
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiobased catalyst-free covalent adaptable networks based on CF3-activated synergistic aza-Michael exchange and transesterification†
Dimitri
Berne
,
Baptiste
Quienne
 ,
Sylvain
Caillol
,
Eric
Leclerc
* and
Vincent
Ladmiral
*
,
Sylvain
Caillol
,
Eric
Leclerc
* and
Vincent
Ladmiral
*
ICGM, Université Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: vincent.ladmiral@enscm.fr; eric.leclerc@enscm.fr
First published on 26th September 2022
Abstract
Recently, fluorine neighboring group activation emerged as a new way of promoting acid–epoxy reaction and transesterification in vitrimer materials. Pursuing this idea, the effect of a CF3 group on aza-Michael addition and aza-Michael exchange was examined and confirmed on model molecules. Following these positive results, a CAN incorporating aza-Michael bonds and CF3-activated ester functions was synthesized and compared to analogous materials deprived of any CF3 groups on the one hand, or of any hydroxyl groups on the other hand. The study of the mechanical properties of these materials highlighted the synergistic effect of the two exchange reactions and the accelerating effect of the fluorinated group. The fluorinated and hydroxylated material was shown to be reprocessable at 100 °C in 1 h under 3 tons whereas the fluorinated only and the hydroxylated only materials were respectively reprocessed at 120 °C and 150 °C. These catalyst-free CANs were synthesized from natural resources further enhancing the sustainability of these materials. This study demonstrates the potential of biobased CANs featuring fluorinated esters as low environmental impact and easily reprocessable materials.
Introduction
Covalent Adaptable Networks (CANs) have emerged in the last two decades as a new polymer family, bridging the gap between the two historical groups of polymers: thermoplastics and thermosets. CANs refer to polymer networks possessing reversible covalent bonds, hence coupling the reshaping capability of thermoplastics with the thermal and chemical resistance of thermosets. The development of CANs is going in the same direction as the current environmental trend of reducing waste and improving the sustainability of materials. Indeed, cross-linked polymers, whose recycling is particularly complex,1 can gain the possibility of being reused instead of only being incinerated if they are converted into CANs. Their recycling through the dynamic character of CANs can then be triggered by different stimuli such as heat,2 pH3 or light.4The area of CANs is in constant development and multiple exchangeable functions have emerged and have been reviewed by several research team.5–8 They have been generally classified according to their mechanism: dissociative or associative.9 Theoretically, in opposition to dissociative CANs, only vitrimers (associative CANs) maintain a constant crosslink density with temperature and can therefore demonstrate an Arrhenius behaviour.10–15 However, numerous dissociative CANs have recently demonstrated Arrhenius behavior in specific ranges of temperature.16–21 Moreover, Dichtel and Elling have been wondering whether the use of associative exchange mechanism alone in cross-link polymer networks was desirable to meet current processing challenges.22 Indeed, as dissociative and associative CANs demonstrated to behave similarly in temperature range usually used for reprocessing, the postulated superiority of vitrimers on dissociative CANs should be reconsidered. Especially since the use of dissociative chemistries could enable to respond to some current CAN challenges (synthesis of high activation energy CANs, adaptation of current thermoplastics reprocessing techniques to CANs, broad CANs application range via the development of new exchange reactions,…).7
Many of the dynamic chemistries mentioned above require catalysts to be activated under thermal stimulus.23,24 However, the use of external catalysts is limiting the panel of applications of CANs. For instance, organic salts25,26 and strong bases,27 used as catalysts in epoxy vitrimers, are usually toxic and corrosive, and may leach out from the materials or undergo ageing.28 Therefore, the concepts of neighbouring group participation (NGP) or internal catalysis inspired from development in organic synthesis have been implemented in CANs.29,30 Several modes of activation have been reported in the literature such as H-bonding,31–33 formation of cyclic intermediate34,35 or tertiary amine catalyst.36,37 Recently, the introduction of fluorinated groups near an ester function allowed to significantly increase the transesterification reaction rate via inductive effects.38–40
Based on these precedents, we wished to explore the possibility of designing a CAN that could relax according to two possible exchange reactions, both sufficiently accelerated by a single activating group to avoid the presence of any external catalyst. This dual mechanism could lead to a network with strong dynamical properties and, coupled to a biosourcing of the monomer and the absence of any additive, it would reinforce the durability and recyclability of such material. We already reported that catalyst-free transesterification vitrimers could be prepared and exhibit vitrimer properties thanks to the presence of vicinal fluorinated groups that accelerate the exchange reaction.38 We wish to present herein a material that associates this transesterification to a much less studied reaction, namely the aza-Michael exchange.41,42 In this synergistic system, both reactions are activated by the same CF3 group positioned on the α-position to the ester. The effect of the CF3 group was first highlighted on the aza-Michael addition and aza-Michael exchange reactions on model compounds. Then, a set of CANs constituted of β-amino esters was synthesized by aza-Michael polyaddition. Bio-based bis-hydroxylated amine and bis-trifluoromethylacrylate were used to synthesize a CAN coupling synergistic and fluorine accelerating effect with a high renewable carbon content. Its non-fluorinated analogue, as well as the one devoid of hydroxy groups were also prepared in order to assess the influence of each group on the mechanical behaviour of the materials. The thermal and stress relaxation behaviours of these materials were evaluated by mechanical and chemical analyses. Finally, these materials were reshaped three times under different conditions and the influence of the reprocessing conditions on the materials was studied. This study highlights how the incorporation of fluorine exchange activator, and synergistic exchange reactions in cross-linked networks, enables to easily synthesize and recycle sustainable materials.
Materials & methods
Materials
Butanediol diglycidyl ether (BDGE), 4,9-dioxadodecanediamine (BDA), 2-methyltetrahydrofurane (Me-THF) and N-benzylmethylamine (BMA) were purchased Sigma-Aldrich Merck (Darmstadt, Germany). Aqueous ammonia (25% NH3) were purchased from VWR International S. A. S (Fontenay-sous-Bois, France). Pripol™ 2033 was kindly provided by Croda (East Cowick, United Kingdom). 2-(Trifluoromethyl)acrylic acid (MAF) (>98%), tert-butyl 2-(Trifluoromethyl)acrylate (MAF-TBE) and methyl 2-(trifluoromethyl)acrylate (MAF-Me) were purchased from SynQuest Labs (Alachua, FL, USA). All materials were used as received. The deuterated NMR solvents (CDCl3 (99.5% isotopic purity), MeOD (99.5% isotopic purity) and DMF-d7 (99.5% isotopic purity)) and were purchased from Eurisotop (Saint-Aubin, France).Characterizations
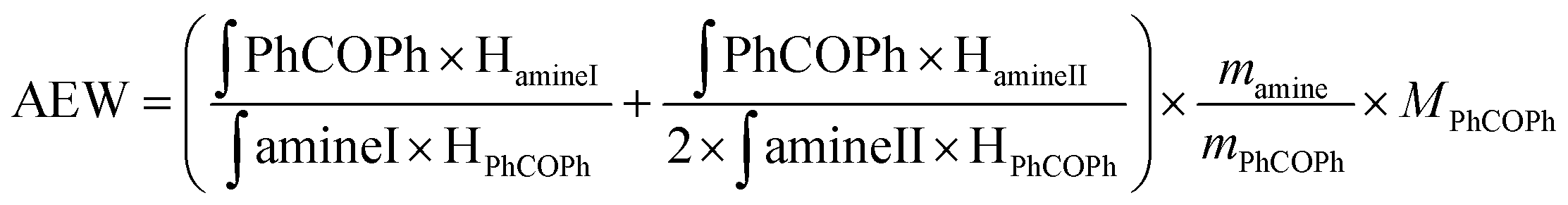 | (1) |
∫PhCOPh: integral of the signal from benzophenone protons; ∫amineI: integral of the signals from protons in α of the primary amine function; ∫amineII: integral of the signals from protons in α position of the secondary amine function; HamineI: number of protons in α position of the primary amine function; HamineII: number of protons in α position of the secondary amine function HPhCOPh: number of benzophenone protons; mamine: mass of amine; mPhCOPh: mass of benzophenone; MPhCOPh: benzophenone molar mass.
Creep recovery experiments were performed at 80 °C and 50 °C by applying 2 kPa shear stress for a duration of 1200 s followed by a recovery period of 1200 s.
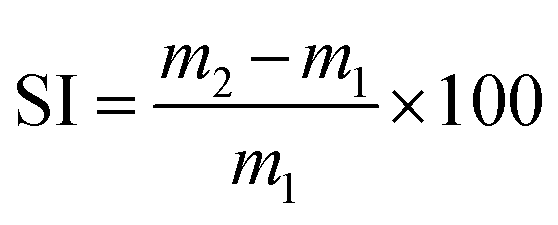 | (2) |
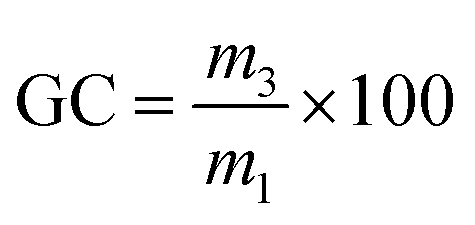 | (3) |
Synthesis
1H-NMR (400 MHz, CDCl3): 1.52 s, O–C–(C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)3, 2.23 (s, N–CH3), 2.69–2.73 (dd, 3J = 12.5 Hz 2J = 4.0 Hz, N–C2–CH), 3.12–3.18 (m, N–CH2–C), 3.26–3.36 (m, N–C2–CH) 3.49–3.64 (m, N–C2–C), 7.25–7.35 (m, Ar) 13C-NMR (101 MHz, CDCl3): 27.9 (O–C–(
3)3, 2.23 (s, N–CH3), 2.69–2.73 (dd, 3J = 12.5 Hz 2J = 4.0 Hz, N–C2–CH), 3.12–3.18 (m, N–CH2–C), 3.26–3.36 (m, N–C2–CH) 3.49–3.64 (m, N–C2–C), 7.25–7.35 (m, Ar) 13C-NMR (101 MHz, CDCl3): 27.9 (O–C–(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3)3), 41.9 (N–H3), 50.3–51.0 (q, 2J = 25.7 Hz, N–CH2–H), 54.0 (q, 3J = 2.6 Hz, N–H2–CH), 62.3 (N–H2–C), 82.5 (O––(CH3)3), 120.3–129.2 (q, F3), 127.2 (CH–CH–H–CH–CH), 128.2 (Ar), 128.9 (H–CH–CH–CH–H), 138.5 (–CH2–N), 166.0 (q, 3J = 3.4 Hz,
H3)3), 41.9 (N–H3), 50.3–51.0 (q, 2J = 25.7 Hz, N–CH2–H), 54.0 (q, 3J = 2.6 Hz, N–H2–CH), 62.3 (N–H2–C), 82.5 (O––(CH3)3), 120.3–129.2 (q, F3), 127.2 (CH–CH–H–CH–CH), 128.2 (Ar), 128.9 (H–CH–CH–CH–H), 138.5 (–CH2–N), 166.0 (q, 3J = 3.4 Hz, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) 19F-NMR (377 MHz, CDCl3): δ/ppm = - 67.3 (CF3).
O) 19F-NMR (377 MHz, CDCl3): δ/ppm = - 67.3 (CF3).
1H-NMR (400 MHz, CDCl3): 2.26 (s, N–CH3), 2.73–2.77 (dd, 3J = 12.6 Hz 2J = 4.2 Hz, N–C2–CH), 3.17–3.22 (m, N–CH2–C), 3.42–3.51 (m, N–C2–CH) 3.49–3.68 (m, N–C2–C), 3.82 (s, O–C3), 7.28–7.38 (m, Ar).
13C-NMR (101 MHz, CDCl3): 42.0 (N–H3), 49.3–50.0 (q, 2J = 26.3 Hz, N–CH2–H), 52.7 (O–H3), 53.7 (q, 3J = 2.7 Hz, N–H2–CH), 62.3 (N–H2–C), 82.5 (O–-(CH3)), 120.0–128.2 (q, 1J = 280.2 Hz, F3), 127.3 (CH–CH–CH–CH–CH), 128.3 (Ar), 128.9 (H–CH–CH–CH–H), 138.3 (–CH2–N), 167.4 (q, 3J = 3.4 Hz, O).
19F-NMR (377 MHz, CDCl3): δ/ppm = −67.10–67.12 (CF3).
1H-NMR (400 MHz, CDCl3): δ/ppm = 1.48 s, O–C–(C3)3, 2.22 (s, N–CH3), 2.45–2.49 (t, 3J = 7.4 Hz, N–CH2–C2), 2.72-2.77 (t, 3J = 7.4 Hz, N–C2–CH2), 3.53 (s, N–C2–C), 7.26–7.34 (m, C–C–C–C–C).
13C-NMR (101 MHz, CDCl3): δ/ppm = 28.1 (O–C–(H3)3), 34.1 (N–CH2–H2), 41.8 (N–H3), 53.1 (N–H2–CH2), 62.1 (N–H2–C), 80.3 (O–-(CH3)3), 127.0 (CH–CH–H–CH–CH), 128.2 (CH–H–CH–H–CH), 129.0 (H–CH–CH–CH–H), 139.1 (–CH2–N), 172.0 (O).
1H-NMR (400 MHz, CD3OD): δ/ppm = 1.67 (m, CH2–C2–CH2), 2.55–2.83 (m, C2–NH–CH2 and C2–NH2), 3.45 (d, CH–C2–O), 3.52 (t, CH2–C2–O), 3.72 (m, C–OH), 3.88 (m, NH–CH2–C–OH).
13C-NMR (101 MHz, CD3OD): δ/ppm = 26.1 (CH2–H2–CH2), 44.4 (H2–NH2), 52.4 (H2–NH–H2 oligomer), 69.0 (CH oligomer), 71.0 (CH2–H2–O), 71.04 (CH), 73.0 ppm (CH–H2–O).
1H-NMR (400 MHz, CDCl3): δ/ppm = 0.89 ppm (m, CH3), 1.02–1.62 ppm (m, CH2–C2–CH2), 1.70 ppm (m, C2–CH2–O), 2.53 ppm (m, CH–C2–CH2), 4.25 ppm (t, C2–O), 6.42 ppm (d, C–C), 6.72 ppm (d, C–C).
13C-NMR (101 MHz, CDCl3): δ/ppm = 14.08 ppm (H3), 22.7 ppm, 25.97 ppm, 28.4–32.02 ppm (CH2–H2–CH2), 28.4 ppm (H2–CH2–O), 31.98 ppm (CH–H2–CH2), 65.96 ppm (t, H2–O), 117.30–125.45 (q, F3), 131.75 ppm (C), 132.46 (q, H2–C), 161.50 ppm (CO).
19F-NMR (377 MHz, CDCl3): δ/ppm = - 65.6 (CF3).
1H-NMR (400 MHz, CDCl3): δ/ppm = 0.89 ppm (m, CH2–C3), 1.02–1.62 ppm (m, CH2–C2–CH2), 1.69 ppm (m, C2–CH2–O), 2.56 ppm (m, CH–C2–CH2), 4.17 ppm (t, C2–O), 5.84 ppm (d, C–C, 2J = 1.5 Hz, 3J = 10.4 Hz), 6.14 ppm (d, C–C–CO, 3J = 17.3 Hz, 10.4 Hz), 6.42 ppm (m, C–C, 2J = 1.5 Hz, 3J = 17.3 Hz).
13C-NMR (101 MHz, CDCl3): δ/ppm = 14.04 ppm (H3), 22.90 ppm, 26.10 ppm, 28.5–31.80 ppm (CH2–H2–CH2), 29.0 ppm (H2–CH2–O), 32.00 ppm (CH–H2–CH2), 64.70 ppm (H2–O), 128.60 (H–C–CO), 130.50 (H2–C), 166.24 ppm (CO).
Results & discussion
Aza-Michael addition
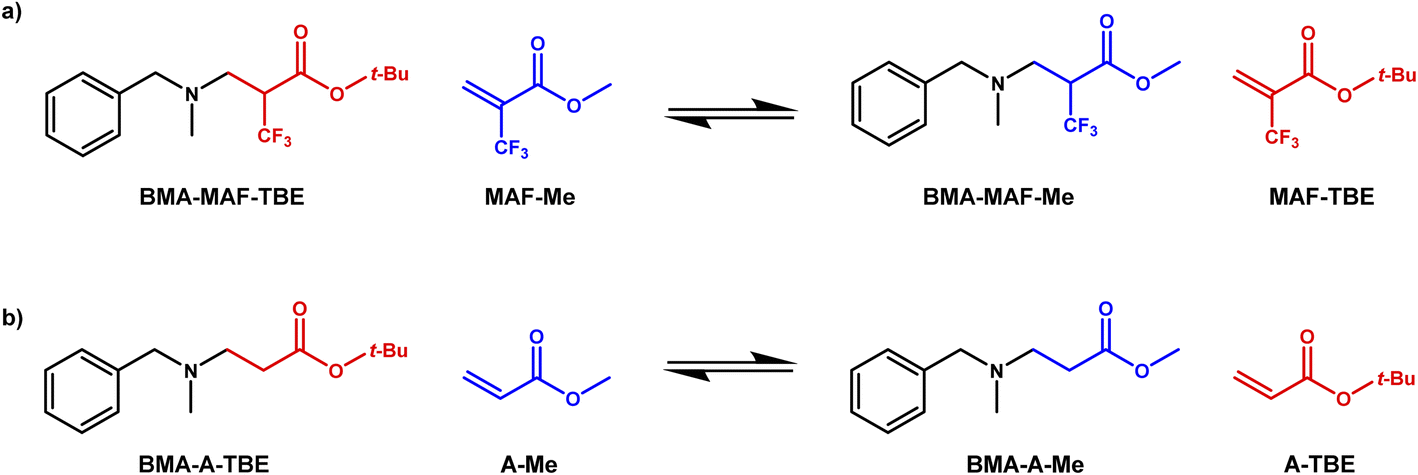
As previously demonstrated with a thia-Michael reaction, the presence of a CF3 group in α position of a Michael acceptor facilitates the addition of Michael donors thanks to the strong electron-withdrawing effect of such a substituent.43,44 Here, the same effect was observed for the aza-Michael reaction with N-benzylmethylamine (BMA) (Scheme 1). Indeed, the reaction with tert-butyl 2-(trifluoromethyl)acrylate (MAF-TBE) was performed at room temperature in 10 minutes in DCM whereas the reaction with tert-butyl acrylate (A-TBE) required a temperature of 70 °C and 24 h to occur under solvent-free conditions. The model molecules BMA-MAF-TBE and BMA-A-TBE (Fig. S1 and S2†) were both obtained in high yields and characterized by 1H-NMR, 13C-NMR, and additionally by 19F-NMR for BMA-MAF-TBE.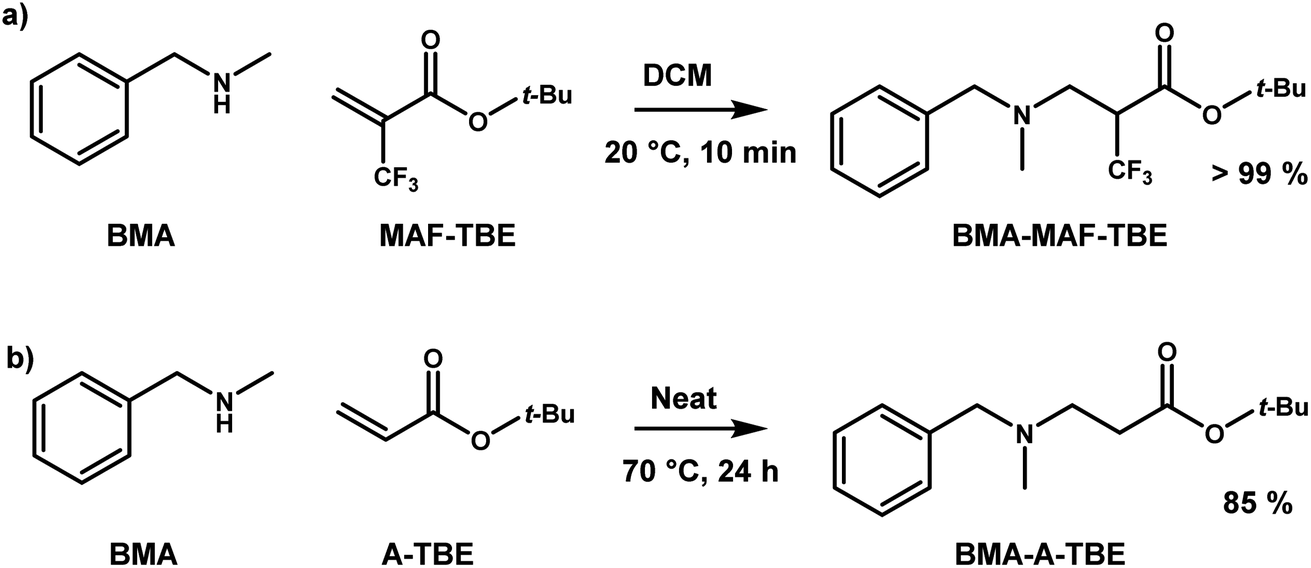 | ||
| Scheme 1 Aza-Michael addition of N-benzylmethylamine on (a) tert-butyl 2-(trifluoromethyl)acrylate (MAF-TBE) and (b) tert-butyl acrylate (A-TBE). | ||
These model molecules were used to specifically evaluate the aza-Michael exchange reaction. For that purpose, BMA-MAF-TBE and BMA-A-TBE were left to react with methyl 2-trifluoromethylacrylate (MAF-Me) or methylacrylate (A-Me) and the formation of respectively BMA-MAF-Me and BMA-A-Me by exchange of Michael acceptor was monitored over time (Scheme 2). BMA-MAF-Me (Fig. S3†) and BMA-A-Me (Fig. S4†) were also separately synthesized to determine their NMR signatures. These reactions were performed using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 BMA-X-TBE:acrylate ratio in DMF at 60 °C and 1 M of each adduct, and were monitored by 19F-NMR and 1H-NMR. No exchange was observed for the non-fluorinated system even after 48 h of reaction, whereas 18% of BMA-MAF-TBE was already converted to BMA-MAF-Me after only 1 h. These results clearly indicate that the presence of a CF3 group drastically accelerate the exchange reaction. Hence, the inductive effects of the CF3 group play a key role on the aza-Michael exchange rate, they activate both the dissociation of β-amino esters, and the addition of amines onto fluorinated double bonds.
:1 BMA-X-TBE:acrylate ratio in DMF at 60 °C and 1 M of each adduct, and were monitored by 19F-NMR and 1H-NMR. No exchange was observed for the non-fluorinated system even after 48 h of reaction, whereas 18% of BMA-MAF-TBE was already converted to BMA-MAF-Me after only 1 h. These results clearly indicate that the presence of a CF3 group drastically accelerate the exchange reaction. Hence, the inductive effects of the CF3 group play a key role on the aza-Michael exchange rate, they activate both the dissociation of β-amino esters, and the addition of amines onto fluorinated double bonds.
 | ||
| Scheme 2 Aza-Michael exchange reactions. | ||
Two additional exchange reactions were examined (Fig. 1): the reaction of BMA-MAF-TBE with MAF-Me and that of BMA-MAF-Me with MAF-TBE. The formation and disappearance of the different MAF adducts were monitored by 19F-NMR over 24 h. The formations of BMA-MAF-TBE and BMA-MAF-Me were characterized by the appearance of two signals for each compound: a doublet at −67.39 and a triplet at −65.76 ppm for BMA-MAF-TBE and a doublet at −67.32 and a triplet at −65.50 for BMA-MAF-Me respectively. Both experiments led to the same final BMA-MAF-TBE/BMA-MAF-Me mixture with a 60/40 mol% ratio (Fig. 1). Hence it appears that BMA-MAF-Me is relatively less stable than BMA-MAF-TBE. These preliminary results highlighted the potential of fluorine-activated aza-Michael as a fast exchange reaction for CAN application. Note that in the materials, the dissociation of the β-aminoesters would likely be more activated (faster) than that of the model compounds due to higher steric hindrance in this preliminary study compared to the following material study.
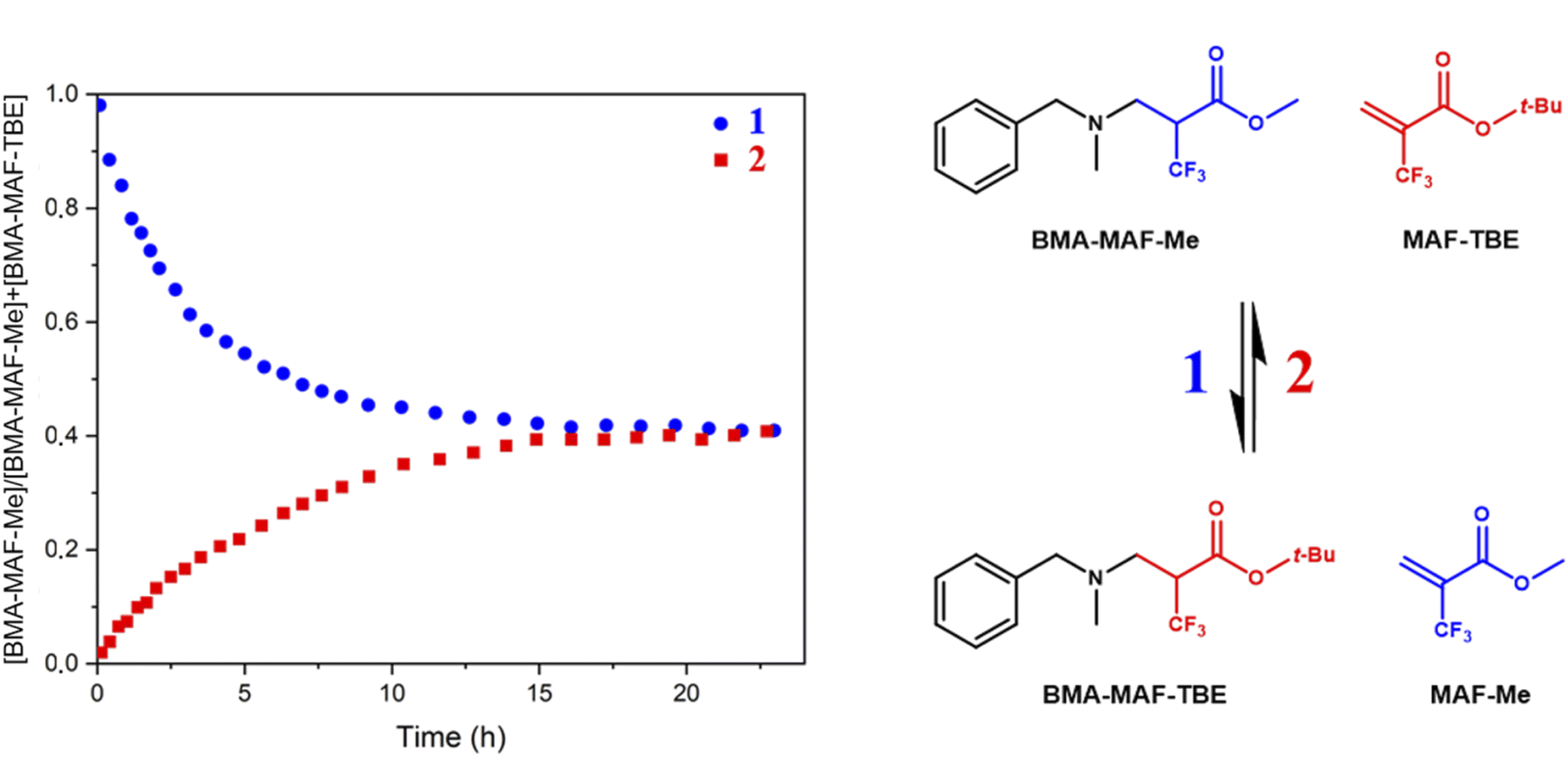 | ||
| Fig. 1 Kinetic monitoring of BMA-MAF-TBE and BMA-MAF-Me exchange reactions at 1 M by 19F-NMR in DMF-d7 at 60 °C. | ||
Material synthesis and characterizations
Following these encouraging results on model molecules, the objective was to prepare β-amino ester (BAE) biobased CANs by aza-Michel polyaddition in order to reduce their impact on the environment. Therefore, biobased bis-acrylate and bis-amine monomers needed to be synthesized from natural resources. Since a primary amine can add onto two acrylates successively, a bis-amine is a potential tetra-functional monomer allowing the formation of a cross-linked network. Moreover, in addition to the exchangeable aza-Michael adducts presented in the network, ester functions could also undergo exchange in the presence of hydroxy functions, leading to a synergistic effect in CANs. Hence, butanediol-di-β-hydroxyamine (BD-β-HA) was used to introduce hydroxy groups in the network.BD-β-HA was synthesized from butanediol diglycidyl ether (BDGE), itself prepared from butanediol and epichlorohydrin, both of which can be biobased. Indeed, butanediol can be obtained from carbohydrates produced by hydrolysis of starch or bacteria fermentation, and epichlorohydrin can be synthesized from glycerol.45,46 Moreover, β-hydroxyamines are known for their facile synthesis, and their high reactivity toward epoxides,47 cyclic carbonates48 and Michael acceptors42 compared to conventional alkylamines due to the H-bonding assistance of the β-OH substituent. BD-β-HA was chosen for three main reasons: its chemical structure enabling the combination of aza-Michael addition and transesterification reaction with its β-OH substituent, its higher reactivity toward Michael acceptors and finally its eco-friendly synthesis. To obtain BD-β-HA, BDGE was reacted with aqueous ammonia in a single step using a sealed reactor and the reaction product was characterized by 1H-NMR and 13C-NMR (Fig. S5†). Since the primary amines formed during the synthesis can still react with the starting epoxide, some BD-β-HA oligomers, are formed. The amount of oligomers was determined by 1H-NMR (signal at 3.88 ppm) to be 16 mol%.
Using a Green Chemistry approach,49 Pripol™ 2033, a fully biobased diol derived from fatty acids, was used to prepare Pripol bis-(trifluoromethyl)acrylate (Pripol-(MAF)2) and Pripol bis-acrylate (Pripol-A2) (Fig. 2). 2-Trifluoromethylacryloyl chloride or acryloyl chloride were added to the diols in the presence of NEt3 at low temperature to prepare the two desired bis-esters. The resulting Pripol-(MAF)2 and Pripol-A2 monomers featured a high biobased carbon content, of respectively 82% and 86%. Pripol-(MAF)2 (Fig. S6†) and Pripol-A2 (Fig. S7†) were both characterized by 1H and 13C-NMR and by 19F-NMR for Pripol-(MAF)2.
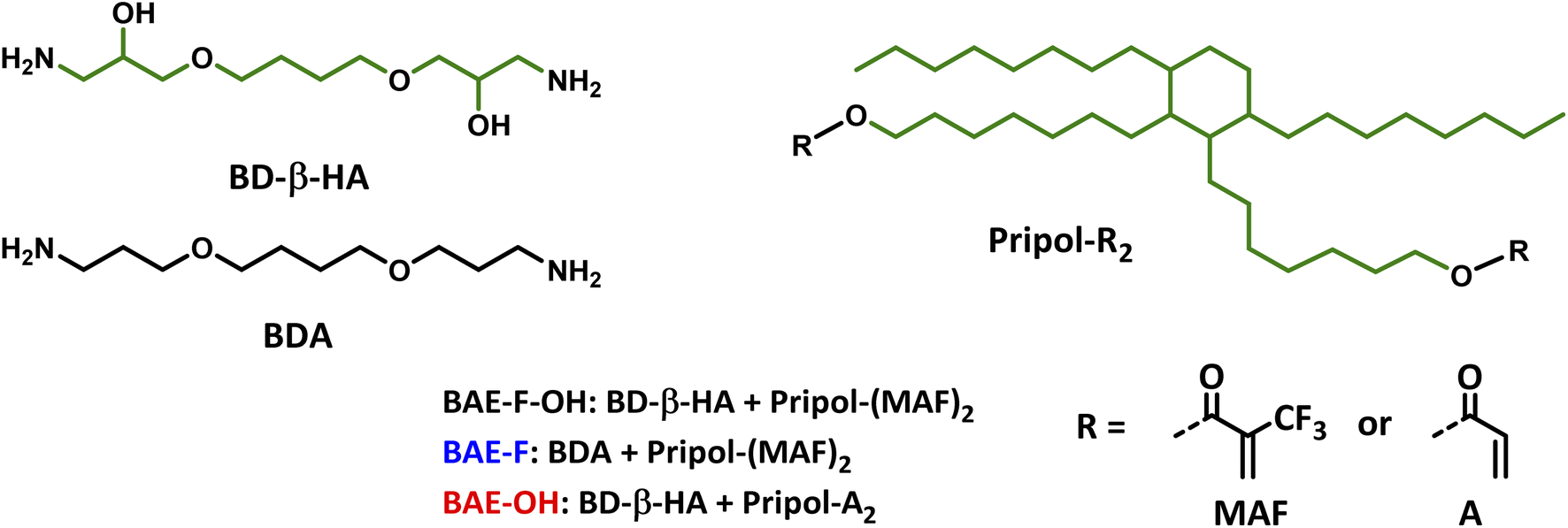 | ||
| Fig. 2 Monomer structures used for the synthesis of BAE networks, the green carbons correspond to biobased carbon. | ||
The activating role of the CF3 group on the aza-Michael exchange that was highlighted at the molecular level could be efficiently implemented in CANs. Three β-amino ester (BAE) thermosets were synthesized via aza-Michael addition (Table 1). BAE–F–OH was prepared by reaction of Pripol-(MAF)2 with BD-β-HA. This material thus contained β-amino-α–CF3–ester moieties able to undergo both CF3-activated aza-Michael exchange reactions and transesterification. BAE–F was prepared from 1,4-bis(3-aminopropoxy)butane (BDA) and Pripol-(MAF)2. This material was thus devoid of hydroxy groups for transesterification reactions, but still fairly prone to aza-Michael exchange via CF3-activation. Finally, BAE–OH was synthesized from Pripol-A2 and BD-β-HA. This third material thus contained free hydroxy groups able to trigger transesterification, but was devoid of CF3 activating groups. The study of the relaxation behaviours of these three thermosets should allow to determine the influence of each NGP on their CAN properties. Note that thanks to the use of biobased monomers, BAE–F–OH, BAE–OH and BAE–F were composed of 84%, 87% and 74% of biobased carbons respectively.
Because of the difference in reactivity between the four monomers, the curing conditions of each thermoset were different, depending on whether the double bond was activated by a CF3 group or not, in agreement with the model reaction study. In the cases of BAE–F–OH and BAE–F, both containing CF3 activating groups, a strong gel was formed after only 1 min of stirring in a SpeedMixer at 25 °C. Therefore, BAE–F–OH and BAE–F were shaped by compression molding at 100 °C and 120 °C for 1 h respectively. In contrast, BAE–OH was cured in an oven at 80 °C for 24 h in a silicon mold. Indeed, without the CF3-activation of the double bond, the crosslinking was much slower and the mixture remained liquid after mixing at 25 °C, highlighting the gap of reactivity compared to BAE–F–OH and BAE–F. In these two materials, the CF3-activation of the Michael acceptors was so strong that the aza-Michael crosslinking was too fast to permit monitoring by rheometry (the gel point could not be determined). Moreover the effect of the hydroxy groups on the aza-Michael additions could not be observed as BAE–F–OH and BAE–F showed the same ultra-fast curing behavior.
No residual exothermy was observed by DSC (Fig. S8†) for any of the materials indicating that they were fully cured under the preparation conditions used. Interestingly, the glass transition temperatures of BAE–F–OH and BAE–OH were identical (−38 °C) suggesting that the CF3 groups had no impact on the chain movement in these materials (Table 2). The Tg of BAE–F was expected to be lower than those of the other materials because of the additional hydrogen bonds provided by the hydroxy functions. However, the presence of BD-β-HA oligomers tends to decrease the Tg by decreasing the AHEW, while commercial BDA is only composed of primary amines. In BAE–F–OH or BAE–OH longer polymer chains are present compared to BAE–F, hence providing more flexibility to these networks and thus explaining the Tg values measured. The gel contents of the BAE networks were comparable (>83%) and confirmed the formation of highly cross-linked networks.
The three BAE networks were also analyzed by DMA as shown in Fig. 3. The storage moduli on the glassy and rubbery plateau of the three materials were of the same order of magnitude. According to the results of the thermogravimetric analyses (Fig. S9†), the thermal stabilities of the fluorinated BAEs (BAE–F and BAE–F–OH) were identical, indicating that the hydroxy group did not significantly affect the material degradation. BAE–OH showed a higher Td5%, possibly because the aza Michael exchange reactions were slower in this material. This phenomenon was already noticed by Du Prez et al.41 who demonstrated that aza-Michael dissociation could be related to thermal degradation. Indeed, as the aza-Michael exchange is assumed to follow a dissociative mechanism, network degradation is dependent on the exchange rate of this reaction and therefore on the temperature.
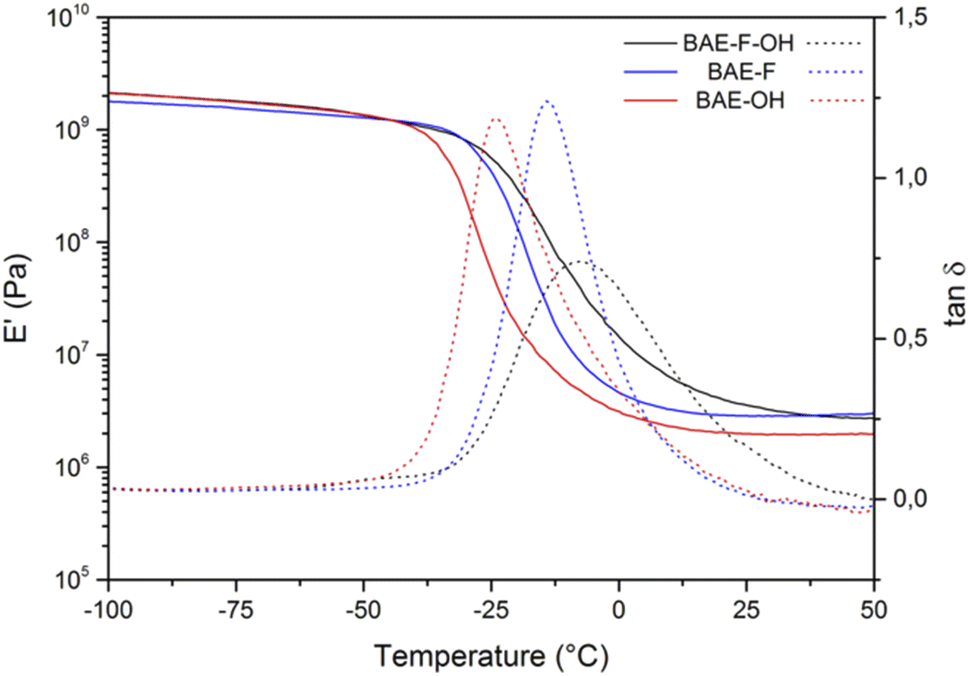 | ||
| Fig. 3 Storage modulus (full curves) and tan(δ) (dotted curves) for BAE–F–OH (black curve), BAE–F (blue curve) and BAE–OH (red curve). | ||
Dynamic properties
In order to study the dynamic character of the three BAE networks, they were subjected to stress-relaxation experiments (Fig. 4 (normalized data) and Fig. S10† (non-normalized data)). A 1% shear strain was applied and the relaxation modulus (G(t)) was monitored as a function of time. As mentioned previously, BAE–F, which can only relax stress via fluorine-activated aza-Michael exchange due to the absence of hydroxyl groups, clearly demonstrated faster relaxation than previously reported CANs based on non-fluorinated aza-Michael exchange. Indeed, even if the comparison is not totally exact because of polymer matrix difference, relaxation times were up to 20 times lower at 130 °C for BAE–F compared to these non-fluorinated CANs.41,42 This result is in good agreement with the preliminary molecular study that highlighted the strong activation of aza-Michael exchange induced by fluorine atoms.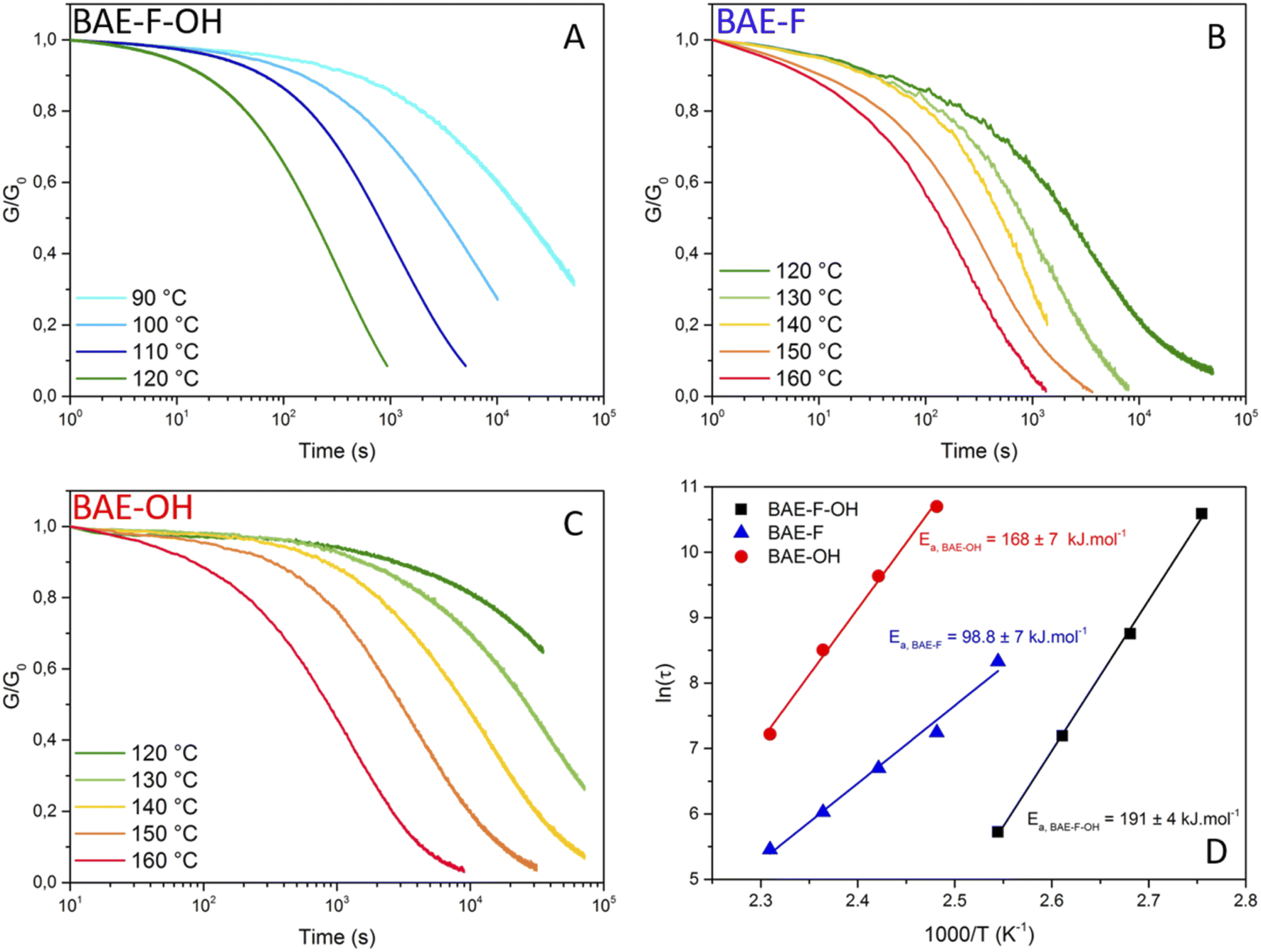 | ||
| Fig. 4 Normalized stress-relaxation curves at different temperatures for a 1% strain (A) for BAE–F–OH, (B) for BAE–F and (C) for BAE–O. (D) Fitting of the relaxation time (τ) vs. temperature data by the Arrhenius equation for BAE–F–OH (black squares), BAE–F (blue triangle) and BAE–OH (red circles). | ||
Recently, CANs having two coexisting mechanisms have gained a lot of interest.50 Some CANs have been based on this concept, for instance disulfide metathesis was combined with transesterification in epoxy vitrimer.51 Du Prez et al. also studied dynamic thiol–yne cross-linking as a new platform for the exchange of thioacetal linkages and demonstrated that associative and dissociative mechanisms occurred concomitantly.52 Following the development of these dual systems previously reported,51,53,54 BAE–F–OH was synthesized to produce a network in which aza-Michael exchange and transesterification can concomitantly occur. Both exchange reactions were activated by the presence of the CF3 group in the alpha and beta positions of the ester function and of the nitrogen atom respectively. Moreover, the polymerization by aza-Michael addition results in the presence of tertiary amines in the network which were previously proven to promote transesterification.37 The comparison of the stress relaxation experiments at 120 °C (Fig. 5A) highlights the interest of using a dual CANs network. Indeed, the fastest relaxation was displayed by BAE–F–OH, in which, both CF3-activated aza-Michael and transesterification reactions can occur, compared to BAE–F for which only fluorine-activated aza-Michael exchange is at play. Finally, the activation effect of the CF3 group was particularly underlined by the comparison of BAE–F–OH and BAE–OH. Indeed, in the absence of CF3 groups in α-position of the ester functions (BAE–OH), the network relaxation was drastically slowed down.
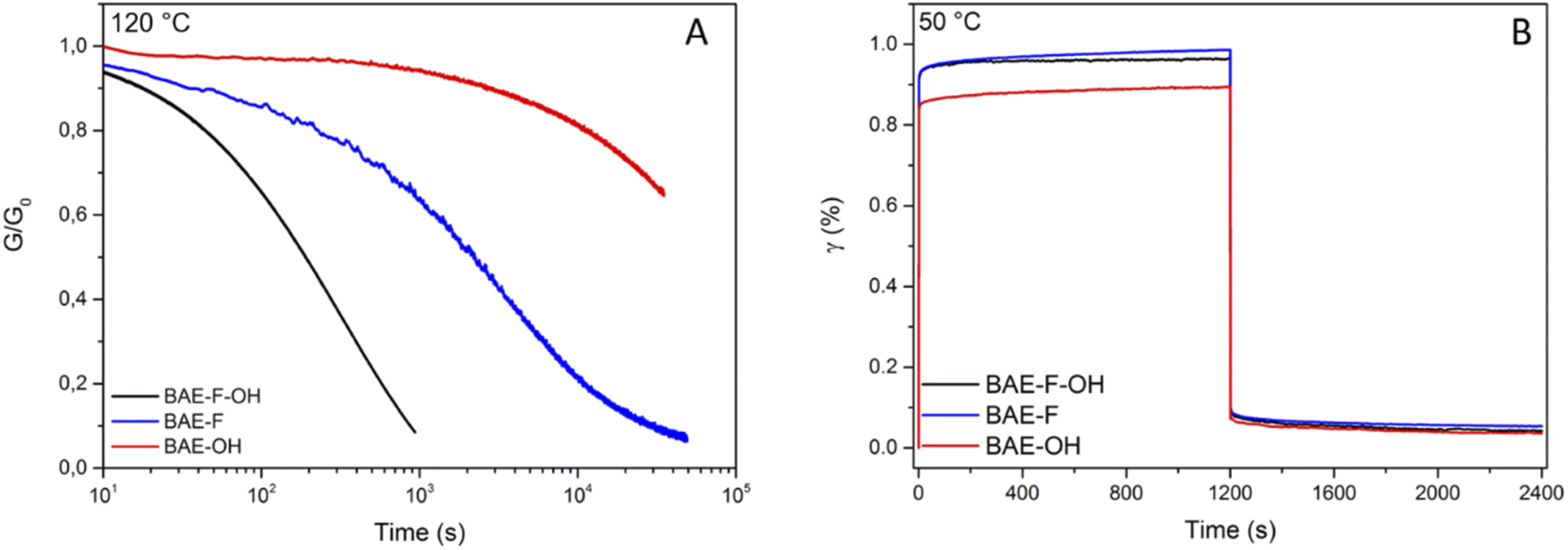 | ||
| Fig. 5 (A) Normalized stress relaxation of BAEs at 120 °C for a 1% strain (B) creep and recovery data for BAEs at 50 °C for an applied stress of 2 kPa. | ||
Each material was studied in a specific temperature range as their relaxation rate were very different: stress-relaxations were performed from 90 to 120 °C for BAE–F–OH (Fig. 4A), from 120 to 160 °C for BAE–F (Fig. 4B) and from 120 to 160 °C for BAE–OH (Fig. 4C). Even if the aza-Michael exchange reaction apparently proceeds via a dissociative mechanism, Arrhenius behaviors were observed for all the BAE networks, and specifically for BAE–F which is based only on aza-Michael exchange. These observations confirm that dissociative CANs can follow an Arrhenius law in a specific range of temperature as recently highlighted by Dichtel and Elling.22Ea,flow of 191 ± 4 kJ mol−1, 98.8 ± 7 kJ mol−1 and 168 ± 7 kJ mol−1 were determined for BAE–F-OH, BAE–F and BAE–OH respectively (Fig. 4D). Even if comparing these Ea,flow values when multiple exchange reactions are at play is complex, the high activation energy displayed by BAE–F–OH is very promising for future industrial applications. Indeed, ideal CANs should not creep at their service temperature, but be able to reshape in a matter of seconds and at moderate temperature to avoid degradation. Therefore the synthesis of CANs endowed with high activation energy and short relaxation times is desirable as these kind of CANs would demonstrate high viscosity-temperature dependence and high creep resistance.
Additionally, creep experiments were performed at 50 °C (Fig. 5B) and 80 °C (Fig. S11†) on the three BAE CANs to further demonstrate the sensitivity of the materials to temperature. After the creep-recovery experiments at 80 °C, BAE–F–OH and BAE–F showed permanent deformations of 0.16% and 0.07% respectively, whereas the deformation of BAE–OH was only of 0.03% (Fig. S11†). Hence, at 80 °C, exchange reactions occurred in BAE–F-OH and BAE–F whereas no exchange reaction took place in BAE–OH, highlighting once again the activating effect of CF3 groups. However, at 50 °C, the three BAEs showed the same small permanent deformation (<0.03%). BAE–F–OH and BAE–F were as creep resistant as BAE–OH at low temperature indicating slow exchange reactions in all three materials. However, these exchange reactions were easily set off at moderate temperature thanks to the CF3-activation, while higher temperatures, favoring degradations, were necessary to trigger these reactions in BAE–OH. These different results are in agreement with previous reports, where high activation flow energy was related to materials with strong creep resistance.29,41,55
In conclusion, the presence of fluorinated groups in these CANs clearly accelerated the aza-Michael exchange reaction and the transesterification. Moreover, a synergistic effect is at play when both aza-Michael and transesterification are complementary used. Finally, biobased CANs with high activation energies were obtained via the combination of CF3-activated (and amine catalyzed) transesterification and aza-Michael exchanges, affording higher creep resistance at low temperature (50 °C) and faster reshaping at moderate to high temperature (100 °C to 150 °C).
Reshaping
In order to demonstrate the reprocessability of the BAE networks, samples were cut into small pieces (approximately 2/2 mm square) and reshaped under different conditions using a hot press (Fig. 6). All samples were subjected to a 1 h treatment under 3 t of pressure, but the reshaping temperature was chosen accordingly to the relaxation analyses performed in the previous section. The reprocessing temperatures were 100 °C, 120 °C and 150 °C for BAE–F–OH, BAE–F and BAE–OH respectively. The three materials were reshaped three times and characterized after each reprocessing (Table S1†).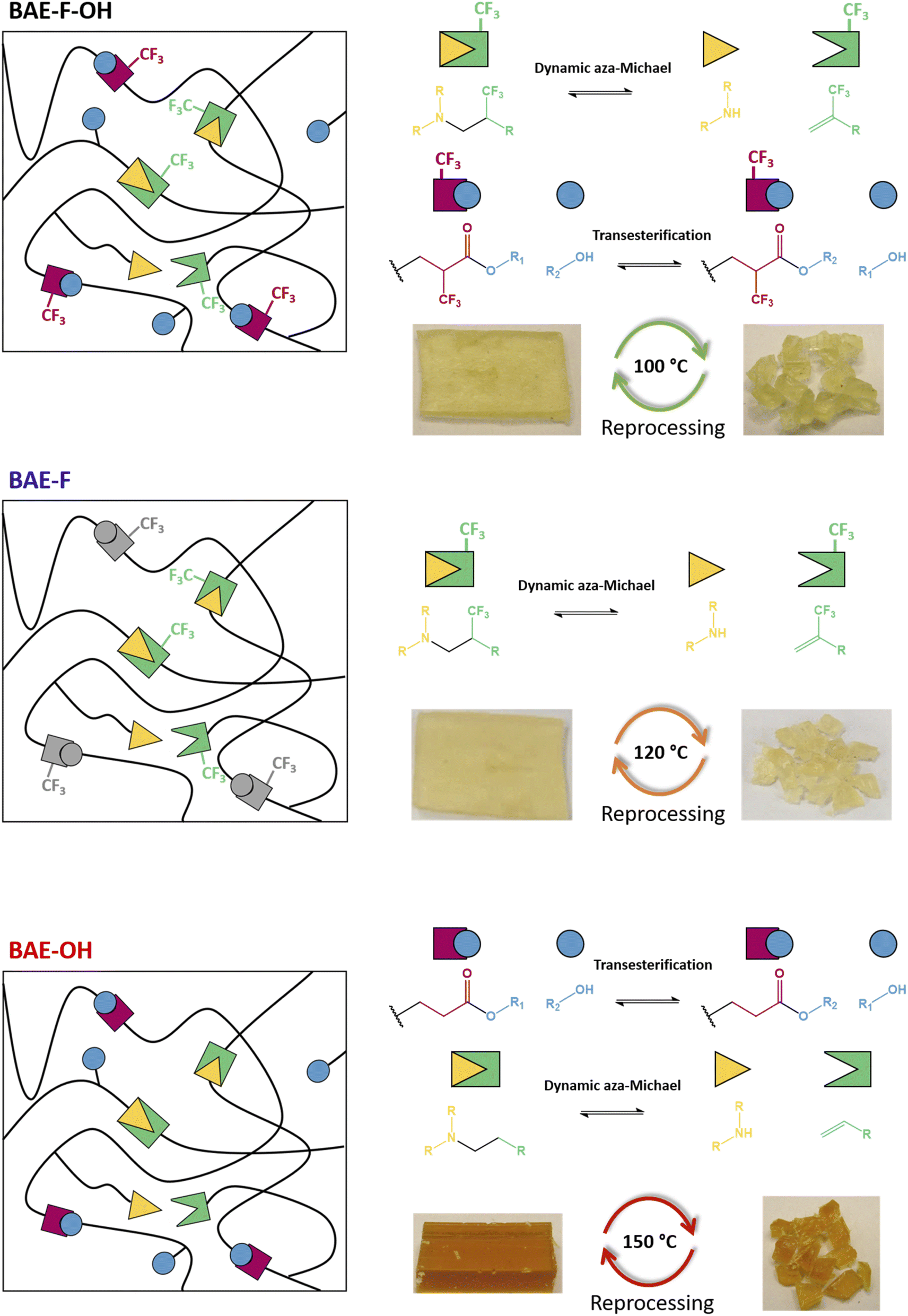 | ||
| Fig. 6 Schematic representations of BAE–F–OH, BAE–F and BAE–OH associated with their respective exchange reactions and reprocessing conditions. | ||
The TGA thermograms of the cured and reshaped samples were almost perfectly superimposable and featured an identical mass loss profile (Fig. S9†), suggesting that the materials were not structurally degraded during the reshaping process. The TGA thermograms also indicated that the thermal degradation temperature of the materials was superior to the reshaping temperature. The chemical integrity of the three networks after reprocessing was confirmed by ATR-FTIR spectroscopy (Fig. S11†). The potential formation of amides resulting from the dissociation of aza-Michael adduct followed by the attack of this amine on an ester was not observed by FTIR. Moreover, the reshaping did not significantly impact the Tg or the Tα of the BAEs networks confirming the conservation of the network structure after reshaping. The swelling index (SI) and gel content (GC) of the materials were also evaluated after each reprocessing cycle. No critical difference was observed for BAE–F–OH and BAE–F whereas the SI and GC of BAE–OH respectively increased and decreased with the number of reshaping cycles. This suggest that the degree of crosslinking decreased slightly in BAE–OH after each reshaping cycle. Finally, DMA analyses (Fig. S12†) showed that the moduli of BAE–F–OH and BAE–F were not significantly impacted by the reprocessing as observed in other CANs materials.56,57 Their values on the glassy and the rubbery plateau remained almost constant. This demonstrates that even though these CANs are partially based on dissociative reaction, there were no loss of crosslinking density upon reshaping. In contrast the moduli of BAE–OH decreased after each reshaping confirming the loss of crosslinking suggested by the evolution of the gel content and swelling index of this material. Although the reasons for this behaviour are not perfectly clear, it may be caused by thermal degradation due to a prolonged treatment at relatively high temperature compared to the others material. The difference of mechanism involved between each materials could potentially also explained this specific behaviour for BAE–OH.
Conclusion
The influence of a CF3 group on the aza-Michael addition and on the reversibility of the reaction was first highlighted on model molecules. Whereas no exchange was observed between aza-Michael adduct and acrylate at 60 °C, the introduction of a CF3 group on the acrylate double bond led to a high exchange rate (18% of exchange in 1 h) under the same conditions.Based on this preliminary molecular study, new biobased monomers derived from Pripol™ 2033 and butanediol diglycidyl ether were synthesized and used to prepare CANs containing up to 87 mol% of renewable carbons. Three CANs based on aza-Michael and/or transesterification exchange reactions were synthesized. The high reactivity of the 2-trifluoromethylacrylate moiety toward aza-Michael addition afforded easy access to catalyst-free networks at room temperature. Moreover, the synergistic effect of the aza-exchange/transesterification and the accelerating effect related to the presence of trifluoromethyl groups were separately evaluated by studying respectively non-hydroxylated and non-fluorinated materials. Indeed, hydroxyl-containing fluorinated material took only 102 s to relax 35% of the initial modulus at 120 °C whereas 912 s and 28842 s were necessary for the fluorinated only and hydroxyl-containing only materials respectively.
The synergistic combination of the two exchange reactions afforded CANs (BAE–F–OH and BAE–OH) with high activation energy reducing their creep at low temperature compared to the mono-exchange reaction material (BAE–F). The presence of fluorinated group on the α-carbon of the ester and on the β-position of the tertiary amine considerably reduced the necessary reshaping temperature from 150 °C to 100 °C, facilitating material reprocessing, without affecting the creep resistance of the material at lower temperature.
An additive-free CAN based on two different exchange reactions, both accelerated by the same activating group, was thus successfully designed and prepared, and showed remarkable reprocessing abilities. This study contributes to enrich the range of application of fluorinated groups as accelerating substituents for CANs, including overlooked reactions such as the aza-Michael exchange, and illustrates the possibility to prepare a catalyst-free CAN from biobased monomers. Such a strategy should open new opportunities to develop more sustainable materials.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was funded by the French National Research Agency ANR (AFCAN project ANR-19-CE06-0014).References
- J. P. Pascault and R. J. J. Williams, Epoxy Polymers: New Materials and Innovations, 2010, DOI:10.1002/9783527628704.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968, DOI:10.1126/science.1212648.
- J. L. Self, N. D. Dolinski, M. S. Zayas, J. Read De Alaniz and C. M. Bates, Brønsted-Acid-Catalyzed Exchange in Polyester Dynamic Covalent Networks, ACS Macro Lett., 2018, 817–821, DOI:10.1021/acsmacrolett.8b00370.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Photoinduced Plasticity in Cross-Linked Polymers, Science, 2005, 308(5728), 1615–1617, DOI:10.1126/science.1110505.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: A Molecular Platform for Designing Functions beyond Chemical Recycling and Self-Healing, Chem. Rev., 2021, 121(3), 1716–1745, DOI:10.1021/acs.chemrev.0c00938.
- Y. Yang, Y. Xu, Y. Ji and Y. Wei, Functional Epoxy Vitrimers and Composites, Prog. Mater. Sci., 2021, 120, 100710, DOI:10.1016/J.PMATSCI.2020.100710.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Vitrimers: Current Research Trends and Their Emerging Applications, Mater. Today, 2021, 51, 586–625, DOI:10.1016/J.MATTOD.2021.07.003.
- N. J. Van Zee and R. V. Nicolaÿ, Permanently Crosslinked Polymers with Dynamic Network Topology, Prog. Polym. Sci., 2020, 104, 101233, DOI:10.1016/J.PROGPOLYMSCI.2020.101233.
- J. M. Winne, L. Leibler and F. E. Du Prez, Dynamic Covalent Chemistry in Polymer Networks: A Mechanistic Perspective, Polym. Chem., 2019, 10(45), 6091–6108, 10.1039/C9PY01260E.
- F. I. Altuna, U. Casado, I. E. Dell'Erba, L. Luna, C. E. Hoppe and R. J. J. Williams, Epoxy Vitrimers Incorporating Physical Crosslinks Produced by Self-Association of Alkyl Chains-SI, Polym. Chem., 2020, 1337–1347, 10.1039/c9py01787a.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Shape Memory Epoxy Vitrimers Based on DGEBA Crosslinked with Dicarboxylic Acids and Their Blends with Citric Acid, RSC Adv., 2016, 6(91), 88647–88655, 10.1039/c6ra18010h.
- M. Hayashi, R. Yano and A. Takasu, Synthesis of Amorphous Low: T g Polyesters with Multiple COOH Side Groups and Their Utilization for Elastomeric Vitrimers Based on Post-Polymerization Cross-Linking, Polym. Chem., 2019, 10(16), 2047–2056, 10.1039/c9py00293f.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Vinylogous Urethane Vitrimers, Adv. Funct. Mater., 2015, 25(16), 2451–2457, DOI:10.1002/adfm.201404553.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Influence of the Polymer Matrix on the Viscoelastic Behaviour of Vitrimers, Polym. Chem., 2020, 11(33), 5377–5385, 10.1039/d0py00114g.
- A. Chao, I. Negulescu and D. Zhang, Dynamic Covalent Polymer Networks Based on Degenerative Imine Bond Exchange: Tuning the Malleability and Self-Healing Properties by Solvent, Macromolecules, 2016, 49(17), 6277–6284, DOI:10.1021/ACS.MACROMOL.6B01443/ASSET/IMAGES/LARGE/MA-2016-01443X_0008.JPEG.
- G. Zhang, Q. Zhao, L. Yang, W. Zou, X. Xi and T. Xie, Exploring Dynamic Equilibrium of Diels–Alder Reaction for Solid State Plasticity in Remoldable Shape Memory Polymer Network, ACS Macro Lett., 2016, 5(7), 805–808, DOI:10.1021/acsmacrolett.6b00357.
- X. Kuang, G. Liu, X. Dong and D. Wang, Correlation between Stress Relaxation Dynamics and Thermochemistry for Covalent Adaptive Networks Polymers, Mater. Chem. Front., 2016, 1(1), 111–118, 10.1039/C6QM00094K.
- D. Pratchayanan, J.-C. Yang, C. L. Lewis, N. Thoppey and M. Anthamatten, Thermomechanical Insight into the Reconfiguration of Diels–Alder Networks, J. Rheol., 2017, 61(6), 1359–1367, DOI:10.1122/1.4997580.
- L. Zhang and S. J. Rowan, Effect of Sterics and Degree of Cross-Linking on the Mechanical Properties of Dynamic Poly(Alkylurea–Urethane) Networks, Macromolecules, 2017, 50(13), 5051–5060, DOI:10.1021/acs.macromol.7b01016.
- M. Hayashi and L. Chen, Functionalization of Triblock Copolymer Elastomers by Cross-Linking the End Blocks via Trans-N -Alkylation-Based Exchangeable Bonds, Polym. Chem., 2020, 11(10), 1713–1719, 10.1039/C9PY01759C.
- P. Chakma, C. N. Morley, J. L. Sparks and D. Konkolewicz, Exploring How Vitrimer-like Properties Can Be Achieved from Dissociative Exchange in Anilinium Salts, Macromolecules, 2020, 53(4), 1233–1244, DOI:10.1021/acs.macromol.0c00120.
- B. R. Elling and W. R. Dichtel, Reprocessable Cross-Linked Polymer Networks: Are Associative Exchange Mechanisms Desirable?, ACS Cent. Sci., 2020, 6(9), 1488–1496, DOI:10.1021/acscentsci.0c00567.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets, J. Am. Chem. Soc., 2012, 134(18), 7664–7667, DOI:10.1021/ja302894k.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, Catalytic Control of the Vitrimer Glass Transition, ACS Macro Lett., 2012, 1(7), 789–792, DOI:10.1021/mz300239f.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. J. Qi, Reprocessing and Recycling of Thermosetting Polymers Based on Bond Exchange Reactions, RSC Adv., 2014, 4(20), 10108–10117, 10.1039/c3ra47438k.
- J. P. Brutman, P. A. Delgado and M. A. P. HillmyerVitrimers, ACS Macro Lett., 2014, 3(7), 607–610, DOI:10.1021/mz500269w.
- Z. Yang, Q. Wang and T. Wang, Dual-Triggered and Thermally Reconfigurable Shape Memory Graphene-Vitrimer Composites, ACS Appl. Mater. Interfaces, 2016, 8, 37, DOI:10.1021/acsami.6b07403.
- T. Liu, B. Zhao and J. Zhang, Recent Development of Repairable, Malleable and Recyclable Thermosetting Polymers through Dynamic Transesterification, Polymer, 2020, 194, 122392, DOI:10.1016/j.polymer.2020.122392.
- M. Guerre, C. Taplan, J. M. Winne and F. E. Du Prez, Vitrimers: Directing Chemical Reactivity to Control Material Properties, Chem. Sci., 2020, 11(19), 4855–4870, 10.1039/D0SC01069C.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Neighboring Group Participation and Internal Catalysis Effects on Exchangeable Covalent Bonds: Application to the Thriving Field of Vitrimer Chemistry, Macromolecules, 2021, 54(9), 3927–3961, DOI:10.1021/acs.macromol.0c02706.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, A Catalyst-Free Epoxy Vitrimer System Based on Multifunctional Hyperbranched Polymer, Macromolecules, 2018, 51(17), 6789–6799, DOI:10.1021/acs.macromol.8b01424.
- T. Liu, S. Zhang, C. Hao, C. Verdi, W. Liu, H. Liu and J. Zhang, Glycerol Induced Catalyst-Free Curing of Epoxy and Vitrimer Preparation, Macromol. Rapid Commun., 2019, 40(7), 1800889, DOI:10.1002/marc.201800889.
- F. I. Altuna, V. Pettarin and R. J. J. Williams, Self-Healable Polymer Networks Based on the Cross-Linking of Epoxidised Soybean Oil by an Aqueous Citric Acid Solution, Green Chem., 2013, 15(12), 3360–3366, 10.1039/c3gc41384e.
- M. Delahaye, J. M. Winne and F. E. Du Prez, Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry, J. Am. Chem. Soc., 2019, 15277–15287, DOI:10.1021/jacs.9b07269.
- M. Delahaye, F. Tanini, J. O. Holloway, J. M. Winne and F. E. Du Prez, Double Neighbouring Group Participation for Ultrafast Exchange in Phthalate Monoester Networks, Polym. Chem., 2020, 11(32), 5207–5215, 10.1039/d0py00681e.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, Silyl Ether as a Robust and Thermally Stable Dynamic Covalent Motif for Malleable Polymer Design, J. Am. Chem. Soc., 2017, 139(42), 14881–14884, DOI:10.1021/jacs.7b08826.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Epoxy Vitrimers with a Covalently Bonded Tertiary Amine as Catalyst of the Transesterification Reaction, Eur. Polym. J., 2019, 113, 297–304, DOI:10.1016/j.eurpolymj.2019.01.045.
- D. Berne, F. Cuminet, S. Lemouzy, C. Joly-Duhamel, R. Poli, S. Caillol, E. Leclerc and V. Ladmiral, Catalyst-Free Epoxy Vitrimers Based on Transesterification Internally Activated by an α–CF 3 Group, Macromolecules, 2022, 55(5), 1669–1679, DOI:10.1021/acs.macromol.1c02538.
- F. Cuminet, D. Berne, S. Lemouzy, E. Dantras, C. Joly-Duhamel, S. Caillol, E. Leclerc and V. Ladmiral, Catalyst-Free Transesterification Vitrimers: Activation via α -Difluoroesters, Polym. Chem., 2022, 8, 5255–5446, 10.1039/D2PY00124A.
- S. Lemouzy, F. Cuminet, D. Berne, S. Caillol, V. Ladmiral, R. Poli and E. Leclerc, Understanding the Reshaping of Fluorinated Polyester Vitrimers by Kinetic and DFT Studies of the Transesterification Reaction, Chem. – Eur. J., 2022, 28(48), e202201135 CrossRef CAS PubMed.
- C. Taplan, M. Guerre and F. E. Du Prez, Covalent Adaptable Networks Using β-Amino Esters as Thermally Reversible Building Blocks, J. Am. Chem. Soc., 2021, 143(24), 9140–9150, DOI:10.1021/jacs.1c03316.
- D. Berne, G. Coste, R. Morales, M. Boursier, J. Pinaud, V. Ladmiral and S. Caillol, Taking Advantage of β-Hydroxy Amine Enhanced Reactivity and Functionality for the Synthesis of Dual Covalent Adaptable Networks, Polym. Chem., 2022, 8, 5255–5446, 10.1039/D2PY00274D.
- T. P. Vasil’eva, A. F. Kolomiets, E. I. Mysov and A. V. Fokin, Comparison of α- and β-Trifluoromethylsubstituted Acrylic Acids in Their Reactions with Thiols, Russ. Chem. Bull., 1997, 46(6), 1181–1183, DOI:10.1007/BF02496227.
- B. Chen, J. Lei and J. Zhao, Michael Addition of Aryl Thiols to 3-(2,2,2-Trifluoroethylidene)Oxindoles under Catalyst-Free Conditions: The Rapid Synthesis of Sulfur-Containing Oxindole Derivatives, J. Chem. Res., 2018, 42(4), 210–214, DOI:10.3184/174751918X15240724383170.
- A. Forte, A. Zucaro, R. Basosi and A. Fierro, LCA of 1,4-Butanediol Produced via Direct Fermentation of Sugars from Wheat Straw Feedstock within a Territorial Biorefinery, Materials, 2016, 9(7), 1–22, DOI:10.3390/MA9070563.
- A. Canela-Xandri, M. Balcells, G. Villorbina, P. Christou and R. Canela-Garayoa, Preparation and Uses of Chlorinated Glycerol Derivatives, Molecules, 2020, 25(11), 2511 CrossRef CAS PubMed.
- A.-S. Mora, R. Tayouo, B. Boutevin, G. David and S. Caillol, Vanillin-Derived Amines for Bio-Based Thermosets, Green Chem., 2018, 20(17), 4075–4084, 10.1039/C8GC02006J.
- B. Quienne, R. Poli, J. Pinaud and S. Caillol, Enhanced Aminolysis of Cyclic Carbonates by β-Hydroxylamines for the Production of Fully Biobased Polyhydroxyurethanes, Green Chem., 2021, 23(4), 1678–1690, 10.1039/D0GC04120C.
- J. Liu, S. Wang, Y. Peng, J. Zhu, W. Zhao and X. Liu, Advances in Sustainable Thermosetting Resins: From Renewable Feedstock to High Performance and Recyclability, Prog. Polym. Sci., 2021, 113, 101353, DOI:10.1016/J.PROGPOLYMSCI.2020.101353.
- M. Podgórski, N. Spurgin, S. Mavila and C. N. Bowman, Mixed Mechanisms of Bond Exchange in Covalent Adaptable Networks: Monitoring the Contribution of Reversible Exchange and Reversible Addition in Thiol–Succinic Anhydride Dynamic Networks, Polym. Chem., 2020, 11(33), 5365–5376, 10.1039/d0py00091d.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, Rapid Stress Relaxation and Moderate Temperature of Malleability Enabled by the Synergy of Disulfide Metathesis and Carboxylate Transesterification in Epoxy Vitrimers, ACS Macro Lett., 2019, 8(3), 255–260, DOI:10.1021/acsmacrolett.9b00015.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–Yne Cross-Linking, Angew. Chem., Int. Ed., 2020, 59(9), 3609–3617, DOI:10.1002/anie.201912902.
- J. O. Holloway, C. Taplan and F. E. Du. Prez, Combining Vinylogous Urethane and β-Amino Ester Chemistry for Dynamic Material Design, Polym. Chem., 2022, 13(14), 2008–2018 RSC.
- Z. Liu, Y. Ma, Z. Zhang, Z. Shi and J. Gao, Rapid Stress Relaxation, Multistimuli-Responsive Elastomer Based on Dual-Dynamic Covalent Bonds and Aniline Trimer, Langmuir, 2022, 38(16), 4812–4819, DOI:10.1021/ACS.LANGMUIR.1C03241.
- L. Li, X. Chen, K. Jin, M. B. Rusayyis and J. M. Torkelson, Arresting Elevated-Temperature Creep and Achieving Full Cross-Link Density Recovery in Reprocessable Polymer Networks and Network Composites via Nitroxide-Mediated Dynamic Chemistry, Macromolecules, 2021, 54(3), 1452–1464, DOI:10.1021/acs.macromol.0c01691.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Catalyst-Free Vitrimers from Vinyl Polymers, Macromolecules, 2019, 52(5), 2105–2111, DOI:10.1021/acs.macromol.8b02477.
- S. Debnath, S. Kaushal and U. Ojha, Catalyst-Free Partially Bio-Based Polyester Vitrimers, ACS Appl. Polym. Mater., 2020, 2(2), 1006–1013, DOI:10.1021/acsapm.0c00016.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta05067f |
| This journal is © The Royal Society of Chemistry 2022 |