
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From green to circular chemistry paved by biocatalysis
Pedro
Lozano
 *a and
Eduardo
García-Verdugo
*b
*a and
Eduardo
García-Verdugo
*b
aDepartamento de Bioquímica y Biología Molecular “B” e Inmunología. Facultad de Química, Universidad de Murcia, E-30.100, Murcia, Spain. E-mail: plozanor@um.es
bDepartamento de Química Inorgánica y Orgánica, Universidad Jaume I, Campus del Riu Sec, Avenida Sos Baynant s/n, E-12071, Castellón, Spain. E-mail: cepeda@uji.es
First published on 4th August 2023
Abstract
Since its origin, green chemistry has headed the best-guiding philosophy for reducing pollution and safeguarding the environment. The twelve Principles of Green Chemistry provide us with enough tools to design sustainable transformations and implement industrial processes by means of renewable feedstocks. All these processes, to avoid or minimise waste production, require not only selective and efficient catalytic transformations with high atom and energy efficiency but also clean separation processes, and the use of non-toxic and safe products. Biocatalysts synthesized by the green chemistry and circular economy principles can constitute the most important and efficient strategy for achieving many of the 17 Sustainable Development Goals set by the United Nations. This will drive our society to a sustainable future by reducing the consumption of resources and drastically minimising the environmental impact of our recalcitrant wastes. This perspective illustrates by a series of selected examples how green and circular chemistry based on biocatalytic processes can pave synergies for sustainable development.
 Pedro Lozano (right) and Eduardo García-Verdugo | Prof. Pedro Lozano (right), born in 1961 in Ceutí (Spain), obtained his Bachelor of Science (Chemistry) at the University of Murcia in 1984, and his PhD in Science (Chemistry) at the same University in 1988. Between 1990 and 1991, he spent two years of postdoctoral training at the “Centre de Transfer en Biotechnologie Gilbert Durand”, Toulouse (France). In 1993, he returned to the Faculty of Chemistry of the University of Murcia (Spain) as Associate Professor of Biochemistry and Molecular Biology, being finally promoted to Full Professor in 2004. Professor Lozano has served as Vice-Dean (1996–2014) and Dean (2014–2022) of the Faculty of Chemistry at the University of Murcia, as well as, Visiting Professor at the Universities of Rennes-France (2002), Bordeaux-France (2013) and Universidade Rio Grande do Sul-Porto Alegre-Brazil (2019). Since 2010, Prof. Lozano leads de Sustainable Chemistry research group at the University of Murcia, and his research activity has always been related to enzyme technology in ionic liquids, supercritical fluids and solventless systems for the development of green and sustainable chemical processes, trying to contribute to a more sustainable future. He is the author of more than 140 publications (i.e. research papers, reviews, book chapters, etc.) and more than 200 scientific contributions in National and International Congresses. Prof. Lozano has been recognized by the 7th IQS-European Award of Enzyme Technology (2003), Fellow of the Royal Society of Chemistry (2017), Spanish ANQUE National Award (2020). Prof. Eduardo García-Verdugo, a passionate advocate of green chemistry, was born in 1972 in Talavera de Reina, Spain. He received his chemistry degree from the University of Valencia in 1995 and went on to complete his PhD in Chemistry at the University Jaume I (Spain) in 2001. Following his doctoral studies, he conducted postdoctoral research at Nottingham University in the UK, working under the guidance of Prof. Martin Poliakoff until 2004. Returning to Spain, he was awarded the Ramon y Cajal fellowship at the University Jaume I, where he made significant contributions until 2009. He then held a permanent position as Cientifico Titular of CSIC at ICP in Madrid from 2009 to 2010. In 2010, he returned to his alma mater, the University Jaume I, where he assumed a permanent academic position. Presently, he continues to contribute to the university's academic community being Full Professor in Organic Chemistry and leading the Supramolecular and Sustainable Research Group. His research turns around integrating various enabling techniques like catalysis, polymeric materials, continuous flow processes, microreactors, bio-catalysis, and neoteric solvents. His goal is to develop efficient and sustainable processes within the scope of Green Chemistry, contributing to a greener and more environmentally friendly future. As a result of his entrepreneurial spirit, he co-founded two spin-off companies, Proyecto Kryptonite SL and Molecular Sustainable Solutions SL. |
1. Green chemistry in the context of the SDGs
Chemistry has been the engine that has allowed humanity to advance. The continuous improvement to the quality of life and life expectancy has permitted us to enjoy levels of comfort that were unimaginable a century ago (such as pharmaceutical drugs to cure diseases, safer and more nutritious foods, clean drinking water, fertilizers and insecticides to improve agricultural productions, fuels, hygiene and beauty products, and materials). Our quality of life depends entirely and positively on chemistry.1In the last 100 years, the world population has increased exponentially from 1.2 to 8 billion people. Simultaneously, our life expectancy has also increased, from 40 to above 80 years old.2 This occurs together with a dominating consumption society model demanding an unprecedented need for resources with the consequent increased residues and waste generation. This creates fundamental doubts about the sustainability of the model. All this unprecedented success has been based on a linear product life cycle management process for product definition, development, and manufacturing (i.e., take-make-use-waste).3 The launch of the 17 Sustainable Development Goals (SDGs) by the United Nations (UN) in 2015 may constitute the most important stimulus to change the course of our model of society, being widely adopted by governments and corporations in an effort to improve the sustainability of our society.4 Most of these goals (e.g. the mitigation and adaptation to climate change, sustainable use and protection of water and marine resources, circular economy, prevention and control of wastes, protection and the recovery of biodiversity and ecosystems, etc.) are fully related with achievements of chemistry.
Green chemistry is a guiding philosophy based on scientific approaches that instigate clean products and processes by the efficient use of nontoxic (principle 1) renewable raw materials (principle 7) and their selective transformation through (bio)catalysis (principle 9), maximizing atom economy (principle 2), avoiding chemical derivatives (principle 8) and eliminating wastes (principle 1), avoiding the use of toxic and hazardous reagents (principle 4), and using safer solvents (principle 5) in the manufacture and application of chemical products, as P. T. Anastas and J. C. Warner summarised in 1998 by the Twelve Principles of Green Chemistry.5 Most of these principles converge directly with many of the 17 SDGs, such as good health and well-being (goal 3), quality education (goal 4), clean water and sanitation (goal 6), affordable and clean energy (goal 7), decent work and economic growth (goal 8), industry, innovation, and infrastructure (goal 9), sustainable cities and communities (goal 11), climate action (goal 13), life below water (goal 14), and life on land (goal 15), as depicted in Fig. 1. In this regard, the UN-SDG 12 “Responsible Consumption and Production” may be considered the most related goal to green chemistry. It emphasizes the key role of “decoupling economic growth from environmental degradation, increasing resource efficiency, and promoting sustainable lifestyles”. This clearly requires and encourages moderation on the use and disposal of the limited resources of our planet, emphasizing the incorporation of recycling wastes as starting materials at the beginning of the industrial production chains.
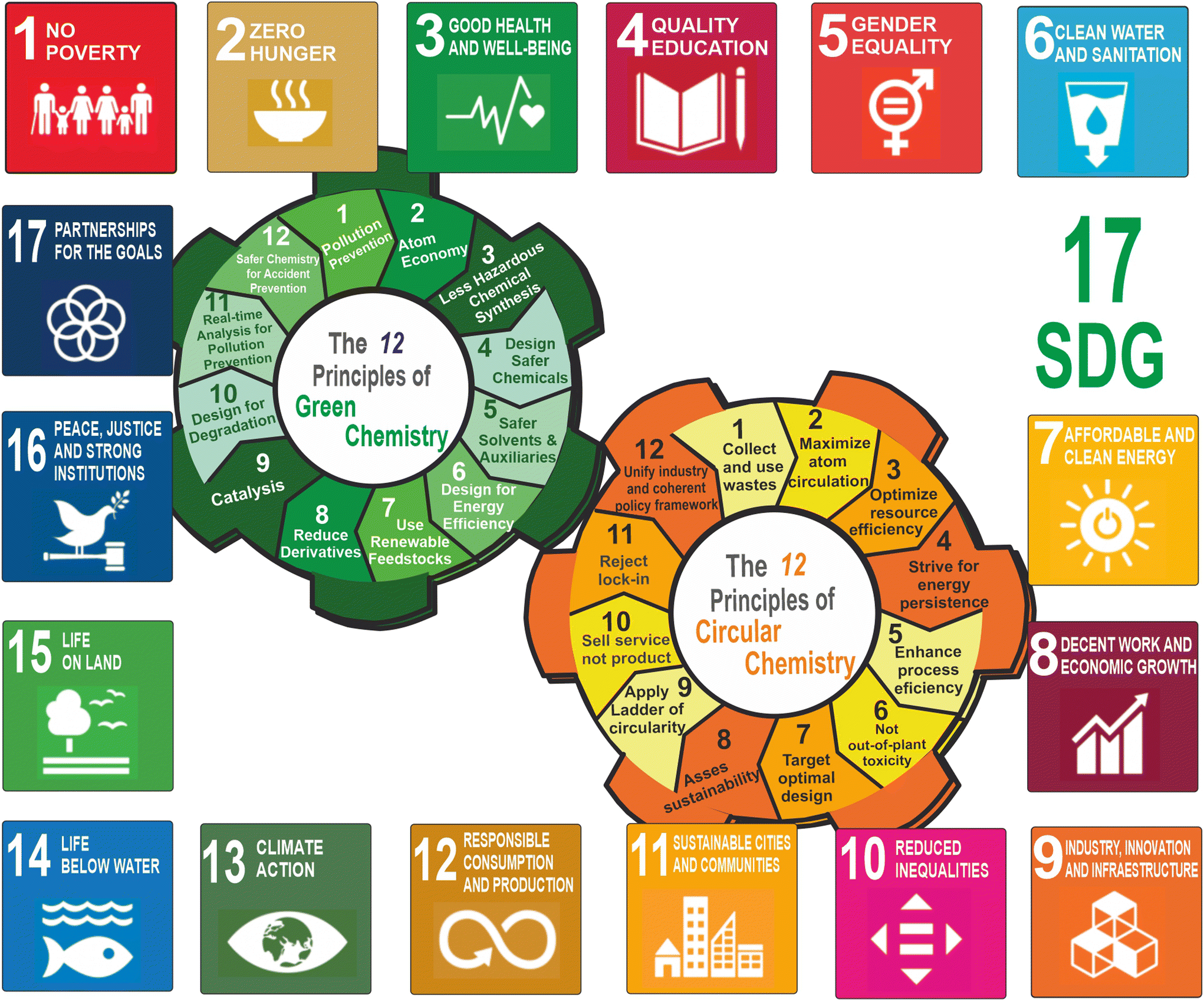 | ||
| Fig. 1 Synergistic coupling between the two “sustainability engines”, represented by the Twelve Principles of Green Chemistry,5 and the Twelve Principles of Circular Chemistry,6 respectively, to achieve the 17 Sustainable Development Goals (SDGs),4 launched by United Nations (UN) in 2015. | ||
On the other hand, by combining the concepts of green chemistry, circular economy, and sustainability, Keijer et al. have proposed the Twelve Principles of Circular Chemistry.6 Most of these principles (such as maximize atom circulation, optimize resource efficiency, strive for energy persistence, and target optimal design) match some of the Principles of Green Chemistry. They also highlight the “3R” cornerstones of circular economy (i.e. recover, recycle, and reuse) during the life cycle of a product involving all the actors at the value chain.7 By this approach, all the wastes resulting from the linear economic model (i.e. take-make-use-waste) should automatically be considered as starting materials for the preparation of alternative products, or for using them as chemical feedstocks in the synthesis of marketable products, achieving the complete recirculation of molecules and materials (see Fig. 2).8
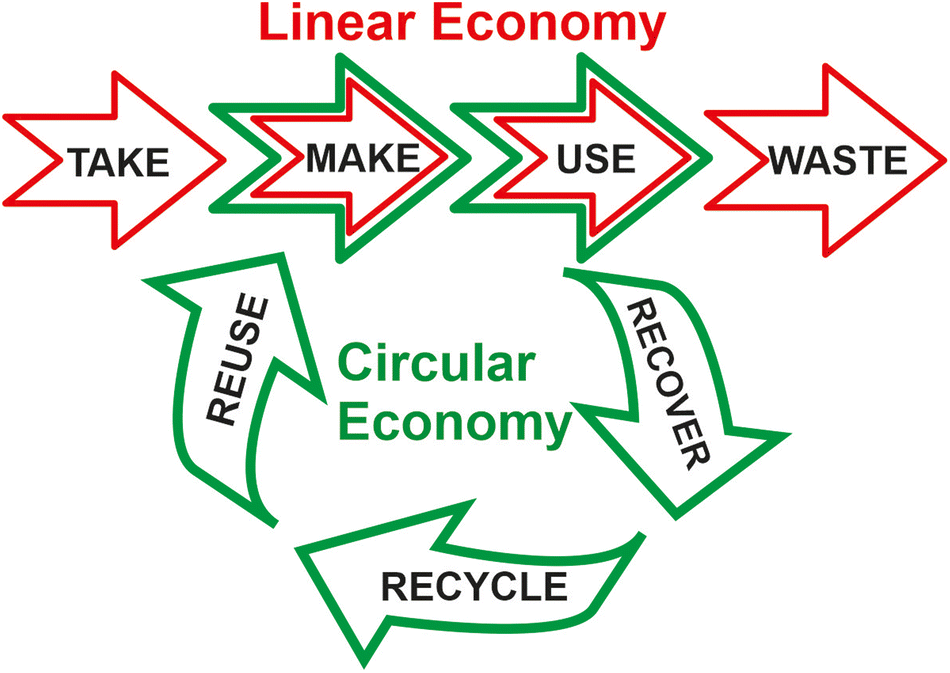 | ||
| Fig. 2 Comparative representation between the linear and circular economic models. | ||
Because of the finite nature of many resources, as well as the limited environmental tolerance of the planet against increasing waste generation, the linear economic model is fully opposite to a sustainable future. An urgent and proper modification of the rules and frameworks of the global economic policy (SDG-16 and SDG-17) must enable a broad circular economic model as soon as possible, and to promote the reduction of the consumption habits of our style of life (SDG-12) through proper education for sustainability (SDG-4).
Waste is a broad concept that usually comprises man-made materials without economic value, which are collected in the trash for landfilled and/or incinerated, or directly “thrown away” in the environment. The introduction of the E-factor in 1992 focussed attention on the problem of waste generation, defined as everything but the desired product, in chemical manufacture and gave rise to a paradigm shift in our concept of efficiency in chemical processes, from one based solely on chemical yield to one that assigns value to eliminating waste.9 Considering the most impacting wastes as those responsible for the daunting global environmental problems (i.e. global warming and the degradation of the natural habitat and its biodiversity), the emission of greenhouse gases into the atmosphere, mainly CO2, and the spreading of recalcitrant plastic materials are urgent issues to address.10 From an overall point of view, both huge problems have a common start-point: the growth of our society at the expense of unbridled use of fossil sources (i.e. coal and oil) that were used for the production of energy, chemicals, plastics, etc. While the 19th century could be named as the “coal century” and the 20th century as the “oil century”, this 21st century should be the “sustainability century”. This global “flag” should be irreversibly linked to two milestones to be progressively reached: (i) the de-fossilization of energy and material (chemicals) sources to be substituted by renewable ones and (ii) the recovery and reuse of the disseminated wastes, mainly those having a fossil origin for their chemical decomposition and/or transformation in new starting materials than to be reintroduced at the industrial production chains.11
In this context, biocatalytic processes offer unique significant advantages since enzymes are derived from inexpensive renewable resources available and are biodegradable and essentially non-toxic, in contrast to precious metal catalysts. They can promote various highly selective (chemo-, regio-, and stereoselective) chemical transformations under mild conditions (pressures and temperatures close to ambient).12 This together with the continuous progress in genomics and directed evolution open the possibility to develop clean biocatalytic processes, including live microbial cells/enzymes, for the transition of chemistry towards a sustainable and circular economy.10,13 This perspective aims to illustrate by several selected examples, the huge possibilities of biocatalysis in combination with other their key enabling tools14 (e.g. ionic liquids, mechanochemistry, chemo- or photocatalytic assisted reactions, etc.) for the transformation of the chemical industries towards a sustainable path, where the selectivity in transformations is maximized, the waste generation is minimized and the recovery and reuse of all the actors of the reaction systems are fully recovered and reused, as claimed by the circular economy criteria.15
2. Sustainable biocatalytic production of biofuels
The transition to renewable energy sources (such as hydroelectric, wind, solar, and geothermal) is absolutely necessary to be able to generate the enormous and growing amounts of energy needed to drive industrial economies.16 Biofuels obtained from renewable biomass should still be considered as another sustainable solution for the overall transition to clean energies. The production of these biofuels should fulfil two key premises: (i) based on waste feedstock from non-edible agro-resources, (ii) obtained by means of fully clean and sustainable transformation/separation processes.17 In addition, the development of several cutting-edge fermentation process (e.g. autotrophic microorganisms, or artificial autotrophs, for biological conversion of CO2 and solar energy to chemicals in third-generation autotrophic biorefineries) and18 clean biocatalytic processes remains as a cornerstone for the sustainability transition of chemistry.9One of the limitations of enzymatic transformations is the enzyme stability. However, when combined with the unsurpassed selectivity of enzymes with the excellent solvent properties of ionic liquids (ILs), an excellent setting for carrying out sustainable chemical transformations in non-aqueous environments is achieved.19 Nevertheless, any implementation of this kind of technology to sustainable industrial chemical processes needs the development of cheap and straightforward protocols suitable for pure product extraction, including the full recovery and recycling of ILs.14,20
Biodiesel and bioethanol are two excellent examples of renewable biofuels that are industrially produced by non-sustainable resources and/or procedures. First, it should be underlined that when any biofuel is obtained from edible resources, its sustainability is clearly compromised because the accessibility and prices on the food market will be conditioned by putting in competition to feed people vs. to “feed” cars. The use of non-edible wastes as substrates to produce biofuel is a unique sustainable way and the most important milestone to be fully achieved. Second, the production of biofuels should be a fully clean process, where not only the highest selectivity in chemical transformations will be reached, but also the full recovery and reuse of all the elements of the reaction systems (i.e. catalysts, solvents, etc.) should be attained.
Biodiesel, commonly named a mixture of fatty acid methyl esters (FAMEs), is a liquid fuel industrially obtained from renewable resources, such as the triacylglycerides and/or free fatty acids, FFAs (e.g. vegetable oils, animal fats, etc.) through a transesterification reaction with methanol carried out by homogeneous alkaline (e.g. KOH, NaOH, etc.) and/or acids (e.g. sulfuric acid), as catalysts, which generate glycerol as a by-product. The non-miscibility between fats and methanol and the low selectivity of these homogeneous catalysts are clear limitations in the sustainability of the process, because not only it is necessary to use large amounts of water for cleaning the biodiesel products, but also the yield is highly dependent on the free fatty acid content, as results of the formation of soaps. Also, the other resulting glycerol by-product should be separated and purified (e.g. by distillation) for any further application, because a dark liquid solution is obtained from this kind of industrial process.21 The lack in the sustainability of biodiesel is clear when using edible sources as raw material, and dirty processes for its synthesis and separation.
As an example of sustainable and clean process for biodiesel, the use of biocatalysts in Sponge-Like Ionic Liquid (SLIL) media can be considered. These media are a type of hydrophobic ILs based on cations with long alkyl side chains (e.g. octadecyl trimethylammonium bis(trifluoromethylsulfonyl)imide ([C18tma][NTf2]); 1-hexadecyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([C16mim][NTf2]), etc.), which behave as sponge-like systems as a result of their temperature switchable ionic liquid/solid phases changes.22
These SLILs have been shown as exceptional reaction media for biocatalytic reactions at temperatures higher than their melting point, as it was demonstrated for the biocatalytic synthesis of biodiesel, e.g. up to 100% yield in 8 h at 60 °C, with exceptional enzyme stability (up to 1370 days half-life time at 60 °C).23 The sustainability of this biocatalytic reaction system was demonstrated by the development of a straightforward and clean approach to extract separately the synthesized biodiesel, the glycerol by-product, and the full recovery of the full SLIL/biocatalytic system for further reuse (see Fig. 3).14,22 Due to the immiscibility of the SLILs with water together with its solid character at room temperature, the addition of a low volume of water (e.g. 20–40% of the overall reaction volume) leads to the precipitation of the SLIL, resulting in a semisolid heterogeneous mixture (see Fig. 3B). By following an iterative cooling/centrifugation protocol (i.e. 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm 1 h at room temperature, 23 and 15 °C, respectively), the mixture was then separated into three phases: an upper IL-free and pure biodiesel phase, a middle IL-free aqueous phase containing glycerol, and a bottom solid containing the SLIL (Fig. 3C). Using this approach, an excellent biocatalytic activity (96–98% biodiesel yield) was obtained. The combined SLIL/biocatalyst system demonstrated complete recovery and was successfully reused, with the activity remaining virtually unchanged throughout 12 consecutive operation cycles. This result highlights the potential for easy scaling up of the process.23 The excellent suitability of this biocatalytic sustainable approach based on SLILs was also demonstrated when using waste cooking oils, even “worsened” by the addition of FFAs (up to 30% w/w), by the addition of FFAs (up to 30% w/w), and/or methanol or solketal as nucleophile acceptors in the transesterification/esterification reactions (i.e. 100% yield of FAMEs and fatty acids solketyl esters, FASEs) in 6 h at 60 °C.
000 rpm 1 h at room temperature, 23 and 15 °C, respectively), the mixture was then separated into three phases: an upper IL-free and pure biodiesel phase, a middle IL-free aqueous phase containing glycerol, and a bottom solid containing the SLIL (Fig. 3C). Using this approach, an excellent biocatalytic activity (96–98% biodiesel yield) was obtained. The combined SLIL/biocatalyst system demonstrated complete recovery and was successfully reused, with the activity remaining virtually unchanged throughout 12 consecutive operation cycles. This result highlights the potential for easy scaling up of the process.23 The excellent suitability of this biocatalytic sustainable approach based on SLILs was also demonstrated when using waste cooking oils, even “worsened” by the addition of FFAs (up to 30% w/w), by the addition of FFAs (up to 30% w/w), and/or methanol or solketal as nucleophile acceptors in the transesterification/esterification reactions (i.e. 100% yield of FAMEs and fatty acids solketyl esters, FASEs) in 6 h at 60 °C.
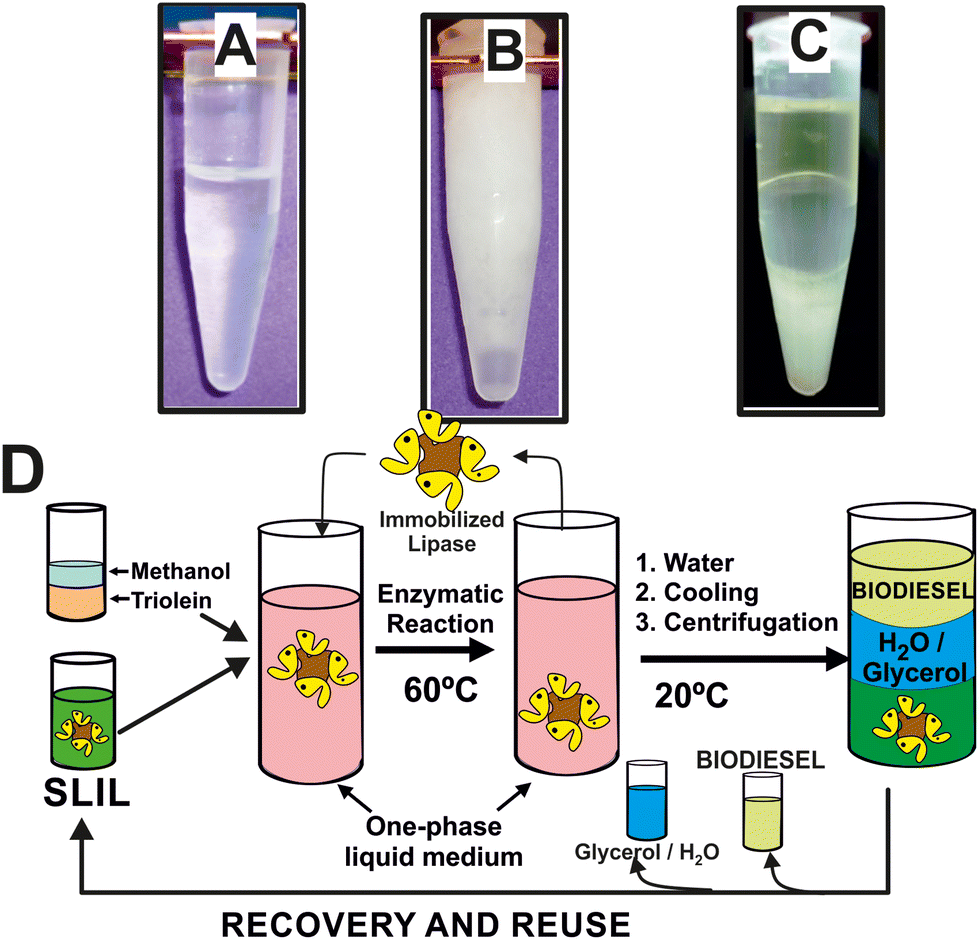 | ||
| Fig. 3 Phase behaviour of the reaction mixture containing both methyl oleate and glycerol products at 60 °C (A), 25 °C after addition of water (B), and after three consecutive centrifugation steps at 15000 rpm (1 h) at room temperature, 23 and 15 °C, respectively (C). Cyclic protocol for the biocatalytic synthesis and purification of biodiesel in sponge-like IL phases, including the full recovery and reuse of the enzyme/IL system (D). Reproduced from ref. 22 with permission from Royal Society of Chemistry, copyright 2015. | ||
The SLIL technology facilitates the straightforward separation of these biofuels by a cooling/centrifugation approach, and the biocatalyst did not show any loss in activity during reuse in these reaction systems after six operation cycles.24 This clean SLIL-based biocatalytic approach was also successfully demonstrated for the direct synthesis and nearly pure separation of terpene flavour esters.25
This technology has also successfully been used for developing one-pot systems suitable for the direct extraction of algal oil from raw material (i.e. Chlorella vulgaris), followed by its biocatalytic transformation to biodiesel and a final biofuel extraction. These mixtures were based on the combination of SLILs with [Bmim][Cl], considering the excellent suitability to carry out the biocatalytic synthesis of biodiesel of the first one, and the ability for dissolving the cellulosic biomass of the second. The extraction of oils was carried out by incubating the dry microalgae in the appropriated IL binary mixture at 110 °C, which after being cooled at 60 °C allowed to be transformed to biodiesel by an immobilized lipase. This resulted in a fast and efficient biodiesel synthesis, with up to 100% yield in 2 h at 60 °C. The subsequent cooling until room temperature and the iterative centrifugation of the resulting semi-solid systems at 20 and 18 °C led to the separation of the liquid algae-based biodiesel from the solid IL mixture, which was recovered and reused for a further operation cycle.26
In conjunction with the non-volatile nature of ILs, the complete insolubility of these SLILs in water, with melting points above room temperature, facilitates their immediate separation through precipitation as solids after cooling the reaction media to room temperature. This characteristic enables a simple and complete ILs recovery. Furthermore, the switchable (liquid–solid) nature of ILs with temperature can potentially aid in mitigating the risk of environmental contamination in the event of an accident during handling. This consideration remains valid, even when considering the justified criticism regarding the non-environmentally friendly characteristics of ILs.22 In this regard, it is crucial to devote increased efforts to the development of ILs that exhibit the desirable characteristics of SLILs, while utilizing cations and anions that are environmentally benign. This approach aims to reach a balance between the desired properties of ILs and their potential impact on the environment.
Another promising biocatalytic approach to valorise waste oils (i.e. cooking oils) is the photoenzymatic decarboxylation of long-chain fatty acids (LCFAs) into the paraffinic unit (C12–C18 hydrocarbon fuel for aviation) by the use fatty acid decarboxylase (FAP) photoenzyme.27,28 This approach presents two advantages in comparison with FAMEs as biofuel: (i) the irreversible nature of the reaction facilitates process design, (ii) the specific heat of combustion of alkanes is ca. 9% higher than that of the corresponding FAMEs. Furthermore, the lower energy demand, especially if sunlight is used making, is comparison with the deoxygenation of LCFAs into paraffins by chemocatalytic methods.29 Hollmann et al. demonstrated that photodecarboxylase from Chlorella variabilis NC64A (CvFAP) can convert a broad range of different fatty acids with turnover numbers up to 8000 (see Fig. 4).30 Furthermore, a cascade reaction combining a lipase (Candida rugosa) and CvFAP pave the way to directly transform oils (i.e. both soybean oil and waste cooking oil) directly into hydrocarbons.31 Alternatively, a multicatalytic route, based on the combination of photo-chemo-enzymatic steps, can be envisioned to produce biofuel from naturally abundant triglycerides. The sequential reaction included the lipase-catalysed hydrolysis of triolein, fatty acid photodecarboxylase (CvFAP)-catalysed decarboxylation of oleic acid, photocatalytic oxidative cleavage of long-chain alkene, and the final decarboxylation of the medium- or short-chain fatty acids catalyzed by CvFAP mutant.32 While the photodecarboxylation process is not currently ready for scaling up, it represents a highly promising approach due to its inherent simplicity. A significant challenge for industrial applications arises from the rapid photoinactivation observed in CvFAP during the process. Different strategies are being evaluated to solve these shortcomings.33 By addressing the issues related to enzyme stability, this process holds potential for future development and eventual implementation on a larger scale. One possible solution is the development of efficient CvFAP mutants by enzyme engineering enabling tuning selectivity towards either long- or short-chain fatty acids and enhancing their stability.34,35 Alternatively, media engineering, including monophasic or bi-phasic systems based on the use of ionic liquids (ILs) or deep eutectic solvents (DESs), can not only be an option to improve the solubility of triacylglycerols and long-chain fatty acids but also contribute to maintain or even enhance the biocatalytic activity for pushing up towards industrial application. This is, however, a seldom explored approach, but initial studies suggest that completely selective C18 decarboxylation over C16 is found when used in a DES. Furthermore, when applying the immobilized enzyme in DES, the yields are >10-fold higher than the ones obtained in aqueous media.36
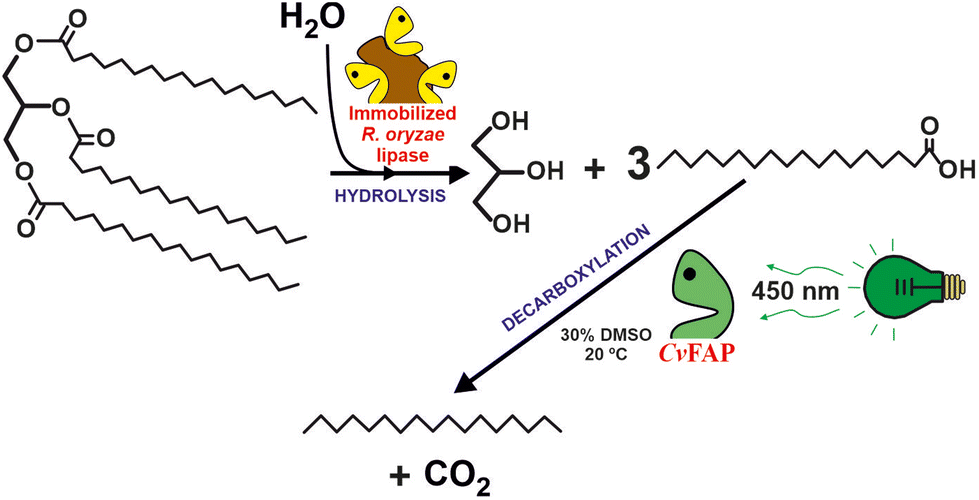 | ||
| Fig. 4 Two-steps biocatalytic cascade to transform natural triglycerides to alkanes by the consecutive actions of Rhizopus oryzae lipase, and the photoactivated fatty acid decarboxylase from Chlorella variabilis NC64A (CvFAP).30,31 | ||
The production of second-generation bioethanol from non-edible biomass (e.g. lignocellulosic biomass) using clean, sustainable, and circular approaches is another great challenge on the research and industrial field.37 Bioethanol production from cellulosic sources consists of three consecutive steps, as follows, the pre-treatment of cellulose to disrupt its highly ordered and rigid structure, hydrolysis of the cellulose to fermentable sugars, and finally, ethanol fermentation by microorganisms.
Although the synergistic action of different biocatalysts (e.g. cellulases, cellobiase, etc.) leads to the full depolymerization of cellulose to its glucose units, the crystalline structure of cellulose, which is supported by multivalent inter- and intramolecular hydrogen bonds, involves high recalcitrance to its enzymatic hydrolysis.38 The ability of certain ILs (e.g. 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]), 1-allyll-3-methylimidazoium chloride ([Amim][Cl]), etc.39) to dissolve cellulose have opened up new opportunities for the industrial valorisation of large amounts of waste cellulose-containing materials (e.g. non-edible biomass wastes,40 cotton clothes wastes,41etc.).
The current pursuits are focused on the less toxic, natural-based, biodegradable, cheaper ILs and enzyme/microorganisms ILs-tolerant, as well as moving towards a one-pot, wash-free process that combines IL pretreatment and saccharification into a single process.42,43 For instance, naturally derived ILs from choline and lysine ([Ch][Lys]) were used in one-pot together with commercial enzyme cocktails Cellic CTec2® and HTec2® for the pretreatment of 30 wt% sorghum.44 The pretreatment at 140 °C was followed by the addition of water and the enzymatic hydrolysis was performed in the presence of 6 wt% [Ch][Lys] leading to glucose yields above 80% and xylose yields above 60%.
On the other hand, different strategies can be used for the development of IL-resistant host strains.45,46 For instance, adaptive laboratory evolution (ALE) has been used to adapt E. coli MG1655-A1 to for 4 different imidazolium ILs ([Emim][Cl], [Emim][Ac], [Bmim][Cl], and [Bmim][Ac]) usually used for biomass pretreatment enabling one-pot biofuel production.47
IL recycling and reuse is a necessary step even for low-cost ILs or one-pot transformations. Several studies reported the recovery rate to be in the range of 85–96%.48 As a representative example, a sustainable and circular process for the enzymatic saccharification of ionic liquid (IL)-pretreated cellulose, in which the IL is fully recovered and recycled, has been developed (Fig. 5).49 Homogeneous cellulose solutions in the IL 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) were used to prepare amorphous cellulose by antisolvent precipitation with water by carefully designed washing step conditions, leading to the recovery of the IL (i.e. up to 99.7% recovery yield) for reuse in further cellulose dissolution/precipitation cyclic processes. Furthermore, the cellulose regenerated in each cycle was an excellent substrate for enzymatic hydrolysis, permitting full hydrolysis to glucose (i.e. up to 98.7% hydrolysis after 4 h at 50 °C; up to 4.9 g glucose per h per L g per enzyme) by the combined action of both cellulase and cellobiase enzymes in batch operations. These biocatalytic systems provide a clear glucose solution that behaves identically to glucose standard solution for metabolizing by Saccharomyces cerevisiae. The scalability of the processes was successfully demonstrated through the utilization of ultrafiltration membrane reactor systems, employing polymeric or ceramic membranes. These systems facilitated the rapid enzymatic saccharification of regenerated cellulose while enabling the complete recovery and reusability of the enzymes. By integrating enzyme saccharification with the filtration process, a clear glucose solution with a concentration of up to 113 mM was obtained, maintaining a constant permeate flow rate of 24.7 L h−1 m−2. The enzymes were successfully reused for nine operation cycles under semi-continuous operation without any loss of enzyme activity. Under continuous operation mode and using ceramic ultrafiltration membranes at different residence times, the enzymatic reactor showed constant profiles in both the permeate flow rate and the glucose concentration, demonstrating the excellent suitability of the proposed approach for the scaling up saccharification of cellulose.50
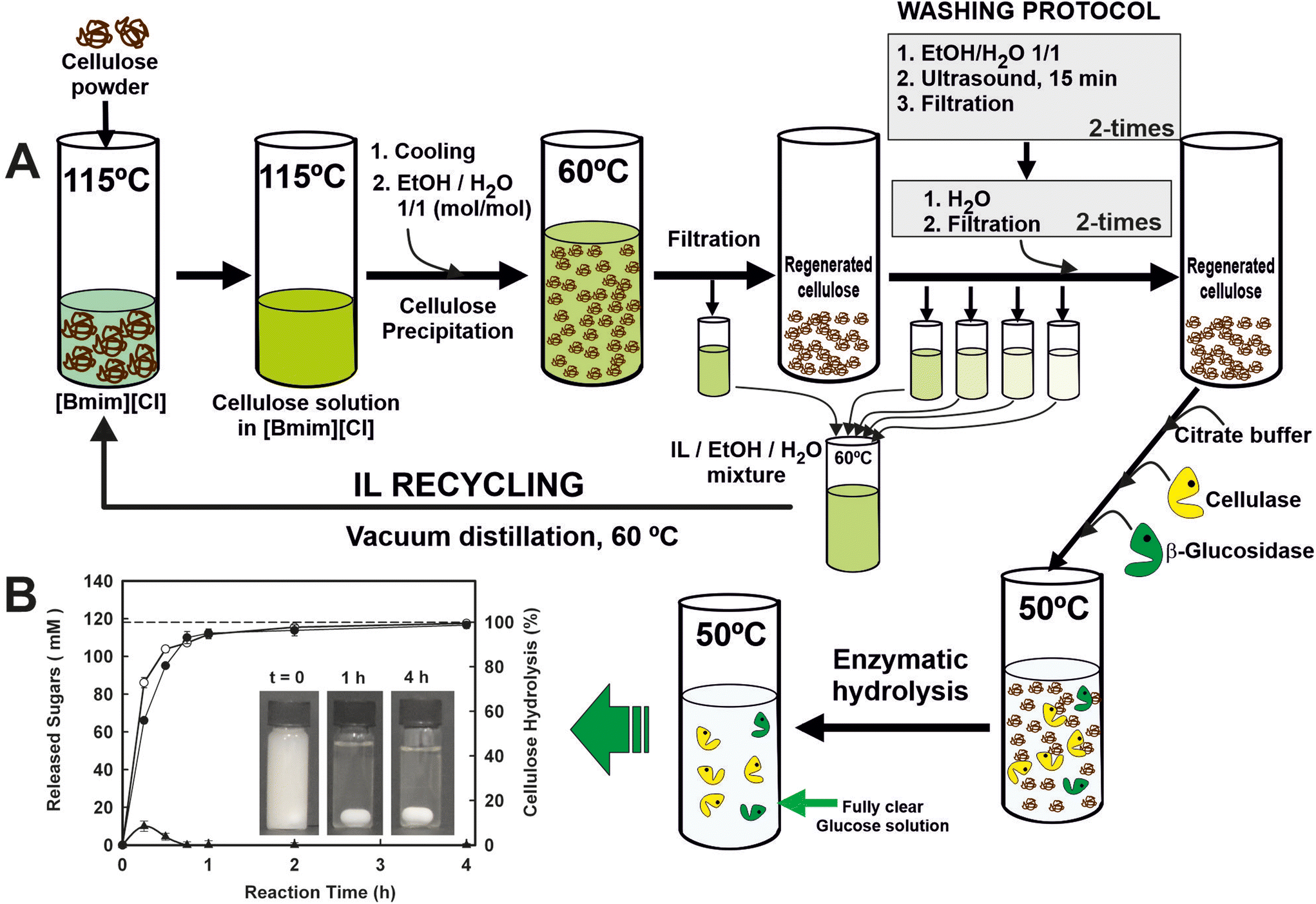 | ||
| Fig. 5 (A) Scheme of the cyclic protocol for the enzymatic saccharification of IL-pre-treated cellulose and the recycling of the ionic liquid 1-butyl-3-methylimidazolium ([Bmim][Cl]). RC: regenerated cellulose. (B) Time-course profiles of total reducing sugars (○), glucose (●) and cellobiose (▲) released by the combined action of cellulase and cellobiase, using an IL-pre-treated crystalline cellulose as substrate. Reproduced from ref. 49 with permission from Royal Society of Chemistry, copyright 2012. | ||
The development of suitable less energy-intensive membrane-based technologies for the recovery of the ILs as an alternative to distillation is needed for the scale-up of IL-based biorefinery technologies. Promising results have been reported with commercially available pervaporation systems with greater than 99.9% [Emim][OAc] (<1 wt% water) recovery from aqueous solution (≤20 wt% IL) and recycled five times.51
Alternative fermentation processes, suitable to obtain other liquid biofuels, like n-butanol by the fermentation of glucose at an industrial scale, will also come. Soucaille et al.52 reported an engineered Clostridium acetobutylicum able to produce n-butanol (i.e. up to 550 g L−1n-butanol production) for the fermentation of high concentration glucose syrup in a continuous process, and applying in situ extraction of n-butanol by distillation under low pressure and high cell density cultures.
