
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous non-invasive gas analysis in artificial photosynthesis reactions using rotational Raman spectroscopy†
Jesper
Schwarz‡
 a,
Aleksandra
Ilic‡
a,
Simon
Kaufhold
a,
Jussi
Ahokas
b,
Pasi
Myllyperkiö
b,
Mika
Pettersson
b and
Kenneth
Wärnmark
*a
a,
Aleksandra
Ilic‡
a,
Simon
Kaufhold
a,
Jussi
Ahokas
b,
Pasi
Myllyperkiö
b,
Mika
Pettersson
b and
Kenneth
Wärnmark
*a
aCentre for Analysis and Synthesis (CAS), Department of Chemistry, Lund University, SE-22100 Lund, Sweden. E-mail: kenneth.wärnmark@chem.lu.se
bDepartment of Chemistry, Nanoscience Center, University of Jyväskylä, FI-40014, Finland
First published on 25th August 2022
Abstract
Optimising reactions in artificial photosynthesis research requires screening of many reaction and operation parameters, which is often resource-intense and time-consuming. In this paper, we demonstrate the use of a rotational Raman-based spectrometer for non-invasive quantification of several gases (H2, O2, N2, CO, CO2) with short analysis times (15 s), enabling high throughput screening. Furthermore, with this device, reaction progress can be monitored in situ, by real-time simultaneous quantification of multiple gases. We have applied this instrument and developed a method to study the O2 dependency of a prototypic light-driven hydrogen evolution reaction, showcasing the value of this approach for the artificial photosynthesis community in general.
The sustainable transformation of our society necessitates the shift from fossil resources to renewable feedstocks. Electrification, as well as a H2- and circular carbon economy, will play a pivotal part in this process. Inspired by nature, researchers in chemical, biological and materials sciences have set out to contribute to this transformation by investigating artificial photosynthesis (AP) approaches.1–3 AP aims to produce high-energy chemicals like H2 (and O2) or e.g. CO, formate or methanol, from abundant resources like H2O, CO2 and light. However, extensive screening for ideal reaction/operation conditions is often necessary due to the complexity of many AP systems. Additionally, synthesis or preparation of the (light-driven) catalysts or the phototropic microorganisms/enzymes is often laborious and resource-intense. Therefore, ways of carrying out these optimisation reactions on a small scale so as to minimise sample consumption are highly sought-after.
Common methods for gas analysis used in AP research labs include (micro) gas chromatography (GC),4 mass spectrometry (MS),5 (Clark-type) electrode sensors6,7 or optical O2 sensors.8 While some of these methods have high sensitivity (ppm range) and fast measurement times (μs–ms), apart from optical sensors,8 they usually require penetration of the reaction vessel to remove a sample volume or bring the sensor into contact with the sample. This holds the risk of leakage and/or contamination of the reaction medium or even consumption of non-negligible amounts of the gas sample, which in turn interferes with fast and reliable analysis and reaction screening. From our observation, Clark-type electrode sensors can face stability issues in reaction media containing organic solvents and additives like amines, thus potentially giving incorrect values. Similarly, for GC in AP, trapping columns are often needed to avoid deterioration of the GC column.
To overcome these constraints, we investigated the use of a device based on rotational Raman spectroscopy for the non-invasive analysis of H2, O2 and N2, and potentially CO and CO2, with 1% relative standard deviation (RSD) at 10s of seconds measurement times. Specifically, we looked at the simultaneous analysis of H2 and O2 in a known light-driven hydrogen evolution reaction (HER) system to exemplify the utility for developing AP systems. Raman spectroscopy of gases9,10 has in recent years been improved through the use of Fibre Enhanced Raman Spectroscopy (FERS)11 and Cavity Enhanced Raman Spectroscopy (CERS).12 These techniques increase the pathlength of the light, thus increasing the signal strength, which has enabled the analysis of gases such as H2 and hydrocarbons down to ppm levels.13 However, the drawback of these systems is also that they require physical sampling of the gaseous analyte. In comparison to absorption-based techniques for gas analysis, such as IR, Raman spectroscopy is still far less common, because the signal strength is orders of magnitudes lower. A distinct advantage of Raman spectroscopy over IR spectroscopy is that IR inactive molecules such as nitrogen can be detected.
A new Raman gas analyser, developed by us, utilises the back-scattering geometry and rotational Raman spectrum of gaseous molecules.14 This geometry enables a simple optical layout where a single lens focuses the incident light to the sample and collects the Raman scattering (Fig. 1). In this way, gases in an optically transparent, closed container – e.g. glass vials – can be analysed non-invasively. Laser light travels from a source (CNI Laser MLL-U-532-200-3) to a primary optical path with mirrors and an edge filter (Semrock 532 nm RazorEdge® ultra-steep). The primary optical path goes through the midpoint of the edge filter. The edge filter reflects the laser light to the lens (focal length 50 mm). The lens focuses the light on a sample and then collects the Raman scattering from the sample before collimating it along the primary optical path. The Stokes part of the Raman scattering passes through the edge filter. The second edge filter is placed perpendicularly to the primary optical axis to filter out the remaining laser light from the Raman scattering. Then the scattered light is focused on the slit (∼100–200 μm) with a lens (focal length 175 mm). A lens (focal length 350 mm) collimates the signal before the volume-phase holographic transmission grating (Wasatch Photonics, 2800 L mm−1). After dispersing the signal, a lens (focal length 350 mm) focuses the signal on a CCD array (Andor iDus 401, 1024 × 127 pixel). The signal is collected with a custom-made software and analysed using library spectra of pure gases and a multicomponent optimisation procedure. The rotational Raman spectrum is rich in information, and many gases of interest in AP research (H2, O2, N2, CO, CO2) possess a rotational spectrum in a narrow spectral range that allows tailoring of a high sensitivity and high-resolution spectrometer in a fixed geometry. The spectra for O2 and H2 are completely separated, which is not a requisite, but is highly advantageous as it facilitates simultaneous measurement of both gases (Fig. 2).
 | ||
| Fig. 1 Left: technical drawing of the Raman-based gas analyser. M = mirror, L = lens, F = edge filter, W = CaF2 window, S = sample. Right: photo from above of the Raman gas analyser with the top cover removed. | ||
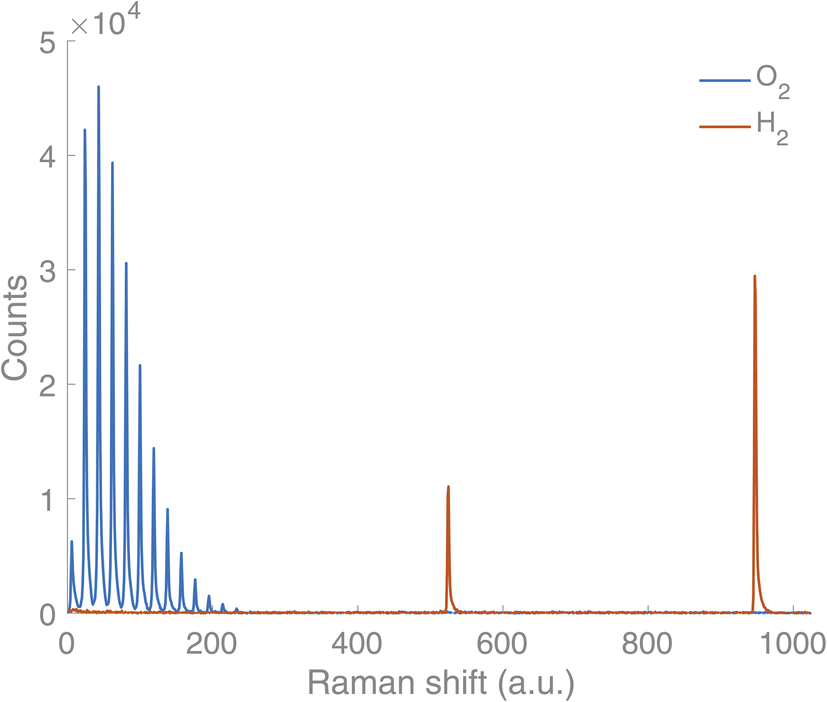 | ||
| Fig. 2 Spectral signature of O2 (blue) and H2 (orange) from the Raman gas analyser. | ||
The analysis of the collected spectrum is based on solving weight factors for the predefined calibration spectra, i.e. solving a set of simultaneous linear equations. In practice, there is also a need for a correction of broad luminescence from the sample vials (low order polynomial), scattering from the air in the light-path inside the instrument, and laser power and wavelength changes during long experiments or between the calibration and sample measurements (linear scaling factors resolved from the internal calibration signal). Those corrections are implemented in the analysis routine. This method allows for simultaneous and precise analysis of gases having strongly overlapping rotational Raman spectra, like for O2, N2, CO and CO2, but also spectrally separated species like H2. The device can analyse gas mixtures where constituents' gas contents vary between 0.5% and 100%. The lower limit is dependent on gas components and experimental conditions. For calibration of the instrument, an argon-filled glass vial (0-point) and a vial filled with the analyte gas (100%-point) were used.
Here, we demonstrate the use of this rotational Raman-based spectrometer for the non-invasive simultaneous real-time monitoring of H2 and O2. The system can quantify gases in small volumes, with each measurement taking 15 s, which enables high throughput screening of many reaction parameters. In order to establish the reliability of the instrument, we recorded calibration curves for H2 and O2. The data for the calibration curves were recorded by flushing a 4.9 mL glass vial with argon (which is Raman silent), followed by the addition of a known quantity of the gas analyte using a gas-tight syringe (Hamilton). The calibration curves showed excellent precision for H2, with R2 > 0.999 for concentrations in the range 40–5000 μM H2 in Ar, and R2 = 0.998 up to 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 μM (Fig. S1–S4†). Furthermore, the accuracy of the instrument was evaluated against reference measurements using conventional Clark-type electrodes (Unisense). This showed that measurements were accurate within 3.5% relative to the Clark-type electrodes. The O2 calibration curves coincided equally well with the Clark-type electrodes (0–8000 μmol L−1, R2 = 0.9994, Fig. S5†). In addition to H2 and O2, N2, CO and CO2 can be measured (Fig. S8–S10†). Quantification of CO and CO2 is of particular interest in CO2-reduction reactions. Although the spectra of these two gases partially overlap, they are sufficiently different so that they can be simultaneously reliably analysed (Fig. S11†). The distance between the spectral lines, as well as the intensities, are different for each gas.15 We have recorded a calibration curve of different concentrations of CO in CO2 (0–6000 μmol L−1, R2 = 0.9985, Fig. S6†) demonstrating that this instrument is potentially applicable to other AP reactions than HERs. In the same manner, O2 and N2 can be separated (Fig. S7†).
000 μM (Fig. S1–S4†). Furthermore, the accuracy of the instrument was evaluated against reference measurements using conventional Clark-type electrodes (Unisense). This showed that measurements were accurate within 3.5% relative to the Clark-type electrodes. The O2 calibration curves coincided equally well with the Clark-type electrodes (0–8000 μmol L−1, R2 = 0.9994, Fig. S5†). In addition to H2 and O2, N2, CO and CO2 can be measured (Fig. S8–S10†). Quantification of CO and CO2 is of particular interest in CO2-reduction reactions. Although the spectra of these two gases partially overlap, they are sufficiently different so that they can be simultaneously reliably analysed (Fig. S11†). The distance between the spectral lines, as well as the intensities, are different for each gas.15 We have recorded a calibration curve of different concentrations of CO in CO2 (0–6000 μmol L−1, R2 = 0.9985, Fig. S6†) demonstrating that this instrument is potentially applicable to other AP reactions than HERs. In the same manner, O2 and N2 can be separated (Fig. S7†).
We then went on to study a conventional light-driven HER to gain further insight into the progression of the reaction and to test the system in a realistic setting. As a model HER, we chose the high turnover three component system developed by Leung et al.16 The system consists of the photosensitiser (PS) [IrIII(dF(CF3)ppy)2(dtbbpy)]PF6 (0.03 mM),17 the proton reduction catalyst (PRC) [CoII(qpy)(OH2)2](ClO4) (qpy = 2,2′:6′,2′′:6′′,2′′′-quaterpyridine)16 (0.6 mM), the sacrificial reductant triethanolamine (TEOA) (0.2 M) and water (5% v/v) in acetonitrile (2 mL). Before irradiation of the solution, it was purged with argon. The reactions were carried out in 4.9 mL glass vials with screw cap septum lids (silicone/PTFE) using LED irradiation (λ = 375 nm, 45 mW) from below the vials. We have also used blue and green LED irradiation (λ = 450 nm and λ = 525 nm), without seeing any influence on the measurements, indicating that light sources placed below the sample do not interfere with the gas analysis. Initial simultaneous measurement of the H2 production by Clark-type electrodes and the Raman gas analyser showed coinciding curves for the H2 content in the headspace (Fig. 3).
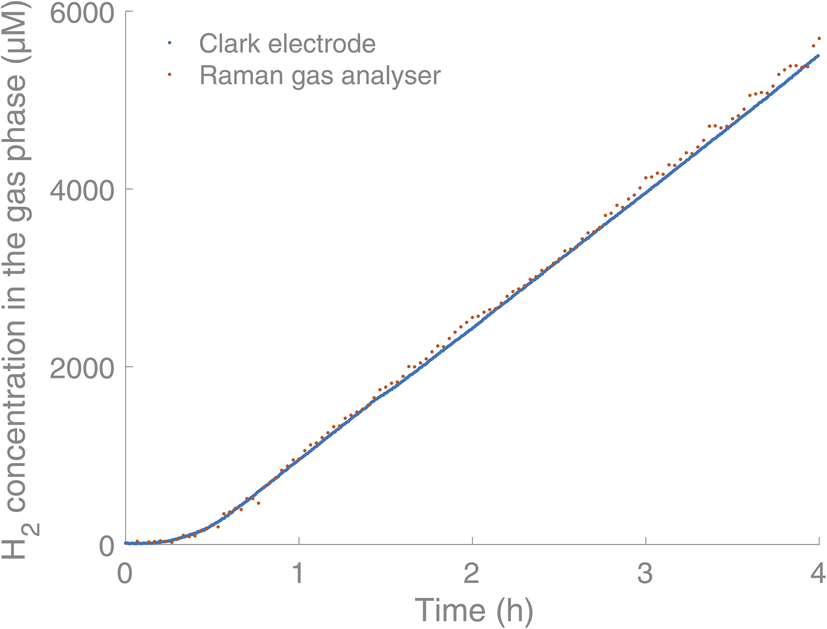 | ||
| Fig. 3 Comparison between the Raman gas analyser (orange) and Clark electrodes (blue, Unisense) in real-time monitoring of H2 in the headspace of a HER. Experimental conditions: [IrIII(dF(CF3)ppy)2(dtbbpy)]PF6 (0.03 mM),17 [CoII(qpy)(OH2)2](ClO4)16 (0.6 mM), TEOA (0.2 M) and water (5% v/v) in acetonitrile (2 mL). | ||
Subsequently, we were interested in exploring the effect of O2 on this HER. We were able to run the reaction and monitor the H2 and O2 content of the headspace in real-time during the irradiation period. In the absence of O2, the H2 formation started after an induction period of 40 minutes. We then proceeded on to add varying amounts of O2, with a gas-tight syringe through the septum, before irradiation. When O2 was added, we saw an initial consumption of O2 before the H2 formation started, and the induction period for H2 formation was increased, from 2 h upon addition of 100 μL of O2 and up to 4 h upon addition of 500 μL of O2 (Fig. 4). H2 formation started once the O2 level fell below 300 μM in the gas phase, while the O2 level continued decreasing down to near zero.
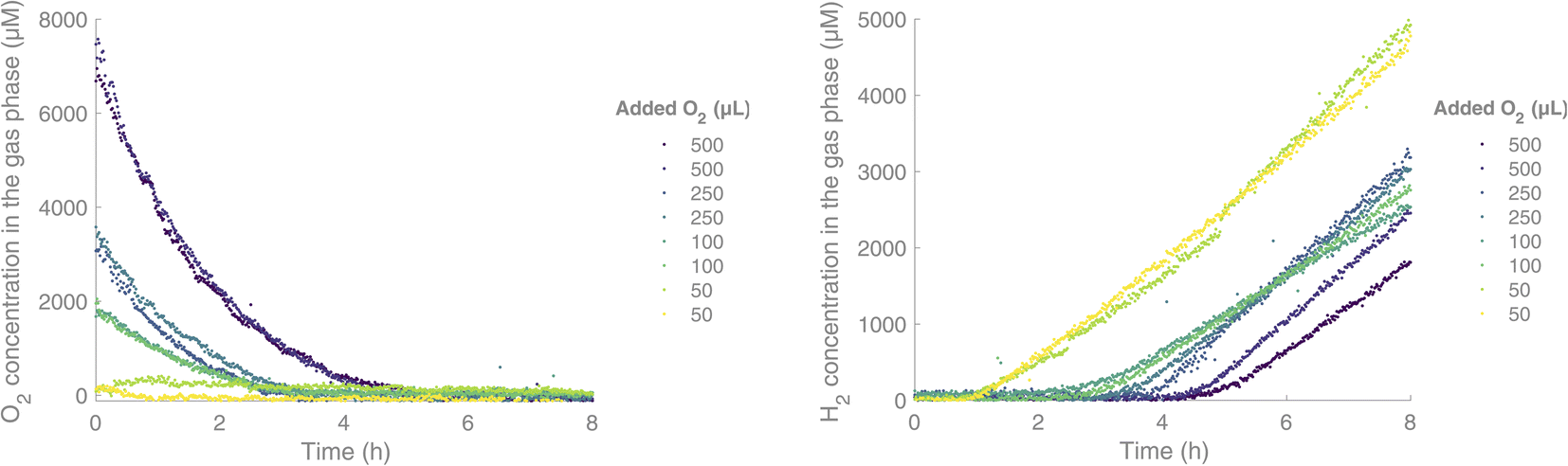 | ||
| Fig. 4 Simultaneous measurement of O2 (left) and H2 (right) in a H2 formation reaction, under Ar (g) with different added amounts of O2 initially. Experimental conditions: [IrIII(dF(CF3)ppy)2(dtbbpy)]PF6 (0.03 mM),17 [CoII(qpy)(OH2)2](ClO4)16 (0.6 mM), TEOA (0.2 M) and water (5% v/v) in acetonitrile (2 mL). | ||
By fitting the O2 concentration to a monoexponential decay, rate constants for the consumption of O2 could be obtained. These showed similar rate constants independent of the initial concentration of O2. For a starting concentration of 7600 μM of O2 in the headspace (addition of 500 μL of O2), the rate constant k was 1.84 × 10−4 s−1. For a starting concentration of 3600 μM and 1900 μM of O2 respectively, rate constants of k = 2.54 × 10−4 s−1 and k = 2.00 × 10−4 s−1 were obtained. The presence of O2 together with a PS with a long-lived triplet exited state is known to induce the formation of reactive oxygen species like singlet oxygen through energy transfer.18,19 Alternatively, the oxygen could be consumed through reduction to H2O or H2O2.20–22 The reactive oxygen species could, in turn, degrade the different components of the catalytic system. At the end of all the HERs performed using this system, with or without added O2, a black suspension/precipitate formed, indicating a reduction of the cobalt catalyst to Co(0) and eventual particle formation. Small changes in this process could also explain the different rates of H2 formation for the high O2-concentration samples. As seen in Fig. 4, the gas analyser provides simultaneous real-time data of both the H2 and O2 concentrations in the headspace of the vial. For both gases, a few outliers can be observed in the curves. In longer measurements (>1000 datapoints) these usually occur a few times, due to cosmic rays hitting the CCD detector.23 They can easily be omitted by fitting the data, but we have chosen to show the raw data including these artefacts for the sake of transparency. From this real-time monitoring of the two gases in parallel, we conclude that no H2 formation is possible until the O2 concentration has reached very low levels.
In addition to enabling the simultaneous quantification of gases, we found that efficient screening of reaction parameters is facile when using this instrument. This can primarily be attributed to two of its inherent advantages, as was shown in the recent development of an HER using an iron PS in combination with different proton reduction catalysts.24 There, a measurement time of 25 s was used and three replicates were recorded, resulting in a total measurement time of less than 1.5 min per sample, while obtaining reliable data for each measurement (RSD = 1%). As a result of the short analysis time, the main factor limiting the number of reactions performed and tracked in parallel when choosing not to trace in situ is the irradiation set-up itself, wherein only a certain number of reaction vials can be irradiated simultaneously, and not the analysis method. Furthermore, the non-invasive measurement by the gas analyser made repeated, continuous sample-taking from the same reaction possible without risking leakage induced by puncturing.
Additionally to being non-invasive, the quantification of the gas content using very small volumes of reaction solution (2 mL) in reaction vessels with a total volume of 4.9 mL (incl. headspace) is possible, both of which result in a highly economic way of optimising reaction conditions. The use of such a non-invasive set-up that allows for small-scale testing of reaction parameters thus leads to minimal wasting of precious materials (PSs, catalysts) in the search for ideal conditions. Finally, the comparably small reaction volumes also result in less organic solvent being required for the individual reactions, which is more cost-efficient and lowers the environmental impact of the optimisation.
In conclusion, we have demonstrated a rotational Raman-based spectrometer as a non-invasive method for real-time simultaneous analysis of gases in small volumes. While all Raman active gases (H2, O2, N2, CO, CO2) can be measured, we have particularly shown the O2-concentration dependency of the onset of a common HER. This serves as one example of how this technique could considerably impact the future investigation and development of AP systems. We envision that this method will enable high throughput screening in AP, resulting in this very promising and impactful field advancing more rapidly. Additionally, the ability to analyse the composition of the gas phase in real time, combined with other in situ techniques, can hopefully provide deeper insight into the kinetics of AP reactions further facilitating optimisation of the studied systems.25
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Jens Uhlig and Sarah Klingler are acknowledged for technical assistance and discussions. KW would like to thank the Swedish Foundation for Strategic Research (SSF, EM16-0067), the Knut and Alice Wallenberg Foundation (KAW, 2018.0074), the Swedish Research Council (VR, 2020-03207), the Swedish Energy Agency (Energimyndigheten), the LMK Foundation and the Sten K Johnson Foundation for financial support. SK acknowledges support from Wenner-Gren Stiftelserna and the Royal Physiographic Society of Lund.References
- S. Berardi, S. Drouet, L. Francàs, C. Gimbert-Suriñach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC
.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946 CrossRef CAS PubMed
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216 RSC
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Inorg. Chem., 2009, 48, 4952 CrossRef CAS PubMed
- H. Park, H.-H. Ou, U. Kang, J. Choi and M. R. Hoffmann, Catal. Today, 2016, 266, 153 CrossRef CAS
- L. Li, L. Duan, F. Wen, C. Li, M. Wang, A. Hagfeldt and L. Sun, Chem. Commun., 2012, 48, 988 RSC
- A. C. Sander, S. Maji, L. Francàs, T. Böhnisch, S. Dechert, A. Llobet and F. Meyer, ChemSusChem, 2015, 8, 1697 CrossRef CAS PubMed
- F. L. Huber, S. Amthor, B. Schwarz, B. Mizaikoff, C. Streb and S. Rau, Sustain. Energy Fuels, 2018, 2, 1974 RSC
- R. W. Wood, Nature, 1929, 123, 279 CrossRef CAS
- D. Schiel and W. Richter, Fresenius' Z. Anal. Chem., 1987, 327, 335 CrossRef CAS
- A. Knebl, D. Yan, J. Popp and T. Frosch, TrAC, Trends Anal. Chem., 2018, 103, 230 CrossRef CAS
- C. Niklas, H. Wackerbarth and G. Ctistis, Sensors, 2021, 21, 1 CrossRef PubMed
- A. Knebl, C. Domes, R. Domes, S. Wolf, J. Popp and T. Frosch, Anal. Chem., 2021, 93, 10546 CrossRef CAS PubMed
-
J. Ahokas and M. Pettersson, WO 2013/079806 A1, 2013
- L. C. Hoskins, J. Chem. Educ., 1975, 52, 568 CrossRef CAS
- C.-F. Leung, S.-M. Ng, C.-C. Ko, W.-L. Man, J. Wu, L. Chen and T.-C. Lau, Energy Environ. Sci., 2012, 5, 7903 RSC
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS
- T. Tachikawa and T. Majima, Langmuir, 2009, 25, 7791 CrossRef CAS PubMed
- B. Tian, W. Gao, X. Ning, Y. Wu and G. Lu, Appl. Catal., B, 2019, 249, 138 CrossRef CAS
- X. Guo, X. Li, X. C. Liu, P. Li, Z. Yao, J. Li, W. Zhang, J. P. Zhang, D. Xue and R. Cao, Chem. Commun., 2018, 54, 845 RSC
- C. Costentin, H. Dridi and J.-M. Savéant, J. Am. Chem. Soc., 2015, 137, 13535 CrossRef CAS PubMed
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800 CrossRef CAS PubMed
- D. Groom, Exp. Astron., 2002, 14, 45 CrossRef
- J. Schwarz, A. Ilic, C. Johnson, R. Lomoth and K. Wärnmark, Chem. Commun., 2022, 58, 5351 RSC
- S. Klingler, J. Hniopek, R. Stach, M. Schmitt, J. Popp and B. Mizaikoff, ACS Meas. Sci. Au, 2022, 2, 157 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2se01119k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |