
Supramolecular assembly of pyrene-tetrathiafulvalene hybrids on graphene: structure–property relationships and biosensing activity†
Marina
Garrido
 a,
Emiliano
Martínez-Periñán
b,
Joaquín
Calbo
c,
Laura
Rodríguez-Pérez
a,
Juan
Aragó
c,
Encarnación
Lorenzo
*bd,
Enrique
Ortí
*c,
Nazario
Martín
*ad and
Ma Ángeles
Herranz
*a
a,
Emiliano
Martínez-Periñán
b,
Joaquín
Calbo
c,
Laura
Rodríguez-Pérez
a,
Juan
Aragó
c,
Encarnación
Lorenzo
*bd,
Enrique
Ortí
*c,
Nazario
Martín
*ad and
Ma Ángeles
Herranz
*a
aDepartamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad Complutense de Madrid, Avda. Complutense s/n, 28040 Madrid, Spain. E-mail: nazmar@ucm.es; maherran@ucm.es
bDepartamento de Química Analítica y Análisis Instrumental, Facultad de Ciencias, Universidad Autónoma de Madrid, C/Francisco Tomás y Valiente 7, 28049 Madrid, Spain. E-mail: encarnacion.lorenzo@uam.es
cInstituto de Ciencia Molecular, Universidad de Valencia, 46980 Paterna, Spain. E-mail: enrique.orti@uv.es
dIMDEA-Nanociencia, c/Faraday 9, Campus Cantoblanco, 28049 Madrid, Spain
First published on 10th July 2021
Abstract
Two different molecular receptors (1 and 2) incorporating one and three pyrene units to promote the π–π interaction with the basal plane of graphene are reported. In order to modulate the electronic properties of graphene, the new receptors are endowed with an electron-donor tetrathiafulvalene moiety (exTTF). The resulting non-covalent hybrids have been characterized by different analytical, spectroscopic and microscopic techniques (TGA, Raman, UV-Vis absorption, TEM and XPS), and the supramolecular interaction of the molecular systems with graphene has been investigated by theoretical calculations. The electrochemical behavior of the pyrene-exTTF hybrids onto distinct graphene-based materials allowed us to determine the differences between both receptors and their affinity for various surfaces. Finally, the non-covalent hybrid formed by a pyrene unit and exTTF has been used for the development of an enzymatic biosensor able to operate at very low potentials.
Introduction
Members of the graphene family of nanomaterials, including single-layer and few-layer graphene (FLG), graphene oxide (GO) and reduced graphene oxide,1 are attracting great attention as ideal components for many applications including electronic devices,2 energy storage3 and sensors,4 due to their unique electrical and mechanical properties.It is well-known that graphene is a zero band-gap semiconductor, which possesses high conductivity and electron mobility that favor charge separation and electron transport.5 However, this singular feature could be a drawback rather than a benefit for its applications in electronics as a semiconductor, since devices made from graphene cannot be switched on and off.6 Therefore, in order to technically exploit graphene, it is necessary to employ non-aggressive techniques to introduce charge carriers—electrons or holes—and/or finite band gaps in the graphene band structure, without disrupting the sp2 hybridization of carbon.7,8
Several functionalization approaches have been developed to modify the surface of graphene, including both covalent and non-covalent modifications.9–13 Non-covalent methods based on π–π stacking are preferable since they preserve the electronic structure and physical properties of graphene. In addition, considering the strong aggregation tendency that graphene presents due to the electrostatic forces and the strong π–π interactions between individual sheets, the supramolecular chemical approach is particularly useful, since it allows the further manipulation and dispersion of graphene.14
Although some molecules do not affect the geometry and electronic configuration of graphene upon adsorption, thus serving as a mere protecting coat of the graphene sheets, some other compounds, for instance, electroactive molecules, may act as n- or p-dopants. In this regard, pyrene and its derivatives are known to feature strong affinities towards the graphene basal plane via π–π stacking.15–17 As such, graphene hybrids have been created using pyrene as the anchoring motif, for example, for graphite exfoliation or as stabilizing agents in water.18,19 Moreover, pyrene covalent dyads, where pyrene is attached to electroactive units such as phthalocyanines,20 porphyrins21 and cyanines22 or fullerene derivatives,23 lead to charge-transfer states upon formation of graphene complexes and photoexcitation.
Dichtel and co-workers investigated the strength of the interaction of pyrene units with graphene by electrochemical methods. By cyclic voltammetry, the behavior of a tripodal pyrene derivative and a monovalent pyrene derivative were compared, and a similar ΔGads value (≈−38 kJ mol−1) was found for both structures. However, the kinetic stabilities extracted from the desorption rate constants proved the formation of more stable monolayers for the tripodal motif under similar conditions.24
Tripodal pyrene binding motifs served to construct graphene nanobuds by making use of the supramolecular π–π interaction of graphene with [60]fullerene endowed with three terminal pyrene units.25 In a more complex system, D’Souza and co-workers described the non-covalent modification of graphene with a multimodular donor–acceptor conjugated system bearing a tripodal motif for the anchoring to graphene.26 Subphthalocyanine acts as the electron-donor moiety, C60 is the electron-acceptor unit and three pyrene units complete the system. The measured redox potentials revealed the influence of graphene on the pyrene and subphthalocyanine units, leaving C60 unperturbed because of its distant position from the graphene surface. A similar scenario was found in the interaction of graphene with the tripodal pyrene anchors covalently linked to porphyrins, as recently reported by some of us.27
Within this context, we decided to investigate the supramolecular modification of graphene with monopodal and tripodal pyrene derivatives of the electron donor 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene (exTTF), and the potential use of these derivatives in biosensing. Traditionally, tetrathiafulvalene (TTF) has been used in combination with oxidoreductase enzymes as a redox mediator in the development of enzyme-based biosensors.28,29 The TTF moiety facilitates the electrical contact between the enzyme redox center and the electrode surface, since the direct contact between the protein redox center and the electrode surface is generally ineffective. In these developments, a crucial step is the immobilization of the TTF on the electrode surface in a disposition that it would be able to interact with the active center of the redox enzyme. As far as we know, exTTF has not previously been used as an enzyme redox mediator. Its similar donor ability compared to TTF30 prompted us to use it as an enzyme redox mediator taking advantage of the great versatility to modify its chemical structure with suitable moieties that facilitate its attachment to the electrode surface.
