
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO reductive oligomerization by a divalent thulium complex and CO2-induced functionalization†
Thomas
Simler
 *a,
Karl N.
McCabe
b,
Laurent
Maron
b and
Grégory
Nocton
*a
*a,
Karl N.
McCabe
b,
Laurent
Maron
b and
Grégory
Nocton
*a
aLCM, CNRS, Ecole Polytechnique, Institut Polytechnique de Paris, Route de Saclay, Palaiseau 91120, France. E-mail: gregory.nocton@polytechnique.edu; thomas.simler@polytechnique.edu
bLPCNO, UMR 5215, Université de Toulouse-CNRS, INSA, UPS, Toulouse, France
First published on 9th May 2022
Abstract
The divalent thulium complex [Tm(Cpttt)2] (Cpttt = 1,2,4-tris(tert-butyl)cyclopentadienyl) reacts with CO to afford selective CO reductive dimerization and trimerization into ethynediolate (C2) and ketenecarboxylate (C3) complexes, respectively. DFT calculations were performed to shed light on the elementary steps of CO homologation and support a stepwise chain growth. The attempted decoordination of the ethynediolate fragment by treatment with Me3SiI led to dimerization and rearrangement into a 3,4-dihydroxyfuran-2-one complex. Investigation of the reactivity of the C2 and C3 complexes towards other electrophiles led to unusual functionalization reactions: while the reaction of the ketenecarboxylate C3 complex with electrophiles yielded new multicarbon oxygenated complexes, the addition of CO2 to the ethynediolate C2 complex resulted in the formation of a very reactive intermediate, allowing C–H activation of aromatic solvents. This original intermolecular reactivity corresponds to an unprecedented functionalization of CO-derived ligands, which is induced by CO2.
Introduction
Owing to environmental and economic reasons, the transformation and valorization of abundant and readily available chemical feedstock such as CO and CO2 are very topical goals.1 Carbon monoxide is a C1 gas that is used in the industrial production of organic feedstock molecules such as methanol2 and acetic acid.3 Synthesis gas (syn-gas: CO/H2) is converted into liquid hydrocarbons and oxygenated molecules on industrial scales by using the Fischer–Tropsch (F–T) process.4 As the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond is one of the strongest chemical bonds (bond dissociation energy of 1075 kJ mol−1),5 this process typically operates under harsh temperature and pressure conditions (200–350 °C, 20–45 bars) using heterogeneous transition metal catalysts (Fe, Co).6 The precise mechanism in the formation of F–T products is still under debate and, to gain more insight into the elementary steps involved, several organometallic complexes have been used as soluble models.7 Besides, recent advances in transition-metal-free F–T chemistry have been reported, describing the formation of growing carbon chains.8
O triple bond is one of the strongest chemical bonds (bond dissociation energy of 1075 kJ mol−1),5 this process typically operates under harsh temperature and pressure conditions (200–350 °C, 20–45 bars) using heterogeneous transition metal catalysts (Fe, Co).6 The precise mechanism in the formation of F–T products is still under debate and, to gain more insight into the elementary steps involved, several organometallic complexes have been used as soluble models.7 Besides, recent advances in transition-metal-free F–T chemistry have been reported, describing the formation of growing carbon chains.8
Although CO homologation has been observed by the coupling between two or more fragments upon insertion of CO into metal–alkyl,9 –aryl,10 –hydride,11 –imide12 or –boryl bonds,13 the direct reductive coupling of CO molecules has only been achieved by a very limited number of systems, typically involving low-valent oxophilic—metallic or non-metallic—elements.14 For example, homogeneous p-,15 and d-block compounds16 have been used to promote CO reductive coupling. Molecular low-valent f-block complexes based on U(III) and Ln(II) metal centers have shown very promising reactivity in the reductive coupling of CO, which can be traced back to their high reducing character and oxophilic nature.16e,17 Detailed experimental17d–f,17j and computational studies17f,18 have been performed by Cloke, Green, Maron and co-workers in order to rationalize the CO reductive coupling reactivity by bulky U(III) sandwich complexes. Depending on the reaction conditions and supporting ligands, selective formation of oxocarbon dianions of the type ethynediolate (A), deltate (B) or squarate (C) was reported (Fig. 1).
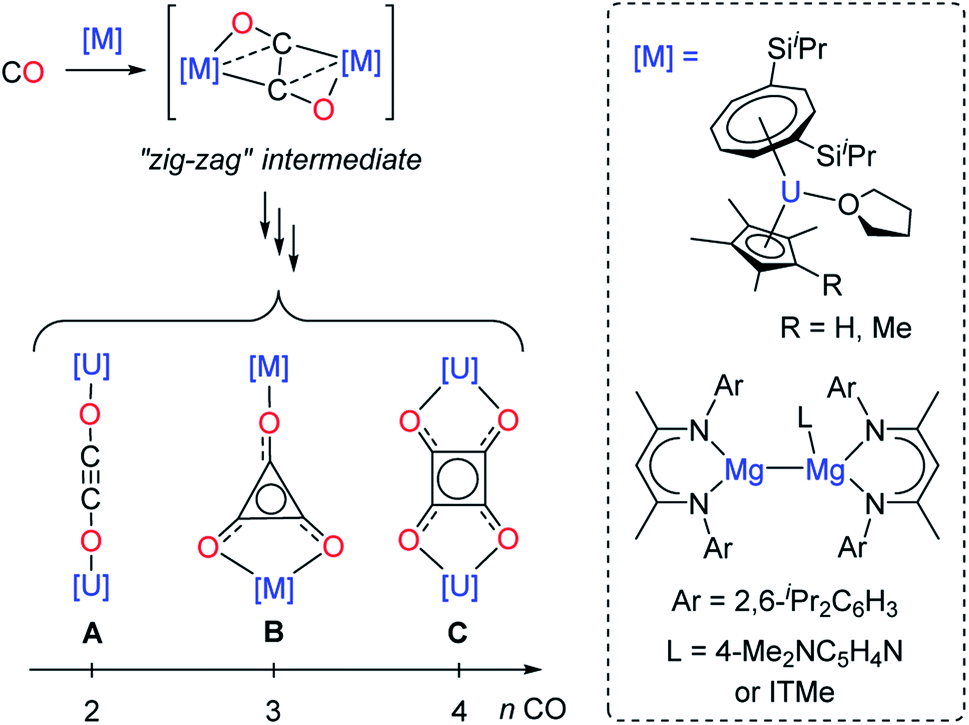 | ||
| Fig. 1 Evolution of the “zig-zag” intermediate into CO oligomerization products upon reaction of selected U(III) and Mg(I) complexes with CO (ITMe = 1,3,4,5-tetramethylimidazol-2-ylidene). | ||
In these examples, DFT calculations pointed to a “zig-zag” intermediate complex featuring a bridging trans-bent [C2O2]2− dianion as a key intermediate in the formation of the different species. A similar intermediate was also recently suggested by Jones and co-workers in the reductive coupling of CO using activated β-diketiminate Mg(I) complexes (Fig. 1).15e,g However, typically, the reactivity of the CO oligomerization products was not further explored. Very recently, Crimmin and co-workers described the sequential formation of C1–C4 chain compounds starting from CO and CO2 feedstocks in the presence of transition metal carbonyl complexes and using β-diketiminate Al(I) reductants.15c,k Other systems allowing sequential chain growth by reductive homologation of CO from isolable intermediates or functionalization reactions of the products are exceedingly rare.15a,l
We have been interested in the use of highly reducing Tm(II)19 and other divalent lanthanide complexes for homogeneous transformations and small molecule activation.20 Herein, we report the reactivity of the Tm(II) complex [Tm(Cpttt)2]21 (Cpttt = 1,2,4-tris(tert-butyl)cyclopentadienyl) towards CO, leading to selective CO reductive di- and trimerization products. The reactivity of the corresponding complexes, which feature central dianionic {CnOn}2− (n = 2,3) oxocarbon moieties, was systematically investigated towards electrophiles (CO2, silylating/alkylating agents). Novel multicarbon oxygenated frameworks were obtained through unusual reactivities, opening new avenues for the functionalization of CO and CO2.
Results and discussion
Synthesis and structure of 1
The divalent thulium complex [Tm(Cpttt)2] (1) was obtained by slightly modifying the original procedure by Nief and co-workers,21 in order to ensure better reproducibility and a higher isolated yield (for details, see ESI†). Crystals of 1 suitable for X-ray diffraction (XRD) studies could be successfully obtained from a concentrated pentane solution of the complex at low temperature (−40 °C). Owing to the very high solubility of 1 in hydrocarbon solvents, only the crystal structure of the less soluble THF adduct was reported in the seminal publication.21a The molecular structure of 1 in the solid state (Fig. 2) confirmed the formation of the base-free complex with two independent molecules of 1 in the asymmetric unit, displaying very similar metrical data. In this divalent lanthanide sandwich complex, the Tm(II) metal center is surrounded by two η5-coordinated Cpttt ligands with Tm–C bonds ranging from 2.652(4) to 2.717(4) Å (in average 2.68 ± 0.02 Å) and Tm–Cp(ctr) (ctr = ring centroid) distances of ca. 2.39 Å (see also Table S4†). In each crystallographically independent molecule, the sum of Tm–Cp(ctr) distances is ca. 4.78 Å, a value only slightly larger than that in the trivalent complex [Tm(Cpttt)2I] (4.74 Å, see Table S4†).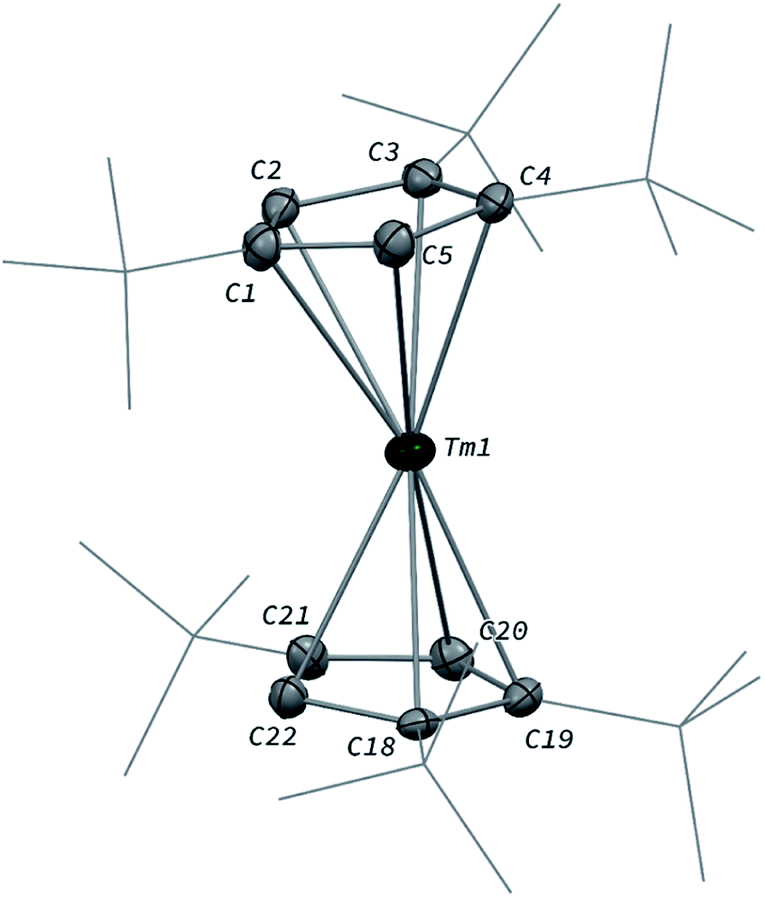 | ||
| Fig. 2 Molecular structure of one of the two independent molecules of 1 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). Only one disordered position for the tBu groups has been depicted and H atoms have been omitted for clarity. Selected bond distances (Å): Tm1–C1 2.712(4), Tm1–C2 2.676(4), Tm1–C3 2.681(4), Tm1–C4 2.664(4), Tm1–C5 2.670(4), Tm1–C18 2.673(4), Tm1–C19 2.677(4), Tm1–C20 2.686(4), Tm1–C21 2.703(4), Tm1–C22 2.658(4). | ||
Although the metal center in 1 is only surrounded by two Cpttt ligands, a slightly bent arrangement is observed in the solid state, with angles between the mean C5 planes in the range 11.0–13.9° and Cp(ctr)–Tm–Cp(ctr) angles of 164.3–167.7°. Similar bent arrangements were observed for the base-free complexes [Ln(Cpttt)2] (Ln = Sm, Eu),22 although 1 presents a Cp(ctr)–Ln–Cp(ctr) arrangement closer to linearity, which may be the result of the smaller ionic radius of Tm(II) compared to Sm(II) (1.03 and 1.17 Å, respectively, in a six-coordinate environment).23 Consistently, the Ln–Cp(ctr) distance is ca. 0.13 Å shorter in 1 in comparison to that in [Sm(Cpttt)2] (see Table S4†).
