
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase transition behaviour and mechanism of 2D TiO2(B) nanosheets through water-mediated removal of surface ligands†
Shirui
Xie
,
Lijing
Fan
,
Yanxin
Chen
,
Jiliang
Cai
,
Fan
Wu
,
Kecheng
Cao
* and
Pengxin
Liu
 *
*
School of Physical Science and Technology, ShanghaiTech University, Shanghai, China. E-mail: caokch@shanghaitech.edu.cn; liupx@shanghaitech.edu.cn
First published on 21st September 2023
Abstract
Phase engineering is a central subject in materials research. The recent research interest in the phase transition behavior of atomically thin 2D materials reveals the important role of their surface chemistry. In this study, we investigated the phase transformation of ultrathin TiO2(B) nanosheets to anatase under different conditions. We found that the convenient transformation in water under ambient conditions is driven by the hydrolysis of surface 1,2-ethylenedioxy groups and departure of ethylene glycol. A transformation pathway through the formation of protonic titanate is proposed. The ultrathin structure and the metastable nature of the precursor facilitate the phase conversion to anatase. Our finding offers a new insight into the mechanism of TiO2(B) phase transition from the viewpoint of surface chemistry and may contribute to the potential application of ultrathin TiO2(B) nanosheets in aqueous environments.
Introduction
Crystalline polymorphism of inorganic compounds offers a practical approach, that is, phase engineering, to tune the basic properties of materials for advanced applications.1–8 For a metastable phase, phase transition is undesirable when the metastable phase outperforms more stable phases, while in other cases, phase transition could be useful for synthesizing expected structures. Evaluating the phase stability of a metastable phase and comprehending the phase transition behaviour are a precondition for materials design, synthesis and application.9–11For example, TiO2(B) is a metastable phase and a naturally occurring polymorph of TiO2.12 When the application scenarios call for maintaining the crystal structure of TiO2(B), phase transition should be avoided. For instance, using TiO2(B) as an anode material of Li-ion batteries to achieve fast charging needs more loosely packed TiO6 octahedra and an open channel along the b-axis of the TiO2(B) structure, which benefits ion intercalation and transportation.13–16 In some application scenarios in which other structures outperform pure TiO2(B) phases, phase transition is a practical method for structure tuning. For instance, TiO2(B)/anatase heterophase junctions facilitate the spatial separation of electron–hole pairs and enhance photocatalytic performances.17,18 Calcining TiO2(B) at different temperatures leads to partial phase transition and the production of heterophase junctions with different mass ratios.19
However, the phase transformation of TiO2(B) is not fully understood. Although transmission electron microscopy (TEM)20 and X-ray diffraction (XRD)17 were used to study the process in situ, it is difficult to use these techniques for water-phase systems (in photocatalytic water splitting, etc.). Besides, the phase transition behaviour of 2D materials is completely different from that of their 3D analogues: it is more sensitive to the surface chemistry.4,21 It is reported that 2D TiO2(B) turns to anatase at modest temperature in water, even as low as 60 °C.22–24 To explain such an unusual phenomenon, several reaction pathways were proposed: through competitive adsorption of water,22 through dissolution/recrystallization23 or through reduced oxides (Ti3O5);24 no consensus was achieved.
Herein, we report that the phase transition of 2D TiO2(B) nanosheets in water occurs even at room temperature. The process is coupled with a surface reaction: hydrolysis of surface 1,2-ethylenedioxy groups (deprotonated ethylene glycol, EG). The removal of surface ligands is vital for phase transformation, both in calcination treatment (through ligand decomposition) and in water-phase treatment (through ligand hydrolysis). Based on our observation, a mechanism from TiO2(B) to anatase via the formation of protonic tetratitanate is proposed.
Experimental section
Synthesis of TiO2(B) nanosheets
TiO2(B) nanosheets were synthesized through a hydrothermal method reported by Wang et al. with slight modification.25 In a typical synthesis, 1 mL of TiCl4 was quickly added into 30 mL of ethylene glycol in a beaker under vigorous stirring. The reaction was allowed to proceed until no HCl fume appeared and the solution turned transparent. Then 1 mL of deionized water was added and mixed. The solution was then transferred into a 50 mL Teflon-lined stainless-steel autoclave and heated at 150 °C for 4 hours. After cooling to room temperature, the product was collected after centrifugation, and repeated washing with water and vacuum drying were carried out.Phase transition behaviour study
The phase transition behaviour of TiO2(B) nanosheets was studied in two ways. In calcination treatment, the precursor was placed in a ceramic crucible and calcined in a muffle furnace at desired temperatures. The ramping rate is 2 degree per minute. The cooled and collected products were denoted as HA-XX K-YY h, in which HA stands for heating in air.In water phase treatment, 150 mg of precursor was dispersed in 30 mL of deionized water in a flask. The flask was then heated in an oil bath at different temperatures (343 K and 373 K) for different durations under stirring. After the reaction, white products were collected through centrifugation, washing and vacuum drying. The collected products were denoted as HW-XX K-YY h, in which HW stands for heating in water.
The pH of the dispersion was adjusted using HCl or NaOH to study the influence of pH on the phase transition behaviour. For H2O2 pre-treatment, 100 mg of the precursor was dispersed in 10 mL of 30% H2O2 in a flask and stirred for 12 hours before collecting.
Characterization
TEM and high-resolution (HRTEM) images were collected using a JEM-2100PLUS and GrandARM300F. Powder XRD was performed on a Bruker D2 PHASER with Cu Kα radiation (λ = 1.54056 Å at 10 mA, 30 kV). Thermogravimetric analysis (TGA) was carried out on a PerkinElmer TGA 4000 using air as the carrier gas. Solid-state Nuclear Magnetic Resonance (ss-NMR) results were collected using a Bruker ADVANCE III HD 400 MHz solid NMR spectrometer at room temperature. 13C magic angle spinning nuclear magnetic resonance (MAS NMR) spectra were recorded in 3.2 mm standard zirconium oxide rotors spinning at 10 kHz with 800 scans. 13C chemical shifts were referenced to adamantane at 38.48 ppm and 29.90 ppm. Fourier transform infrared spectroscopy (FT-IR) was carried out on a Bruker V70. The specific surface areas were determined with an Autosorb-iQ-MP-AG using the Brunauer–Emmett–Teller (BET) method. Raman spectra were recorded on an ANDOR (SR-500I-D2-1F1) instrument with a laser source of 532 nm wavelength.Quantification
The phase content was calculated based on the Raman spectrum.26,27 By using Lorentzian peak profile fitting, the mass ratio can be obtained with the following equation: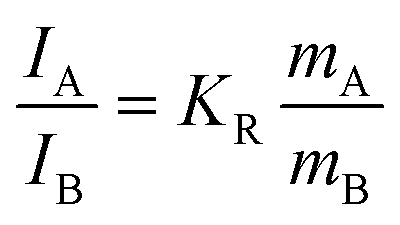 | (1) |
In the equation, mA and mB are the relative masses of anatase and TiO2(B). IA and IB stand for the scattering intensities of the peak at 144 cm−1 assigned to anatase and the peak at 256 cm−1 assigned to TiO2(B), respectively. KR = 6.57, which is the quotient of the scattering factors of the two peaks obtained by calculation.
The amount of surface ligands was calculated based on TGA and specific surface area measurements. The TGA method can only show an overall weight loss (WL). The weight loss has three sources of contribution: due to the decomposition/combustion of EG, due to the dehydration reaction between surface –OH groups, and due to further dehydration as the surface area decreased after heating to 450 °C. We have the following equation:
| mH2O + mEG = WL | (2) |
| nTi-OH + nTi-EG = k × ΔS | (3) |
To determine the value of k, we used HW-373 K-3 h as a reference, and all EG ligands were removed and replaced by –OH. Considering that two –OH groups yield one H2O molecule, we have the following equation:
| mH2O = 9nTi-OH | (4) |
Previous studies showed that deprotonated EG is a bidentate ligand on the surface of TiO2(B) nanosheets, forming a –Ti–OCH2CH2O–Ti– structure.28,29 After calcination, –Ti–O–Ti– is the most likely formed structure. Eqn (5) is as follows:
| mEG = 44nTi-EG/2 | (5) |
Combining eqn (2)–(5), we can estimate the amount of both EG and –OH ligands in a given sample (Table S1†).
Results and discussion
TiO2(B) nanosheets and their surface chemistry
The as-prepared TiO2 nanosheets had a sheet-like morphology with wrinkles and stacking due to their ultrathin structure (Fig. 1a and Fig. S1†), similar to previous studies.25 The combined HRTEM images, fast Fourier transform (FFT) pattern (Fig. 1b) and powder XRD pattern (Fig. 1c) showed that the nanosheets were in the TiO2(B) phase with the [010] facet exposed. The diffraction peaks of the nanosheets were well assigned to TiO2(B) (JCPDS 04-008-7931). The (020) diffraction peak at 48.6° was relatively strong, in comparison with the predominant (001) diffraction at 14.2° of the standard JCPDS card, which confirms the presence of more [010] facets and the preferentially oriented structure of TiO2(B) nanosheets. Besides, a strong diffraction peak at 7.9° that did not belong to the TiO2(B) phase was observed. The large d-spacing (1.12 nm) was best explained as an interlayer distance of stacked TiO2 nanosheets, which is commonly observed for other 2D-structured materials (strong diffraction in the small angle region).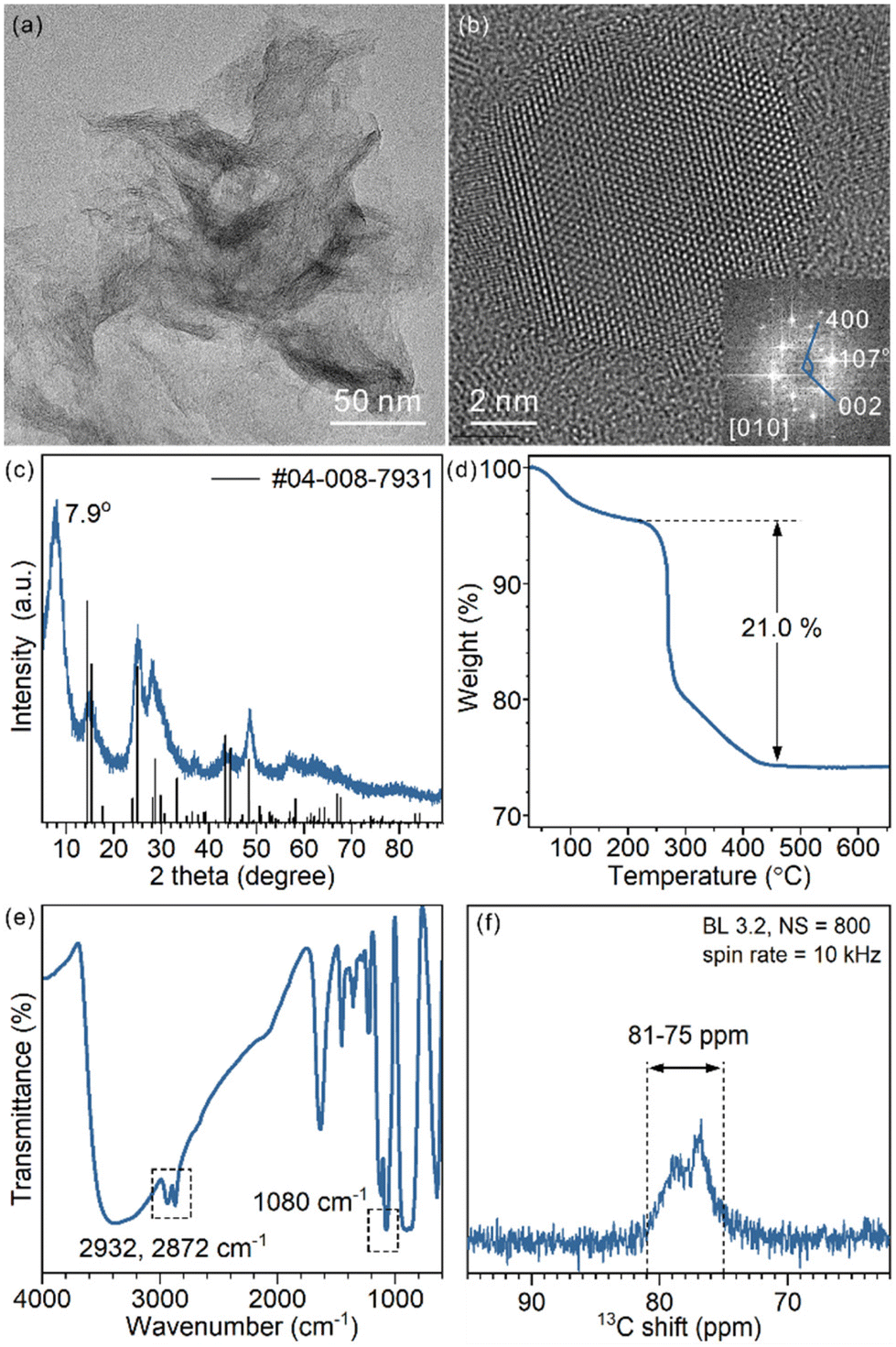 | ||
| Fig. 1 (a) Representative TEM image, (b) HRTEM image and FFT pattern, (c) XRD pattern, (d) TGA curve (e) FT-IR spectrum and (f) 13C ss-NMR spectrum of the as-prepared TiO2(B) nanosheets. | ||
To analyse the composition of TiO2(B) nanosheets, TGA was conducted (Fig. 1d). The weight loss below 220 °C is best explained as the removal of water that physically adsorbed on the surface and in the interlayer space. The abrupt weight loss started at 220 °C and ended at 450 °C, giving an overall 21.0% weight loss during the heating process. Such a weight loss was mainly a result of thermal decomposition and calcination of surficial organic ligands (incomplete combustion led to carbonaceous deposition and a black product).
To identify the surface species, FT-IR and ss-NMR spectra were recorded. Adsorbed EG was observed in the FT-IR spectra (Fig. 1e): CH2 vibration modes at 2932 cm−1 and 2872 cm−1 and the C–O stretching mode at 1080 cm−1. The presence of EG was also proved by 13C ss-NMR (Fig. 1f). Signals ranging from 75 ppm to 81 ppm assigned to CH2 groups were observed. The chemical shift was slightly higher than that of free ethylene glycol (62.5 ppm) (Fig. S2†), which implies a surface-bound structure.30 EG was introduced in the synthesis as a reagent, solvent and also protecting agent to stabilize the product. The EG ligands were chemically adsorbed, as they cannot be removed through repeated washing. Moreover, physically adsorbed EG is easily removed upon heating, unlike what was observed in TGA, considering the boiling point of EG being 195 °C (Fig. S3†). As an X-type bidentate ligand, the most possible coordination structure of the ligand on the surface should be –Ti–(OCH2)2 or –Ti–OCH2CH2O–Ti–, depending on the number of Ti sites each ligand is coordinated with.22 Alternatively, it can be viewed as surficial 1,2-ethylenedioxy groups (denoted as EG ligands for short).