3. Capture and valorisation of CO2 for chemicals
Currently one of the biggest challenges for society is to combat global warming, which requires the implementation of CO2 mitigation strategies. Within a circular economy framework, CO2 should be considered waste material produced in different processes that must be upgraded into added-value products. In this context, two main areas of work are being developed CO2 to power, or CO2 to chemicals. Several recent review articles have summarized the blossoming novel chemical, photochemical, and electrochemical processes to achieve sustainable decarbonization.53 Alternatively, enzymatic CO2 conversion represents a promising technology by itself or in combination with other green tools that can lead to green processes moving forwards to a CO2 circular economy. In vitro and in vivo enzymes can play key roles in this field being able not only to activate and transform CO2 into synthetically relevant target molecules and energetic vectors but also to contribute to CO2 capture technologies.54,55The simpler transformation of CO2 by an enzyme is the direct conversion CO2 to bicarbonate (HCO3−). Carbonic anhydrase (CA), which is a common enzyme that regulates CO2 in living organisms, catalyses this transformation at a high turnover rate, up to 106 per second. Thus, CA is an obvious enzymic alternative for CO2 capture, sequestration, and post-utilization. However, the relatively harsh conditions in the absorption processes (i.e. up to 60 °C for the absorption while above 100 °C for stripping, with common solvents at strong alkaline conditions, trace contaminants sulfur oxides (SOx), nitrous oxides (NOx), etc.) can cause the denaturation of the enzyme and reduce the enzyme activity/stability. In this regard, protein engineering techniques (i.e. directed evolution, rational design methods, etc.) and different immobilization strategies have been revealed as suitable tools to overcome such limitations, allowing CA to be applied even in relevant industrial conditions.56,57 It should be highlighted that in addition the protein engineering techniques de novo protein design has also been envisioned for the design of hyper-stable structures.58 Although the levels of activity of natural CA have not been reached so far (hydration efficiencies 500-fold to 1400-fold lower than the fastest human CA), there are significantly faster than small-molecule CA mimics enhancement factors of up to ∼104.59
On the other hand, the immobilization strategy (i.e. adsorption, entrapment, cross-linking, or covalent bonding) together with the nature and shape of the support not only contributes to improving the enzyme stability and facilitates the separation and the reuse in multiple cycles but also addresses limitations related to the low solubility of CO2 and the mass transfer.60 Many efforts have been devoted in this field with a wide variety of inorganic and organic supports used.61 Among them, the use of metal–organic frameworks (MOFs), which are extended porous network materials assembled by a bottom-up building block approach from metal-based nodes and organic linkers, can be highlighted.62 MOFs are excellent candidates for the immobilization of the CO2 active enzymes as they present high surface area, tunable pore size, and low heat capacity, which provide them various properties, such as CO2 adsorption.63 For instance, zeolitic imidazolate frameworks (ZIFs) have been reported to immobilize the CA into ZIF nanoparticles.64 The composite exhibits CA-improved thermal stability, maintaining a relative activity of 134% after six cycles of reuse. Furthermore, a 1.5-fold higher catalytic activity for CO2 absorption than their free CA is also observed, which can be attributed to the synergistic enhancement of CO2 adsorption by highly crystalline and porous ZIF support. Thus, CA/MOF can be a good candidate to promote CO2 capture in industrial applications.65
Membrane technologies for carbon capture is another promising field where the immobilization of CA or in general CO2-active enzymes can have a significant impact as it can lead to low energy consumption, high processability, and lower maintenance costs methodology for the adsorption and separation of CO2 from gas mixtures. Significant advances have been reported in this field.66 For instance, Jiang, Brinker et al. developed an extremely selective for CO2 by nanoconfining CA in the nanopores achieving a highly confined locally concentrated CA leading to high enzyme concentrations.67 The CO2 is captured and dissolved to form HCO3−, which is further regenerated as pure CO2 at the hydrophobic surface of the membrane. At atmospheric pressure, pH 7 and at a rate of 2600 GPU (Gas Permeation Units), the selectivity of the enzymatic membrane with CO2/N2 and CO2/H2 were as high as 788 and 1500, respectively, times greater than can be achieved in an aqueous solution, and stable over three month.
CO 2 Solutions by Saipem is a post-combustion CO2 capture technology based on the solvent washing of the flue gas by means of a water-based solution containing potassium carbonate salt and a commercial low-cost carbonic anhydrase.68 The enzyme is an engineering strain of carbonic anhydrase (CA) through a directed evolution process being highly tolerant to flue gas contaminants, such as SOx and NOx.69 The CA significantly accelerates the conversion of CO2 into bicarbonate, during absorption, and the reverse reaction (bicarbonate to CO2) during solvent regeneration. The enzyme is used at very low concentrations, and it is exceptionally selective to catalyze the reactions only with CO2. Enzyme-catalyzed aqueous salt solutions can be deployed with a variety of gas-scrubbing equipment configurations to replace costly and environmentally challenged amine solvents. The technology has been demonstrated to capture the CO2 emissions from cement clinker production. A gas/liquid CO2-packed column absorption catalyzed by CA is used and HCO3− subsequent is used to produce limestone (CaCO3). The CaCO3 minerals are physically/chemically stable preventing the release of CO2 back into the atmosphere naturally and are also raw materials for the fabrication of Portland cement, or as building-block materials enabling the circularity of the process.
Other promising enzymes for CO2 valorization are formate dehydrogenases (FDHs), which are heterogeneous groups of enzymes that catalyze the oxidation of formic acid to carbon dioxide. Under certain conditions, FDHs can exhibit reverse activity, reducing CO2 to formate with high selectivity and under mild reaction conditions. The transformation of CO2-to-formate is not only important by itself, but also for the possibility of designing a biorefinery from in vitro and vivo CO2/formate biocatalytic systems inspired by the CO2 metabolic process in cells. Thus, different approaches are being developed based on FDHs.70 Noteworthy, in vitro cascade multi-enzymatic CO2 transformations have been developed to produce target fuels and chemicals. In this cascade, the product of a catalytic transformation is transferred to an additional enzyme to be further transformed into a more complex product.71 These cascades also help to increase the efficiency of the transformation by shifting the reaction equilibrium to the desired direction.
A key parameter to be considered in these in vitro cascade enzymatic processes for the transformation of CO2 is the recycling of the cofactors. Indeed, it needed sustainable approaches for NADH/NAD+ regeneration to achieve efficient and economically viable enzymatic CO2 transformation into chemicals.72 For instance, CO2 can be transformed into methanol by the CO2 reduction into formate by FDH, followed by the reduction of formate to formaldehyde catalyzed by formaldehyde dehydrogenase (FaldDH). Finally, methanol is obtained from the further reduction of formaldehyde by alcohol dehydrogenase (ADH). All these reductions are mediated by NADH as a cofactor, generating the two protons and two electrons required for the reduction. Hence, the so-called “second enzymes” (i.e. glucose dehydrogenase,73 glutamate dehydrogenase,74 or phosphite dehydrogenase75) are needed to transfer a proton and an electron from sacrificial substrates to NAD+ closing the loop for the cofactor regeneration.76 These methodologies present the shortcoming that usually requires sacrificial donors, including organic substrates (e.g. glucose, glutamic acid, and lactate) or inorganic salts (e.g. sodium dithionite, sodium borohydride, and phosphite); therefore, reducing the atom economy and greenness of the process as a substantial amount of waste is co-generated, tarnishing the suitability for scaling up.
As mentioned in the case of CA, a great deal of effort has been also focused on the immobilization of the cocktail of enzymes to facilitate their use and practical application.77 Although the immobilization of the enzyme usually contributes to better performance in terms of activity and stability, it should emphasize the importance of positioning sequential enzymes to overcome substrate diffusion limitations.78 In general, co-immobilized enzymes have been found to ensure proper substrate/product channeling and improve the activity, stability, and reusability in comparison with the soluble enzymes.
The efficiency of these biocatalytic cascades is also limited by the low solubility of CO2 in the aqueous media.79 To overcome this limitation, the enzymatic reaction is accompanied by the addition of CA, which can increase the concentration of HCO3− in the media. An alternative strategy relies on the use of biocompatible ILs composed of choline and amino acids (i.e. [CH][Glu], [CH][Pro], [CH][Gly], and [CH][His]). The ILs as co-solvents were evaluated in the biocatalytic membrane reactor by passing a mixture of CO2, IL, and cofactor through the enzyme-loaded membrane (see Fig. 6).80 An aqueous mixture 20% [CH][Glu] in which the CO2 concentration of the methanol was around 15 times higher than in the control Tris-HCl buffer solution. The molecular simulation dynamics suggest that CO2 stays for a longer time in the vicinity of the active site of the enzyme due to the presence of the ILs phase. Longer retention times may therefore result in faster CO2 conversion. Similar findings were obtained by Huang, Zhang et al. a 143-fold increase in CO2 conversion over CbFDH using “ionozyme” based on ionic liquids (ILs) as a solvent and enzyme stabilizer.81 The remarkable performance is attributed to stabilization of the enzyme structure with increased solvation structure and shortening the distance (3.9 Å) between NADH and CO2 to favour the hydride transfer by facilitating their relative orientation and forming new hydrogen bonds at the active sites.
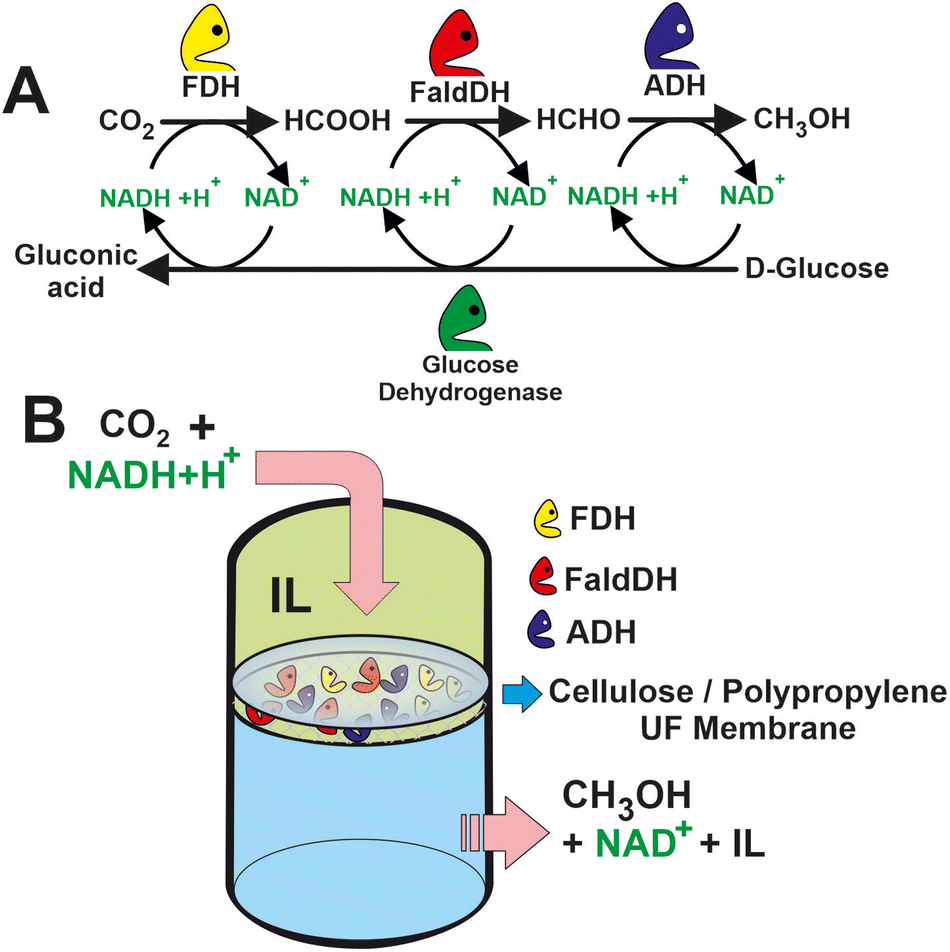 | ||
| Fig. 6 (A) Scheme of a multi-enzyme system for methanol synthesis from CO2 with in situ regeneration of NADH. (B) Set-up of the proposed IL-based multi-enzymatic membrane reactor for CO2 transformation to methanol.80 | ||
Chemo-enzymatic process integrating biocatalytic transformation with chemo-, photo-, and/or electrocatalytic steps offers multiple benefits of different types, including enhanced selectivity, high activity, and tolerance towards diverse reaction conditions. In this regard, there are different examples in which the enzymatic CO2 transformations are enhanced when coupled with other non-enzymatic steps. For instance, the use of photocatalysis to reduce NAD+ to NADH is a promising solution to boost the application of CO2-enzyme transformation that exploits limitless solar energy avoiding the use of “second enzymes” for cofactor regeneration. Photosensitizers are selected from different organic dyes (e.g. fluorescein, eosin Y, and their derivatives) and porphyrin-based materials (e.g. porphyrin and its single metal element derivatives) or alternatively semiconductors (e.g. CdS, TiO2, g-C3N4, carbon dot, and BiVO4). Different advances are reported in the development of photosensitizers for the reduction of NAD+.82,83 For instance, Zhang and Pinelo et al. have designed an ionic porphyrin as a photosensitizer for the in situ solar-driven reduction of NAD+ to NADH and cascade reduction of CO2 to methanol induced by the cascade action of FDH from Candida boidinii, FaldDH from Pseudomonas sp. and ADH.84 Compared with the free system, methanol concentration was increased sevenfold when a membrane was used as a support to integrate cascade enzymatic reaction and NADH regeneration. Liu et al. have also reported the three co-immobilized dehydrogenases on hollow fiber membranes but using commercial pristine TiO2 as simple UV/TiO2 photocatalytic and clean H2O as an electron donor making the process green and sustainable.85 With the in situ regeneration of NADH, methanol yield could reach 38.6% after 5 h, which was 3.81 times that of the single enzyme-catalyzed system.
Electro-enzyme coupling systems, where an electricity-driven CO2 reaction is catalyzed by free or immobilized enzymes at the cathode chamber of the bioelectrochemical systems, also offer attractive advantages.86 The electrocatalytic conversion of CO2 can take place in the performances of NADH-independent and NADH-dependent oxidoreductases in the form of direct electron transfer (DET) and mediated electron transfer (MET) with or without the addition of natural or artificial cofactors. For instance, Minteer et al. have reported the reduction of CO2 to formate by a molybdenum-dependent FDH, which was immobilized at an electrode surface with a low-potential redox polymer, employing cobaltocene to mediate electrons to Mo-FDH.87 Formate production was confirmed by conducting a secondary enzymatic assay in addition to NMR spectroscopy, and a faradaic efficiency of 99 ± 5% confirmed formate to be the only product of CO2 reduction by this bioelectrode.
Zhang et al.88 have evaluated the use of different natural deep eutectic solvents (NADES), including glutamate glycerol (GluGly), serine glycerol (SerGly), arginine glycerol (ArgGly), and histidine glycerol (HisGly), as the co-electrolyte for the electro-enzymatic conversion of CO2 to methanol using the three dehydrogenases (FDH, FaldDH, and ADH) in their free forms with the electrochemical regeneration of NADH. The NADES provides a biocompatible media with improved CO2 solubility and good electric conductivity leading to a methanol yield two times higher than that in the Tris-HCl buffer (0.22 mM) and 16-times higher than the control reaction.
Microorganisms can also play a crucial role in harnessing the capabilities of the metabolic pathways and enzymatic activities of microbial cell factories to convert CO2 into value-added products. For instance, by integrating heterologous genes for lactic and itaconic acid synthesis, a synthetic autotrophic strain of Komagataella phaffii (formerly known as Pichia pastoris) can serve as a versatile platform for the production of value-added chemicals using CO2 as the primary feedstock. Experiments using 13C labeling demonstrated that these engineered strains have the capability to assimilate CO2 as the sole carbon source and produce organic acids, with yields reaching up to 600 mg L−1 of lactic acid or ∼2 g L−1 of itaconic acid.89 This highlights the potential of using different microorganisms to convert CO2 into various fuels and chemicals, including ethanol, fatty acids, proteins, and biofuels, all in a carbon-neutral manner.18 Thus, utilizing microorganisms to convert CO2 into fuels and chemicals is a promising avenue for utilizing CO2 as a feedstock and further advancing the concept of a circular carbon economy.
Less explored are, however, the combination chemo enzymatic cascade process where the CO2 is coveted by chemo catalyst and the resulting product is transformed by biocatalyst in the desired final product. In this context, a sustainable chemo-enzymatic process for producing both glycerol carbonate acrylate (GCA) and glycerol carbonate methacrylate (GCMA), as useful monomers for the preparation of biodegradable plastic materials, was reported. The process consisted of two consecutive catalytic steps, which can be carried out by either sequential or one-pot experimental approaches. Glycidyl (meth)acrylate was first synthesized by the enzymatic transesterification of (meth)acrylate vinyl ester with glycidol in SLILs22 as the reaction medium (100% yield after 6 h at 60 °C, see Fig. 7). SLILs not only provided a suitable reaction medium but also allowed the simple isolation of the resulting glycidyl esters as an IL-free pure fraction through a straightforward cooling/centrifugation protocol. The second catalytic step consisted of the synthesis of GCA, or GCMA, as the outcome of the cycloaddition of CO2 to the obtained glycidyl (meth)acrylate catalysed by a covalently attached 1-decyl-2-methylimidazolium moiety (supported ionic liquid-like phase, SILLP)90 in a solvent-free system and under mild conditions (60 °C, 1 bar), leading to up to 100% yield after 6 h. The components of the reaction system (biocatalyst/SLIL/SILLP) can be fully recovered and reused for at least 6 cycles with unchanged catalytic performance.91
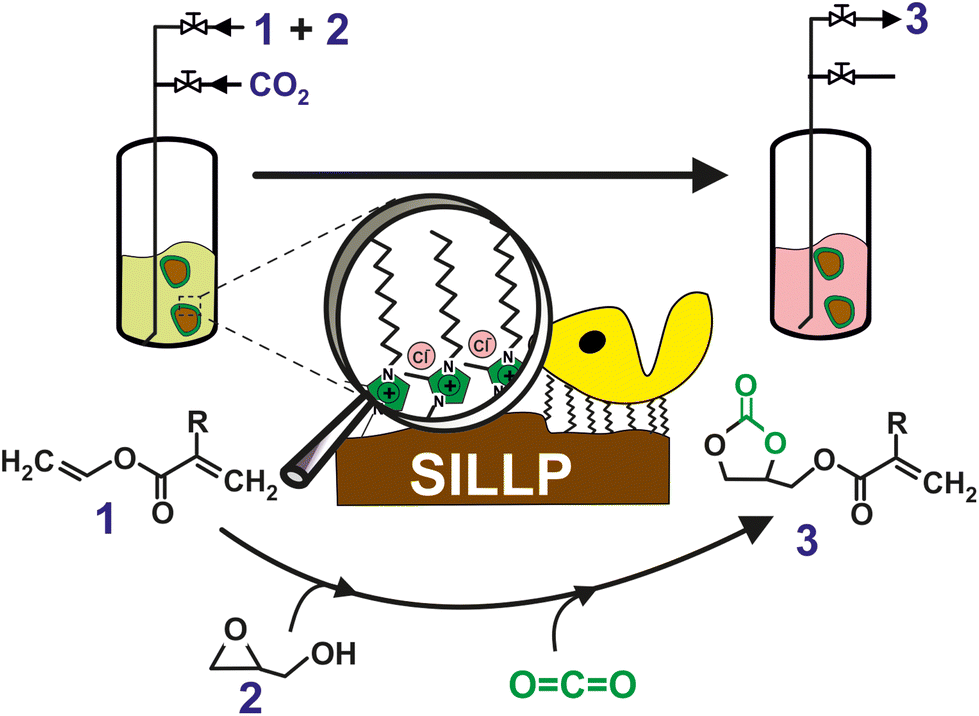 | ||
| Fig. 7 One-pot chemo-enzymatic synthesis of GCA or GCMA (3) by means of two consecutive reactions carried out by the immobilization of CALB onto 1-alkyl-3-methylimidazolium-based SILLPs, as a dual catalyst, under solvent-free conditions. Vinyl (meth)acrylate (1); glycidol (2). Vinyl acrylate (R = –H); vinyl methacrylate (R = –CH3). Reproduced from ref. 91 with permission from Royal Society of Chemistry, copyright 2021. | ||
The transformation of CO2 to other bulk chemicals by means of CO2 and sustainable and cheap energy can be achieved by hybrid systems with in vitro and in vivo enzymatic systems.92 Indeed, formatotrophic microbes can economically convert CO2/formate into bulk chemicals, such as fuels, other value-added products (e.g. solvents, plastic, monomers, pigments), and even protein. In the same context, it is also noteworthy that the living Quantum Dot (QD)-bacterial nanobiohybrid reported by Nagpal et al. that can provide large turnover numbers and frequencies along with high quantum efficiency for the direct conversion of CO2 and light into MKs, BDO, H2, IPA, NH3, FA, and PHB using a range of different light-absorbing QDs and targeted enzymes in different bacterial strains. The system presents broad applicability by simply suspending them in buffered water and bubbling air and/or CO2 demonstrating the potential and possible application of the proposed method for bioenergy supply and carbon assimilation. This type of integrated platform paves the way for other photo/electro enzymatic reaction systems that enable efficient and sustainable production of a broad range of chemicals and fuels under mild conditions from renewable sources of energy.
4. (Bio)catalytic depolymerisation of plastic materials
Since its invention in the early 20th century, plastics have surpassed most other human-made materials with the rapid production growth of nearly 200-fold from 2 million tons (Mt) in 1950 to 359 Mt in 2018. Overall, nearly 60% of plastics that have ever been made are estimated to be landfilled or discarded in the environment without proper treatment, which will persist for centuries with very slow degradation.93 Although the transition from plastics based on petroleum derivatives towards biodegradable polymeric materials based on renewable resources should be carried out as soon as possible by reducing the use of fossil resources, the development of sustainable approaches for depolymerizing petrochemical plastic wastes is another urgent necessity. Thus, it is imperative to develop novel methodologies fulfilling the green chemistry principles that allow plastic waste management to move towards a circular economy. They should not only consider plastic recycling but also aim polymer upcycling.94 First introduced by Gunter Pauli, the term “upcycling” refers to any process enabling the transformation of by-products, undesired, unwanted, or waste products into new products with increased “value”.95Emerging technologies for the chemical recycling of waste plastics have attracted significant attention from academia and industry. Green innovations in chemical and biological catalyst design and reaction engineering are key for promoting the development of useful approaches for plastic recycling and upgrading by overcoming the kinetic and thermodynamic limitations of depolymerization reactions.96 This is a huge challenge as the recalcitrant character of plastic wastes is enhanced by its crystallinity, or by the presence of other formulation components (e.g. metals, dyes, pigments, fillers, antioxidants, plasticizers, etc.) that provide further barriers to the interfacial (bio)catalytic action, or even its inhibition.97,98
In this context, the use of microorganisms or enzymes by themselves or in combination with chemocatalysts can be envisioned as a greener method to develop novel recycling or upcycling processes. In principle, the use of biocatalysts enables polymer transformations at lower temperatures and without the need for any toxic reagents. However, the efficiency of the biocatalytic treatment is highly dependent on the nature of the bonds present in the polymeric backbone. Thus, there are hydrolyzable bonds in backbone plastics, such as ester, amide or carbamate bonds, as shown in poly(ethylene terephthalate) [PET], Nylon, and polyurethane [PU]. In this case, enzymes, such as lipases, esterases, ureases, and cutinases, are among those with a certain ability to perform depolymerization of this kind of plastic.99 On the other hand, there are non-hydrolyzable C–C backbone plastics (i.e. polyethylene [PE], polypropylene [PP], polyvinyl chloride [PVC], and polystyrene [PS], or expanded polystyrene [EPS]) where the C–C bond is generally more recalcitrant to be hydrolysed by the direct enzymatic action.100 As a representative example, the Trametes versicolor IFO 6482 fungi was able to reduce the PE elongation by 20% in 3 days, reducing the Mw from 242000 to 28300 Da.101
Recent studies have pushed forward the biological recycling of hydrolyzable bond backbone plastics, such as polyester, using enzymes for the depolymerization step. Indeed, biocatalytic approaches based on either the whole cells (e.g. bacterial, fungi, etc.) or enzymes (e.g. laccases, peroxidases, alkane hydroxylases, etc.) have been shown as suitable systems for PE depolymerization.102 More than 24 different enzymes with PET degrading ability have been identified. All of these enzymes are hydrolases, catalysing the breaking of the PET polymer into terephthalic acid (TPA), ethylene glycol (EG), bis(2-hydroxyethyl) terephthalate (BHE), and (mono-(2-hydroxyehyl)terephthalic acid (MHET).103 Interestingly, a comparison of terephthalic acid (TA) production from petrol to that from enzymatic PET depolymerization revealed a 69% lower energy requirement and 17% lower greenhouse gas emissions for the latter, thus encouraging the development of biocatalytic recycling processes.104
As a representative example of a fully sustainable industrial approach for the biocatalytic depolymerization of PET from waste plastic bottles, the process developed by Marty et al. at the French company CARBIOS, should be underlined. By using computer-aided protein engineering techniques, this group has produced a PET depolymerase variant, useful for PET based on high aromatic terephthalate units, that achieves, over 10 hours, a minimum of 90% CARBIOS has developed a PET hydrolysis process that achieves a high productivity rate of 15.5 grams of terephthalate per liter per hour. This corresponds to approximately 200 grams per kilogram of PET suspension, with an enzyme concentration of 2 milligrams per gram of PET. Importantly, this hydrolysis process has been demonstrated to be cost-effective, with an increased cost of only around 4% compared to the virgin polymer, as shown in Fig. 8. Furthermore, CARBIOS has successfully demonstrated the polymerization of purified PET monomers, further advancing the readiness level of their technology. As a result, CARBIOS is regarded as the leading company with the closest system to being proven in an operational environment to reach a technology readiness level of 9.105
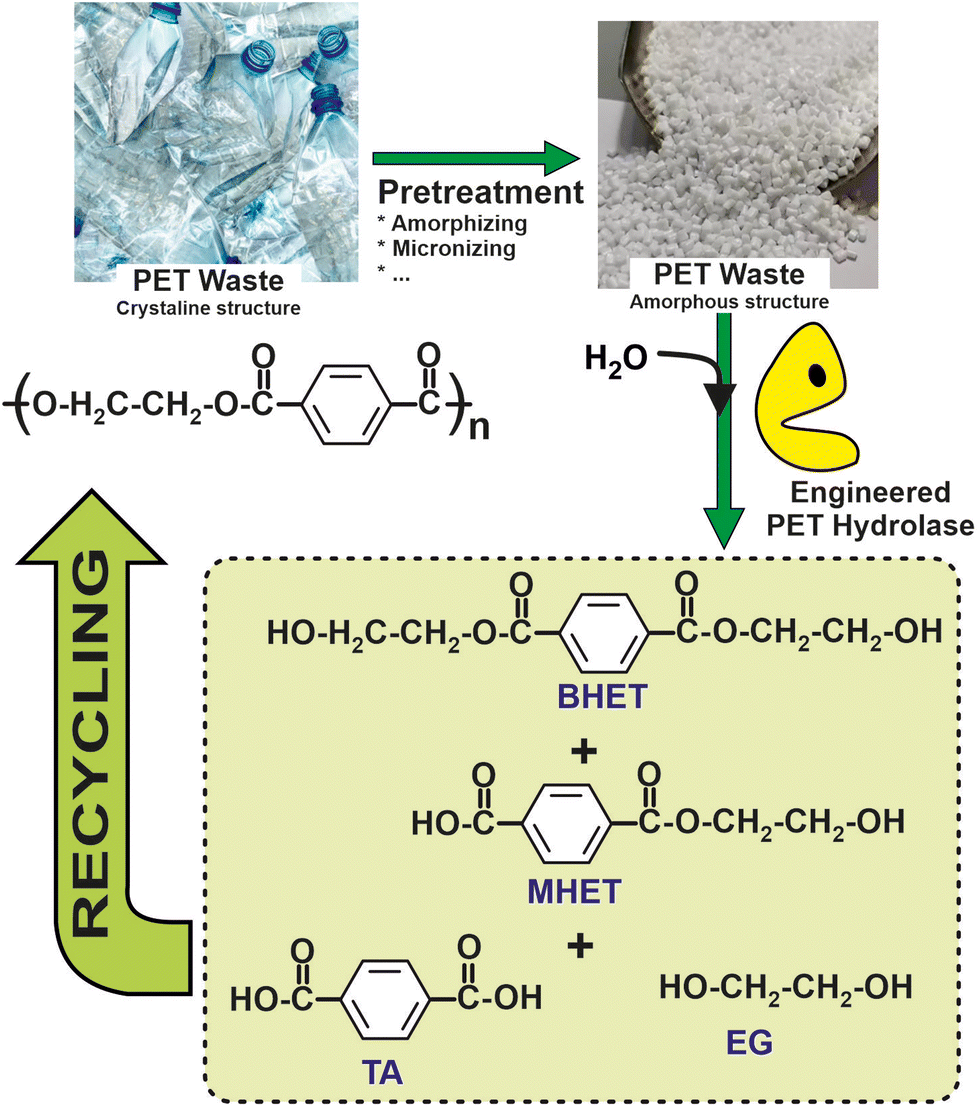 | ||
| Fig. 8 Schematic representation of a circular chemistry process for recovery and reuse of polyethylenepterephtalate (PET), based on the enzymatic hydrolysis by means of engineered biocatalysts.105 | ||
Since its beginning in 2011, CARBIOS has made significant advances in its technology for PET depolymerization. Starting at a scale of 20 kg PET in a 150 L reactor, CARBIOS has achieved a remarkable 97% depolymerization rate within a reaction time of 16 hours at a pilot scale of 1000 L in 2022.106 This substantial progress paves the way for the envisioned future, where CARBIOS aims to annually recycle over 2 million tons of PET by 2030.107 By doing so, their solution can help prevent more than 1.8 million tons of plastic from ending up in landfills or being incinerated each year, also reducing CO2 emissions by approximately 2 million tons.
The main challenge to depolymerise high-quality PET feedstocks is that it requires an energy-intensive melt-amorphization step ahead of enzymatic treatment. Different efforts are being pursued to achieve more efficient and/or stable biocatalytic systems aiming either to get new insights into the structure–function relationship of PET hydrolases beneficial for the PET depolymerization performance,108 or developing novel strains of PET hydrolases.109–113 Alternatively, the use of moist–solid reaction mixtures under milling can be envisioned as a suitable alternative to traditional dilute solutions enabling the direct enzymatic depolymerization of high-crystallinity PET, including post-consumer packaging, and even mixed plastics, while avoiding the energy-expensive melt-amorphization step currently considered necessary for efficient enzymatic PET depolymerization.114
The enzymatic degradation of lower-grade PET waste, including discarded fabrics, textiles, and waste carpets (polyester fibers), also presents an opportunity for the application of enzyme-based systems for textile-to-textile recycling under mild conditions (e.g. aqueous reaction media, atmospheric pressure, and temperature up to 65–70 °C). Furthermore, in the case of complex fibre mixtures, the high substrate specificity provided by the enzyme eliminates the need for fibre separation. For instance, Kaabel et al. reported that the direct enzymatic hydrolysis of highly crystalline PET textiles mechanoenzymatic PET hydrolysis by the commercial enzyme is unaffected by common contaminants in PET recycling streams, such as polypropylene, colourants, and cotton, and is also not hindered by the simultaneous saccharification of cotton in the solid state. PET/cotton textiles could be directly and selectively depolymerized to terephthalic acid (TPA) by using a commercial cutinase from Humicola insolens under moist–solid reaction conditions, affording up to 30 ± 2% yield of TPA. Besides, the simultaneous or sequential application of cellulase enzymes CTec2® renders to the cotton codepolymerization providing up to 83 ± 4% yield of glucose without any negative influence on the TPA yield.115
A potential renewable alternative to PET is 100% bio-based polyethylene furandicarboxylate (PEF). PEF is produced by polymerizing furandicarboxylic acid (FDCA), obtained from sugars, with bio-based mono-ethylene glycol. PEF is considered a renewable-based solution with superior performance properties compared to the widely used fossil-based counterpart PET.116 PEF large-scale production through Avantium's YXY Technology was validated at pilot scale and was announced to be ready for scaling up to a flagship plant in 2023. PEF price is a major constraint in its full penetration into the market. However, owing to PEF superior barrier properties, its production and commercialization can be boosted and can benefit from specific niche markets.117 In this context, biocatalysis offers a promising approach for both the synthesis and recycling of PEF.118 By utilizing enzymes, such as CALB Novozyme 435, the polycondensation of dimethyl 2,5-furandicarboxylate with aliphatic diols can be catalysed in ILs and DESs, providing an alternative to traditional polymerization methods. This enzymatic approach allows the production PEF without the formation of undesired by-products that can cause colouration.119 Moreover, biocatalytic processes have been investigated for the depolymerization and potential recycling of PEF. Thus, enzymes capable of digesting PET, such as the newly discovered PETase, obtained through engineering, can also efficiently break down both PEF, offering a promising approach for the degradation and potential recycling of these polymers.120 In addition, the hydrolysis of bio-based polymer blends, such as poly(ethylene 2,5-furandicarboxylate)/polyglycolide acid, can be achieved using porcine pancreas lipase in aqueous saline media.121
Polyurethane (PU) is another plastic product with huge environmental impact. PU is produced per year on a global scale at 23.89 million metric tons in 2022 and with an increasing rate of 5% per year.122 Since many PU types have a thermoset nature with covalent crosslinking, their recycling is still extremely challenging. As a result of the absence of reuse procedures, a large amount of PU waste produced in the European Union goes to landfill (up to 45%) or incineration (up to 33%), which means a dramatic impact on the environment, while only 5% is mechanically recycled. Every year, over 40 million mattresses are discarded in the European Union, representing a stacked pile of 904-times higher than the Mount Everest, and this amount should be increased by the post-production of wastes (e.g. mattress trimmings) up to 10% of total PUF production.123 Despite a common name, PUs are a very heterogeneous group of compounds obtained by the polyaddition reaction of a wide variety of polyisocyanates and polyols. This structural diversity not only influences the characteristics of the resulting polymer but also their possible recycling strategy. Thus, special attention should be put to the specific PU composition as directly correlates with the recycling/upgrading response. Polyester-polyurethane (PS-PU) obtained from polyester-based alcohol is much more prone to hydrolytic degradation due to the presence of hydrolysable ester groups than ether-polyurethane (PE-PU) that presents on the polymeric backbone ether and urethane bonds with a strong resistance to be hydrolysed by biocatalyst. Therefore, in most cases, esterases are enzymes usually reported for the degradation of PS-PU being able to break PUs to hydrolyse the ester bonds in polyester-PUs but not cleaving the stronger urethane bonds.124 Although several studies have reported enzymatic hydrolysis of the urethane bounds of carbamate compounds,125 the biocatalytic degradation of the PUs backbone have not been reported. The enzymatic degradation of PUs remains mainly limited to polyester-PU mixtures (e.g. Impranil®) by means of the concerted action of extracellular and cytoplasmic esterase and urethane-cleaving activities of Alicycliphilus denitrificans BQ1.126 Also, Magnin et al. reported the use of Candida antarctica lipase B for the hydrolysis of PU foams derived from 2,4-toluene diisocyanate (TDI) and polycaprolactone (PCL) diols leading to 6-hydroxycaproic acid (6-HCA) and a short diacid containing TDI linked with urethane linkage to two 6-HCA units.127 The recovered compounds can be used, in principle, as components for the synthesis of new polymers in an open-loop upcycling strategy.
A possible solution for the recovery of the polyether-polyurethane foam PUs is to develop a two-step chemo-enzymatic, where a chemocatalytic process breaks down the polymers degrading the PUs into suitable reagents for a further enzymatic treatment. In this regard, Branson et al. developed a recycling procedure consisting of glycolysis followed by enzymatic hydrolysis, allowing both the polyether polyols and the aromatic diamines to be recovered from polyether-polyurethane foams.128 The process consists of the glycolysis of the polymer at 200 °C using an excess of diethylene glycol (DEG) containing 1% (w/w) of tin(II)-2-ethylhexanoate as a catalyst. The glycolysis is followed by the enzymatic hydrolysis of the resulting low molecular weight dicarbamate by a metagenome-derived urethanase releasing the glycol (DEG), carbon dioxide, and the aromatic diamine (TDA) and opening this strategy to broadly diverse polyether-polyurethane wastes. The discovery of new urethanases was achieved by isolated DNA from soil collected from a site that had been exposed to polyurethanes and produced a metagenome library. This library was screened for urethanase activity allowing the identification of active urethanases.
To upgrade plastic, or even mixed plastic waste, into valuable products, a few promising multicatalytic approaches pairing chemical and biological catalysts have been envisioned recently. In this context, Diao et al. described a simple chemical–biological hybrid method for upcycling PET by cascading the alkaline hydrolysis of PET and the upgrading of the resulting monomers into high-value chemicals.129 The process allows the conversion of both terephthalic acid (TPA) and ethylene glycol (EG) from waste plastic (PET at $1.00 kg−1) to high-value chemicals (e.g. lycopene valued at several hundreds to thousands of $ per kilogram). The rational metabolic engineering of Rhodococcus jostii strain PET (RPET), which can directly use PET hydrolysate as a sole carbon source, improves the lycopene production by more than 500-fold over that of the wild type reaching a production of lycopene of 1300 mg L−1. The catabolic pathways of TPA and EG support the cell growth of the RPET strain. TPA is converted to acetyl-CoA and succinate to fuel the TCA cycle via the beta-ketoadipate pathway.
Sullivan et al. developed a two-stage oxidation and biological funnelling approach that can break down individual and mixed polymers (PS, HDPE, and PET) by chemical oxidation and reform them, in the second step, into various platform or speciality chemicals (i.e. β-ketoadipate or polyhydroxyalkanoates) by adjusting the metabolic engineering pathways of a robust soil bacterium, such as Pseudomonas putida.130 The initial metal-catalyzed oxidation (Co(II), Mn(II) and acetic acid) of polymers offers an agnostic feedstock approach to deconstruct the mixed-polymer waste by autoxidative depolymerization into oxygenated small molecules (i.e. terephthalic acid, benzoic acid and dicarboxylic acids). These water-soluble compounds can be the feedstocks for biological funnelling, wherein an engineered microbe converts diverse chemicals to a single product. Two strains of Pseudomonas putida were used to convert the mixture to polyhydroxyalkanoates, a natural polyester with growing industrial applications or alternatively to convert benzoate and terephthalate to β-ketoadipate, a monomer for performance advantaged polymers.
Following a similar methodology, Rabot et al. generated a distribution of diacids from the catalytic oxidative depolymerization of a series of polyethylenes.131 These diacids are rapidly isolated and upgraded by the engineered strains of Aspergillus nidulans into structurally complex and pharmacologically active compounds (i.e. perbenzaldehyde, citreoviridin, and mutilin) expanding the catalogue of products to which PE can be upcycled. Indeed, engineered strains of the filamentous fungus Aspergillus nidulans have also been reported for the biosynthetically conversion of benzoic acid, which is obtained by the oxidative depolymerization of PS, to the structurally diverse pharmacologically active secondary metabolites, such as ergothioneine, pleuromutilin, and mutilin.132
These multi(bio)catalytic approaches, where different green tools provide synergies, pave the way to the upgrading of mixed polymeric wastes into value-added products within the framework of the circular economy, while applying the green chemistry concepts to meet the major challenges of the fight against climate change.
5. Conclusions
The 17 SDGs are an important individual and collective wake-up call for a necessary transition of our model of society. The limited resources of our planet, and our indiscriminate use of them, urgently need us to change both our production systems and our consumption habits, and to implement new technological tools to mitigate the effects of global warming, and to protect our environment and the biodiversity of our planet. And the best weapon to face these challenges is scientific knowledge through which we can educate and train our young people, who are aimed to build our future. An online study (N = 529) on psychological factors influencing preferences for three types of plastic bottles (i.e. conventional fossil-based PET bottle, visually identical bio-based PEF bottle, and a visually distinct bio-based PEF bottle with a paper outer layer) reported positive attitudes towards bio-based plastic, as well as their willingness to pay more for it, and, irrespective of being observed, overwhelmingly preferred the bio-based bottles (96.8%).133Chemistry is the science that has contributed the most to improving our quality of life. The molecules and materials created by chemists, later implemented in their industrial production, have allowed us to reach levels of comfort unimaginable a century ago, and chemistry is called to continue to play a leading role towards solving all these great challenges of humanity by providing appropriate solutions based on sustainability criteria.
The development of a circular chemical industry, built on the principles of green chemistry, is probably the definite path to a sustainable future. The examples presented in this paper show that it is possible, not only to design clean chemical processes, but also to recover and reuse wastes, and reintroduce them back into industrial production chains. Biocatalysts are playing and will play an essential role, and their technological potential is undoubtedly one of our greatest weapons. The amazing and efficient synergies found through the combination of biocatalysts and other key enabling technologies are opening important paths for the creation of new sustainable circular chemical processes to produce chemicals and upgraded waste, based on sustainable processes and the circularity of materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been partially supported by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021- 124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN –European Union Next Generation EU-PRTR (TED2021-129626B-C21/C22), and Fundación SENECA (21884/PI/22) grants.Notes and references
- P. Lozano, Mini-Rev. Org. Chem., 2023, 20, 3–4, DOI:10.2174/1570193X19666220221105712.
- United Nations report, https://www.un.org/en/global-issues/population.
- M. Kirschner, Adv. Sustainable Syst., 2022, 6, 2100046 CrossRef.
- United Nations, Transforming our world. The 2030 Agenda for Sustainable Development. A/RES/70/1, New York, 2015. https://sdgs.un.org/es/goals Search PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Nat. Chem., 2019, 11, 190–195 CrossRef CAS PubMed.
- H. Mutlu and L. Barner, Macromol. Chem. Phys., 2022, 223, 2200111 CrossRef CAS.
- E. A. Olivetti and J. M. Cullen, Science, 2018, 360, 1396–1398 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2023, 25, 1704–1728 RSC.
- R. A. Sheldon and D. Brady, ChemSusChem, 2022, 15, e202102628 CrossRef CAS PubMed.
- J. Garcia-Martinez, Angew. Chem., Int. Ed., 2021, 60, 4956–4960 CrossRef CAS PubMed.
- Biocatalysis in Green Solvents, ed. P. Lozano, Academic Press-Elsevier London, 2022. ISBN: 9780323913065 Search PubMed.
- Enzymes for Solving Humankind's Problems: Natural and Artificial Systems in Health, Agriculture, Environment and Energy, ed. J. J. G. Moura, I. Moura and L. B. Maia, Springer Nature Switzerland AG, 2021. ISBN: 9783030583149 Search PubMed.
- R. Villa, E. Alvarez, R. Porcar, E. Garcia-Verdugo, S. V. Luis and P. Lozano, Green Chem., 2019, 21, 6527–6544 RSC.
- K. N. Ganesh, D. Zhang, S. J. Miller, K. Rossen, P. J. Chirik, M. C. Kozlowski, J. B. Zimmerman, B. W. Brooks, P. E. Savage, D. T. Allen and A. M. Voutchkova-Kostal, Environ. Sci. Technol., 2021, 55(13), 8459–8463 CrossRef CAS PubMed.
- T. S. Genc and S. Kosempel, Energies, 2023, 16, 2965, DOI:10.3390/en16072965.
- B. F. Pfleger and R. Takors, Curr. Opin. Biotechnol., 2023, 80, 102913 CrossRef CAS PubMed.
- (a) Z. H. Liu, K. Wang, Y. Chen, T. W. Tan and J. Nielsen, Nat. Catal., 2020, 3, 274–288 CrossRef CAS; (b) S. Cestellos-Blanco, J. M. Kim, N. G. Watanabe, R. R. Chan and P. D. Yang, iScience, 2021, 24, 102952 CrossRef CAS PubMed.
- (a) T. Itoh, Chem. Rec., 2023, e202200275, DOI:10.1002/tcr.202200275; (b) T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS PubMed.
- Z. Li, Q. Han, K. Wang, S. Y. Song, Y. J. Xue, X. L. Ji, J. L. Zhai, Y. H. Huang and S. J. Zhang, Catal. Rev., 2022 DOI:10.1080/01614940.2022.2074359.
- G. Santori, G. Di Nicola, M. Moglie and F. Polonara, Appl. Energy, 2010, 92, 109–132 CrossRef.
- P. Lozano, J. M. Bernal, E. Garcia-Verdugo, G. Sanchez-Gomez, M. Vaultier, M. I. Burguete and S. V. Luis, Green Chem., 2015, 17, 3706–3717 RSC.
- P. Lozano, J. M. Bernal, G. Sanchez-Gómez, G. Lopez-Lopez and M. Vaultier, Energy Environ. Sci., 2013, 6, 1328–1338 RSC.
- P. Lozano, C. Gomez, A. Nicolas, R. Polo, S. Nieto, J. M. Bernal, E. Garcia-Verdugo and S. V. Luis, ACS Sustainable Chem. Eng., 2016, 4, 6125–6132 CrossRef CAS.
- (a) P. Lozano, J. M. Bernal and A. Navarro, Green Chem., 2012, 14, 3026–3033 RSC; (b) E. Alvarez, J. Rodriguez, R. Villa, C. Gomez, S. Nieto, A. Donaire and P. Lozano, ACS Sustainable Chem. Eng., 2019, 13307–13314 CrossRef CAS.
- P. Lozano, J. M. Bernal, C. Gomez, E. Alvarez, B. Markiv, E. Garcia-Verdugo and S. V. Luis, Catal. Today, 2020, 346, 87–92 CrossRef CAS.
- X. B. Guo, A. Xia, W. Y. Zhang, Y. Huang, X. Q. Zhu, X. Zhu and Q. Liao, Bioresour. Technol., 2023, 128232 CrossRef CAS PubMed.
- T. Iqbal, S. Chakraborty, S. Murugan and D. Das, Chem. – Asian J., 2022, 17, 202200105 Search PubMed.
- B. S. Chen, Y. Y. Zeng, L. Liu, L. Chen, P. G. Duan, R. Luque, R. Ge and W. Y. Zhang, Renewable Sustainable Energy Rev., 2022, 158, 112178 CrossRef CAS.
- M. M. E. Huijbers, W. Zhang, F. Tonin, F. Hollmann, M. M. E. Huijbers, W. Zhang, F. Tonin and F. Hollmann, Angew. Chem., Int. Ed., 2018, 57, 13648–13651 CrossRef CAS PubMed.
- (a) Y. J. Ma, X. Z. Zhang, W. Y. Zhang, P. L. Li, Y. R. Li, F. Hollmann and Y. H. Wang, ChemPhotoChem, 2020, 4, 39–44 CrossRef CAS; (b) H. T. Duong, Y. Q. Wu, A. Sutor, B. O. Burek, F. Hollmann and J. Z. Bloh, ChemSusChem, 2021, 14, 1053–1056 CrossRef CAS PubMed.
- W. H. Xu, K. H. Mou, H. N. Zhou, J. Xu and Q. Wu, Green Chem., 2022, 24, 6589–6598 RSC.
- T. M. Hedison, D. J. Heyes and N. S. Scrutton, Curr. Res. Chem. Biol., 2022, 2, 100017 CrossRef CAS.
- W. H. Xu, Y. Chen, D. Y. Li, Z. G. Wang, J. Xu and Q. Wu, Mol. Catal., 2022, 524, 112261 CrossRef CAS.
- P. Santner, L. K. Szabo, S. N. Chanquia, A. H. Merrild, F. Hollmann, S. Kara and B. E. Eser, ChemCatChem, 2021, 13, 4038–4046 CrossRef CAS.
- S. N. Chanquia, F. V. Benfeldt, N. Petrovai, P. Santner, F. Hollmann, B. E. Eser and S. Kara, ChemBioChem, 2022, 23, e202200482 CrossRef CAS PubMed.
- P. R. Yaashikaa, P. S. Kumar and S. Varjani, Bioresour. Technol., 2022, 343, 126126 CrossRef CAS PubMed.
- T. Kuthiala, K. Thakur, D. Sharma, G. Singh, M. Khatri and S. K. Arya, Int. J. Biol. Macromol., 2022, 209, 1956–1974 CrossRef CAS PubMed.
- (a) R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed; (b) H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC.
- (a) Z. Usmani, M. Sharma, P. Gupta, Y. Karpichev, N. Gathergood, R. Bhat and V. K. Gupta, Bioresour. Technol., 2020, 304, 123003 CrossRef CAS PubMed; (b) S. Periyasamy, V. Karthik, P. S. Kumar, J. B. Isabel, T. Temesgen, B. M. Hunegnaw, B. B. Melese, B. A. Mohamed and D. V. N. Vo, Environ. Chem. Lett., 2022, 20, 1129–1152 CrossRef CAS.
- M. Mazotto, J. D. Silva, L. A. A. de Brito, N. U. Rocha and A. D. Soares, Environ. Technol. Innovation, 2021, 23, 101760 CrossRef.
- C. Lehmann, F. Sibilla, Z. Maugeri, W. R. Streit, P. D. de Maria, R. Martinez and U. Schwaneberg, Green Chem., 2012, 14, 2719–2726 RSC.
- J. K. Zhao, M. R. Wilkins and D. H. Wang, Bioresour. Technol., 2022, 364, 128045 CrossRef CAS PubMed.
- E. Sundstrom, J. Yaegashi, J. P. Yan, F. Masson, G. Papa, A. Rodriguez, M. Mirsiaghi, L. Liang, Q. He, D. Tanjore, T. R. Pray, S. Singh, B. Simmons, N. Sun, J. Magnuson and J. Gladden, Green Chem., 2018, 20, 2870–2879 RSC.
- J. Grewal, S. K. Khare, L. Drewniak and K. Pranaw, J. Mol. Liq., 2022, 362, 119796 CrossRef CAS.
- M. D. Portillo and A. Saadeddin, Crit. Rev. Biotechnol., 2015, 35, 294–301 CrossRef CAS PubMed.
- S. Z. Wang, G. Cheng, J. Dong, T. Tian, T. S. Lee, A. Mukhopadhyay, B. A. Simmons, Q. P. Yuan and S. W. Singer, ACS Sustainable Chem. Eng., 2019, 7, 1457–1463 CrossRef CAS.
- J. X. Zhang, D. Z. Zou, S. Singh and G. Cheng, Sustainable Energy Fuels, 2021, 5, 1655–1667 RSC.
- P. Lozano, B. Bernal, I. Recio and M. P. Belleville, Green Chem., 2012, 14, 2631–2637 RSC.
- P. Lozano, B. Bernal, A. G. Jara and M. P. Belleville, Bioresour. Technol., 2014, 151, 159–165 CrossRef CAS PubMed.
- J. Sun, J. Shi, N. V. S. N. M. Konda, D. Campos, D. J. Liu, S. Nemser, J. Shamshina, T. Dutta, P. Berton, G. Gurau, R. D. Rogers, B. A. Simmons and S. Singh, Biotechnol. Biofuels, 2017, 10, 154 CrossRef PubMed.
- N. P. T. Nguyen, C. Raynaud, I. Meynial-Salles and P. Soucaille, Nat. Commun., 2018, 9, 3682 CrossRef PubMed.
- Y. A. Alli, P. O. Oladoye, O. Ejeromedoghene, O. M. Bankole, O. A. Alimi, E. O. Omotola, C. A. Olanrewaju, K. Philippot, A. S. Adeleye and A. S. Ogunlaja, Sci. Total Environ., 2023, 68, 161547 CrossRef PubMed.
- I. Bernhardsgrutter, G. M. Stoffel, T. E. Miller and T. J. Erb, Curr. Opin. Biotechnol., 2021, 67, 80–87 CrossRef PubMed.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123(9), 5702–5754 CrossRef CAS PubMed.
- C. Bernal, K. Rodriguez and R. Martinez, Biotechnol. Adv., 2018, 36, 1470–1480 CrossRef CAS PubMed.
- D. Maciel, P. Christakopoulos, U. Rova and I. Antonopoulou, Chemosphere, 2022, 299, 134419 CrossRef PubMed.
- K. J. Koebke, T. B. J. Pinter, W. C. Pitts and V. L. Pecoraro, Chem. Rev., 2022, 122, 12046–12109 CrossRef CAS PubMed.
- V. M. Cangelosi, A. Deb, J. E. Penner-Hahn and V. L. Pecoraro, Angew. Chem., Int. Ed., 2014, 53, 7900–7903 CrossRef CAS PubMed.
- X. Zhu, C. X. Du, B. Gao and B. He, J. Environ. Manage., 2023, 332, 117370 CrossRef CAS PubMed.
- H. Rasouli, K. Nguyen and M. C. Iliuta, Sep. Purif. Technol., 2022, 296, 121299 CrossRef CAS.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C. K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- Y. L. Yuan, F. F. Wang, H. Li, S. Su, H. Gao, X. L. Han and S. Z. Ren, Process Biochem., 2022, 122, 214–223 CrossRef CAS.
- S. H. Zhang, M. N. Du, P. J. Shao, L. D. Wang, J. X. Ye, J. Chen and J. M. Chen, Environ. Sci. Technol., 2018, 52, 12708–12716 CrossRef CAS PubMed.
- P. J. Shao, Y. Shen, J. X. Ye, J. K. Zhao, L. D. Wang and S. H. Zhang, Sep. Purif. Technol., 2023, 315, 123683 CrossRef CAS.
- Y. M. Zhang, J. Y. Zhu, J. W. Hou, S. L. Yi, B. V. der Bruggen and Y. T. Zhang, J. Membr. Sci. Lett., 2022, 2, 100031 CrossRef.
- Y. Q. Fu, Y. B. Jiang, D. Dunphy, H. F. Xiong, E. Coker, S. Chou, H. X. Zhang, J. M. Vanegas, J. G. Croissant, J. L. Cecchi, S. B. Rempe and C. J. Brinker, Nat. Commun., 2018, 9, 990 CrossRef PubMed.
- L. Fradette, S. Lefebvre and J. Carley, Energy Procedia, 2017, 114, 1100–1109, DOI:10.1016/j.egypro.2017.03.1263.
- O. Alvizo, L. J. Nguyen, C. K. Savile, J. A. Bresson, S. L. Lakhapatri, E. O. P. Solis, R. J. Fox, J. M. Broering, M. R. Benoit, S. A. Zimmerman, S. J. Novick, J. Liang and J. J. Lalonde, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16436–16441 CrossRef CAS PubMed.
- H. Chen, Y. Huang, C. Sha, J. M. Moradian, Y. C. Yong and Z. Fang, Renewable Sustainable Energy Rev., 2023, 178, 113271 CrossRef CAS.
- J. F. Shi, Y. J. Jiang, Z. Y. Jiang, X. Y. Wang, X. L. Wang, S. H. Zhang, P. P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC.
- V. K. Sharma, J. M. Hutchison and A. M. Allgeier, ChemSusChem, 2022, 15, e202200888 CAS.
- X. J. Yu, D. Niks, X. Ge, H. Z. Liu, R. Hille and A. Mulchandani, Biochemistry, 2019, 58, 1861–1868 CrossRef CAS PubMed.
- B. El-Zahab, D. Donnelly and P. Wang, Biotechnol. Bioeng., 2008, 99, 508–514 CrossRef CAS PubMed.
- M. Baccour, A. Lamotte, K. Sakai, E. Dubreucq, A. Mehdi, K. Kano, A. Galarneau, J. Drone and N. Brun, Green Chem., 2020, 22, 3727–3733 RSC.
- K. Bachosz, J. Zdarta, M. Bilal, A. S. Meyer and T. Jesionowski, Sci. Total Environ., 2023, 868, 161630 CrossRef CAS PubMed.
- T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef.
- N. C. Dubey and B. P. Tripathi, ACS Appl. Bio Mater., 2021, 4, 1077–1114 CrossRef CAS PubMed.
- Q. Y. Liao, W. F. Liu and Z. H. Meng, Biotechnol. Adv., 2022, 60, 108024 CrossRef CAS PubMed.
- Z. B. Zhang, J. Muschiol, Y. H. Huang, S. B. Sigurdardottir, N. von Solms, A. E. Daugaard, J. Wei, J. Q. Luo, B. H. Xu, S. J. Zhang and M. Pinelo, Green Chem., 2018, 20, 4339–4348 RSC.
- X. L. Ji, Y. J. Xue, Z. Li, Y. R. Liu, L. Liu, P. K. Busk, L. Lange, Y. H. Huang and S. J. Zhang, Green Chem., 2021, 23, 6990–7000 RSC.
- V. K. Sharma, J. M. Hutchison and A. M. Allgeier, ChemSusChem, 2022, 15, e202200888 CAS.
- Y. Y. Zhang, Y. J. Zhao, R. Li and J. Liu, Sol. RRL, 2020, 5, 2000339 CrossRef.
- Z. B. Zhang, J. H. Tong, X. L. Meng, Y. J. Cai, S. S. Ma, F. Huo, J. Q. Luo, B. H. Xu, S. J. Zhang and M. Pinelo, ACS Sustainable Chem. Eng., 2021, 9, 11503–11511 CrossRef CAS.
- Q. Y. Liao, M. L. Guo, M. L. Mao, R. Gao, Z. H. Meng, X. L. Fan and W. F. Liu, Process Biochem., 2023, 129, 44–55 CrossRef CAS.
- Y. M. Guo, X. M. Hong, Z. M. Chen and Y. Q. Lv, J. Energy Chem., 2023, 80, 140–162 CrossRef CAS.
- M. W. Yuan, S. Sahin, R. Cai, S. Abdellaoui, D. P. Hickey, S. D. Minteer and R. D. Milton, Angew. Chem., Int. Ed., 2018, 57, 6582–6586 CrossRef CAS PubMed.
- Z. B. Zhang, H. Wang, Y. Nie, X. P. Zhang and X. Y. Ji, Front. Chem., 2022, 10, 894106 CrossRef CAS PubMed.
- M. Baumschabl, O. Ata, B. M. Mitic, L. Lutz, T. Gassler, C. Troyer, S. Hann and D. Mattanovich, Proc. Natl. Acad. Sci. U. S. A., 2023, 19, e2211827119, DOI:10.1073/pnas.2211827119.
- E. Garcia-Verdugo, B. Altava, M. I. Burguete, P. Lozano and S. V. Luis, Green Chem., 2015, 17, 2693–2713 RSC.
- R. Villa, R. Porcar, S. Nieto, A. Donaire, E. Garcia-Verdugo, S. V. Luis and P. Lozano, Green Chem., 2021, 23, 4191–4200 RSC.
- X. Fang, S. Kalathil and E. Reisner, Chem. Soc. Rev., 2020, 49, 4926–4952 RSC.
- C. Y. Wang, Y. Liu, W. Q. Chen, B. Zhu, S. Qu and M. Xu, J. Ind. Ecol., 2021, 25, 1300–1317 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- G. Pauli and J. F. Hartkemeyer, UpCycling, Chronik Verlag im Bertelsmann LEXIKON Verlag GmbH. 1999 Search PubMed.
- (a) L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Roman-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS; (b) H. Q. Li, H. A. Aguirre-Villegas, R. D. Allen, X. L. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. Garcia, I. Hermans, M. S. Kim, J. Z. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. S. Castillo, K. L. Sanchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Y. Wu, V. M. Zavala, P. Z. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- E. Nikolaivits, B. Pantelic, M. Azeem, G. Taxeidis, R. Babu, E. Topakas, M. B. Fournetm and J. Nikodinovic-Runic, Front. Bioeng. Biotechnol., 2021, 9, 696040 CrossRef PubMed.
- O. Garcia-Depraect, S. Bordel, R. Lebrero, F. Santos-Beneit, R. A. Borner, T. Borner and R. Munoz, Biotechnol. Adv., 2021, 53, 107772 CrossRef CAS PubMed.
- S. Lee, Y. R. Lee, S. J. Kim, J. S. Lee and K. Min, Chem. Eng. J., 2023, 454, 140470 CrossRef CAS.
- M. Fujisawa, H. Hirai and T. Nishida, J. Polym. Environ., 2001, 9, 103–108 CrossRef CAS.
- M. E. E. Temporiti, L. Nicola, E. Nielsen and S. Tosi, Microorganisms, 2022, 10, 1180 CrossRef CAS PubMed.
- R. P. Magalhaes, J. M. Cunha and S. F. Sousa, Int. J. Mol. Sci., 2021, 22, 11257 CrossRef CAS PubMed.
- A. Singh, N. A. Rorrer, S. R. Nicholson, E. Erickson, J. S. DesVeaux, A. F. T. Avelino, P. Lamers, A. Bhatt, Y. M. Zhang, G. Avery, L. Tao, A. R. Pickford, A. C. Carpenter, J. E. McGeehan and G. T. Beckham, Joule, 2021, 5, 2479–2503 CrossRef CAS.
- (a) V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guemard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. Andre, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed; (b) V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. Andre, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed; (c) https://www.carbios.com/ .
- S. Thiyagarajan, E. Maaskant-Reilink, T. A. Ewing, M. K. Julsing and J. van Haveren, RSC Adv., 2022, 12, 947–970 RSC.
- Carbios 2022 annual results, https://www.carbios.com/en/carbios-presents-its-2022-annual-results/.
- M. M. Aboelnga and S. Kalyaanamoorthy, ACS Sustainable Chem. Eng., 2022, 10, 15857–15868 CrossRef CAS.
- B. Y. Deng, Y. Yue, J. Yang, M. J. Yang, Q. Xing, H. Peng, F. Wang, M. Li, L. X. Ma and C. Zhai, Commun. Biol., 2023, 6, 39 CrossRef CAS PubMed.
- M. F. M. White and S. Wallace, Angew. Chem., Int. Ed., 2023, 62, e202216963 CrossRef CAS PubMed.
- L. X. Shi, P. Liu, Z. J. Tan, W. Zhao, J. F. Gao, Q. Gu, H. W. Ma, H. F. Liu and L. L. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202218390 CrossRef CAS PubMed.
- E. Erickson, J. E. Gado, L. Avilan, F. Bratti, R. K. Brizendine, P. A. Cox, R. Gill, R. Graham, D. J. Kim, G. Konig, W. E. Michener, S. Poudel, K. J. Ramirez, T. J. Shakespeare, M. Zahn, E. S. Boyd, C. M. Payne, J. L. DuBois, A. R. Pickford, G. T. Beckham and J. E. McGeehan, Nat. Commun., 2022, 13, 7850 CrossRef CAS PubMed.
- Y. Lu, D. J. Diaz, N. J. Czarnecki, C. Z. Zhu, W. T. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef PubMed.
- S. Kaabel, J. P. D. Therien, C. E. Deschenes, D. Duncan, T. Friscic and K. Auclair, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026452118 CrossRef CAS PubMed.
- S. Kaabel, J. Arciszewski, T. H. Borchers, J. P. D. Therien, T. Friscic and K. Auclair, ChemSusChem, 2023, 16, e202201613 CrossRef CAS PubMed.
- P. Stegmann, T. Gerritse, L. Shen, M. Londo, A. Puente and M. Junginger, J. Cleaner Prod., 2023, 395, 136426 CrossRef CAS.
- E. de Jong, H. A. Visser, A. S. Dias, C. Harvey and G.-J. M. Gruter, Polymers, 2022, 14, 943, DOI:10.3390/polym14050943.
- K. Loos, R. Y. Zhang, I. Pereira, B. Agostinho, H. Hu, D. Maniar, N. Sbirrazzuoli, A. J. D. Silvestre, N. Guigo and A. F. Sousa, Front. Chem., 2020, 8, 585, DOI:10.3389/fchem.2020.00585.
- F. Silvianti, D. Maniar, L. Boetje, A. J. J. Woortman, J. van Dijken and K. Loos, ACS Polym. Au, 2023, 3, 82–95 CrossRef CAS.
- P. Austin, M. D. Allen, B. S. Donohoe, N. A. Rorrer, F. L. Kearns, R. L. Silveira, B. C. Pollard, G. Dominick, R. Duman, K. El Omari, V. Mykhaylyk, A. Wagner, W. E. Michener, A. Amore, M. S. Skaf, M. F. Crowley, A. W. Thorne, C. W. Johnson, H. L. Woodcock, J. E. McGeehan and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4350–E4357, DOI:10.1073/pnas.1718804115.
- W. Q. Han and X. Liao, J. Appl. Polym. Sci., 2023, 140, e53698, DOI:10.1002/app.53698.
- J. Chow, P. Perez-Garcia, R. Dierkes and W. R. Streit, Microb. Biotechnol., 2023, 16, 195–217 CrossRef CAS PubMed.
- EUROPUR (European Association of Flexible Polyurethane Foam Blocks Manufacturers), https://www.isopa.org/media/2763/EoL_Brochure_2021_EUROPUR.pdf.
- W. Liu, J. He, R. Xue, B. Xu, X. J. Qian, F. X. Xin, L. M. Blank, J. Zhou, R. Wei, W. L. Dong and M. Jiang, Biotechnol. Adv., 2021, 48, 107730 CrossRef PubMed.
- X. R. Jin, J. X. Dong, X. F. Guo, M. Z. Ding, R. Bao and Y. Z. Luo, Polym. Int., 2022, 71, 1384–1392 CrossRef CAS.
- J. Fuentes-Jaime, M. Vargas-Suarez, M. J. Cruz-Gomez and H. Loza-Tavera, Biodegradation, 2022, 33, 389–406 CrossRef CAS PubMed.
- A. Magnin, L. Entzmann, A. Bazin, E. Pollet and L. Averous, ChemSusChem, 2021, 4234–4241 CrossRef CAS PubMed.
- Y. Branson, S. Soltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jager and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- J. J. Diao, Y. H. Hu, Y. X. Tian, R. Carr and T. S. Moon, Cell Rep., 2023, 42, 111908 CrossRef CAS PubMed.
- P. Sullivan, A. Z. Werner, K. J. Ramirez, L. D. Ellis, J. R. Bussard, B. A. Black, D. G. Brandner, F. Bratti, B. L. Buss, X. Dong, S. J. Haugen, M. A. Ingraham, M. O. Konev, W. E. Michener, J. Miscall, I. Pardo, S. P. Woodworth, A. M. Guss, Y. Roman-Leshkov, S. S. Stahl and G. T. Beckham, Science, 2022, 378, 207–211 CrossRef PubMed.
- C. Rabot, Y. H. Chen, S. Bijlani, Y. M. Chiang, C. E. Oakley, B. R. Oakley, T. J. Williams and C. C. C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202214609 CAS.
- C. C. C. Wang, T. J. Williams, C. Rabot, Y. H. Chen, S. Y. Lin, B. Miller, Y. M. Chiang, C. E. Oakley and B. R. Oakley, J. Am. Chem. Soc., 2023, 145(9), 5222–5230 CrossRef PubMed.
- V. Zwicker, C. Brick, G. J. M. Gruter and F. van Harrevel, Sustain. Prod. Consum., 2023, 35, 173–183 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |