
In vivo stealthified molecularly imprinted polymer nanogels incorporated with gold nanoparticles for radiation therapy†
Yukiya
Kitayama
 *ab,
Takuya
Yamada
a,
Kentaro
Kiguchi
a,
Aoi
Yoshida
a,
Shuhei
Hayashi
a,
Hiroaki
Akasaka
c,
Kazunori
Igarashi
d,
Yuya
Nishimura
b,
Yu
Matsumoto
d,
Ryohei
Sasaki
c,
Eri
Takano
a,
Hirobumi
Sunayama
a and
Toshifumi
Takeuchi
*a
*ab,
Takuya
Yamada
a,
Kentaro
Kiguchi
a,
Aoi
Yoshida
a,
Shuhei
Hayashi
a,
Hiroaki
Akasaka
c,
Kazunori
Igarashi
d,
Yuya
Nishimura
b,
Yu
Matsumoto
d,
Ryohei
Sasaki
c,
Eri
Takano
a,
Hirobumi
Sunayama
a and
Toshifumi
Takeuchi
*a
aGraduate School of Engineering, Kobe University, 1-1, Rokkodai-cho, Nada-ku, Kobe, 657-8501, Japan. E-mail: kitayama@omu.ac.jp; takeuchi@gold.kobe-u.ac.jp
bGraduate School of Engineering, Osaka Prefecture University, 1-1, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan
cGraduate School of Medicine, Kobe University, Chuo Ku, 7-5-1, Kusunoki Cho, Kobe, Hyogo 650-0017, Japan
dGraduate School of Medicine and Faculty of Medicine, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan
First published on 27th May 2022
Abstract
Radiation therapy is a representative therapeutic approach for cancer treatment, wherein the development of efficient radiation sensitizers with low side effects is critical. In this study, a novel stealth radiation sensitizer based on Au-embedded molecularly imprinted polymer nanogels (Au MIP-NGs) was developed for low-dose X-ray radiation therapy. Surface plasmon resonance measurements reveal the good affinity and selectivity of the obtained Au MIP-NGs toward the target dysopsonic protein, human serum albumin. The protein recognition capability of the nanogels led to the formation of the albumin-rich protein corona in the plasma. The Au MIP-NGs acquire stealth capability in vivo through protein corona regulation using the intrinsic dysopsonic proteins. The injection of Au MIP-NGs improved the efficiency of the radiation therapy in mouse models of pancreatic cancer. The growth of the pancreatic tumor was inhibited even at low X-ray doses (2 Gy). The novel strategy reported in this study for the synthesis of stealth nanomaterials based on nanomaterial–protein interaction control shows significant potential for application even in other approaches for cancer treatment, diagnostics, and theranostics. This strategy paves a way for the development of a wide range of effective nanomedicines for cancer therapy.
Introduction
The development of efficient therapeutic agents for cancer treatment is highly sought after in current medical frontiers because cancer is among the most common causes of death even in developed nations. Engineered nanomaterials, such as micelles,1–3 liposomes,4,5 nanoparticles,6,7 and nanogels,8,9 exhibit significant potential as materials for nanocarriers in drug delivery systems, realizing continuous and efficient anti-cancer agent delivery toward tumors and reducing the side effects in non-diseased tissues. To date, a wide range of nanomaterial-based medicines has been developed specifically for cancer treatment procedures, which are currently dominated by chemotherapy,10–12 surgical therapy,13 and radiation therapy.14,15 Nanomaterial-assisted chemotherapy, wherein molecular drugs are utilized for targeted cancer treatment, has been widely used. Accordingly, this next-generation chemotherapy procedure is established as an effective cancer therapeutic approach.16–19 Radiation therapy is also a potent and efficient cancer treatment approach. In this procedure, minimally invasive, outpatient-based cancer treatments can be performed. However, surrounding normal tissues and/or organs can be damaged during this treatment, which severely limits the application of radiation therapy. Therefore, to enhance the efficiency of this method and to minimize the side effects toward normal tissues, the radiation dose employed can be reduced using radiation-sensitizing agents. However, compared to chemotherapy, radiation sensitization for cancer treatment is not yet well-developed. Furthermore, nanomaterial-assisted radiation therapy remains a challenge.To achieve the efficient intravenous delivery of radiation sensitizers, the stealth capability of nanomaterials is necessary to overcome the immunological response, that is, to prevent their elimination from the reticuloendothelial systems, induced by the non-specific adsorption of antibodies and their complements as components in the bloodstream. Stealth nanocarriers are commonly synthesized through surface modifications using hydrophilic polymers, such as poly(ethylene glycol),20,21 zwitterionic polymer,22 and poly(phosphoester). These hydrophilic polymers suppress the non-specific binding of the opsonic proteins, thereby realizing long periods of blood circulation. This modification strategy has been found to be effective for a wide range of nanomaterials. However, recent reports showed that the delivery efficacy of systems that utilize multiple injections of polymer-coated nanomaterials was reduced owing to accelerated blood clearance phenomena.23–25 Therefore, the development of alternative methods to confer stealth capability to nanomaterials are essential.
Recently, the use of the protein corona concept to impart stealth capability to nanomaterials has been reported. In this approach, intrinsic proteins are adsorbed on the injected nanomaterials, which subsequently forms a new interface in the blood stream.26–29 The protein corona composition critically affects the immunological response and non-specific adsorption of opsonic proteins, which triggers the immunological cascade. To achieve efficient drug delivery and long periods of blood circulation, the opsonic proteins must be eliminated from the protein corona on the nanocarriers. In our previous study, a novel strategy to obtain stealth nanomaterials with immune escape capability against reticuloendothelial systems was developed based on the in situ control of the protein corona using intrinsic dysopsonic proteins.30–32 In this strategy, the in situ regulation of the protein corona was achieved by conferring dysopsonic protein recognition capability on the nanomaterials. Molecularly imprinted polymer nanogels (MIP-NGs), which are promising materials for artificial antibodies, were prepared. Molecular imprinting, which is a templated polymerization method, is a promising approach for the synthesis of artificial antibodies.33–47 The MIP-NGs bound intrinsic human serum albumin (HSA) in the blood vessels, which led to the prolonged blood circulation. Furthermore, the passive tumor-targeting property of the prepared MIP-NGs through the enhanced permeability and retention (EPR) effect was also confirmed. Therefore, this material can be used as a novel stealth nanocarrier for cancer therapy.
In this study, novel stealth radiation sensitizers for nanomaterial-assisted radiation therapy based on gold nanoparticle (Au NP)-incorporated MIP-NGs (Au MIP-NGs) were synthesized. Herein, Au NPs were used owing to their excellent radio sensitizing effect and high biocompatibility.48–51 To fabricate Au MIP-NGs, the monomer containing the disulfide linkage was copolymerized in the MIP-NGs; the formation of the Au NPs occurred in situ inside the MIP-NGs. The Au NPs were also used as tracers of the nanogels in vivo; the in vivo distribution of the Au MIP-NGs was investigated by calculating the accumulated amount of Au in the organs of laboratory mice. Lastly, a demonstration of the radiation therapy using Au MIP-NGs was performed in mouse models of pancreatic cancer. To the best of our knowledge, this is the first report on the fabrication of stealth radiosensitizers for cancer treatment based on an in situ functionalization strategy.
Results and discussion
Novel stealth radiation sensitizers based on nano-sized Au MIP-NGs were prepared through a three-step procedure: (i) synthesis of the MIP-NGs with disulfide linkages, (ii) reduction of the disulfide linkages to form the free thiol groups, and (iii) in situ formation and trapping of the Au NPs inside the MIP-NGs (Scheme 1).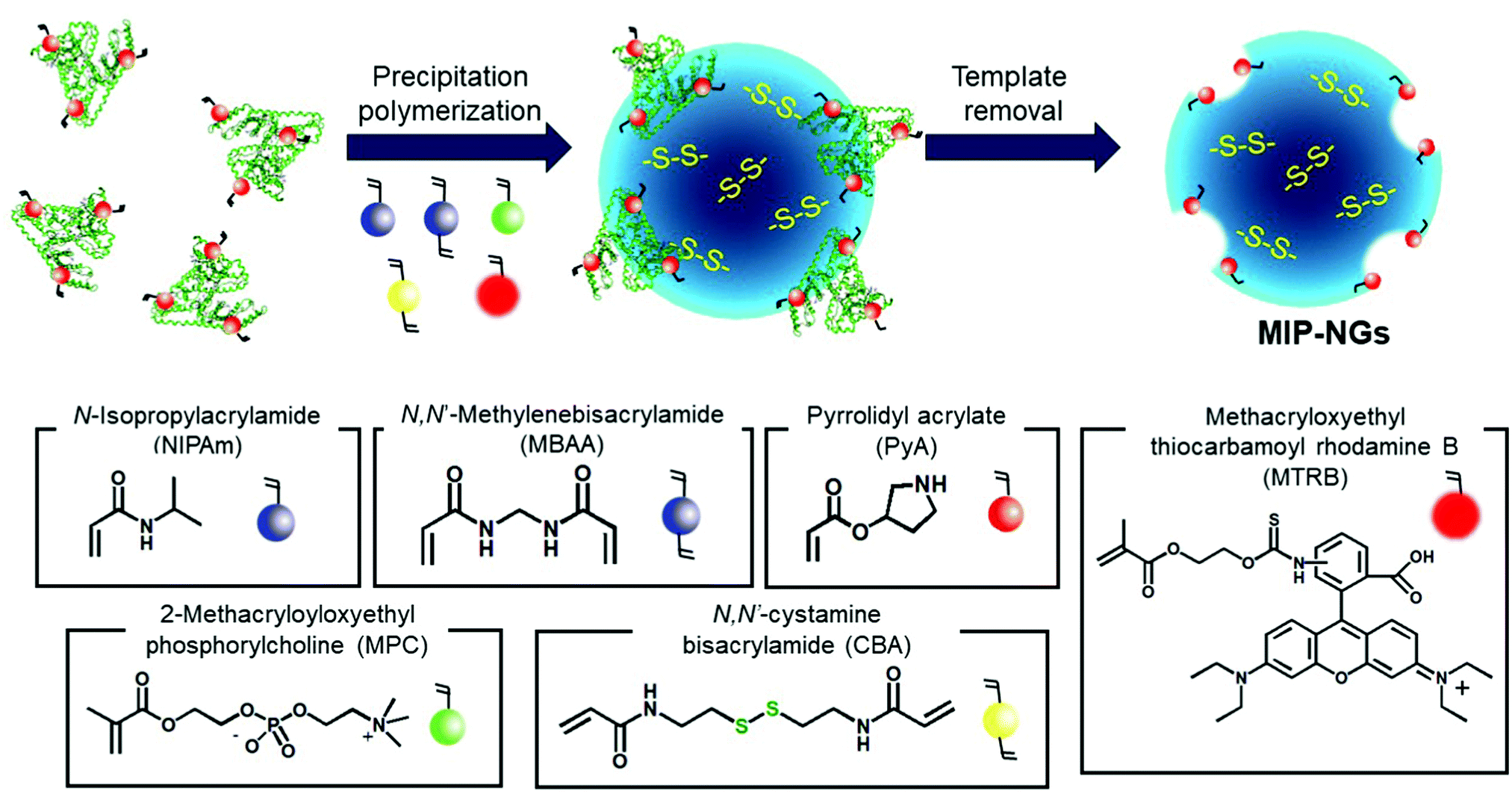 | ||
| Scheme 1 Synthetic route of the Au NP-incorporated MIP-NGs (Au MIP-NGs) MIP-NGs: molecularly imprinted polymer nanogels. | ||
In this study, MIP-NGs containing disulfide linkages were prepared through emulsifier-free precipitation polymerization using HSA as the template in a phosphate-buffered saline (PBS) (pH 7.4). N-Isopropyl acrylamide (NIPAm, comonomer) and N,N′-methylenebisacrylamide (MBAA, crosslinking agent) were used to form the polymeric nanogel platforms during precipitation polymerization. Pyrrolidyl acrylate (PyA) was used as an effective functional monomer for HSA imprinting.52 2-Methacryloyloxyethyl phosphorylcholine (MPC), which is a highly hydrophilic biocompatible monomer, was copolymerized to reduce the particle size and increase the lower critical solution temperature, which prevent the in vivo coagulation of the resulting nanogels.53–55 To trap the Au NPs formed in the MIP-NGs, N,N′-cystamine bisacrylamide (CBA) was also copolymerized; the disulfide linkages were selected owing to their low susceptibility toward free radical attack. To monitor the blood circulation profile, methacryloyloxyethyl thiocarbamoyl rhodamine B (MTRB) was used for the fluorescent labeling of the MIP-NGs in intravenous confocal laser scanning microscopy (IVCLSM). To remove the remaining monomers/initiator, water-soluble oligomers, and HSA template, the obtained MIP-NGs were purified through size-exclusion and ion-exchange chromatography.56 The obtained MIP-NGs were approximately 122 nm (PDI: 0.22) in size (Fig. S1, ESI†). Thereafter, the purified MIP-NGs were treated using an aqueous solution of tris(2-carboxyethyl) phosphine (TCEP). This mild reducing agent was used to generate the thiol groups from the disulfide linkage inside the nanogels. Using a 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) assay, the thiol content in the thiolated MIP-NGs was estimated to be 0.53 μmol mg−1; 5-thio-(2-nitrobenzoic acid) (λmax = 412 nm) was generated from the reaction between the DTNB and free thiol groups (Fig. 1a). The thiolated MIP-NGs were incubated with HAuCl4·3H2O. Thereafter, NaBH4 was added to reduce the Au3+ ions to Au0 and in turn form the Au NPs. The Au NPs produced through the reduction of HAuCl4·3H2O without the MIP-NGs were highly agglomerated. In contrast, the Au NPs synthesized in the presence of the thiolated MIP-NGs were stable, resulting in the formation of the nano-sized Au MIP-NGs with an average nanoparticle size of approximately 49 nm (PDI: 0.28) (Fig. 1b). The smaller MIP-NG particles following Au NP generation are possibly ascribable to polymer shrinking due to network formation through the Au NPs generated in the nanogels. The transmission electron microscopy (TEM) images in Fig. 1c reveal the incorporation of the Au NPs into the MIP-NGs. Furthermore, UV-vis measurements were performed to measure the absorbance of the Au NPs (Fig. 1d). Through inductively coupled plasma (ICP) spectrometry, the Au content in the Au MIP-NGs was estimated to be 12.0 wt%. To evaluate the molecular recognition capability of the Au MIP-NGs, non-imprinted polymer nanogels with Au NPs (Au NIP-NGs) were synthesized through a synthesis procedure similar to that of the Au MIP-NGs without using the HSA template. From dynamic light scattering (DLS) and ICP measurements, the nanoparticle size of the Au NIP-NGs was approximately 61 nm (PDI: 0.30), which is smaller than that of the NIP-NGs prior to Au NPs generation (146 nm; PDI: 0.22), and its Au content (12.6 wt%) was relatively similar to that of Au MIP-NGs (Fig. S1 and S2, ESI†).
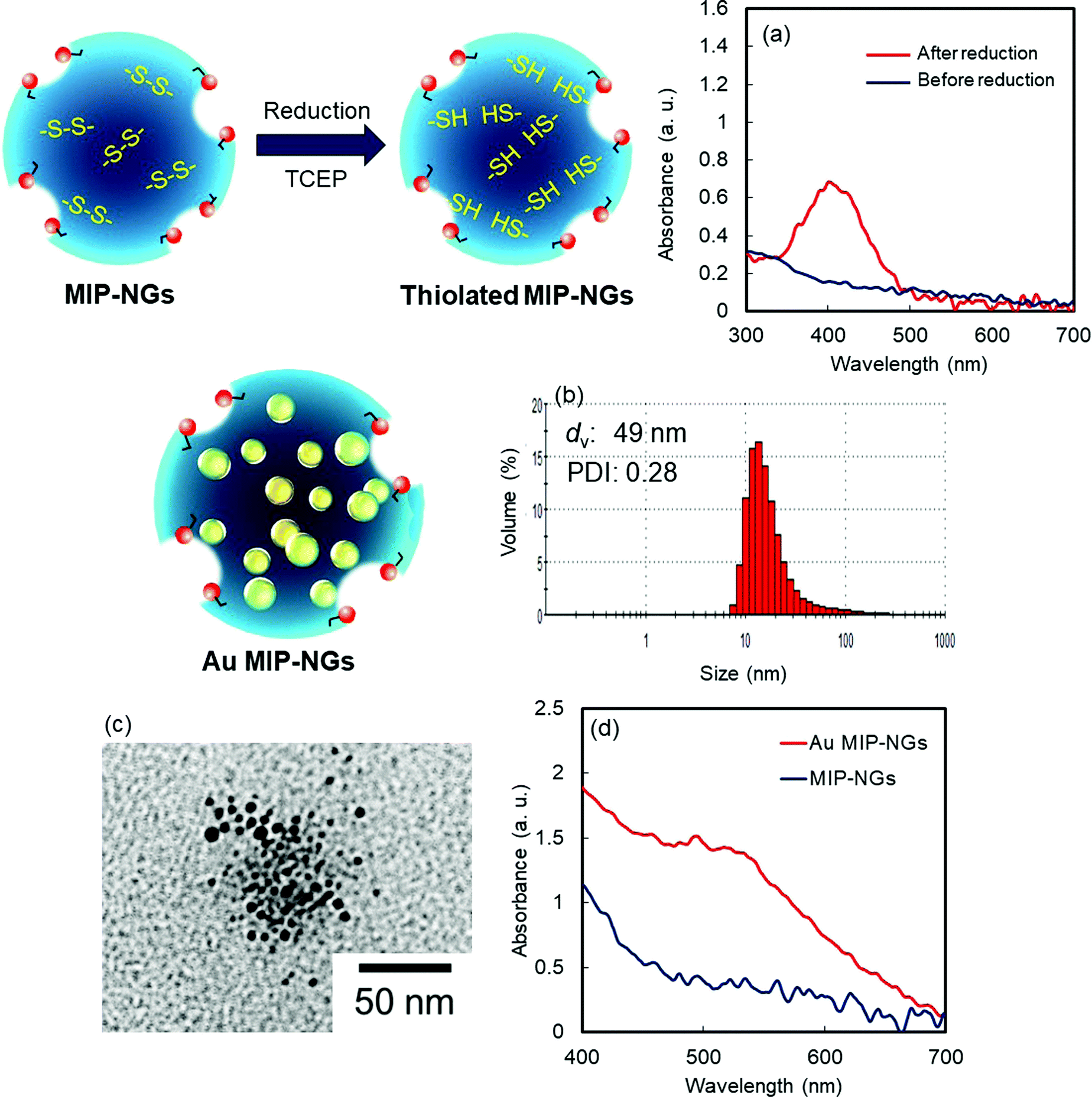 | ||
| Fig. 1 (a) DTNB assay for MIP-NGs before (blue) and after (red) the reduction of MIP-NGs using TCEP. (b) Particle size distribution measured using DLS and (c) TEM image of Au MIP-NGs. (d) UV-Vis spectra of the MIP-NGs before (blue) and after (red) the generation of Au NPs. TCEP: tris(2-carboxyethyl) phosphine; DLS: dynamic light scattering; TEM: transmittance electron microscopy; MIP-NGs: molecularly imprinted polymer nanogels; Au NPs: gold nanoparticles. | ||
The molecular recognition properties of the Au MIP-NGs were studied through surface plasmon resonance (SPR) using protein-immobilized sensor chips; the reference proteins used were immunoglobulin G (IgG), fibrinogen (Fib), and HSA. The value of the resonance unit change (ΔRU) of the Au MIP-NGs for the target protein HSA gradually increased with the increasing particle concentration. Furthermore, the ΔRU values of the Au MIP-NGs were significantly higher than those of the reference Au NIP-NGs (Fig. 2a). Lastly, the amounts of the Au MIP-NGs bound to the IgG- and Fib-immobilized sensor chips were significantly lower than those bound to the HSA-immobilized chip, thereby revealing the high selectivity of the Au MIP-NGs toward HSA (Fig. 2b).