Phase transition behaviour of TiO2(B) nanosheets
The phase transition behaviour of the TiO2(B) nanosheets was studied either through calcination or heating in water dispersion, and in both cases the anatase phase was formed. For schematic illustration, the structural model of EG-protected TiO2(B) nanosheets was adopted from our previous study (Fig. 2a).28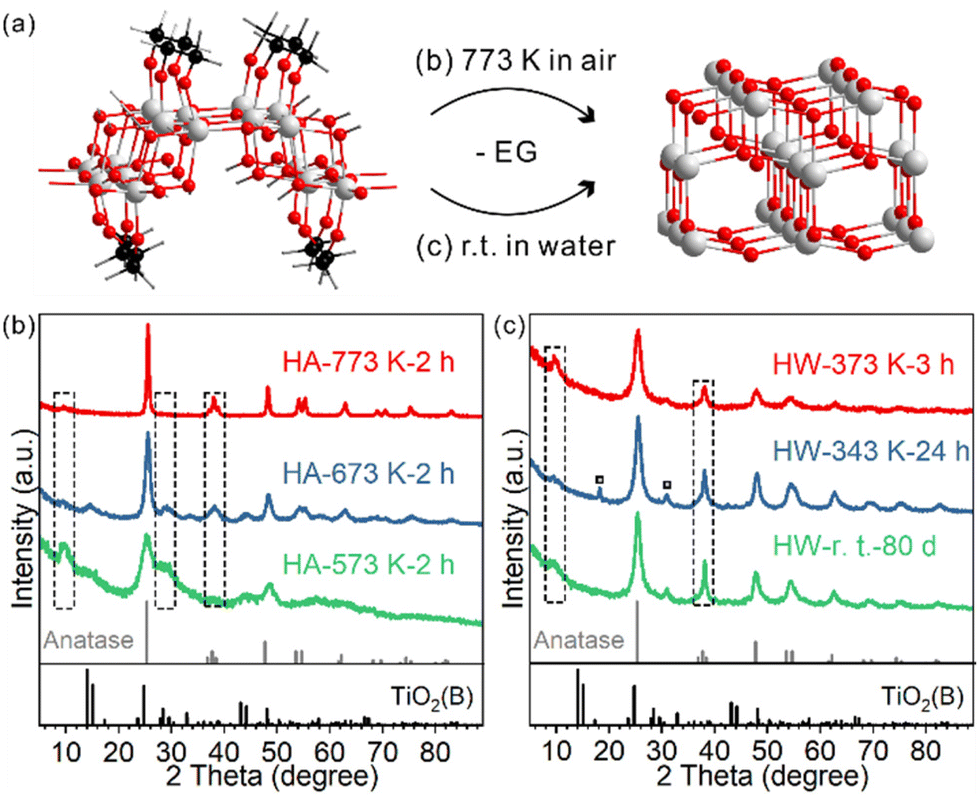 | ||
| Fig. 2 (a) Scheme showing the phase transformation from EG-protected TiO2(B) nanosheets to anatase either through heating in air or in water. Colour code: O (red), Ti (grey), C (black), and H (white). XRD patterns of the samples heated at different temperatures (b) in air and (c) in water. | ||
For TiO2(B) nanosheets calcined at different temperatures for two hours, the phase transition started to occur for HA-573 K-2 h and completed for HA-773 K-2 h (Fig. 2b and Fig. S4†). The diffraction peak at 28.7° indexed to TiO2(B) were observed for lower temperature-calcined samples and was negligible for HA-773 K-2 h. In contrast, the diffraction peak at 37.8° indexed to the anatase (004) facet emerged and increased in intensity as the calcination temperature increased. The peak at 7.9° seen for the precursor gradually disappeared as more anatase phase formed, which indicates a loss of the stacked layer structure, probably due to the surface roughness observed in TEM images (Fig. S5†).
The temperature range (573 K–773 K) for phase transition happens to fall in the major temperature range of weight loss in TGA. Meanwhile, the FT-IR spectra of the calcined samples showed a correlation between the extent of phase transition and the amount of ligand residue (Fig. S6†). Hence, the calcination-induced phase transition is coupled with EG combustion. Because of the ultrathin structure of the nanosheets, the phase transition temperature range is lower than those of other TiO2(B) materials (typically completed at over 973 K).17,31,32
In water dispersion, the phase transition is much easier. To our surprise, the phase transition is a spontaneous process even at room temperature (Fig. 2c). After storing the water dispersion of TiO2(B) nanosheets on a bench for 80 days, no detectable TiO2(B) phase could be observed from XRD; the phase transition towards the anatase phase was complete, as evidenced by the characteristic diffraction peak at 37.8° indexed to anatase. The stacking feature was maintained, as shown by the diffraction peak at 7.9°. Such a low temperature is about 500 degrees lower than those of calcination systems. The process can be accelerated when the dispersion is heated and stirred: 24 hours for 343 K and 3 hours for 373 K. For HW-343 K-24 h, two small peaks marked with a square symbol indicate a possible reaction intermediate, which will be discussed later. TEM and HRTEM images were collected to characterize the morphology and crystal structure details for the anatase phase product.
For HW-373 K-3 h, the original sheet-like morphology disappeared, replaced by rod-shaped and rhombic nanoparticles (Fig. S7†). These nanoparticles were approximately 10 nm in size and stacked to some extent. The HRTEM image showed that the nanoparticles were well crystallized anatase. The FFT results indicate that the exposed facet of some anatase nanoparticles is (101). Other exposed facets may also exist, but it is difficult to observe them due to stacking. The specific surface area of HW-373 K-3 h was 214.9 m2 g−1, lower than that of TiO2(B) nanosheets (341.5 m2 g−1), but much larger than those of calcined anatase products (53.2 m2 g−1, Fig. S8†), which is consistent with the observed small particle size of the water-phase product.
The phase transition behaviour is sensitive to the chemical environment around the TiO2(B) nanosheets. Both the low turning temperature and distinct morphology indicate a quite different reaction mechanism when TiO2(B) nanosheets were heated in water, in comparison with heating in air. We reasoned that the surface state of TiO2(B) nanosheets should determine how the transformation occurs, given the atomic-layer thickness nature of the precursor.33 To understand the molecular mechanism and elementary steps, we focused on studying how water interacts with the nanosheets and how water alters the surface chemistry.