Here, we describe the synthesis of monopodal and tripodal pyrene receptors endowed with an exTTF unit and their non-covalent interaction with graphene. The expected enhancement in stability and orientation control due to the multivalent effect obtained with three pyrene units vs. the monopodal molecule is discussed. Furthermore, it is likely that these systems offer robust monolayers that display the active functionality away from the graphene surface, enabling the exTTF moieties to interact with other species in solution, or with enzymes such as horseradish peroxidase (HRP) immobilized on the electrode surface for hydrogen peroxide sensing as described below.
Results and discussion
Synthesis of pyrene–exTTF receptors
To ensure strong binding of the exTTF units to the graphene basal plane, two pyrene receptors 1 and 2, containing one and three pyrene units, respectively, have been synthesized (Scheme 1). | ||
| Scheme 1 Synthetic procedure for the synthesis of 2 and molecular structure of 1. | ||
The common step in the synthesis of 1 and 2 is the Horner–Wadsworth–Emmons reaction between dimethyl-(1,3-dithiol-2-yl)-phosphonate and an anthraquinone derivative.31 The exTTF derivative 1 was obtained, following the previously reported procedure (1H-NMR in Fig. S1, ESI†),32 by an esterification reaction between 4-(1-pyrenyl)butyric acid and 2-(hydroxymethyl)-9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene in the presence of 1,3-dicyclohexylcarbodiimide (DCC) and (dimethylamino) pyridine (DMAP). The synthesis of 2 was carried out following the sequence of reactions shown in Scheme 1. The terminal alkyne 333 and the iodo-exTTF derivative 434 were synthesized in turn according to the previously described procedures. Compound 5 was obtained by Sonogashira coupling between these two precursors in the presence of Pd(PPh3)4, CuI and triethylamine. The treatment of molecule 5 with BBr3 allowed trisphenol 6 to be obtained, which was isolated and subsequently reacted without further purification in an etherification process with 4-(1-pyrenyl)-1-butanol tosylate 724 to afford the desired final product 2. The 1H-NMR spectrum of 2 is shown in Fig. S8 (ESI†). The signals corresponding to the aliphatic chains and to the pyrene units can be observed in addition to the exTTF characteristic signals, confirming the formation of the desired final product. Moreover, products 2, 5 and 6 were fully characterized by 1H- and 13C-NMR, FTIR, mass spectrometry and UV-Vis absorption spectroscopy, see the ESI† (Fig. S2–S10) for details.
Supramolecular functionalization of graphene
Before accomplishing the non-covalent functionalization of graphene with compounds 1 and 2, their interaction in solution was investigated by using UV-Vis absorption titrations. Initial studies were carried out with 4-(1-pyrenyl)-1-butanol and exTTF as references. A dispersion of FLG (ca. 0.06 mg mL−1) was obtained after 2.5 h sonication of graphite in N-methylpyrrolidone (NMP) and centrifugation at 500 rpm for 45 minutes. For each titration, different behaviors were observed. In the case of 4-(1-pyrenyl)-1-butanol (Fig. S11, ESI†), spectral changes associated with the addition of FLG were noticed. The pyrene bands with maxima at 329 and 345 nm decreased in intensity, and two pseudo-isosbestic points appear at 313 and 351 nm, respectively. These changes suggest the interaction of 4-(1-pyrenyl)-1-butanol with FLG. However, in the titration experiment with exTTF (Fig. S12, ESI†), no changes were observed in the spectra, indicating the absence of appreciable interactions with FLG at that concentration.Once the studies were carried out with the reference 4-(1-pyrenyl)-1-butanol and exTTF molecules, evaluation of the interactions between 1 or 2 with FLG was accomplished. For 1, a similar behavior to that observed for 4-(1-pyrenyl)-1-butanol is noticed (Fig. 1). A decrease in the intensity of the pyrene bands, as well as for the exTTF characteristic absorption band at 435 nm, is registered with the addition of FLG. These spectral changes are accompanied by the appearance of several pseudo-isosbestic points at 323, 351, 412 and 445 nm (see insets in Fig. 1), which suggest the interaction between FLG and the whole molecule.35–37 On the other hand, in the titration experiments with 2, an increase of the absorbance in the region between 400–600 nm is observed, but no significant change for the absorption band of exTTF was noticed (Fig. S13–14, ESI†). A hypochromic effect was observed for pyrene absorption bands (λ1 = 315 and λ2 = 345 nm) and, again, two pseudo-isosbestic points appear at 311 and 351 nm, respectively. All these data suggest that, in this case, only the pyrene units are interacting with FLG.
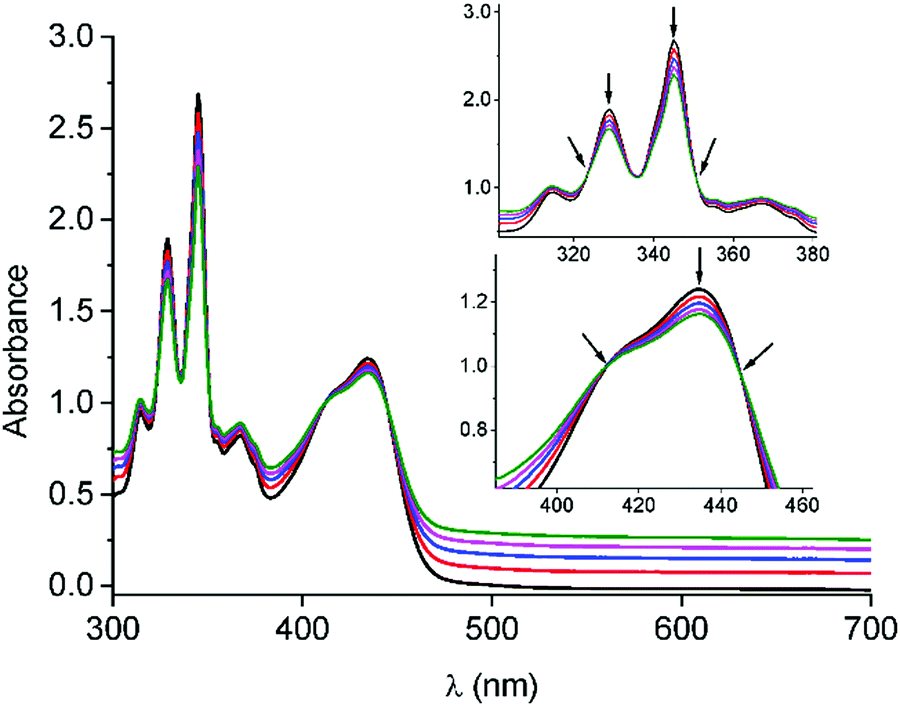 | ||
| Fig. 1 UV-Vis absorption spectra obtained upon titration of 1 (5.87 × 10−5 M) with FLG. Each addition corresponds to 100 μL. The arrows point to the decrease in the intensity of the pyrene bands and the appearance of pseudo-isosbestic points upon FLG addition. | ||
To corroborate the interaction between FLG and the pyrene moieties of molecules 1 and 2, the systems were further investigated by fluorescence titrations.38 In the fluorescence measurements for 1, an emission quenching is noticed with the increasing addition of FLG (Fig. S15, ESI†). This emission is attenuated by 40% in relation to its original intensity. In the case of 2, the experiments evidenced a similar trend (Fig. S16, ESI†). Moreover, the maximum emission of the pyrene excimer slightly shifts to lower wavelengths from 486 to 483 nm with the addition of increasing quantities of FLG. This shift could be explained by the disruption of the non-covalent interaction between the pyrene rings when the molecule is interacting with FLG.
Once the interactions between 1 and 2 with FLG were verified, the synthesis of the supramolecular hybrids was carried out. The desired receptor was suspended in a FLG dispersion previously exfoliated in NMP. The mixture was sonicated for 30 minutes, and subsequently filtered and washed with dichloromethane to remove the excess of molecules 1 or 2 until transparency of the filtrate. The solids obtained were characterized by several techniques to confirm the non-covalent functionalization of FLG with the monopodal or tripodal exTTF receptors.
UV-Vis absorption spectroscopy is very useful to determine whether supramolecular modification takes place. The spectrum of the complex 1/FLG (Fig. S17, left, ESI†) shows the characteristic pyrene peaks at 329 and 345 nm, respectively. Moreover, a broad band is observed at 432 nm due to the exTTF units. The peak positions match with the maxima of 1, thus confirming the presence of the receptor in the complex. In the case of complex 2/FLG (Fig. S17, right, ESI†), the spectrum shows similar trends to that of 1/FLG with the characteristic two peaks of the pyrene moieties. However, the absorption band of the exTTF moiety is less intense for the tripodal derivative 2, which makes the assignment of this band difficult for complex 2/FLG.
In order to evaluate in a quantitative manner the functionalization degree of FLG with the monopodal and tripodal receptors, TGA analyses under air conditions were performed with the obtained supramolecular complexes.39 Complex 1/FLG undergoes a 35% weight loss, which corresponds approximately to a ratio of one molecule of 1 per 107 carbon atoms. Besides, the thermogram of 1 presents a maximum in its first derivative curve at 300 °C, which matches with the desorption in the supramolecular complex (Fig. S18, ESI†). In the case of complex 2/FLG, the observed weight loss is around 32%, which means that there is around one molecule of 2 per 264 carbon atoms. This lower functionalization degree can be attributed to the greater size of 2vs.1, requiring molecule 2 to have more space than the monovalent derivative 1 in order to interact with FLG. Again, the desorption of the supramolecular complex 2/FLG coincides with the first derivative maximum of 2 (Fig. S19, ESI†). As expected, both supramolecular complexes maintain the decomposition pattern of the monopodal and tripodal exTTF-based receptors, although with a small increase of the temperature decomposition maxima, which indicates thermal stabilization of the whole system due to the non-covalent binding to FLG.
Raman spectroscopy was employed to determine how the non-covalent functionalization influences the properties and structure of FLG. In Fig. 2, the Raman spectra for the supramolecular complexes of both 1 and 2 with FLG are shown. At first glance, the rate of defects (D band) does not increase after the functionalization, which is an expected result considering that no covalent bond is formed upon supramolecular functionalization. The shape of the 2D band is quite affected by the supramolecular complexation. We used this band to quantitatively estimate the thickness of graphene flakes.40 For FLG, the mean number of layers is 7, whereas for complexes 1/FLG and 2/FLG, the mean number of layers is 4 and 5, respectively. Regarding the G band, this is shifted to lower frequencies in the complexes when compared to pristine FLG, suggesting that the interaction of FLG with the pyrene–exTTF receptors causes its n-doping.41,42 The shift of the G band is slightly greater for complex 1/FLG (9 cm−1) than for complex 2/FLG (7 cm−1), which suggests different interactions for each organic molecule. Furthermore, the I2D/IG ratio also varies with the doping degree,43 being smaller for 1, which implies a slightly higher doping degree (greater shift), as can be observed in the inset of Fig. 2.
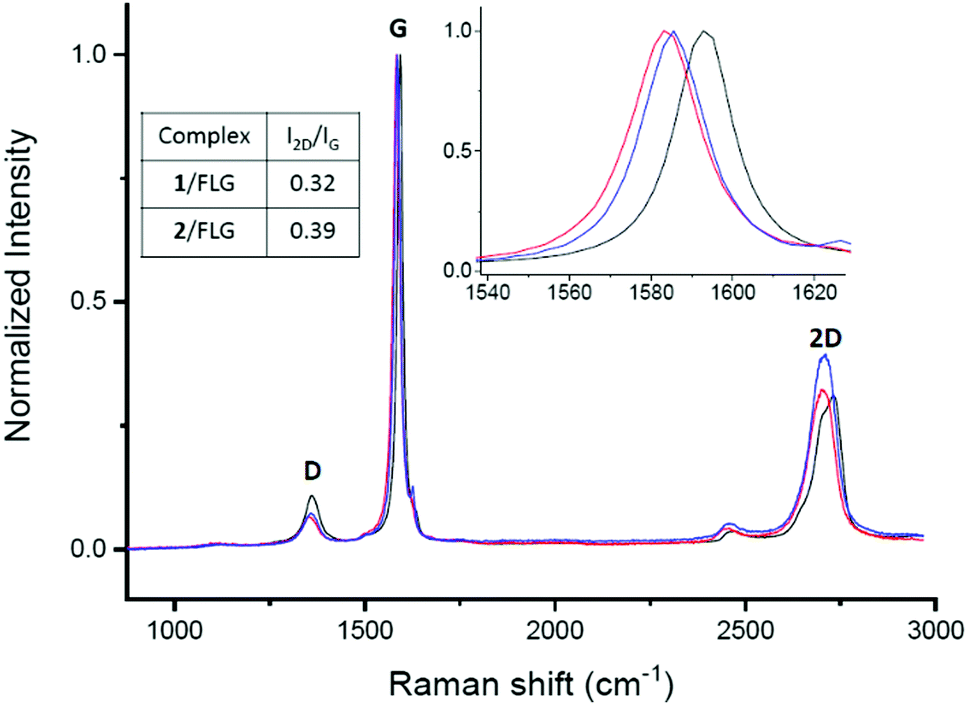 | ||
| Fig. 2 Raman spectra (λexc = 532 nm) of FLG (black) and its complexes with 1 (red) and 2 (blue). | ||
The morphology of the supramolecular complexes was investigated using transmission electron microscopy (TEM) analysis. The re-aggregation of graphene flakes after the functionalization and washing process is prevented by the incorporation of 1 or 2, since TEM analysis reveals the nanomaterial disintegrated and with regular flakes that are randomly stacked onto each other (Fig. 3). The images are quite different from the FLG obtained after the exfoliation in NMP, where more layers can be observed (Fig. S20, ESI†).
 | ||
| Fig. 3 TEM images of FLG after supramolecular functionalization with 1 (left) and 2 (right). | ||
To supplement the structural characterization, X-ray photoelectron spectroscopy (XPS) was carried out to determine the elemental composition of the complexes and molecules 1 and 2, by relating the obtained binding energies with the different elements, their electronic states and the type of hybridization.44 In the XPS survey spectra of complexes 1/FLG and 2/FLG, in addition to the core level contributions of C 1s (284.6 eV) and O 1s (532.6 eV), photoelectrons collected from the S 2p (around 160 eV) are observed (Fig. 4 and Fig. S21, ESI†). In the inset spectra shown in Fig. 4, the peak corresponding to the S 2p in the 1/FLG complex appears shifted to lower binding energies compared with 1. This shift suggests a charge transfer from 1 to FLG.45 The S 2p peak is also observed for complex 2/FLG, but in this case the value of the binding energy is not significantly shifted when compared with 2 (Fig. S21, ESI†). This means that the exTTF in the tripodal derivative does not seem to interact with the graphene surface, maybe because of its distant position from it, which agrees with the results of the UV-Vis absorption titrations and the theoretical calculations described below.
 | ||
| Fig. 4 XPS analysis of FLG (black), derivative 1 (red) and 1/FLG complex (blue). Detailed inset of the S 2p band of 1 (upper graph) compared to complex 1/FLG (lower graph). | ||
Theoretical calculations were performed to confirm the disposition of receptors 1 and 2 over FLG, and to shed light onto the non-covalent interactions giving rise to the resulting supramolecular nanohybrids. Molecular mechanics/molecular dynamics simulations were performed for monopodal (1) and tripodal (2) derivatives onto a graphene sheet in THF solution along 200 ns using the NPT ensemble by means of the NAMD package46 (see the ESI† for full computational details). Control simulations were carried out for the constituting exTTF and pyrene compounds. Calculations predict that the interaction between exTTF and graphene is not strong enough to promote a stable supramolecular assembly, leading to sequential adsorption/desorption processes along the dynamics (Fig. S22, ESI†). In contrast, the pyrene molecule is able to bind graphene permanently through π–π interactions, with an intermolecular pyrene-to-graphene distance of ca. 3.5 Å (Fig. S23, ESI†). Moving to 1, the monopodal structure allows for strong binding between both pyrene and exTTF moieties and the graphene sheet, with no observation of any disassembling process along the whole simulation. The exTTF and pyrene moieties are able to either self-interact weakly, leading to a folded disposition (Fig. 5a), or remain far apart in an extended configuration (Fig. S24a and S25, ESI†). These two regimes coexist along the simulations and are both expected in the experiments. Eventually, the exTTF moiety can detach from the graphene surface, with exTTF–graphene distances up to 14 Å (Fig. S26, ESI†), and partially interact with the π-surface of the pyrene unit (exTTF–pyrene distance of ca. 6 Å, Fig. S25, ESI†). This conformational state is however occasional and does not remain in time (Fig. S25 and S26, ESI†).
 | ||
| Fig. 5 Representative snapshots (side and top views) of the molecular dynamics simulation for monopodal 1 (a) and tripodal 2 (b) assembled with graphene in solution. | ||
In the interaction of 2 with graphene, the tripodal configuration allows for a strong supramolecular recognition towards graphene through the three pyrene legs (Fig. 5b), the resulting assembly being persistent along the 200 ns simulation time. The intramolecular distance between the pyrene moieties varies in a wide range between 10 and 30 Å, depending on the relative position of the pyrene legs (Fig. S24b and S27, ESI†), but they remain parallel to the graphene surface with pyrene–graphene intermolecular distances of around 3.5 Å (Fig. S28, ESI†). In contrast, the exTTF head stays in an apical disposition, away from the carbon nanomaterial, thus preventing any kind of exTTF–graphene interaction. This orientation is maintained along the dynamics simulation, with an average exTTF–graphene intermolecular distance of ca. 12.5 Å (Fig. S29 and S30, ESI†).
In order to identify and quantify the non-covalent interactions stabilizing the supramolecular nanohybrids between 1–2 and graphene, representative snapshots of the molecular dynamics (Fig. 5a and b for 1 and 2, respectively) were extracted, and their geometries were optimized using the MM3 force field,47 keeping the atoms of the graphene sheet frozen and in vacuum conditions (see the ESI† for further details). The minimum-energy geometry of 1 (see Fig. S31a, ESI†) indicates stabilizing π–π interactions between pyrene and graphene at ca. 3.5 Å, together with short CH⋯π (2.5 Å) and O⋯π (3.4 Å) contacts between the aliphatic chain and graphene. Interestingly, the exTTF unit interacts with graphene by means of a mixed supramolecular interaction involving π–π, CH⋯π and S⋯π forces, which is found to be the most stable arrangement for the exTTF–graphene complexation.30 Theoretical calculations predict an interaction energy (Eint) of −58 kcal mol−1 for the graphene complex with 1. This value is slightly larger than the sum of the individual pyrene and exTTF contributions (−23.4 and −26.1 kcal mol−1, respectively), indicating a small but active role of the aliphatic chain (through CH⋯π and O⋯π interactions) in the final stability of the nanohybrid.
In the case of the graphene complex with 2 (Fig. S31b, ESI†), theoretical calculations predict short π–π interactions of ca. 3.5 Å between the three pyrene units and graphene, similar to that calculated for the analogous monopodal complex. In addition, CH⋯π and O⋯π contacts are calculated at 2.4–2.5 and 3.2 Å, respectively, between the leg connectors and graphene. In contrast to the monopodal 1 nanohybrid, the exTTF moiety is in the apical position, thus far away from the graphene sheet, and preventing any stabilizing interaction between both entities. Theoretical calculations predict an interaction energy between 2 and graphene of −104.2 kcal mol−1. This Eint is significantly larger than three times the pyrene–graphene stabilization of −23.4 kcal mol−1, thus pointing to the extra stability conferred by the saturated chain linker and the triphenoxymethyl platform.
Electrochemical studies and biosensing evaluation
The electrochemical behavior of 1 and 2 onto graphene surfaces was also studied (Fig. S32, ESI†). The study was carried out at three different graphene/glassy carbon modified electrodes: graphene oxide/glassy carbon electrode (GO/GC), electrochemically reduced graphene oxide/glassy carbon electrode (GO-ER/GC) and pristine graphene/glassy carbon electrode (FLG/GC). The non-covalent functionalization of these electrodes was carried out by immersing them in a solution of 1 or 2 (0.36 mM in THF) for 15 hours. Then, the electrodes were washed with THF to remove the molecules non-directly adsorbed on the electrode surface. The cyclic voltammograms (CVs) of these electrodes were recorded using an electrochemical cell containing clean electrolyte (0.1 M tetrabutylammonium perchlorate, TBAP, in DMF) (Fig. S33, ESI†). In all cases, the redox couple associated with the oxidation/reduction of the exTTF moiety is evident, meaning that the molecules are adsorbed onto the electrode surface. The exTTF molecule was employed as a reference to compare with the electrodes modified with 1 and 2. The differences observed in the current intensity can be explained by the interactions between the organic compounds and the surface of the electrodes. GO (Fig. S33A, ESI†) is a hydrophilic material due to the oxygenated groups present on its surface, so the affinity of the molecules coated with pyrenes is smaller than the one of the exTTF. On the other hand, FLG and electrochemically reduced graphene oxide are more hydrophobic because of the higher π-conjugated surface, which favors the π–π interactions between the pyrene units and the electrodes (Figs S33B, C and Table S1, ESI†). In fact, for these two electrodes almost no signal was observed for exTTF, so the adsorption takes place by the pyrene moieties present in 1 and 2.Laviron analyses were performed to determine the differences in the electron-transfer rates between both molecules.48 The values obtained for the transfer coefficient (α) and the heterogeneous charge-transfer rate constant (k°) are summarized in Table S2 (ESI†). In all cases, the value of k° is higher for the monovalent derivative, which suggests that the tripodal derivative is adsorbed through the pyrene units with the exTTF moiety far away from the electrode surface in comparison with the monovalent molecule, making electron transfer between them difficult. These results agree well with the theoretical predictions discussed above and with previously reported results on the heterogeneous rate constants of electron transfer in monopodal and tripodal pyrene systems containing redox-active Co(tpy) complexes.49 In addition, the dependence with time of the adsorption process was investigated by determining the surface coverage (Γ) of the electrodes. The modified electrodes were immersed in the solutions of exTTF derivatives 1 or 2 for increasing times. Fig. 6 depicts the surface coverage as a function of the adsorption time for each modified electrode. The surface coverage is noticeably higher for the GO-ER/CG electrode than for the others, although no significant differences are observed for the two pyrene receptors (Fig. S34, ESI†). The kinetic stability of the adsorbed molecules was also evaluated. Due to the higher surface coverage of the GO-ER/CG electrode, subsequent experiments were carried out with this electrode. The modified electrode was transferred to a clean electrolyte solution, and several CVs were successively applied. Adsorption isotherms of 1 and 2 on GO-ER/GC were carried out fixing 15 hours as the proper time to reach an equilibrium of the adsorption process of 1 and 2 on the electrode surface (Fig. S35 and Table S3, ESI†). They were fitted to Langmuir and Freundlich models. For both 1 and 2, the data fit better with the Langmuir model, which points to the adsorption as a monolayer of the electroactive molecule onto the nanomaterial. In the case of the FLG/GC electrodes, only the adsorption isotherms of 2 fit the Langmuir model. This fact indicates that a single monolayer of 2 is adsorbed onto FLG, due to the tripodal disposition. For 1, the isotherm does not fit this model, suggesting a complex adsorption of the molecule on the nanomaterial, as predicted above by theoretical calculations. In Fig. S36 (ESI†), the desorption behavior for molecules 1 and 2 is shown. The presence of three pyrene units in the tripodal derivative 2 allows for a stronger interaction with the electrode surface and, subsequently, to a lower desorption rate than in the case of the monopodal derivative 1, giving a stable layer with a high surface coverage along with time.
 | ||
| Fig. 6 Electrode surface coverage as a function of the adsorption time for 1 (black) and 2 (red) for (a) GO/GC, (b) GO-ER/GC and (c) FLG/GC electrodes. | ||
In addition to the fundamental electrochemical characterization of the modified electrodes, the development of an enzymatic biosensor was achieved. 1/GO-ER/GC electrodes were used to prepare the biosensor, since these electrodes exhibit the highest surface coverage and a better electrochemical response in aqueous media. Horseradish peroxidase (HRP) was immobilized by cross-linking with glutaraldehyde50 onto the 1/GO-ER/GC electrode. Once the enzyme was immobilized, cyclic voltammetry measurements were carried out in a 0.1 M phosphate buffer solution (PBS) (pH = 6.5) with increasing amounts of H2O2. As observed in Fig. 7b, the intensity of the reduction peak increases and that of the oxidation peak decreases at higher concentrations of H2O2, which is characteristic of an electrocatalytic redox process. These measurements were repeated in the absence of the enzyme and no response was obtained, indicating that 1 acts as a redox mediator, facilitating the electrical contact between the enzyme redox center and the electrode surface. In the case of peroxidases, it has been shown that both one- and two-electron mediators can be employed. As depicted in Fig. 7a, upon addition of H2O2, 1 is able to reduce the oxidized form of the enzyme (HRPox) generated in the enzymatic reaction to HRPred, giving rise to the oxidized form of the redox mediator, which in turn is rapidly reduced back at the electrode surface, resulting in the electrocatalytic signal. This signal is proportional to the concentration of H2O2. Chronoamperometric measurements (Fig. S37A, ESI†) demonstrate that the biosensor presents a wide linear response (Fig. S37C, ESI†) to H2O2 and operates at very low potentials with a detection limit of 1.4 μM, calculated using the criteria 3σ/m; where σ is the standard deviation of the blank (signal obtained in the absence of H2O2) and m is the slope of the calibration line. This value of the limit of detection is comparable to that of other previously reported peroxide biosensors.51–53
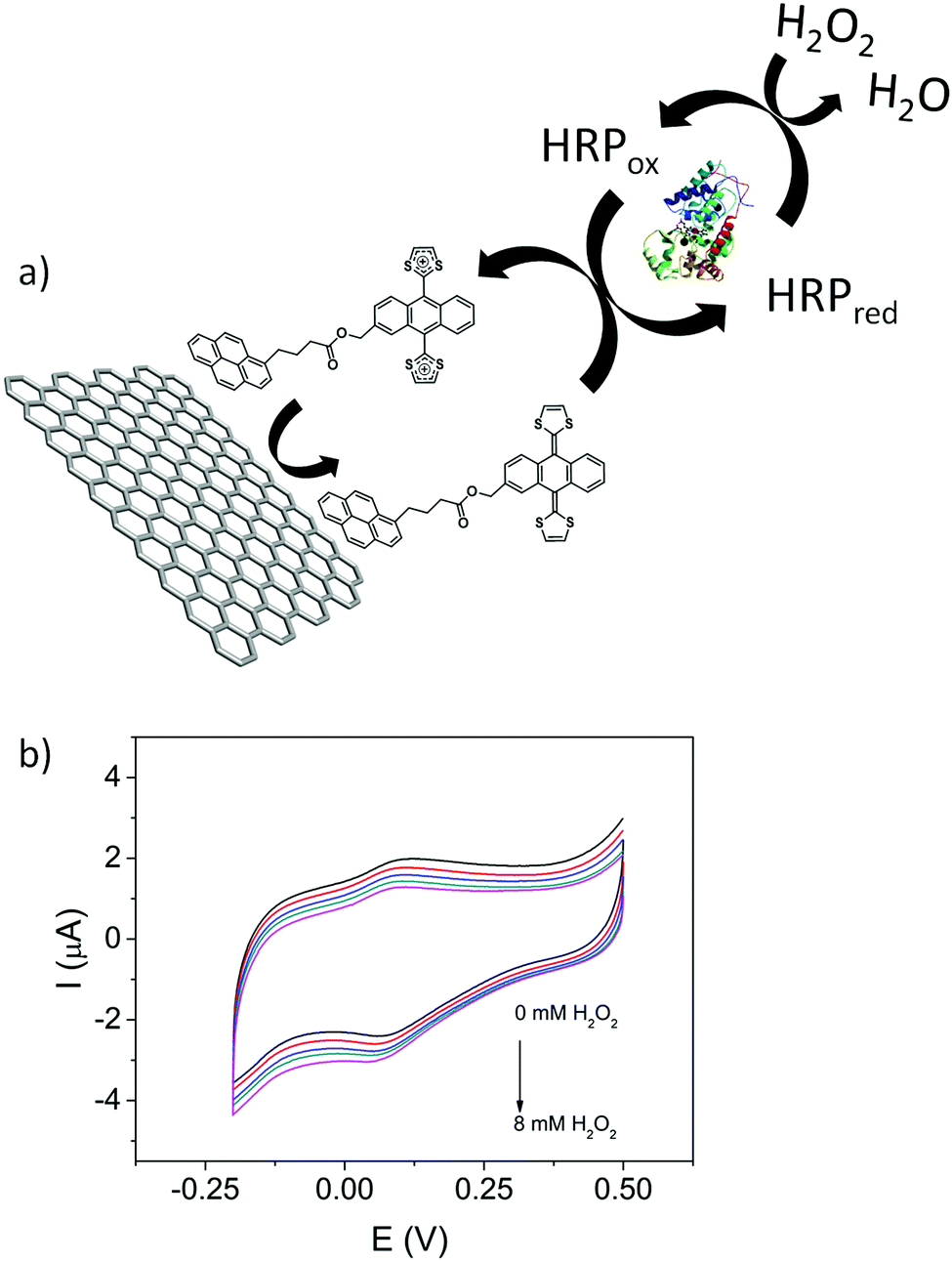 | ||
| Fig. 7 (a) Scheme of the bioelectrocatalytic process. (b) Cyclic voltammograms of HRP/1/GO-ER/GC in 0.1 M PBS (pH = 6.5) with increasing amounts of H2O2 (from 0 mM to 8 mM). Scan rate is 10 mV s−1. | ||
These properties make HRP/1/GO-ER/GC a good sensing platform, which can be expanded to biosensors that couple other enzymes to generate H2O2, as for example lactate oxidase or glucose oxidase.
Conclusions
The research developed in this work has provided valuable information on the construction of supramolecular aggregates between the molecular receptors 1 and 2, incorporating an exTTF electron donor unit, and FLG. The synthesis of the supramolecular complexes 1/FLG and 2/FLG was confirmed by UV-Vis absorption spectroscopy, TGA and Raman, suggesting that the interaction of the different systems with the basal plane of graphene modifies its electronic properties through π–π stacking interactions. Moreover, XPS spectroscopy determined that in the case of 1/FLG the peak corresponding to S 2p in complex 1/FLG is shifted to lower binding energies, suggesting a charge transfer from the exTTF unit to FLG.Theoretical calculations demonstrated that the most probable conformations for both complexes are those that maximize the π–π interactions between the whole molecule and the basal plane of graphene. For 1, both the pyrene and the exTTF units interact with the graphene surface, whereas in the tripodal derivative 2 this maximization implies the approach of the three pyrene units to the graphene surface whereas the exTTF remains far away from it in apical position.
The electrochemical behavior of 1 and 2 was studied in three different graphene-based modified glassy carbon (GC) electrodes. The heterogeneous charge-transfer rate constant is greater for the monopodal derivative 1, supporting the fact that for 2 the exTTF unit is located far away from the electrode surface. In addition, the kinetic stability highlights that, in spite of its higher size and the lower surface coverage, the tripodal derivative gives rise to more stable layers due to the presence of the three pyrene units. Finally, 1/GO-ER/GC modified electrodes were used to develop an enzymatic biosensor, which is able to operate at very low potentials with a wide linear response.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Spanish Ministry of Science and Innovation (MICINN) (projects CTQ2017-83531-R, CTQ2017-84327-P, PGC2018-099568-B-I00, and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the CAM (QUIMTRONIC, Project Y2018/NMT-4783), the Generalitat Valenciana (PROMETEO/2020/077), and European Feder funds (PGC2018-099568-B-I00) is acknowledged. We dedicate this article to Professors Concepció Rovira and Jaume Veciana for their outstanding career and remarkable contributions to the field of materials for molecular electronics and magnetism.Notes and references
- G. Bottari, M. A. Herranz, L. Wibmer, M. Volland, L. Rodríguez-Pérez, D. M. Guldi, A. Hirsch, N. Martín, F. D’Souza and T. Torres, Chem. Soc. Rev., 2017, 46, 4464–4500 RSC.
- X. Yu, H. Cheng, M. Zhang, Y. Zhao, L. Qu and G. Shi, Nat. Rev. Mater., 2017, 2, 17046 CrossRef CAS.
- Z. Wang, H. Gao, Q. Zhang, Y. Liu, J. Chen and Z. Guo, Small, 2019, 15, 1803858 CrossRef.
- S. O. Woo, J. Froberg, Y. Pan, S. Tani, B. R. Goldsmith, Z. Yang and Y. Choi, ACS Appl. Electron. Mater., 2020, 2, 913–919 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS PubMed.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- A. Ciesielski and P. Samorì, Adv. Mater., 2016, 28, 6030–6051 CrossRef CAS.
- A. Criado, M. Melchionna, S. Marchesan and M. Prato, Angew. Chem., Int. Ed., 2015, 54, 10734–10750 CrossRef CAS PubMed.
- S. Eigler and A. Hirsch, Angew. Chem., Int. Ed., 2014, 53, 7720–7738 CrossRef CAS PubMed.
- L. Rodríguez-Pérez, M. A. Herranz and N. Martín, Chem. Commun., 2013, 49, 3721–3735 RSC.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- T. R. B. Ramakrishna, M. Mathesh, Z. Liu, C. Zhang, A. Du, J. Liu, C. J. Barrow, M. Chen, M. J. Biggs and W. Yang, Langmuir, 2020, 36, 13575–13582 CrossRef CAS PubMed.
- D. Parviz, S. Das, H. S. T. Ahmed, F. Irin, S. Bhattacharia and M. J. Green, ACS Nano, 2012, 6, 8857–8867 CrossRef CAS.
- S. Bailey, D. Visontai, C. J. Lambert, M. R. Bryce, H. Frampton and D. Chappell, J. Chem. Phys., 2014, 140, 054708 CrossRef.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- J. Malig, C. Romero-Nieto, N. Jux and D. M. Guldi, Adv. Mater., 2012, 24, 800–805 CrossRef CAS PubMed.
- G. Katsukis, J. Malig, C. Schulz-Drost, S. Leubner, N. Jux and D. M. Guldi, ACS Nano, 2012, 6, 1915–1924 CrossRef CAS PubMed.
- A. Roth, M.-E. Ragoussi, L. Wibmer, G. Katsukis, G. de la Torre, T. Torres and D. M. Guldi, Chem. Sci., 2014, 5, 3432–3438 RSC.
- B. Limburg, J. O. Thomas, G. Holloway, H. Sadeghi, S. Sangtarash, I. C.-Y. Hou, J. Cremers, A. Narita, K. Müllen, C. J. Lambert, G. A. D. Briggs, J. A. Mol and H. L. Anderson, Adv. Funct. Mater., 2018, 28, 1803629 CrossRef.
- A. Roth, C. Schierl, A. Ferrer-Ruiz, M. Minameyer, L. Rodríguez-Pérez, C. Villegas, M. A. Herranz, N. Martín and D. M. Guldi, Chem, 2017, 3, 164–173 CAS.
- S. Qu, M. Li, L. Xie, X. Huang, J. Yang, N. Wang and S. Yang, ACS Nano, 2013, 7, 4070–4081 CrossRef CAS PubMed.
- J. A. Mann, J. Rodríguez-López, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 17614–17617 CrossRef CAS PubMed.
- M. Garrido, J. Calbo, L. Rodríguez-Pérez, J. Aragó, E. Ortí, M. A. Herranz and N. Martín, Chem. Commun., 2017, 53, 12402–12405 RSC.
- C. B. Kc, G. N. Lim and F. D’Souza, Angew. Chem., Int. Ed., 2015, 54, 5088–5092 CrossRef CAS.
- M. Garrido, M. K. Volland, P. W. Münich, L. Rodríguez-Pérez, J. Calbo, E. Ortí, M. A. Herranz, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2020, 142, 1895–1903 CrossRef CAS PubMed.
- L. Bifulco, C. Cammaroto, J. D. Newman and A. P. F. Turner, Anal. Lett., 1994, 27, 1443–1452 CrossRef CAS.
- A. Kausaite-Minkstimiene, V. Mazeiko, A. Ramanaviciene, Y. Oztekin, A. O. Solak and A. Ramanavicius, Electroanalysis, 2014, 26, 1528–1535 CrossRef CAS.
- F. G. Brunetti, J. L. López, C. Atienza and N. Martín, J. Mater. Chem., 2012, 22, 4188–4205 RSC.
- A. J. Moore and M. R. Bryce, Synthesis, 1991, 26–28 CrossRef CAS.
- M. A. Herranz, C. Ehli, S. Campidelli, M. Gutiérrez, G. L. Hug, K. Ohkubo, S. Fukuzumi, M. Prato, N. Martín and D. M. Guldi, J. Am. Chem. Soc., 2008, 130, 66–73 CrossRef PubMed.
- P. D. Jones and T. E. Glass, Tetrahedron, 2004, 60, 11057–11065 CrossRef CAS.
- M. C. Díaz, B. M. Illescas, C. Seoane and N. Martín, J. Org. Chem., 2004, 69, 4492–4499 CrossRef PubMed.
- F. G. Brunetti, H. Isla, J. Aragó, E. Ortí, E. Pérez and N. Martín, Chem. – Eur. J., 2013, 19, 9843–9848 CrossRef CAS.
- E. M. Pérez, L. Sánchez, G. Fernández and N. Martín, J. Am. Chem. Soc., 2006, 128, 7172–7173 CrossRef PubMed.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- A. T. Haedler, H. Misslitz, C. Buehlmeyer, R. Q. Albuquerque, A. Köhler and H.-W. Schmidt, ChemPhysChem, 2013, 14, 1818–1829 CrossRef CAS PubMed.
- J. Liu, G. Chen and M. Jiang, Macromolecules, 2011, 44, 7682–7691 CrossRef CAS.
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O’Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O’Brien, E. K. McGuire, B. Mendoza Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624–630 CrossRef CAS.
- R. Voggu, B. Das, C. S. Rout and C. N. R. Rao, J. Phys.: Condens. Matter, 2008, 20, 472204 CrossRef.
- X. Dong, D. Fu, W. Fang, Y. Shi, P. Chen and L.-J. Li, Small, 2009, 5, 1422–1426 CrossRef CAS.
- A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V. Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C. Ferrari and A. K. Sood, Nat. Nanotechnol., 2008, 3, 210–215 CrossRef CAS PubMed.
- M. E. Lipińska, S. L. H. Rebelo, M. F. R. Pereira, J. A. N. F. Gomes, C. Freire and J. L. Figueiredo, Carbon, 2012, 50, 3280–3294 CrossRef.
- C. Urban, D. Écija, Y. Wang, M. Trelka, I. Preda, A. Vollmer, N. Lorente, A. Arnau, M. Alcamí, L. Soriano, N. Martín, F. Martín, R. Otero, J. M. Gallego and R. Miranda, J. Phys. Chem. C, 2010, 114, 6503–6510 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- N. L. Allinger, Y. H. Yuh and J. H. Lii, J. Am. Chem. Soc., 1989, 111, 8551–8566 CrossRef CAS.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- J. Rodríguez-López, N. L. Ritzert, J. A. Mann, C. Tan, W. R. Dichtel and H. D. Abruña, J. Am. Chem. Soc., 2011, 133, 6224–6236 Search PubMed.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and C. Caldron, Biotechniques, 2014, 37, 790–802 CrossRef PubMed.
- A. M. López Marzo, C. C. Mayorga-Martínez and M. Pumera, Biosens. Bioelectron., 2020, 151, 111980 CrossRef.
- D. A. Centeno, X. H. Solano and J. J. Castillo, Bioelectrochemistry, 2017, 116, 33–38 CrossRef CAS PubMed.
- F. A. Gutiérrez, M. D. Rubianes and G. A. Rivas, Anal. Chim. Acta, 2019, 1065, 12–20 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details including synthesis and characterization, theoretical calculations and electrochemical studies. See DOI: 10.1039/d1tc01442k |
| This journal is © The Royal Society of Chemistry 2021 |