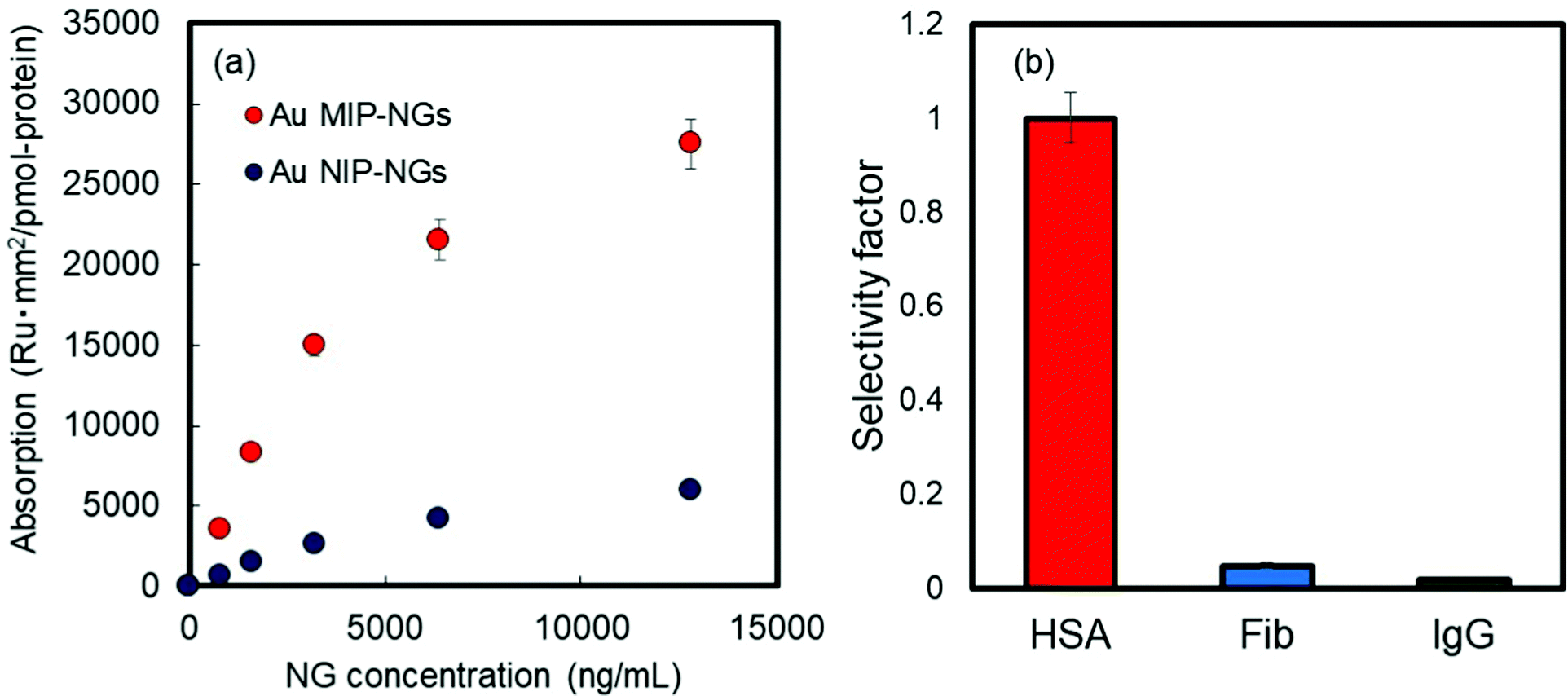 | ||
| Fig. 2 (a) Binding isotherms of the HSA measured by SPR on the Au MIP-NGs (red) and Au NIP-NGs (blue). (b) Selectivity test on Au MIP-NGs using HSA and reference proteins (Fib and IgG). Au MIP-NGs: Au NP-incorporated molecularly imprinted polymer nanogels; Au NIP-NGs: Au NP-incorporated non-imprinted polymer nanogels; HSA: human serum albumin; Fib: fibrinogen; IgG: immunoglobulin G; SPR: surface plasmon resonance. | ||
The protein corona formed on the Au MIP-NGs was analyzed. Au MIP-NGs were incubated in human serum. Thereafter, the protein corona compositions of Au MIP-NGs and Au NIP-NGs were analyzed through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) after the desorption of the protein from the Au MIP-NGs and Au NIP-NGs samples using a urea–thiourea buffer. The protein bands observed in both nanogels can be attributed mainly to HSA and its dimers; the other bands corresponding to other proteins were negligible (Fig. 3a). Furthermore, the amounts of HSA (Fig. 3b) and its dimers (Fig. 3c) bound to the Au MIP-NGs were approximately 2.4 and 4.0 times higher than those bound to the reference nanogels, respectively. These results reveal the formation of an albumin-rich protein corona layer on the Au MIP-NGs in human serum and indicate the higher albumin concentrations on the imprinted nanogels than those on the reference Au NIP-NGs. Furthermore, the HSA dimer/HSA monomer band intensity ratio on the original human serum was significantly smaller than that of the protein corona layer on the Au MIP-NGs (Fig. S3, ESI†). Therefore, the Au MIP-NGs-bound HSA dimers exhibit high affinity owing to multivalent effects and/or the locally concentrated dimerization of HSA monomers bound on the Au MIP-NGs.
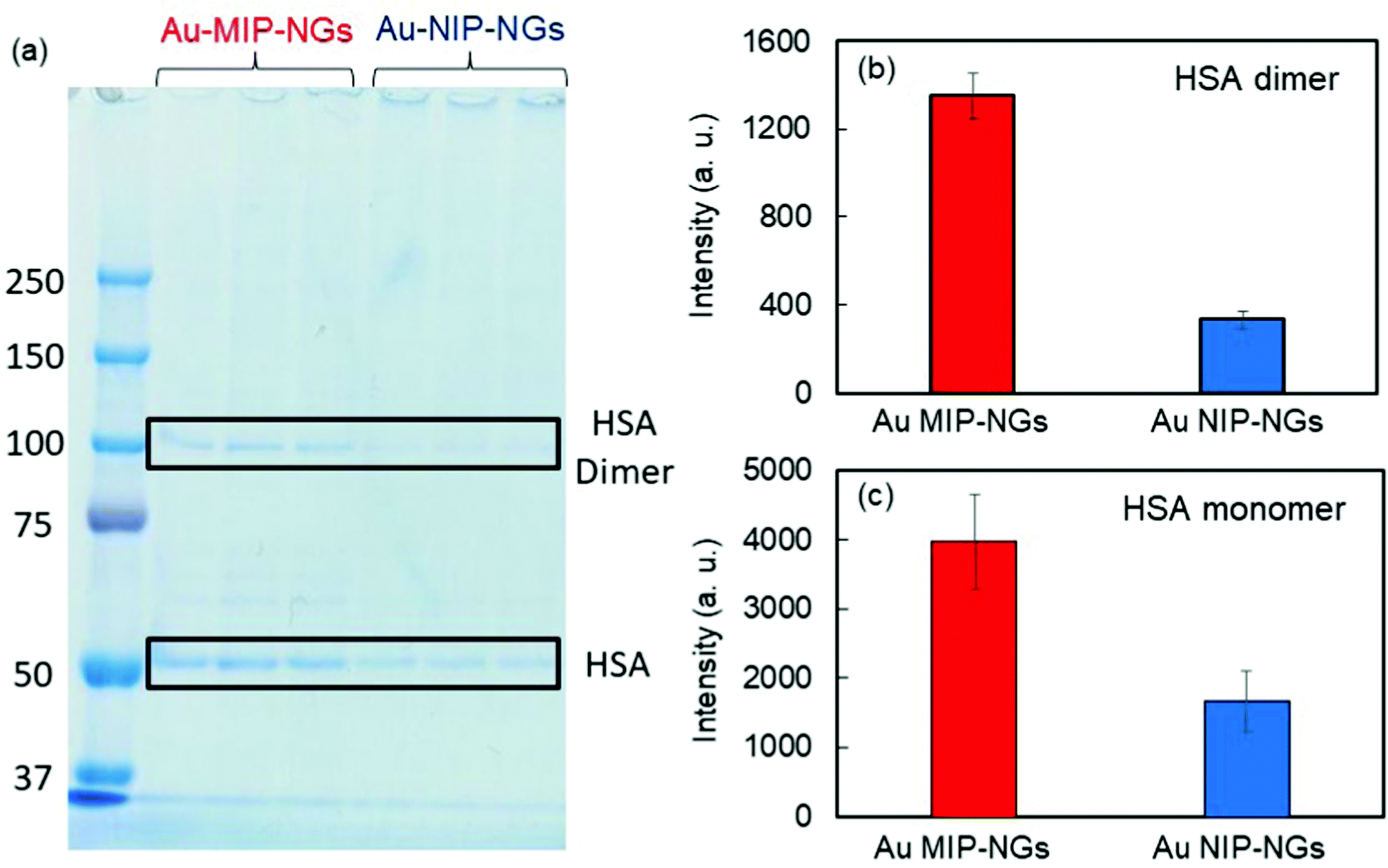 | ||
| Fig. 3 (a) Protein bands derived from the protein corona formed on the Au MIP-NGs and Au NIP-NGs analyzed through SDS-PAGE. Band intensities derived from (b) HSA monomer and (c) HSA dimer on Au MIP-NGs (red) and Au NIP-NGs (blue). Au MIP-NGs: Au NP-incorporated molecularly imprinted polymer nanogels; Au NIP-NGs: Au NP-incorporated non-imprinted polymer nanogels; HSA: human serum albumin; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis. | ||
Prior to in vitro and in vivo applications of the nanogels, the cytotoxicity of the Au MIP-NGs in normal fibroblast NIH-3T3 cells was evaluated using the cell counting kit-8 (CCK-8) assay. High cell viability and negligible cytotoxicity were observed at all the tested concentrations of the Au MIP-NGs (as high as 120 μg mL−1) (Fig. S4, ESI†). Furthermore, the cancer-targeting properties of the Au MIP-NGs were also studied. The accumulation of Au in each organ revealed that the Au MIP-NGs can be traced in vivo. Accordingly, the biodistribution of the Au MIP-NGs in mouse models of pancreatic cancer was investigated; the amount of Au that accumulated in each organ after the intravenous injection of the Au MIP-NGs was investigated through ICP spectrometry. In particular, the accumulation of Au in the liver and spleen was monitored. The liver contains immune-related cells, such as Kupffer and dendritic cells; the spleen has reticular cells. Therefore, these organs are related to the reticuloendothelial system for immunological responses. Therefore, to achieve an efficient and highly targeted cancer treatment, the accumulation of the nanogels in the liver and spleen should be suppressed. The amount of the Au MIP-NGs that accumulated in the liver and spleen was lower than that of the Au NIP-NGs (Fig. 4a and b). The concentrations of different biomarkers, such as leucine aminopeptidase (LAP), alkaline phosphatase (ALP), and cholinesterase (ChE), in the blood of mice models were determined to evaluate the in vivo toxicity due to the accumulation of Au MIP-NGs in the liver. The concentrations of LAP, ALP, and ChE are related to liver damage (Fig. S5, ESI†). The blood concentrations of these three proteins in mice injected with the Au MIP-NGs were similar to those in control mice injected with the buffered solution. Therefore, the Au MIP-NGs are nontoxic and highly biocompatible. The in vivo confocal laser scanning microscopy revealed that longer blood circulation times were achieved when Au MIP-NGs were used (Fig. S6, ESI†). The Au MIP-NGs circulate in blood for much longer times than the previously reported HSA-imprinted nanogels30 and Fc-domain imprinted nanogels.31 This observation is possibly due to the Au MIP-NG particles being of suitable size for blood circulation, since the particles of the previously reported nanogels are much smaller (HSA-imprinted nanogels: ∼35 nm,30 Fc-domain imprinted nanogels: ∼16 nm31) than the Au MIP-NG particles (∼61 nm). The effect of particle size on blood circulation will be investigated in detail in the future. Furthermore, the increasing concentration of Au in the pancreatic tumor over time reveals that the Au MIP-NGs gradually accumulated in the tumor (Fig. 4c). Significantly, the amount of Au MIP-NGs that accumulated in the tumor was higher than that of the Au NIP-NGs (Fig. 4d). As discussed above, the Au MIP-NGs showed a higher HSA affinity and formed more HSA-rich protein coronas. The HSA rich protein corona facilitates Au MIP-NG cloaking, leading to its escape from the immunological response. Consequently, the Au MIP-NGs circulated longer in the blood and accumulated less in the liver and spleen. Therefore, the Au MIP-NGs exhibited an in vivo stealth capability and effectively escaped the immune response. Maeda and co-workers reported that tumors have leaky blood vessels compared to normal tissue.57 Furthermore, tumors lack lymphatic systems. Consequently, nanocarriers less than 100 nm in size reportedly accumulate selectively in tumors (EPR effect).58 The Au MIP-NG particles are less than 100 nm in size on average, as shown in Fig. 1b, and they show long blood circulation profiles due to in situ stealthification. Thus, they accumulate in tumors through their passive tumor-targeting properties that are based on the EPR effect; hence, the radiation sensitizer was successfully delivered to the pancreatic cancer tumor through the in situ stealthified Au MIP-NGs nanocarrier.
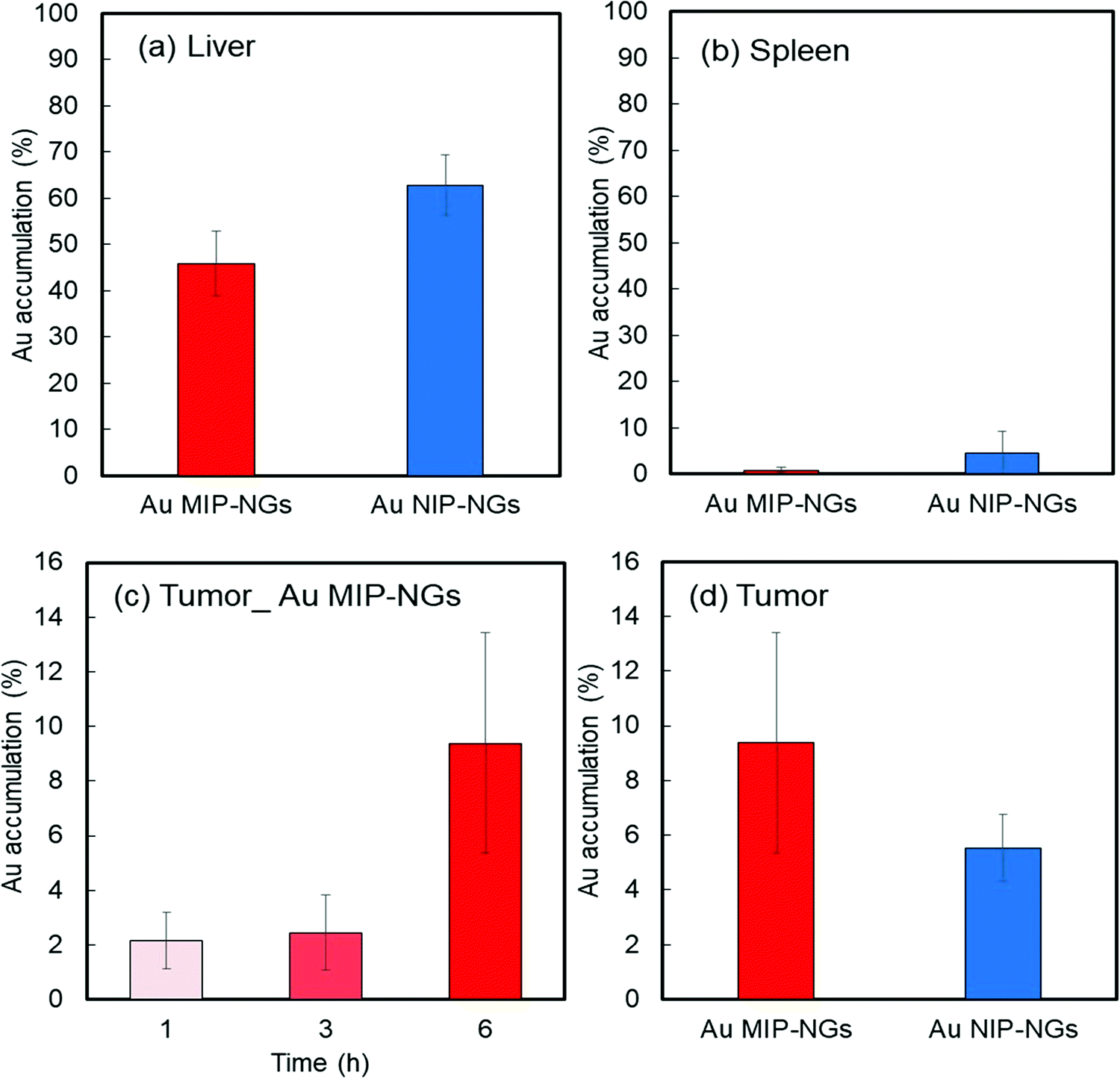 | ||
| Fig. 4 Accumulation of Au in (a) liver and (b) spleen of mouse models of pancreatic cancer after 1 h of injection with Au MIP-NGs (red) and Au NIP-NGs (blue). (c) Time course of the accumulation of Au MIP-NGs in pancreatic tumor. (d) Accumulation of Au in tumor of mouse models after 6 h of injection with Au MIP-NGs (red) and Au NIP-NGs (blue). Au MIP-NGs: Au NP-incorporated molecularly imprinted polymer nanogels; Au NIP-NGs: Au NP-incorporated non-imprinted polymer nanogels. | ||
Finally, radiation therapy for pancreatic cancer was demonstrated using the novel stealth Au MIP-NGs as the radiation sensitizers. The mouse models of pancreatic cancer were irradiated (2 or 4 Gy) after the intravenous injection of Au MIP-NGs or the control PBS. In general, the tumor volume gradually increased. For the PBS-injected mice, tumor growth was effectively suppressed by the X-ray irradiation; increasing the X-ray irradiation dose further inhibited the growth of the tumor (Fig. 5a). These results imply that the radiation therapy was effective on the mouse models of pancreatic cancer. Under the same irradiation intensity, the growth of the tumor was suppressed more effectively in the Au MIP-NG-injected mice than in the reference PBS-injected model. Furthermore, the tumor growth suppression efficiencies in the Au MIP-NG-injected mice obtained after the irradiation with 2 or 4 Gy was almost similar; therefore, radiation therapy was effective even at a lower X-ray irradiation dose (2 Gy). At 2 Gy, the tumor volume in the Au MIP-NG-injected mice was significantly smaller than that in the reference PBS-injected model (P < 0.05) (Fig. 5b). Moreover, similar to the observations on the PBS-injected model, the tumor volume in the Au MIP-NG-injected mice was larger without X-ray irradiation. This implies that the Au MIP-NGs did not exhibit cancer treatment potential without X-ray irradiation (Fig. S7, ESI†). Furthermore, under different conditions, the body weights of the mice models were similar. Therefore, the intravenous injection of the Au MIP-NGs and the X-ray irradiation did not cause considerable toxicity in the mice (Fig. S8, ESI†). These results prove that the novel stealth Au MIP-NGs produced in this study are promising sensitizers for highly targeted radiation cancer treatments.
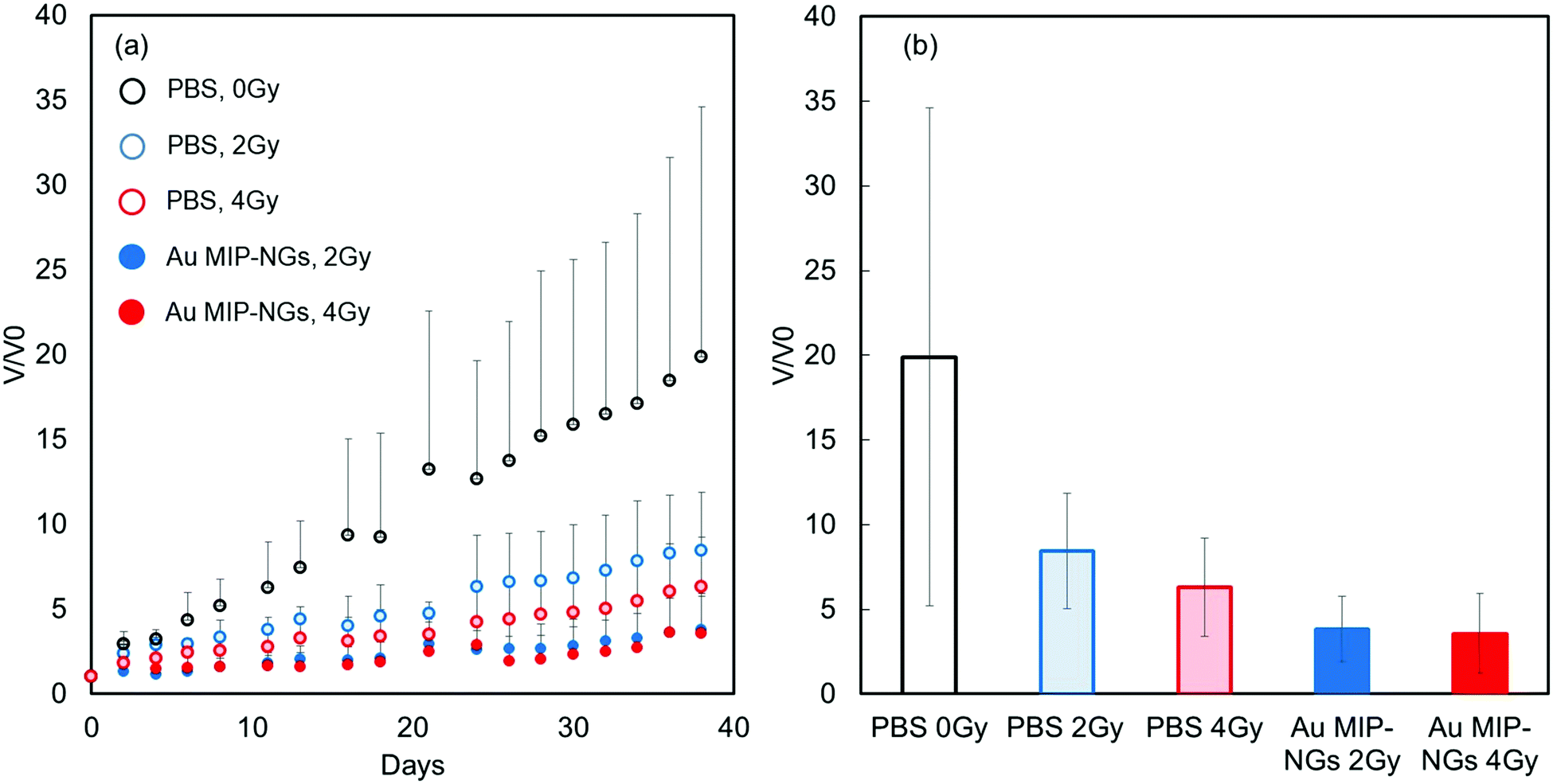 | ||
| Fig. 5 (a) Tumor volume changes in X-ray irradiated mouse models of pancreatic cancer injected with Au MIP-NGs (closed circles) and control phosphate-buffered saline (PBS) (open circles): X-ray irradiation dose: 0 (black), 2 (blue), and 4 Gy (red). Au MIP-NGs: Au NP-incorporated molecularly imprinted polymer nanogels; Au NIP-NGs: Au NP-incorporated non-imprinted polymer nanogels. | ||
Conclusions
In this study, novel radiation sensitizers with in vivo stealth properties were fabricated using Au MIP-NGs. The Au MIP-NGs were formed through the in situ reduction of Au3+ and trapping of the Au NPs in the thiolate MIP-NGs with HSA-imprinted nanocavities. The Au MIP-NGs exhibited high selectivity toward HSA and formed an albumin-rich protein corona in the human serum. Results of the biodistribution test confirmed the passive targeting property of the Au MIP-NGs toward refractory pancreatic cancer based on the EPR effect. Using the same X-ray irradiation dose, the growth of the tumor was suppressed more effectively in the Au MIP-NGs-injected mice than in the reference PBS-injected model. Furthermore, using the Au MIP-NGs sensitizer, the radiation therapy was successful even at a low X-ray irradiation dose (2 Gy). This study paves a way for the development of different stealth nanomedicines for cancer treatment based on the molecular imprinting-based strategy and in situ formation of the dysopsonic protein-rich corona.Author contributions
Y. K. and T. T. progressed this project totally. T. Y., K. K., A. Y., and S. H. and Y.K. synthesized the Au MIP-NGs. The IVCLSM was performed with K. I. and Y. M. Radiation therapy for cancer model mice was performed with Y. N., A. H. and R. S. H. S. and E. T. helped manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (18H05398) and the Foundation for Biomedical Research and Innovation at Kobe (Japan). We thank Professor Hideto Minami of Kobe University (Japan) for his valuable suggestions.References
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6(12), 815–823 CrossRef CAS PubMed
.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47(1), 113–131 CrossRef CAS PubMed
- V. P. Torchilin, Pharm. Res., 2007, 24(1), 1–16 CAS
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65(1), 36–48 CrossRef CAS PubMed
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4(2), 145–160 CrossRef CAS PubMed
- R. Singh and J. W. Lillard Jr., Exp. Mol. Pathol., 2009, 86(3), 215–223 CrossRef CAS PubMed
- O. Veiseh, J. W. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62(3), 284–304 CrossRef CAS PubMed
- M. Hamidi, A. Azadi and P. Rafiei, Adv. Drug Delivery Rev., 2008, 60(15), 1638–1649 CrossRef CAS PubMed
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33(4), 448–477 CrossRef CAS
- L. Kelland, Nat. Rev. Cancer, 2007, 7(8), 573–584 CrossRef CAS PubMed
- J. H. Schiller, D. Harrington, C. P. Belani, C. Langer, A. Sandler, J. Krook, J. Zhu and D. H. Johnson, N. Engl. J. Med., 2002, 346(2), 92–98 CrossRef CAS PubMed
- D. J. Slamon, B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajamonde, T. Fleming, W. Eiermann, J. Wolter, M. Pegram, J. Baselga and L. Norton, N. Engl. J. Med., 2001, 344(11), 783–792 CrossRef CAS PubMed
- A. L. Vahrmeijer, M. Hutteman, J. R. Van Der Vorst, C. J.-H. Van De Velde and J. V. Frangioni, Nat. Rev. Clin. Oncol., 2013, 10(9), 507–518 CrossRef CAS PubMed
- R. Timmerman, R. Paulus, J. Galvin, J. Michalski, W. Straube, J. Bradley, A. Fakiris, A. Bezjak, G. Videtic, D. Johnstone, J. Fowler, E. Gore and H. Choy, JAMA, 2010, 303(11), 1070–1076 CrossRef CAS PubMed
- T. J. Whelan, J. P. Pignol, M. N. Levine, J. A. Julian, R. MacKenzie, S. Parpia, W. Shelley, L. Grimard, J. Bowen, H. Lukka, F. Perera, A. Fyles, K. Schneider, S. Gulavita and C. Freeman, N. Engl. J. Med., 2010, 362(6), 513–520 CrossRef CAS PubMed
- R. De Souza, P. Zahedi, C. J. Allen and M. Piquette-Miller, Drug Delivery, 2010, 17(6), 365–375 CrossRef CAS PubMed
- Q. Hu, W. Sun, C. Wang and Z. Gu, Adv. Drug Delivery Rev., 2016, 98, 19–34 CrossRef CAS PubMed
- T. Iwamoto, Biol. Pharm. Bull., 2013, 36(5), 715–718 CrossRef CAS PubMed
- M. Sui, W. Liu and Y. Shen, J. Controlled Release, 2011, 155(2), 227–236 CrossRef CAS PubMed
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55(3), 403–419 CrossRef CAS PubMed
- J. Milton Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2(3), 214–221 CrossRef PubMed
- Y. Zhu, H. S. Sundaram, S. Liu, L. Zhang, X. Xu, Q. Yu, J. Xu and S. Jiang, Biomacromolecules, 2014, 15(5), 1845–1851 CrossRef CAS PubMed
- E. T.-M. Dams, P. Laverman, W. J.-G. Oyen, G. Storm, G. L. Scherphof, J. W.-M. Van Der Meer, F. H.-M. Corstens and O. C. Boerman, J. Pharmacol. Exp. Ther., 2000, 292(3), 1071–1079 CAS
- T. Ishida, M. Ichihara, X. Wang, K. Yamamoto, J. Kimura, E. Majima and H. Kiwada, J. Controlled Release, 2006, 112(1), 15–25 CrossRef CAS PubMed
- T. Ishida and H. Kiwada, Int. J. Pharm., 2008, 354(1-2), 56–62 CrossRef CAS PubMed
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(38), 14265–14270 CrossRef CAS PubMed
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3(1-2), 40–47 CrossRef CAS
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M.-V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8(7), 543–557 CrossRef CAS PubMed
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8(10), 772–781 CrossRef CAS PubMed
- T. Takeuchi, Y. Kitayama, R. Sasao, T. Yamada, K. Toh, Y. Matsumoto and K. Kataoka, Angew. Chem., Int. Ed., 2017, 56(25), 7088–7092 CrossRef CAS PubMed
- T. Morishita, A. Yoshida, N. Hayakawa, K. Kiguchi, C. Cheubong, H. Sunayama, Y. Kitayama and T. Takeuchi, Langmuir, 2022, 36(36), 10674–10682 CrossRef PubMed
- N. Hayakawa, Y. Kitayama, K. Igarashi, Y. Matsumoto, E. Takano, H. Sunayama and T. Takeuchi, ACS Appl. Mater. Interfaces, 2022, 14(14), 16074–16081 CrossRef CAS PubMed
- Z. Bie, Y. Chen, J. Ye, S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54(35), 10211–10215 CrossRef CAS PubMed
- G. Pan, Y. Zhang, Y. Ma, C. Li and H. Zhang, Angew. Chem., Int. Ed., 2011, 50(49), 11731–11734 CrossRef CAS PubMed
- H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 45(15), 2392–2396 CrossRef CAS PubMed
- J. L. Urraca, A. J. Hall, M. C. Moreno-Bondi and B. Sellergren, Angew. Chem., Int. Ed., 2006, 45(31), 5158–5161 CrossRef CAS PubMed
- L. Chen, S. Xu and J. Li, Chem. Soc. Rev., 2011, 40(5), 2922–2942 RSC
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100(7), 2495–2504 CrossRef CAS PubMed
- B. Sellergren and C. J. Allender, Adv. Drug Delivery Rev., 2005, 57(12), 1733–1741 CrossRef CAS PubMed
- D. A. Spivak, Adv. Drug Delivery Rev., 2005, 57(12), 1779–1794 CrossRef CAS PubMed
- G. Wulff, Chem. Rev., 2002, 102(1), 1–27 CrossRef CAS PubMed
- R. Horikawa, H. Sunayama, Y. Kitayama, E. Takano and T. Takeuchi, Angew. Chem., Int. Ed., 2016, 55(42), 13023–13027 CrossRef CAS PubMed
- S. Ichikawa, N. Shimokawa, M. Takagi, Y. Kitayama and T. Takeuchi, Chem. Commun., 2018, 54(36), 4557–4560 RSC
- K. Mori, M. Hirase, T. Morishige, E. Takano, H. Sunayama, Y. Kitayama, S. Inubushi, R. Sasaki, M. Yashiro and T. Takeuchi, Angew. Chem., Int. Ed., 2019, 58(6), 1612–1615 CrossRef CAS PubMed
- T. Takeuchi, T. Mori, A. Kuwahara, T. Ohta, A. Oshita, H. Sunayama, Y. Kitayama and T. Ooya, Angew. Chem., Int. Ed., 2014, 53(47), 12765–12770 CrossRef CAS PubMed
- T. Takeuchi, H. Sunayama, E. Takano and Y. Kitayama, Adv. Biochem. Eng./Biotechnol., 2015, 150, 95–106 CrossRef CAS PubMed
- K. Takimoto, E. Takano, Y. Kitayama and T. Takeuchi, Langmuir, 2015, 31(17), 4981–4987 CrossRef CAS PubMed
- S. Jain, D. G. Hirst and J. M. O'Sullivan, Br. J. Radiol., Suppl., 2012, 85(1010), 101–113 CrossRef CAS PubMed
- D. B. Chithrani, S. Jelveh, F. Jalali, M. Van Prooijen, C. Allen, R. G. Bristow, R. P. Hill and D. A. Jaffray, Radiat. Res., 2010, 173(6), 719–728 CAS
- X. D. Zhang, D. Wu, X. Shen, J. Chen, Y. M. Sun, P. X. Liu and X. J. Liang, Biomaterials, 2012, 33(27), 6408–6419 CrossRef CAS PubMed
- J. F. Hainfeld, D. N. Slatkin and H. M. Smilowitz, Phys. Med. Biol., 2004, 49(18), N309–N315 CrossRef CAS PubMed
- Y. Inoue, A. Kuwahara, K. Ohmori, H. Sunayama, T. Ooya and T. Takeuchi, Biosens. Bioelectron., 2013, 48, 113–119 CrossRef CAS PubMed
- W. Feng, S. Zhu, K. Ishihara and J. L. Brash, Langmuir, 2005, 21(13), 5980–5987 CrossRef CAS PubMed
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J., 1990, 22(5), 355–360 CrossRef CAS
- T. Moro, Y. Takatori, K. Ishihara, T. Konno, Y. Takigawa, T. Matsushita, U. I.-L. Chung, K. Nakamura and H. Kawaguchi, Nat. Mater., 2004, 3(11), 829–836 CrossRef CAS PubMed
- C. Cheubong, A. Yoshida, Y. Mizukawa, N. Hayakawa, M. Takai, T. Morishita, Y. Kitayama, H. Sunayama and T. Takeuchi, Anal. Chem., 2020, 92(9), 6401–6407 CrossRef CAS PubMed
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 4(6), 6387–6392 Search PubMed
- I. Brigger, C. Dubernet and P. Couvreur, Adv. Drug Delivery Rev., 2002, 54(5), 631–651 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: The particle size distribution of Au NIP-NGs, SDS-PAGE analysis of human serum, cytotoxicity, biomarker protein concentrations, and effect of Au MIP-NGs on tumor volume change without X-ray irradiation are available free of charge via the internet. See DOI: https://doi.org/10.1039/d2tb00481j |
| This journal is © The Royal Society of Chemistry 2022 |