical inertness. The Ni–Ru/AC catalyst was synthesized by incipient wetness impregnation. XPS analysis revealed the BE of Ru 3d5/2 increased by 0.2 eV and the BE of Ni decreased. This electron transfer from Ru to Ni caused an increase of interaction between Ni and Ru. TEM and TEM-DES found the particle size of bulk particles to range between 8–14 nm and large particles to range between 20–22 nm. The average particle size was 10 nm. SEM-EDX analysis revealed that the Ni and Ru particles were evenly and separately distributed across the surface of the AC support. Catalytic experiments were conducted at 120 °C, under 0.5 MPa H2 for 2 h with BPE as an analogue of the α-O-4 bond in lignin. The 10% Ni–1% Ru/AC catalyst was able to fully convert BPE selectively to toluene (96.8%) and phenol (90.3%) with minimal side products (benzene 3.4%, methylcyclohexane 2.7%, and cyclohexanol 5.5%). This is greatly improved from 10% Ni/AC and 1% Ru/AC on their own. Increasing pressure of H2 from 0.2 MPa to 1.1 MPa resulted in decreased selectivity toward aromatic products, furthering the hypothesis that increased hydrogen pressures are detrimental for preventing ring hydrogenation. Increasing reaction temperature from 80 °C to 160 °C resulted in increased conversion of BPE and increased selectivity toward arenes up to 120 °C where full conversion of BPE occurred. Past 120 °C, arene selectivity no longer increased. Fig. 20 compares the recyclability of 10% Ni–1% Ru/AC with that of 10% Ni/AC. As shown, 10% Ni–1% Ru/AC is less susceptible to deactivation compared to 10% Ni/AC. Investigating this further, TEM images were obtained of the pre-reaction catalyst and spent catalyst. Minimal agglomeration of the bimetallic catalyst was observed compared to the monometallic catalysts, which helps explain the greater reusability of the bimetallic catalyst. Catalytic experiments were conducted using other lignin model compounds. Chosen compounds included DPE as a model of the 4-O-5 bond and phenethyl phenyl ether (PPE) as a model of the β-O-4 bond. The conversion of DPE using 10% Ni–1% Ru/AC was 86% with the product mixture containing benzene (50%), phenol (17%), and cyclohexanol (51%). This is an improvement from 10% Ni/AC which has a low (15%) conversion of DPE to benzene (7%), phenol (trace), and cyclohexanol (7.2%). PPE was fully converted to toluene (98%) using the bimetallic catalyst compared to the much lower 20% conversion from 10% Ni/AC. The product selectivity was also improved compared to that of 1% Ru/AC which had a 78% conversion of PPE to benzene (43%), phenol (10%), and cyclohexanol (55%). This shows that the bimetallic catalyst greatly benefits from the synergistic effects of combining hydrogenation metals with metals with high oxophilicity.
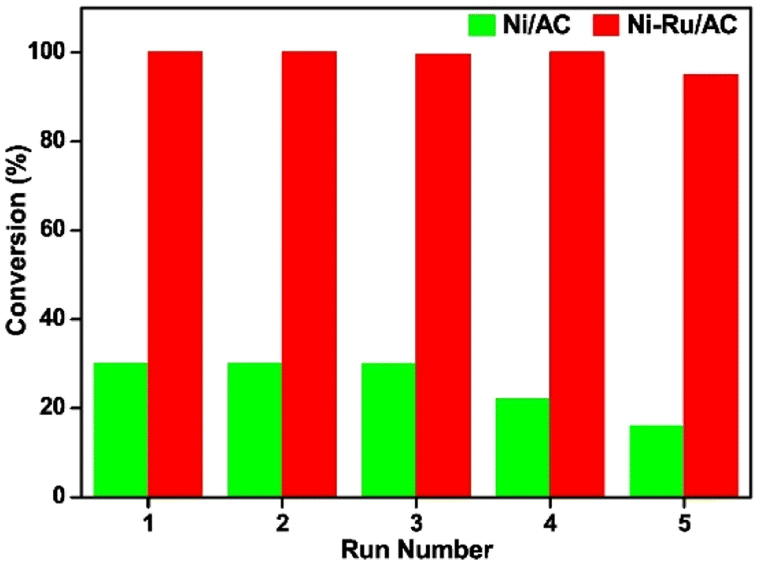 | ||
| Fig. 20 Reusability of 10% Ni/AC (green) and 10% Ni–1% Ru/AC in BPE conversion (red). Reproduced from ref. 83 with permission from Elsevier, copyright 2020. | ||
2.5 Reductive catalytic fractionation
While the focus of this review is the selective HDO of lignin derived compounds, it is important to consider the selectivity of depolymerization methods to produce lignin derived aromatic compounds. For over five decades, there have been reports on several methods used in fractionation of lignocellulose with more attention on cellulose fraction than lignin fraction.3,87 In fact, lignin components are disposed of at a low cost, mainly for combustion and in the manufacturing of wood finishing materials. The earliest fractionation methods: pyrolysis, hydrocracking, hydrogenolysis, oxidation, and hydrolysis are mainly thermo-catalytic non selective processes.88 The non-selectivity of these earliest strategies prevented robust utilization of lignin biofuels and useful chemicals.89,90 The growing interest in biofuels for greener energy coupled with need for increased generation of other platform chemicals of phenolic functionalities has led to recent development in biomass fractionation capturing lignin components as the main target in the fractionation of lignocellulosic biomass.10 The challenges associated with earliest developed fractionation methods is the irreversible condensation of lignin during its isolation.10,90 To tackle these challenges, reductive catalytic fractionation (RCF) of biomass was more recently introduced.91,92 Reductive catalytic fractionation utilizes the principle of “lignin first” strategy which aims at selectively cleaving the lignin β-O-4 ether bond to depolymerize lignin and then preventing the condensation of the broken bonds by reduction of the depolymerized chemical structures.88,91 In this strategy, more attention is focused on the lignin extraction and its depolymerization to monomers and dimers before cellulose valorization. Mechanistically, RCF is carried out in two main steps: (1) solvolytic extraction of lignin and (2) lignin depolymerization coupled with the stabilization of the phenolic monomers through reduction routes. The solid residue obtained in this process is the cellulose and hemicellulose components, making RCF an efficient pathway through which complete extraction of biomass components can be achieved.91 The ability to stabilize the reactive phenolic monomers is its major merit over other lignocellulose fractionation processes.8The effectiveness of the RCF process can be accessed by taken into account the extent of delignification, removal of hemicellulose and retention of cellulose component.93 Solvents used and the catalyst applied can affect lignin-first delignification efficiency (LFDE). The Sels group has investigated the effect of solvents used on these lignin-first efficiency parameters and found that out that methanol and ethylene glycol performed the best.3 In addition, both basic and acidic additives have been reported to create changes in the delignification process.10,94 For example, additions of phosphoric acid led to increased monomer yield when compared to no additive. On the other hand, the NaOH additive has been reported to cause repolymerization and loss of the carbohydrate fractions. Reductive catalytic fractionation has also been studied using many plant materials classified based on the nature of their identifiable biomass content. Three groups of biomass by which these plant materials fall in are hardwood, softwood, and herbaceous.3 Based on monomer yield, hardwood has proven to be more promising. These feedstocks when passed through RCF process selectively yield different phenol derivatives; for instance on cracking woody biomass, ethyl-, propanol-, methoxypropyl-, propyl- and propenyl substituted derivatives are generated.95,96 Moreover, these derivatives solely depend on the abundance of guaicyl (G), syringyl (S) and p-hydroxyphenyl (H) monomer available in the lignin oil. When herbaceous plants are used as feedstock during RCF process, carboxylic acids or esters of methoxyphenols are generated based on the solvent used.3,91
Tungstate-zirconia (WZr) support has found unique application in RCF due to its large Brønsted acid site which enhances the dehydration step in bio-oil upgrading.97 Mechanistically, oxygen removal in deoxygenation processes and conversion of deoxygenated molecules to alkanes is facilitated by the metal-tungstate-zirconia bifunctionality.97 Its effectiveness in RCF and ability to prevent coke formation invariably promotes the catalyst reusability.98,99 Large coke formation leads to deactivation of heterogenous catalyst used in bio-oil refining.8 The bifunctionality of metal–WZr in the RCF process is seen in the synergistic activation of the polymer by WZr and hydrogenation of the reactive enolates by the hydrogen carrying metal. Ha et al. utilized this unique property of WZr in combination with hydrogen adsorption feature of Ni, Co, Pd and Ru in the reductive catalytic fractionation of a lignocellulose biomass feedstock, Mongolian oak (MO).100 Performance of RCF is rated using monomer yields, selectivity, and catalyst reusability.90 As highlighted by the group, the lignin oil is abundant in guaicyl (G) and syringyl (S) monomers with less p-hydroxyphenyl or holocellulose (H) units (Fig. 26). In most reported RCF processes, the liquid fraction is mostly G and S units as most cellulosic fractions are found in the solid residue.91,92,94,101,102 WZr support showed some level of activity at the extraction level but at the depolymerization stage it could only cause cracking of holocellulose and not guaiacyl or syringyl. Introduction of a catalytic metal to the WZr support increased the yield of all lignin monomer and dimer at temperatures above 150 °C (Fig. 21). Among the studied metals, Ni, Pd and Ru showed increased monomer yields compared to the support alone, but Ru and Ni gave the best monomer yields of 18.6 and 16.8%. An insight into the synergistic activities of the metal and WZr using CO adsorption studies revealed large metal dispersion for the Ru and Ni deposited on WZr. The higher activities of these Ni and Ru catalysts were determined as a function of their acidity which leads to more active sites. The group followed this using NaOH titration and Ru proved to have more active sites than the rest while the introduction of Co to WZr decreased the acidity. The monomers of MO lignin oil extracted in this work contained mainly propyl- and allyl-branched guaiacols and syringols but the distribution of these components in the cracked oil fractions is affected by the catalyst and the reaction conditions applied. Co/WZr, Ni/WZr and Ru/WZr caused the cleavage of β-O-4 bond in the lignin monomer and gave propyl-substituted guaiacols and syringols. There has been several reports on the use of Ru and Pd catalyst for hydrogenation and hydrodeoxygenation of lignin molecules.57,103–106 The work of Shinyoung et al. corroborated this finding, the Ru and Pd are better hydrogenation catalysts compared to Ni and Co. Evidenced by the Ru and Pd performing complete hydrogenation of allyl groups during lignin depolymerization, where allyl groups were observed in the product mixtures when Co and Ni catalysts were used.
