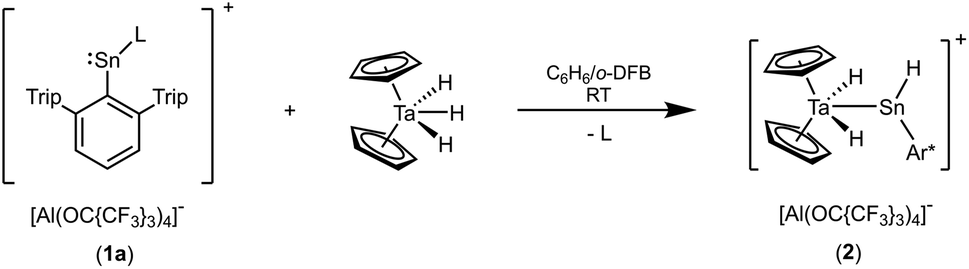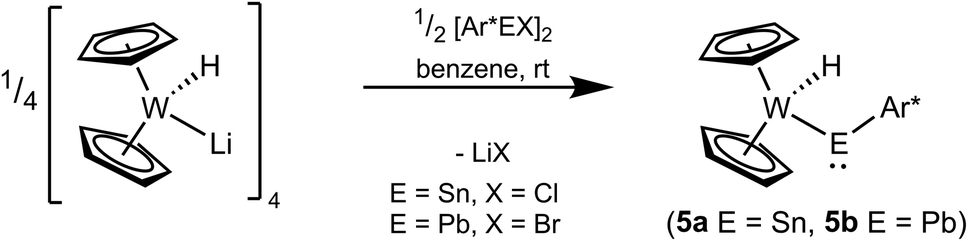DOI:
10.1039/D2SC00297C
(Edge Article)
Chem. Sci., 2022,
13, 3999-4009
Hydridotetrylene [Ar*EH] (E = Ge, Sn, Pb) coordination at tantalum, tungsten, and zirconium†
Received
17th January 2022
, Accepted 21st February 2022
First published on 22nd February 2022
Introduction
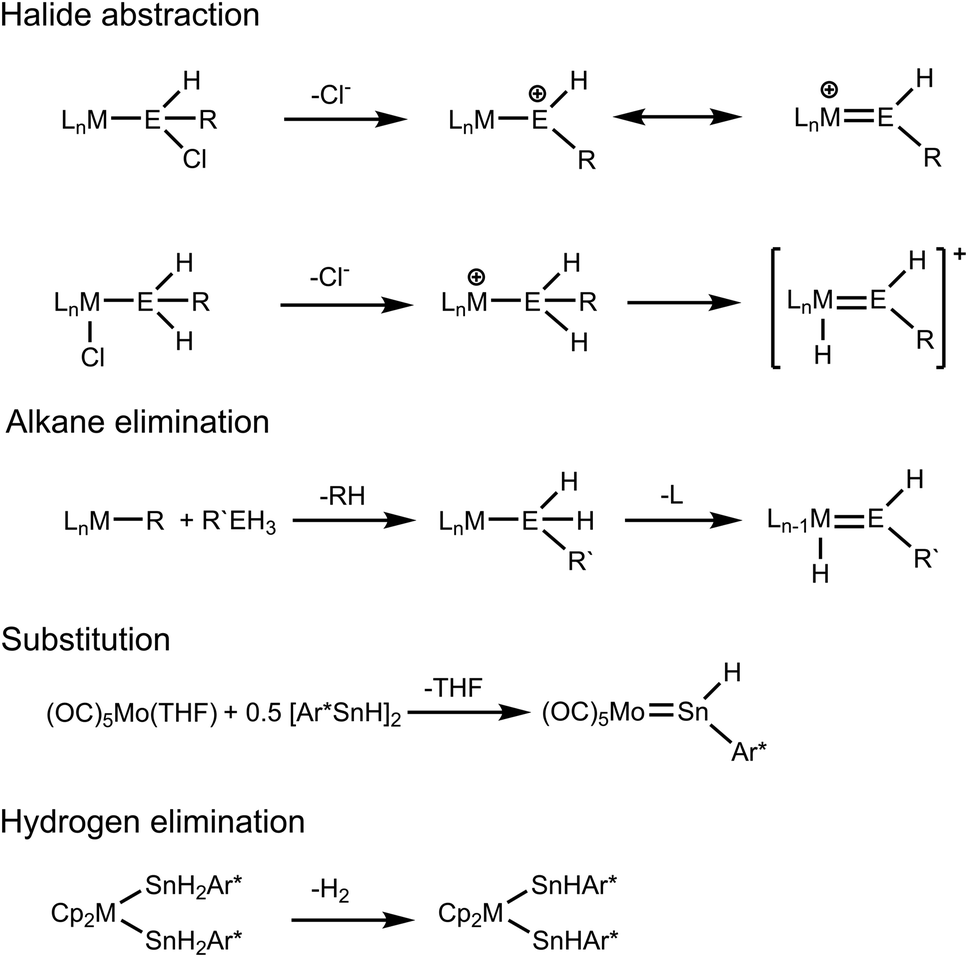
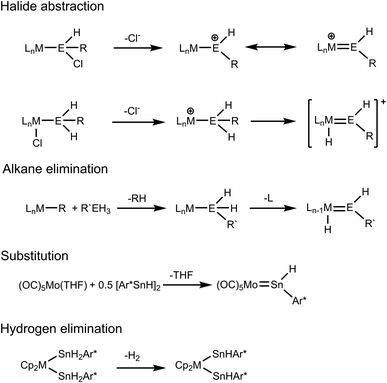
The coordination chemistry of tetrylenes [ER2; E = Si, Ge, Sn, Pb; R = aryl, alkyl, amide, alkoxide, silyl] has been studied successfully over the last fifty years.1–12 While numerous examples of tetrylene transition metal complexes and reports on their chemistry have been published, there is only a small number of hydridotetrylene coordination compounds in the literature. Hydridotetrylene complexes were synthesized by chloride abstraction from chlorosilyl or germyl ligands and by chloride abstraction from transition metal substituted chloride complexes followed by a 1,2-H migration (Scheme 1).13–15 The research groups of Tilley and Tobita developed the alkane elimination route for the syntheses of hydridotetrylene coordination compounds by reacting organometallic alkyl complexes with organoelement trihydrides of Si, Ge, and Sn (Scheme 1). After transfer of the tetryl group to the transition metal, an open coordination site allows 1,2-H migration to give the hydridotetrylene ligand.16–28 Recently Power et al. presented an example for a nucleophilic substitution using the low valent tin hydride Ar*SnH [Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl] as a ligand to replace a coordinating THF (Scheme 1).29 This research group investigated the coordination chemistry of anions [Ar*GeH2]− and [Ar*SnH2]− in reaction with group 4 metallocene dihalides. Interestingly after substitution of the halides, the stannyl complexes spontaneously eliminate hydrogen to give the bis(hydridostannylene) coordination compounds (M = Zr, Hf).30 Stabilized as N-heterocyclic carbene adducts, dihydrogermylene and stannylene were synthesized and coordinated as ligands at tungsten pentacarbonyl fragments.31,32
 |
| | Scheme 1 Reported synthetic methods for hydridotetrylene complexes. | |
Low-coordinate cations of heavy Group 14 elements are attractive compounds for reactivity studies.33 Due to their high sensitivity, preparation of these cations was realized using bulky ligands.34,35 Cp* ligands as well as terphenyl derivatives are prominent substituents in this context and cations like [Cp*E]+ (E = Ge, Sn, Pb) and [Ar*E–L]+ (E = Sn, L = NHC, PtBu3; E = Pb, L = toluene) were reported.34,36–40 Furthermore, amides provided with sterically demanding groups stabilize Group 14 element cations, which exhibit further contacts with flanking aryl or imine groups.41–43 Chelating ligands like diketiminates or aminotroponiminates have also been employed for synthesis of low-coordinate cations of Ge, Sn and Pb.44,45 So far, reactivity studies of those low valent cations focus on studies of oxidative additions.33,35,46 Thus, Tobita and co-workers studied insertion of a cationic metallogermylene into E–H bonds (E = H, B, and Si).35 We present in this publication reactions of low-valent Group 14 cations of Sn and Pb with bis(cyclopentadienyl) hydrides of zirconium, tantalum and tungsten resulting in the formation hydridotetrylene coordination compounds. The metallocene hydrides were chosen because of their good accessibility and different electron configurations. In the case of germanium, a reversible interconversion between a coordinated hydridogermylene and hydrido-metallogermylene is presented.
Results and discussion
Both starting materials, the low valent cations of tin and lead, were synthesized straightforwardly as salts of weakly coordinating anions. Low valent hydrides of tin and lead [(Ar*EH)2] (E = Sn, Pb)47–49 stabilized by a bulky terphenyl substituent Ar* [Ar* = 2,6-Trip2C6H3, Trip = 2,4,6-triisopropylphenyl] were reacted with the hydride abstraction reagent [Ph3C][Al(OC{CF3}3)4] to give yellow [Ar*Sn–L][Al(OC{CF3}3)4] (1a) and orange [Ar*Pb–L][Al(OC{CF3}3)4] (1b) as crystalline material (L = solvent benzene).37,39,47–51
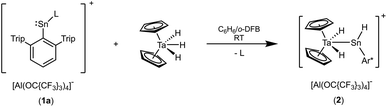
Dissolved in 1,2-difluorobenzene (o-DFB) the cations were reacted with one equivalent of a benzene solution of tantalocene trihydride Cp2TaH3. In the case of the tin cation a product was selectively formed (Scheme 2) and in the lead case a mixture of undefined reaction products was obtained. The tantalum complex 2 was isolated by crystallization and in Fig. 1 the molecular structure of the cation of 2 is depicted.
 |
| | Scheme 2 Reaction of the low valent tin cation of 1a with Cp2TaH3 (L = C6H6). | |
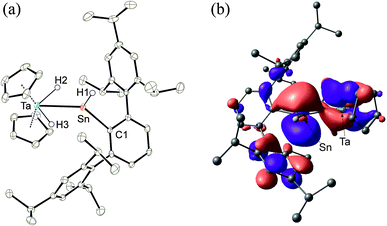 |
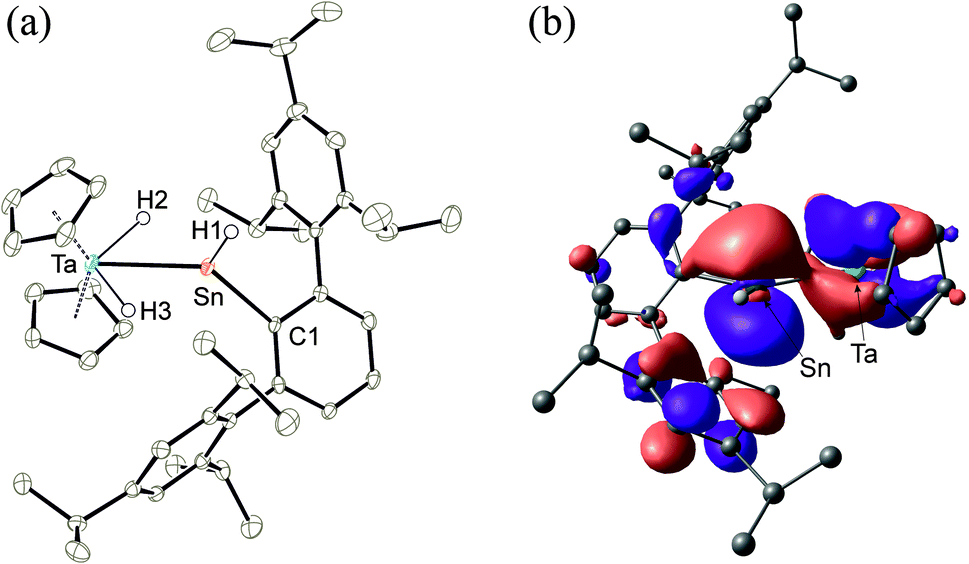
| | Fig. 1 Left: ORTEP of the molecular structure of the cation of 2. Thermal ellipsoids are shown at 50% probability level. The anion [Al(OC{CF3}3)4]− as well as co-crystallised 1,2-difluorobenzene have been omitted. Selected interatomic distances [Å] and angles [°]: Ta–Sn 2.7207(2), Sn–C1 2.161(3), Ta–H3 1.770(19), Ta–H2 1.81(3), Sn–H1 1.68(4), Sn–H2 2.18(3), Ta–Cp 2.372(3)–2.402(3), Ta–Sn–H1 115.7(14), Ta–Sn–C1 133.3(1); right (mirrored perspective): LUMO of the cation of 2. | |
In the cationic tantalum complex 2 a Ta–Sn bond is formed, and a hydride is transferred from the tantalum atom to the tin atom resulting in coordination of hydridoorgano stannylene [Ar*SnH]. The Ta–Sn bond length of 2.7207(2) Å can be compared with the Ta–Sn bond [2.752(1) Å] found in complex Cp2TaH2(SnCl2CH3).52 The LUMO of cation 2 shown in Fig. 1 exhibits to a larger extent an empty p-orbital at the tin atom. In the 1H NMR for the Ta–H units a doublet was found at −3.75 ppm (3JHH = 1.4 Hz), also showing coupling with the tin atom (2JSnH = 300 Hz). The signal for the Sn–H unit (t, 3JHH = 1.7 Hz) was found at a relatively high frequency, 15.55 ppm, showing a large 1JSn–H coupling constant (1JSnH = ca. 1040 Hz). Hydridostannylene coordination at osmium exhibits a comparable signal in the 1H NMR spectrum [Cp*(iPr3P)(H)Os![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Sn(H)trip, 1H NMR 19.4 ppm (SnH), 1JSnH = 775 Hz, 119Sn NMR 786 ppm].19 The 119Sn NMR signal of 2 at 1161 ppm (dt, 1JSnH = 1047 Hz, 2JSnH = 308 Hz) can be compared with signals found for triply coordinate stannylium cations [R3Sn]+ (R = Trip 714 ppm, R = Dur 720 ppm, R = tBu2MeSi 2653 ppm) (Trip = 2,4,6-iPr3C6H2, Dur = 2,3,5,6-Me4C6H).53–57 To rationalize the 119Sn NMR data of 2, NMR chemical shift calculations have been carried out using ORCA and ADF program packages.58–60 The calculated chemical shift of the tin atom of 2 amounts to 981 ppm (exp. 1161 ppm). Furthermore, in the optimized structure of 2, contacts between one of the aryl moieties of the terphenyl substituent and the tin atom were found. Thus, complex 2 can be regarded as a hydridostannylene complex with a stannylene acting as σ-donor ligand exhibiting no π-acceptor character. This results in an empty p-orbital at the tin atom and the triply coordinate tin atom can be regarded as a stannylium cation. Furthermore, a 119Sn NMR signal at 1161 ppm lies in the range of known triply coordinate stannylium cations (vide supra) and therefore interactions of the TaH units with the stannylium cation of 2 can be excluded.
Sn(H)trip, 1H NMR 19.4 ppm (SnH), 1JSnH = 775 Hz, 119Sn NMR 786 ppm].19 The 119Sn NMR signal of 2 at 1161 ppm (dt, 1JSnH = 1047 Hz, 2JSnH = 308 Hz) can be compared with signals found for triply coordinate stannylium cations [R3Sn]+ (R = Trip 714 ppm, R = Dur 720 ppm, R = tBu2MeSi 2653 ppm) (Trip = 2,4,6-iPr3C6H2, Dur = 2,3,5,6-Me4C6H).53–57 To rationalize the 119Sn NMR data of 2, NMR chemical shift calculations have been carried out using ORCA and ADF program packages.58–60 The calculated chemical shift of the tin atom of 2 amounts to 981 ppm (exp. 1161 ppm). Furthermore, in the optimized structure of 2, contacts between one of the aryl moieties of the terphenyl substituent and the tin atom were found. Thus, complex 2 can be regarded as a hydridostannylene complex with a stannylene acting as σ-donor ligand exhibiting no π-acceptor character. This results in an empty p-orbital at the tin atom and the triply coordinate tin atom can be regarded as a stannylium cation. Furthermore, a 119Sn NMR signal at 1161 ppm lies in the range of known triply coordinate stannylium cations (vide supra) and therefore interactions of the TaH units with the stannylium cation of 2 can be excluded.
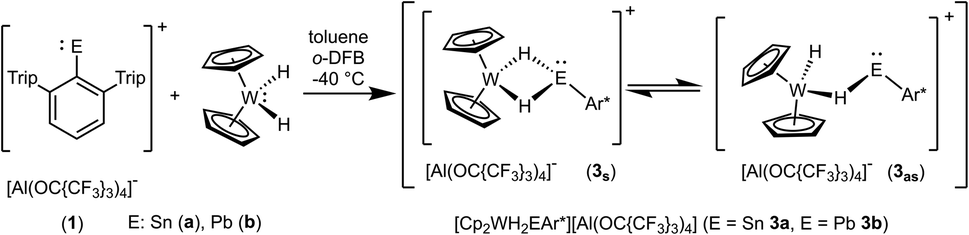
In contrast to tantalocene trihydride Cp2TaH3 the transition metal in tungstenocene dihydride Cp2WH2 is not in the maximum possible oxidation state but in state four and has two valence electrons. Reacting the cations [Ar*Sn]+1a and [Ar*Pb]+1b (see ESI for synthetic details†) with Cp2WH2 at low temperature (−40 °C) resulted in highly coloured reaction mixtures [E: Sn (3a) pink; Pb (3b) violet] (Scheme 3). Unlike complex 2, the resulting products 3a and 3b do not show any hydride transfer to the main group metal, instead, coordination of the Cp2WH2 fragment via the hydride substituents at the electrophilic Group 14 element was found.
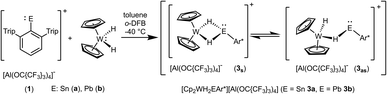 |
| | Scheme 3 Reaction of Cp2WH2 with low valent tin 1a and lead 1b cations. | |
In the 1H NMR spectrum both complexes 3a and 3b exhibit at rt a broad resonance for the hydride substituents [−10.83 (3a), −7.55 ppm (3b)]. Below −20 °C two different isomers are observable, a symmetric one (3s): 3as −10.86 ppm, JW–H 78 Hz; 3bs −7.49 ppm, JW–H 75 Hz; and an asymmetric one (3as): 3aas −11.13 ppm, JW–H 51 Hz, −8.57 ppm, JW–H 77 Hz; 3bas −12.31 ppm, JW–H 52 Hz, −4.30 ppm, JW–H 76 Hz (signal at higher frequency corresponds to the bridging W–H–E unit). Only in the 1H NMR spectra of 3a183W satellites were unambiguously observed. Due to shorter relaxation times of the 119Sn nucleus, tin satellites are expected to be broadened beyond recognition, however 1J119Sn–H coupling was observed in the 119Sn NMR spectrum. These isomers have also been characterized by an 1H–183W-HMQC-NMR experiment at −40 °C featuring cross peaks for the symmetric and asymmetric isomer (see ESI for spectra†). Rocchigian, Bochmann and Hrobárik et al. reported recently on the different coordination modes of Cp2WH2 at Au(I)/Au(III) cations.61 In the case of asymmetric hydride coordination a comparable 1H NMR spectrum was observed: −11.4 ppm, JW–H 48 Hz, −9.6 ppm, JW–H 61 Hz.61 Furthermore, chemical exchange of all hydrides in cations 3 was proven by 1H–1H EXSY NMR studies. For the characterization of the isomers by 119Sn, 207Pb and 183W NMR spectroscopy see Table 1 and the ESI.† The chemical shift of the heteroatoms tin and lead in the hydride bridged complexes 3a (119Sn: 1786, 1735 ppm) and 3b (207Pb: 7986 ppm, 207Pb NMR signal of other isomer was not observed) can be compared with the values found for the rhodium complexes showing a comparable structural motif [(Ph3P)2RhH2EAr*] [E = Sn 1728 ppm, E = Pb 8195 ppm].62 The 183W chemical shifts of 3a and 3b (Table 1) lie in the range known for metallocene hydride complexes of tungsten.63–65 The 183W chemical shift is more affected by the isomerism, symmetric or asymmetric (3asvs.3aas, 3bsvs.3bas: ≈300–400 ppm), than the nature of the tetrel, tin or lead (3asvs.3bs, 3aasvs.3bas: ≈35–80 ppm).
Table 1 Characteristic data of the presented tantalum and tungsten complexes 2–9
| Compound (E) |
1H NMR M–H [ppm] |
J
183W–H [Hz] |
1H NMR E–H [ppm] |
J
E–H (E: 119Sn/207Pb) [Hz] |
119Sn/207Pb NMR [ppm] |
183W NMR |
d M–E [Å] |
|
Not isolated, observed in solution as intermediate.
|
|
2 (Sn) |
−3.75 |
|
15.55 |
1040 |
1161 |
|
2.7207(2) |
|
3a (Sn) |
|
|
|
|
|
|
|
|
3a
s
|
−10.86 |
78 |
|
ca. 270 |
1786 |
−3910 |
|
|
3a
as
|
−11.13, −8.57 |
51, 77 |
|
|
1735 |
−4309 |
|
|
3b (Pb) |
|
|
|
|
|
|
|
|
3b
s
|
−7.49 |
75 |
|
|
7986 |
−3994 |
|
|
3b
as
|
−12.31, −4.30 |
52, 76 |
|
|
|
−4259 |
|
|
4a (Sn) |
−12.57 |
67 |
15.13 |
1193 (1J), 129 (2J) |
1057 |
−3629 |
2.6221(2) |
|
4b
(Pb) |
−13.67 |
66 |
42.13 |
ca. 530 |
|
|
|
|
4c (Ge) |
−11.08 |
69 |
11.30 |
|
|
−3585 |
|
|
5a (Sn) |
−12.37 |
90 |
|
|
2883 |
−4182 |
2.7589(4)/2.7488(4) |
|
5b (Pb) |
−16.15 |
91 |
|
|
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 534 534 |
−2772 |
2.8226(2) |
|
5c (Ge) |
−11.03 |
92 |
|
|
|
−4079 |
|
|
6 (Sn) |
−13.82 |
63 |
6.33 |
1344 (1J), 165 (2J) |
−231 |
−4378 |
|
|
7
|
−12.91 |
66 |
5.12 |
1274, 1216 |
−236 |
−3921 |
2.7582(2) |
|
8
|
−12.51 |
68 |
|
132 (2J) |
1223 |
−3526 |
|
|
9
|
|
|
10.04 |
|
|
−3096 |
|
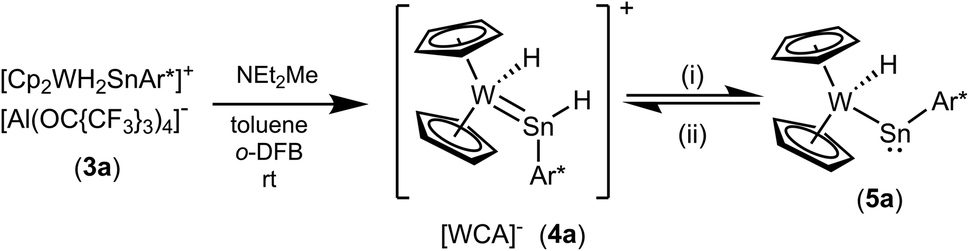
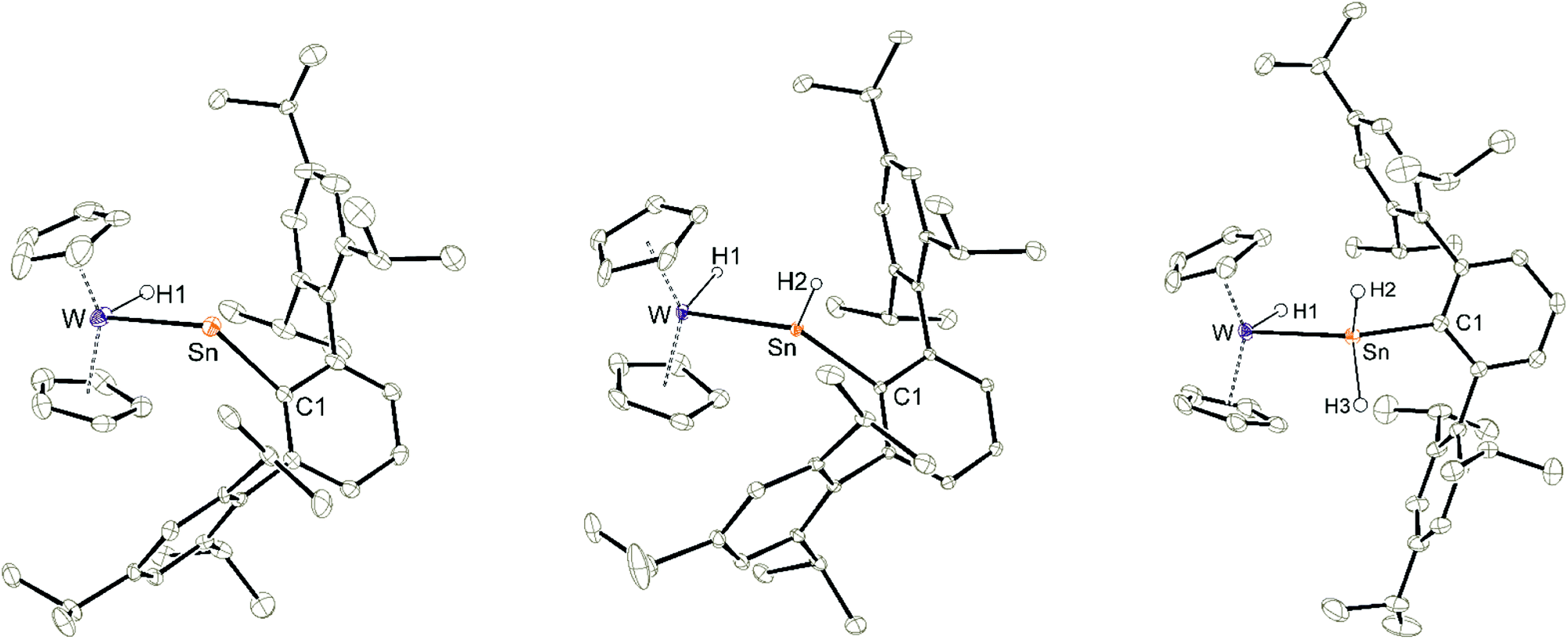
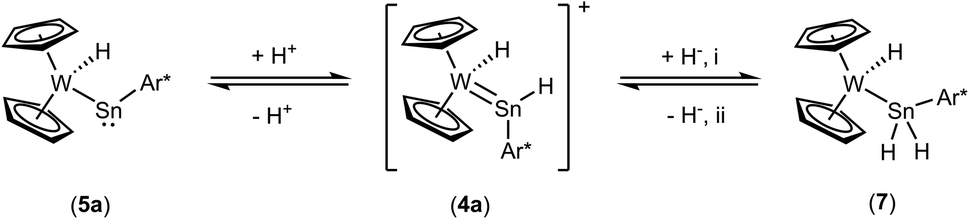
Using a slight excess of Cp2WH2 in the synthesis of 3a (E: Sn), the deep pink solution decolourises to orange to form the cationic hydridotungsten hydridostannylene complex 4a. Presumably, mediated by the basic Cp2WH2, as a proton-shuttle, a tungsten-bound proton of 3a migrates to Sn and a W–Sn bond is formed.66 The model of a proton-migration mediated by a Bronsted-basic Cp2WH2 is corroborated by the observation that addition of amine-bases such as diethyl methylamine to isolated 3a equally enables the isomerization (Scheme 4). The molecular structure of the cation of 4a is shown in Fig. 2. The W–Sn bond length of 2.6221(2) Å is shorter than typical single bonds (2.68–2.81 Å)2,67–71 between these elements and longer than the W–Sn triple bond (2.46–2.50 Å).72,73 In quantum chemical calculations π-donation from tungsten to the empty p-orbital at tin atom was found in addition to a W–Sn σ-bond (see ESI† for details of DFT calculations using ORCA and NBO analysis).58,59,74 The short W–Sn bond in 4a together with the slight π-donation from W to the empty p-orbital at tin might be interpreted as an indicator for partial double bond character between tin and tungsten. In the 1H NMR spectrum the signals of the hydride units in 4a (W–H −12.73 ppm, Sn–H 15.13 ppm) were found at frequencies typical for Cp2WH-fragments and hydridostannylene coordination.19,22,30,61 The 1JSn–H coupling constant of 1193 Hz lies close to the values found in 2 and in the literature examples [Cp2M(Ar*SnH)2] M: Ti 1250 Hz, Zr 1125 Hz, Hf 1060 Hz.30 In the 119Sn NMR spectrum the signal was found at 1057 ppm corroborating stannylene coordination at transition metals.19,75 Deprotonation of 4a was carried out in reaction with a N-heterocyclic carbene 1,3,4,5-tetramethylimidazol-2-ylidene (MeNHC) at 80 °C in toluene/1,2-difluorobenzene (Scheme 4). The colour of the solution changes from orange to yellow to green and metallostannylene 5a was isolated after crystallization in a yield of 44%. The molecular structure of 5a is presented in Fig. 2. The W–Sn bond length of 2.7589(4) Å is a short single bond for a metallostannylene of tungsten (vide supra).76,77 Comparing the W–Sn bond lengths in 4a [2.6221(2) Å] and 5a [2.7488(4) Å], due to the partial double bond character, found for 4a, the protonated derivative 4a exhibits a shorter W–Sn length. In the 119Sn{1H} NMR spectrum of 5a a signal at high frequency of 2883 ppm indicates a metallostannylene.76 The signal for the W–H hydride of 5a was found in the 1H NMR spectrum in the typical range for transition metal hydrides at −12.37 ppm. Reprotonation of metallostannylene 5a at the tin atom was achieved in reaction with the acid [H(Et2O)2][BArF]78 to give the cationic hydridostannylene complex 4a, now with the weakly coordinating anion [BArF]− (Scheme 4). Thus, protonation of metallostannylene complexes is an alternative route for the synthesis of hydridotetrylene coordination compounds.
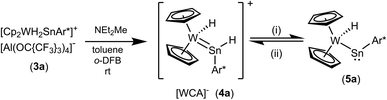 |
| | Scheme 4 (i) MeNHC, toluene/C6H4F2, 70 °C, 3 d, – [MeNHC–H][Al(OC{CF3}3)4], (ii) [H(Et2O)2][BArF] toluene/o-DFB. {[B(3,5-{CF3}2C6H3)4]− = [BArF]−}. | |
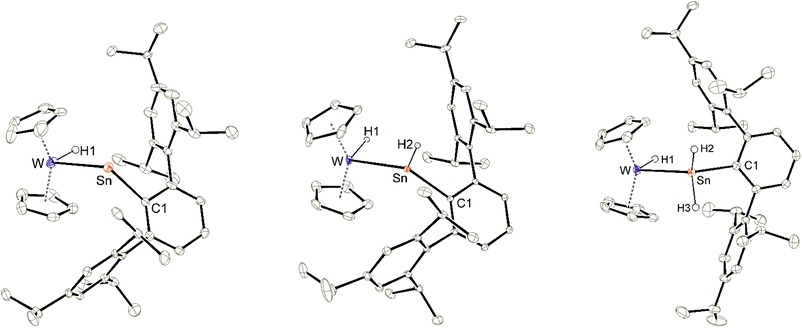 |
| | Fig. 2 ORTEP of the molecular structures of 5a, 4a and 7. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms except the W–H and Sn–H units and the anion of 4a have been omitted. Selected interatomic distances [Å] and angles [°]: 5a, W–Sn 2.7589(4), W–Cp 2.266(5)–2.330(5), Sn–C1 2.264(4), W–Sn–C1 108.3(1); 4a,W–Sn 2.6221(2), Sn–H2 1.60(5), W–Cp 2.282(4)–2.330(4), Sn–C1 2.144(2), W–Sn–C1 138.64(6), H1–W–Sn 90.0(18), W–Sn–H2 119.5(18), H2–Sn–C1 101.8(18); 7, W–Sn 2.7582(2), Sn–C1 2.196(3), W–Cp 2.247(3)–2.357(3), W–H1 1.69(5), Sn–H2 1.76(3), Sn–H3 1.76(3), W–Sn–C1 134.1(1), H1–W–Sn 87.0(15), W–Sn–H2 108.5(14), W–Sn–H3 112.8(10), H2–Sn–C1 100.1(14), H3–Sn–C1 97.6(10). | |
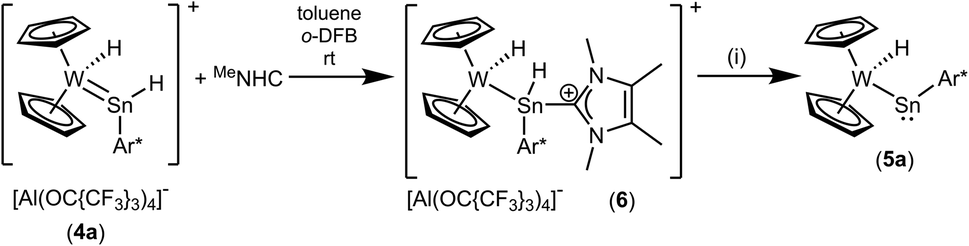
Deprotonation of the cation 4a was achieved with MeNHC at 80 °C (Scheme 4). Reacting the cation with the carbene at rt an adduct (Scheme 5, 6) of the N-heterocyclic carbene at the tin was isolated and characterized by NMR spectroscopy. This adduct 6 is also the product of the reaction between 3a and MeNHC. Heating this adduct 6 to 80 °C gives the deprotonation product 5a. Tobita et al. studied the NHC-induced conversion of hydrido(hydrogermylene) complexes into a germylyne coordination compound. In this stepwise procedure NHC-adduct formation was found followed by deprotonation of a W–H unit by the NHC.79,80 The reactivity of the cationic arylhydridostannylene complex 4a was investigated in reaction with LiAlH4 (Scheme 6) and styrene (Scheme 7). Nucleophilic attack of a hydride anion at the tin atom of cation 4a was observed in reaction with LiAlH4. The aryldihydridostannyl ligand coordinates at the tungsten atom in complex 7 with a W–Sn bond length of 2.7582(2) Å.2,67–71 The reverse reaction, hydride abstraction from complex 7 was also investigated in reaction with the trityl salt [Ph3C][Al(OC{CF3}3)4] to give the salt 4a in moderate yield of 71% based on results of NMR spectroscopy (Scheme 6). Direct addition or elimination of hydrogen (to transfer of 5a into 7 or 7 into 5a) was not observed (heating or photochemical activation of 7, addition of hydrogen to 5a, 1–3.5 atm). However, stepwise addition and abstraction of protons and hydride ligands is a possible pathway for the reversible conversion of 5a into 7 (Scheme 4 and 6).
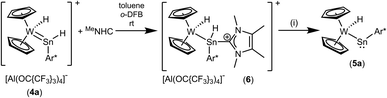 |
| | Scheme 5 (i): Toluene/o-DFB, 80 °C, 3d, – [MeNHC–H][Al(OC{CF3}3)4]. | |
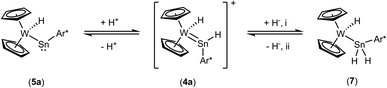 |
| | Scheme 6 Addition and abstraction of protons and hydride ligands. (i): rt, LiAlH4, Et2O; (ii): rt, [Ph3C][Al(OC{CF3}3)4], o-DFB. | |
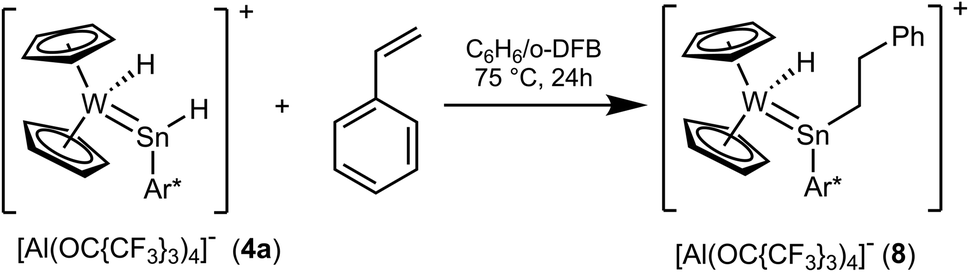
With an excess of styrene, hydride complex 4a exhibits hydrostannylation of the olefin at 75 °C (Scheme 7). Insertion of olefines and carbon dioxide into the Sn–H bond of low valent organotinhydrides was studied by Power et al. and for a coordinated organohydrido germylene another example of styrene insertion is presented in the literature.14,29,81
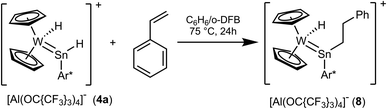 |
| | Scheme 7 Insertion of styrene into a Sn–H bond. | |
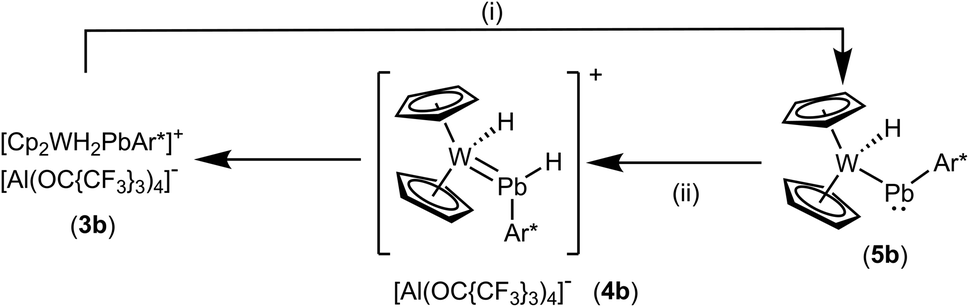
In the case of the low valent lead cation the tungstenocene dihydride adduct 3b does not exhibit a hydrogen atom migration from tungsten to lead as in the case of the tin compound 3a. Lead adduct 3b was readily deprotonated at rt by MeNHC to yield the plumbylene complex 5b (Scheme 8). Evidence for the formation of an NHC adduct comparable to 6 was not found. Plumbylene 5b was crystallized from a pentane solution at −40 °C (see Fig. 3 for ORTEP of the molecular structure). The W–Pb distance of 2.8226(2) Å found in 5b is shorter than the W–Pb distance in the plumbylene [Cp(CO)3W–PbAr*, W–Pb 3.006(1) Å].82 Furthermore the 207Pb NMR resonance of 5b was observed at a very high frequency of 10534 ppm [Cp(CO)3W–PbAr* 207Pb NMR 9374 ppm].82 Protonation of 5b was carried out at −40 °C using [H(Et2O)2][Al(OC{CF3}3)4] as the proton source (Scheme 8). The cold reaction mixture was directly investigated by low temperature 1H NMR spectroscopy. Besides signals of the starting material 5b, and the product 3b two new signals at very low (−13.67 ppm) and very high (42.13 ppm) frequencies were detected. Based on a 1H–1H–COSY-LR-NMR experiment (see ESI†) both signals belong to the same compound. Furthermore, the signal at high frequency exhibits both 207Pb- (530 Hz) and 183W-satellites (39 Hz). In the case of the low frequency signal 183W-satellites (66 Hz) were observed. Due to comparable spectroscopic data, we speculate about the structure of this intermediate 4b and suggest a hydridoplumbylene motive like in the tin case 4a. Based on this molecular structure of 4a the tin atom was exchanged against a lead atom and this structure was optimized using DFT calculations (see ESI for details†).58,59 This optimized structure was used for chemical shift calculations of the protons with the ADF program package.60 The results of these calculations [PbH exp. 42.13 ppm, calc. 39.3 ppm and WH exp. −13.67 ppm, calc. −11.5 ppm] lie close to the experimentally observed signals. This extreme high frequency chemical shift of the PbH unit at 42.13 ppm can be explained with the spin–orbit coupling of the heavy atom with the light atom (SO-HALA effect).83 This effect was already observed for low valent lead hydride exhibiting a 1H NMR signal at 35.61 ppm.49 However, 4b is only an intermediate in the protonation of 5b and the isolation was not possible, which could be due to the weak Pb–H bond. With these NMR data of 4b we present first indications for a hydridoplumbylene ligand as the missing piece of Group 14 hydridotetrylene coordination chemistry.
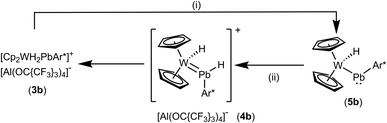 |
| | Scheme 8 (i): + MeNHC, toluene/o-DFB, rt, 3 h, − [MeNHC–H][Al(OC{CF3}3)4], (ii): + [H(Et2O)2][Al(OC{CF3}3)4], toluene/C6H4F2, rt, – 2 Et2O. | |
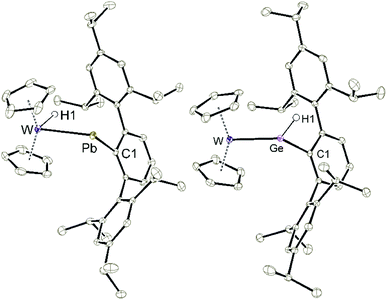 |
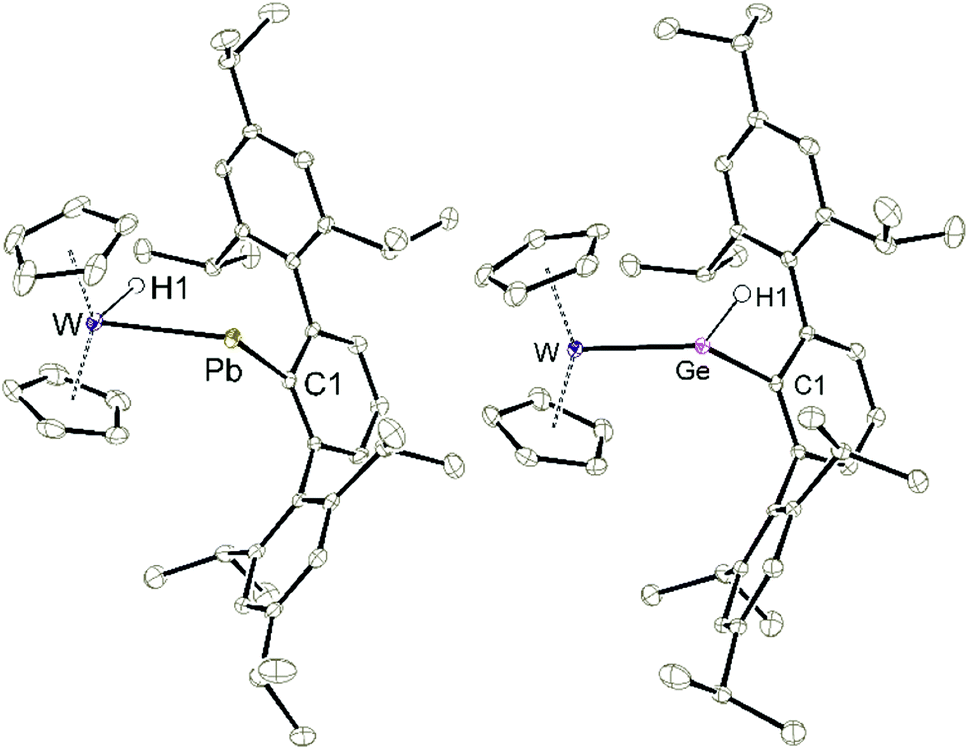
| | Fig. 3 ORTEP of the molecular structures of 5b and 9. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms except the W–H and Ge–H units have been omitted. Selected interatomic distances [Å] and angles [°]: 5b, W–Pb 2.8226(2), W–Cp 2.260(3)–2.330(3), Pb–C1 2.353(3), W–Pb–C1 110.1(1); 9, W–Ge 2.4271(3), W–Cp 2.248(2)–2.310(2), Ge–C1 1.984(2), Ge–H 1.53(2), W–Ge–C1 134.7(1). | |
An alternative protocol for the synthesis of tungstenocenyl substituted tetrylenes 5a and 5b starts with deprotonated tungstenocene dihydride [Cp2W(H)Li]4 (Scheme 9). In reaction of the anionic hydride with the respective low valent aryltetrylene halides, the tetrylenes of tin and lead were straightforwardly synthesized. Based on NMR spectroscopy the products were formed nearly quantitatively and after crystallization isolated in moderate yield.
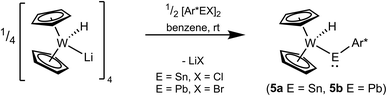 |
| | Scheme 9 Synthesis of the tetrylenes of tin 5a and lead 5b with [Cp2W(H)Li]4 as starting material. | |
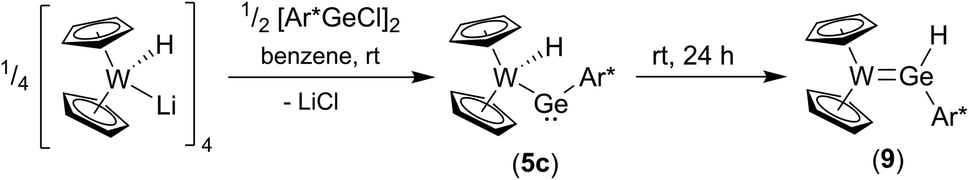
To synthesize the homologous tungstenocenyl substituted germylene 5c the tetrameric anion [Cp2W(H)Li]4 was reacted with arylgermanium chloride (Scheme 10). After five minutes stirring at rt in the dark green solution, a mixture of products was identified by 1H NMR spectroscopy: two signals exhibiting 183W-satellites were found at −11.03 ppm (1JW–H = 92 Hz) and +10.02 ppm (1JW–H = 35 Hz). The signal at low frequency together with the 1JW–H coupling constant is an indicator for a W–H unit and fits in the series of tetrylene tungsten hydride complexes [Cp2W(H)-EAr*]: E = Pb (5b) −16.15 ppm, (1JW–H = 91 Hz), E = Sn (5a) −12.37 ppm, (1JW–H = 90 Hz), E = Ge (5c) −11.03 ppm (Table 1). After stirring the product mixture for further 24 h the signal at low frequencies indicating 5c vanished and only the hydride signal at high frequency remains. Therefore, we suggest germylene 5c being the kinetically controlled product. Crystallization at −40 °C yields dark green crystals of the arylhydridogermylene complex 9 in moderate yield (53%). The molecular structure of 9 is shown in Fig. 3. Probing the electronic structure of complex 9 DFT calculations together with NBO analysis were performed. Representative NLMOs for the σ- and π-bond were placed in the ESI.† The WGe distance of 2.427(3) Å found in 9 can be compared with hydridogermylene coordination at a CpW(CO)2 fragment synthesized by Tobita et al. [2.4289(8) Å].21
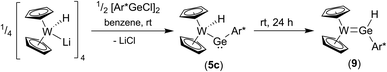 |
| | Scheme 10 Reaction of the anion [Cp2W(H)Li]4 with low valent arylgermanium chloride. | |
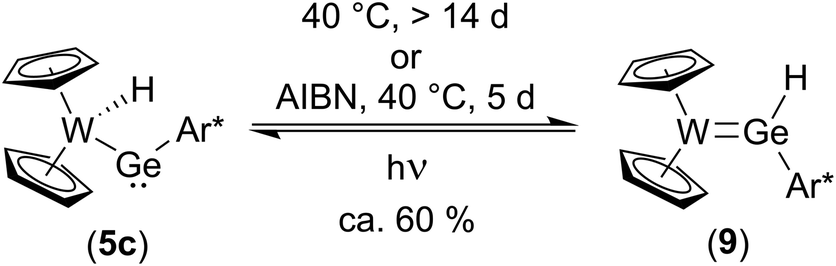
To further investigate the transformation of the kinetically controlled product 5cvia a 1,2-H-shift into the thermodynamically controlled product 9, a solution of a crystalline sample of 9 was treated with light from a mercury-vapor lamp. A mixture between 9 and 5c (40%/60%) was observed (Scheme 11). Interestingly, from this mixture transformation of 5c to 9 is much slower (14 d at 40 °C) than in the original reaction mixture (Scheme 10). The faster 1,2-H-shift in the original reaction mixture is probably caused by side products resulting from the synthesis. After addition of AIBN to this mixture (crystalline sample) a much faster conversion of 5c to 9 was observed making a radical mechanism feasible. With the reversible conversion of 5c to 9 a rare example for a reversible 1,2-H-shift together with the direct observation of both species, the hydrido-metallogermylene (5c) and hydridogermylene (9) is presented. DFT calculations confirm both germanium isomers 5c and 9 being close in energy (see ESI for further details of DFT calculations†). However, in the case of the homologous tin compounds DFT calculations clearly indicate higher stability of the hydrido-metallostannylene isomer (5a) compared to a hypothetical arylhydridostannylene-coordination (analogous to Ge-compound 9). Tilley and co-workers investigated the 1,2-hydrogen migration of heavy group 14 element hydrides coordinated at transition metal centres and have drawn the conclusion, that for tin the hydrido-metallotetrylenes are favoured, in contrast to silicon and germanium, both preferring the hydridotetrylene coordination.14,19,20,22,84,85 Green et al. published a reversible 1,2-H-shift equilibrium between a hydridocarbene transition metal complex and a methyl ligand.86 In the case of metallostannylene 5a our results are in line with the findings made by Tilley et al.84,85 The germanium isomers 9 and 5c are remarkably close in energy and therefore we were able to characterize both products of 1,2-H shift (Scheme 11).
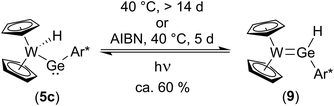 |
| | Scheme 11 Reversible 1,2-H-shift. | |
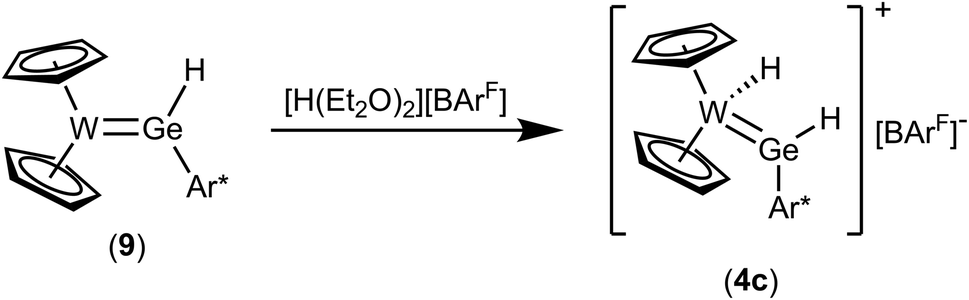
Protonation of complex 9 was studied in reaction with acid [H(Et2O)2][BArF] in a mixture of pentane and 1,2-difluorobenzene (Scheme 12). Yellow crystalline material was isolated in moderate yield and based on the NMR data (Table 1) the protonation of the tungsten atom was found. This type of reaction was already presented in the literature in the case of a germylene complex of ruthenium, which was protonated at the transition metal in reaction with a strong Brønsted acid.14 Cation 4c is a homologue of the isolated tin cation 4a and the intermediately formed plumbylene 4b. The 1H NMR data (signals of GeH and WH, Table 1) of 4c are in line with the data of the heavier homologues.
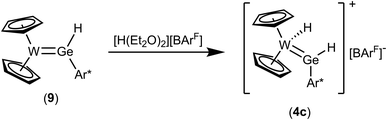 |
| | Scheme 12 Protonation of the hydridogermylene complex 9. | |
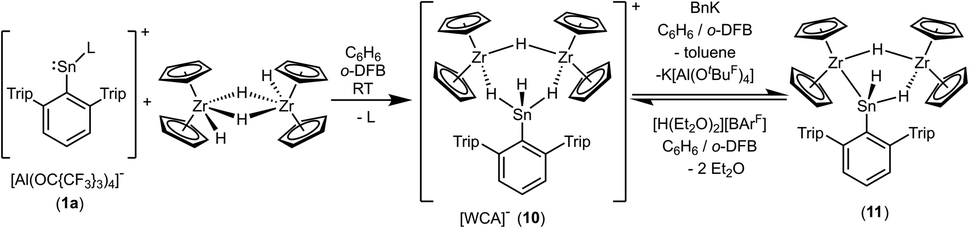
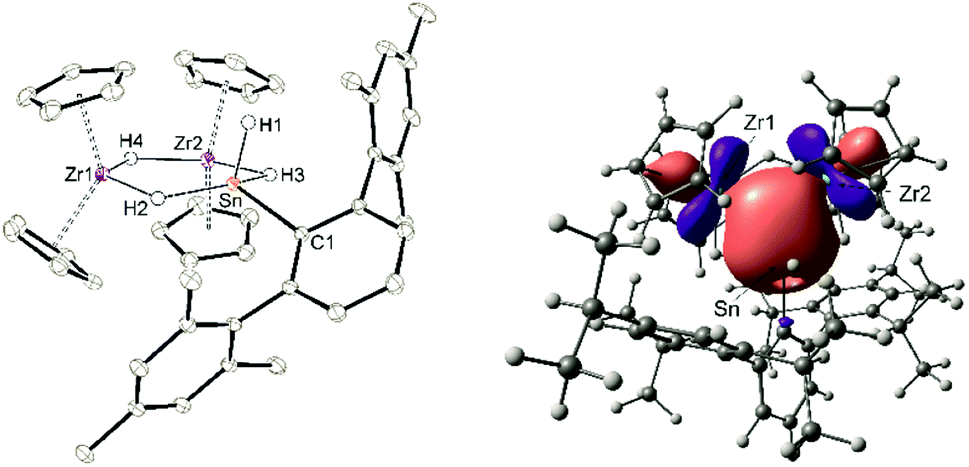
Both cations, the low valent lead and tin cation (1a, 1b), were also reacted with zirconocene dihydride [Cp2ZrH2]2, but only in the case of the tin salt reaction products could be isolated (Scheme 13).87–89 Salt 10 was selectively formed no matter which stoichiometry was chosen. After stirring an equimolar mixture of 1a and dimeric dihydride followed by crystallization from 1,2-diflourobenzene at −40 °C yellow crystals of 10 were isolated in a yield of 69%. The molecular structure in the solid state is depicted in Fig. 4. The dimeric motif of the zirconocene dihydride remains intact. The tin cation coordinates at two Zr–H units forming two hydride bridges showing almost similar Zr–Sn distances [3.0185(2), 3.0325(2) Å]. Due to the hydride bridge the Zr–Sn distances are much longer than bond lengths found for typical stannylene coordination [2.794(1)–2.872(1) Å].30,75,90,91 One hydride ligand is transferred to the tin atom as a terminal coordinating hydride. The hydride atoms inside the six membered ring were found in the final difference Fourier map and the [–Zr–H–Sn–H–Zr–H–] ring shows an almost planar structural motif. This type of dimeric zirconium–hydride complex chelating a main group element fragment was also described by Bulychev et al. for [(Cp2ZrH)2H(AlCl2)].92,93 In the 1H NMR spectrum the Sn–H hydride ligand exhibits a signal at 9.06 ppm (1JSn–H = 1344 Hz), which can be compared with values observed for Ar*SnH coordination at group 4 metallocene fragments.30 The signals for the bridging hydride ligands were observed at lower frequencies Zr–H–Sn (−0.06 ppm) and Zr–H–Zr (−6.24 ppm), lying in the region typical for zirconium hydrides.92 The 119Sn-NMR signal was observed at relatively low frequency (440 ppm) for low valent tin but can be explained with the high coordination number at the tin atom. The electronic situation was investigated using DFT calculations together with NBO analysis. The lone pair of the tin atom is shared between the two Zr (24, 23%) and the Sn (48%) atoms featuring a three centre two electron bond (see Fig. 4 for NLMO). Both Zr–H units (H2, H3) donate roughly 10% of the two electrons of the bond into the empty p-orbital of tin (see ESI for NLMO†). We interpret compound 10 as a low valent aryl tin hydride Ar*SnH chelated by the cation [(Cp2ZrH)2H]+via two hydride bridges.
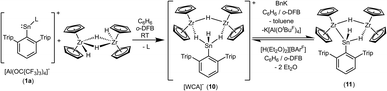 |
| | Scheme 13 Synthesis of the hydride bridged trinuclear SnZr2–cation and deprotonation to give 11 (L = C6H6). | |
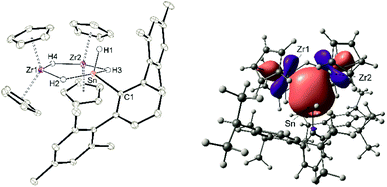 |
| | Fig. 4 ORTEP and NLMO exhibiting the three centre two electron bond of cation [(Cp 2ZrH)2HSnHAr*]+ of 10. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms except the Zr–H and Sn–H units, methyl groups and the anion of 10 have been omitted. Selected interatomic distances [Å] and angles [°]: Zr1–Sn 3.0185(2), Zr2–Sn 3.0325(2), Zr1–Zr2 3.577(2), Zr1–Cp 2.4845(14) – 2.5328(15), Zr2–Cp 2.4760(14) – 2.5162(15), Sn–C1 2.1678(12), Sn–H1 1.57(3), Sn–H2 2.07(3), Sn–H3 1.98(3), Zr1–H2 1.84(3), Zr1–H4 2.00(2), Zr2–H3 1.89(3), Zr2–H4 1.97(2), Zr1–Sn–Zr2 72.5(1), C1–Sn–H1 95.8(10), H2–Sn–H3 144.7(11), H2–Zr1–H4 121.6(11), H3–Zr2–H4 118.3(10). | |
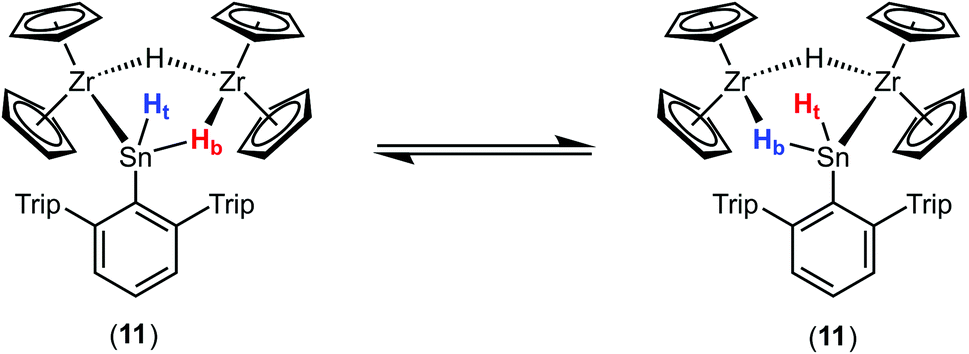
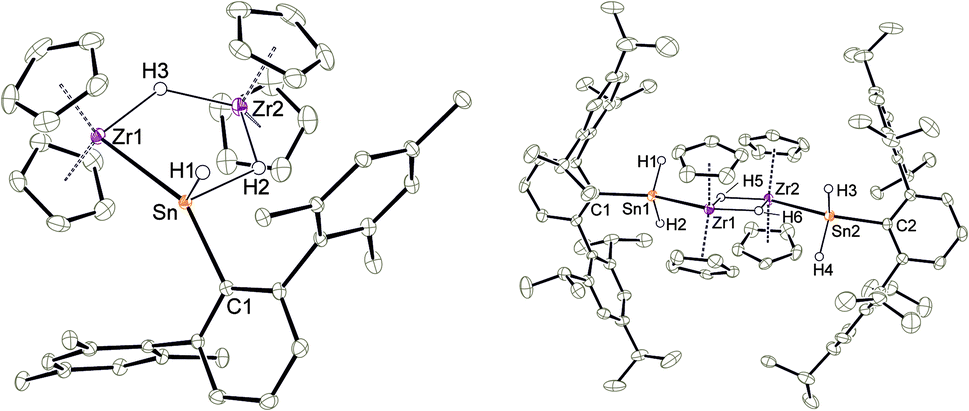
Deprotonation of the cationic hydride 10 was successful with the strong base benzyl potassium to give the trinuclear hydride [(Cp2Zr)2H2SnHAr*] 11, which was crystallized from hexane solution at −40 °C (Scheme 13, Fig. 5 and ESI† for molecular structure and NLMOs of 11). The deprotonation reduced the zirconium atoms to oxidation state III. Reprotonation of 11 was achieved at −40 °C with [H(Et2O)2][BArF] in 1,2-difluorobenzene as solvent (Scheme 13). Hydride 11 features a Zr–Sn bond of 2.8597(5) Å, which can be compared with stannylene coordination at zirconocene fragments [2.79–2.87 Å].75,90,91 Complex 11 was also investigated using DFT calculations and NLMOs representing the Zr–Sn σ-bond and the three centre Zr–Sn–Zr two electron bond are presented in ESI.† In the 1H NMR spectrum recorded at rt only broad signals were observed. At −20 °C however for each Cp–ligand and the hydride substituents sharp signals were found [Sn–H: 7.19 ppm 1JSn–H ≈ 860 Hz, Zr–H–Zr −12.90 ppm, Zr–H–Sn −3.08 ppm]. Via1H–1H-EXSY-NMR an exchange between the terminal Sn–H and the bridging Zr–H–Sn hydride ligands was detected. The exchange of these hydride ligands (Scheme 14) should be responsible for the broadening of the signals in the rt NMR spectrum.
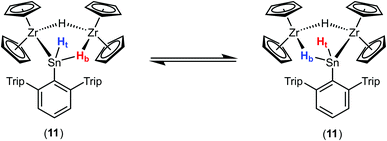 |
| | Scheme 14 Dynamic interplay of hydride substituents. | |
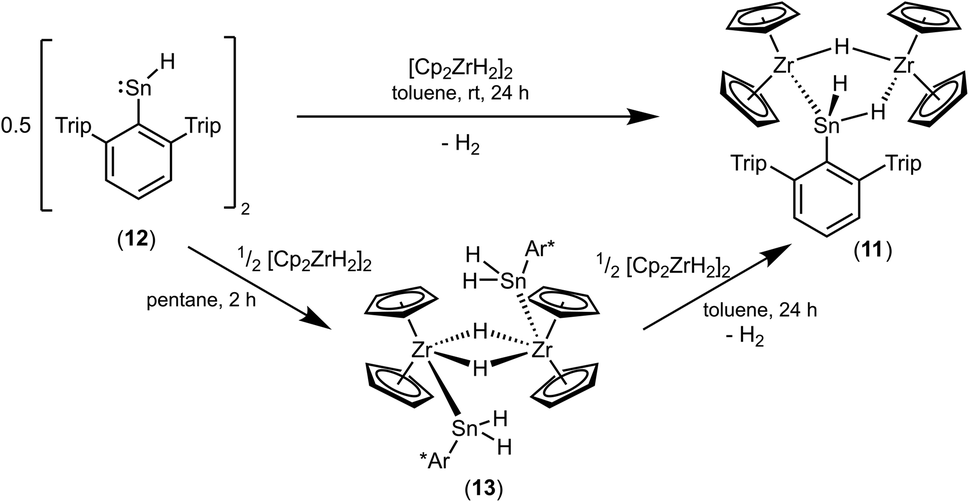
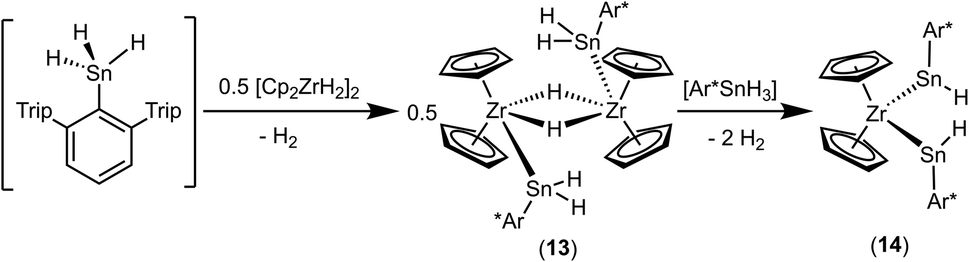
An alternative synthetic pathway for the formation of complex 11 was found with the reaction of Power's low valent tin hydride 12 with zirconocene dihydride in toluene at room temperature (Scheme 15, ratio Sn/Zr: 1/2).47 After a spontaneous liberation of dihydrogen, product complex 11 was isolated in high yield (92%) and, based on NMR spectroscopy, in high purity. Crystallization from hexane affords 11 as black/brown crystals. Interestingly, the stepwise formation of 11 was found by stirring a pentane suspension of the zirconium hydride [Cp2ZrH2]2 and low valent tin hydride 12 (ratio Sn/Zr: 1/1) at rt for 2 h. Double insertion of the tin moieties into the terminal Zr–H units was found and the bis(aryldihydridostannyl) complex 13 (Scheme 15, Fig. 5 for molecular structure) was isolated in a yield of 82%. After addition of a further equivalent of zirconocene dihydride to a toluene solution of 13, liberation of dihydrogen together with formation of 11 was found (Scheme 15).
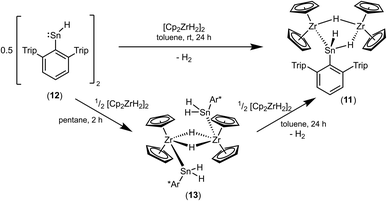 |
| | Scheme 15 Alternative route for the synthesis of 11. | |
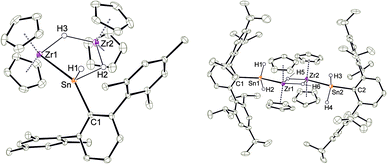 |
| | Fig. 5 Molecular structures of 11 and 13. Thermal ellipsoids are shown at 50% probability level. Hydrogen atoms except the Zr–H and Sn–H units and methyl groups of 11 have been omitted. Selected interatomic distances [Å] and angles [°]: 11, Zr1–Sn 2.8597(5), Zr2–Sn 3.2228(6), Zr1–Cp 2.478(2)–2.551(2), Zr2–Cp 2.494(2)–2.520(2), Sn–C1 2.2257(18), Sn–H1 1.67(3), Sn–H2 2.14(4), Zr1–H3 2.05(3), Zr2–H2 1.77(4), Zr2–H3 1.96(3), Zr1–Sn–C1 152.19(5), Zr1–Sn–H1 109.9(12), Zr1–Sn–H2 99.8(12), H2–Zr2–H3 118.4(16), H3–Zr1–Sn 88.9(9); 13, Sn1–Zr1 2.9813(4), Sn2–Zr2 2.9794(4), Sn1–C1 2.211(3), Sn2–C2 2.216(3), Zr1–Zr2 3.4468(4), Zr1–Cp 2.480(4)–2.538(4), Zr2–Cp 2.475(4)–2.526(4), Sn1–H1 1.65(4), Sn2–H3 1.66(5), Sn2–H4 1.72(6), Zr1–H5 2.04(4), Zr1–H6 1.97(4), Zr2–H5 1.95(4), Zr2–H6 2.01(4), C1–Sn1–Zr1 139.9(1), C2–Sn2–Zr2 137.8(1). | |
Complex 13 was also formed in reaction of Ar*SnH3 and 0.5 [Cp2ZrH2]2. Addition of further equivalents of Ar*SnH3 yields together with liberation of hydrogen the hydridostannylene complex [Cp2Zr(SnHAr*)2] (14) (Scheme 16).30 The formation of the Zr–Sn bonds in 13 and 14 is an example for dehydrocoupling reactions between Zr–H and Sn–H species via a σ-bond metathesis. This type of M–Sn bond formation was discussed as part of the mechanism in dehydropolymerizations of secondary stannanes catalyzed by zirconocene and hafnocene derivatives.94–96
 |
| | Scheme 16 Reductive elimination hydrogen and formation of the bis(hydridostannylene) complex 14. | |
Conclusions
To conclude, hydridotetrylene formation was found reacting the low valent tin cation [Ar*Sn]+ with tantalocene trihydride, tungstenocene dihydride and the dimeric zirconocene dihydride. In the tantalum and tungsten case, coordination of the low valent hydride [Ar*SnH] at the metal was found and, in the zirconium compound, the tin hydride moiety is chelated by two Zr–H units. The homologous lead cation forms with tungstenocene dihydride an adduct [Cp2WH2PbAr*]+ (3b). After a sequence of deprotonation and reprotonation of this lead adduct, first indications for hydridoplumbylene coordination at a transition metal were observed by low temperature 1H NMR spectroscopy. The cationic hydridostannylene complex of tungstenocenehydride [Cp2W(H)Sn(H)Ar*]+ (4a) shows reactivity at the tin atom: deprotonation results in formation of the hydrido-tungstenostannylene complex [Cp2W(H)SnAr*] (5a), hydride addition gives a dihydridostannyl complex and in reaction with styrene hydrostannylation was observed.
Metallotetrylene complexes of tungstenocene [Cp2W(H)EAr*] (E = Ge, Sn, Pb) were also synthesized in reaction of the deprotonation product [Cp2W(H)Li]4 with aryltetrylene halides of germanium, tin, and lead. Only in the case of germanium a reversible transformation of the hydrido-tungstenogermylene [Cp2W(H)GeAr*] (5c) to the hydridogermylene [Cp2WGeHAr*] (9) was observed. In comparison to the 1,2-H shift at 40 °C (14 d), after addition of AIBN product formation (40 °C) was completely finished after 5 days. Likely, this 1,2-H shift from the tungsten to the germanium atom is catalyzed by radical species. Under the influence of light, a 1,2-H transfer back to the tungsten atom was found. Therefore, interconversion between metallogermylene (5c) and hydridogermylene (9) represents a system that exhibits a directly observable and reversible 1,2-H-migration. Corroborated by DFT calculations the 1,2-H shift between 5c and 9 is almost an isenthalpic reaction.
Dimeric zirconocene dihydride [Cp2ZrH2]2 was reacted with the low valent tin cation [Ar*Sn]+ and the low valent tin hydride [Ar*SnH]2. Whereas the cationic product was deprotonated to give the dimeric Zr(III) complex [({Cp2Zr}2{μ-H})(μ-H)Sn(H)Ar*] (11), this complex together with hydrogen resulted directly from the reaction with [ArSnH]2. Aryltintrihydride Ar*SnH3 reacts with dimeric zirconium dihydride to give, as a product of dehydrocoupling reactions via σ-bond metathesis, the bis(hydridostannylene) complex of zirconocene [Cp2Zr(SnHAr*)2].
Data availability
Full experimental and computational details are provided as part of the ESI.†
Author contributions
Investigations, writing, original draft preparation, review M. W.; preparation of 1b S. J.; improved synthesis of 1a and scientific discussion M. A.; special NMR experiments K. E.; discussion of X-ray measurements H. S.; DFT calculation, manuscript review C. P. S.; supervision, funding acquisition, DFT calculation, manuscript writing and review L. W.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. A. thanks the Fonds der Chemischen Industrie for a scholarship. We acknowledge support of the state of Baden-Württemberg through bwHPC and the German research Foundation (DFG) through grant no INST 40/575-1 FUGG (Justus 2 cluster).
Notes and references
- M. F. Lappert and P. P. Power, Dalton Trans., 1985, 51–57 RSC.
- W. Petz, Chem. Rev., 1986, 86, 1019–1047 CrossRef CAS.
- M. F. Lappert and R. S. Rowe, Coord. Chem. Rev., 1990, 100, 267–292 CrossRef CAS.
- J. Baumgartner and C. Marschner, Rev. Inorg. Chem., 2014, 34, 119 CAS.
- L. Álvarez-Rodríguez, J. A. Cabeza, P. García-Álvarez and D. Polo, Coord. Chem. Rev., 2015, 300, 1–28 CrossRef.
- J. A. Cabeza, P. García-Álvarez and D. Polo, Eur. J. Inorg. Chem., 2016, 10–22 CrossRef CAS.
- B. Blom, M. Stoelzel and M. Driess, Chem.–Eur. J., 2013, 19, 40–62 CrossRef CAS PubMed.
- J. Y. Corey, Chem. Rev., 2016, 116, 11291–11435 CrossRef CAS PubMed.
- G. Frenking, R. Tonner, S. Klein, N. Takagi, T. Shimizu, A. Krapp, K. K. Pandey and P. Parameswaran, Chem. Soc. Rev., 2014, 43, 5106–5139 RSC.
- R. J. Somerville and J. Campos, Eur. J. Inorg. Chem., 2021, 2021, 3488–3498 CrossRef CAS PubMed.
- M. S. Nechaev, Organometallics, 2021, 40, 3408–3423 CrossRef CAS.
- A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- R. S. Simons, J. C. Gallucci, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2002, 654, 224–228 CrossRef CAS.
- M. E. Fasulo and T. D. Tilley, Chem. Commun., 2012, 48, 7690–7692 RSC.
- P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640–13641 CrossRef CAS PubMed.
- J. D. Feldman, J. C. Peters and T. D. Tilley, Organometallics, 2002, 21, 4065–4075 CrossRef CAS.
- T. Watanabe, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2004, 43, 218–221 CrossRef CAS PubMed.
- M. Ochiai, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2007, 46, 8192–8194 CrossRef CAS PubMed.
- P. G. Hayes, C. W. Gribble, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 4606–4607 CrossRef CAS PubMed.
- P. G. Hayes, R. Waterman, P. B. Glaser and T. D. Tilley, Organometallics, 2009, 28, 5082–5089 CrossRef CAS.
- H. Hashimoto, T. Tsubota, T. Fukuda and H. Tobita, Chem. Lett., 2009, 38, 1196–1197 CrossRef CAS.
- H.-J. Liu, J. Guihaumé, T. Davin, C. Raynaud, O. Eisenstein and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 13991–13994 CrossRef CAS PubMed.
- M. C. Lipke, F. Neumeyer and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 6092–6102 CrossRef CAS PubMed.
- T. P. Dhungana, H. Hashimoto, M. Ray and H. Tobita, Organometallics, 2020, 39, 4350–4361 CrossRef CAS.
- P. W. Smith and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 3880–3883 CrossRef CAS PubMed.
- B. V. Mork, T. D. Tilley, A. J. Schultz and J. A. Cowan, J. Am. Chem. Soc., 2004, 126, 10428–10440 CrossRef CAS PubMed.
- P. G. Hayes, C. Beddie, M. B. Hall, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 428–429 CrossRef CAS PubMed.
- R. C. Handford, M. A. Nesbit, P. W. Smith, R. D. Britt and T. D. Tilley, J. Am. Chem. Soc., 2022, 144, 358–367 CrossRef CAS PubMed.
- Q. Zhu, J. C. Fettinger and P. P. Power, Dalton Trans., 2021, 50, 12555–12562 RSC.
- J.-J. Maudrich, M. Widemann, F. Diab, R. H. Kern, P. Sirsch, C. P. Sindlinger, H. Schubert and L. Wesemann, Chem.–Eur. J., 2019, 25, 16081–16087 CrossRef CAS PubMed.
- S. M. I. Al-Rafia, A. C. Malcolm, S. K. Liew, M. J. Ferguson and E. Rivard, J. Am. Chem. Soc., 2011, 133, 777–779 CrossRef CAS PubMed.
- E. Rivard, Chem. Soc. Rev., 2016, 45, 989–1003 RSC.
- A. Rit, R. Tirfoin and S. Aldridge, Angew. Chem., Int. Ed., 2016, 55, 378–382 CrossRef CAS PubMed.
- P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y.-H. Tsay, Chem. Ber., 1980, 113, 757–769 CrossRef CAS.
- K. Inomata, T. Watanabe, Y. Miyazaki and H. Tobita, J. Am. Chem. Soc., 2015, 137, 11935–11937 CrossRef CAS PubMed.
- P. Jutzi, R. Dickbreder and H. Nöth, Chem. Ber., 1989, 122, 865–870 CrossRef CAS.
- S. Hino, M. Brynda, A. D. Phillips and P. P. Power, Angew. Chem., Int. Ed., 2004, 43, 2655–2658 CrossRef CAS PubMed.
- C. P. Sindlinger, F. S. W. Aicher and L. Wesemann, Inorg. Chem., 2017, 56, 548–560 CrossRef CAS PubMed.
- F. Diab, F. S. W. Aicher, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, Chem.–Eur. J., 2019, 25, 4426–4434 CrossRef CAS PubMed.
- C. P. Sindlinger, F. S. W. Aicher, H. Schubert and L. Wesemann, Angew. Chem., Int. Ed., 2017, 56, 2198–2202 CrossRef CAS PubMed.
- J. Li, C. Schenk, F. Winter, H. Scherer, N. Trapp, A. Higelin, S. Keller, R. Pöttgen, I. Krossing and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 9557–9561 CrossRef CAS PubMed.
- A. Schäfer, W. Saak, D. Haase and T. Müller, Chem.–Eur. J., 2009, 15, 3945–3950 CrossRef PubMed.
- A. Hinz, Chem.–Eur. J., 2019, 25, 3267–3271 CrossRef CAS PubMed.
- M. J. Taylor, A. J. Saunders, M. P. Coles and J. R. Fulton, Organometallics, 2011, 30, 1334–1339 CrossRef CAS.
- H. V. R. Dias and Z. Wang, J. Am. Chem. Soc., 1997, 119, 4650–4655 CrossRef CAS.
- R. J. Mangan, A. Rit, C. P. Sindlinger, R. Tirfoin, J. Campos, J. Hicks, K. E. Christensen, H. Niu and S. Aldridge, Chem.–Eur. J., 2020, 26, 306–315 CrossRef CAS PubMed.
- B. E. Eichler and P. P. Power, J. Am. Chem. Soc., 2000, 122, 8785–8786 CrossRef CAS.
- S. Weiß, H. Schubert and L. Wesemann, Chem. Commun., 2019, 55, 10238–10240 RSC.
- J. Schneider, C. P. Sindlinger, K. Eichele, H. Schubert and L. Wesemann, J. Am. Chem. Soc., 2017, 139, 6542–6545 CrossRef CAS PubMed.
- I. Krossing, H. Brands, R. Feuerhake and S. Koenig, J. Fluorine Chem., 2001, 112, 83–90 CrossRef CAS.
- J. D. Queen, J. C. Fettinger and P. P. Power, Chem. Commun., 2019, 55, 10285–10287 RSC.
- T. M. Arkhireeva, B. M. Bulychev, A. N. Protsky, G. L. Soloveichik and V. K. Bel'sky, J. Organomet. Chem., 1986, 317, 33–40 CrossRef CAS.
- I. Zharov, B. T. King, Z. Havlas, A. Pardi and J. Michl, J. Am. Chem. Soc., 2000, 122, 10253–10254 CrossRef CAS.
-
T. Müller, in Advances in Organometallic Chemistry, ed. R. West, A. F. Hill and F. G. A. Stone, Academic Press, 2005, vol. 53, pp. 155–215 Search PubMed.
- J. B. Lambert and B. Kuhlmann, Chem. Commun., 1992, 931–932, 10.1039/C39920000931.
- A. Sekiguchi, T. Fukawa, V. Y. Lee and M. Nakamoto, J. Am. Chem. Soc., 2003, 125, 9250–9251 CrossRef CAS PubMed.
- A. Schäfer, F. Winter, W. Saak, D. Haase, R. Pöttgen and T. Müller, Chem.–Eur. J., 2011, 17, 10979–10984 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- ADF2019.3, Vrije Universiteit Amsterdam, 2019, http://www.scm.com.
- L. Rocchigiani, W. T. Klooster, S. J. Coles, D. L. Hughes, P. Hrobárik and M. Bochmann, Chem.–Eur. J., 2020, 26, 8267–8280 CrossRef CAS PubMed.
- M. Widemann, K. Eichele, H. Schubert, C. P. Sindlinger, S. Klenner, R. Pöttgen and L. Wesemann, Angew. Chem., Int. Ed., 2021, 60, 5882–5889 CrossRef CAS PubMed.
-
J. Mason, Multinuclear NMR, Plenum Press, New York, NY 1987 Search PubMed.
-
R. K. Harris and B. E. Mann, NMR and the periodic table, Academic Press, London, 1978 Search PubMed.
- T. A. Mobley, R. Gandour, E. P. Gillis, K. Nti-Addae, R. Palchaudhuri, P. Rajbhandari, N. Tomson, A. Vargas and Q. Zheng, Organometallics, 2005, 24, 3897–3906 CrossRef CAS.
- M. P. Johnson and D. F. Shriver, J. Am. Chem. Soc., 1966, 88, 301–304 CrossRef CAS.
- M. S. Holt, W. L. Wilson and J. H. Nelson, Chem. Rev., 1989, 89, 11–49 CrossRef CAS.
- M. L. McCrea-Hendrick, C. A. Caputo, J. Linnera, P. Vasko, C. M. Weinstein, J. C. Fettinger, H. M. Tuononen and P. P. Power, Organometallics, 2016, 35, 2759–2767 CrossRef CAS.
- S. M. Mansell, R. H. Herber, I. Nowik, D. H. Ross, C. A. Russell and D. F. Wass, Inorg. Chem., 2011, 50, 2252–2263 CrossRef CAS PubMed.
- A. Y. Khalimon, K. Y. Dorogov, A. V. Churakov, L. G. Kuzmina, D. A. Lemenovskii, J. A. K. Howard and G. I. Nikonov, Dalton Trans., 2007, 2440–2449 RSC.
- P. Kircher, G. Huttner, B. Schiemenz, K. Heinze, L. Zsolnai, O. Walter, A. Jacobi and A. Driess, Chem. Ber., 1997, 130, 687–699 CrossRef CAS.
- A. C. Filippou, P. Portius, A. I. Philippopoulos and H. Rohde, Angew. Chem., Int. Ed., 2003, 42, 445–447 CrossRef CAS PubMed.
- A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339–3341 CrossRef CAS.
-
E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018.
- H. Arp, J. Baumgartner, C. Marschner, P. Zark and T. Müller, J. Am. Chem. Soc., 2012, 134, 10864–10875 CrossRef CAS PubMed.
- B. E. Eichler, A. D. Phillips, S. T. Haubrich, B. V. Mork and P. P. Power, Organometallics, 2002, 21, 5622–5627 CrossRef CAS.
- Y. N. Lebedev, U. Das, G. Schnakenburg and A. C. Filippou, Organometallics, 2017, 36, 1530–1540 CrossRef CAS.
- M. Brookhart, B. Grant and A. F. Volpe, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- T. Fukuda, H. Hashimoto and H. Tobita, J. Organomet. Chem., 2017, 848, 89–94 CrossRef CAS.
- T. Fukuda, T. Yoshimoto, H. Hashimoto and H. Tobita, Organometallics, 2016, 35, 921–924 CrossRef CAS.
- S. Wang, M. L. McCrea-Hendrick, C. M. Weinstein, C. A. Caputo, E. Hoppe, J. C. Fettinger, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2017, 139, 6586–6595 CrossRef CAS PubMed.
- L. Pu, P. P. Power, I. Boltes and R. Herbst-Irmer, Organometallics, 2000, 19, 352–356 CrossRef CAS.
- J. Vícha, J. Novotný, S. Komorovsky, M. Straka, M. Kaupp and R. Marek, Chem. Rev., 2020, 120, 7065–7103 CrossRef PubMed.
- H.-J. Liu, C. Landis, C. Raynaud, O. Eisenstein and T. D. Tilley, J. Am. Chem. Soc., 2015, 137, 9186–9194 CrossRef CAS PubMed.
- P. W. Smith, R. C. Handford and T. D. Tilley, Organometallics, 2019, 38, 4060–4065 CrossRef CAS.
- N. J. Cooper and M. L. H. Green, Dalton Trans., 1979, 1121–1127 RSC.
- D. G. Bickley, N. Hao, P. Bougeard, B. G. Sayer, R. C. Burns and M. J. McGlinchey, J. Organomet. Chem., 1983, 246, 257–268 CrossRef CAS.
- S. B. Jones and J. L. Petersen, Inorg. Chem., 1981, 20, 2889–2894 CrossRef CAS.
- B. D. James, R. K. Nanda and M. G. H. Walbridge, Inorg. Chem., 1967, 6, 1979–1983 CrossRef CAS.
- R. M. Whittal, G. Ferguson, J. F. Gallagher and W. E. Piers, J. Am. Chem. Soc., 1991, 113, 9867–9868 CrossRef CAS.
- J. Bareš, P. Richard, P. Meunier, N. Pirio, Z. Padělková, Z. Černošek, I. Císařová and A. Růžička, Organometallics, 2009, 28, 3105–3108 CrossRef.
- A. I. Sizov, T. M. Zvukova, A. V. Khvostov, V. K. Belsky, A. I. Stash and B. M. Bulychev, J. Organomet.
Chem., 2003, 681, 167–173 CrossRef CAS.
- A. I. Sizov, T. M. Zvukova, V. K. Belsky and B. M. Bulychev, J. Organomet. Chem., 2001, 619, 36–42 CrossRef CAS.
- T. Imori, V. Lu, H. Cai and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 9931–9940 CrossRef CAS.
- T. Imori and T. D. Tilley, Chem. Commun., 1993, 1607–1609 RSC.
- J. Guihaumé, C. Raynaud, O. Eisenstein, L. Perrin, L. Maron and T. D. Tilley, Angew. Chem., Int. Ed., 2010, 49, 1816–1819 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full characterization, NMR spectra, DFT details and XYZ coordinates. CCDC 2142080–2142089. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2sc00297c |
|
| This journal is © The Royal Society of Chemistry 2022 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Lars
Wesemann
*b and
Lars
Wesemann