Despite the established high reducing character of Tm(II) complexes,19,24 no reaction of 1 was observed with either N2 or H2 and no traces of decomposition could be noticed upon heating a C6D6 solution of the complex at 80 °C for several days. This unusual stability contrasts with the reported reactivity of base-free samarocene [Sm(Cp*)2] (Cp* = η5-C5Me5) with N2.25 The bulky nature of the Cpttt ligand is vital to kinetically stabilize the Tm(II) complex and avoid thermal decomposition, as observed when using the smaller Cp* or disubstituted 1,2-R2C5H3 (R = tBu, SiMe3) ligands.26 Despite the steric protection imposed by the Cpttt ligands, coordination of a Lewis base to the metal center has been achieved in related [Ln(Cpttt)2] complexes (Ln = Yb and Sm).22a,b We therefore reasoned than the high reducing character of the Tm(II) center19,27 in 1 in addition to the oxophilic nature of f-elements28 may lead to interesting reactivity towards the gaseous oxocarbons CO and CO2.
Reactivity of 1 towards CO and CO2
Addition of CO to a degassed solution of 1 in toluene led to an immediate color change from deep purple to light brown, indicating prompt consumption of the highly colored Tm(II) complex 1. Depending on the stoichiometry in CO, two different complexes were isolated (Scheme 1).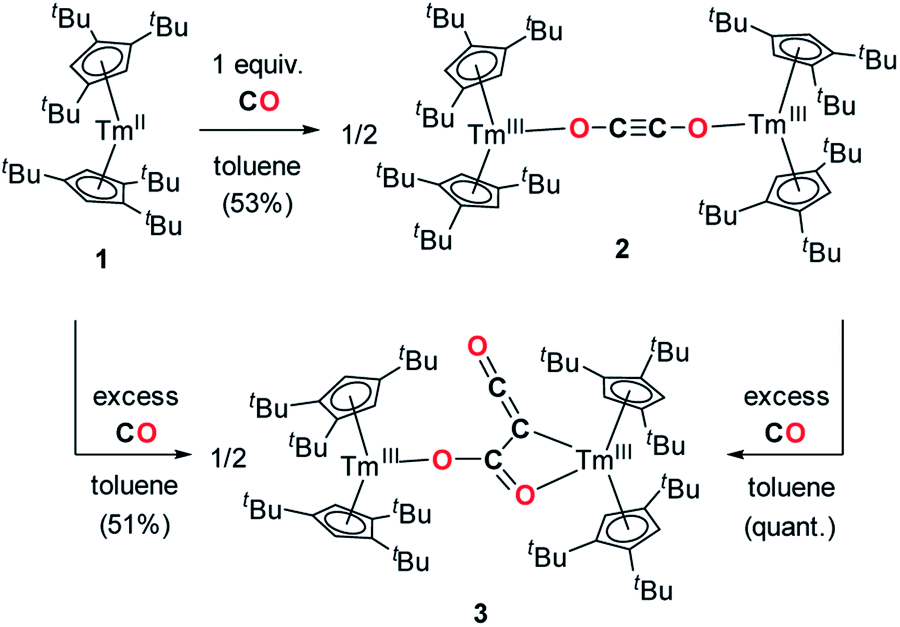 | ||
| Scheme 1 Synthesis of the CO dimerization and trimerization products 2 and 3. | ||
When the reaction was performed using one molar equivalent of CO per 1, the CO dimerization product 2 was isolated in 53% crystalline yield. The molecular structure of 2 in the solid state (Fig. 3) was unambiguously confirmed by XRD studies and revealed the formation of the dinuclear complex [Tm(Cpttt)2]2(μ-κ(O):κ(O′)-C2O2) featuring a bridging ethynediolate ligand between the two oxidized Tm(III) metal centers. The C–C bond distance in the C2O22− moiety of 1.226(10) Å is within the typical range for related ethynediolate bridged dimers of f-block elements,16e,17f–j,17l and is consistent with a CC triple bond. The Tm–O–C angles of 146.7(5) and 151.9(5)° depart from linearity and the Tm–O bond distances of 2.066(5) and 2.078(5) Å are at the long end of the range reported for Tm(III) complexes featuring terminal alkoxy ligands (from ca. 1.96 to 2.09 Å).29 In line with the oxidation of the thulium metal center from the +II to the +III oxidation state, the sum of the two Tm–Cp(ctr) separations for each metal center in 2, in the range 4.69–4.71 Å, is ca. 0.08 Å shorter than that in 1.
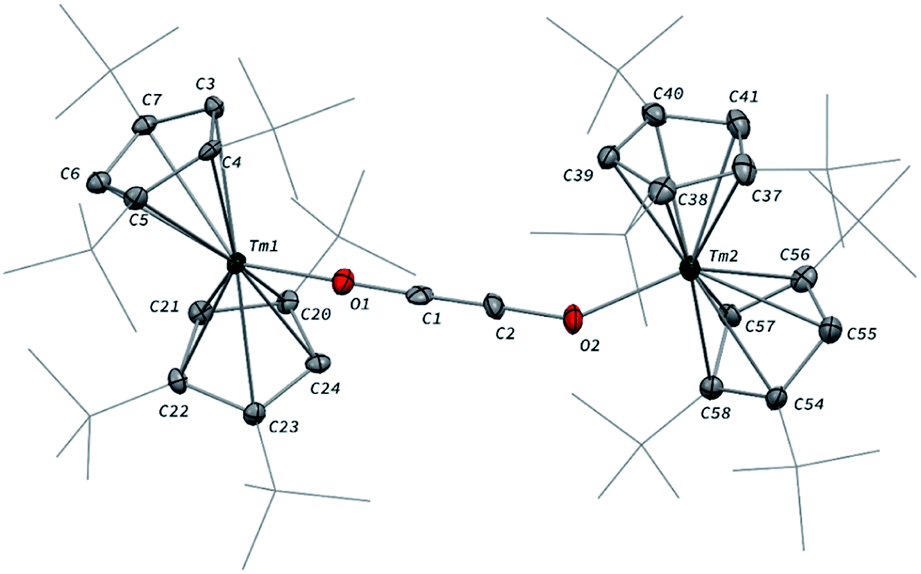 | ||
| Fig. 3 Molecular structure of 2 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). H atoms have been omitted for clarity. Selected bond distances (Å) and angles [°]: C1–C2 1.226(10), C1–O1 1.265(9), C2–O2 1.296(9), Tm1–O1 2.066(5), Tm2–O2 2.078(5); C2–C1–O1 178.4(8), C1–C2–O2 178.1(9), C1–O1–Tm1 151.9(5), C2–O2–Tm2 146.7(5). | ||
Analysis of the 1H NMR spectrum of 2 in toluene-d8 revealed only very broad resonances at room temperature, possibly resulting from a fluxional behavior of the Cpttt ligands.20a,22b A better resolved spectrum featuring three broad resonances at δ ≈ 171, 32 and 2 ppm was obtained upon recording the spectrum at 80 °C (see Fig. S2–S3†), and is consistent with free rotation of the Cpttt ligands leading to an overall D2h symmetric species in solution at high temperature. In the IR spectrum of 2, no significant absorption band was observed in the range 1500–2800 cm−1, all the more so in the expected region for CC triple bonds, which can be explained by the IR selection rules (lack of change in the molecular dipole). The isotopically labelled complex [Tm(Cpttt)2]2(μ-κ(O):κ(O′)-13C2O2) (2-13C) was obtained by the same procedure using 13C-enriched CO. As expected, the IR spectra of 2 and 2-13C are nearly identical (see Fig. S37†), one only notable difference being the band at 1329 cm−1 in 2 that is red-shifted to 1304 cm−1 upon 13C labelling. The corresponding absorption band can be confidently assigned to the C–O stretching vibration, as the experimental ratio between these numbers, 1.019, compares well with the theoretical value of 1.023 obtained from reduced mass considerations. Owing to the highly paramagnetic nature of Tm(III) complexes, no 13C NMR resonances could be detected, even for the isotopically enriched 2-13C.
The formation of ethynediolate fragments upon reductive coupling of CO has been reported for a handful of low-valent f-block complexes, mostly based on U(III) metal centers,17f–j and recently by Evans and co-workers upon treatment of in situ generated Ln(II) species or [K(18-c-6)2][Tm{N(SiMe3)2}3] with CO.16e,17l The sensitivity of the corresponding ethynediolate products to thermolysis was found to be ligand dependent.17g–i In the case of 2, no traces of decomposition could be detected upon heating a toluene solution of the complex at 100 °C for several days.
In contrast to the formation of 2, the reaction of 1 with excess CO led to the CO trimerization product [Tm(Cpttt)2]2(μ-κ(O):κ2(C,O′)-C2O3) (3) isolated in 51% crystalline yield (Scheme 1). The isotopically labelled complex [Tm(Cpttt)2]2(μ-κ(O):κ2(C,O′)-13C2O3) (3-13C) was obtained analogously using 13C-enriched carbon monoxide. The X-ray structure of 3 confirmed the formation of a ketenecarboxylate (O2C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO)2− fragment bridging the two Tm(III) centers (Fig. 4). The reductive trimerization of CO into such a motif has previously only been described in two reports by Evans and co-workers using reducing lanthanide complexes based on Cp* ligands.17a,b The Tm–C bond distances in 3, ranging from 2.601(6) to 2.769(6) Å (in average 2.67 ± 0.06 Å) (see also Table S5†) are slightly longer than those observed in 2, which may be the result of the larger steric demand of the (μ-κ(O):κ2(C,O′)-C2O3)2− bridging ligand. The latter is coordinated to the two Tm metal centers via a bridging carboxylate unit (Tm2–O2 2.307(5), Tm1–O3 2.098(5) Å) and further coordinated to Tm2 with a Tm2–C2 bond distance of 2.473(7) Å. The sum of the bonding angles around C2 (359.6°) is pointing to a planar arrangement for C1, C2, C3 and Tm2. In the ketenecarboxylate fragment, the bond distances within the ketene moiety (C1–C2 1.284(10) Å, C1–O1 1.181(9) Å) are indicative of double bonds while the adjacent C2–C3 bond is much longer (1.430(9) Å) and in the range of single C–C bonds. The carboxylate C3–O2 and C3–O3 bond distances of 1.261(8) and 1.296(8) Å, respectively, are very similar, supporting a delocalized negative charge over the carboxylate moiety. Overall, the metrical data within the ketenecarboxylate fragment are in good agreement with those previously reported by Evans and co-workers, the only major structural difference lying in the coordination mode of the trimerized CO unit.17a,b While the (O2C–CCO)2− fragment is only bridging two Tm(Cpttt)2 subunits in 2, further dimerization and association into tetranuclear clusters were observed when using the smaller Cp* ligands.17a,b
CO)2− fragment bridging the two Tm(III) centers (Fig. 4). The reductive trimerization of CO into such a motif has previously only been described in two reports by Evans and co-workers using reducing lanthanide complexes based on Cp* ligands.17a,b The Tm–C bond distances in 3, ranging from 2.601(6) to 2.769(6) Å (in average 2.67 ± 0.06 Å) (see also Table S5†) are slightly longer than those observed in 2, which may be the result of the larger steric demand of the (μ-κ(O):κ2(C,O′)-C2O3)2− bridging ligand. The latter is coordinated to the two Tm metal centers via a bridging carboxylate unit (Tm2–O2 2.307(5), Tm1–O3 2.098(5) Å) and further coordinated to Tm2 with a Tm2–C2 bond distance of 2.473(7) Å. The sum of the bonding angles around C2 (359.6°) is pointing to a planar arrangement for C1, C2, C3 and Tm2. In the ketenecarboxylate fragment, the bond distances within the ketene moiety (C1–C2 1.284(10) Å, C1–O1 1.181(9) Å) are indicative of double bonds while the adjacent C2–C3 bond is much longer (1.430(9) Å) and in the range of single C–C bonds. The carboxylate C3–O2 and C3–O3 bond distances of 1.261(8) and 1.296(8) Å, respectively, are very similar, supporting a delocalized negative charge over the carboxylate moiety. Overall, the metrical data within the ketenecarboxylate fragment are in good agreement with those previously reported by Evans and co-workers, the only major structural difference lying in the coordination mode of the trimerized CO unit.17a,b While the (O2C–CCO)2− fragment is only bridging two Tm(Cpttt)2 subunits in 2, further dimerization and association into tetranuclear clusters were observed when using the smaller Cp* ligands.17a,b
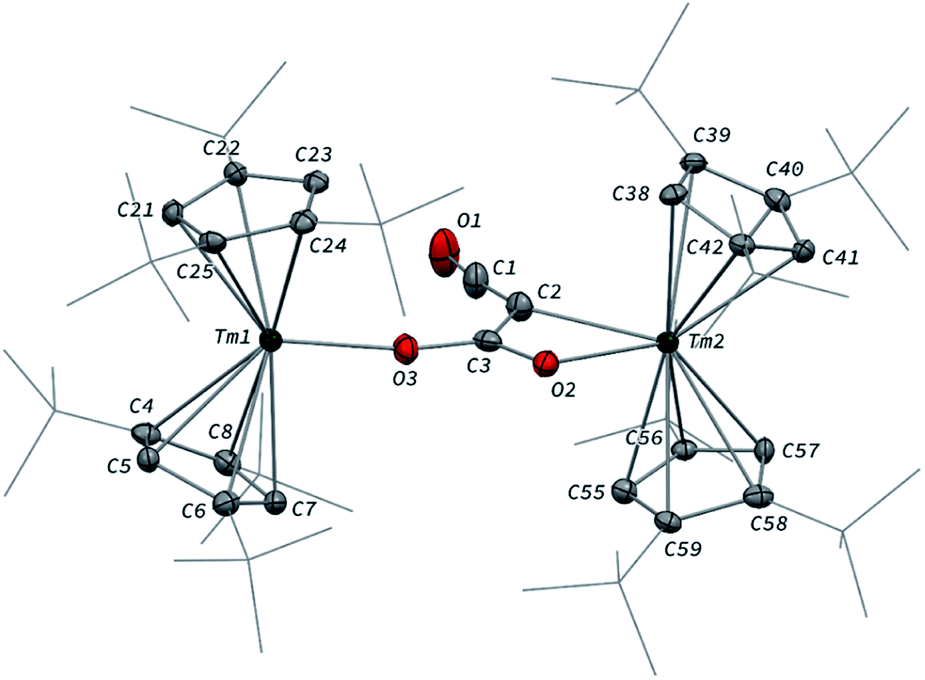 | ||
| Fig. 4 Molecular structure of 3 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). H atoms have been omitted for clarity. Selected bond distances (Å) and angles [°]: O1–C1 1.181(9), O2–C3 1.261(8), O3–C3 1.296(8), C1–C2 1.284(10), C2–C3 1.430(9), Tm1–O3 2.098(5), Tm2–O2 2.307(5), Tm2–C2 2.473(7); C1–C2–C3 128.6(7), C3–O2–Tm2 99.2(4), C3–O3–Tm1 170.8(4), O1–C1–C2 172.2(8), O2–C3–C2 116.0(6), O3–C3–C2 123.5(6), C1–C2–Tm2 143.5(5), C3–C2–Tm2 87.5(4). | ||
The 1H NMR spectrum of 3 at room temperature revealed several broad resonances in the range δ −100 to 400 ppm, as a result of the paramagnetic nature of the complex and restricted free rotation of the Cpttt ligands.20a Upon heating to 80 °C (see Fig. S6†), a simpler baseline was observed featuring four main and relatively broad (Δν1/2 ≈ 1500–3200 Hz) resonances at δ 185, 153, 32 and −33 ppm, assigned to the tBu groups. The number of signals and the corresponding integrations support free rotation of the Cpttt ligands at high temperature, resulting in an overall Cs symmetry in solution. It should be noted that complex 3 was found to be thermally stable at least up to 110 °C in toluene solution. As a result of the highly paramagnetic nature of the complex, no signals could be observed in the 13C{1H} NMR spectra of both 3 and 3-13C. In the IR spectrum of 3, a very strong absorption band was observed at 2066 cm−1 in the ketene region, shifting to 2003 cm−1 in 3-13C upon 13CO isotope labelling (see Fig. S38†). In order to confidently assign the corresponding absorption band, the vibrational spectra of 3 and 3-13C were calculated at the DFT level of theory (B3PW91, see details in the ESI†). In the computed spectra (Fig. S66–S68†), the asymmetric stretch of the O1–C1–C2 ketene fragment gives rise to an intense absorption band at 2190 cm−1 in 3 and 2121 cm−1 in 3-13C. Although the (uncorrected) computed values are ca. 120 cm−1 larger than the experimental ones, the ratios between the wavenumbers of two isotopologues (1.031 experimentally and 1.033 computationally) are almost identical, confirming the assignment of the band.
The overall reaction leading to the (O2C–CCO)2− fragment involves a reductive trimerization of CO, more precisely the net transfer of two electrons from two Tm(II) centers to three molecules of CO. Since the central carbon atom (C2, Fig. 4) in 3 presents no C–O connectivity, complete cleavage of one CO triple bond has occurred. Although CO cleavage and further homologation is thought to occur in heterogeneous Fischer–Tropsch systems, such reactivity is not common in homogeneous systems.4c,7b,14a,17a,17b
The observed CO reductive homologation induced by the Tm(II) complex 1, leading to the selective formation of either the ethynediolate complex 2 or the ketenecarboxylate complex 3, is the result of a fine balance between steric protection and coordinative unsaturation. A similar observation was already reported in the case of U(III) mixed-sandwich complexes reported by Cloke and co-workers, which allowed possible reductive CO di-, tri- or tetramerization depending on the steric demand of the supporting ligands and amount of CO.17j Since selective formation of the CO dimerized product 2 is observed in the presence of one molar equivalent of CO per Tm(II) center, we further examined whether 2 may be an intermediate in the formation of 3. In a control experiment, the reaction of isolated 2 in toluene solution with excess CO was monitored by paramagnetic 1H NMR spectroscopy (see Fig. S7†), revealing clean and quantitative transformation into 3. This reaction is relatively slow at room temperature, requiring ca. 1–2 days to reach completion, which suggests a relatively high activation barrier. To gain further insights in the elementary steps leading to the formation of 2 and 3, i.e. the overall mechanism of this unusual reaction, DFT calculations were performed and confirmed that 2 appears indeed as an intermediate in the formation of 3 (see below). To the best of our knowledge, this result is unprecedented since all reported ethynediolate complexes have been either synthesized under excess of CO,15j,17g–i,17l or the isolated products were found inert towards external CO.17f To date, systems allowing controlled carbon chain growth by sequential insertions of CO on isolated intermediates are exceedingly rare. Such examples include CO addition on the boron–boron triple bond of a diboryne compound described by the group of Braunschweig,15a and recent work by Crimmin and co-workers on systems based on transition metal carbonyls together with aluminum(I) reductants.15c,k
Finally, to investigate whether other reaction conditions have an influence on the selectivity, temperature and solvent effects were examined. Performing the addition of CO at low temperature (−78 °C) or in different solvents (pentane, benzene, Et2O and THF) led to similar results signifying that the stoichiometry in CO is the only parameter controlling the selectivity in this reaction. It should be however noted that the reaction proceeds much more slowly in coordinating solvents such as THF, requiring several days until complete consumption of 1 (see Fig. S4†), which can be traced back to competitive coordination of THF and CO to the metal center and a relatively high activation barrier (see below). An inhibitory role of THF in the reduction of organic substrates by Sm(II) complexes has previously been observed in the literature.30 Interestingly, in the U(III) systems reported by Arnold and co-workers, THF coordination was found to completely stop the reactivity towards CO.17g,h Such a different behavior may originate in the steric demand of the Cpttt ligands that induces weak and reversible coordination of the usually strongly bound THF donor.21a A greater degree of covalency of U–L vs. Ln–L bonds (L = THF or other donor ligands) may also contribute to decrease the L lability and suppress reactivity for U(III) systems in donor solvents.31 The unique steric properties of the Cpttt ligand may also explain the different reactivity of [Sm(Cpttt)2] and [Sm(Cp*)2] towards CO. Although both SmII complexes have very similar redox potentials associated with the SmIII/SmII couple (ca. −2.12 and −2.10 V vs. ferrocenium/ferrocene in THF, respectively),22b,32 [Sm(Cpttt)2] is inert towards CO,20a while the Cp* analog leads to CO reductive trimerization.17a
The reactivity of the Tm(II) complex 1 was also investigated towards CO2 as divalent lanthanide complexes have been reported to yield oxalate and/or carbonate complexes upon reduction of CO2.33 The reaction of 1 with excess CO2 led to the formation of the carbonate complex 4 isolated in 72% yield. Its IR spectrum features a strong absorption band at 1430 cm−1, assigned to C–O stretching in the coordinated carbonate ligand.34 The X-ray structure of the complex (see Fig. S44†) confirmed the presence of a bridging carbonate ligand with a μ-κ(O):κ2(O′,O′′) coordination mode between the two TmIII(Cpttt)2 fragments, as previously observed for the Sm(III) analogue [Sm(Cpttt)2(CO3)].20a A more detailed description of the structure can be found in the ESI.†
Mechanistic insights by DFT calculations
The formation of complex 3 has been investigated at the DFT level (B3PW91) and the computed enthalpy profile at room temperature is depicted in Fig. 5. The reaction begins with the side-on coordination of CO to 1. This coordination induces the reduction of CO and the oxidation of Tm(II) to Tm(III), the radical on CO being stabilized by η2 coordination to the metal center. This step is computed to be endothermic by 16.3 kcal mol−1, which fits well with immediate changes in the color and 1H NMR spectrum of the solution in toluene-d8. Unlike the calculated pathway for CO reductive coupling at U(III) mixed-sandwich complexes,18b the CO moiety is not further reduced by the coordination of a second molecule of 1 but rather a radical coupling between two Tm(III)(CO˙−) fragments is observed. This step also contrasts with that calculated for CO reductive oligomerization on Mg(I) dimers where the addition of CO occurs sequentially (see additional details in the ESI†).15e,g,15j It can be noted that a radical pathway for CO oligomerizations has been recently suggested on the basis of fragmentation reactions by mass spectrometry and computational quantum calculations on squarate species, i.e. CO tetramerization products.35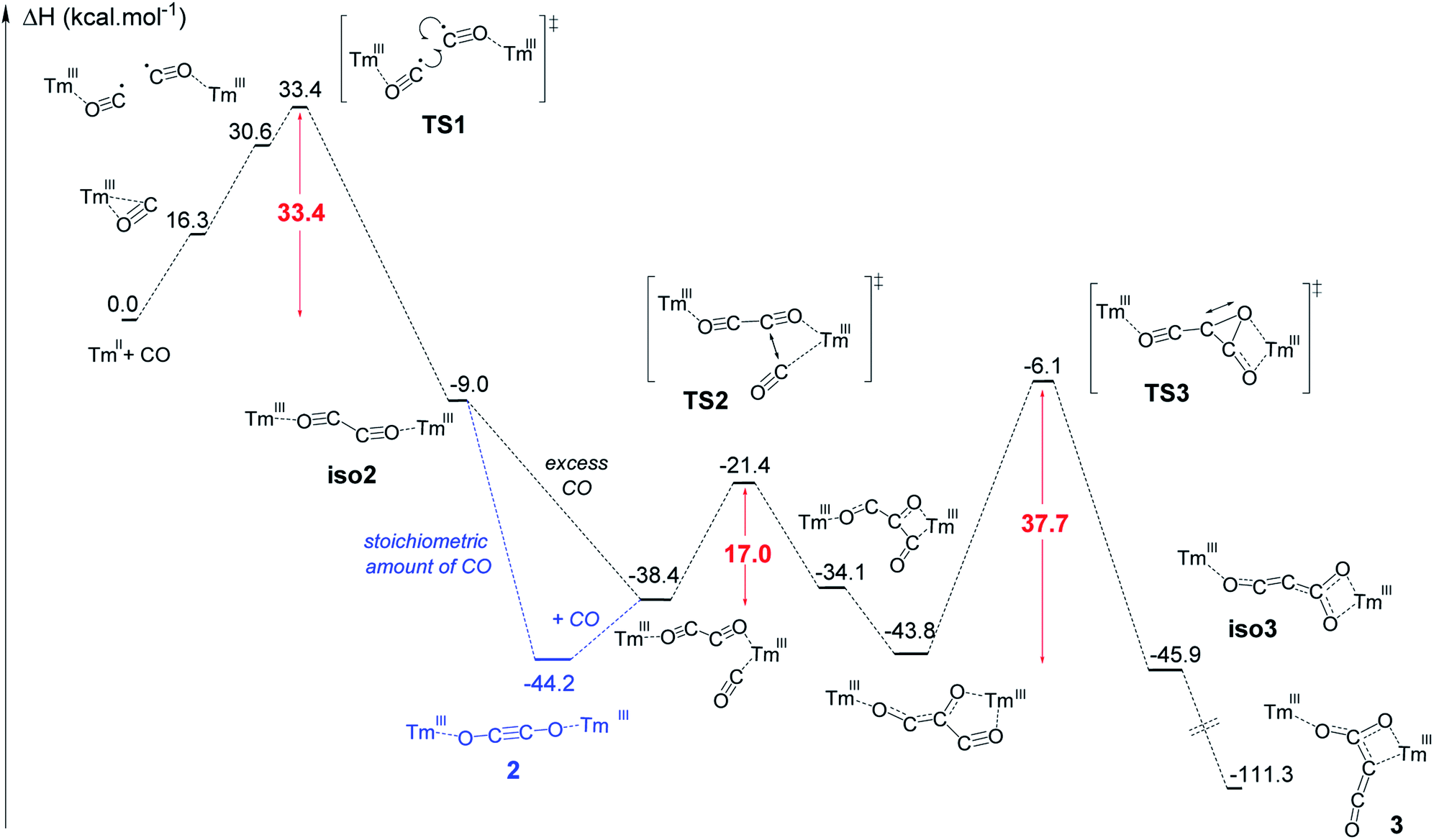 | ||
| Fig. 5 Computed enthalpy profile at room temperature for the formation of 2 and 3. | ||
In addition, the possible formation of a lanthanide complex featuring a reduced (CO)˙− ligand has already been reported in the case of highly reducing and transient Y(II) and Lu(II) complexes,16e,17l which supports the generation of a related (CO)˙− intermediate upon reaction of 1 with CO. The associated transition state (TS1) has been located on the Potential Energy Surface (PES). The radical coupling barrier is 33.4 kcal mol−1 from 1 (but only 17.1 kcal mol−1 from the Tm(III) intermediate complex), in line with a kinetically accessible but relatively slow reaction. Accordingly, slow evolution in the 1H NMR spectrum of the reaction between 1 and CO (1 equiv.) in toluene-d8 was observed over several hours at room temperature. The radical nature of the TS is highlighted by the computed unpaired spin densities (see Fig. S56†) where 0.6 unpaired electron is located on the two carbons that couple. Following the intrinsic reaction coordinate, the formation of the ethynediolate complex iso2 is favorable (−9.0 kcal mol−1 with respect to 1). This complex displays a “zig-zag” geometry which is the key intermediate in this reactivity (see Fig. 1) and is similar to the “zig-zag” intermediate suggested in previous studies.15e,g,17f,g,18b This intermediate can either yield the more stable linear structure, 2, which is what happens with a stoichiometric amount of CO, or bind another CO molecule, as obtained for a CO excess. In our case, these two possibilities are thermodynamically competitive (the CO coordination is exothermic by 29.4 kcal mol−1 from iso2 while the linear ethynediolate complex 2 is 35.2 kcal mol−1 more stable than the “zig-zag” one). The formation of the stable CO adduct is easily explained by the nature of the HOMO of iso2, that can easily overlap with the π* orbital of CO (see Fig. S57†). It is interesting to note that the optimized geometry of a CO adduct on 2 yields the same structure, so that complex 2 is also linked to the computed reaction profile with an excess of CO. From the CO adduct, a C–C coupling TS has been located and corresponds to a nucleophilic attack of the ethynediolate to the π system of CO (see the HOMO at the TS in Fig. S58†). The associated barrier is low (17.0 kcal mol−1), yielding a stable ketene-type intermediate (−43.8 kcal mol−1). This ketene intermediate can isomerize with a barrier of 37.7 kcal mol−1 (TS3) to form complex 3 (see details in the ESI†), which is the most stable complex of the entire profile (−111.3 kcal mol−1). The relatively high activation barrier calculated for TS3 (ca. 38 kcal mol−1) is consistent with a relatively slow (1–2 days) transformation of 2 to 3 in the presence of excess CO at room temperature. It should be noted that an activation barrier of up to ca. 40 kcal mol−1 is not unprecedented for a reaction involving a lanthanide metallocene which is occurring slowly at room temperature.36
Additional single-point calculations using a larger basis set or other functionals (wB97xd and M062X) (see ESI and Fig. S63–S65†) showed slightly lower but still relatively high activation barrier energies. Despite this barrier being at the upper limit of that typically accepted for a reaction occurring at room temperature, it should be noted that, from a strictly thermodynamic point of view, 3 is much more stable than 2 (67.1 kcal mol−1 lower in energy), which drives the reaction towards formation of the CO trimerization product 3.
To further investigate the validity of the DFT calculation results, the reaction between 2 and excess 13CO was performed. The IR spectrum of the corresponding product (Fig. S40†) shows an intense absorption band at 2004 cm−1, very similar to that of 3-13C (2003 cm−1), indicating that scrambling of 13C labels occurred in the presence of excess 13CO. A possible mechanism to explain the 13C scrambling step, consistent with the computed energetic profile, can be found in the ESI (Scheme S1†).
Reactivity of 3 towards electrophiles
Having complex 3 at hand with a reproducible procedure and in decent isolated yield, we investigated its reactivity towards electrophiles. Indeed, the ketenecarboxylate complex 3 presents a polarized lanthanide–carbon bond, which is prone to further functionalization or insertion reactions.33,37 It should be noted that, for the two previously reported ketenecarboxylate complexes based on Ln(Cp*)2 (Ln = La, Sm) fragments,17a,b no study of their reactivity was described.The reaction of 3 with CO2 in toluene-d8 proceeded instantaneously and cleanly at room temperature, as evidenced by paramagnetic 1H NMR monitoring of the reaction. The corresponding product 5 was isolated in 82% yield (Scheme 2) and crystallographically characterized. Analysis of the molecular structure of 5 in the solid state (Fig. 6) revealed the insertion of CO2 in the Tm–C bond of 3, yielding a nearly planar bridging μ-κ2(O):κ2(O)-ketenedicarboxylate ligand. The two Tm(III) centers are each surrounded by a chelating carboxylate group and two η5-coordinated Cpttt ligands. The C–C bond distances between the ketene and carboxylate groups are identical within experimental error (C1–C2 1.460(9) Å and C2–C3 1.461(10) Å) resulting in an overall and non-crystallographically imposed C2v symmetry (not taking into account the substitution patterns on the Cpttt ligands). The C2–C4 and O5–C4 bond distances, 1.343(10) and 1.142(10) Å, respectively, are consistent with double bonds and a ketene fragment, and are similar to the corresponding distances in 3 (C1–C2 and O1–C1 bond distances of 1.284(10) and 1.181(9) Å, respectively). The sums of the two Tm–Cp(ctr) separations for both metal center in 5 are 4.735 and 4.746 Å, with Tm–C bond distances ranging from 2.618(7) to 2.733(7) Å (in average 2.66 ± 0.04 Å) (See also Table S5†).
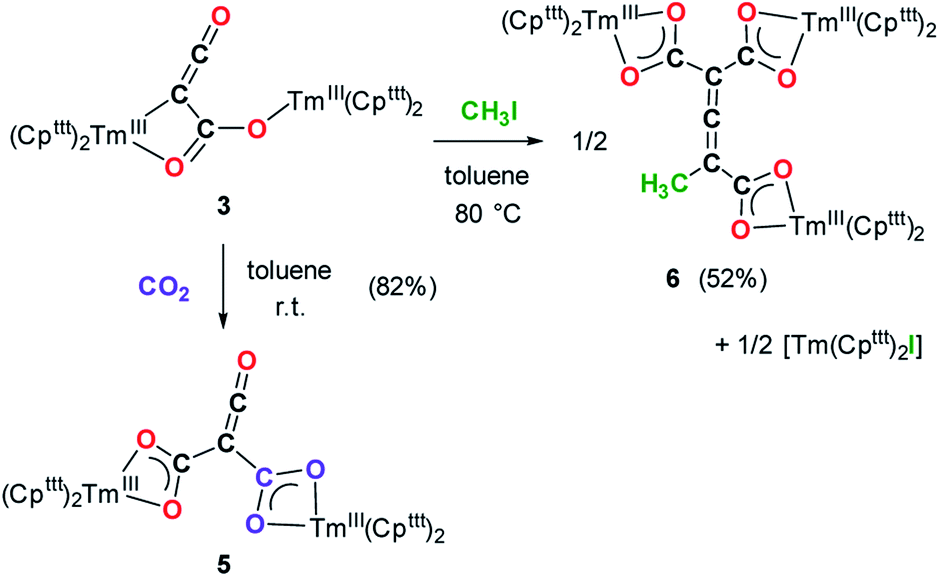 | ||
| Scheme 2 Reactivity of 3 towards CO2 and MeI. | ||
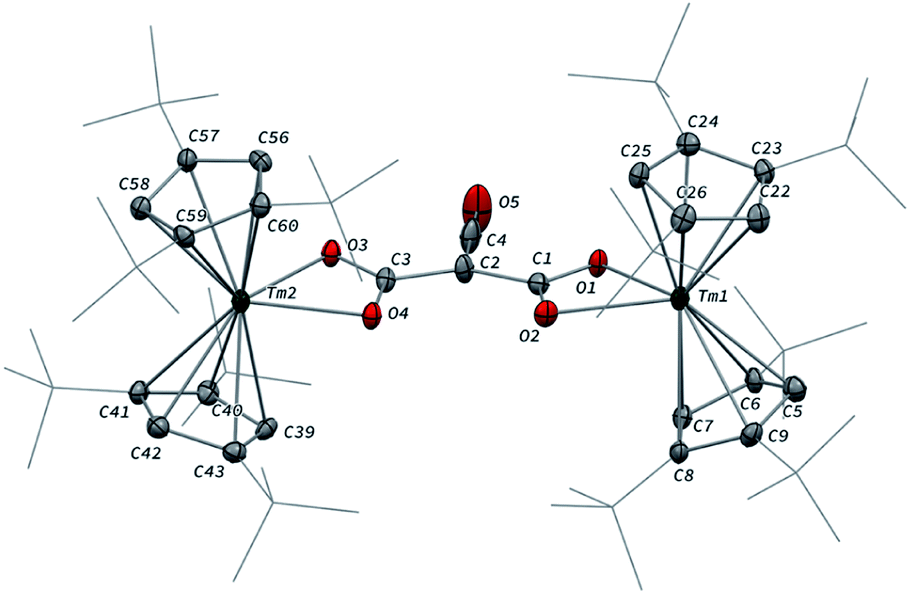 | ||
| Fig. 6 Molecular structure of 5 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). H atoms have been omitted for clarity. Selected bond distances (Å) and angles [°]: Tm1–O1 2.282(5), Tm1–O2 2.354(5), Tm2–O3 2.305(5), Tm2–O4 2.318(5), O1–C1 1.283(8), O2–C1 1.263(8), O3–C3 1.273(8), O4–C3 1.267(8), O5–C4 1.142(10), C1–C2 1.460(9), C2–C3 1.461(10), C2–C4 1.343(10); O1–Tm1–O2 56.9(2), O3–Tm2–O4 57.0(2), O1–C1–C2 116.0(6), O2–C1–O1 120.3(6), O2–C1–C2 123.6(6), C1–C2–C3 131.7(6), C1–C2–C4 113.0(7), C3–C2–C4 115.3(6), O3–C3–C2 118.1(6), O3–C3–O4 120.5(6), O4–C3–C2 121.4(6), O5–C4–C2 178.5(10). | ||
The 1H NMR spectrum of 5 in toluene-d8 revealed six main broad and paramagnetically shifted resonances at room temperature (see Fig. S11 and S12†), resulting from restricted rotation of the Cpttt ligands around the metal center. Upon heating to 60 °C, the spectrum evolved into three very broad (Δν1/2 ≈ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–17000 Hz) resonances at δ −16, 51 and 197 ppm, which is in agreement with freely rotating Cpttt ligands and an overall C2v symmetry in solution. In the IR spectrum of 5, the two bands were detected at 2157 and 2146 cm−1, assigned to the asymmetric ketene stretching vibration,38 along with an intense absorption band at 1572 cm−1 corresponding to the asymmetric stretching of the coordinated carboxylate groups.39
000–17000 Hz) resonances at δ −16, 51 and 197 ppm, which is in agreement with freely rotating Cpttt ligands and an overall C2v symmetry in solution. In the IR spectrum of 5, the two bands were detected at 2157 and 2146 cm−1, assigned to the asymmetric ketene stretching vibration,38 along with an intense absorption band at 1572 cm−1 corresponding to the asymmetric stretching of the coordinated carboxylate groups.39
Although 3 was found to react with Me3SiCl upon heating to 60 °C, no crystals suitable for XRD studies could be obtained to unambiguously assign the nature of the formed products. When the reaction was performed using MeI as electrophile (Scheme 2), no reaction occurred at room temperature but slow conversion of 3 was observed in toluene-d8 upon heating the reaction mixture at 80 °C for 3 days. Paramagnetic 1H NMR monitoring of the reaction revealed the formation [Tm(Cpttt)2I], identified by comparison with the 1H NMR spectrum of an authentic sample, along with a new set of signals (see Fig. S15†). After evaporation of the solvent, the crude product was washed several times with pentane to remove [Tm(Cpttt)2I]. Recrystallization of the remaining solid from hot toluene afforded crystals of 6 suitable for XRD studies. Its molecular structure (Fig. 7) features a trinuclear Tm(III) complex in which each thulium center is coordinated by a chelating carboxylate group and two η5-coordinated Cpttt ligands. The C2–C4 and C4–C5 bond distances in 6 of 1.300(6) and 1.325(5) Å, respectively, together with the almost linear C2–C4–C5 arrangement (bonding angle of 172.2(4)°), are consistent with CC double bonds in an allene-type structure. In contrast, the C1–C2, C2–C3, C5–C6 and C5–C7 bonds, with distances from 1.486(6) to 1.512(5) Å, are in the typical range for C–C single bonds. The carbon atoms C2 and C5 present a trigonal planar geometry (sum of the bonding angles around C2 and C5 of 360.0 and 359.3°), consistent with sp2 hybridization. The Tm–O bond distances in 6, in the range 2.299(3)–2.333(3) Å, are similar to those observed in the dicarboxylate complex 5 (from 2.282(5) to 2.354(5) Å).
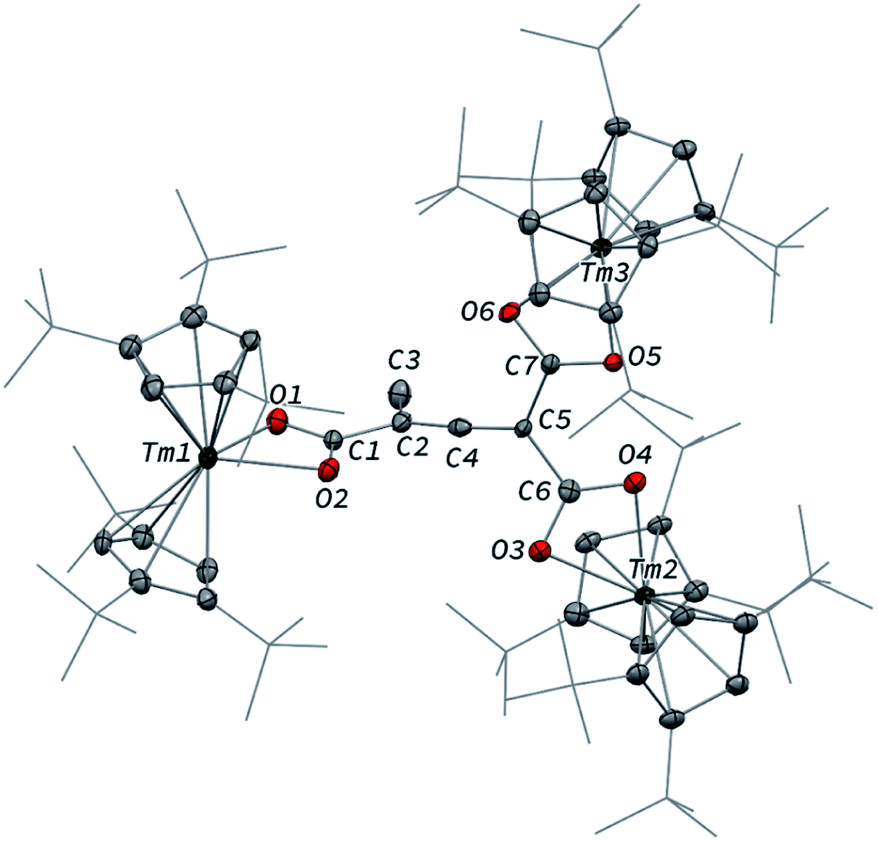 | ||
| Fig. 7 Molecular structure of 6 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). H atoms have been omitted for clarity. Selected bond distances (Å) and angles [°]: Tm1–O1 2.299(3), Tm1–O2 2.315(3), Tm2–O3 2.319(3), Tm2–O4 2.307(3), Tm3–O5 2.304(3), Tm3–O6 2.333(3), O1–C1 1.274(5), O2–C1 1.268(5), O3–C6 1.284(5), O4–C6 1.269(5), O5–C7 1.271(4), O6–C7 1.268(4), C1–C2 1.486(6), C2–C3 1.501(6), C2–C4 1.300(6), C4–C5 1.325(5), C5–C6 1.512(5), C5–C7 1.492(5); O1–C1–C2 118.1(4), O1–C1–O2 119.6(4), O2–C1–C2 122.2(4), C1–C2–C3 116.6(4), C1–C2–C4 121.1(4), C3–C2–C4 122.3(4), C2–C4–C5 172.2(4), C4–C5–C6 117.6(3), C4–C5–C7 119.4(3), C6–C5–C7 122.3(3), O3–C6–C5 118.2(4), O3–C6–O4 121.8(4), O4–C6–C5 119.9(3), O5–C7–C5 119.7(3), O5–C7–O6 121.4(4), O6–C7–C5 119.0(3). | ||
The 1H NMR spectra of 6 in the 20–80 °C temperature range display a complex pattern of paramagnetically shifted signals (Fig. S14†), as a result of the non-symmetric nature of the complex. In the IR spectrum of 6, the intense absorption band at 1583 cm−1 corresponds to the asymmetric stretching band of the coordinated carboxylate groups, similarly to the absorption band observed in 5. In addition, a weak band is observed at 1938 cm−1 and assigned to the asymmetric CCC stretching vibration.39
Although the mechanism for the formation of 6 is elusive to date and involves several rearrangement steps, the observed reactivity finds its origin in the reactive Tm–C bond in 3. It should be stressed that the stoichiometry of the reaction is strictly respected as one equivalent of both 6 and [Tm(Cpttt)2I] are formed from two equivalents of 3, in other words, one tris-anionic {MeC3(CO2)3}3− framework and one iodide ligand are the result of the reaction of two di-anionic {C3O3}2− fragments with one molecule of MeI. Overall, this reaction corresponds to an unprecedented oligomerization and functionalization of CO through formation of an allene–tricarboxylate complex in which the {C6O6} core exclusively arises from six CO molecules.
Reactivity of 2 towards electrophiles
Intrigued by the reactivity of 3 towards electrophiles, we examined the reactivity of 2 towards CO2 and silyl electrophiles. The reactivity of 2 was investigated towards silylating agents to evaluate the possibility of decoordination and liberation of the anionic oxocarbon ligand. It should be noted that decoordination of CO reduction products from oxophilic U(IV) centers has previously been achieved upon treatment with silyl electrophiles.17i,j,40 No reaction occurred upon addition of excess Me3SiCl to 2, even upon heating to 80 °C. In contrast, the addition of Me3SiOTf to a solution of 2 in toluene-d8 led to a complete conversion after 15 h at room temperature (Scheme 3). Analysis of the reaction mixture by paramagnetic 1H NMR spectroscopy revealed resonances corresponding to [Tm(Cpttt)2OTf] (8), identified by comparison with the 1H NMR spectrum of an authentic sample of 8 (see ESI and Fig. S18†), along with a new set of paramagnetically shifted signals. Crystallization from a concentrated pentane solution afforded yellow crystals of 7 suitable for XRD studies. Its molecular structure (Fig. 8) showed an unusual CO tetramerization product featuring a deprotonated 3,4-dihydroxyfuran-2-one moiety bridging two Tm(III) metal centers.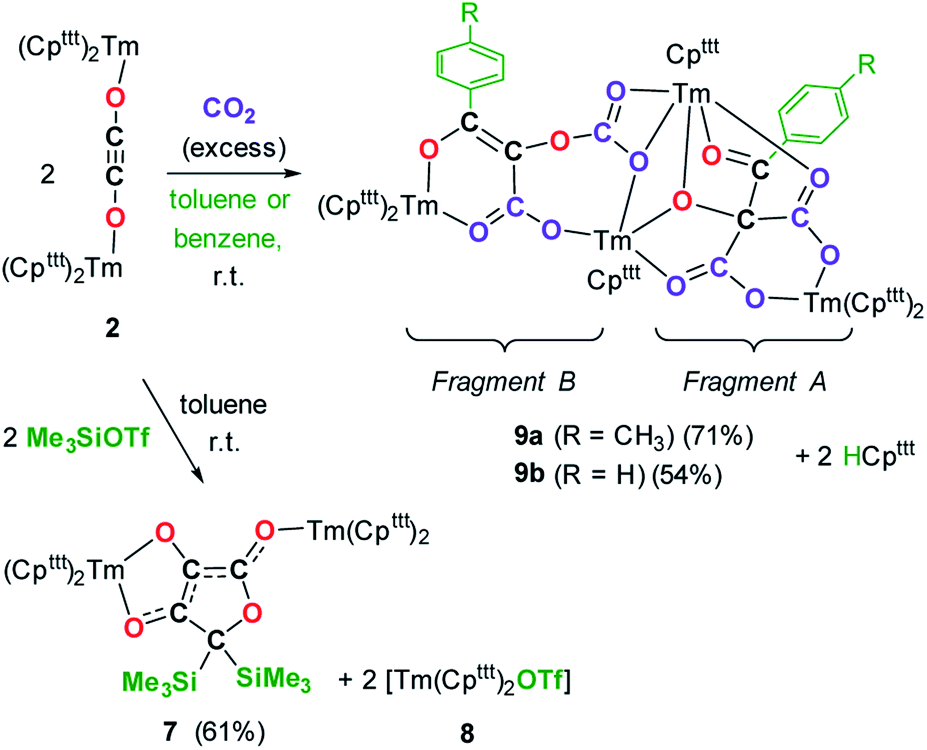 | ||
| Scheme 3 Reaction of 2 with CO2 and Me3SiOTf. | ||
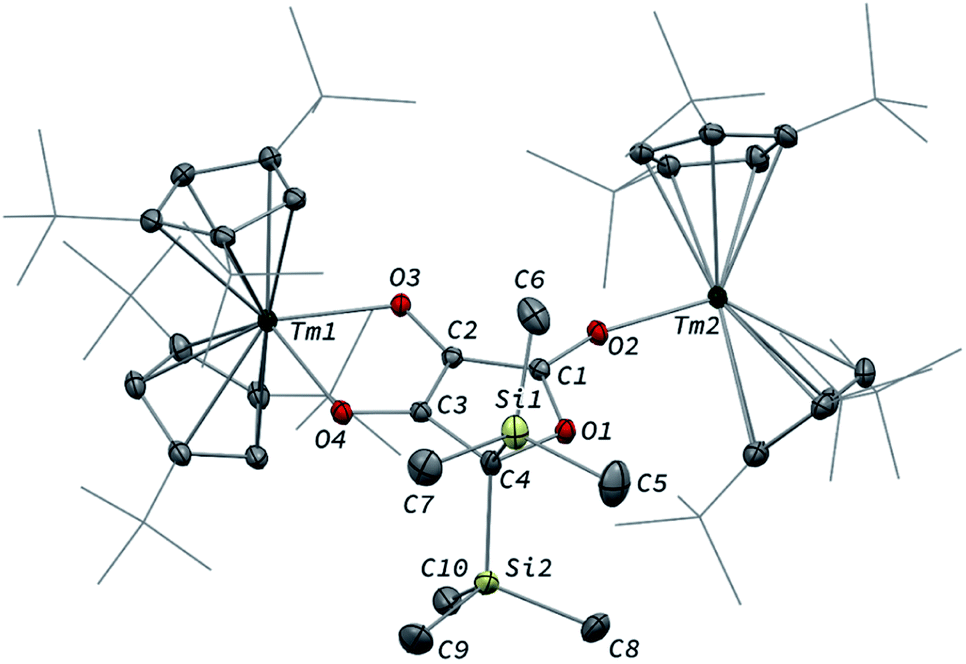 | ||
| Fig. 8 Molecular structure of 7 in the solid state with thermal ellipsoids at the 40% probability level (except for the tBu groups depicted in wireframe). H atoms have been omitted for clarity. Selected bond distances (Å) and angles [°]: Tm1–O3 2.200(2), Tm1–O4 2.333(2), Tm2–O2 2.146(2), O1–C1 1.368(3), O1–C4 1.486(3), O2–C1 1.280(3), O3–C2 1.339(3), O4–C3 1.290(3), C1–C2 1.397(3), C2–C3 1.396(3), C3–C4 1.494(3); O3–Tm1–O4 74.78(6), C1–O2–Tm2 163.1(2), C2–O3–Tm1 112.88(13), C3–O4–Tm1 109.86(14). | ||
The bond distances within the oxygen heterocycle are consistent with a conjugated system and delocalization of one negative charge over O2 and O4, similar to the case in β-diketonate ligands. As a result, the C1–C2 and C2–C3 bond distances are almost identical (1.397(3) and 1.396(3) Å, respectively), as are the O2–C1 and O4–C3 bond distances (1.280(3) and 1.290(3) Å, respectively). In contrast, the unconjugated O3–C2 bond is longer with a distance of 1.339(3) Å. The Tm–O bond distances are lying in the range 2.200(2)–2.333(2) Å. The paramagnetic 1H NMR spectrum of 7 features a complex baseline over the temperature range 20–80 °C (see Fig. S17†), which results from the low symmetry of the complex in solution. In the IR spectrum, the coordinated β-diketonate group gives rise to an intense absorption band at 1639 cm−1.
Interestingly, reaction with Me3SiOTf did not lead to decoordination of the ethynediolate ligand, contrary to the reactivity observed by Liddle and co-workers upon treatment of a related tris-amido U(IV) ethynediolate complex with a similar strong silyl electrophile.17i In this previous study, the reaction with Me3SiI led to the silylation of the hydroxy groups and liberation of the ethynediolate fragment, which subsequently dimerized and rearranged into a spectroscopically identified furanone product. Indeed, acetylene diether compounds have been found to be stable at room temperature only in the presence of bulky substituents,41 or η2-coordinated to transition metals.16b The X-ray authenticated complex 7 unambiguously shows the formation of a similar furanone moiety in which the two trimethylsilyl groups are attached at the C4 position of the heterocycle rather than the O3 and O4 oxygen atoms, the latter remaining coordinated to the metal center. The mechanism leading to the formation of 7 is thought to involve electrophilic activation of the ethynediolate by the silyl electrophile, which contrasts with the proton-induced transformation reported earlier.17i The exploration of other functionalization agents to allow the liberation of the ethynediolate moiety is currently in progress.
We next evaluated the reactivity of 2 towards CO2 (Scheme 3). The addition of CO2 (1.3 bar, excess) to a solution of 2 in toluene led to an immediate reaction at room temperature. After work-up, red crystals of 9a suitable for XRD studies were obtained in good yield (71%) from a concentrated pentane solution. The molecular structure of the complex is depicted in Fig. 9 and reveals an unexpected product in which C–H activation at the para position of the toluene solvent occurred, yielding an unprecedented polyoxygenated ligand framework. In the resulting tetranuclear Tm(III) complex, only six Cpttt ligands are present, suggesting that two Cpttt ligands have been released in their protonated form. The formation of HCpttt was confirmed by 1H NMR analysis of the volatiles of the reaction (see details in the ESI and Fig. S23†). The thulium centers are hold together via two isomeric bridging {μ3-C7H7(C4O6)}2− fragments. Fragment A (see Scheme 3) is best described by a μ3-κ2(O1,O3):κ2(O2,O5):κ3(O4,O5,O6)–C7H7(C4O6) ligand based on a 2-hydroxy-2-(p-toluoyl)malonate framework while fragment B corresponds to a μ3-κ2(O7,O8):κ2(O7,O10):κ2(O11,O12)–C7H7(C4O6) ligand derived from 2-(carboxyoxy)-3-hydroxy-3-(p-tolyl)acrylic acid (a simplified view in which the Cpttt ligands have been omitted is depicted in Fig. S52†). Both Tm1 and Tm4 are coordinated by two η5–Cpttt ligands and two oxygen atoms with Tm–O bond distances in the range 2.195(3)–2.273(3) Å. In contrast, Tm2 and Tm3 feature half-sandwich arrangements, both surrounded by one η5–Cpttt ligand along with four and five oxygen donors, respectively. The corresponding Tm–O bond distances are spanning a larger range (2.156(3)–2.643(3) Å), as a result of the different types of oxygen donors, namely alkoxide, ketone, carboxylate and carbonate groups. The tetrasubstituted carbon C2 in fragment A features C–C bond distances of 1.550(5)–1.567(5) Å corresponding to single bonds, whereas the C13–C14 and C14–C15 bond distances in fragment B of 1.426(6) and 1.384(6) Å, respectively, are more consistent with a delocalized double bond within the 3-hydroxyacrylate moiety. Accordingly, the sum of the bonding angles around C14 and C15 of 360.0° is indicative of sp2 hybridization.
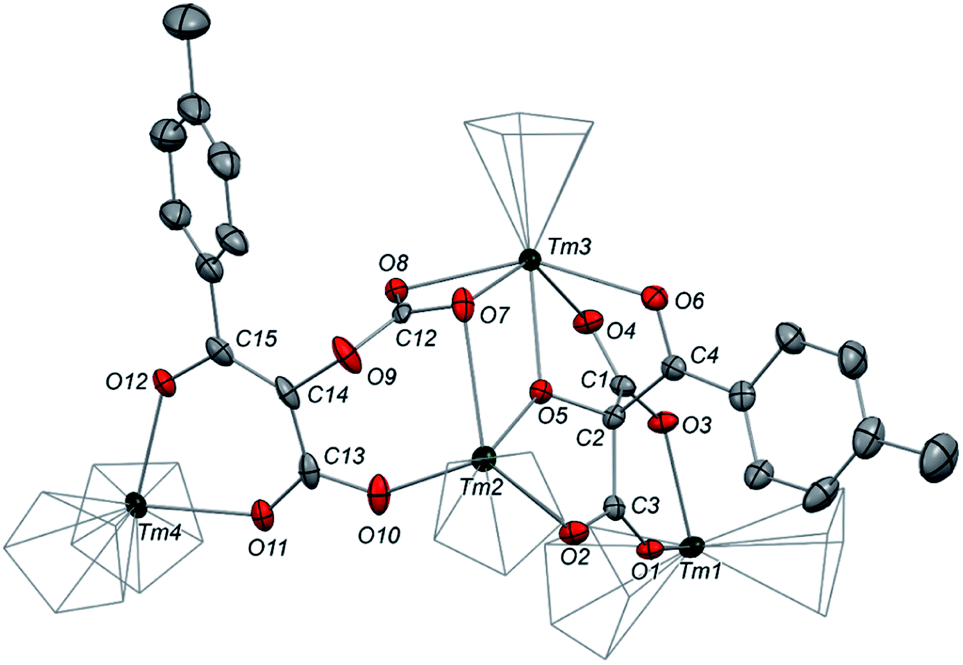 | ||
| Fig. 9 Molecular structure of 9a in the solid state with thermal ellipsoids at the 40% probability level (except for the Cp groups depicted in wireframe). H atoms, tBu groups and non-coordinating solvent molecules have been omitted for clarity. Selected bond distances (Å): Tm1–O1 2.273(3), Tm1–O3 2.242(3), Tm2–O2 2.266(3), Tm2–O5 2.263(3), Tm2–O7 2.643(3), Tm2–O10 2.156(3), Tm3–O4 2.261(3), Tm3–O5 2.294(3), Tm3–O6 2.386(3), Tm3–O7 2.428(3), Tm3–O8 2.333(3), Tm4–O11 2.234(3), Tm4–O12 2.195(3), O1–C3 1.244(4), O2–C3 1.252(5), O3–C1 1.248(4), O4–C1 1.259(4), O5–C2 1.402(4), O6–C4 1.235(5), O7–C12 1.271(4), O8–C12 1.231(5), O9–C12 1.338(5), O9–C14 1.428(5), O10–C13 1.291(5), O11–C13 1.260(5), O12–C15 1.300(5), C1–C2 1.565(5), C2–C3 1.550(5), C2–C4 1.567(5), C13–C14 1.426(6), C14–C15 1.384(6). | ||
The 1H NMR spectrum of 9a in toluene-d8 (Fig. S20†) shows a very complex pattern with several paramagnetically shifted resonances in the range δ −360 to +300 ppm, as a result of the C1 symmetric nature of 9a. The signals corresponding to the p-tolyl fragments could be successfully assigned by comparison with the spectrum of the partly deuterated 9a-2H (see Fig. S21–S22†), the latter prepared using the same procedure but in toluene-d8 instead of protio-toluene.
The IR spectrum of 9a features a very intense absorption band at 1638 cm−1, consistent with a coordinated ketone group, along with strong absorption bands at 1585 and 1552 cm−1, which can be assigned to the carboxylate groups or delocalized CC bond in the newly formed ligand. Performing the same reaction in benzene instead of toluene as solvent led to the isolation of complex 9b which X-ray structure revealed identical bonding and substitution patterns as observed in 9a (see Fig. S53 and S54†).
A detailed study of the elementary steps leading to the formation of 9a–9b is outside the scope of this publication and will be disclosed in due course. A possible simplified mechanism to account for the formation of the two polyoxygenated fragments is depicted in Scheme 4. In both cases, it would begin with an electrophilic aromatic substitution on the toluene or benzene solvent induced by the interaction of the ethynediolate complex 2 with CO2. The acidic proton in the Wheland intermediate is trapped by one Cpttt ligand, resulting in the release of HCpttt, which was observed spectroscopically. This first step could therefore be seen as a Friedel–Crafts-type reaction leading to the functionalization of toluene or benzene and formation of a deprotonated form of 2,3-dihydroxy-3-(p-tolyl)acrylate. This polyoxygenated intermediate further reacts with one more equivalent of CO2 either at the nucleophilic carbon atom or at the non-conjugated anionic oxygen atom, yielding fragments A or B, respectively. It should be noted that this reaction proceeds smoothly at room temperature and contrasts with the thermolysis reactivity of some uranium(IV) ethynediolate complexes that were found to undergo ligand degradation or intramolecular C–H activation at elevated temperature, leading to ethenediolate-type complexes.17h,i
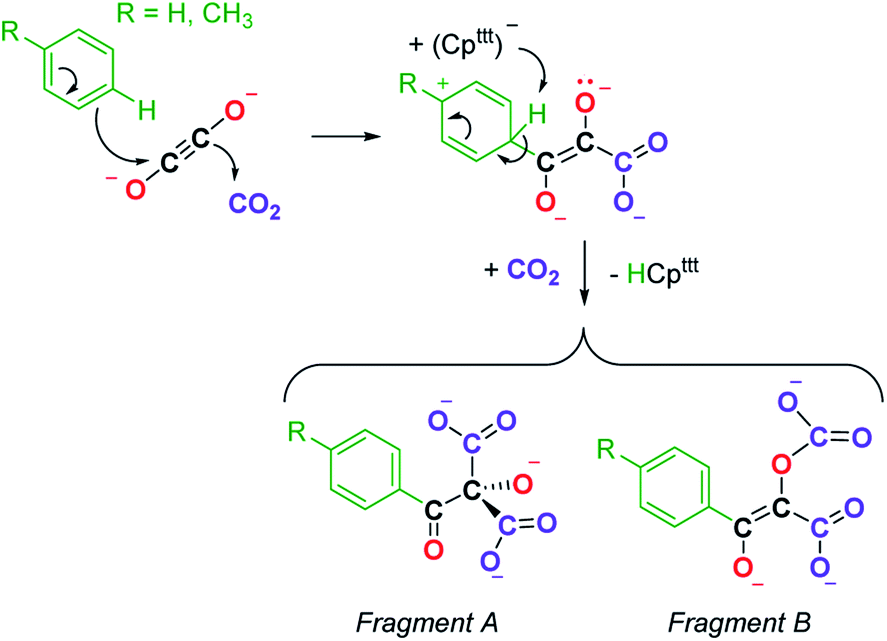 | ||
| Scheme 4 Possible simplified mechanism in the formation of 9a–9b leading to the formation of fragments A and B. | ||
The observed C–H activation of toluene and benzene in 9a and 9b, respectively, is reminiscent of the C–H activation at the coordinated 4-dimethylaminopyridine (DMAP) ligand that has been described upon addition of CO to an activated Mg(I) dimer complex (see Fig. 1).15g In this case, DFT calculations suggested that the C–H activation is induced by a “zig-zag” {C2O2}2− intermediate similar to iso2 (Fig. 5).
The major difference in the case of 2 in comparison to other systems lies in its lack of reactivity in toluene in the absence of CO2, even at elevated temperatures (up to 100 °C). Only upon addition of CO2 is a highly reactive system formed that allows intermolecular C–H activation on toluene or benzene.
Conclusions
In conclusion, the divalent thulium complex [Tm(Cpttt)2] exhibits a rich reactivity towards CO and CO2 allowing the selective formation of carbonate (C1), ethynediolate (C2) and ketenecarboxylate (C3) complexes. The reactivity of the CO dimerization product 2 with external CO at room temperature is remarkable and proves the intermediacy of 2 in the formation of the ketenecarboxylate complex 3, which was also supported by DFT calculations. The reactivity of 2 and 3 was systematically investigated towards electrophiles (see Scheme 5 for a summary) leading to the following observations: (a) while 3 features an expected nucleophilic reactivity towards CO2, the addition of methyl iodide yields a C7 allene–tricarboxylate complex in which the C6O6 core is exclusively built from CO molecules; (b) the ethynediolate complex 2 shows unexpected reactivity towards CO2 and silylating agents. In the case of CO2, a highly reactive species is formed, which is able to induce an intermolecular C–H activation on the solvent. This novel reactivity opens new avenues for the formation of multicarbon oxygenated compounds by small molecule activation and the functionalization of CO and CO2 to value-added chemicals. The exploration of new procedures to enable decoordination of the oxygenated products and further catalytic applications are currently in progress in our laboratory.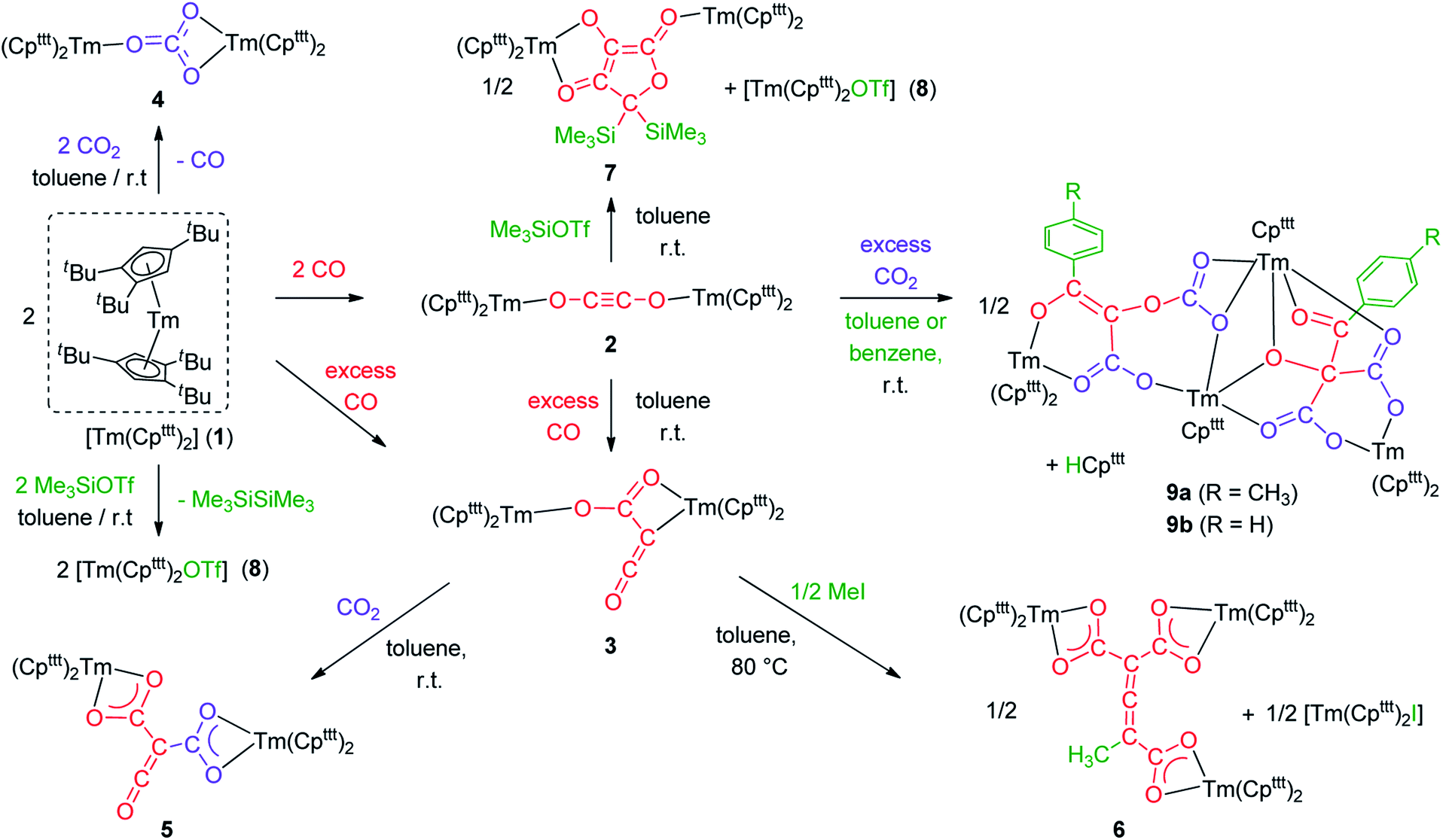 | ||
| Scheme 5 Summary scheme of all the reactivity described in this study starting from [Tm(Cpttt)2] (1). | ||
Data availability
Crystallographic data for 1–7, 9a and 9b have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2118417–2118424, 2128950) and can be obtained from https://www.ccdc.cam.ac.uk/structures. Other primary data (NMR and IR spectra) can be downloaded from the Zenodo repository under DOIs: https://doi.org/10.5281/zenodo.6545548 (for 1), https://doi.org/10.5281/zenodo.6545724 (for 2), https://doi.org/10.5281/zenodo.6545746 (for 3), https://doi.org/10.5281/zenodo.6545760 (for 4), https://doi.org/10.5281/zenodo.6545770 (for 5), https://doi.org/10.5281/zenodo.6545778 (for 6), https://doi.org/10.5281/zenodo.6545826 (for 7), https://doi.org/10.5281/zenodo.6545851 (for 8), https://doi.org/10.5281/zenodo.6545875 (for 9a), https://doi.org/10.5281/zenodo.6545898 (for 9b).Author contributions
TXS: conceptualization, experimental data collection, and analysis; KNM: computational analysis; LM: computational analysis, validation; GN: conceptualization, project administration, data analysis, validation, funding acquisition. All authors contributed to the writing, editing, and revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union’ Horizon H2020 research program (grant agreement No. 716314). CNRS and Ecole Polytechnique are thanked for funding. LM is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.Notes and references
- Activation of Small Molecules: Organometallic and Bioinorganic Perspectives, ed. W. B. Tolman, Wiley-VCH Verlag, Weinheim, 2006, pp. 1–363 Search PubMed.
- (a) K. Klier, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1982, vol. 31, pp. 243–313 Search PubMed; (b) P. M. Jones, J. A. May, J. B. Reitz and E. I. Solomon, Inorg. Chem., 2004, 43, 3349–3370 CrossRef CAS PubMed.
- J. H. Jones, Platinum Met. Rev., 2000, 44, 94–105 CAS.
- (a) F. Fischer and H. Tropsch, Brennst.-Chem., 1926, 7, 97–104 CAS; (b) C. Masters, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Academic Press, 1979, vol. 17, pp. 61–103 Search PubMed; (c) N. M. West, A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Coord. Chem. Rev., 2011, 255, 881–898 CrossRef CAS; (d) A. de Klerk, in Kirk-Othmer Encyclopedia of Chemical Technology, Wiley-VCH, Weinheim, 2013, pp. 1–20 Search PubMed.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981–8995 CrossRef CAS PubMed.
- (a) C. K. Rofer-DePoorter, Chem. Rev., 1981, 81, 447–474 CrossRef CAS; (b) A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef CAS PubMed.
- (a) P. T. Wolczanski and J. E. Bercaw, Acc. Chem. Res., 1980, 13, 121–127 CrossRef CAS; (b) W. A. Herrmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 117–130 CrossRef; (c) R. Whyman, A. P. Wright, J. A. Iggo and B. T. Heaton, J. Chem. Soc., Dalton Trans., 2002, 771–777 RSC; (d) J. A. Buss and T. Agapie, Nature, 2016, 529, 72–75 CrossRef CAS PubMed; (e) J. A. Labinger, J. Organomet. Chem., 2017, 847, 4–12 CrossRef CAS; (f) J. A. Buss, G. A. Bailey, J. Oppenheim, D. G. VanderVelde, W. A. Goddard and T. Agapie, J. Am. Chem. Soc., 2019, 141, 15664–15674 CrossRef CAS PubMed; (g) J. Ohata, A. Teramoto, H. Fujita, S. Takemoto and H. Matsuzaka, J. Am. Chem. Soc., 2021, 143, 16105–16112 CrossRef CAS PubMed.
- (a) A. Phanopoulos, S. Pal, T. Kawakami and K. Nozaki, J. Am. Chem. Soc., 2020, 142, 14064–14068 CrossRef CAS PubMed; (b) M. Xu, Z.-w. Qu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2021, 143, 634–638 CrossRef CAS PubMed.
- (a) W. J. Evans, A. L. Wayda, W. E. Hunter and J. L. Atwood, J. Chem. Soc., Chem. Commun., 1981, 706–708 RSC; (b) T. J. Marks, Science, 1982, 217, 989–997 CrossRef CAS PubMed; (c) K. G. Moloy and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 7051–7064 CrossRef CAS.
- H. R. Sharpe, A. M. Geer, L. J. Taylor, B. M. Gridley, T. J. Blundell, A. J. Blake, E. S. Davies, W. Lewis, J. McMaster, D. Robinson and D. L. Kays, Nat. Commun., 2018, 9, 3757 CrossRef PubMed.
- (a) W. J. Evans, J. W. Grate and R. J. Doedens, J. Am. Chem. Soc., 1985, 107, 1671–1679 CrossRef CAS; (b) C. C. Cummins, G. D. Van Duyne, C. P. Schaller and P. T. Wolczanski, Organometallics, 1991, 10, 164–170 CrossRef CAS; (c) E. L. Werkema, L. Maron, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 2529–2541 CrossRef CAS PubMed; (d) R. Lalrempuia, C. E. Kefalidis, S. J. Bonyhady, B. Schwarze, L. Maron, A. Stasch and C. Jones, J. Am. Chem. Soc., 2015, 137, 8944–8947 CrossRef CAS PubMed; (e) M. D. Anker, C. E. Kefalidis, Y. Yang, J. Fang, M. S. Hill, M. F. Mahon and L. Maron, J. Am. Chem. Soc., 2017, 139, 10036–10054 CrossRef CAS PubMed; (f) S. Hu, T. Shima and Z. Hou, J. Am. Chem. Soc., 2020, 142, 19889–19894 CrossRef CAS PubMed.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897–4901 CrossRef CAS PubMed.
- B. Wang, G. Luo, M. Nishiura, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16967–16973 CrossRef CAS PubMed.
- (a) R. Y. Kong and M. R. Crimmin, Dalton Trans., 2020, 49, 16587–16597 RSC; (b) U. Rosenthal, Chem.–Eur. J., 2020, 26, 14507–14511 CrossRef CAS PubMed; (c) S. Fujimori and S. Inoue, Commun. Chem., 2020, 3, 175 CrossRef CAS; (d) S. Fujimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef CAS PubMed.
- (a) H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025–1028 CrossRef CAS PubMed; (b) M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 8746–8750 CrossRef CAS PubMed; (c) R. Y. Kong and M. R. Crimmin, J. Am. Chem. Soc., 2018, 140, 13614–13617 CrossRef CAS PubMed; (d) Y. Wang, A. Kostenko, T. J. Hadlington, M.-P. Luecke, S. Yao and M. Driess, J. Am. Chem. Soc., 2019, 141, 626–634 CrossRef CAS PubMed; (e) K. Yuvaraj, I. Douair, A. Paparo, L. Maron and C. Jones, J. Am. Chem. Soc., 2019, 141, 8764–8768 CrossRef CAS PubMed; (f) A. V. Protchenko, P. Vasko, D. C. H. Do, J. Hicks, M. Á. Fuentes, C. Jones and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 1808–1812 CrossRef CAS PubMed; (g) K. Yuvaraj, I. Douair, D. D. L. Jones, L. Maron and C. Jones, Chem. Sci., 2020, 11, 3516–3522 RSC; (h) Y. Xiong, S. Yao, T. Szilvási, A. Ruzicka and M. Driess, Chem. Commun., 2020, 56, 747–750 RSC; (i) A. Paparo, K. Yuvaraj, A. J. R. Matthews, I. Douair, L. Maron and C. Jones, Angew. Chem., Int. Ed., 2021, 60, 630–634 CrossRef CAS PubMed; (j) H.-Y. Liu, R. J. Schwamm, S. E. Neale, M. S. Hill, C. L. McMullin and M. F. Mahon, J. Am. Chem. Soc., 2021, 143, 17851–17856 CrossRef CAS PubMed; (k) R. Y. Kong, M. Batuecas and M. R. Crimmin, Chem. Sci., 2021, 12, 14845–14854 RSC; (l) M. Batuecas, R. Y. Kong, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2022, e202202241 CAS.
- (a) P. A. Bianconi, I. D. Williams, M. P. Engeler and S. J. Lippard, J. Am. Chem. Soc., 1986, 108, 311–313 CrossRef CAS; (b) E. M. Carnahan, J. D. Protasiewicz and S. J. Lippard, Acc. Chem. Res., 1993, 26, 90–97 CrossRef CAS; (c) B. B. Wayland, A. E. Sherry and V. L. Coffin, J. Chem. Soc., Chem. Commun., 1989, 662–663 RSC; (d) T. Watanabe, Y. Ishida, T. Matsuo and H. Kawaguchi, J. Am. Chem. Soc., 2009, 131, 3474–3475 CrossRef CAS PubMed; (e) M. Fang, J. H. Farnaby, J. W. Ziller, J. E. Bates, F. Furche and W. J. Evans, J. Am. Chem. Soc., 2012, 134, 6064–6067 CrossRef CAS PubMed.
- (a) W. J. Evans, J. W. Grate, L. A. Hughes, H. Zhang and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 3728–3730 CrossRef CAS; (b) W. J. Evans, D. S. Lee, J. W. Ziller and N. Kaltsoyannis, J. Am. Chem. Soc., 2006, 128, 14176–14184 CrossRef CAS PubMed; (c) B. Wayland and X. Fu, Science, 2006, 311, 790–791 CrossRef CAS PubMed; (d) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829–831 CrossRef CAS PubMed; (e) O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602–9603 CrossRef CAS PubMed; (f) A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed; (g) S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed; (h) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC; (i) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed; (j) N. Tsoureas, O. T. Summerscales, F. G. N. Cloke and S. M. Roe, Organometallics, 2013, 32, 1353–1362 CrossRef CAS; (k) R. Yadav, T. Simler, M. T. Gamer, R. Köppe and P. W. Roesky, Chem. Commun., 2019, 55, 5765–5768 RSC; (l) A. J. Ryan, J. W. Ziller and W. J. Evans, Chem. Sci., 2020, 11, 2006–2014 RSC.
- (a) G. Aitken, N. Hazari, A. S. P. Frey, F. G. N. Cloke, O. Summerscales and J. C. Green, Dalton Trans., 2011, 40, 11080–11088 RSC; (b) D. McKay, A. S. P. Frey, J. C. Green, F. G. N. Cloke and L. Maron, Chem. Commun., 2012, 48, 4118–4120 RSC.
- F. Nief, Dalton Trans., 2010, 39, 6589–6598 RSC.
- (a) M. Xémard, V. Goudy, A. Braun, M. Tricoire, M. Cordier, L. Ricard, L. Castro, E. Louyriac, C. E. Kefalidis, C. Clavaguéra, L. Maron and G. Nocton, Organometallics, 2017, 36, 4660–4668 CrossRef; (b) V. Goudy, A. Jaoul, M. Cordier, C. Clavaguéra and G. Nocton, J. Am. Chem. Soc., 2017, 139, 10633–10636 CrossRef CAS PubMed; (c) M. Xémard, M. Cordier, E. Louyriac, L. Maron, C. Clavaguéra and G. Nocton, Dalton Trans., 2018, 47, 9226–9230 RSC; (d) D. Wang, J. Moutet, M. Tricoire, M. Cordier and G. Nocton, Inorganics, 2019, 7, 58 CrossRef CAS PubMed.
- (a) F. Jaroschik, F. Nief and L. Ricard, Chem. Commun., 2006, 426–428 RSC; (b) F. Jaroschik, F. Nief, X.-F. Le Goff and L. Ricard, Organometallics, 2007, 26, 3552–3558 CrossRef CAS.
- (a) F. Weber, M. Schultz, C. D. Sofield and R. A. Andersen, Organometallics, 2002, 21, 3139–3146 CrossRef CAS; (b) G. Nocton and L. Ricard, Dalton Trans., 2014, 43, 4380–4387 RSC; (c) H. Sitzmann, T. Dezember, O. Schmitt, F. Weber, G. Wolmershäuser and M. Ruck, Z. Anorg. Allg. Chem., 2000, 626, 2241–2244 CrossRef CAS.
- (a) R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef; (b) M. D. Walter, G. Wolmershäuser and H. Sitzmann, J. Am. Chem. Soc., 2005, 127, 17494–17503 CrossRef CAS PubMed.
- (a) M. N. Bochkarev, Coord. Chem. Rev., 2004, 248, 835–851 CrossRef CAS; (b) J. Moutet, J. Schleinitz, L. La Droitte, M. Tricoire, F. Pointillart, F. Gendron, T. Simler, C. Clavaguéra, B. Le Guennic, O. Cador and G. Nocton, Angew. Chem., Int. Ed., 2021, 60, 6042–6046 CrossRef CAS.
- W. J. Evans, T. A. Ulibarri and J. W. Ziller, J. Am. Chem. Soc., 1988, 110, 6877–6879 CrossRef CAS.
- (a) W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7927–7928 CrossRef CAS PubMed; (b) I. L. Fedushkin, F. Girgsdies, H. Schumann and M. N. Bochkarev, Eur. J. Inorg. Chem., 2001, 2001, 2405–2410 CrossRef; (c) W. J. Evans, N. T. Allen and J. W. Ziller, Angew. Chem., Int. Ed., 2002, 41, 359–361 CrossRef CAS; (d) F. Nief, D. Turcitu and L. Ricard, Chem. Commun., 2002, 1646–1647 RSC; (e) F. Nief, B. T. de Borms, L. Ricard and D. Carmichael, Eur. J. Inorg. Chem., 2005, 2005, 637–643 CrossRef.
- (a) W. J. Evans, Organometallics, 2016, 35, 3088–3100 CrossRef CAS; (b) M. T. Trinh, J. C. Wedal and W. J. Evans, Dalton Trans., 2021, 50, 14384–14389 RSC.
- S. Cotton, in Lanthanide and Actinide Chemistry, John Wiley & Sons, Ltd, Chichester (U.K.), 2006, pp. 1–263 Search PubMed.
- (a) L. J. Bowman, K. Izod, W. Clegg and R. W. Harrington, Organometallics, 2007, 26, 2646–2651 CrossRef CAS; (b) T. J. Boyle, L. A. M. Ottley, S. D. Daniel-Taylor, L. J. Tribby, S. D. Bunge, A. L. Costello, T. M. Alam, J. C. Gordon and T. M. McCleskey, Inorg. Chem., 2007, 46, 3705–3713 CrossRef CAS PubMed; (c) T. Sanden, M. T. Gamer, A. A. Fagin, V. A. Chudakova, S. N. Konchenko, I. L. Fedushkin and P. W. Roesky, Organometallics, 2012, 31, 4331–4339 CrossRef CAS; (d) T. J. Boyle, M. L. Neville, J. M. Sears, R. E. Cramer, M. A. Rodriguez, T. M. Alam and S. P. Bingham, Polyhedron, 2016, 118, 52–60 CrossRef CAS.
- (a) M. Janjetovic, A. M. Träff, T. Ankner, J. Wettergren and G. Hilmersson, Chem. Commun., 2013, 49, 1826–1828 RSC; (b) T. V. Chciuk, G. Hilmersson and R. A. Flowers, J. Org. Chem., 2014, 79, 9441–9443 CrossRef CAS PubMed.
- (a) G. R. Choppin, J. Alloys Compd., 2002, 344, 55–59 CrossRef CAS; (b) M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- J. M. Veauthier, E. J. Schelter, C. N. Carlson, B. L. Scott, R. E. D. Re, J. D. Thompson, J. L. Kiplinger, D. E. Morris and K. D. John, Inorg. Chem., 2008, 47, 5841–5849 CrossRef CAS PubMed.
- U. Bayer and R. Anwander, Dalton Trans., 2020, 49, 17472–17493 RSC.
- A. M. Greenaway, T. P. Dasgupta, K. C. Koshy and G. G. Sadler, Spectrochim. Acta, Part A, 1986, 42, 949–954 CrossRef.
- J. S. Jestilä and E. Uggerud, J. Org. Chem., 2019, 84, 14005–14014 CrossRef PubMed.
- L. Maron, E. L. Werkema, L. Perrin, O. Eisenstein and R. A. Andersen, J. Am. Chem. Soc., 2005, 127, 279–292 CrossRef CAS PubMed.
- (a) X. Zhou and M. Zhu, J. Organomet. Chem., 2002, 647, 28–49 CrossRef CAS; (b) V. Goudy, M. Xémard, S. Karleskind, M. Cordier, C. Alvarez Lamsfus, L. Maron and G. Nocton, Inorganics, 2018, 6, 82 CrossRef; (c) T. Simler, T. J. Feuerstein, R. Yadav, M. T. Gamer and P. W. Roesky, Chem. Commun., 2019, 55, 222–225 RSC; (d) U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef CAS PubMed.
- G. Maier, M. Heider and C. Sierakowski, Tetrahedron Lett., 1991, 32, 1961–1962 CrossRef CAS.
- R. M. Silverstein, F. X. Webster and D. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, Hoboken, NJ, 7th edn, 2005, pp. 1–512 Search PubMed.
- A. S. P. Frey, F. G. N. Cloke, M. P. Coles, L. Maron and T. Davin, Angew. Chem., Int. Ed., 2011, 50, 6881–6883 CrossRef CAS PubMed.
- (a) F. Serratosa, Acc. Chem. Res., 1983, 16, 170–176 CrossRef CAS; (b) R. Gleiter and D. B. Werz, Chem. Rev., 2010, 110, 4447–4488 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, 1H NMR and IR spectra, X-ray crystallographic details and DFT calculations. CCDC 2118417–2118424, 2128950. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01798a |
| This journal is © The Royal Society of Chemistry 2022 |