
Sacrifice and valorization of biomass to realize energy exploitation and transformation in a photoelectrochemical way
Daobin
Tang
 ,
Jianguo
Liu
*,
Xinghua
Zhang
,
Lungang
Chen
,
Longlong
Ma
and
Qi
Zhang
*
,
Jianguo
Liu
*,
Xinghua
Zhang
,
Lungang
Chen
,
Longlong
Ma
and
Qi
Zhang
*
Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing, 210096, China. E-mail: zhangqiseu@seu.edu.cn; liujg@seu.edu.cn
First published on 25th August 2023
Abstract
Photoelectrochemistry (PEC) transfers photoexcited electrons from a photocatalyst to a cathode through a wire, to avoid, to a considerable extent, the recombination of charge carriers in solitary photocatalysis. The moderate bias also consumes less energy than electrocatalysis, rendering PEC a green and promising transformation approach. PEC biomass conversion can be performed at a lower bias than PEC water splitting, and biomass holds great potential for valorization as the world's largest renewable resource. In recent years, there has been escalating interest in the study of PEC valorization from the sacrifice of biomass, but only a few reviews have systematically and specifically addressed it. This review discusses the conversion mechanism of organics, the catalyst and system configuration, and the effect of other variables in detail based on different structures and types of biomass substrates. A brief outlook on the development of the field is outlined in the end as well, and PEC is expected to be an alternative low-carbon, energy-saving, multi-objective route to biomass valorization.
1. Introduction
Except for a few resource-based countries, there is a burgeoning concern about the reserves of non-renewable fossil resources around the world. The waste and harmful gases generated by the consumption of fossil resources threaten the global ecological environment significantly. The search for alternative green raw materials and more environmentally sound and efficient technological conditions is currently a global trend. In sustainable development, solar energy is regarded as a key technology due to its abundance and worldwide availability for collection and conversion.1,2 Biomass, derived from plant photosynthesis, is an energy carrier of solar energy and is considered one of the most promising alternatives to combat the energy crisis and environmental problems of fossil fuels.3 Carbohydrates and/or aromatic ring-rich fractions in biomass can be converted into biofuels or chemicals by fermentation.4Biomass is mainly composed of cellulose, hemicellulose, and lignin, and the relative content of each component varies with the type of biomass. Taking lignocellulose, about 100 billion tons of production per annum globally, as an example, the content of cellulose and hemicellulose is about 40–50 wt% and 20–30 wt%, respectively, and this is mainly a C5–C6 carbohydrate polymer. In contrast, the content of lignin is slightly lower, about 15–25 wt%.5 The structures also differed to some extent between the different components (Fig. 1). Cellulose is a homopolymer composed of β-D-glucopyranose units linked by a β-glycosidic bond that can be deconstructed into glucose monomers.6 Hemicelluloses are amorphous branched polymers with a low polymerization degree (∼200). Hemicellulose is more readily hydrolyzed than cellulose, which allows it to be removed under more benign reaction conditions. Hemicellulose contains mainly pentoses, hexoses, and glyoxylates. Pentoses (xylose and arabinose) and hexoses (glucose, galactose, mannose, rhamnose, and fucose) are sources of sugars that can be valorized to biofuels and renewable chemicals.6 Lignin, on the other hand, contains the world's largest renewable aromatic monomer and is an integral part of the cell wall. However, the utilization of lignin is not satisfactory. The pulp and paper industry produces about 5 million metric tons of lignin each year.7 However, 95% of lignin remains abandoned or is burned during the biorefining process due to its complex and irregular structure.7,8 Lignin has an amorphous three-dimensional network structure and the different aromatic monomers are interconnected by aliphatic C–C and C–O bonds. The polymers contain many hydroxyl and methoxy groups and the three major monomeric units of lignin can be distinguished according to the number of methoxy groups on the benzene ring: p-coumaryl alcohol, coniferyl alcohol, and sinapyl alcohol.9 The abundant aromatic content and the variety of oxygen-containing functional groups enable lignin to have gigantic potential for upgrading.
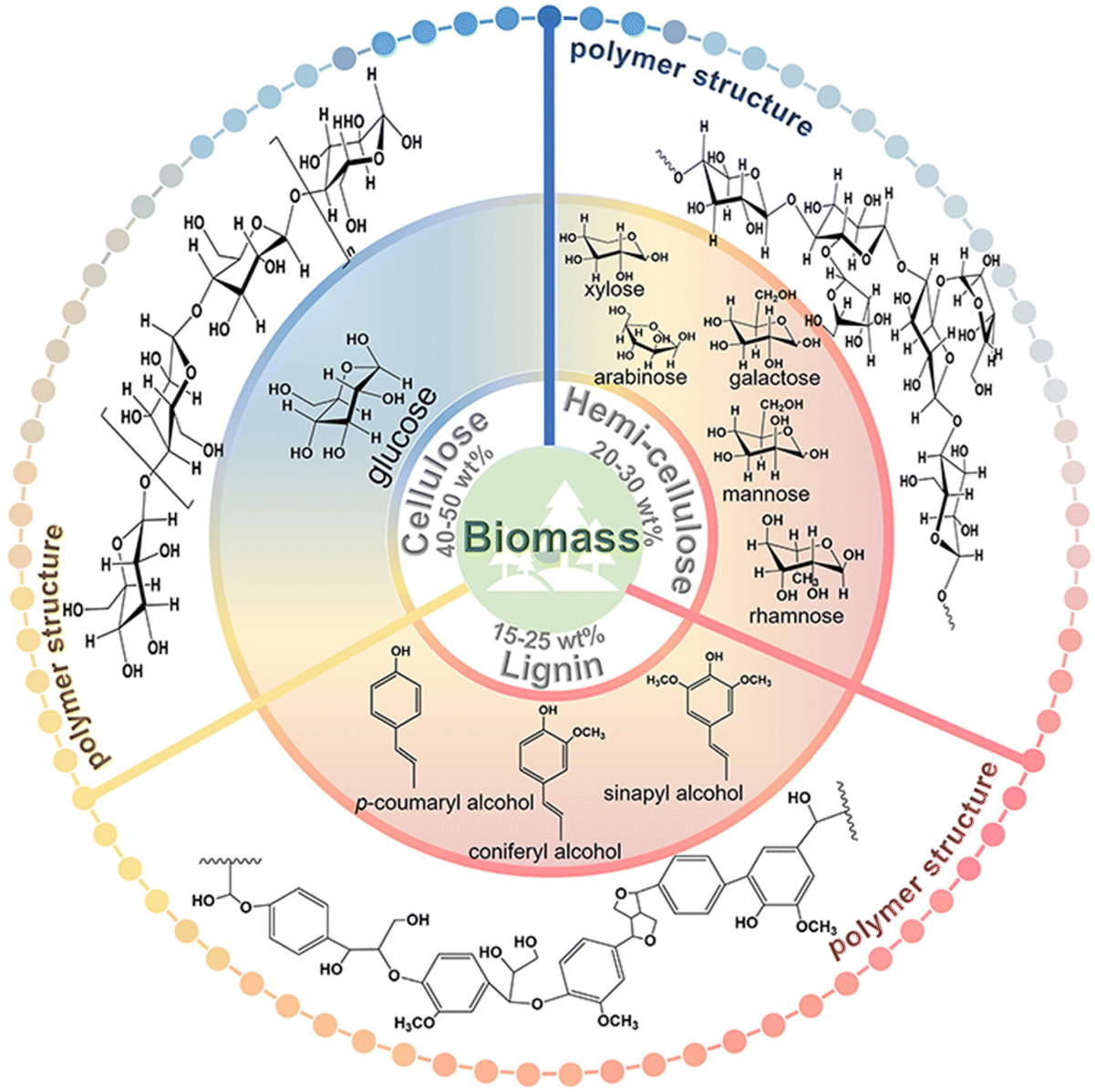 | ||
| Fig. 1 The polymer structures and depolymerized monomers/monomeric units of the three major components of biomass. | ||
Biomass feedstock is commonly treated as a “fuel” source and converted into a fuel form through pyrolysis, saccharification, and fermentation, such as H2, alcohols, and polysaccharides.10 These transforming processes burden the overall process and lead to unwanted energy consumption, so it would be better to utilize (hemi-)cellulose or lignin directly as a “fuel”.11 This purpose can be achieved through photocatalytic, electrocatalytic, and photoelectrochemical (PEC) systems under mild conditions (Fig. 2). For simple photocatalysis, illumination of light with a specific wavelength range or visible light (collectively referred to as incident light) onto a semiconductor catalyst excites electrons (e−) in the valence band (VB) to the conduction band (CB), while the same number of holes (h+) is generated in the VB. h+ is strongly oxidizing and can carry out the oxidation of the substrate at the VB. Correspondingly, reduction reactions can be performed at the CB. Shang et al.12 reviewed in detail the study of bifunctional photocatalytic reaction systems in which photogenerated electrons and holes can be exploited simultaneously, with the organics acting as sacrificial reagents to eventually produce high value-added products. However, the notable drawbacks of photocatalysis are low reaction rates and low quantum efficiency due to the recombination of photogenerated charges.13 At the same time, the insolubility of biomass hinders the supply of “fuel” in the liquid phase. The effective photodegradation of solid substrates is also challenging since a large contact area between the substrate and the photocatalyst is essential in photocatalysis.
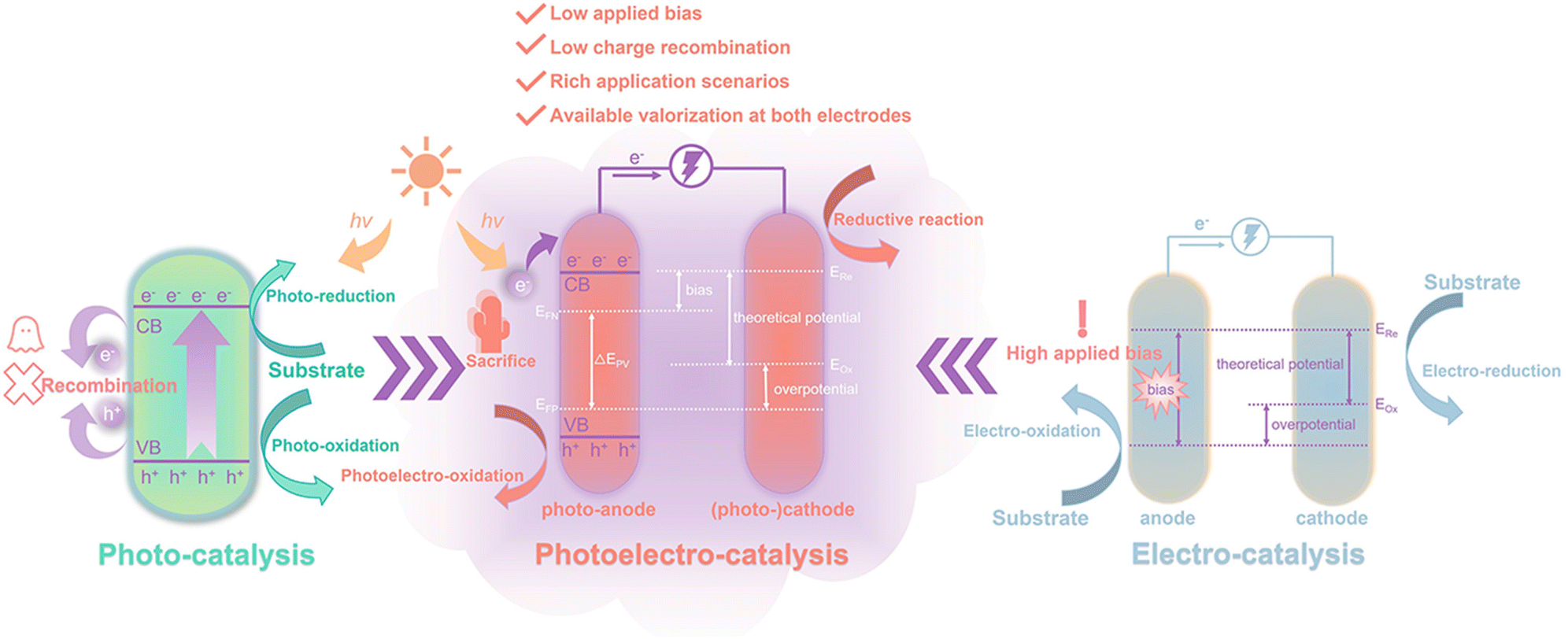 | ||
| Fig. 2 Comparison of photo-, electro-, and photoelectro-catalysis. EFP and EFN are the quasi Fermi levels of holes and electrons, respectively; EOx and ERe are the oxidative and reductive potentials of the substrate, respectively. | ||
Electrocatalysis is able to efficiently convert electrical energy into chemical energy and the electrical energy can be driven by sustainable energy sources (e.g. solar photovoltaic, wind, and hydrogen).14,15 In an electrocatalytic system, all electrons are induced by an external bias, which determines the current density of the circuit. The actual applied voltage is often higher than the theoretical energy requirement to compensate for overpotential or other energy losses.16 In contrast, semiconductor-based PEC systems that absorb and convert solar energy are capable of alleviating the dependence on pure electricity.
2. Working configurations of PEC systems
Photoelectrochemical cells use solar and electrical energy to promote target chemical reactions. In 1972, Honda et al.17 achieved PEC water splitting by coupling a crystalline n-TiO2 photoanode and a platinum cathode under ultraviolet (UV) radiation. Since then, tremendous applied advancements in the field of PEC using a range of photoanodes have been emerging.18 Numerous studies have spotlighted the application of water splitting, which is a thermodynamically uphill reaction.19,20 During water splitting, the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) occur simultaneously at the cathode and anode, respectively. The reaction kinetics of the OER is slower than that of the HER because the OER is a four-electron transfer process accompanied by the formation of O–O bonds, whereas the HER is only a two-electron transfer process. In other words, the OER is the rate-limiting step for water cleavage.21–23 The oxidation of biomass and its derivatives is a promising alternative to the OER with a low oxidative potential.24–26 The substrate being oxidized, which can be photoconverted into high-value organic products, is usually waste or pollutants left over from industrial processes and production.27 The whole process is simple and efficient; hence PEC decomposition of biomass-derived waste/pollutants is also recognized as one of the most promising solutions.20,28–322.1. Reaction mechanism of PEC
As shown in Fig. 2, photogenerated electrons on the VB are excited to the CB under illumination, and the same number of photogenerated holes is generated in the VB. Due to the presence of an external electric field, the highly oxidizing holes are transferred to the semiconductor surface to oxidize the substrate, and the photogenerated electrons are transferred along the external circuit to the cathode for the reduction reaction. The photogenerated carriers transported in the circuit create the photocurrent. The photovoltage generated at the photoanode can be determined by the potential difference (ΔEPV) between the quasi Fermi energy levels of electrons and holes. For a particular photoelectrochemical reaction and semiconductor, the photocurrent density of the PEC system normally depends on the incident light intensity and the applied external bias. In terms of the operation mechanism, the photogenerated charge resulting from light illumination onto the catalyst is the essential reason for the catalytic activity of the catalytic system. Therefore, the incident light intensity plays a prominent role and the maximum current density of the system depends on it. If the incident light intensity is constant, the current density will increase with a higher bias.16During the PEC oxidation of biomass derivatives, the active species driving the substrate in the reaction show more variations compared to water splitting. In addition to the direct contact of photogenerated holes with organic substrates, the hydroxyl radicals (˙OH) generated by the splitting of water and the superoxide radicals (˙O2−) generated by the reduction of soluble oxygen exhibit equally strong oxidative properties, although the oxidation selectivity of ˙OH is relatively low.5 Whether a single reactive species or multiple reactive species play a decisive role in PEC activity needs to be analyzed on the basis of the specific catalytic system. Therefore, the investigation of the specific active species in biomass PEC systems seems to be valuable and indispensable.
2.2. Comparison of PEC, photocatalysis, and electrocatalysis
It is hardly possible to make a photocatalyst undergo both oxidation and reduction half-reactions under the premise of reduced recombination of photogenerated electrons and holes, but the PEC system can be employed for separate studies of cathodes and anodes and allows better testing and rational development of catalysts for oxidation reactions.33 For both electrocatalysis and PEC, electrocatalysis can produce a higher current density than PEC at a high bias. In the case of water splitting, light illumination of several sun powers is required for PEC if a current density of 100 mA cm−2 is to be produced.34 Oxidation reactions of many organics are thermodynamically and kinetically easier to be initiated than those of water, so PEC systems are capable of achieving higher current densities at a lower bias than electrocatalysis.35 A common characteristic of photocatalysis and electrocatalysis is the generation of electrons and the following participation and promotion of subsequent chemical reactions. In addition to photons and electrons induced by photocatalytic and electrocatalytic processes, there are a number of generated radical intermediates that are other common active components, such as carbon-centered radicals, ˙OH, ˙O2−, and chemically generated reactive oxygen species. These components are also applicable in the PEC process, and for more details on the radical induction mechanism, please refer to the review by Wu et al.5As the applications of PEC systems continue to expand, more and more studies are shifting from water splitting to the exploration of the valorization of biomass and its derivatives as sacrificial agents. The oxidation of biomass at the anode builds the bridge of energy transfer for the whole system as a process of electron extraction. The biomass itself is either consumed at the anode or escalated simultaneously with the cathode to valorized products, which is of great research interest. However, a review of the literature that systematically summarizes and explores this research area is currently unavailable. This work will survey the PEC high-value applications of (hemi-)cellulose, lignin, and their derivatives in the last five years. Meanwhile, future research perspectives are proposed based on the discussion on targets of valorization, designs of systems, catalysts, and other impact factors.
3. Valorization application in PEC systems through biomass-based substrates
The majority of current bio-organic substrate conversions take place employing photoelectro-oxidation. Based on the lower oxidative potential of biomass-based substrates compared to water, a large number of different types of biomass and their derived monomers are used for a wide variety of high-value purposes. Cathode sides are being tried for more and more reductive applications beyond efficient H2 production. From the perspective of substrate conversion, the valorization applications at the anode and cathode will be explored separately in detail.3.1. Photoelectro-oxidation at the anode
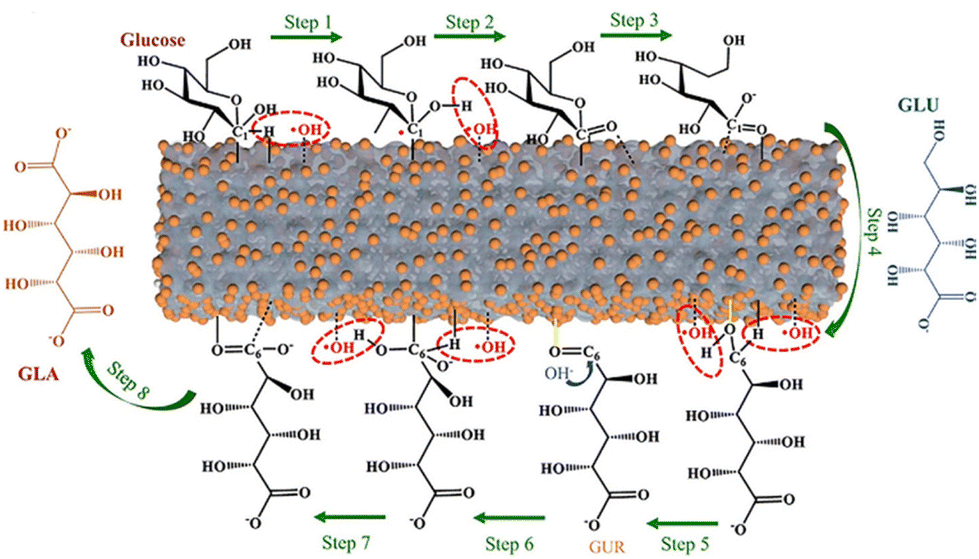
The oxidation performance can be characterized using line scan voltammetry (LSV) curves. The photooxidation of organic substrates can be visualized with lower onset potentials and surging current densities compared to catalytic systems without organic substrates (sometimes can be directly interpreted as water splitting systems), proving that the PEC of organic compounds is thermodynamically and dynamically preferable. The photocurrent can be enhanced to varying degrees by different types of organic compounds.37 However, in contrast to (hemi-)cellulose, lignin has a more complex structure and darker color, which makes direct photocatalysis of the liquid phase system more difficult. Therefore, there are also some differences in the PEC application for (hemi-)cellulose and lignin at the anode.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.40,41 The C5O bond is cleaved by hydrolysis and desorbed from the photoanode to produce gluconic acid.40 The C6-position is finally oxidized through the highly selective oxidation of the primary alcohol by Pt and the binding of the ˙OH radical to yield L-guluronic acid. The C6O bond and re-formed C6–OH activated by holes as well as ˙OH will be regarded as the C1-position and follow the same pathway to eventually produce glucaric acid. The conversion of gluconic acid into L-guluronic acid is the rate-controlling step and Pt plays a key role in the reaction with adsorbed ˙OH radicals.38 The nanostructured semi-transparent WO3 photoanode is capable of oxidizing glucose to gluconic acid and glucaric acid with acidic sulfate under simulated sunlight irradiation. The 2e− oxidation pathway of glucose in this system is a direct transfer of photogenerated holes to the adsorbed organics, followed by the injection of a single-electron reaction intermediate to the CB to form gluconic acid.42 Pan et al.,43 on the other hand, exploited the NiOOH/α-Fe2O3 photoanode to oxidize glucose to formate (FA), where aldose is the main intermediate in the conversion process (Fig. 5a). Glucose was first oxidized to arabinose and FA by C1–C2 bond cleavage. Arabinose underwent similar successive C1–C2 bond ruptures to form the corresponding aldose and finally FA. This catalyst is also highly versatile for real biomass substrates. The PEC performance of the substrates obeys the following order: glucose > (hemi)cellulose > raw biomass.43
O bonds.40,41 The C5O bond is cleaved by hydrolysis and desorbed from the photoanode to produce gluconic acid.40 The C6-position is finally oxidized through the highly selective oxidation of the primary alcohol by Pt and the binding of the ˙OH radical to yield L-guluronic acid. The C6O bond and re-formed C6–OH activated by holes as well as ˙OH will be regarded as the C1-position and follow the same pathway to eventually produce glucaric acid. The conversion of gluconic acid into L-guluronic acid is the rate-controlling step and Pt plays a key role in the reaction with adsorbed ˙OH radicals.38 The nanostructured semi-transparent WO3 photoanode is capable of oxidizing glucose to gluconic acid and glucaric acid with acidic sulfate under simulated sunlight irradiation. The 2e− oxidation pathway of glucose in this system is a direct transfer of photogenerated holes to the adsorbed organics, followed by the injection of a single-electron reaction intermediate to the CB to form gluconic acid.42 Pan et al.,43 on the other hand, exploited the NiOOH/α-Fe2O3 photoanode to oxidize glucose to formate (FA), where aldose is the main intermediate in the conversion process (Fig. 5a). Glucose was first oxidized to arabinose and FA by C1–C2 bond cleavage. Arabinose underwent similar successive C1–C2 bond ruptures to form the corresponding aldose and finally FA. This catalyst is also highly versatile for real biomass substrates. The PEC performance of the substrates obeys the following order: glucose > (hemi)cellulose > raw biomass.43
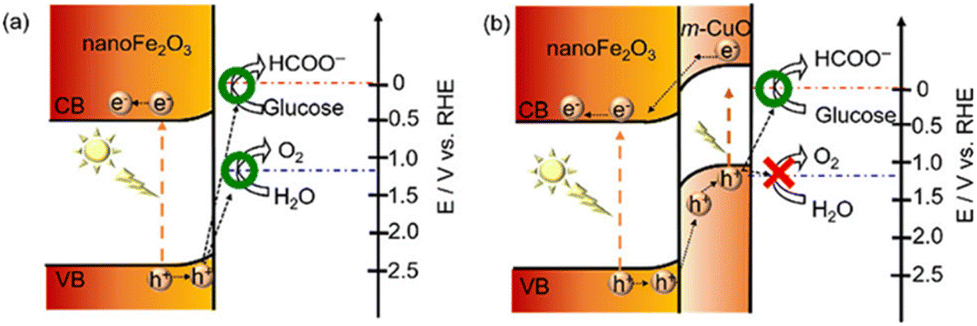 | ||
| Fig. 3 Schematic representation of the electronic band structures of (a) nanoFe2O3 and (b) nanoFe2O3|m-CuO. Adapted from ref. 36 with permission from the Royal Society of Chemistry, copyright 2022. | ||
 | ||
| Fig. 4 Possible reaction pathway from glucose to glucaric acid over the Pt/def-TiO2 photoanode. Adapted from ref. 38 with permission from Springer Nature, copyright 2023. | ||
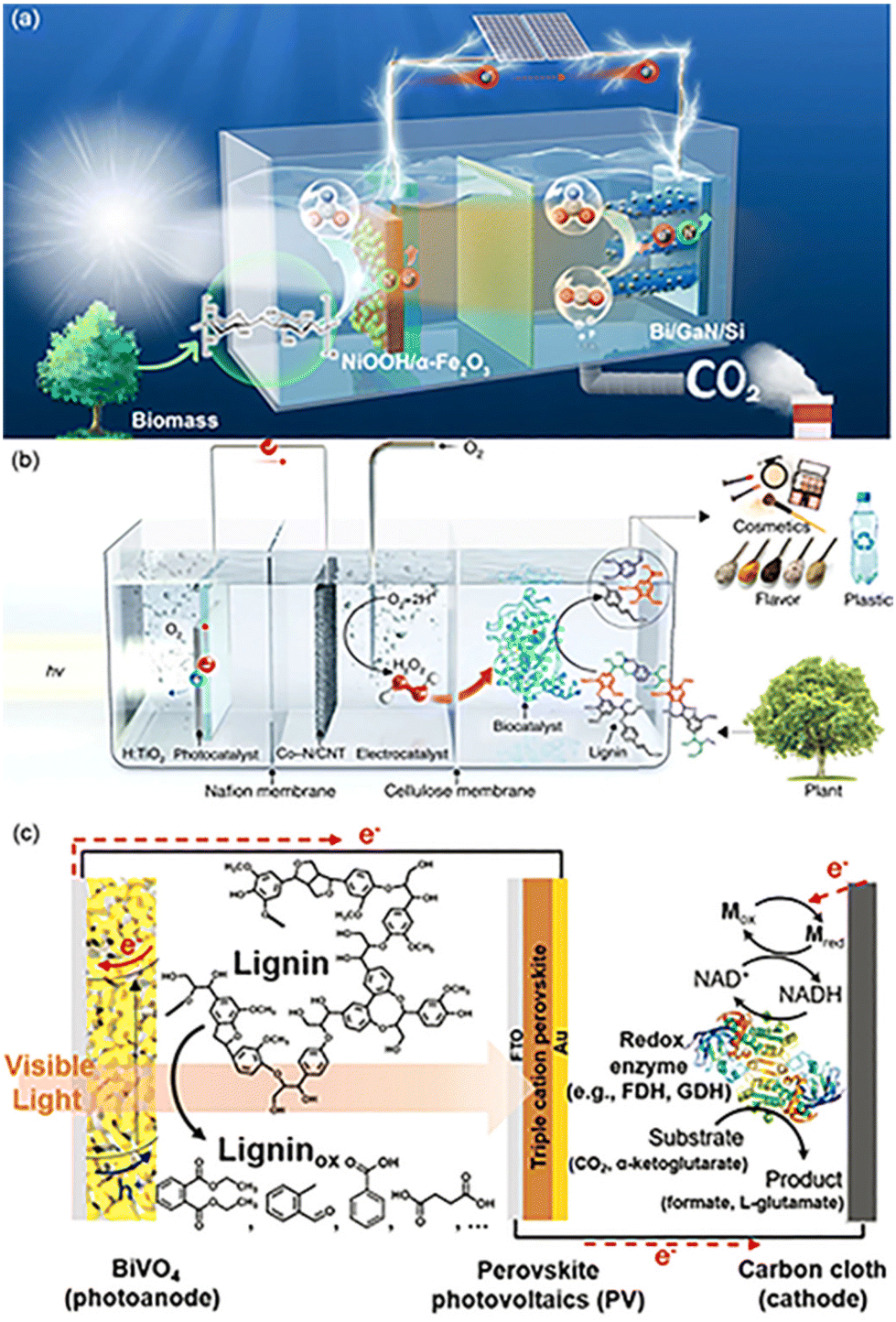 | ||
| Fig. 5 Several designs of PEC configurations. (a) Schematic illustration of the tandem PEC cell for FA production from biomass and CO2. The PEC reactor consists of two compartments of photoanodic biomass oxidation (left-hand side) and photocathodic CO2 reduction (right-hand side) for concurrent FA production. Adapted from ref. 43 with permission from Springer Nature, Copyright 2023. (b) Schematic illustration of the three-compartment photo-electro-biochemical reactor. The photo-electro-biochemical reactor consists of three separate compartments. The first compartment (left-hand side) is the anodic part of the photo-electrochemical cell where water oxidation takes place. The photoanode is connected to the Co–N/CNT cathode through a copper wire for in situ H2O2 generation in the second compartment (middle part). A Nafion proton exchange membrane separates the anodic and cathodic parts of the photo-electrochemical cell. The schematic of the second and third compartments shows the gradual increase in the concentration of photogenerated H2O2 and continuous flow to the biocatalytic system (third compartment), which are ideal conditions for selective lignin conversion. Adapted from ref. 47 with permission from Springer Nature, copyright 2019. (c) Schematic illustration of the lignin-fueled photoelectrochemical platform for solar-driven redox biotransformations. The BiVO4 photoanode promotes the photoelectrochemical oxidation of lignin. The triple-cation perovskite PV forms a tandem structure with the BiVO4 photoanode, providing supplementary photovoltage to drive reactions under zero external bias. The carbon-cloth cathode reduces NAD+ to NADH for the reductive conversion of substrates using redox enzymes. Adapted from ref. 48 with permission from the Royal Society of Chemistry, copyright 2020. | ||
The other common biomass-derived substrate is glycerol. Glycerol is a by-product of the biodiesel production process and comprises roughly one-tenth of the biodiesel mass.44 Glycerol is considered a promising sacrificial agent due to its non-toxic and trihydroxy structure.45 In addition to being sacrificed to CO2 for hydrogen evolution at the cathode, glycerol can be converted into many high-value chemicals such as dihydroxyacetone (DHA), glyceric acid (GLA), glycolic acid (GLCA), lactic acid, acetic acid, and FA.46 The photo-oxidation of glycerol occurs through two main pathways:
(i) Reaction with photogenerated holes directly:
| R1 + nh+ → nH+ + Rox (oxidized product) | (1) |
The photogenerated holes can activate glycerol molecules and efficiently cleave the C–C bonds of C2 and C3 intermediates. Consequently, highly selective FA50,51 and other C1 products are easily available through this catalytic pathway. For example, in the photoelectro-transformation of biomass for hydrogen production, most of the C is eventually converted into CO2.52,53 The N-doped WO3 photoanode, however, is able to reform C from glycerol to CO, since CO itself is an important fuel and chemical that is widely exploited in the synthesis of olefins and aromatics.54,55 During the oxidation of this system, glyceraldehyde is the main liquid product, accompanied by the other liquid product FA. Aldehydes prefer to produce CO, while FA mainly liberates CO2. Glycerol is first directly oxidized by photogenerated holes to glyceraldehyde, which is further de-carbonylated to CO and glycolaldehyde. Glycolaldehyde is then de-carbonylated to CO and formaldehyde. In contrast, aldehyde intermediates such as formaldehyde are readily over-oxidized to acids thereby leading to CO2 (Fig. 6c).49
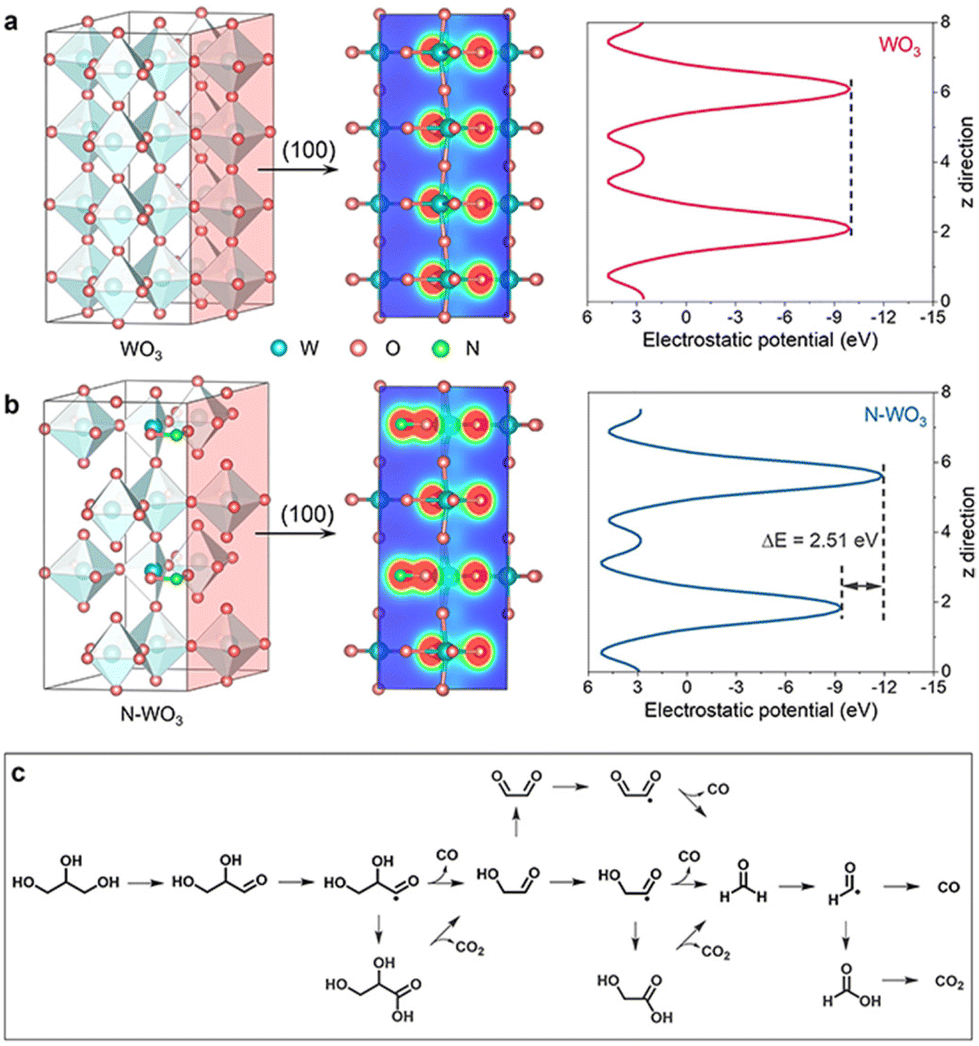 | ||
| Fig. 6 Photoelectronic properties. Electron density isosurface (100) and electrostatic potentials of (a) WO3 and (b) N–WO3. (c) Proposed reaction route for PEC glycerol reforming to CO. Adapted from ref. 49 with permission from John Wiley and Sons, copyright 2022. | ||
(ii) Reaction with some oxidizing species indirectly, taking ˙OH as an example:
| R1 + ˙OH → Rox (oxidized product) | (2) |
DHA has been widely applied as a high-value raw material in the cosmetic, pharmaceutical, fine chemical, and food industries. The price of DHA is $150 per kg, while the price of raw and purified glycerol is only $0.11 and $0.66 per kg, respectively, which makes the catalytic production of DHA economically attractive.56,57 The Bi2O3/TiO2 photoanode was able to PEC oxidize glycerol to DHA with 75.4% selectivity, and the conversion of the system could reach 50%, which is 160 times higher than that by photocatalysis under the same light conditions and 7 times higher than that by electrocatalysis at the same voltage (1 V vs. RHE). The PEC glycerol oxidation is an ˙OH radical-mediated process, and photogenerated holes play an essential role in the activation of H2O by electron spin resonance (ESR), isotope labeling, fluorescent probe experiments, liquid chromatography-mass spectrometry (LC-MS), and free radical quenching experiments. PEC glycerol oxidation is accompanied by a lattice oxygen oxidation process.39 Hence, the catalytic system follows two main parallel reaction mechanisms: (i) oxidation of glycerol by electrophilic ˙OH generated by water oxidation and (ii) dehydrogenation of glycerol by lattice oxygen followed by oxidation of lattice hydroxyl groups by photogenerated holes. The PEC configuration integrated with the Pt-perovskite photocathode and the Cu30Pd70 oxidation catalyst for the transition of three model compounds namely glycerol, ethylene glycol, and glucose revealed selectivities of 79 ± 4%, 95 ± 2%, and 65 ± 7% for glyceric acid, glycolic acid, and gluconic acid, respectively.59 The slightly lower selectivity of gluconic acid may be due to the base degradation, isomerization and other side reactions of glucose over time.60
Tartaric acid (C4H6O6), a biomass derivative rich in hydroxyl and carboxyl groups, is not only adopted as an organic fuel but is also able to complex with many metal ions, such as Bi3+, Cu2+, Fe2+, etc.61,62 Thus, tartaric acid is able to establish a strong complexation with BiVO4, and this complexation also serves as a bridge for charge and energy transfer (Fig. 7b). It not only improves the photoelectric conversion efficiency of BiVO4 but also effectively converts the chemical energy of biomass into H2.58 A current density of 13.54 mA cm−2 can be achieved at 1.23 V vs. RHE, which is 1.8 times the theoretical maximum current density of BiVO4. Organic fuels containing only hydroxyl or carboxyl groups do not maximize PEC performance, while hydroxy-carboxyl-based biomass with other strong complexation capabilities can also contribute to PEC performance. Besides, Lv et al.37 found FA, diethanolamine and triethanolamine as hole sacrificial agents, while methanol, glucose, and urea were ˙OH radical sacrificial agents after PEC oxidation of different kinds of organic compounds using Mn-doped CdS quantum dot-supported TiO2 nanotube arrays (NTAs). Incident photon-to-current efficiency (IPCE) tests indicate a current doubling effect for methanol, where the ˙OH radical (which generates the first photoelectron) leads to a highly reduced α-hydroxymethyl radical.63 The radical then injects a second electron into the photoanode to form formaldehyde.63–65 Each photon produces two photoelectrons, so the theoretical IPCE is up to 200%. In practice, only 130% is achieved at 320 nm due to the limitation of net charge separation and transfer. The three alcohol moieties of triethanolamine show the same current doubling effect as an effective electron donor, with about 160% at 320 nm. 5-Hydroxymethylfuran (HMF) was oxidized to 2,5-furandicarboxylic acid (FDCA) by a 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)-mediated photo-electronic system, which consumed a charge of 55 C. HMF was ultimately oxidized to FDCA through the intermediates of furandialdehyde and furfuraldehyde acid. The yield and FE of FDCA were 100% at 0.7 V vs. RHE.66
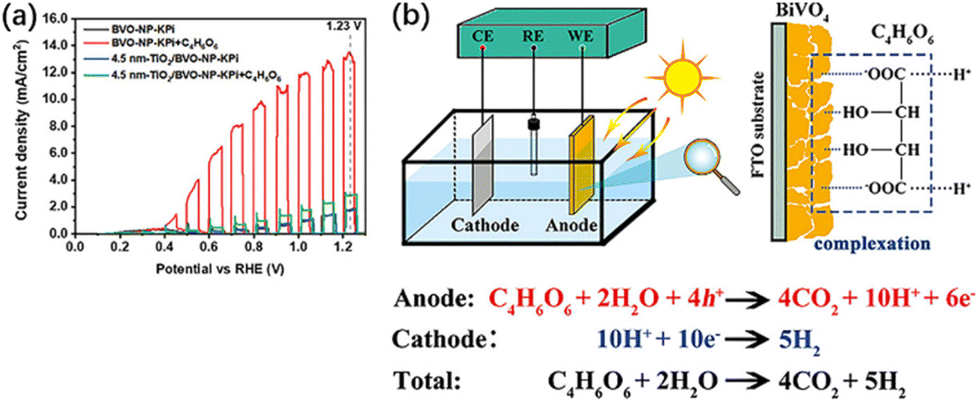 | ||
| Fig. 7 (a) Current density versus potential curves of BVO-NP and 4.5 nm TiO2/BVO-NP photoanodes in potassium phosphate (KPi) and KPi + 0.5 M C4H6O6 solutions under AM 1.5G illumination or in the dark. (b) Schematic illustration of the detailed reaction process in the BiVO4 PEC fuel cell system. Adapted from ref. 58 with permission from John Wiley and Sons, copyright 2022. | ||
In addition to model compounds, PEC oxidation of biomass feedstocks is being progressively explored. Cellulose films deposited on porous TiO2 are used as a fuel and configured with the Pt cathode to decompose cellulose to CO2.11 Carbonyl carbohydrate chains such as gluconic acid, oxalic acid, acetic acid, and FA are intermediate products dissolved in the liquid phase during PEC. In the decomposition process, cellulose is first cleaved to short carboxylic acids, which are further decomposed to CO2. Pre-treated polyethylene terephthalate (PET) powder, real-life commercial PET bottles, and microcrystalline cellulose similarly underwent PEC conversion using the Cu30Pd70 catalyst with the Pt-perovskite photocathode. After 10 h in both compartments, the selectivities of glycolic acid from PET powder and PET bottles were 94% and 91%, respectively, and that of gluconic acid from microcrystalline cellulose was 67%.59 Microalgal biomass can generate renewable energy because it does not need to occupy arable land for growth and is characterized by high lipid yields per unit, short growth cycles, and high environmental adaptability.67 However, the thick cell wall and selectively permeable cell membrane structure make the extraction of lipids difficult.68 Based on this, the PEC system consisting of the N-doped TiO2 nanotube photoanode, a bipolar membrane, and the P–Pd cathode utilizes eventually ˙OH radicals, which are produced from the H2O2 generated from H2 and O2 at both electrodes69,70 by the strong adsorption and activation ability of Pd-based catalysts, to realize this purpose:71
| H2 + Pd0 → Pd0˙H2 → Pd0[H]2 | (3-1) |
| Pd0[H]2 + Pd0˙O2 → Pd0˙H2O2 + Pd0 | (3-2) |
| H2O2 + e− → ˙OH + OH− | (4) |
˙OH radicals can attack the hydrogen bonds among the carbon chains between cellulose and pectin molecules in the algal cell wall.72 ˙OH radicals can also attack diacylglycerides in the cell membrane, leading to peroxidation that breaks carbon chains of unsaturated fatty acids as well as lipids from saturated fatty acids.73 After the cell wall and cell membrane rupture, organic solvents are then allowed to diffuse into the cell to extract the lipids. At the same time, ˙OH also breaks single and double bonds in chlorophyll, which contributes to the decolorization of crude oil.74
Although there are many studies on the PEC conversion of β-O-4 bonds, different conversion outcomes can be achieved. There are four main conversion pathways as follows: (i) oxidation of β-O-4 alcohol to β-O-4 ketone; (ii) Cα–Cβ cleavage of the β-O-4 bond; (iii) CAr–Cα cleavage of the β-O-4 bond; and (iv) Cβ–O cleavage of the β-O-4 bond (Fig. 8). Pathway (i) can be achieved by dye-sensitized photoelectrochemical (DSPEC) systems. The C–O bond of β-O-4 can significantly be weakened after alcohol oxidation of the lignin skeleton, which contributes to the subsequent cleavage.76,77 Kim et al.78 explored the oxidative reaction species of Kraft lignin using α-Fe2O3 (hematite) photoanodes and found that photogenerated holes play a key role in lignin depolymerization. Lignin depolymerization can be accomplished by oxidation of Cα–OH to CαO and subsequent oxidative cleavage of Cα–Cβ. It can be expected that the depolymerization proceeds through pathway (i) and pathway (ii) successively. BiVO4 facilitates the direct transfer of photogenerated holes to lignin, thus weakening the C–C bond and causing the decomposition as well as oxidation of lignin to organic acids.79 Path (ii) is the most common way of breaking the β-O-4 bond and the typical product is the formation of benzaldehyde types of molecules. β-O-4 bonds are successfully converted by depositing V2O5 on the BiVO4 photoanode under a bias of 2 V. The depolymerization products are mainly p-anisaldehyde, phenol, and FA.80 The presence of hydroxymethyl at the β-position of β-O-4 significantly increases the decomposition activity, and the presence of methoxy at the para-position of both benzene rings further improves the conversion. Route (iii) was also carried out by a DSPEC system, where the active hydrogen atom transfer (HAT) mediator was 4-acetamido-2,2,6,6-tetramethylpiperidine-1-oxyl (ACT). Oxidation of HAT-DSPEC was able to afford 2,6-dimethoxybenzoquinone and the corresponding other cleavage product after CAr–Cα bond severing (Fig. 8 Route iii). Model compounds that do not contain p-hydroxyl groups are unable to undergo C–C bond fracture.81 The selectivity of the reaction depends on the reaction time and the presence of light. The gradual decrease of the anodic current with prolonged illumination time is associated with the irreversible depletion of the β-O-4 model compound and degradation of the photocatalyst.82 Route (iv) can be achieved by a two-step one-pot method of β-O-4 bonds (Fig. 8 Route iv and Fig. 11). This method is subject to electrocatalytic oxidation followed by photocatalytic cleavage to finally obtain cracked ketones and guaiacol.83 The whole catalytic system does not strictly belong to the scope of PEC, which is a combination of photocatalysis and electrocatalysis but has a strong significance. The reactions were performed on coniferyl- and sinapyl-derived dimers, and even natural lignin with good yields, and the investigation of lignin depolymerization revealed that the electrocatalytic oxidation–photocatalytic cleavage process not only cleaves the β-O-4 bond efficiently but also leaves the other components intact with high selectivity. The extraction of benzyl hydrogen during the reaction may be the rate- or turnover-determining step.
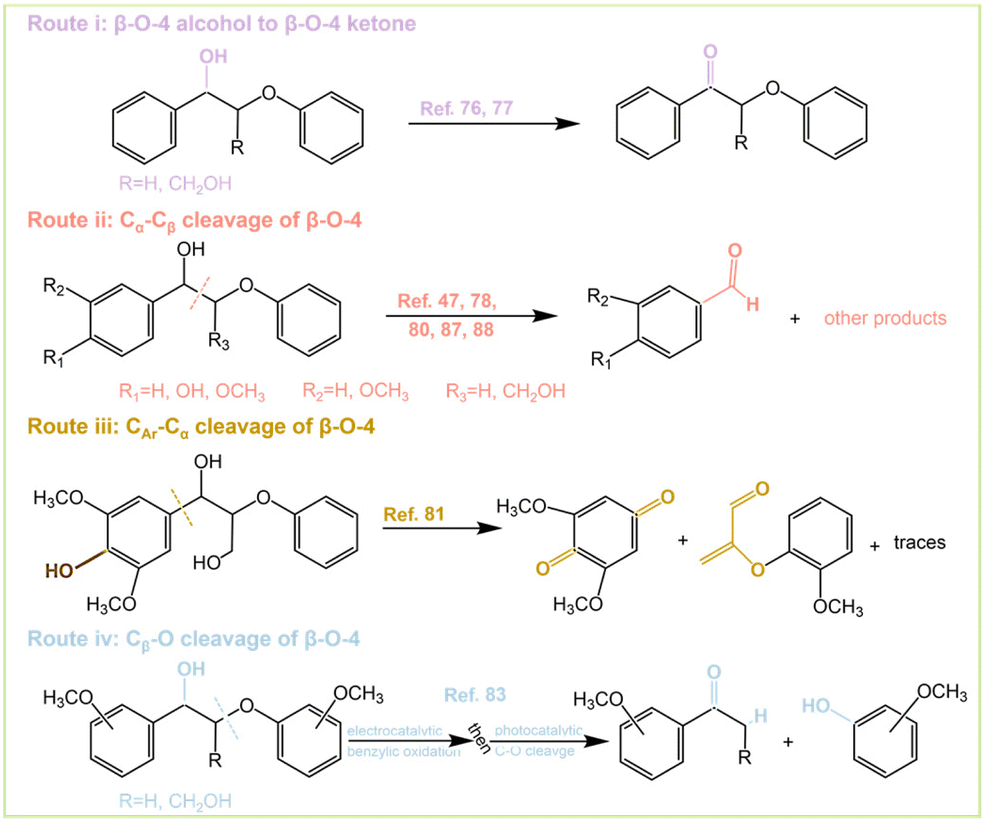 | ||
| Fig. 8 Four routes of PEC β-O-4 transformation. | ||
Oxidation of lignin occurred more readily than water oxidation when using a tandem structure of the BiVO4 photoanode with a perovskite PV for the conversion of real biomass feedstocks, but other non-lignin biocomponents (e.g., cellulose and xylose) had negligible effects on photocurrent generation.48 Oxidation of lignin occurs mainly at bonds with lower dissociation energies such as β-O-4 (54–72 kcal mol−1) and β-5 (54–63 kcal mol−1) rather than at bonds with higher dissociation energies such as β–β (81 kcal mol−1).84 Further extension of the overoxidation time enables decomposition to the simplest form, i.e. CO2. The phosphomolybdic acid (PMA) PEC biomass oxidation system also proved to have a more outstanding performance than PEC water oxidation. Compared to the high voltage (1.5–1.6 V vs. RHE) required to reach 10 mA cm−2 for the aqueous oxidation system,85,86 the biomass system required only 0.95 V vs. RHE at the same current density.87 Comparison of the relative contents of β-O-4, β-5, and β–β structures of lignin before and after the reaction demonstrated that the main selective rupture of the oxidation reaction was that of the β-O-4 bond. The main products of lignin depolymerization are vanillin and CO (Fig. 9c), where CO is mainly formed due to the presence of dissolved oxygen molecules. According to Arrhenius kinetics, the activation energies for PMA reduction by oxidation of lignin, cellulose, hemicellulose, and lignocellulosic biomass are 24, 102, 79, and 47 kJ mol−1 (0.249, 1.057, 0.819, and 0.487 eV), respectively, indicating that lignin is the main source of electrons and is more readily depolymerized than other components.88 Lignin was selectively removed from the lignocellulosic biomass after reacting with PMA, resulting in the formation of a hierarchical porous microstructure. The general reaction mechanism is as follows:
| Lignin + PMA3− → Ligninox + PMA5− | (5-1) |
| Lignin → Ligninox + 2H+ + 2e− | (5-2) |
| PMA3− + 2H+ + 2e− → PMA5− | (5-3) |
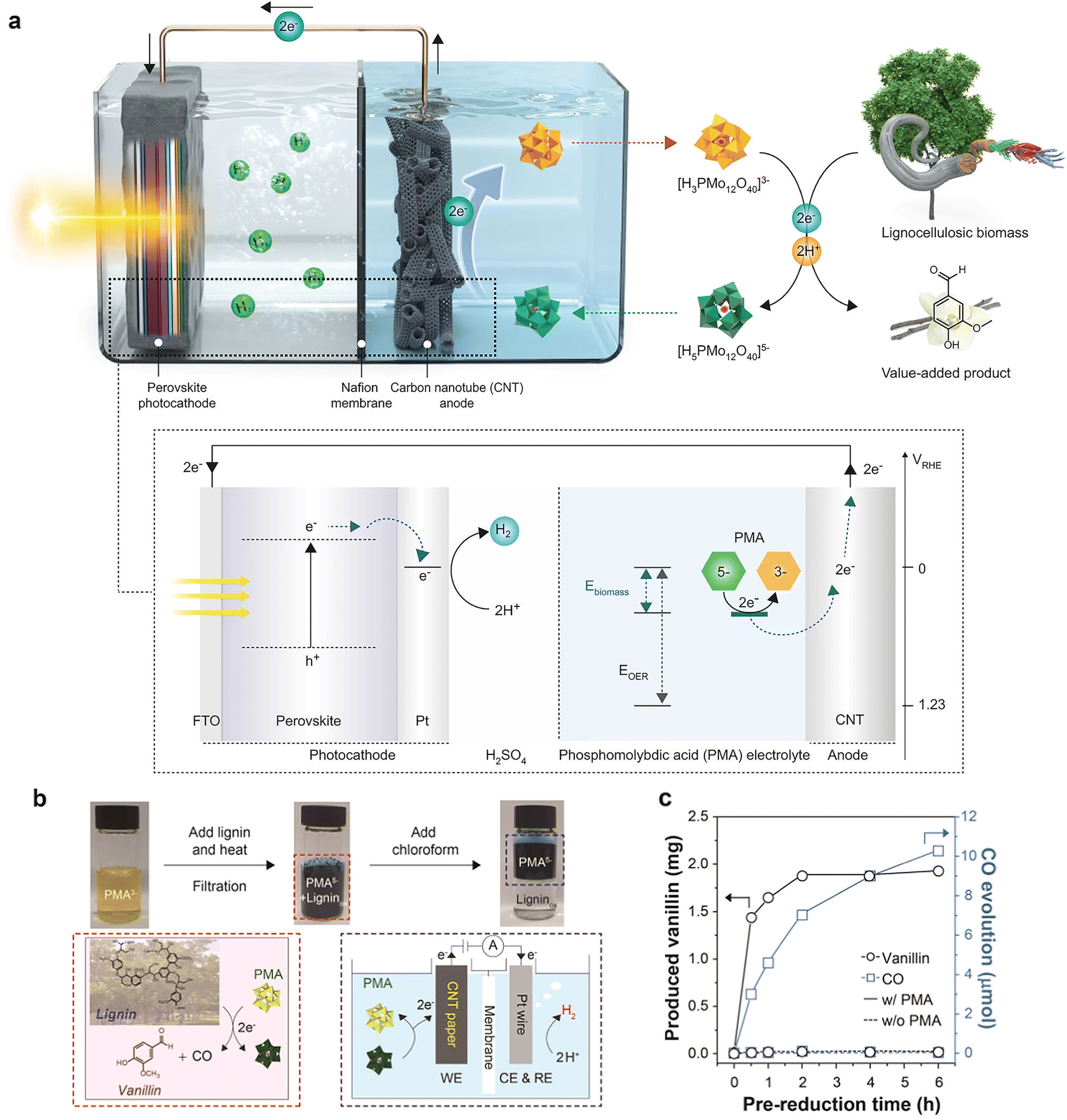 | ||
| Fig. 9 (a) The perovskite photocathode with the Pt catalyst and the electron extraction system from biomass are combined for bias-free hydrogen evolution. Adapted from ref. 88 with permission from Springer Nature, copyright 2022. (b) Photographs and reaction scheme showing the prereduction of PMA upon lignin oxidation and separation of oxidized by-products from lignin using chloroform. (c) Concentration profile of CO and vanillin upon oxidation of lignin using PMA at 50 °C. Adapted from ref. 87 with permission from the American Chemical Society, copyright 2020. | ||
In addition, there is the PEC phenol oxidation process catalyzed by SnO2 and Sb photoanodes coated on TiO2 nanotubes in order to provide electrons for electrochemical CO2 reduction. The current density under visible light irradiation is 12 times higher than that under dark conditions when phenol is added to Na2SO4 solution.88 The reactions at the anode are as follows:
| C6H5OH + 28˙OH → 6CO2 + 17H2O + 28e− | (6-1) |
| 2H2O → O2 + 4H+ + 4e− | (6-2) |
100% selective benzyl alcohol to benzaldehyde conversion was achieved by the Bi2MoO6@TiO2 NTA photoanode and the C/Cu2ONW photocathode.90 Benzaldehyde is a raw material that is widely employed in pharmaceutical, chemical, printing, and dyeing areas.91,92 The estimated flat-band potential (EFB) of the photoanode is −0.34 V, which is more electronegative than that of O2/˙O2− (−0.28 V vs. NHE).93 The photoanode can adsorb O2via photogenerated electrons and further activate it to the ˙O2− radical. Benzyl alcohol can be directly accelerated and converted into benzaldehyde through such radicals. Therefore, O2 is very critical for the solution system.94 The conversion of benzyl alcohol into benzaldehyde is a 2e− process, and the conversion of benzaldehyde into benzoic acid is also a 2e− process. The conversion of benzyl alcohol into benzaldehyde is a little easier, but the conversion of benzaldehyde into CO2 is more difficult because the reaction is a 30e− process. In the investigation for the effect of different substituents (e.g., –OCH3, –CH3, –F, and –NO2) on the conversion of benzyl alcohol at the para-position, it was found that *C6H5–CH2OH+ is the major intermediate. –OCH3 has both an electron-withdrawing induction effect and an electron-donating conjugation effect. However, –OCH3 is an electron-donating functional group because the lone pair of oxygen plays a dominant role in the electron-donating conjugation effect. The electron-donating functional groups are more helpful for the formation of *C6H5–CH2OH+ intermediates, so the conversion of 4-methoxybenzyl alcohol is higher than that of benzyl alcohol. In contrast, the strong electron-withdrawing induction effect of the –NO2 group reduces the electron cloud density of benzyl intermediates. Therefore, the higher the electron-donating ability, the higher the conversion. The electron-donating ability of different substituents is: –OCH3 > –CH3 > –H > –F > –NO2. Benzoic acid is a precursor of phenol, nylon, and benzoate plasticizers in the chemical industry, and is also known to be applied as a food preservative, mold inhibitor, and pharmaceutical additive.95,96 After oxidation by the nanowire-structured Mo–BVO–TiCoNO–CoFeOxy photoanode, benzyl alcohol was oxidized to benzoic acid (97.7% yield) in water, accompanied by an H2 yield of 136.8 μmol cm−2.97 Benzyl alcohol adsorbed on the photoelectrode is oxidized by photogenerated h+, where the alcohol hydroxyl group reacts with hydroxide in the electrolyte to form H2O, which is stripped off from the substrate, leaving the remaining carbonyl radicals. Subsequently, the carbonyl radical interacts with the hydroxyl radical in the electrolyte to form benzoic acid.
Through oxidation in the anode cell, many valuable chemicals or fuels are delivered from the biomass derivatives. But what has to be mentioned is that many substrates are oxidized to low-value CO2 in the anode cell in order to garner hydrogen or other valorized products efficiently in the cathode cell. The simultaneous valorization at both cathode and anode is also an ideal concept.
3.2. Reductive valorization at the cathode
When electrons accumulated on the CB at the anode are transferred to the cathode, a large amount of H+ can be reduced to H2. H2 is green source of energy and is recognized as one of the most promising alternatives among the existing forms of energy (coal, petroleum, fossil fuels) in society.98,99 Therefore, for most PEC conversions in order to achieve efficient hydrogen production at the cathode side, biomass-derived waste is often used as a hole sacrificial agent, and the performance of PEC for H2 production depends entirely on the photoelectro-transformation of the photo-electrode.100 The oxidation of PEC furfuryl alcohol with the TiO2 nanotube anode and the Ni foam cathode yielded only a small amount of furfural, attributed to the competitive reaction between furfural and water oxidation. The O2 from H2O oxidation diffuses to the cathode, which in turn affects the rate of hydrogen production.101 Kong et al.49 achieved an H2 evolution rate of 237 mmol m−2 h−2 with purity >99.99% using the N-doped WO3 photoanode and the Pt cathode. When PMA is utilized as an oxidation catalyst, the p-type photocathode Pt-black silicon can be used for efficient hydrogen production.87 After the replacement of Pt-black silicon by a perovskite photocathode, a maximum current density of 19.8 mA cm−2 was achieved without bias under 1 sun illumination. The hydrogen production efficiency reached 512 μmol cm−2 h−1, which is a record-breaking high value for the solar hydrogen production system reported so far, regardless of the type of device. At the same time, the system permits the continuous production of high-value chemicals through biomass depolymerization at night and solar hydrogen production conversion in the daytime.88There is a relative shortage of research on photocatalytic CO2 reduction, and Shang in his review had great expectations for the reduction of CO2.12 The application of CO2 reduction based on the sacrifice of organics in PEC systems has also been developed in recent years. The system for PEC phenol oxidation starts from electrochemical CO2 reduction using a Cu nanowire cathode to reduce CO2 to CO, accompanied by the production of H2 and small amounts of carbohydrates such as CH4, C2H4, and C2H6.89 The amount of available CO can be increased when the system uses NaHCO3 instead of Na2SO4, because the bicarbonate increases the CO2 concentration in the solution thus facilitating the reduction of CO2.102 PEC along with biocatalysis similarly achieved the CO2 to FA conversion process. CO2 was reduced to FA using nicotinamide cofactor analog (NADH)-dependent formate dehydrogenase from Thiobacillus sp. (TsFDH). The synthesis rate of FA was 4.5 times higher than that based on water oxidation.48 The concentration of lignin at the anode was proportional to the formation rate of FA. The oxidation of lignin, during biocatalytic CO2 reduction, is the rate-controlling step. This system also achieved selective amination of α-ketoglutarate to L-glutamate using L-glutamate dehydrogenase in addition to CO2 reduction (Fig. 5c).
Moreover, the application of PEC in combination with biocatalysis seems to be even broader. The regenerated NADHs activate ene-reductases of the old yellow enzyme family, which can catalyze the enantioselective reduction of α,β-unsaturated carbohydrates. The conversion of 2-methyl-2-cyclohexen-1-one into enantiopure (R)-2-methylcyclohexanone was achieved together with a silicon photovoltaic mesoporous indium tin oxide (Si/mesoITO) photocathode.78 Lignin peroxidase isozyme H8 (LiPH8) from white rot bacteria is capable of excellent selective splitting of β-O-4.103 However, the high concentration of H2O2 is decisive for the system's stability. Photocatalytic production of H2O2 is a straightforward, clean, and ambient operable process, exploiting photogenerated electrons for O2 reduction to produce H2O2. Based on this idea, divided three-compartment photo-electro-biochemical systems were designed for the valorization of lignin without additional bias and sacrificial agents (Fig. 5b). H2O is oxidized by photogenerated holes on the VB to O2, and electrons are transferred to the cathodic cell for the reduction of O2 to harvest H2O2. H2O2 penetrates through the size-selective cellulose membrane and is then bio-catalytically valorized for lignin.47 The main product of β-O-4 cleavage is 3,4-dimethoxybenzaldehyde, the conversion of which is mainly limited by the concentration of H2O2. The bioenzyme was replaced with the horseradish peroxidase (HRP) biocatalyst in the presence of H2O2 for biopolymer synthesis as well. The conversion and biopolymer yield can be up to 98.3% and 73.3%, respectively, using coniferyl alcohol as the feedstock. The polymers consisted of common bonding types like β-O-4, β–β, and β-5. The number average molecular weight was 1103. Chitin is the second largest source of carbohydrates for the production of biofuels and chemicals due to its efficient and sustainable transformations. But destroying the crystalline structure similar to cellulose and producing soluble oligosaccharides and monosaccharides is one of the challenges in the development of chitin.104 An inorganic biohybrid platform by integrating the Si photocathode and lytic polysaccharide monooxygenase (LPMO)-based enzyme systems can cascade catalyze α-chitin to soluble oxidized oligosaccharides. 2,6-Dimethyl-1,4-benzoquinone was chosen as a connecting agent among a host of quinones due to the best electrochemical reversibility and suitable redox potential. In the presence of light, the quinone mediator is reduced on a silicon photocathode. Capitalizing on the reducing power provided by the mediator, α-chitin is then degraded to soluble products by oxygen activation on LPMO. At the same time, the mediator is recovered to its initial state to complete the cycle. All components are indispensable.104
Whether in the anodic or cathodic cell, there are many more well-known model compounds, bonding types, and even pristine biomass in the field of PEC biomass that can be explored, accompanied by conversion mechanisms that are challenging to fulfill by photocatalysis, electrocatalysis, and even thermo-catalysis. These exciting results cannot be achieved without the design and preparation of catalysts. Intelligent design of catalysts at both electrodes combined with appropriate catalytic configurations can lead to endless possibilities for PEC biomass conversion.
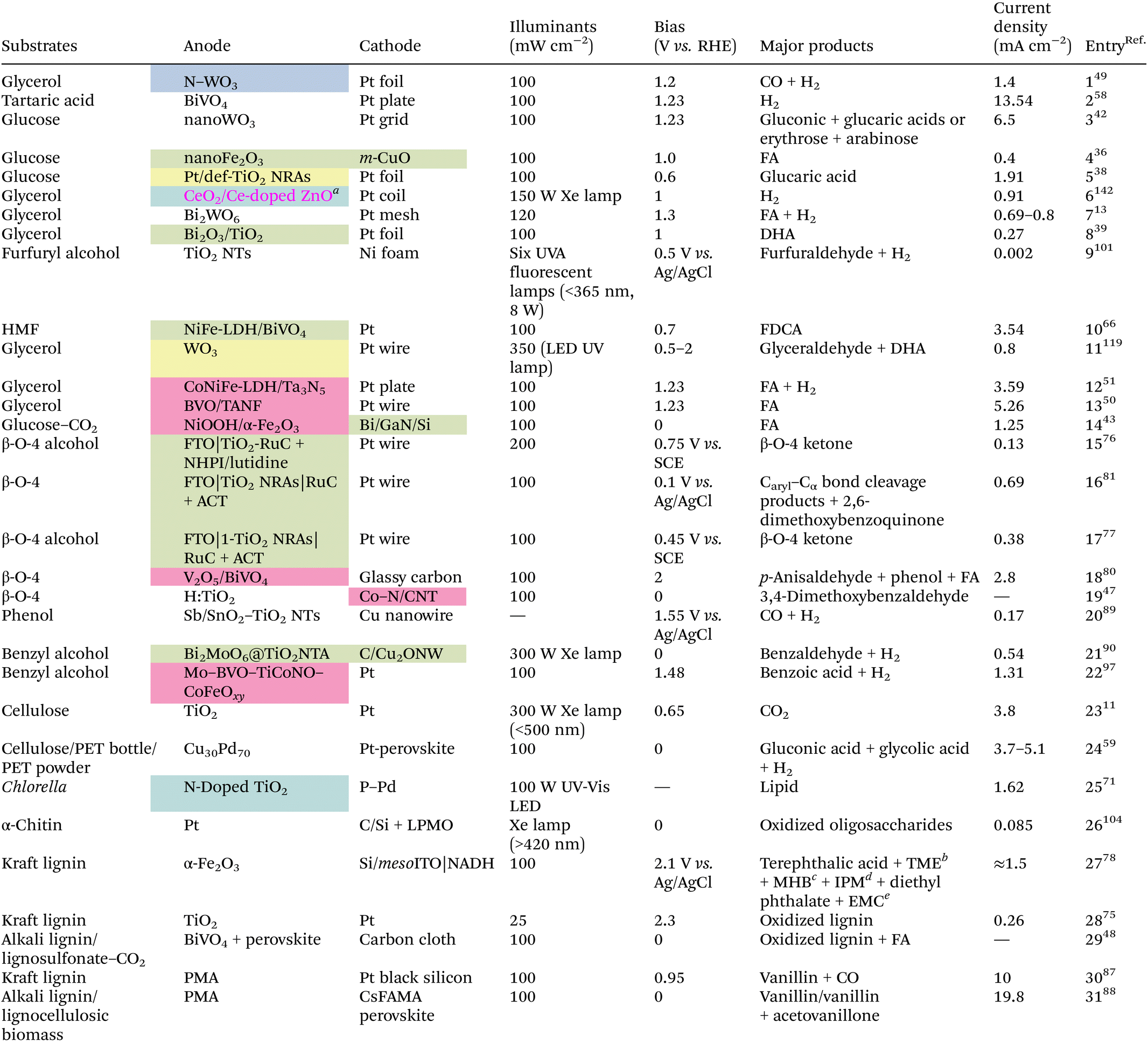
4. Design and exploitation of catalysts and PEC configurations
Similar to plain photocatalysis, a prerequisite for a PEC system to function well is the formation of an internal electric field (IEF) in the semiconductor photocatalyst/system. Aside from the direct application of acquainted photocatalysts, there are many ways to increase the catalytic activity, for example by creating defects such as oxygen vacancies, atomic doping, and atomic or compound modifications (Table 1). Nevertheless, a unique configuration is particularly vital when special conversion outcomes need to be achieved or the applied bias of the whole system needs to be brought down.| Several catalyst preparation methods: oxygen vacancy defect (yellow), atom doping (blue), element/compound decoration (pink), or heterojunction construction (green).a The catalyst was prepared by both CeO2 decoration and Ce doping.b TME = 1-(2,3,4,5-tetramethoxy-6-methylphenyl)ethenone.c MHB = methyl 2-hydroxybenzoate.d IPM = (4-isopropylphenyl)methanol.e EMC = enantiopure (R)-2-methylcyclohexanone. |
|---|
|
4.1. The modulation of catalysts
The main types of semiconductor catalysts utilized in biomass PEC systems are currently centered on TiO2-based, WO3-based, BiVO4-based, perovskite, Fe2O3-based, and Si-based catalysts. Most types are already well known in photocatalysis, and there are discrepancies in the conversion from catalysts due to different band gaps.O by mesoporous TiO2 combined with (bis-2,2′-bipyridine)(2,2′-bipyridine-4,4′-dicarboxylic acid) Ru(II) (RuC) as a catalyst and N-hydroxyphthalimide (NHPI)/lutidine as a co-catalyst.76 RuC could catalyze the oxidation of NHPI to the N-oxy radical (PINO) form. The presence of lutidine is necessary to facilitate the charge transfer from photoexcited RuC* to NHPI. Beforehand, Bosque et al.83 achieved the scission of the ether bond in a one-pot method using the electro-oxidation of β-O-4 by NHPI/lutidine followed by photocatalysis. The one-pot is featured in the fact that photocatalysis could be directly performed after the oxidation with the simple addition of the [Ir(ppy)2(dtbbpy)](PF6) photocatalyst, diisopropylethylamine (DIPEA), and FA to the reaction vessel. The photocatalytic performance is guaranteed even in the presence of additives from electrocatalytic oxidation in the system. NHPI is first oxidized to PINO at the anode, which can be facilitated by the proton-coupled electron transfer process of 2,6-dimethylpyridine (Fig. 11). HAT regenerates NHPI from the substrate and produces α-hydroxy radical 1′ intermediates that should be prone to β-O-4 fracture. Polypyridine Ru(II) complexes have been widely applied in water splitting and lignin depolymerization due to their wide visible absorbance and durable excited state lifetimes.109,110 After replacing the mesoporous TiO2 grown on the surface of fluorine-doped tin oxide (FTO) electrodes with TiO2 NRAs (denoted as FTO|t-TiO2|RuC, t is the thickness of TiO2 nanorods (μm)), it was found that the length of TiO2 NRAs has a significant effect on the photocatalytic performance; the shorter the NRAs the better the performance. The >99% yield of β-O-4 2° alcohol to ketone conversion and 88% FE could be achieved via the FTO|1-TiO2|RuC anode with the shortest NRAs on the surface after 4 h of reaction.77 ACT served as a photocatalyst to efficiently quench RuC* emission, indicating the charge transfer from photoexcited RuC* to ACT. This quenching efficiency is 3 times more amplified than that of the NHPI/base pair and does not require a base.81
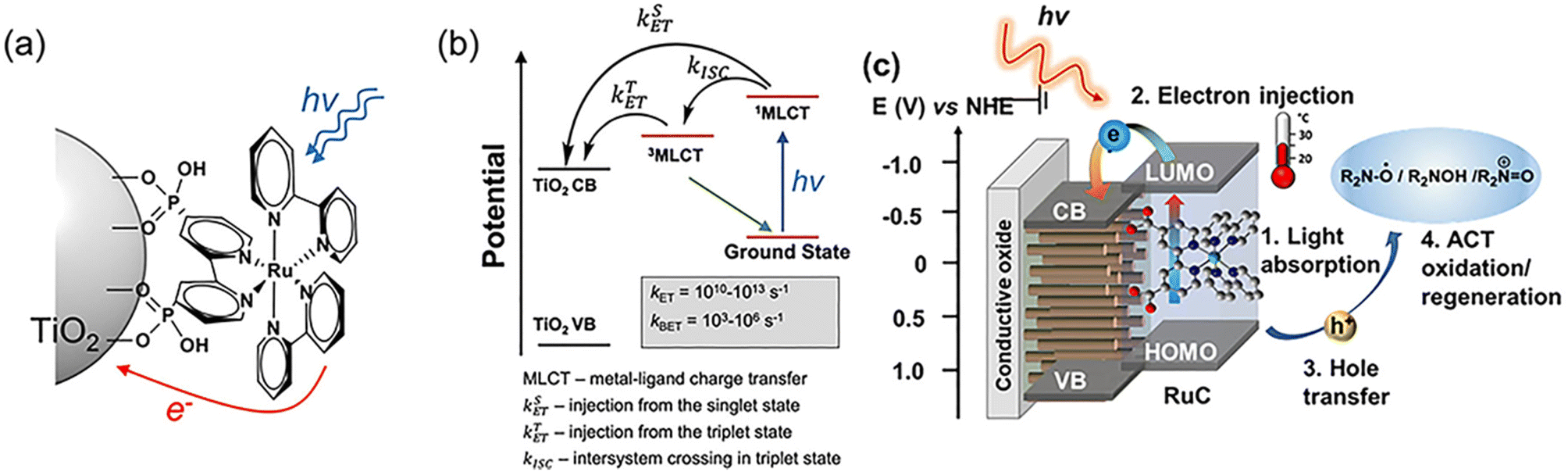 | ||
| Fig. 10 (a) Illustration of a Ru-based chromophore and semiconductor assembly. (b) Photodynamics of the Ru(II) chromophore on a TiO2 surface, electron injection from a singlet state and a triplet state into the conduction band, and back electron transfer to oxidized Ru3+. (c) Schematic for the visible light induced charge transfer between surface bound RuC and ACT in a PEC system. Adapted from ref. 105 with permission from the Royal Society of Chemistry, copyright 2023. | ||
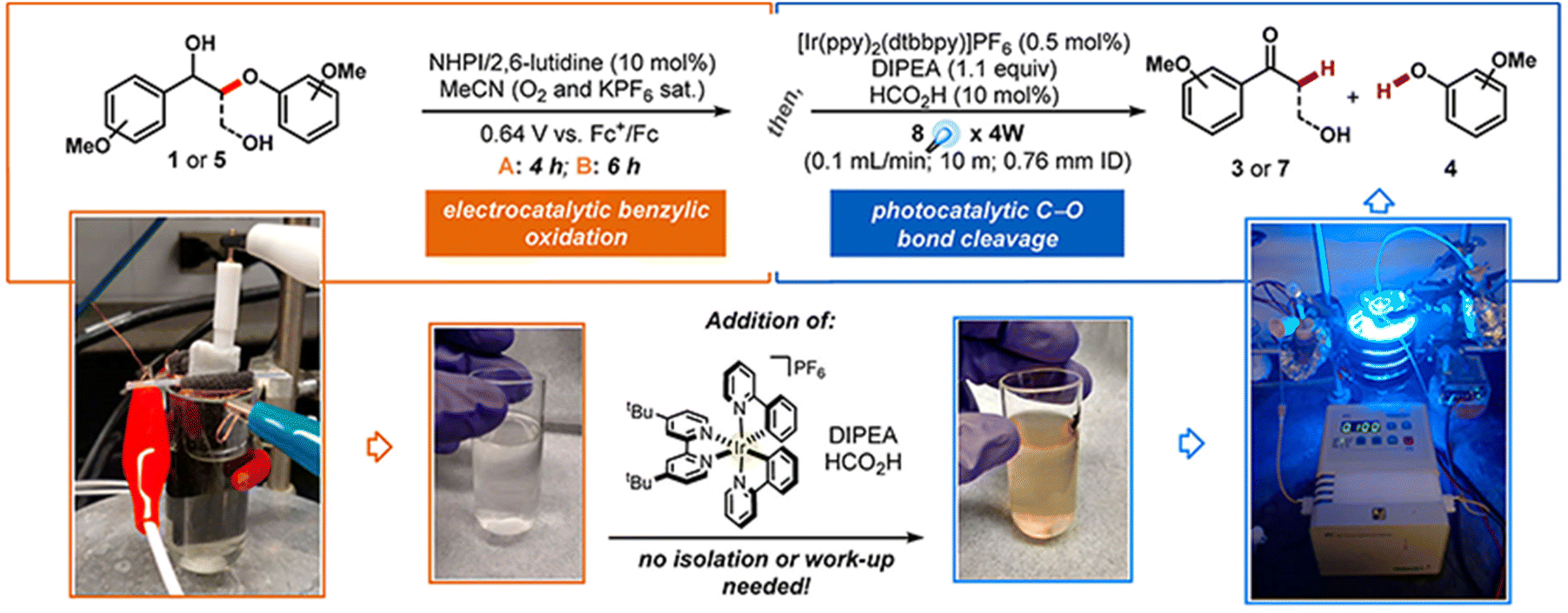 | ||
| Fig. 11 Schematic for the one-pot electrocatalytic oxidation–reductive cleavage of β-O-4. Adapted from ref. 83 with permission from the American Chemical Society, copyright 2017. | ||
Single-atom anchored defective TiO2 NRAs as photoanodes can selectively oxidize glucose to high-value glucaric acid.38 The oxygen vacancy-induced defect structure can modulate the charge carrier dynamics and energy band structure. The band gap of def-TiO2 generated by oxygen vacancy defects drops from 2.99 eV to 2.70 eV. The narrower band gap is attributed to: (i) additional transitions due to incremental Ti3+ states; (ii) oxygen vacancies; (iii) hydroxyl functional groups; and (iv) other defects at different energy levels.111 The prepared Pt/def-TiO2 NRA catalyst is a core–shell structure made up of a rutile crystalline core with a disordered shell layer. The disordered shell layer furnishes a large number of active sites for the anchoring of single-atom Pt. The loading of Pt improves the selectivity for glucaric acid and the facilitation of Pt induces fewer holes to be involved in C–C bond breaking. In the system of PEC glycerol oxidation to DHA with 75.4% selectivity using the Bi2O3/TiO2 photoanode, the p–n junction is formed by the Fermi energy level potential difference between the n-type semiconductor TiO2 and the p-type semiconductor Bi2O3,112 which generates a strong IEF. This p–n junction effectively inhibits the recombination of photogenerated carriers.39 Bi2O3 prefers to be in contact with the middle –OH of glycerol, which facilitates the selective oxidation of the substrate. At the same time, DHA, whereas, is tough to be adsorbed on Bi2O3, avoiding further oxidation and improving selectivity. An analogous case is the Bi2MoO6@TiO2 NTA photoanode and the C/Cu2ONW photocathode, which are n-type and p-type semiconductors, respectively. The Fermi level of the Bi2MoO6@TiO2 NTA photoanode is more negative than that of the C/Cu2ONW photocathode, which causes an internal bias and drives the electrons of the photoanode to transfer and combine with the holes of the photocathode.90 The tubular structure of the TiO2 NTA can further enhance the refraction of light and improve light utilization. Native quantum dots (QDs) are very prone to recombination, and combining QDs with wide band gap acceptors into type-II heterojunctions can effectively extract charge and prevent recombination.113 In the TiO2 NTA supported Mn-doped CdS QD photoanode, Mn doping increases the photoelectron lifetime of the composite photoanode and is more effective in enhancing the oxidation of the ˙OH radical sacrificial agent, and there may be a correlation between both of them.37 The nominal doping of Mn at 15% has the longest photoelectron charge lifetime. Further doping results in the dominance of doping-induced defects and conduces the recombination of photoelectron charges.
Apart from the exploitation of UV light directly, a variety of methods to interact with TiO2 to extend the light absorption of the catalytic system appear to be a more advisable way out for TiO2-based catalysts. Its cost and performance advantages will keep it from being obsolete for a long time.
C asymmetric hydrogenation.
4.2. The design of PEC configurations
Apart from the fabrication of highly active catalysts or complexes by designing vacancy defects, atomic doping, and elemental or compound modifications, the synergistic settlement of catalysts at both photoanodes can likewise fulfill specific conversion targets. In the NiOOH/α-Fe2O3–Si/mesoITO complex system, the long wavelength photons (>600 nm) passing through the front α-Fe2O3 photoanode are available to the rear Si-based photocathode, which allows the hybrid apparatus to encompass most of the spectrum43 (Fig. 5a). There is also a Pt-perovskite photocathode with an electrocatalyst CuPd alloy for the PEC system. This system is a highlight in photocatalysis as it applies dark mixed waste solution directly on the anode side without blocking the light in the cathode cell. A similar system is the three-compartment photo-electro-biochemical system (Fig. 5b). In order to avoid the underperformance from the dark lignin in conjunction with photocatalysis, the lignin is arranged in the biocatalytic cell for transformation. Since the lignin and photocatalyst are separated, the light absorption efficiency of the dark lignin is not decreased.47 To resolve the contact problem between the reactants and the catalyst, cellulose films are deposited directly on the TiO2 photoanode. The original porous and particulate TiO2 surface is completely renewed to a uniform and smooth surface after cellulose support. The PEC reaction takes place near the interface of TiO2 and cellulose.11 Under solar illumination, nano-Fe2O3 can drive both water and glucose oxidation. Yet the FE of oxidation of glucose is only 60.8 ± 1.5% at 1.0 V vs. RHE and the selectivity of FA is not satisfactory. m-CuO could not only act as an electrocatalyst, but the p–n heterojunction composed of m-CuO and nanoFe2O3 could modulate the oxidation of photoinduced holes. The valence band edge of m-CuO is similar to its flat-band potential (1.15 eV), so the photogenerated holes of this heterojunction can only oxidize glucose. As shown in Fig. 6, photoelectric water oxidation is almost eliminated by the passivation of m-CuO. Moreover, the photocurrent of nanoFe2O3|m-CuO containing glucose at 1.23 V vs. RHE is 15 times higher than that of glucose-free nanoFe2O3|m-CuO.36With the aim of minimizing the bias, it is common to employ n-type and p-type semiconductors for the anode and cathode, respectively, apart from preparing catalysts with a high light absorption range and high activity, or by means of bio-catalysis.90 Compensation for the potential difference can also be implemented by the series structure of the photoanode and PV. In the PEC lignin oxidation system with BiVO4 and cathodic biocatalytic hydrogenation, there are three major components: (i) the BiVO4 photoanode for PEC lignin oxidation; (ii) the tandem structure of the tri-cation perovskite PV with BiVO4 and providing supplementary photovoltage to drive the redox reaction at 0 bias; and (iii) the carbon-cloth cathode to reduce NAD+ to NADH for enzyme redox transformation.48 The tandem structure compensates for the potential difference through Z-schematic adsorption, resulting in a significant decrease in the molecular weight of lignin (Fig. 5c). Luo et al.39 established a self-powered PEC system based on a two-electrode setup, consisting of a PV plate (∼2 V) that provides a constant bias and a homemade PEC flow cell comprising a Bi2O3/TiO2 photoanode with a 7 cm2 irradiated area and a Pt wire cathode. A DHA yield of 1.04 mg cm−2 h−1 (selectivity >70%) and an H2 yield of 0.32 mL cm−2 h−1 were obtained for this system.
The semiconductor materials with suitable band gaps could be prepared by modulating the catalyst structure with abundant elements and designing suitable PEC systems. The techniques combining the advantages of electrocatalysis or bio-catalysis could ultimately realize low-bias set-up and high-value conversion. This is the main feature of the current PEC area but it still holds infinite research potential. The flexibility of photoelectric conversion, which allows simultaneous valorized conversion at both anode and cathode sides or even in multiple compartments, is its greatest advantage. During the implementation of the system, some components or variables can also have an impact on the final catalytic performance.
5. The effect of related variables on PEC systems
There is no doubt that a suitable preparation of the photocatalyst and a smart configuration can directly determine the specific PEC conversion. At the same time, some factors such as solution pH, pretreatment, polarization, membrane, and substrate concentration can affect the optimal operating conditions or the selectivity of the target product. A summary of some representative works is presented here.(i) Solution pH: The nanocrystalline Bi2WO6 photoanode is capable of converting glycerol into FA at different pH values, but the performance of the transformation varies greatly. In acidic media, the selectivity increases to 88% but the photocurrent decreases to 50%. In alkaline media, glycerol oxidation is more convenient and the photocurrent increases to 0.8 mA cm−2, but the selectivity of the product decreases.13 The reason for this is that at lower pH, there is stronger adsorption of glycerol to the photoanode, making the reaction from photogenerated holes to further oxidation easier, and thereby more selective for formic acid. Benzyl alcohol can be oxidized to benzoic acid in strong bases without organic solvents, while it is only oxidized to benzaldehyde in neutral organic-free solvents or just in organic solvent systems.97 Except that, a huge pH difference at both sides of the membrane even leads to the leakage of polluting ions.75
(ii) Pretreatment: For polymers, the structural properties of the substrates vary substantially depending on the processing methods in the industry. In the system of PEC microalgal biomass cleavage for lipid extraction, PEC pretreated Chlorella achieved a lipid extraction efficiency of 96%, roughly 10 times higher than that of non-pretreated Chlorella.71 The pretreatment step of biomass in the PEC biomass depolymerization system plays a key role in FA production. The FA yields of untreated samples were one to two orders of magnitude lower than those of pretreated ones.43 The PEC conversion of PET powder, real-life commercial PET bottles, and microcrystalline cellulose by the CuPd alloy was likewise found to achieve even negligible selectivity of glycolic acid and gluconic acids from non-pretreated feedstocks.59 The oxidation product of β-O-4 by PMA is mainly vanillin, while those of lignocellulose are mainly vanillin and acetovanillone. The differences in the outputs may be attributed to distinctions in the molecular structure of the various raw materials and diverse chemical treatment methods.88 The tandem structure of BiVO4 with a perovskite is able to degrade the molecular weight of alkali lignin from 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mw to 15000 Mw, and the molecule keeps its aromatic ring intact. In contrast, the aromatic rings are converted into carbonyl or carboxyl groups from the decomposition of lignosulfonate. The properties of lignin oxidation can be differentiated from various sources and pretreatment methods.48
000 Mw to 15000 Mw, and the molecule keeps its aromatic ring intact. In contrast, the aromatic rings are converted into carbonyl or carboxyl groups from the decomposition of lignosulfonate. The properties of lignin oxidation can be differentiated from various sources and pretreatment methods.48
(iii) Polarization: The production of C2 and C1 chemicals was retarded and the production of C3 was simulated under a sufficiently high potential (1.5 V vs. Ag/AgCl) in the PEC glycerol oxidation system by monoclinic WO3, as the polarized oxygen evolution reaction consumes ˙OH, further avoiding the excessive oxidation of ˙OH.119 In the system with Kraft lignin as a reactant, the J–V curve from the voltage scanning from −1 to 3 V laid above that from 3 to −1 V. This hysteresis is attributed to the low ion transfer rate and the long distance between the electrodes, bringing about polarization near the electrode surface.75
(iv) Membranes: For a given PEC system, membranes are capable of selectively permeating target ions or components. Different types of membranes have different functions and should be treated precisely. In a three-compartment photo-electro-biochemical system, Nafion and cellulose membranes are used to separate the anode cell, the cathode cell, and the biocatalytic cell, whereby the bioenzyme system is protected from reactive oxygen species (˙OH and ˙O2−), shear stress from oxygen bubbles, high concentrations of hydrogen peroxide, and UV radiation47 (Fig. 5b).
(v) Substrate concentration: Adequate contact between the reactants and the catalyst surface is one of the key factors for efficient PEC conversion. A moderate range of substrate concentrations ensure the uninterrupted contact between the catalytically active site and organic molecules, thereby maintaining the reaction rate and current density. Instead, a too high substrate concentration would affect the adsorption on the electrode surface due to diffusion limitation or agglomeration, and eventually, the current density would drop abruptly.
6. Conclusion and prospect
To be frank, the energy utilization efficiency of biomass electrocatalysis is high, and many of these systems could elevate the FE of organic substrates to about 100%. Inevitably, the selectivity of products in some fine chemistry areas is cut down because certain sensitive or highly active reaction processes are too vigorous. The applied potential itself that sustains electrocatalysis is a form of energy that needs to be injected into the system at the beginning of the reaction. Photocatalysis takes advantage of specific wavelengths of light or sunlight to perform selective conversion of substrates with the benefit of semiconductor catalysts. The PEC system is capable of transferring electrons to the cathode through wires to avoid charge recombination and to realize valuable applications with smooth parallel oxidation and reduction reactions. Biomass, the world's largest renewable resource, has the merits of degradation at a lower bias than water. By preparing suitable semiconductor catalysts and setting up a subtle photoelectrode device and a system model, biomass can be sacrificed for a range of valorizations.Currently, however, most studies have concentrated on the conversion of glucose, glycerol, and β-O-4. These substrates are derived compounds that best represent the structure of the biomass. Upon attack by h+, ˙OH, or other reactive species, the substrates are consumed or decomposed into small molecule products. The generation of glucaric acid and the CAr–Cα bond cleavage of β-O-4 exemplify the extremely strong oriented transformation ability of the PEC system. Mechanistic investigation of the conversion and exploration of energy formats for more types of model compounds are not clear yet, which is detrimental to the adequate utilization of biomass components. The combination of PEC valorization pathways in multiple realms is also not merely circumscribed to electrocatalysis and biocatalysis. The flexibility of PEC systems leads to more possibilities for valorization of biomass. More and more studies are gradually looking at electrocatalysis and photocatalysis for inspiration and attempting breakthroughs as well as innovations in PEC. Beyond improving the selectivity of the aimed products, the modulation of semiconductors with different band gaps alters the redox intensity, while the arrangement of configurations concerning cathodes and anodes reduces the bias and achieves multi-path conversion. It is noteworthy that there is a lack of reductive valorization pathways for biomass substrates at the cathode and photoreduction of biomass will make PEC more universal like other catalytic approaches.
The development of PEC, similar to pure photocatalysis, is still at a theoretical level. The low optical transmittance and insolubility of polymeric raw materials are the main challenges. The upgrading of PEC needs to go through a procedure like structural clarification of biomass feedstock, efficiency improvement of energy harnessing, scale-up of reaction apparatus, and cost economization of catalysts and systems. These problems are being addressed step by step with the cooperation of multidisciplinary scientific research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Key R&D Program of China (2022YFE0207200), the National Natural Science Foundation of China (52236010), the Fundamental Research Funds for the Central Universities (No. 2242022R10058), and the Academician and Expert Workstation of Yunnan Province, China (No. 202205AF150024).References
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- Q. Sun, B. W. J. Chen, N. Wang, Q. He, A. Chang, C.-M. Yang, H. Asakura, T. Tanaka, M. J. Hülsey, C.-H. Wang, J. Yu and N. Yan, Angew. Chem., Int. Ed., 2020, 59, 20183–20191 CrossRef CAS PubMed.
- Z. Xu, P. Lei, R. Zhai, Z. Wen and M. Jin, Biotechnol. Biofuels, 2019, 12, 32 CrossRef PubMed.
- K. Wu, M. Cao, Q. Zeng and X. Li, Green Energy Environ., 2023, 8, 383–405 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- M. Wang and F. Wang, Adv. Mater., 2019, 31, 1901866 CrossRef PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- Z.-H. Liu, R. K. Le, M. Kosa, B. Yang, J. Yuan and A. J. Ragauskas, Renewable Sustainable Energy Rev., 2019, 105, 349–362 CrossRef CAS.
- L. J. R. Nunes, T. P. Causer and D. Ciolkosz, Renewable Sustainable Energy Rev., 2020, 120, 109658 CrossRef.
- Y. Kageshima, T. Yoshimura, S. Koh, M. Mizuno, K. Teshima and H. Nishikiori, ChemCatChem, 2021, 13, 1530–1537 CrossRef CAS.
- W. Shang, Y. Li, H. Huang, F. Lai, M. B. J. Roeffaers and B. Weng, ACS Catal., 2021, 11, 4613–4632 CrossRef CAS.
- L. L. Nascimento, J. Z. Marinho, A. L. R. dos Santos, A. M. de Faria, R. A. C. Souza, C. Wang and A. O. T. Patrocinio, Appl. Catal., A, 2022, 646, 118867 CrossRef CAS.
- S. S. Wong, R. Y. Shu, J. G. Zhang, H. C. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Green Chem., 2020, 22, 7233–7264 RSC.
- F. Yang, Z. Ke, Z. Li, M. Patrick, Z. Abboud, N. Yamamoto, X. Xiao and J. Gu, ChemSusChem, 2020, 13, 3391–3403 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- Y. Kuang, T. Yamada and K. Domen, Joule, 2017, 1, 290–305 CrossRef CAS.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
- J.-Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- K. Li and Y. Sun, Chem. – Eur. J., 2018, 24, 18258–18270 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- T. Uekert, C. M. Pichler, T. Schubert and E. Reisner, Nat. Sustain., 2021, 4, 383–391 CrossRef.
- S. Mandal, S. Adhikari, P. Shengyan, M. Hui and D.-H. Kim, Appl. Surf. Sci., 2019, 498, 143840 CrossRef CAS.
- D. K. Lee, D. Lee, M. A. Lumley and K.-S. Choi, Chem. Soc. Rev., 2019, 48, 2126–2157 RSC.
- Y. He, T. Hamann and D. Wang, Chem. Soc. Rev., 2019, 48, 2182–2215 RSC.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- E. Antolini, J. Environ. Chem. Eng., 2019, 7, 103241 CrossRef CAS.
- W. Yang, R. R. Prabhakar, J. Tan, S. D. Tilley and J. Moon, Chem. Soc. Rev., 2019, 48, 4979–5015 RSC.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed.
- L. M. Reid, T. Li, Y. Cao and C. P. Berlinguette, Sustainable Energy Fuels, 2018, 2, 1905–1927 RSC.
- P.-C. Chuang and Y.-H. Lai, Catal. Sci. Technol., 2022, 12, 6375–6383 RSC.
- X. Lv, C. Hu, J. Shang, P. H. L. Sit, F. L. Y. Lam and W. Y. Teoh, Catal. Today, 2019, 335, 468–476 CrossRef CAS.
- Z. Tian, Y. Da, M. Wang, X. Dou, X. Cui, J. Chen, R. Jiang, S. Xi, B. Cui, Y. Luo, H. Yang, Y. Long, Y. Xiao and W. Chen, Nat. Commun., 2023, 14, 142 CrossRef CAS PubMed.
- L. Luo, W. Chen, S.-M. Xu, J. Yang, M. Li, H. Zhou, M. Xu, M. Shao, X. Kong, Z. Li and H. Duan, J. Am. Chem. Soc., 2022, 144, 7720–7730 CrossRef CAS PubMed.
- Y.-L. Men, P. Liu, Y. Liu, X.-Y. Meng and Y.-X. Pan, Ind. Eng. Chem. Res., 2022, 61, 4300–4309 CrossRef CAS.
- T. Rafaïdeen, S. Baranton and C. Coutanceau, Appl. Catal., B, 2019, 243, 641–656 CrossRef.
- K. Jakubow-Piotrowska, B. Witkowski and J. Augustynski, Commun. Chem., 2022, 5, 125 CrossRef CAS PubMed.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao, M. Shen, Z. Li, Z. Zou, R. Xiao and S. Chu, Nat. Commun., 2023, 14, 1013 CrossRef CAS PubMed.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- V. Kumaravel and M. Kang, Catalysts, 2020, 10, 492 CrossRef CAS.
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2021, 9, 2000804 CrossRef CAS.
- M. Ko, L. T. M. Pham, Y. J. Sa, J. Woo, T. V. T. Nguyen, J. H. Kim, D. Oh, P. Sharma, J. Ryu, T. J. Shin, S. H. Joo, Y. H. Kim and J.-W. Jang, Nat. Commun., 2019, 10, 5123 CrossRef PubMed.
- D. Wang, S. H. Lee, S. Han, J. Kim, N. V. T. Trang, K. Kim, E.-G. Choi, P. Boonmongkolras, Y. W. Lee, B. Shin, Y. H. Kim and C. B. Park, Green Chem., 2020, 22, 5151–5160 RSC.
- F. Kong, H. Zhou, Z. Chen, Z. Dou and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210745 CrossRef CAS PubMed.
- Y. Han, M. Chang, Z. Zhao, F. Niu, Z. Zhang, Z. Sun, L. Zhang and K. Hu, ACS Appl. Mater. Interfaces, 2023, 15, 11678–11690 CrossRef CAS PubMed.
- Q. Wang, X. Ma, P. Wu, B. Li, L. Zhang and J. Shi, Nano Energy, 2021, 89, 106326 CrossRef CAS.
- M. F. Kuehnel and E. Reisner, Angew. Chem., Int. Ed., 2018, 57, 3290–3296 CrossRef CAS PubMed.
- A. V. Puga, Coord. Chem. Rev., 2016, 315, 1–66 CrossRef CAS.
- K. Cheng, W. Zhou, J. Kang, S. He, S. Shi, Q. Zhang, Y. Pan, W. Wen and Y. Wang, Chem, 2017, 3, 334–347 CAS.
- F. Jiao, J. Li, X. Pan, J. Xiao, H. Li, H. Ma, M. Wei, Y. Pan, Z. Zhou, M. Li, S. Miao, J. Li, Y. Zhu, D. Xiao, T. He, J. Yang, F. Qi, Q. Fu and X. Bao, Science, 2016, 351, 1065–1068 CrossRef CAS PubMed.
- G. S. Kumar, Y. Wee, I. Lee, H. J. Sun, X. Zhao, S. Xia, S. Kim, J. Lee, P. Wang and J. Kim, Chem. Eng. J., 2015, 276, 283–288 CrossRef CAS.
- S. Bagheri, N. M. Julkapli and W. A. Yehye, Renewable Sustainable Energy Rev., 2015, 41, 113–127 CrossRef CAS.
- Z. Wang, Y. Guo, M. Liu, X. Liu, H. Zhang, W. Jiang, P. Wang, Z. Zheng, Y. Liu, H. Cheng, Y. Dai, Z. Wang and B. Huang, Adv. Mater., 2022, 34, 2201594 CrossRef CAS PubMed.
- S. Bhattacharjee, V. Andrei, C. Pornrungroj, M. Rahaman, C. M. Pichler and E. Reisner, Adv. Funct. Mater., 2022, 32, 2109313 CrossRef CAS.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- S. Tang, Z. Wang, D. Yuan, C. Zhang, Y. Rao, Z. Wang and K. Yin, J. Cleaner Prod., 2020, 268, 122253 CrossRef CAS.
- P. Morandi, V. Flaud, S. Tingry, D. Cornu and Y. Holade, J. Mater. Chem. A, 2020, 8, 18840–18855 RSC.
- C.-y. Wang, R. Pagel, D. W. Bahnemann and J. K. Dohrmann, J. Phys. Chem. B, 2004, 108, 14082–14092 CrossRef CAS.
- X. Gong and W. Y. Teoh, J. Catal., 2015, 332, 101–111 CrossRef CAS.
- W. Y. Teoh, L. Mädler and R. Amal, J. Catal., 2007, 251, 271–280 CrossRef CAS.
- L. Zheng, P. Xu, Y. Zhao, Z. Bao, X. Luo, X. Shi, Q. Wu and H. Zheng, Appl. Catal., B, 2023, 331, 122679 CrossRef CAS.
- C. S. K. Lin, L. A. Pfaltzgraff, L. Herrero-Davila, E. B. Mubofu, S. Abderrahim, J. H. Clark, A. A. Koutinas, N. Kopsahelis, K. Stamatelatou, F. Dickson, S. Thankappan, Z. Mohamed, R. Brocklesby and R. Luque, Energy Environ. Sci., 2013, 6, 426–464 RSC.
- G.-S. Ha, M. M. El-Dalatony, D.-H. Kim, E.-S. Salama, M. B. Kurade, H.-S. Roh, A. El-Fatah Abomohra and B.-H. Jeon, Bioresour. Technol., 2020, 302, 122809 CrossRef CAS PubMed.
- S. Lee and Y.-M. Chung, Mater. Lett., 2019, 234, 58–61 CrossRef CAS.
- F. Li, Q. Shao, M. Hu, Y. Chen and X. Huang, ACS Catal., 2018, 8, 3418–3423 CrossRef CAS.
- Y. Wu, W. Xiang, L. Li, H. Liu, N. Zhong, H. Chang and B. E. Rittmann, Chem. Eng. J., 2021, 420, 130517 CrossRef CAS.
- L. Hua, L. Guo, M. Thakkar, D. Wei, M. Agbakpe, L. Kuang, M. Magpile, B. P. Chaplin, Y. Tao, D. Shuai, X. Zhang, S. Mitra and W. Zhang, Bioresour. Technol., 2016, 203, 112–117 CrossRef CAS PubMed.
- R. J. Keller, R. P. Sharma, T. A. Grover and L. H. Piette, Arch. Biochem. Biophys., 1988, 265, 524–533 CrossRef CAS PubMed.
- K.-i. Takamiya, T. Tsuchiya and H. Ohta, Trends Plant Sci., 2000, 5, 426–431 CrossRef CAS PubMed.
- J. King and S. S. C. Chuang, Catal. Commun., 2021, 149, 106219 CrossRef CAS.
- S. Li, Z.-J. Li, H. Yu, M. R. Sytu, Y. Wang, D. Beeri, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Energy Lett., 2020, 5, 777–784 CrossRef CAS.
- S. Li, E. W. Shuler, D. Willinger, H. T. Nguyen, S. Kim, H. C. Kang, J.-J. Lee, W. Zheng, C. G. Yoo, B. D. Sherman and G. Leem, ACS Appl. Mater. Interfaces, 2022, 14, 22799–22809 CrossRef CAS PubMed.
- J. Kim, Y. Um, S. Han, T. Hilberath, Y. H. Kim, F. Hollmann and C. B. Park, ACS Appl. Mater. Interfaces, 2022, 14, 11465–11473 CrossRef CAS PubMed.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- T. Li, J. Y. Mo, D. M. Weekes, K. E. Dettelbach, R. P. Jansonius, G. M. Sammis and C. P. Berlinguette, ChemSusChem, 2020, 13, 3622–3626 CrossRef CAS PubMed.
- S. Li, S. Kim, A. H. Davis, J. Zhuang, E. W. Shuler, D. Willinger, J.-J. Lee, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Catal., 2021, 11, 3771–3781 CrossRef CAS.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- Y. Lin, Z. Tian, L. Zhang, J. Ma, Z. Jiang, B. J. Deibert, R. Ge and L. Chen, Nat. Commun., 2019, 10, 162 CrossRef PubMed.
- H. Oh, Y. Choi, C. Shin, T. V. T. Nguyen, Y. Han, H. Kim, Y. H. Kim, J.-W. Lee, J.-W. Jang and J. Ryu, ACS Catal., 2020, 10, 2060–2068 CrossRef CAS.
- Y. Choi, R. Mehrotra, S.-H. Lee, T. V. T. Nguyen, I. Lee, J. Kim, H.-Y. Yang, H. Oh, H. Kim, J.-W. Lee, Y. H. Kim, S.-Y. Jang, J.-W. Jang and J. Ryu, Nat. Commun., 2022, 13, 5709 CrossRef CAS PubMed.
- C. Guo, P. He, R. Cui, Q. Shen, N. Yang and G. Zhao, Adv. Energy Mater., 2019, 9, 1900364 CrossRef.
- Z. Zhou, Y.-N. Xie, W. Zhu, H. Zhao, N. Yang and G. Zhao, Appl. Catal., B, 2021, 286, 119868 CrossRef CAS.
- J. Zheng, X. Chen, X. Zhong, S. Li, T. Liu, G. Zhuang, X. Li, S. Deng, D. Mei and J.-G. Wang, Adv. Funct. Mater., 2017, 27, 1704169 CrossRef.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- K. Jing, W. Ma, Y. Ren, J. Xiong, B. Guo, Y. Song, S. Liang and L. Wu, Appl. Catal., B, 2019, 243, 10–18 CrossRef CAS.
- B. Zhang, J. Li, Y. Gao, R. Chong, Z. Wang, L. Guo, X. Zhang and C. Li, J. Catal., 2017, 345, 96–103 CrossRef CAS.
- P. Xu, P. López-Rojas and T. Ritter, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- K. Danchana, P. Jitthiang, K. Uraisin and V. Cerdà, Food Chem., 2021, 361, 130086 CrossRef CAS PubMed.
- P.-d. Wu, L. Li, K. Wang, H. Li and Z. Fang, Green Chem., 2023, 25, 2771–2781 RSC.
- W.-K. Wang, J.-J. Chen, Z.-Z. Lou, S. Kim, M. Fujitsuka, H.-Q. Yu and T. Majima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18827–18833 CrossRef CAS PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- X. Li, G. Wang, L. Jing, W. Ni, H. Yan, C. Chen and Y.-M. Yan, ChemComm, 2016, 52, 2533–2536 RSC.
- C. M. Pecoraro, M. Bellardita, V. Loddo, D. Virtù, F. Di Franco and M. Santamaria, Appl. Catal., A, 2023, 650, 118987 CrossRef CAS.
- N. C. Deb Nath, S. Y. Choi, H. W. Jeong, J.-J. Lee and H. Park, Nano Energy, 2016, 25, 51–59 CrossRef CAS.
- V. Sáez-Jiménez, J. Rencoret, M. A. Rodríguez-Carvajal, A. Gutiérrez, F. J. Ruiz-Dueñas and A. T. Martínez, Biotechnol. Biofuels, 2016, 9, 198 CrossRef PubMed.
- H. Zhang, Y. Zhao, H. Zhang, H. Zhou, H. Wang, X. Zong, H. Yin and C. Li, Appl. Catal., B, 2020, 265, 118558 CrossRef CAS.
- S. Li, S. Park, B. D. Sherman, C. G. Yoo and G. Leem, ChemComm, 2023, 59, 401–413 RSC.
- J. R. Swierk and T. E. Mallouk, Chem. Soc. Rev., 2013, 42, 2357–2387 RSC.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- G. Leem, B. D. Sherman and K. S. Schanze, Nano Convergence, 2017, 4, 37 CrossRef PubMed.
- G. Leem, B. D. Sherman, A. J. Burnett, Z. A. Morseth, K.-R. Wee, J. M. Papanikolas, T. J. Meyer and K. S. Schanze, ACS Energy Lett., 2016, 1, 339–343 CrossRef CAS.
- Z. Tian, D. Wang, K. Bu, J. Lin, S. Zhang, W. Zhao, P. Qin and F. Huang, Adv. Funct. Mater., 2018, 28, 1803328 CrossRef.
- K. He, T. Tadesse Tsega, X. Liu, J. Zai, X.-H. Li, X. Liu, W. Li, N. Ali and X. Qian, Angew. Chem., Int. Ed., 2019, 58, 11903–11909 CrossRef CAS PubMed.
- J. Schneider, A. Vaneski, G. R. Pesch, A. S. Susha, W. Yang Teoh and A. L. Rogach, APL Mater., 2014, 2, 126102 CrossRef.
- X. Wang, G. Hai, B. Li, Q. Luan, W. Dong and G. Wang, Chem. Eng. J., 2021, 426, 130822 CrossRef CAS.
- D. Kong, X. Ruan, J. Geng, Y. Zhao, D. Zhang, X. Pu, S. Yao and C. Su, Int. J. Hydrogen Energy, 2021, 46, 28043–28052 CrossRef CAS.
- R. Yang, Z. Zhu, C. Hu, S. Zhong, L. Zhang, B. Liu and W. Wang, Chem. Eng. J., 2020, 390, 124522 CrossRef CAS.
- Y. Wang, K. Ding, R. Xu, D. Yu, W. Wang, P. Gao and B. Liu, J. Cleaner Prod., 2020, 247, 119108 CrossRef CAS.
- S. Corby, L. Francàs, A. Kafizas and J. R. Durrant, Chem. Sci., 2020, 11, 2907–2914 RSC.
- J. Yu, J. González-Cobos, F. Dappozze, F. J. López-Tenllado, J. Hidalgo-Carrillo, A. Marinas, P. Vernoux, A. Caravaca and C. Guillard, Appl. Catal., B, 2022, 318, 121843 CrossRef CAS.
- Q. Mi, A. Zhanaidarova, B. S. Brunschwig, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 5694–5700 RSC.
- J. C. Hill and K.-S. Choi, J. Phys. Chem. C, 2012, 116, 7612–7620 CrossRef CAS.
- R. Solarska, C. Santato, C. Jorand-Sartoretti, M. Ulmann and J. Augustynski, J. Appl. Electrochem., 2005, 35, 715–721 CrossRef CAS.
- M. Jadwiszczak, K. Jakubow-Piotrowska, P. Kedzierzawski, K. Bienkowski and J. Augustynski, Adv. Energy Mater., 2020, 10, 1903213 CrossRef CAS.
- Y. Hu, Y. Pan, Z. Wang, T. Lin, Y. Gao, B. Luo, H. Hu, F. Fan, G. Liu and L. Wang, Nat. Commun., 2020, 11, 2129 CrossRef CAS PubMed.
- A. M. Asiri, T. Nawaz, M. B. Tahir, N. Fatima, S. B. Khan, K. A. Alamry, S. Y. Alfifi, H. M. Marwani, M. M. Al-Otaibi and S. Chakraborty, Int. J. Hydrogen Energy, 2021, 46, 39058–39066 CrossRef CAS.
- B. Zhang, X. Huang, Y. Zhang, G. Lu, L. Chou and Y. Bi, Angew. Chem., Int. Ed., 2020, 59, 18990–18995 CrossRef CAS PubMed.
- K. Zhang, B. Jin, C. Park, Y. Cho, X. Song, X. Shi, S. Zhang, W. Kim, H. Zeng and J. H. Park, Nat. Commun., 2019, 10, 2001 CrossRef PubMed.
- H. Zhang, H. Li, Z. Wang, Z. Zheng, P. Wang, Y. Liu, X. Zhang, X. Qin, Y. Dai and B. Huang, Appl. Catal., B, 2018, 238, 586–591 CrossRef CAS.
- Y. W. Lee, P. Boonmongkolras, E. J. Son, J. Kim, S. H. Lee, S. K. Kuk, J. W. Ko, B. Shin and C. B. Park, Nat. Commun., 2018, 9, 4208 CrossRef PubMed.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- A. G. Curto, T. H. Taminiau, G. Volpe, M. P. Kreuzer, R. Quidant and N. F. van Hulst, Nat. Commun., 2013, 4, 1750 CrossRef PubMed.
- T. Hu, Y. Wang, H. Xiao, W. Chen, M. Zhao and J. Jia, ChemComm, 2018, 54, 13363–13366 RSC.
- Z. Chen, Y.-C. He, J.-H. Chen, X.-Z. Fu, R. Sun, Y.-X. Chen and C.-P. Wong, J. Phys. Chem. C, 2018, 122, 8976–8983 CrossRef CAS.
- J.-W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 7447 CrossRef PubMed.
- J. Kim, Y. W. Lee, E.-G. Choi, P. Boonmongkolras, B. W. Jeon, H. Lee, S. T. Kim, S. K. Kuk, Y. H. Kim, B. Shin and C. B. Park, J. Mater. Chem. A, 2020, 8, 8496–8502 RSC.
- M. D. Symes and L. Cronin, Nat. Chem., 2013, 5, 403–409 CrossRef CAS PubMed.
- Y. Xiao, C. Feng, J. Fu, F. Wang, C. Li, V. F. Kunzelmann, C.-M. Jiang, M. Nakabayashi, N. Shibata, I. D. Sharp, K. Domen and Y. Li, Nat. Catal., 2020, 3, 932–940 CrossRef CAS.
- Y. Pihosh, T. Minegishi, V. Nandal, T. Higashi, M. Katayama, T. Yamada, Y. Sasaki, K. Seki, Y. Suzuki, M. Nakabayashi, M. Sugiyama and K. Domen, Energy Environ. Sci., 2020, 13, 1519–1530 RSC.
- J. Seo, H. Nishiyama, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2018, 57, 8396–8415 CrossRef CAS PubMed.
- L. Wang, X. Zhou, N. T. Nguyen, I. Hwang and P. Schmuki, Adv. Mater., 2016, 28, 2432–2438 CrossRef CAS PubMed.
- J. Kegel, I. M. Povey and M. E. Pemble, Nano Energy, 2018, 54, 409–428 CrossRef CAS.
- S. Kim, E. An, I. Oh, J. B. Hwang, S. Seo, Y. Jung, J.-C. Park, H. Choi, C. H. Choi and S. Lee, Catal. Sci. Technol., 2022, 12, 5517–5523 RSC.
- S. Rajendran, M. M. Khan, F. Gracia, J. Qin, V. K. Gupta and S. Arumainathan, Sci. Rep., 2016, 6, 31641 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |