
Defect-stabilized and oxygen-coordinated iron single-atom sites facilitate hydrogen peroxide electrosynthesis†
Taotao
Gao
ab,
Lu
Qiu
bc,
Minghao
Xie
d,
Zhaoyu
Jin
 e,
Panpan
Li
*a and
Guihua
Yu
*d
e,
Panpan
Li
*a and
Guihua
Yu
*d
aCollege of Materials Science and Engineering, Sichuan University, Chengdu, 610065, P. R. China. E-mail: panpanli@scu.edu.cn
bInstitute for Advanced Study, Chengdu University, Chengdu, 610106, P. R. China
cCollege of Chemical Engineering, Sichuan University, Chengdu, 610065, P. R. China
dMaterials Science and Engineering Program and Walker Department of Mechanical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: ghyu@austin.utexas.edu
eInstitute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China, Chengdu, 610054, P. R. China
First published on 2nd August 2023
Abstract
The selective two-electron electrochemical oxygen reduction reaction (ORR) for hydrogen peroxide (H2O2) production is a promising and green alternative method to the current energy-intensive anthraquinone process used in industry. In this study, we develop a single-atom catalyst (CNT-D-O-Fe) by anchoring defect-stabilized and oxygen-coordinated iron atomic sites (Fe–O4) onto porous carbon nanotubes using a local etching strategy. Compared to O-doped CNTs with vacancy defects (CNT-D-O) and oxygen-coordinated Fe single-atom site modifying CNTs without a porous structure (CNT-O-Fe), CNT-D-O-Fe exhibits the highest H2O2 selectivity of 94.4% with a kinetic current density of 13.4 mA cm−2. Fe–O4 single-atom sites in the catalyst probably contribute to the intrinsic reactivity for the two-electron transfer process while vacancy defects greatly enhance the electrocatalytic stability. Theoretical calculations further support that the coordinated environment and defective moiety in CNT-D-O-Fe could efficiently optimize the adsorption strength of the *OOH intermediate over the Fe single atomic active sites. This contribution sheds light on the potential of defect-stabilized and oxygen-coordinated single-atom metal sites as a promising avenue for the rational design of highly efficient and selective catalysts towards various electrocatalytic reactions.
 Guihua Yu | We would like to express our sincerest congratulations to Materials Horizons on its remarkable 10-year anniversary. Our partnership with the journal dates back to our first publication in 2021 and has grown stronger ever since. It brings us great pride to be part of this prestigious journal's journey, as the journal has made impactful contributions to the field of materials science. Let us celebrate the continued success and productivity of Materials Horizons for the next decade! |
New conceptsThe production of hydrogen peroxide (H2O2) through the electrocatalytic oxygen reduction reaction (ORR) requires catalytic materials with robust catalytic activity and high two-electron selectivity. The micro-environments of the catalytic sites play a crucial role in determining the catalytic performance. In this study, we present a novel approach to construct a unique oxygen-coordinated iron single-atom site (Fe–O4) modified with vacancy defects on carbon nanotubes (CNT-D-O-Fe) using iron oxide-induced local etching. The oxygen-coordinated environment enhances the two-electron intrinsic reactivity for iron single-atom sites, while the vacancy defects greatly improve the electrocatalytic stability. As a result, CNT-D-O-Fe exhibits exceptional H2O2 selectivity, kinetic current density, and durable stability. Theoretical calculations further reveal that the oxygen-coordinated environment and the defective moiety of CNT-D-O-Fe efficiently optimize the adsorption strength of the *OOH intermediate over the Fe single atomic active sites. This work provides an in-depth understanding of the effects of the microenvironmental structure on the reaction path and catalytic performance. The unique local coordinated environment regulation concept at the atomic scale paves a new way for the design of highly efficient transition-metal single-atom catalysts. |
Introduction
With the rapid growth of the economy, the demand for hydrogen peroxide (H2O2) has been increasing significantly in various industries, such as bleaching, chemical synthesis, wastewater treatment, and disinfection, with an expected need of 6 million tons by 2024.1–3 However, the current industrial H2O2 production primarily relies on the anthraquinone process, which is associated with inherent drawbacks, especially high energy consumption, which hinders green and sustainable development.4,5 Therefore, it is crucial to explore eco-friendly and efficient alternative approaches for H2O2 production. The electrocatalytic oxygen reduction reaction (ORR), powered by renewable energy, provides an attractive H2O2 synthetic strategy through a two-electron reaction pathway. However, the ORR involves complex O–O bond cleavage mechanisms, resulting in blend products of H2O2 or H2O together with a slow kinetic rate.6 Thus, it is vital to design efficient catalysts to improve the selectivity and activity of H2O2 electrosynthesis. The intrinsic performance is determined by the electronic structure of active sites, which influences the adsorption strength and activation state of reactants and intermediates during the electrocatalysis process.7–13 In particular, the optimal binding energy between the reaction intermediate (*OOH) and active sites can promote the preservation of the O–O bond to inhibit over-reduction and the formation of H2O.14–18 Therefore, the rational modulation of the electronic structure enables efficient activity and high H2O2 selectivity of the active sites.19Single-atom catalysts (SACs) offer remarkable advantages, including greatly increased atomic utilization and highly tunable electronic structures compared with bulk and nanocatalysts.20–26 Specifically, the electronic structure of carbon-supported and nonmetallic heteroatom-coordinated metal-based SACs (such as a metal–nitrogen–carbon structure) can be tailor-tuned via a series of achievable strategies such as the adjustment of coordination environments, metal centers, and the density of the single atomic sites, with the aim of improving the catalytic performance.4,6,12,23 Although some studies have confirmed that Fe–N–C SACs display efficient catalytic activity for the ORR, the catalytic reaction pathway mainly focuses on the four-electron reduction process producing H2O rather than H2O2.22,27–30 Given this, the regulation of the micro-environment of Fe single-atom sites, such as nonmetallic coordination sites and the structure of the carbon substrate can be utilized to alter the reduction path and thus selectivity, while understanding the effects of the coordinated environment on the ORR activity of Fe single-atom sites is still lacking.
Herein, the oxygen-coordinated Fe single atomic site modifying multiwalled carbon nanotubes (CNTs) with abundant vacancy defects (CNT-D-O-Fe) is achieved by a local etching strategy, exhibiting the desired ORR catalytic performance. Meanwhile, the ORR catalytic activity, stability, and H2O2 selectivity of CNT-D-O-Fe greatly surpass those of O-doped CNTs with vacancy defects (CNT-D-O) and oxygen-coordinated Fe single-atom site modifying CNTs without porous structures (CNT-O-Fe). Density functional theory (DFT) calculations further reveal that synergistic effects between the oxygen-coordinated environment and vacancy defects substantially improve the adsorption strength of the *OOH intermediate at the Fe single-atom active sites to enhance the two-electron ORR catalytic activity. This work provides an in-depth understanding of the effects of the coordination environment and vacancy defects on the reaction path and catalytic activity of the metal single-atom sites, which would benefit the precise design of single-atom catalysts with predictable performance.
Results and discussion
Materials synthesis and characterization
As shown in Fig. 1, the oxygen-coordinated iron single-atom sites (Fe–O4) are anchored on the vacancy defect-rich CNTs (CNT-D-O-Fe) via a distinctive iron oxide-induced local etching strategy. Before the etching treatment, the pore-forming reagents (iron-based compounds) are evenly loaded on the CNTs with a smooth surface (Fig. 2a) to obtain the CNT-Fe-OH (Fig. S1, ESI†). After high-temperature etching, the generated sample displays abundant porous structures (Fig. S2, ESI†) and iron-based compounds transform into Fe2O3 nanoparticles according to the XRD pattern in Fig. S3 (ESI†). Then CNT-D-O-Fe is obtained via further acid treatment to thoroughly remove the residual Fe2O3. The thermal gravity (TG) and differential thermal gravity (DTG) curves in Fig. S4 (ESI†) show that the carbon species in CNT-Fe-OH start to break down at ca. 470 °C and the weight decreases rapidly at ca. 565 °C. The corresponding temperatures of the original CNT (ca. 510 °C and ca. 603 °C, respectively) are significantly higher than CNT-Fe-OH. Therefore, the local carbon species in CNT-Fe-OH lose high thermal stability due to the induction of iron-based compounds, which facilitates the formation of defect structures in the obtained CNT-D-O-Fe.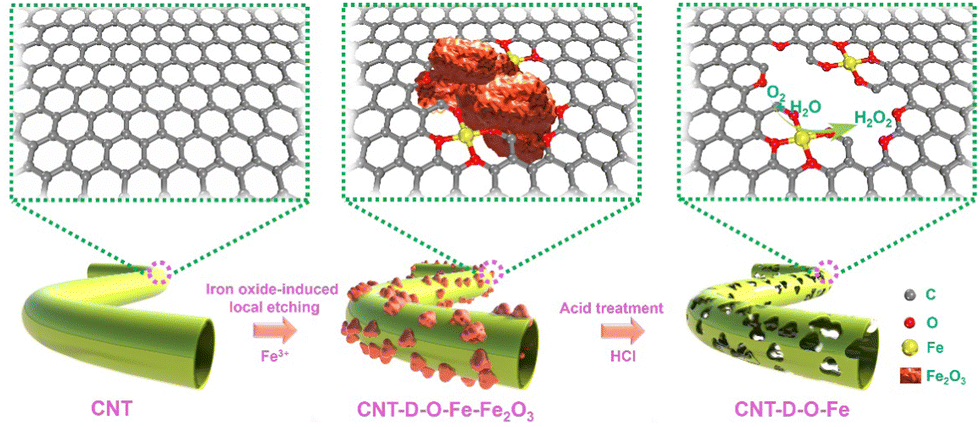 | ||
| Fig. 1 Schematic of the preparation process of CNT-D-O-Fe. | ||
 | ||
| Fig. 2 (a) SEM image of CNTs; (b) SEM and (c) TEM images of CNT-D-O-Fe; (d) HRTEM image of CNT-D-O-Fe and the corresponding fast Fourier transform (FFT) patterns (the inset) of the local CNT-D-O-Fe around the vacancy defects; (e) HAADF-STEM image of CNT-D-O-Fe; (f) Raman spectra of the samples. | ||
Differing from the CNT with a smooth surface (Fig. 2a), CNT-D-O-Fe based on the optimal preparation conditions demonstrates abundant defective structures over the CNT (Fig. 2b and Fig. S5, ESI†). The transmission electron microscope (TEM) image (Fig. 2c) of CNT-D-O-Fe further confirms the presence of pores. Furthermore, the high-resolution transmission electron microscope (HRTEM) image and the local fast Fourier transform (FFT) patterns near the porous structures (Fig. 2d and the inset) show the breakage and the increase in disorder regarding the lattice fringes of CNTs. This result suggests that the formation of porous structures also induces a reduced degree of graphitization for carbon. The high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image of CNT-D-O-Fe in Fig. 2e exhibits a vacancy defect around the porous structure. The isolated bright spots with a diameter of ca. 0.1 nm in the defect structure can be assigned to the isolated iron single atoms, revealing the successful introduction of iron single-atom sites during the etching process.
The control sample, CNT-O-Fe (0.03 wt% for iron content, Table S1, ESI†) only containing iron sites and without pore structure, is prepared via heating CNT-Fe-OH in an argon environment and the subsequent acid treatment. Similar to the CNT, the CNT-O-Fe shows a smooth surface (Fig. S6, ESI†), but iron sites are uniformly introduced in the CNT-O-Fe based on the element mapping of Fe in Fig. S7 (ESI†). In contrast, a limited number of porous structures are observed over CNT-D-O after a similar treatment of CNT-D-O-Fe except for the addition of Fe compounds (Fig. S8 and S9, ESI†). This result demonstrates the pivotal role of iron-based compounds in making defective porous structures of CNT.
Pore diameter distribution and (Brunauer–Emmett–Teller) BET-specific surface area of CNT, CNT-O-Fe, CNT-D-O, and CNT-D-O-Fe are analyzed based on the nitrogen adsorption–desorption curves (Fig. S10a, ESI†). The presence of lots of porous structures makes CNT-D-O-Fe show a higher BET-specific surface area (Table S2, 195.2 m2 g−1, ESI†), superior to CNT-O-Fe (82.1 m2 g−1), CNT-D-O (116.9 m2 g−1), and CNT (81.7 m2 g−1). Fig. S10b (ESI†) also displays the larger pore size distributions of CNT-D-O-Fe compared with CNT-D-O, CNT-O-Fe, and CNT. The higher specific surface area and bigger pore structure suggest a better mass transfer for CNT-D-O-Fe during electrocatalysis. To investigate the degree of graphitization of carbon in these samples, the Raman tests are conducted and shown in Fig. 2f. The ratio of the peak intensities of D and G peaks (1342 cm−1 and 1570 cm−1, respectively) for CNT-D-O-Fe is 1.045, higher than those of CNT-D-O (0.744), CNT-O-Fe (0.703) and CNT (0.700). This result demonstrates that the abundant porous structures make CNT-D-O-Fe have a lower degree of graphitization of carbon with more vacancy defects.31–33 These characteristics are beneficial to the exposure of the active sites and the improvement of adsorption strength for the reactants.34–36
The X-ray photoelectron spectroscopy (XPS) survey spectrum of CNT-D-O-Fe in Fig. S11a (ESI†) reveals the presence of Fe, O, and C. The O 1s XPS peaks at ca. 531.7 eV (Fig. S11c, ESI†) can be assigned to the oxygen species in C–O–Fe.37–40 However, the low Fe content in CNT-D-O-Fe (0.15 wt%, Table S1, ESI†) makes it difficult to confirm the chemical valence state and the corresponding fine structure of the iron single atoms only based on the XPS analysis. Therefore, the XAFS spectroscopy is further used to precisely explore the fine structure of CNT-D-O-Fe including the coordination environment and chemical state of the iron site. According to the Fe K-edge X-ray absorption near edge structure (XANES) of CNT-D-O-Fe, Fe2O3, and Fe foil in Fig. 3a, the oxidation state of iron in CNT-D-O-Fe is about +3 based on the similar rising-edge position (ca. 7120.6 eV) and the pre-edge peak (ca. 7115.6 eV) for CNT-D-O-Fe compared with Fe2O3. However, the unique planar geometry of the oxygen-coordinated single atomic iron sites (Fe–O4) makes CNT-D-O-Fe show a different first derivative of the Fe K-edge XANES signal in comparison to Fe2O3 (Fig. S13, ESI†).37 Besides, the peak at ca.1.52 Å in the Fourier transform of the Fe K-edge extended X-ray absorption fine structure (FT-EXAFS) (Fig. 3b) reveals that the iron atom in CNT-D-O-Fe is mainly bonded to O atoms instead of other Fe atoms (ca. 2.20 Å for Fe–Fe bond). The Fe–O bond length in CNT-D-O-Fe (ca. 1.52 Å) differs from that in Fe2O3 (ca. 1.46 Å). Additionally, Fe2O3 exhibits an observable peak at ca. 2.76 Å, attributed to the Fe-Fe interaction in the second coordination shell, while such a signal is absent in CNT-D-O-Fe due to its unique single-atom coordination environment. According to the above analysis, the vacancy defect modifying oxygen-coordinated iron single atomic site (Fe–O4) is used as the model structure (the inset of Fig. 3c) to fit the FT-EXAFS spectrum. The fitting results in k and R spaces are well matched with the experimental results (Fig. 3c and Fig. S14, ESI†). The fitting model structure based on the experimental data shows the Fe–O coordination number of ca. 3.60 with a bonding length of ca. 2.06 Å (Table S3, ESI†). Besides, the wavelet transform (WT) plots (Fig. 3d–f) of Fe foil, Fe2O3, and CNT-D-O-Fe are performed according to the values of k and R spaces to better visualize the corresponding EXAFS spectra. With regard to the CNT-D-O-Fe, only one main peak at 1.3–2.0 Å and 4–7 Å−1 is assigned to the Fe–O bond, which is obviously different from the Fe–O bond (1.2–1.6 Å and 3–7 Å−1) in Fe2O3. Besides, another strong peak for Fe2O3 can be attributable to the Fe–Fe shell in oxides.41,42 The above results involving the composition and fine structure fully demonstrate that the in situ partial etching functionalization strategy induced by iron oxide successfully prepares the oxygen-coordinated single-atom iron site on defects-rich CNT (CNT-D-O-Fe).
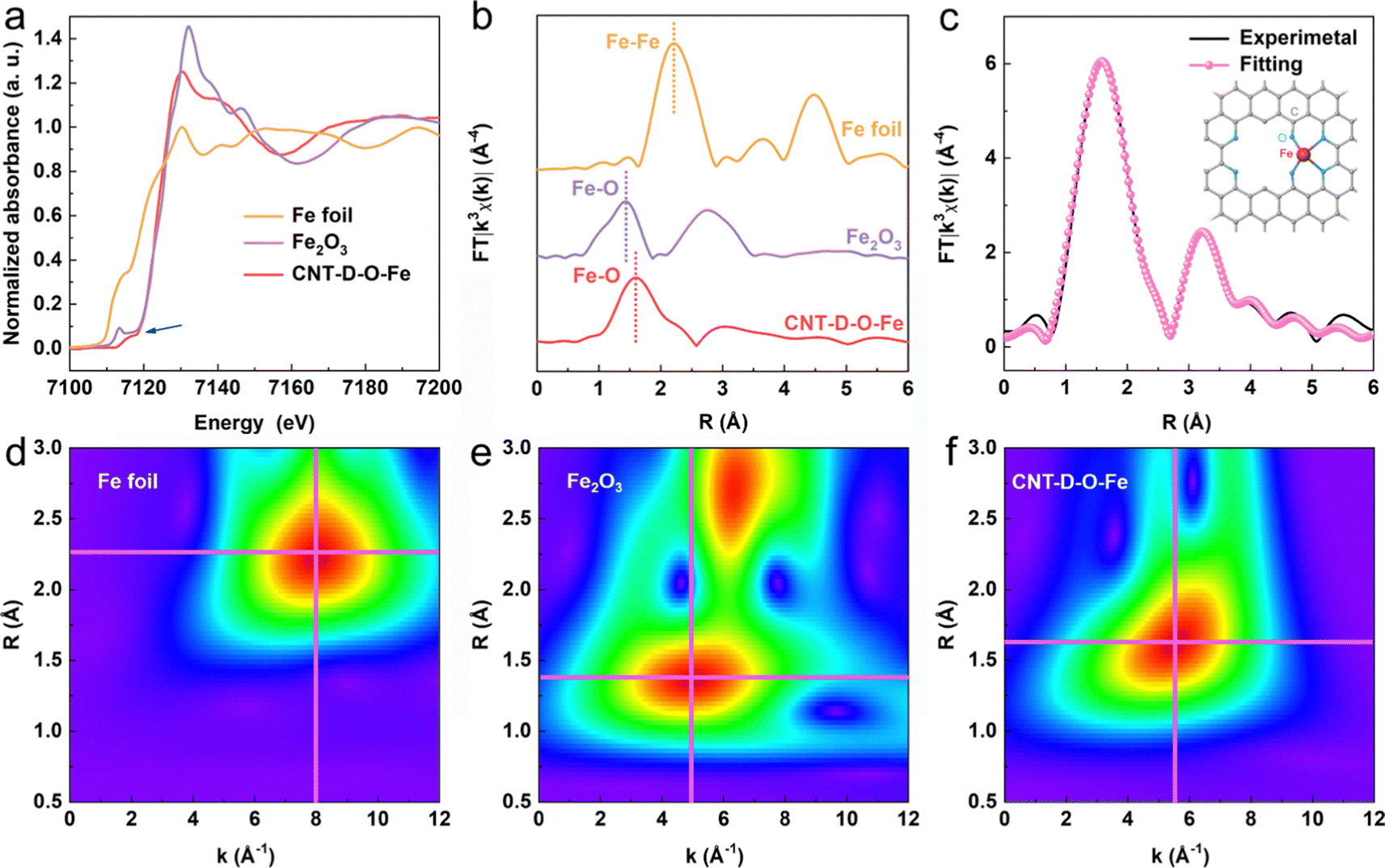 | ||
| Fig. 3 (a) XANES and (b) FT-EXAFS of Fe foil, Fe2O3, and CNT-D-O-Fe, the intensity of Fe foil is halved. (c) The fitting curves of Fe K-edge EXAFS spectra of the CNT-D-O-Fe in R space and the optimized Fe–O4 structure with defects (the inset, gray ball: C atom, blue ball: O atom, red ball: Fe atom). WT plots of (d) Fe foil, (e) Fe2O3, and (f) CNT-D-O-Fe based on the EXAFS. | ||
Electrocatalytic 2e−-ORR performance
The ORR catalytic performances toward H2O2 production for CNT-D-O-Fe and the as-prepared control materials (CNT-D-O, CNT-O-Fe, and CNT) are evaluated in the O2-saturated 0.1 M KOH solution (Fig. S15, ESI†) via the rotating ring disk electrode (RRDE). The linear sweep voltammograms (LSV) recorded at a rotation speed of 1600 rpm are shown in Fig. 4a. The pristine CNT only displays an unfavorable cathodic current density (−2.20 mA cm−2 at 0.550 V vs. reversible hydrogen electrode (RHE)) and the low onset potential of 0.771 V vs. RHE (determined at the current density of 0.150 mA cm−2) among the group of curves.4 After introducing abundant vacancy defects and oxygen-coordinated single-atom iron sites, the ORR reduction current density of CNT-D-O-Fe greatly increases (−2.72 mA cm−2 at 0.550 V vs. RHE) with the high onset potential of 0.823 V vs. RHE. However, the catalysts with the modification of iron sites (CNT-O-Fe, −2.36 mA cm−2 at 0.550V vs. RHE, 0.776 V vs. RHE) or oxygen-induced vacancy defects (CNT-D-O, -2.48 mA cm−2 at 0.550 V vs. RHE, 0.803 V vs. RHE) both show inferior reduction current density and lower onset potential compared with the CNT-D-O-Fe. The Tafel plots in Fig. 4b exhibit similar slopes (approximately 80 mV dec−1) for the samples in this study, suggesting an identical reaction mechanism with the first electron transfer to O2 being the rate-determining step under Temkin adsorption isotherm conditions.43 Additionally, the kinetic current density of CNT-D-O-Fe at 0.550 V vs. RHE is 13.4 mA cm−2 (Fig. 4c). This value is higher than those of other catalysts, including CNT-D-O (12.1 mA cm−2), CNT-O-Fe (9.62 mA cm−2), and CNT (8.52 mA cm−2), further highlighting the superior intrinsic catalytic performance of CNT-D-O-Fe. The results imply that the defective structure and oxygen-coordinated Fe single-atom sites could efficiently contribute to the ORR kinetic rate.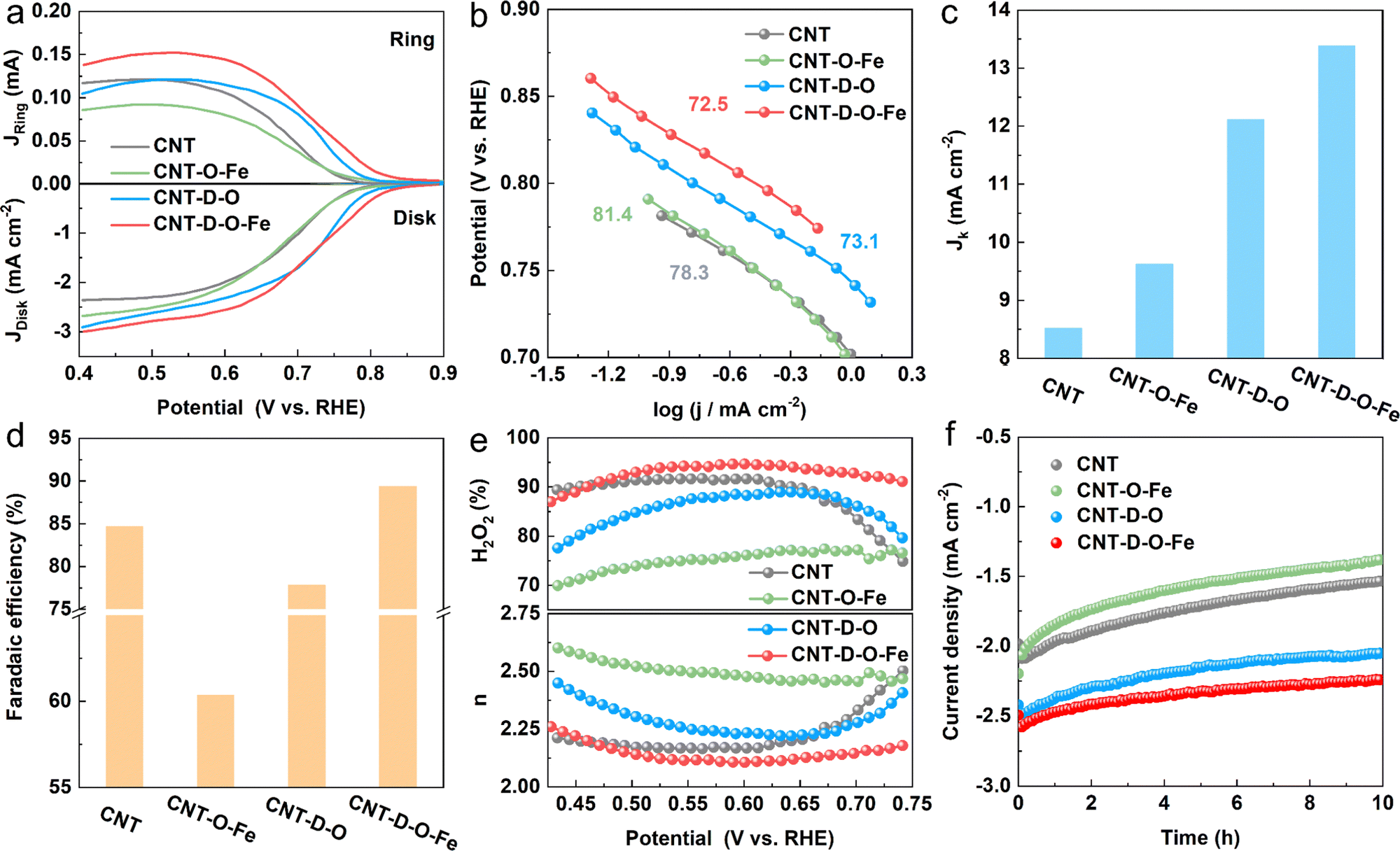 | ||
| Fig. 4 (a) LSV curves (ring current and disk current density, respectively), (b) the Tafel plots of CNT, CNT-O-Fe, CNT-D-O, and CNT-D-O-Fe; (c) Jk and (d) faradaic efficiency for H2O2 at 0.550 V vs. RHE, (e) electron transfer number and selectivity for H2O2; (f) stability test of the catalytic materials at 0.550 V vs. RHE. | ||
The faradaic efficiency, selectivity of H2O2, and the electron transfer number were calculated based on the corresponding ring and disk current. As shown in Fig. 4 and Fig. S16 (ESI†), CNT-D-O-Fe displays high faradaic efficiency (89.4%) and selectivity for H2O2 (94.4%) at 0.550 V vs. RHE, which is better than CNT-D-O, CNT-O-Fe, CNT and some previously reported catalysts (Table S4, ESI†). Furthermore, compared with CNT-D-O, CNT-O-Fe, and CNT, CNT-D-O-Fe is more likely to undergo ORR via the two-electron transfer pathway as shown in Fig. 4e.
In addition, although CNT-O-Fe and CNT-D-O display higher ORR reduction current densities, their H2O2 selectivity (75.3% and 87.5%, respectively) is lower than that of the CNT (91.7%). This difference suggests that the individual introduction of vacancy defects or iron single-atom sites would not enhance the H2O2 production efficiency. Nevertheless, the synergistic combination of oxygen-coordinated iron single-atom sites (Fe–O4) and vacancy defects in CNT-D-O-Fe greatly increases the ORR activity (higher reduction current density) while promoting the two-electron pathway to improve the H2O2 selectivity. Chronoamperometry (at 0.550 V vs. RHE for 600 min) was conducted to evaluate the catalytic stability. The current density of CNT-D-O-Fe shows little noticeable fluctuation and can retain about 90.2% of the initial current density after the electrolysis for 10 h. But the current densities of CNT-O-Fe and CNT-D-O are only 84.7% and 63.0% of the initial value, which reveals that the existence of defects remarkably enhances the catalytic stability of iron single-atom sites in CNT-D-O-Fe. It is postulated that the introduction of the vacancy defect could increase the unsaturation of the carbon substrate, thereby enhancing the interaction between single atomic sites and the substrate. This effectively mitigated the dissolution and aggregation of metal atoms during the catalytic process.44,45 So the CNT-D-O-Fe shows high catalytic stability.
Theoretical insights into the electrocatalyst for the ORR mechanism
To investigate the mechanism of the advanced two-electron ORR catalytic performance of CNT-D-O-Fe in depth, the optimized model structures (Fig. 5a) including CNT-D-O-Fe, CNT-O-Fe, CNT-D-O, CNT-O, and CNT-D were constructed to perform the density functional theory (DFT) calculations. Furthermore, Fig. 5b and Fig. S17 (ESI†) provide the side and top views of the optimal theoretical models (CNT-D-O-Fe and CNT-O-Fe) with and without adsorption of O2 and *OOH species, respectively.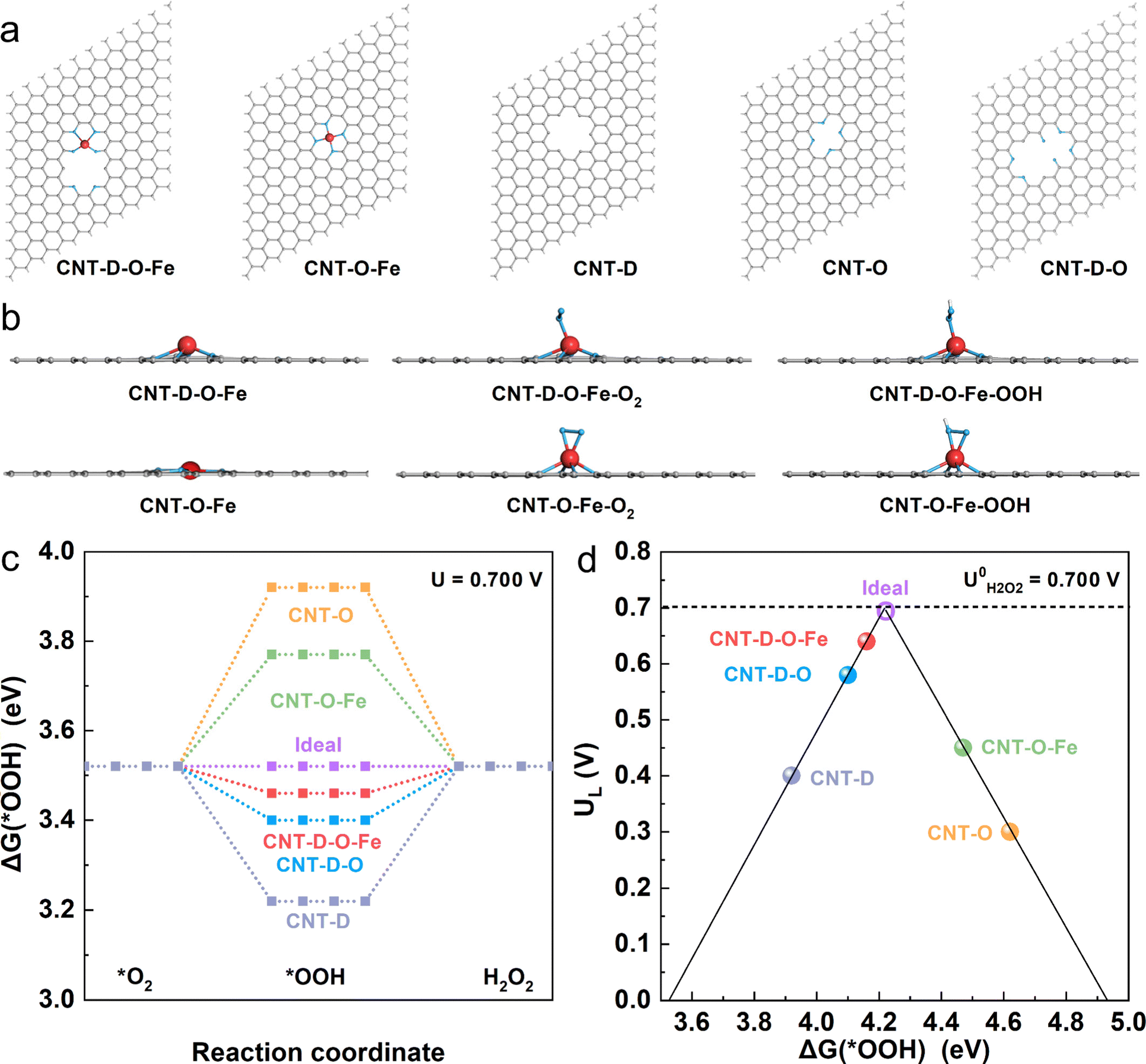 | ||
| Fig. 5 (a) The optimized structures for CNT-D-O-Fe, CNT-O-Fe, CNT-D, CNT-O, and CNT-D-O; (b) the side view of the optimal theoretical models (CNT-D-O-Fe, CNT-O-Fe) with and without adsorption of O2 and *OOH species (gray ball: C atom, blue ball: O atom, red ball: Fe atom, white ball: H atom). (c) Free-energy diagram for O2-to-H2O2 on these optimal model structures at the equilibrium potential (U = 0.700 V). (d) Catalytic activity volcano plot with the calculated limiting potential (UL) as the y-axis and the calculated free energy of *OOH as the x-axis. The standard redox potential of the two-electron ORR (0.700 V) is displayed via the horizontal dashed line. | ||
Clearly, the introduction of vacancy defects alters the configuration of the iron single atomic site and subsequently affects the adsorption state of both O2 and *OOH species. This fundamental alteration serves as the underlying reason for the observed disparity in the catalytic activity. In principle, the two-electron ORR first undergoes one-electron reduction (from O2 to *OOH), and then *OOH is further reduced to H2O2. Since both processes involve *OOH intermediates, the free energy of adsorbed *OOH species (ΔG(*OOH)) serves as a critical descriptor for evaluating the catalytic activity of the two-electron ORR that produces H2O2.46–49 To this end, ΔG(*OOH) of all model structures at the equilibrium potential (0.700 V vs. RHE) was calculated and is shown in Fig. 5c. Obviously, the ΔG(*OOH) of CNT-D-O-Fe (3.46 eV) is very close to the ideal value (3.52 eV) and indicates a moderate adsorption strength for *OOH. In contrast, the adsorption strength of *OOH on CNT-O (3.92 eV) and CNT-O-Fe (3.77 eV) is too weak, while it is too strong for CNT-D-O (3.40 eV) and CNT-D (3.22 eV). The limiting potential (UL) is defined as the maximum potential at which both two one-electron reduction steps (from O2 to *OOH and subsequent reduction from *OOH to H2O2) become thermodynamically favorable.50 Therefore, the activity volcano plots based on the UL and ΔG(*OOH) are summarized to further underline the intrinsic activity of the catalyst models (Fig. 5d). The most efficient activity with zero overpotential is the vertex value of the volcano at the equilibrium potential (0.700 V vs. RHE).47,50,51 Notably, the CNT-D-O-Fe exhibits the highest catalytic activity for the two-electron reduction of O2 to H2O2 with an overpotential of merely 0.06 V. This value is essentially lower than those of other theoretical models, including CNT-O-Fe (0.25 V), CNT-D-O (0.12 V), CNT-O (0.40 V), and CNT-D (0.30 V). The computational results suggest that the synergy between vacancy defect and oxygen-coordinated environment effectively improves the adsorption strength of *OOH intermediates on single atomic iron sites, resulting in robust ORR catalytic activity and high H2O2 selectivity (Fig. 4).
Conclusions
In summary, we have successfully designed and synthesized CNT-D-O-Fe, featuring oxygen-coordinated iron single-atom sites (Fe–O4) modified with vacancy defects. CNT-D-O-Fe exhibits highly efficient ORR catalytic activity and remarkable H2O2 selectivity when compared to other catalysts such as CNT, CNT-D-O, and CNT-O-Fe, with the presence of vacancy defects contributing to its robust catalytic stability. Theoretical calculations further confirm the enhancement of ΔG(*OOH) for CNT-D-O-Fe, resulting from the synergistic effect between vacancy defects and oxygen-coordinated environments. Our work provides a revealing example of defect-stabilized and oxygen-coordinated metal single-atom materials as advanced catalysts for the ORR to produce H2O2. Furthermore, it offers a deeper insight into the roles of coordination environments and defects on catalytic performance, and also opens up new possibilities for designing unique single-atom catalysts with high activity and selectivity. We believe that our findings could contribute significantly to the development of versatile and low-cost electrocatalysts for more important reactions beyond oxygen reduction to hydrogen peroxide.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P.L. acknowledges the funding support from the National Nature Science Foundation of China (No. 52202372), the Sichuan Science and Technology Program (2023NSFSC0436 and 2023NSFSC0089), and the Fundamental Research Funds for the Central Universities (YJ2021151). G.Y. acknowledges the funding support from the Welch Foundation Award F-1861, the Norman Hackerman Award in Chemical Research by the Welch Foundation, and the Camille Dreyfus Teacher-Scholar Award. T.G. acknowledges the Chengdu University new faculty start-up funding (No. 2081920074). The authors would like to thank Xie Han from Shiyanjia Lab (www.shiyanjia.com) for the XPS tests.Notes and references
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- H. Gong, Z. Wei, Z. Gong, J. Liu, G. Ye, M. Yan, J. Dong, C. Allen, J. Liu, K. Huang, R. Liu, G. He, S. Zhao and H. Fei, Adv. Funct. Mater., 2022, 32, 2106886 CrossRef CAS.
- K. H. Koh, A. H. Bagherzadeh Mostaghimi, Q. Chang, Y. J. Kim, S. Siahrostami, T. H. Han and Z. Chen, EcoMat, 2022, 5, e12266 Search PubMed.
- Y. Wu, Y. Ding, X. Han, B. Li, Y. Wang, S. Dong, Q. Li, S. Dou, J. Sun and J. Sun, Appl. Catal., B, 2022, 315, 121578 CrossRef CAS.
- Z. Deng and X. Wang, Nano Res., 2022, 15, 4599–4605 CrossRef CAS.
- P. Wang, Z. Jin, P. Li and G. Yu, Chem. Catal., 2022, 2, 1277–1287 CrossRef CAS.
- T. Gao, C. Zhou, X. Chen, Z. Huang, H. Yuan and D. Xiao, J. Mater. Chem. A, 2020, 8, 18367–18375 RSC.
- W. Qiu, X. Chen, Y. Liu, D. Xiao, P. Wang, R. Li, K. Liu, Z. Jin and P. Li, Appl. Catal., B, 2022, 315, 121548 CrossRef CAS.
- W. Qiu, M. Xie, P. Wang, T. Gao, R. Li, D. Xiao, Z. Jin and P. Li, Small, 2023, 2300437 CrossRef CAS PubMed.
- Z. Jin, Anal. Chem., 2023, 95, 6477–6489 CrossRef CAS PubMed.
- T. Gao, S. Yu, Y. Chen, X. Li, X. Tang, S. Wu, B. He, H. Lan, S. Li, Q. Yue and D. Xiao, J. Colloid Interface Sci., 2023, 642, 120–128 CrossRef CAS PubMed.
- Q. Yang, W. Xu, S. Gong, G. Zheng, Z. Tian, Y. Wen, L. Peng, L. Zhang, Z. Lu and L. Chen, Nat. Commun., 2020, 11, 5478 CrossRef CAS PubMed.
- E. Jung, H. Shin, W. Hooch Antink, Y. E. Sung and T. Hyeon, ACS Energy Lett., 2020, 5, 1881–1892 CrossRef CAS.
- Z. Wang, Q. K. Li, C. Zhang, Z. Cheng, W. Chen, E. A. McHugh, R. A. Carter, B. I. Yakobson and J. M. Tour, ACS Catal., 2021, 11, 2454–2459 CrossRef CAS.
- M. Melchionna, P. Fornasiero and M. Prato, Adv. Mater., 2019, 31, 1802920 CrossRef PubMed.
- Y. Jia, Z. Xue, J. Yang, Q. Liu, J. Xian, Y. Zhong, Y. Sun, X. Zhang, Q. Liu, D. Yao and G. Li, Angew. Chem., Int. Ed., 2022, 61, e202110838 CAS.
- J. H. Kim, Y. T. Kim and S. H. Joo, Curr. Opin. Electrochem., 2020, 21, 109–116 CrossRef CAS.
- P. Li, Z. Fang, Z. Jin and G. Yu, Chem. Phys. Rev., 2021, 2, 041305 CrossRef CAS.
- Z. Jin, P. Li, Z. Fang and G. Yu, Acc. Chem. Res., 2022, 55, 759–769 CrossRef CAS PubMed.
- T. Gao, X. Tang, X. Li, S. Wu, S. Yu, P. Li, D. Xiao and Z. Jin, ACS Catal., 2022, 13, 49–59 CrossRef.
- Z. Jin, P. Li, Y. Meng, Z. Fang, D. Xiao and G. Yu, Nat. Catal., 2021, 4, 615–622 CrossRef CAS.
- Z. Jin and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12651 CrossRef CAS PubMed.
- P. Li, R. Li, Y. Liu, M. Xie, Z. Jin and G. Yu, J. Am. Chem. Soc., 2023, 145, 6471–6479 CrossRef CAS PubMed.
- P. Li, L. Liao, Z. Fang, G. Su, Z. Jin and G. Yu, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2305489120 CrossRef PubMed.
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li, Q. Jia, S. Mukerjee, R. Yang, Z. Jiang, D. Su, C. Liu and W. Xing, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng, C. Yang, Z. Li, X. Wang, S. Hao, Y. Chen and M. Liu, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- H. Yang, Z. Li, S. Kou, G. Lu and Z. Liu, Appl. Catal., B, 2020, 278, 119270 CrossRef CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cançado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1290 RSC.
- T. Gao, C. Zhou, Y. Zhang, Z. Jin, H. Yuan and D. Xiao, J. Mater. Chem. A, 2018, 6, 21577–21584 RSC.
- T. Gao, X. Li, X. Chen, C. Zhou, Q. Yue, H. Yuan and D. Xiao, Chem. Eng. J., 2021, 424, 130416 CrossRef CAS.
- T. Gao, S. Wu, X. Li, C. Lin, Q. Yue, X. Tang, S. Yu and D. Xiao, Inorg. Chem. Front., 2022, 9, 3598–3608 RSC.
- R. Li, T. Gao, P. Wang, W. Qiu, K. Liu, Y. Liu, Z. Jin and P. Li, Appl. Catal., B, 2023, 331, 122677 CrossRef CAS.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- X. Li, Y. Guo, T. Gao, P. Li, Z. Jin and D. Xiao, ACS Appl. Mater. Interfaces, 2021, 13, 57411–57421 CrossRef CAS PubMed.
- Z. Mou, J. Peng, R. Yan, Q. Yang, B. Zhao and D. Xiao, Carbon, 2023, 203, 11–20 CrossRef CAS.
- Z. Mou, R. Yan, J. Peng, Y. Li, Z. Huang, Z. Wang, B. Zhao and D. Xiao, Chem. Eng. J., 2023, 465, 142986 CrossRef CAS.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- J. M. Vega and H. Kamin, J. Biol. Chem., 1977, 252, 896–909 CrossRef CAS PubMed.
- N. Ramaswamy and S. Mukerjee, J. Phys. Chem. C, 2011, 115, 18015–18026 CrossRef CAS.
- Y. Zhang, L. Guo, L. Tao, Y. Lu and S. Wang, Small Methods, 2019, 3, 1800406 CrossRef.
- P. Zhu, P. Song, W. Feng, D. Zhao, T. Liu, J. Zhang and C. Chen, J. Mater. Chem. A, 2020, 10, 17948 RSC.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- H. Li, P. Wen, D. S. Itanze, Z. D. Hood, S. Adhikari, C. Lu, X. Ma, C. Dun, L. Jiang, D. L. Carroll, Y. Qiu and S. M. Geyer, Nat. Commun., 2020, 11, 3928 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Nat. Commun., 2020, 11, 2178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00882g |
| This journal is © The Royal Society of Chemistry 2023 |