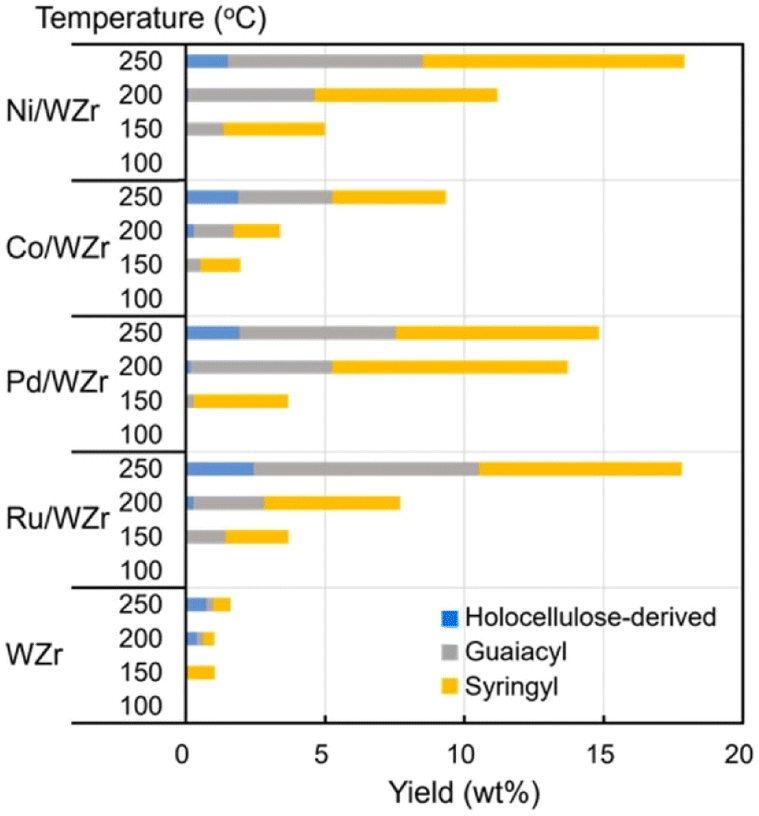 | ||
| Fig. 21 Catalytic performance of different metals on WZr support on the Mongolian oak showing product distribution from GC-FID. Reproduced from ref. 96 with permission from ACS, copyright 2016. | ||
The more deoxygenated a lignin derived fuel, the more efficient the use in bioenergy generation. This is the origin of recent studies on the selective hydrodeoxygenation of these lignin derived molecules.16,40,78 Herbaceous biomass has been found to contain less lignin monomers compared to woody biomass.91,95,107 However, there are great availabilities of herbaceous biomass due to increased agricultural waste emanating from multiple season harvest of most herbaceous crops.108,109 Ebikade et al. have explored the RCF of herbaceous biomass into useful biofuels and platform chemicals.110 The preliminary kinetic studies by the group investigated the possibilities of depolymerization in the absence of catalyst and feed hydrogen. Looking at the effect of solvent in Fig. 22, alcohol solvents with the addition of water exhibited greater depolymerization than nonaqueous solvents. The researchers showed that larger yields of saturated monomers compared to unsaturated monomers can be obtained upon addition of a Ru catalyst.110 It has been reported that selective hydrogenation of the lignin monomers' side-chain CC double bonds will afford a non-repolymerization process while preserving the benzene ring;111 which will yield higher energy value products and chemicals of greater utility than the ring hydrogenated derivatives. From Fig. 22, higher conversion of the unsaturated lignin oils to saturated lignin oils was observed by the authors upon pressurizing their system with molecular hydrogen. The products of RCF of herbaceous biomass showed high selectivity towards alkyl phenols and identified products are guaiacols, syringols and hydroxyphenols, though in lesser yields than reported values in woody biomass.91 The authors also investigated the effects of this ash content on Ru/alumina pellets and Ru/C catalysts used in the reductive catalytic fractionation of several forms of herbaceous biomass.110 It was found by the group that the ash content negatively affected the lignin oil yield but not selectivity of the lignols. Future works call on catalyst design that will be able to handle this challenge.
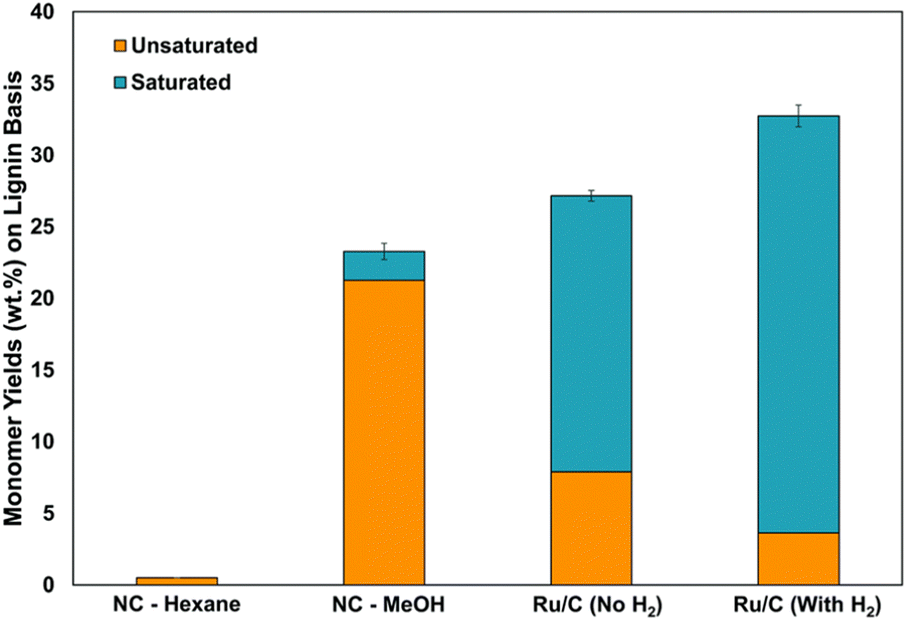 | ||
| Fig. 22 Effects of pressurized hydrogen on monomer yields in reductive catalytic fractionation of herbaceous biomass. Reproduced from ref. 109 with permission from ACS, copyright 2018. | ||
One of the most considered concepts in sustainable energy advancement is the techno-economic feasibility of the process being studied.112 Efficient use of the feedstock is a measure of this techno-economic process, and this is mainly viewed through the level of theoretical maximum yield of lignin-derived phenolic monomers (LDPMs).113 Majority of the RCF processes involving Ru, Pd, Pt based catalysts uses 5 wt% loading of these catalysts despite their high cost.91,102,114 Synergistic activity of low-cost Zn and high cost Pd metal can be a way of going for reduction in cost of using 100% noble metal.92 For instance, in RCF of poplar ZnPd/C with 5 wt% loading resulted in 54% monomer yield which is more than what has been obtained in 5 wt% Pd/C (49% monomer yield).92 This is a sign that cutting the cost of catalyst or replacing costly noble metals does not have to translate to lower monomer yield. Another approach involving complete avoidance of the noble metals by using late transition metal Ni/C catalyst on poplar wood, which gave a yield of 17.2% monomer.115 Modifications of the Ni based RCF catalysts and reaction conditions have been found to have afforded high yields of phenolic monomers.101,116 In order to explore a more economical and effective approach towards RCF, Park et al. have proposed a design with noble metal loadings as low as 0.25 wt%.113 Here the design involves the stabilization of the Pd single-atom on nitrogen-doped graphene (Pd/CNx). This is expected to afford the Pd a more activity than carbon support which is the popularly used support. The authors reported more than 50% yield of the lignin derived phenolic monomers upon using the new catalyst on birchwood and less than 50% yield was recorded on oak wood, pine wood and miscanthus substrate.113 The new modified Pd catalyst, when compared to other modifications involving additives such as 5 wt% Pd/C + YbIII-triflate (55% monomer yield), did not performed better on birchwood.88 However, in the area of cost and less loading of the noble metal, it has shown to be a better techno-economic approach than other Pd/C modifications involving higher loadings. A similar 0.25 wt% Pd loading on activated carbon support gave less than 35% monomer yield on birchwood substrate.113 The excellent performance of the Pd025/CNx in this case can be linked to the CNx support. Fig. 23 shows behaviors of different catalyst loadings on the RCF of birchwood in which increases in catalyst loading did not translate to increases in monomer yield and selectivity. Size of the catalyst depends on the preparation procedure and from the XRD plots shown in Fig. 24. The Pdx/CNx catalysts were prepared in single atom scale with the Pd peaks not being visible due to the size of the atoms/clusters being lower than the diffraction limit. It is not certain what should be responsible for lower performance of Pd1 and Pd0.5 than Pd0.25, although the authors hypothesized that this is connected to the greater number of uncoordinated Pd in Pd1 and Pd0.5 seeing that all the Pd precursors were exposed to 1 g of CNx each. From the selectivity of the phenolic monomers, the more exposed Pd0.25 with higher coordinated Pd atoms showed more generation of the highly hydrodeoxygenated species, propyl syringol and propyl guaiacol. This is expected to be due to high exposure to hydrogen, hence, more adsorbed hydrogen for the hydrogenation of the propenyl syringol or guaiacol was available.
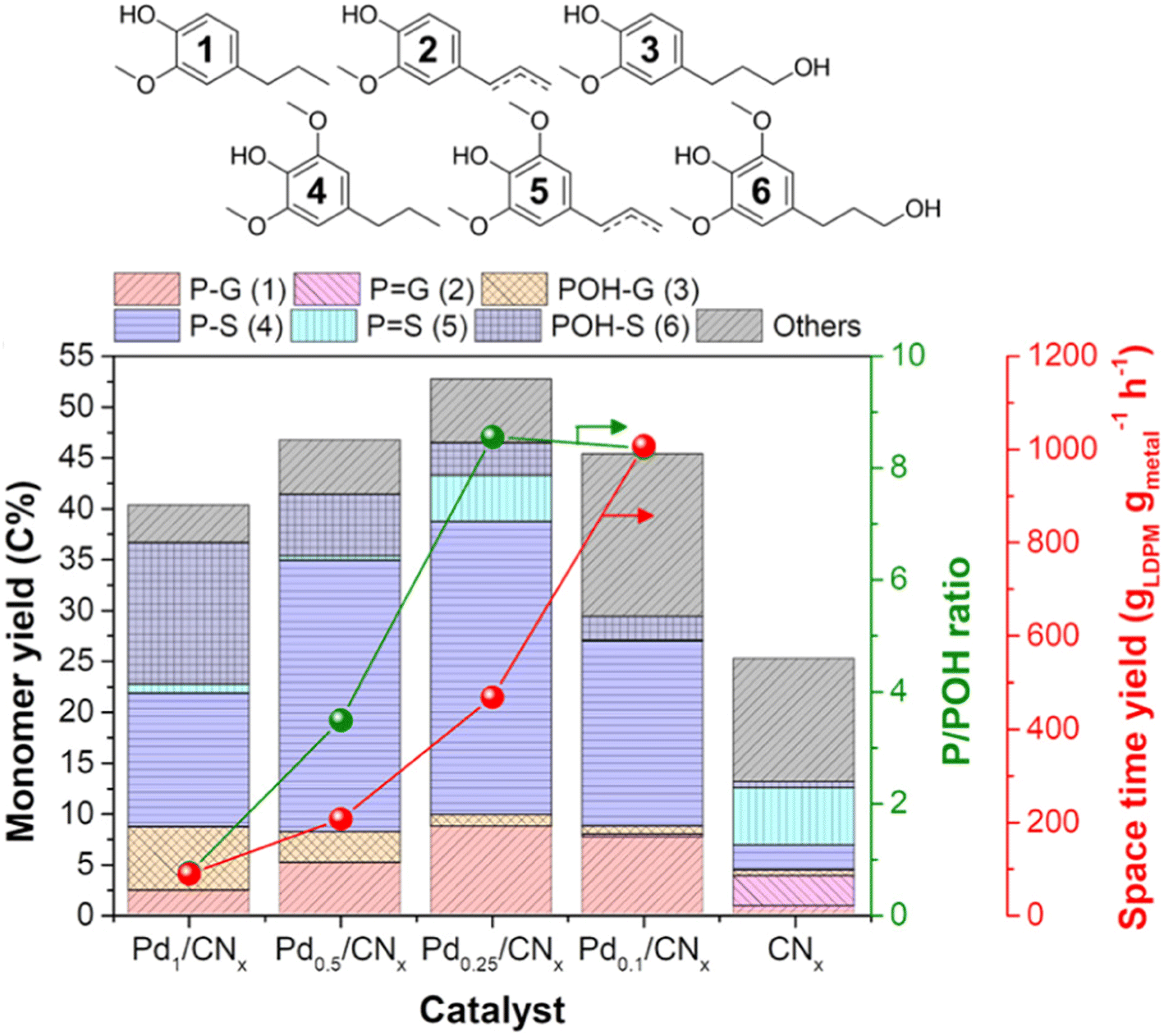 | ||
| Fig. 23 Selectivity and yield of phenolic monomers of Birch Wood derived lignin. Reproduced from ref. 112 with permission from American Association for the Advancement of Science, copyright 2020. | ||
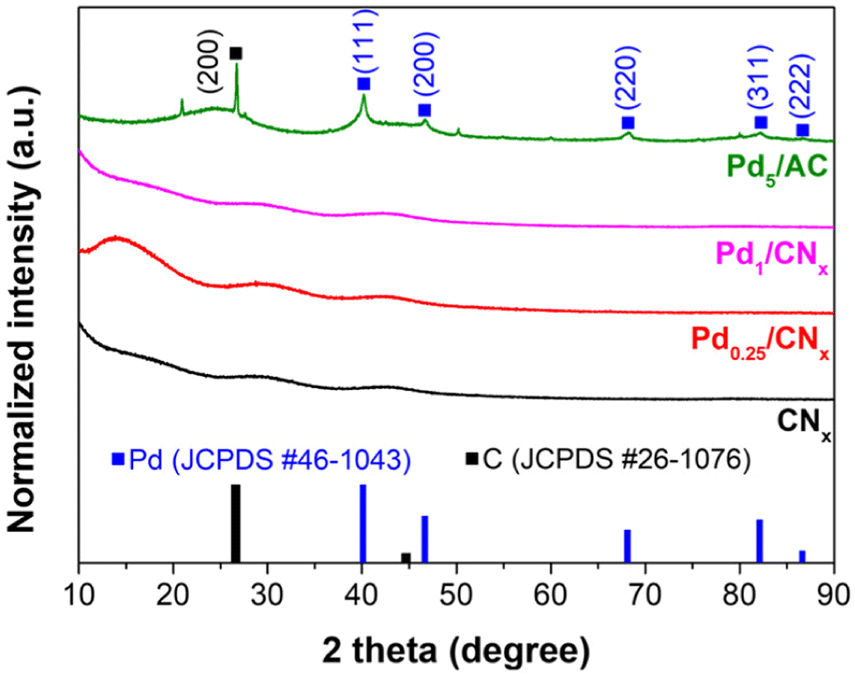 | ||
| Fig. 24 XRD patterns of different Pd ratios on activated carbon and N-doped carbon supports. Reproduced from ref. 112 with permission from American Association for the Advancement of Science, copyright 2020. | ||
The design of structurally controlled metal catalysts is one of the recent approaches towards synthesizing catalysts with atomically dispersed or clustered active sites.117,118 This has led to more attention being paid to the catalyst synthetic procedure that could accommodate certain features for envisaged activities of the prepared catalysts. The advancement into shape, size, and electronic properties regulations in metal nanoparticles is due to the less activity encountered from the traditional atomically smooth large nanoparticles and high cost encountered due to more loading of less active catalysts.119 Some works have reported that Ru can exercise maximum turnover frequency at particle size up to 4 nm, while larger particle sizes will lead to poor performance.120 Lack of the contributions of the support, atom arrangement, or effects of the Ru precursors used in the catalyst synthesis in the account of the activity have made these claims not substantial enough to base the activities of catalysts on particle size; however, this concept has been utilized in some synthetic processes such as ammonia decomposition and methanation.119,121 More recent works on reductive catalytic fractionation of lignocellulosic biomass and hydrodeoxygenation of lignin monomers using heterogenous approach have been fashioned to utilize particle size and properties of the catalyst support in activity and selectivity control. Naseeb et al. combined these two features in their design of metal–acid nanoparticle-supported ultrafine Ru nanoclusters for the purpose of reductive catalytic fractionation of lignocellulose biomass of pine wood.38 Using scanning transmission electron microscopy (STEM) gives proper visualization of the atom's arrangement and dispersion pattern. A clear understanding of this concept can be seen in the reported Ru nanocluster shown in Fig. 25.38 From the high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM – Fig. 25d) the clear view of what appeared as lumps of honeycombs in SEM (a) and TEM (b) is observed and the Ru atoms are well seen as they appeared on the support.
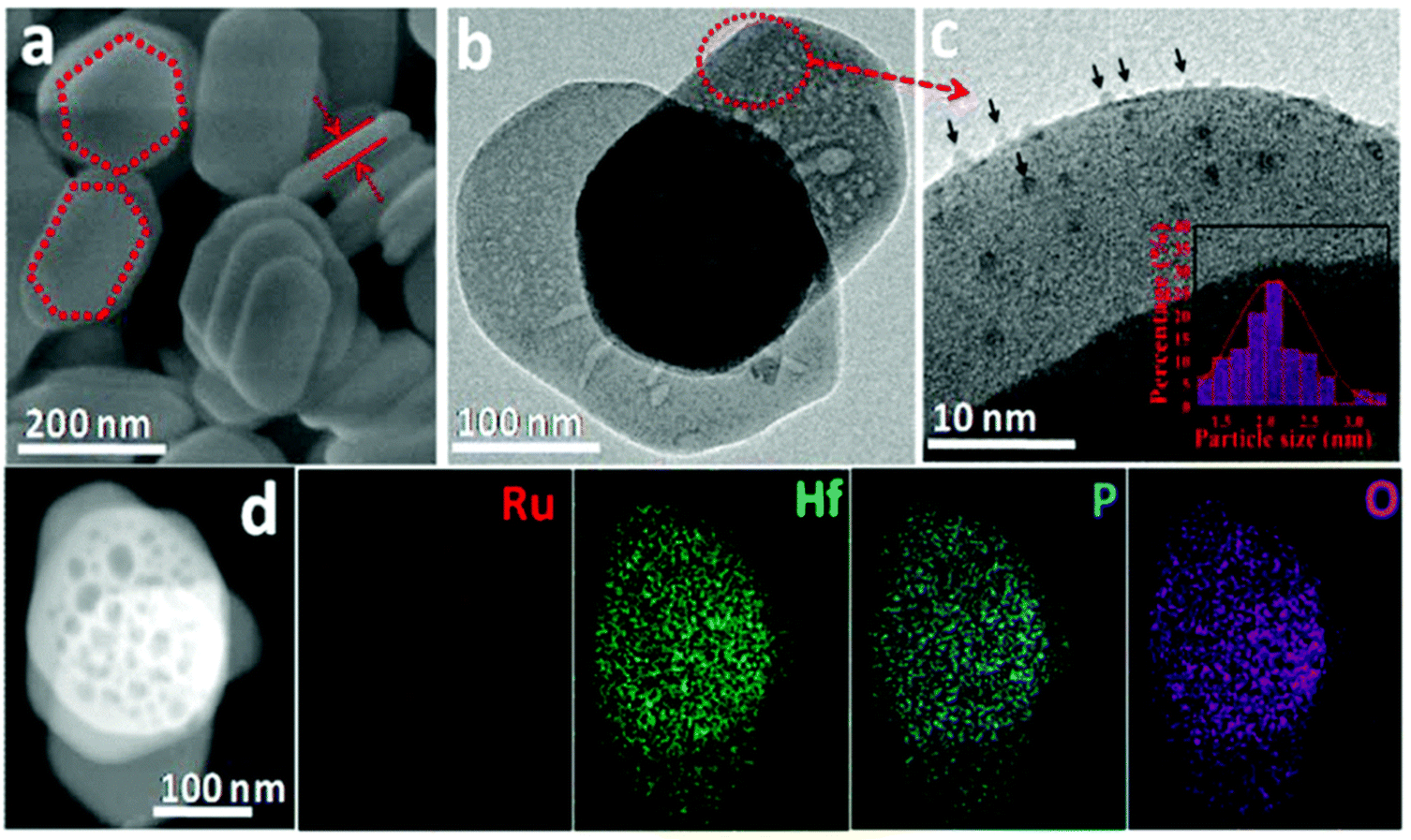 | ||
| Fig. 25 Solid state imaging ((a): SEM; (b and c): TEM; (d): HAADF-STEM) and elemental mapping of active Ru/α-HfP catalyst. Reproduced from ref. 38 with permission from RSC, copyright 2019. | ||
The authors evaluated the activities of the Ru nanoclusters using pine wood biomass and technical lignin. The question on whether the size, shape and electronic property of the catalyst were responsible for the recorded performance (yield and selectivity) of the catalyst is answered in several ways. The catalyst support was able to degrade ether and ester lignocellulose linkages but could not conduct depolymerization. In reductive catalytic fractionation, depolymerization occurs in the presence of reductive hydrogen which is activated by the metal catalyst. The presence of Ru influenced the depolymerization of the pine lignin into lignin monomers comprising mainly propyl guaiacol in higher yield and selectivity than other aromatics (Fig. 26 top).38 Extending the reaction time gave fully hydrodeoxygenated cyclo hexyls (Fig. 26 bottom). The ultrafine size of the nanoclusters and the acidity of the support were cited by the authors as the major factors that promoted both the depolymerization and valorization of the lignin monomers to hydrodeoxygenated compounds.38 The stability of the Ru nanoclusters was tested and it shows high stability up to fourth cycle in hydrogenation of lignin biomass and there is no difference in the crystallinity and morphology of the used catalyst and fresh catalyst. However, lack of further use beyond the recorded reusability is attributed to decrease in recovery of the used catalysts as the number of reaction cycles increases.38
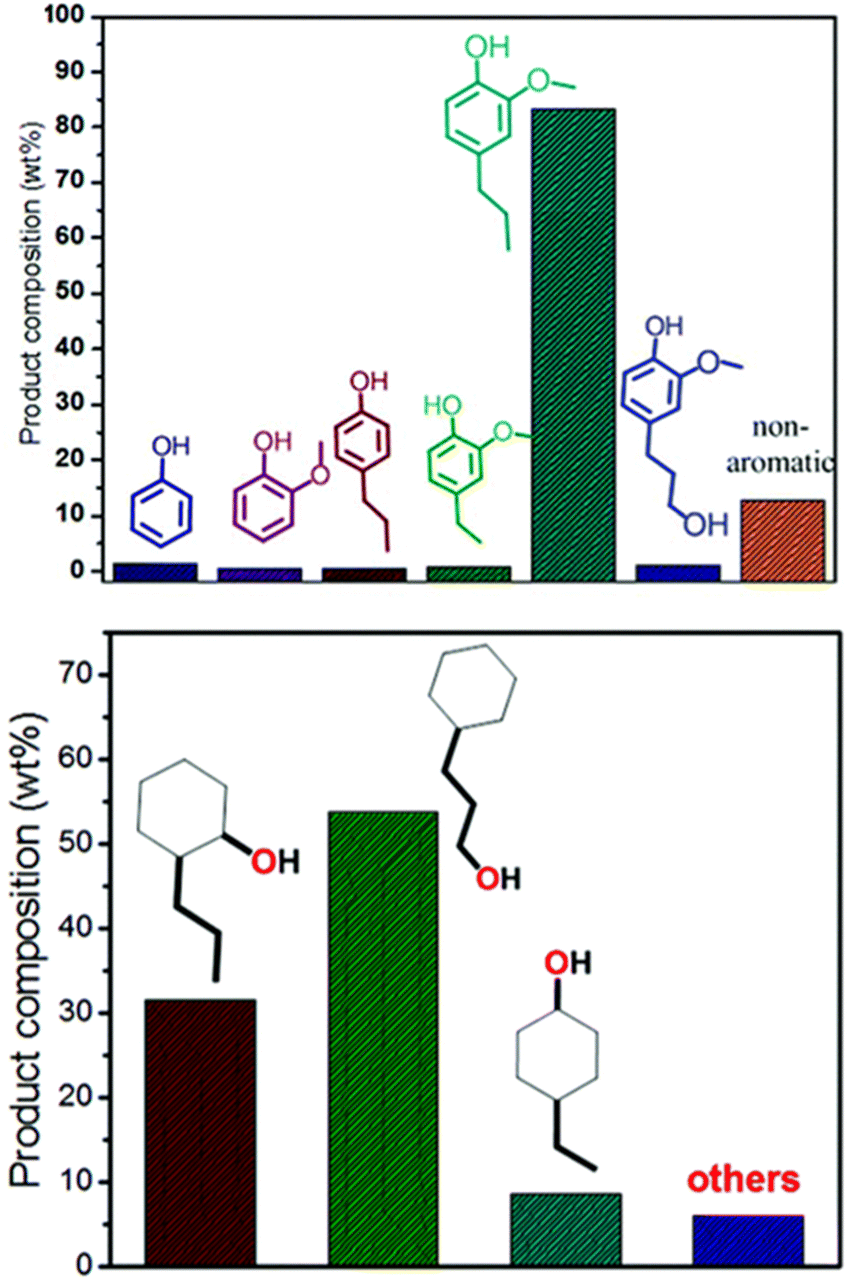 | ||
| Fig. 26 Product distribution of extracted (top) and fractionated (bottom) native lignin oil. Reproduced from ref. 38 with permission from RSC, copyright 2019. | ||
The most popularly used catalyst support for RCF is carbon.3,93 In most of the reported works it has been used with Pd or Ru. Recently, Pd/C was employed in the reductive catalytic fractionation of hemp.122 The mostly underutilized hemp hurd was valorized by Muangmeesri et al. using Pd/C in the presence of formic acid as the hydrogen donor and high yield of monophenolic compounds (>33%) with high S-lignin derivatives was obtained by the group.122 The activities of Pd/C was positively influenced by paratoluensulfonic acid (p-TSA) hydrolysis catalyst and phenolic monomer yields increased to >38%.122 The acid as a hydrogen donor plays important role in stabilization of the generated phenolic monomers. In this report, the RCF process in the absence of hydrogen donor saw a decrease in stable phenolic monomer yield to <16%.122 However, in the work of the Sels group, molecular hydrogen was used as direct source of hydrogen in the RCF of Pine wood using Pd/C.123 The phenolic monomer yield in this case is 34% and the focus was on the elucidation of the structure of the phenolic monomer products of the RCF process. Using chromatographic and spectroscopic techniques, the group was able to assign several inter-unit linkages and this shows absence of C–C linkages showing the efficiency of RCF over the traditional earlier methods for biomass depolymerization.123 Zhang et al. have employed Pd/C in the reductive catalytic fractionation of bamboo sawdust biomass.124 The optimum yield of 47.7 wt% of phenolic monomers mainly comprising of hydroxycinnamic and guaiacol/syringol which are core β-O-4 units.124 Molecular hydrogen was employed here as the hydrogen source for the optimum yield while an absence of hydrogen led to reduction in phenolic monomer yield down to 13.2 wt%.124 Ru/C have also been employed in the reductive catalytic fractionation of different biomass materials in recent works.3,93 The interest in Ru based catalyst is mainly due its hydrogen carrying ability and it has been applied in many other works both as molecular or heterogeneous catalysts in hydrogenation processes. Chen et al. have compared the reactivities of Ru/C and Rh@HCS catalysts towards reductive catalytic fractionation of birch sawdust using molecular hydrogen as the hydrogen source.125 From their findings, the catalyst supports produce relatively equal product yield (∼47 wt%) but noticeable differences in the selectivity of the products (n-propyl alcohol and n-propyl monomers).125 The selectivity towards the former was favored when Rh@HCS was used and the Ru/C gave lower yields. For the latter, more selectivity was achieved with Pd/C than Rh@HCS. The difference in acidity of the supports was reported to be responsible for this. Luterbacher group, on the other hand, have performed RCF on different endocarp biomass of different nut shells and fruit kernels where different activities and monomer yields were obtained using Ru/C.126 From the reported works employing Pd/C and Ru/C, it will be difficult to compare their activities seeing that they have been used on different biomass which have different lignin oil components.
3. Summary and outlook
The selective catalytic conversion of lignin derived compounds is essential for the advancement of utilizing biomass as a renewable energy and commodity chemical source. The phenolic polymer structure of lignin offers the ability to generate high value aromatic products from biomass. Depolymerization routes, such as reductive catalytic fractionation highlighted in this review, can generate large amounts of oxygenated aromatic monomers and dimers. These products, however, are both poor fuels and difficult to safely store due to the high oxygen content making the molecules peroxide formers. Therefore, selective hydrodeoxygenation of these monomer and dimer products is an important step towards the formation of aromatic commodity compounds. Selective cleavage of C–O bonds of lignin derived compounds without hydrogenation of the aromatic rings is critical because H2 use would be maximized, aromatic compounds are higher value chemicals compared to alkanes, and the by-product of the reaction would often just be water. As this review highlights, many recent reports have focused on the selective deoxygenation of lignin derived or inspired compounds, yet more advancements are necessary to truly achieve high or complete selectivity.This review focused on metal catalysts that are commonly used to perform deoxygenation of lignin derived compounds, Ni, Ru, and Pd, but recent reports using Cd,127 Co,128,129 Mo,130 Cu,131 and Ir132,133 have also been reported. For any of the metal catalysts used, the majority of the reports utilized heterogeneous catalysts on solid supports. These catalysts are nanoparticles ranging in size from 2–20 nm and can be single metal or bimetallic systems. General selectivity towards hydrodeoxygenation is achieved in the studies reported in this review, however, complete selectivity towards deoxygenation over ring hydrogenation has still rarely been achieved. Within this review, reactivity trends with respect to selective deoxygenation have become apparent. Aspects that affect the product distribution during catalytic hydrodeoxygenation reactions include: the H2 pressure, reaction temperature, the choice of solvent, reaction time, and the identity of the metal in the catalyst.
The most observed trend in the recent reports reviewed here within, and from earlier reports, is that H2 pressure has a large effect on the ratio between aromatic and ring hydrogenated products. Nearly uniformly across all reports, the higher the H2 pressure during the hydrodeoxygenation reaction, the more ring hydrogenated products were formed. This trend is attributed to an excess of activated hydrogen atoms being formed during the reaction which leads to ring hydrogenation in addition to deoxygenation of the lignin derived substrates.30 It is obvious then, that researchers moving forward with the goal of selective deoxygenation while maintaining substrate aromaticity must use lower pressures of H2 during the catalytic reactions. An optimization of H2 pressure, to ensure enough active sites to promote deoxygenation while limiting an excess of active sites that may lead to ring hydrogenation, should be an essential part of each catalytic study. In this review, the reports that obtained some of the highest selectivity for deoxygenation over ring hydrogenation kept H2 pressure at or below 10 atm. Furthermore, some reports opted for not adding H2 and instead using alcohol additives or solvent to perform transfer hydrogenation reactions. While this approach appeared to limit ring hydrogenation reactions, solvent interference in the reaction was observed in many cases as discussed further below. A couple exceptions to this general H2 pressure trend have, however, been reported. Molecular palladium catalysts showed no change in reaction selectivity towards deoxygenation over a H2 pressure range from 1 to 20 atm.17 This may be due to the nature of the molecular catalyst, which lacks an extended surface for the aromatic ring to bind, as compared to metallic nanoparticle catalysts. A second example involved a Pd on carbon catalyst in a packed-bed flow reactor system in the gas phase. Despite the high H2 pressure of the reaction, the short time-on-stream of the substrate may have prevented ring hydrogenation reactions, and this study reported remarkable deoxygenation selectivity.81
Reaction temperature also played a role when examining product distribution trends. Due to the high bond dissociation energies of C–O bonds found in lignin derived compounds (>100 kcal mol−1), elevated temperatures of ≥200 °C are typically required to promote deoxygenation reactions. While higher temperatures could lead to unwanted side reactions such as ring hydrogenation or solvent reactivity, no discernible trend between reaction temperature and deoxygenation selectivity over ring hydrogenation was observed in this review. Some reports did show that selective deoxygenation was achieved at lower temperatures of ≤150 °C.17,32,79 These reports, however, were only able to achieve C–O bond activation for highly activated benzylic C–O bonds (BDE ≈85 kcal mol−1), and no reported cleavage of phenolic C–O bonds, which are more common in lignin derived molecules, were reported.
Solvent can play a large role in hydrodeoxygenation reactivity and selectivity. For reactions performed under a pressure of H2 gas, the solvents' main role is to solubilize the substrates and the ratio of aromatic to ring hydrogenated products is still dominated by the H2 pressure. However, some reports show that solvents may be incorporated into products mixtures. For example, instead of performing deoxygenation of alcohols, isopropanol solvent led to the formation of benzoic acids from veratryl alcohol80 and ethyl acetate solvent lead to the formation of benzyl acetate from benzyl alcohol.17 Thus it is important for researchers to perform a carbon balance with each reaction to ensure that solvent reactivity is not consuming substrate in unwanted reactions. Researchers have also used solvents as hydrogen atom sources instead of using added H2. In these reports, the selectivity of deoxygenation products over ring hydrogenation was generally greater than reports which utilized high H2 pressures, if solvent reactivity was avoided. Gas-phase, solvent-free reactions have also shown promise for increasing deoxygenation selectivity.81
In the recent reports in this review, the majority perform catalytic reactions in batch reactors and at high substrate conversion. These reaction setups may be detrimental to product selectivity as substrate consumption may lead to unwanted side reactions, such as ring hydrogenation of the deoxygenated products. In fact, reports have shown that complete selectivity to deoxygenated products over ring hydrogenation can be achieved in batch reactors if substrate conversion is kept below 25%.17 In that report, when the reactions were run to 95%+ substrate conversion, ring hydrogenated products were observed. This highlights the need for researchers to either (1) during batch reactions, track the product formation with reaction time to determine optimal conversion/selectivity ratios, or (2) perform reactions in flow reactors to minimize the time products can be in contact with the catalyst surface. Either one of these options may allow for reactions to be performed under higher temperatures and pressures to increase activity while maintaining selectivity for the desired deoxygenated aromatic products.
With respect to catalyst identity, two reactivity trends arise: (1) smaller nanoparticle catalysts tend to lead to less ring hydrogenated products, and (2) bimetallic catalysts tend to exhibit higher reactivity than monometallic counterparts. Throughout this review, many reports directly compared smaller versus larger nanoparticle catalysts and in each case, the smaller catalysts exhibited less ring hydrogenation reactivity. Furthermore, complete selectivity for hydrodeoxygenation over ring hydrogenation was reported.16,17,40,81 In these reports either a molecular catalyst was utilized, or surface modification of a heterogeneous catalyst was used to block most of the surface area of the nanoparticle catalyst. These results point towards a future rational design process for catalytic hydrodeoxygenation activity. In the nanoparticle surface modification study, the authors hypothesized that the blocked surface sites prevent the aromatic rings from interacting with the catalyst, thus preventing ring hydrogenation. For the molecular catalysts, which could be considered a form of a “single-site” catalyst,134 no extended catalyst surface exists for the ring to interact with, thus no ring hydrogenation was observed. In both cases, however, the catalysts were not stable to elevated temperature ≥200 °C, and thus deoxygenation of inactivated C–O bonds, such as phenolic bonds, was not observed. These results suggest, though, that researchers in the field of selective deoxygenation move towards smaller, or even single-site, catalysts in order to prevent interaction between the aromatic rings and the catalysts. While single-atom heterogeneous catalysts may not be able to efficiently activate H2 for hydrodeoxygenation reactions, exceedingly small nanoparticles or metal clusters may provide the proper balance to achieve selective hydrodeoxygenation over ring hydrogenation.134
On the topic of activating H2 for hydrodeoxygenation reactions, bimetallic catalysts have been utilized to combine metals that are efficient for H2 activation with oxophilic metals that are efficient in C–O bond activation. Many metals, such as Pd, that are efficient H2 activators also commonly lead to ring hydrogenation products. Conversely, more oxophilic metals, such as Fe, may not be able to effectively activate H2, but can promote selective C–O bond cleavage over ring hydrogenation. Combining the reactivity of these two classes of metal catalysts has the potential to generate an active and selective catalyst for deoxygenation reactions. In fact, all of the bimetallic catalyst papers discussed in this review reported that the bimetallic catalysts were more active and more selective in hydrodeoxygenation reactions than the analogous single metal catalysts.
Overall, progress on the selective conversion of lignin derived biomass to aromatic products has been achieved over the past few years. Advances in reductive catalytic fractionation and other lignin depolymerization techniques are offering a growing feedstock of oxygen-containing aromatic compounds. Selective conversion of these feedstocks into deoxygenated aromatics such as benzene, toluene, and propyl benzene offer a route towards the development of biorefineries.105 As researchers continue to develop the reactivity trends and insights from the reports highlighted here, rational design of catalysts will follow and lead towards complete selectivity of high value aromatic commodity chemicals from lignin biomass.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank the National Science Foundation under Grant No. 1954850 for financial support.References
- M. Ragnar, G. Henriksson, M. E. Lindström, M. Wimby, J. Blechschmidt and S. Heinemann, in Ullmann's Encyclopedia of Industrial Chemistry, 2014, pp. 1–92, DOI:10.1002/14356007.a18_545.pub4.
- B. Hahn-Hägerdal, M. Galbe, M. F. Gorwa-Grauslund, G. Lidén and G. Zacchi, Trends Biotechnol., 2006, 24, 549–556 CrossRef PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- H. Luo and M. M. Abu-Omar, Green Chem., 2018, 20, 745–753 RSC.
- A. Kumar, M. Jindal, S. Maharana and B. Thallada, Energy Fuels, 2021, 35, 16965–16994 CrossRef CAS.
- M. W. Nolte and B. H. Shanks, Energy Technol., 2017, 5, 7–18 CrossRef CAS.
- E. Leng, Y. Guo, J. Chen, S. Liu, J. E and Y. Xue, Fuel, 2022, 309, 122102 CrossRef CAS.
- H. Zhang, S. Fu, X. Du and Y. Deng, ChemSusChem, 2021, 14, 2268–2294 CrossRef CAS PubMed.
- D. Bourbiaux, J. Pu, F. Rataboul, L. Djakovitch, C. Geantet and D. Laurenti, Catal. Today, 2021, 373, 24–37 CrossRef CAS.
- T. Renders, S. Van den Bosch, S. F. Koelewijn, W. Schutyser and B. F. Sels, Energy Environ. Sci., 2017, 10, 1551–1557 RSC.
- P. S. Chauhan, R. Agrawal, A. satlewal, R. Kumar, R. P. Gupta and S. S. V. Ramakumar, Int. J. Biol. Macromol., 2022, 197, 179–200 CrossRef CAS PubMed.
- Y. Rong, N. Ji, Z. Yu, X. Diao, H. Li, Y. Lei, X. Lu and A. Fukuoka, Green Chem., 2021, 23, 6761–6788 RSC.
- Y. Li, Z. Dang and P. Gao, Nano Sel., 2021, 2, 847–864 CrossRef CAS.
- S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS PubMed.
- H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2018, 8, 148–187 CrossRef CAS.
- C.-H. Lien and J. W. Medlin, J. Phys. Chem. C, 2014, 118, 23783–23789 CrossRef CAS.
- N. A. DeLucia, A. Jystad, K. Vander Laan, J. M. M. Tengco, M. Caricato and A. K. Vannucci, ACS Catal., 2019, 9, 9060–9071 CrossRef CAS.
- F. Yang, D. Liu, H. Wang, X. Liu, J. Han, Q. Ge and X. Zhu, J. Catal., 2017, 349, 84–97 CrossRef CAS.
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS PubMed.
- V. S. Prabhudesai, L. Gurrala and R. Vinu, Energy Fuels, 2022, 36, 1155–1188 CrossRef CAS.
- M. Lang and H. Li, ACS Sustainable Chem. Eng., 2022, 10, 13208–13243 CrossRef CAS.
- H. Wei, Z. Wang and H. Li, Green Chem., 2022, 24, 1930–1950 RSC.
- J. Zhang, J. Sun and Y. Wang, Green Chem., 2020, 22, 1072–1098 RSC.
- S. De, A. S. Burange and R. Luque, Green Chem., 2022, 24, 2267–2286 RSC.
- Y. Geng and H. Li, ChemSusChem, 2022, 15, e202102495 CrossRef CAS PubMed.
- N. Ji, J. Yin, Y. Rong, H. Li, Z. Yu, Y. Lei, S. Wang and X. Diao, Catal. Sci. Technol., 2022, 12, 3751–3766 RSC.
- Y. Guo, Y. Jing, Q. Xia and Y. Wang, Acc. Chem. Res., 2022, 55, 1301–1312 CrossRef CAS PubMed.
- X. Yu and C. T. Williams, Catal. Sci. Technol., 2023, 13, 802–825 RSC.
- A. M. Robinson, J. E. Hensley and J. W. Medlin, ACS Catal., 2016, 6, 5026–5043 CrossRef CAS.
- X. Wang, M. Arai, Q. Wu, C. Zhang and F. Zhao, Green Chem., 2020, 22, 8140–8168 RSC.
- T. Cordero-Lanzac, R. Palos, I. Hita, J. M. Arandes, J. Rodríguez-Mirasol, T. Cordero, J. Bilbao and P. Castaño, Appl. Catal., B, 2018, 239, 513–524 CrossRef CAS.
- J. G. Tillou and A. K. Vannucci, J. Organomet. Chem., 2021, 944, 121848 CrossRef CAS.
- T. Nimmanwudipong, R. C. Runnebaum, D. E. Block and B. C. Gates, Energy Fuels, 2011, 25, 3417–3427 CrossRef CAS.
- I. T. Ghampson, R. Canales and N. Escalona, Appl. Catal., A, 2018, 549, 225–236 CrossRef CAS.
- I. T. Ghampson, C. Sepúlveda, R. García, J. L. G. Fierro and N. Escalona, Catal. Sci. Technol., 2016, 6, 4356–4369 RSC.
- W. Jin, L. Pastor-Pérez, D. Shen, A. Sepúlveda-Escribano, S. Gu and T. Ramirez Reina, ChemCatChem, 2019, 11, 924–960 CrossRef CAS.
- Z. Luo, Z. Zheng, L. Li, Y.-T. Cui and C. Zhao, ACS Catal., 2017, 7, 8304–8313 CrossRef CAS.
- N. Ullah, A. H. Odda, K. Liang, M. A. Kombo, S. Sahar, L.-B. Ma, X.-X. Fang and A.-W. Xu, Green Chem., 2019, 21, 2739–2751 RSC.
- F. Yang, D. Liu, Y. Zhao, H. Wang, J. Han, Q. Ge and X. Zhu, ACS Catal., 2018, 8, 1672–1682 CrossRef CAS.
- N. A. DeLucia, N. Das, S. Overa, A. Paul and A. K. Vannucci, Catal. Today, 2018, 302, 146–150 CrossRef CAS.
- Y. Jing, L. Dong, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2020, 13, 4181–4198 CrossRef CAS PubMed.
- L. Jiang, H. Guo, C. Li, P. Zhou and Z. Zhang, Chem. Sci., 2019, 10, 4458–4468 RSC.
- H. Ma, H. Li, W. Zhao, X. Li and J. Long, Energy Procedia, 2019, 158, 370–375 CrossRef CAS.
- F. Lin, Y. Ma, Y. Sun, X. Men, Y. Zhu, T. Gao and K. Zhao, Appl. Catal., A, 2020, 598, 117552 CrossRef CAS.
- G. Kim, J. Seo, J.-W. Choi, J. Jae, J.-M. Ha, D. J. Suh, K.-Y. Lee, J.-K. Jeon and J.-K. Kim, Catal. Today, 2018, 303, 130–135 CrossRef CAS.
- Z. Luo, J. Kong, B. Ma, Z. Wang, J. Huang and C. Zhao, ACS Sustainable Chem. Eng., 2020, 8, 2158–2166 CrossRef CAS.
- Z. Jia, N. Ji, X. Diao, X. Li, Y. Zhao, X. Lu, Q. Liu, C. Liu, G. Chen, L. Ma, S. Wang, C. Song and C. Li, ACS Catal., 2022, 12, 1338–1356 CrossRef CAS.
- W. Song, Y. He, S. Lai, W. Lai, X. Yi, W. Yang and X. Jiang, Green Chem., 2020, 22, 1662–1670 RSC.
- B. Shumeiko and D. Kubička, ChemSusChem, 2022, 15, e202102099 CrossRef CAS PubMed.
- B. Chen, C. He, M. Cao, X. Qiu, X. Ouyang and Y. Qian, Green Chem., 2022, 24, 846–857 RSC.
- R. Shu, R. Li, B. Lin, C. Wang, Z. Cheng and Y. Chen, Biomass Bioenergy, 2020, 132, 105432 CrossRef CAS.
- J. Guo, Y. L. Ma, J. Y. Yu, Y. J. Gao, N. X. Ma and X. Y. Wu, BMC Chem., 2019, 13, 36 CrossRef CAS PubMed.
- B. M. Matsagar, T.-C. Kang, Z.-Y. Wang, T. Yoshikawa, Y. Nakasaka, T. Masuda, L.-C. Chuang and K. C. W. Wu, React. Chem. Eng., 2019, 4, 618–626 RSC.
- C. Zhu, J.-P. Cao, X.-Y. Zhao, T. Xie, J. Ren and X.-Y. Wei, J. Energy Inst., 2019, 92, 74–81 CrossRef CAS.
- C. Chen, D. Wu, P. Liu, J. Li, H. Xia, M. Zhou and J. Jiang, Green Chem., 2021, 23, 3090–3103 RSC.
- J. Long, Q. Zhang, T. Wang, X. Zhang, Y. Xu and L. Ma, Bioresour. Technol., 2014, 154, 10–17 CrossRef CAS PubMed.
- T. N. Phan, Y.-K. Park, I.-G. Lee and C. H. Ko, Appl. Catal., A, 2017, 544, 84–93 CrossRef CAS.
- H. Wang, M. Feng and B. Yang, Green Chem., 2017, 19, 1668–1673 RSC.
- D. Liu, Y. M. López-De Jesús, J. R. Monnier and C. T. Williams, J. Catal., 2010, 269, 376–387 CrossRef CAS.
- H. Tan, X. Li, H. Ma, X. Yang, T. Zhan, W. Xie, S. Wang and J. Zhu, Sustainable Energy Fuels, 2022, 6, 2745–2754 RSC.
- H. Liu, T. Jiang, B. Han, S. Liang and Y. Zhou, Science, 2009, 326, 1250 CrossRef CAS PubMed.
- Y. Xu, Z. Peng, Y. Yu, D. Wang, J. Liu, Q. Zhang and C. Wang, New J. Chem., 2020, 44, 5088–5096 RSC.
- C. Chen, D. Wu, P. Liu, H. Xia, M. Zhou, X. Hou and J. Jiang, React. Chem. Eng., 2021, 6, 559–571 RSC.
- C. Chen, P. Liu, H. Xia, M. Zhou and J. Jiang, J. Chin. Chem. Soc., 2021, 68, 582–591 CrossRef CAS.
- F. Lin, Y. Ma, Y. Sun, Z. Song, X. Men, Y. Wu, Y. Zhu, T. Gao and Y. Zhong, Renewable Energy, 2022, 189, 1278–1291 CrossRef CAS.
- B. Luo, R. Li, R. Shu, C. Wang, J. Zhang and Y. Chen, Ind. Eng. Chem. Res., 2020, 59, 17192–17199 CrossRef CAS.
- S. Li, B. Liu, J. Truong, Z. Luo, P. C. Ford and M. M. Abu-Omar, Green Chem., 2020, 22, 7406–7416 RSC.
- S. Salakhum, K. Saenluang and C. Wattanakit, Sustainable Energy Fuels, 2020, 4, 1126–1134 RSC.
- X. Zhang, H. Yan, L. Zhu, T. Li and S. Wang, Adv. Sustainable Syst., 2020, 4, 1900136 CrossRef CAS.
- M. Garedew, D. Young-Farhat, S. Bhatia, P. Hao, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2020, 4, 1340–1350 RSC.
- M. A. Hossain, T. K. Phung, M. S. Rahaman, S. Tulaphol, J. B. Jasinski and N. Sathitsuksanoh, Appl. Catal., A, 2019, 582, 117100 CrossRef CAS.
- R. Shu, R. Li, B. Lin, B. Luo and Z. Tian, Fuel, 2020, 265, 116962 CrossRef CAS.
- M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- Y. Xin, L. Dong, Y. Guo, X. Liu, Y. Hu and Y. Wang, J. Catal., 2019, 375, 202–212 CrossRef CAS.
- J.-P. Cao, T. Xie, X.-Y. Zhao, C. Zhu, W. Jiang, M. Zhao, Y.-P. Zhao and X.-Y. Wei, Fuel, 2021, 284, 119027 CrossRef CAS.
- L. Hu, X.-Y. Wei, Y.-H. Kang, X.-H. Guo, M.-L. Xu and Z.-M. Zong, J. Energy Inst., 2021, 96, 269–279 CrossRef CAS.
- W. Jiang, J.-P. Cao, J.-X. Xie, L. Zhao, C. Zhang, X.-Y. Zhao, Y.-P. Zhao and J.-L. Zhang, Energy Fuels, 2021, 35, 19543–19552 CrossRef CAS.
- N. A. DeLucia, A. Jystad, K. V. Laan, J. M. M. Tengco, M. Caricato and A. K. Vannucci, ACS Catal., 2019, 9, 9060–9071 CrossRef CAS.
- S. Alijani, S. Capelli, C. Evangelisti, L. Prati, A. Villa and S. Cattaneo, Catal. Today, 2021, 367, 220–227 CrossRef CAS.
- H. Zhang, Y. Liu, S. Fu and Y. Deng, Int. J. Biol. Macromol., 2021, 169, 274–281 CrossRef CAS PubMed.
- C. Liu, C. Zhou, Y. Wang, X. Liu, L. Zhu, H. Ma, Z. Zhou and F. Qi, Proc. Combust. Inst., 2021, 38, 4345–4353 CrossRef CAS.
- X. Gao, S. Zhu and Y. Li, Mol. Catal., 2019, 462, 69–76 CrossRef CAS.
- Y.-P. Zhao, F.-P. Wu, Q.-L. Song, X. Fan, L.-J. Jin, R.-Y. Wang, J.-P. Cao and X.-Y. Wei, J. Energy Inst., 2020, 93, 899–910 CrossRef CAS.
- C. Zhu, J.-P. Cao, X.-Y. Zhao, T. Xie, M. Zhao and X.-Y. Wei, Fuel Process. Technol., 2019, 194, 106126 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- M. S. Zanuttini, B. O. Dalla Costa, C. A. Querini and M. A. Peralta, Appl. Catal., A, 2014, 482, 352–361 CrossRef CAS.
- J. M. Martínez, J. M. Granado, D. Montané, J. Salvadó and X. Farriol, Bioresour. Technol., 1995, 52, 59–67 CrossRef.
- X. Huang, O. M. Morales Gonzalez, J. Zhu, T. I. Korányi, M. D. Boot and E. J. M. Hensen, Green Chem., 2017, 19, 175–187 RSC.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- M. L. Stone, E. M. Anderson, K. M. Meek, M. Reed, R. Katahira, F. Chen, R. A. Dixon, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2018, 6, 11211–11218 CrossRef CAS.
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC.
- T. Parsell, S. Yohe, J. Degenstein, T. Jarrell, I. Klein, E. Gencer, B. Hewetson, M. Hurt, J. I. Kim, H. Choudhari, B. Saha, R. Meilan, N. Mosier, F. Ribeiro, W. N. Delgass, C. Chapple, H. I. Kenttämaa, R. Agrawal and M. M. Abu-Omar, Green Chem., 2015, 17, 1492–1499 RSC.
- X. Liu, F. P. Bouxin, J. Fan, V. L. Budarin, C. Hu and J. H. Clark, ChemSusChem, 2020, 13, 4296–4317 CrossRef CAS PubMed.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef CAS.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2016, 4, 6940–6950 CrossRef CAS.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Liu, H. I. Kenttämaa and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 2316–2322 CrossRef CAS.
- O. G. Marin-Flores, A. M. Karim and Y. Wang, Catal. Today, 2014, 237, 118–124 CrossRef CAS.
- D. G. Barton, S. L. Soled and E. Iglesia, Top. Catal., 1998, 6, 87–99 CrossRef CAS.
- I. Kim, A. A. Dwiatmoko, J.-W. Choi, D. J. Suh, J. Jae, J.-M. Ha and J.-K. Kim, J. Ind. Eng. Chem., 2017, 56, 74–81 CrossRef CAS.
- S. Oh, S. Gu, J.-W. Choi, D. J. Suh, H. Lee, C. S. Kim, K. H. Kim, C.-J. Yoo, J. Choi and J.-M. Ha, J. Environ. Chem. Eng., 2022, 10, 108085 CrossRef CAS.
- X. Liu, H. Li, L.-P. Xiao, R.-C. Sun and G. Song, Green Chem., 2019, 21, 1498–1504 RSC.
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC.
- C. E. J. J. Vriamont, T. Chen, C. Romain, P. Corbett, P. Manageracharath, J. Peet, C. M. Conifer, J. P. Hallett and G. J. P. Britovsek, ACS Catal., 2019, 9, 2345–2354 CrossRef CAS.
- K. Wu, X. Li, W. Wang, Y. Huang, Q. Jiang, W. Li, Y. Chen, Y. Yang and C. Li, ACS Catal., 2022, 12, 8–17 CrossRef CAS.
- T. H. Parsell, B. C. Owen, I. Klein, T. M. Jarrell, C. L. Marcum, L. J. Haupert, L. M. Amundson, H. I. Kenttämaa, F. Ribeiro, J. T. Miller and M. M. Abu-Omar, Chem. Sci., 2013, 4, 806–813 RSC.
- C.-H. Lien and J. W. Medlin, J. Catal., 2016, 339, 38–46 CrossRef CAS.
- S. Li, W. Li, Q. Zhang, R. Shu, H. Wang, H. Xin and L. Ma, RSC Adv., 2018, 8, 1361–1370 RSC.
- A. U. Buranov and G. Mazza, Ind. Crops Prod., 2008, 28, 237–259 CrossRef CAS.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 4464–4480 CrossRef CAS.
- E. O. Ebikade, N. Samulewicz, S. Xuan, J. D. Sheehan, C. Wu and D. G. Vlachos, Green Chem., 2020, 22, 7435–7447 RSC.
- W. Lan and J. S. Luterbacher, Chimia, 2019, 73, 591 CrossRef CAS PubMed.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, Science, 2020, 367, 1385–1390 CrossRef CAS PubMed.
- J. Park, H. S. Cahyadi, U. Mushtaq, D. Verma, D. Han, K.-W. Nam, S. K. Kwak and J. Kim, ACS Catal., 2020, 10, 12487–12506 CrossRef CAS.
- X. Ouyang, X. Huang, J. Zhu, M. D. Boot and E. J. M. Hensen, ACS Sustainable Chem. Eng., 2019, 7, 13764–13773 CrossRef CAS.
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef CAS.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC.
- K. An and G. A. Somorjai, ChemCatChem, 2012, 4, 1512–1524 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- A. M. Karim, V. Prasad, G. Mpourmpakis, W. W. Lonergan, A. I. Frenkel, J. G. Chen and D. G. Vlachos, J. Am. Chem. Soc., 2009, 131, 12230–12239 CrossRef CAS PubMed.
- W. Raróg-Pilecka, E. Miśkiewicz, D. Szmigiel and Z. Kowalczyk, J. Catal., 2005, 231, 11–19 CrossRef.
- A.-X. Yin, W.-C. Liu, J. Ke, W. Zhu, J. Gu, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2012, 134, 20479–20489 CrossRef CAS PubMed.
- S. Muangmeesri, N. Li, D. Georgouvelas, P. Ouagne, V. Placet, A. P. Mathew and J. S. M. Samec, ACS Sustainable Chem. Eng., 2021, 9, 17207–17213 CrossRef CAS PubMed.
- K. Van Aelst, E. Van Sinay, T. Vangeel, E. Cooreman, G. Van den Bossche, T. Renders, J. Van Aelst, S. Van den Bosch and B. F. Sels, Chem. Sci., 2020, 11, 11498–11508 RSC.
- K. Zhang, H. Li, L.-P. Xiao, B. Wang, R.-C. Sun and G. Song, Bioresour. Technol., 2019, 285, 121335 CrossRef PubMed.
- L. Chen, A. P. van Muyden, X. Cui, Z. Fei, N. Yan, G. Laurenczy and P. J. Dyson, JACS Au, 2021, 1, 729–733 CrossRef CAS PubMed.
- J. Behaghel de Bueren, F. Héroguel, C. Wegmann, G. R. Dick, R. Buser and J. S. Luterbacher, ACS Sustainable Chem. Eng., 2020, 8, 16737–16745 CrossRef CAS.
- H. Yoo, M.-W. Lee, S. Lee, J. Lee, S. Cho, H. Lee, H. G. Cha and H. S. Kim, ACS Catal., 2020, 10, 8465–8475 CrossRef CAS.
- S. Liu, L. Bai, A. P. van Muyden, Z. Huang, X. Cui, Z. Fei, X. Li, X. Hu and P. J. Dyson, Green Chem., 2019, 21, 1974–1981 RSC.
- W. Wu, H. Liu, H. Wu, B. Zheng, S. Han, K. Zhang, X. Mei, C. Xu, M. He and B. Han, ACS Sustainable Chem. Eng., 2021, 9, 11862–11871 CrossRef CAS.
- E. Blanco, D. A. Aguirre-Abarca, J. N. Díaz de León and N. Escalona, New J. Chem., 2020, 44, 12027–12035 RSC.
- H.-R. Tian, Y.-W. Liu, Z. Zhang, S.-M. Liu, T.-Y. Dang, X.-H. Li, X.-W. Sun, Y. Lu and S.-X. Liu, Green Chem., 2020, 22, 248–255 RSC.
- X. Li, B. Zhang, X. Pan, J. Ji, Y. Ren, H. Wang, N. Ji, Q. Liu and C. Li, ChemSusChem, 2020, 13, 4409–4419 CrossRef CAS PubMed.
- W. Yao, S. Das, N. A. DeLucia, C. Boudreaux, F. Qu, A. K. Vannucci and E. T. Papish, Organometallics, 2019, 39, 662–669 CrossRef.
- X. Liang, N. Fu, S. Yao, Z. Li and Y. Li, J. Am. Chem. Soc., 2022, 144, 18155–18174 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00232b |
| This journal is © The Royal Society of Chemistry 2023 |