Surface speciation in water phase transition
Time-tracking experiments were performed at 373 K and monitored by Raman spectroscopy (Fig. 3a) and XRD (Fig. S9†). The Raman spectrum of the precursor showed several peaks at 256, 382, 433, 477, 553, 632 and 828 cm−1, which belonged to the TiO2(B) phase. Some typical vibrations below 210 cm−1 were not observed,26,27 probably due to the precursor's two-dimensional structure. As the treatment duration prolonged, the characteristic peaks of TiO2(B) at 256 and 828 cm−1 gradually decreased in intensity and the peak of anatase at 144 cm−1 increased (Fig. 3a). For 1h- and 2h-treated samples, both phases coexisted. The contributions of anatase and TiO2(B) for each sample were quantified using peak fitting and are shown in Fig. 3d. The relative mass ratios of TiO2(B) were 85.1% and 50.1% for 1h- and 2h-treated samples, respectively. The 3h-treated sample was pure anatase.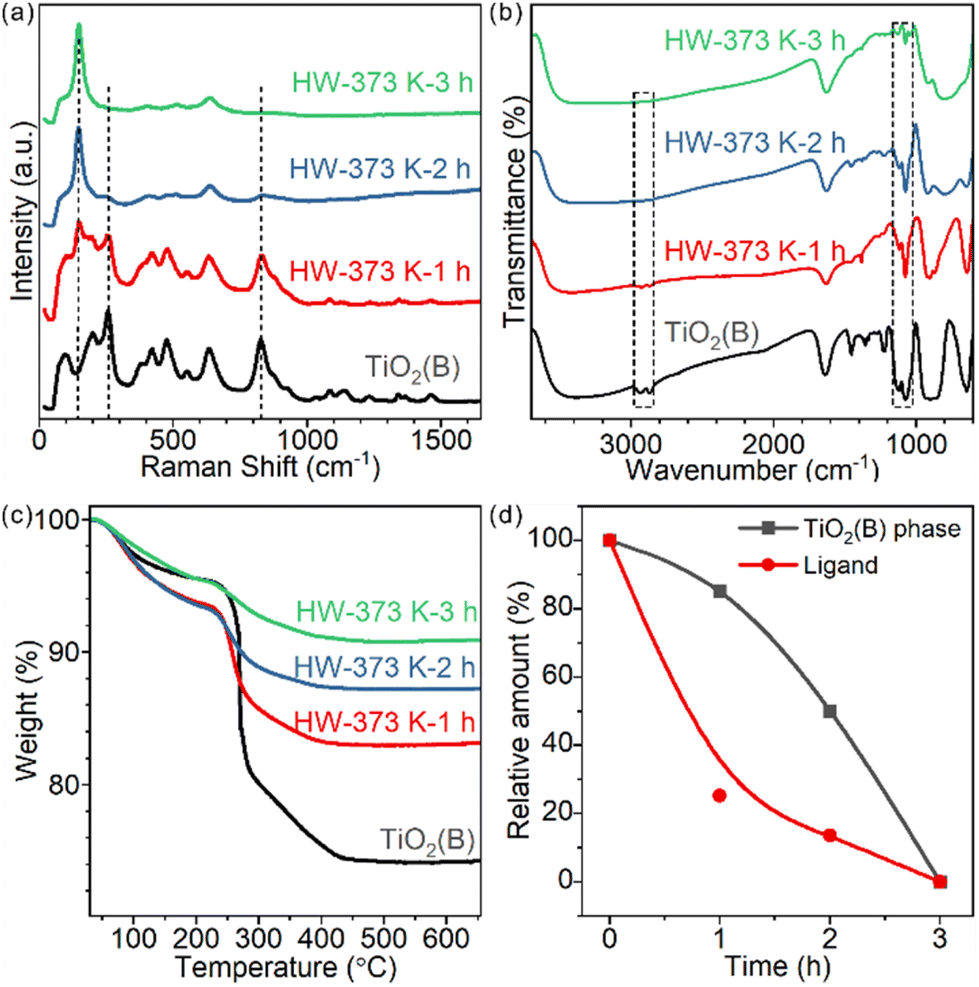 | ||
| Fig. 3 (a) Raman and (b) FT-IR spectra and (c) TGA results of the TiO2(B) nanosheets and samples obtained after heating in water at 373 K for 1, 2 and 3 hours; and (d) calculated mass ratio of the TiO2(B) phase and residual EG ligand. | ||
For these partially transformed heterophase materials, surface species were studied by FT-IR spectroscopy (Fig. 3b). The amount of chemically adsorbed EG decreased with prolongation of the treatment duration. Similar to the calcination-induced phase transition process, the phase transition in water is also accompanied by the removal of EG ligands. Chemically adsorbed EG ligands formed strong Ti–O covalent bonds with the surface Ti cations of TiO2 nanosheets. The most likely reaction to break the Ti–O bonds in water is hydrolysis: protonation of the bidentate 1,2-ethylenedioxy groups and hydroxylation of the surface Ti sites. We testified such a hypothesis by detecting the composition of the supernatant with 1H solution NMR, finding the released EG (Fig. S10†).
To quantify the amount of EG on each sample, TGA measurements were conducted (Fig. 3c). For products treated for 1, 2 and 3 hours, the weight loss was 10.5%, 6.1% and 4.5%, respectively. The shape of the curves also changed, indicating the different compositions of surface species.
To further understand how ligand departure and phase transition correlate, we estimated the EG ligand amount for each sample based on TGA and surface area measurements (Fig. 3d and Table S1,† Experimental section). Clearly, the departure of the EG ligand was faster than the phase transformation from TiO2(B) to anatase. Time-tracking experiments at 343 K were performed to confirm the observations (Fig. S11†) and the trend was the same.
Ligand removal-induced phase transition
So far, we have shown that heating TiO2(B) nanosheets in water has two consequences: (1) EG ligand removal from the surface and (2) phase transition towards the anatase phase. It is not clear yet whether the two consequences only occur in succession or have a direct causal connection between them. In other words, whether the removal of the EG ligand benefits the nanosheet's phase transformation from TiO2(B) to anatase is unknown.We investigated how the pH of the aqueous dispersion affected the phase transition behaviour, considering that the surface chemical reaction in EG removal is a hydrolysis reaction (Fig. 4a). After treatment in a neutral environment, the product HW-343 K-12 h was a mixture of TiO2(B) and anatase phases. When the temperature and duration were kept consistent, we found that the phase transition was boosted in an acidic environment (pH = 1), while being suppressed in a basic environment (pH = 12). The product after acid treatment was the pure anatase phase. Almost no phase transformation was observed for the base-treated sample.
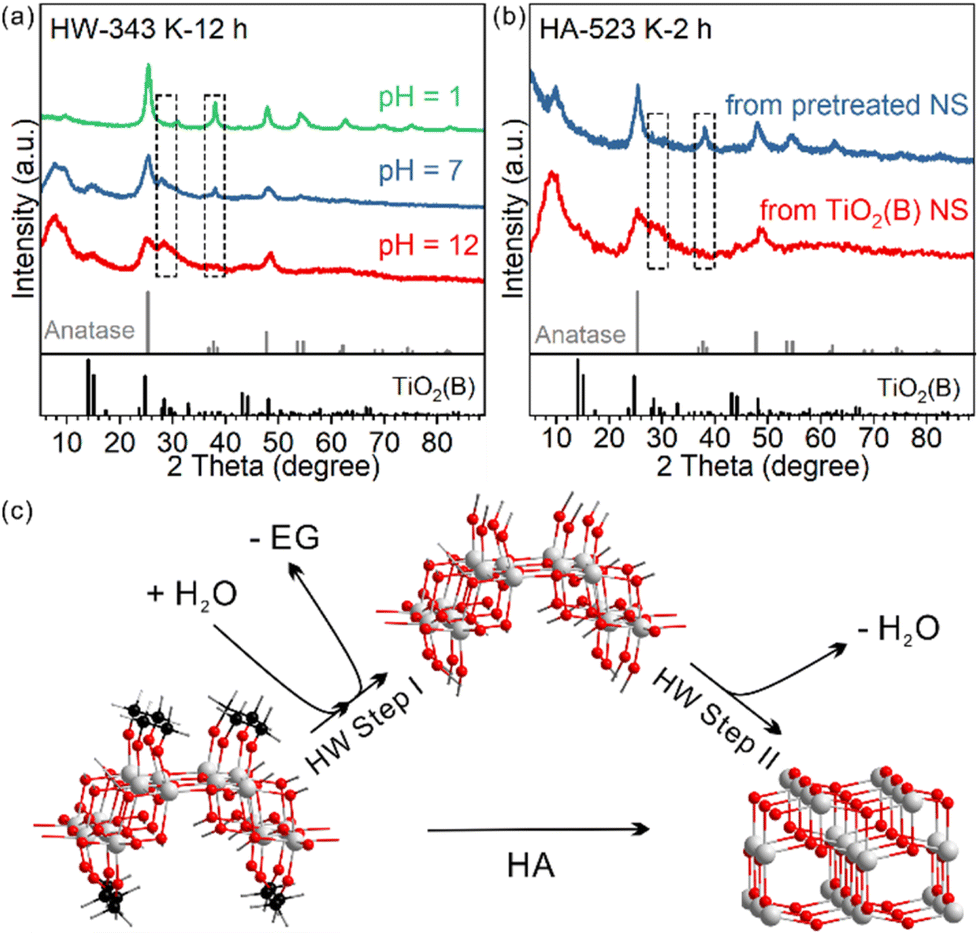 | ||
| Fig. 4 XRD patterns of the samples obtained (a) after heating TiO2(B) nanosheets in water dispersion of different pH values at 343 K for 12 hours and (b) after heating the pre-treated and as-prepared TiO2(B) nanosheets in air at 523 K for 2 hours. (c) Scheme showing the surface speciation in the phase transition. | ||
To know which process is affected by acidity, ligand removal or new crystal phase formation, TGA measurements were conducted to detect the amount of EG ligand residue after the treatment (Fig. S12†). We found that EG ligands are more likely to leave the surface in the acid than in neutral water, much more than that in the base.
This result not only confirmed that ligand removal was a hydrolysis reaction and could be accelerated by the protonic acid, but also indicated that the removal of EG from the surface of TiO2(B) nanosheets was the first step in the observed water phase transition (step I in Fig. 4c). Only by removing the EG ligands first could the phase transition occur. It should be noted that the step I product in Fig. 4c may not exist. The structure is just for schematic representation to show that surface EG ligands were replaced by hydroxyl groups.
We further confirmed the protective role of EG in calcination systems. We intentionally removed a part of surface EG ligands before calcination, to see how it affects the phase transition behaviour. After 12-hour treatment at 343 K in water, the TiO2(B) precursor partly converted to anatase and EG ligands were partly removed (Fig. S11†). The sample was then calcined at 523 K for 2 hours. The phase transition to anatase was systematic, in sharp contrast to that in the direct calcined sample without water pre-treatment (Fig. 4b). We also used H2O2 to remove the EG ligands (Fig. S13 and 14†).34 The amorphous product after H2O2 treatment completely converted to well-crystallized anatase after calcination at 573 K for 2 hours, while the directly calcined sample without H2O2 pre-treatment was still TiO2(B) dominated (Fig S4).
Overall, these results show that: (1) EG ligands protect and prevent TiO2(B) from converting to anatase and (2) in both water-phase and air-phase heating cases, the removal of EG ligands triggers the phase transformation.
A possible intermediate or a side product
For HW-343 K-24 h, apart from the diffraction pattern of the anatase phase, some peaks not belonging to anatase or TiO2(B) were observed (Fig. 5a). We found that these peaks were best indexed to a H4Ti4O10 phase or H2Ti4O9·H2O (Fig. 5b), a layered protonic tetratitanate compound.35–37 The structure is composed of corrugated ribbons of four edge-sharing TiO6 octahedra.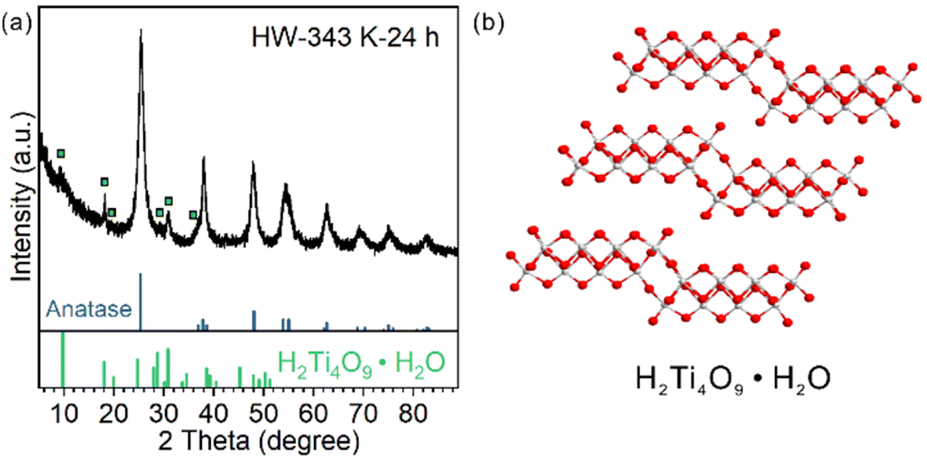 | ||
| Fig. 5 (a) XRD patterns of the samples obtained after heating TiO2(B) nanosheets in water dispersion at 343 K for 24 hours; peaks belonging to H4Ti4O10 are marked with squares. (b) Crystal structure of H4Ti4O10. | ||
These peaks were reported before in other research studies, vaguely described as the intermediate transition phase.22,24 Ye et al. assigned these signals to the Ti3O5 phase,24 in accordance with an in situ TEM study.20 However, Ti3O5 is an oxygen-deficient compound. It is possible to be formed by heating H2Ti3O7 nanofibers under vacuum (TEM chamber) as reported.20 It is not likely to be formed in the water phase with abundant oxygen atom donors, especially when the precursor was ultrathin nanosheets but not bulk materials so that the mass transportation of oxygen atom donors (H2O molecules) would not be a problem.
Protonic tetratitanate could be an intermediate transition phase, based on these facts: (1) structural similarities: both the precursor TiO2(B) nanosheets and protonic tetratitanate have a two-atomic-layered two-dimensional structure; (2) calcining hydrogen titanate is a commonly used synthetic method to obtain TiO2 materials in the TiO2(B) phase, anatase phase or their heterophase junctions; and (3) the transformation from protonic tetratitanate to anatase through dehydration condensation in liquid media is known.38 More investigation is needed to know if protonic tetratitanate is the intermediate or a side product.
Conclusions
The phase stability of ultrathin TiO2(B) nanosheets and their phase transformation to anatase were investigated under different conditions. Such a transition can be achieved by either heating in air above 673 K or in an aqueous phase below 373 K. Surprisingly, the transition also occurred at room temperature in water. In both methods, the removal of surface EG ligands, either through decomposition and combustion or through hydrolysis, is the first step of phase transition. Due to the ultrathin structure, the TiO2(B) nanosheets are unstable without EG ligands on its surface and convert to anatase easily. Our work shows the important role of surface ligands in the phase transition and phase stability of 2D nanomaterials.Author contributions
P. L. conceived the project and supervised the research. S. X. performed most of the preparation and characterization. L. F., Y. C. and F. W. performed part of sample characterization. J. C. and K. C. performed the TEM and HRTEM characterization. All the authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Start-up Funding and the Double First-Class Initiative Fund of ShanghaiTech University (SYLDX0072022 and SYLDX0142022). P. L. acknowledges the Shanghai Pujiang Talent Program (No. 21PJ1410400).References
- H. Li and X. Wang, Acc. Chem. Res., 2019, 52, 780 CrossRef CAS PubMed.
- M. S. Sokolikova and C. Mattevi, Chem. Soc. Rev., 2020, 49, 3952 RSC.
- J. Liu, J. Huang, W. Niu, C. Tan and H. Zhang, Chem. Rev., 2021, 121, 5830 CrossRef CAS PubMed.
- W. Li, X. Qian and J. Li, Nat. Rev. Mater., 2021, 6, 829 CrossRef CAS.
- Y. Chen, Z. C. Lai, X. Zhang, Z. X. Fan, Q. Y. He, C. L. Tan and H. Zhang, Nat. Rev. Chem., 2020, 4, 243 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494 CrossRef CAS PubMed.
- S. Lu, J. Liang, H. Long, H. Li, X. Zhou, Z. He, Y. Chen, H. Sun, Z. Fan and H. Zhang, Acc. Chem. Res., 2020, 53, 2106 CrossRef CAS PubMed.
- L. G. Li, C. Liu, S. H. Liu, J. Wang, J. J. Han, T. S. Chan, Y. Y. Li, Z. W. Hu, Q. Shao, Q. B. Zhang and X. Q. Huang, ACS Nano, 2022, 16, 14885 CrossRef CAS PubMed.
- V. V. Brazhkin, Phys.-Usp., 2006, 49, 719 CrossRef CAS.
- B. Patra, S. Jana, L. A. Constantin and P. Samal, J. Phys. Chem. C, 2021, 125, 4284 CrossRef CAS.
- A. Richard and F. Corà, J. Phys. Chem. C, 2023, 127, 10766 CrossRef CAS PubMed.
- A. Vittadini, M. Casarin and A. Selloni, J. Phys. Chem. C, 2009, 113, 18973 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 2286 CrossRef CAS PubMed.
- G. Armstrong, A. R. Armstrong, J. Canales and P. G. Bruce, Chem. Commun., 2005, 2454 RSC.
- A. G. Dylla, G. Henkelman and K. J. Stevenson, Acc. Chem. Res., 2013, 46, 1104 CrossRef CAS PubMed.
- X. Hua, Z. Liu, M. G. Fischer, O. Borkiewicz, P. J. Chupas, K. W. Chapman, U. Steiner, P. G. Bruce and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 13330 CrossRef CAS PubMed.
- D. He, H. Su, X. Q. Li, H. J. Yu, M. Zubair, L. Wang, S. C. Mao and J. S. Wang, Sci. Bull., 2018, 63, 314 CrossRef CAS PubMed.
- Z. Wang, Y. Wang, W. Zhang, Z. Wang, Y. Ma and X. Zhou, J. Phys. Chem. C, 2019, 123, 1779 CrossRef CAS.
- T. Hongo and A. Yamazaki, Microporous Mesoporous Mater., 2011, 142, 316 CrossRef CAS.
- Y. Lei, J. Li, Z. Wang, J. Sun, F. Chen, H. Liu, X. Ma and Z. Liu, Nanoscale, 2017, 9, 4601 RSC.
- E. D. Hanson, L. M. Lilley, J. D. Cain, S. Q. Hao, E. Palacios, K. Aydin, C. Wolverton, T. Meade and V. P. Dravid, Mater. Chem. Phys., 2019, 225, 219 CrossRef CAS.
- G. L. Xiang, Y. G. Wang, J. Li, J. Zhuang and X. Wang, Sci. Rep., 2013, 3, 1411 CrossRef PubMed.
- Y. X. Wang, C. H. Wang, X. T. Zhang, P. P. Sun, L. N. Kong, Y. A. Wei, H. Zheng and Y. C. Liu, Appl. Surf. Sci., 2014, 292, 937 CrossRef CAS.
- H. Jin, W. You, K. Tian, E. Kong, X. Ye, Y. Wang and J. Ye, Langmuir, 2022, 38, 15282 CrossRef CAS PubMed.
- G. Xiang, T. Li, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 6801 RSC.
- T. Beuvier, M. Richard-Plouet and L. Brohan, J. Phys. Chem. C, 2009, 113, 13703 CrossRef CAS.
- A. G. Dylla and K. J. Stevenson, J. Mater. Chem. A, 2014, 2, 20331 RSC.
- P. X. Liu, Y. Zhao, R. X. Qin, S. G. Mo, G. X. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. D. Zang, B. H. Wu, G. Fu and N. F. Zheng, Science, 2016, 352, 797 CrossRef CAS PubMed.
- G. Xiang, Y. Tang, Z. Liu, W. Zhu, H. Liu, J. Wang, G. Zhong, J. Li and X. Wang, Nano Lett., 2018, 18, 7809 CrossRef CAS PubMed.
- J. De Roo, Chem. Mater., 2023, 35, 3781 CrossRef CAS.
- Y. L. Wang, W. Zhang, Z. H. Wang, Y. M. Cao, J. M. Feng, Z. L. Wang and Y. Ma, Chin. J. Catal., 2018, 39, 1500 CrossRef CAS.
- C. Wang, X. Zhang and Y. Liu, Nanoscale, 2014, 6, 5329 RSC.
- W. B. Li, X. F. Qian and J. Li, Nat. Rev. Mater., 2021, 6, 829 CrossRef CAS.
- Z. Wei, D. Liu, W. Wei, X. Chen, Q. Han, W. Yao, X. Ma and Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15533 CrossRef CAS PubMed.
- T. Sasaki, M. Watanabe, Y. Komatsu and Y. Fujiki, Inorg. Chem., 1985, 24, 2265 CrossRef CAS.
- M. R. Allen, A. Thibert, E. M. Sabio, N. D. Browning, D. S. Larsen and F. E. Osterloh, Chem. Mater., 2010, 22, 1220 CrossRef CAS.
- M. Catti, I. Pinus and A. Scherillo, J. Solid State Chem., 2013, 205, 64 CrossRef CAS.
- S. Yin, S. Uchida, Y. Fujishiro, M. Aki and T. Sato, J. Mater. Chem., 1999, 9, 1191 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, TGA, TEM, BET and FT-IR results. See DOI: https://doi.org/10.1039/d3dt02752j |
| This journal is © The Royal Society of Chemistry 2023 |