
Mass transport-enhanced electrodeposition of Ni–S–P–O films on nickel foam for electrochemical water splitting†
Raul A.
Marquez-Montes
 a,
Kenta
Kawashima
a,
Yoon Jun
Son
b,
Jason A.
Weeks
a,
H. Hohyun
Sun
b,
Hugo
Celio
c,
Víctor H.
Ramos-Sánchez
d and
C. Buddie
Mullins
*abcef
a,
Kenta
Kawashima
a,
Yoon Jun
Son
b,
Jason A.
Weeks
a,
H. Hohyun
Sun
b,
Hugo
Celio
c,
Víctor H.
Ramos-Sánchez
d and
C. Buddie
Mullins
*abcef
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: mullins@che.utexas.edu
bMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
cTexas Materials Institute, The University of Texas at Austin, Texas 78712, USA
dFacultad de Ciencias Químicas, Universidad Autónoma de Chihuahua, Chihuahua 31125, Mexico
eCenter for Electrochemistry, The University of Texas at Austin, Austin, Texas 78712, USA
fH2@UT, The University of Texas at Austin, Texas 78712, USA
First published on 26th February 2021
Abstract
Electrochemical water splitting is one of the most promising approaches for sustainable energy conversion and storage toward a future hydrogen society. This demands durable and affordable electrocatalysts for the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). In this study, we report the preparation of uniform Ni–P–O, Ni–S–O, and Ni–S–P–O electrocatalytic films on nickel foam (NF) substrates via flow cell-assisted electrodeposition. Remarkably, electrodeposition onto 12 cm2 substrates was optimized by strategically varying critical parameters. The high quality and reproducibility of the materials is attributed to the use of a 3D-printed flow cell with a tailored design. Then, the as-fabricated electrodes were tested for overall water splitting in the same flow cell under alkaline conditions. The best-performing sample, NiSP/NF, required relatively low overpotentials of 93 mV for the HER and 259 mV for the OER to produce a current density of 10 mA cm−2. Importantly, the electrodeposited films underwent oxidation into amorphous nickel (oxy)hydroxides and oxidized S and P species, improving both HER and OER performance. The superior electrocatalytic performance of the Ni–S–P–O films originates from the unique reconstruction process during the HER/OER. Furthermore, the overall water splitting test using the NiSP/NF couple required a low cell voltage of only 1.85 V to deliver a current density of 100 mA cm−2. Overall, we demonstrate that high-quality electrocatalysts can be obtained using a simple and reproducible electrodeposition method in a robust 3D-printed flow cell.
Introduction
Electrocatalytic water splitting offers encouraging benefits for a clean and sustainable energy transition from fossil fuels.1,2 This process allows high-purity hydrogen (H2) production at the cathode through the hydrogen evolution reaction (HER), while the oxygen evolution reaction (OER) occurs at the anode.3,4 However, despite numerous improvements, the sluggish kinetics of both reactions impose challenging overpotential constraints, which entail the use of noble metals (e.g., Pt, Pd) and noble metal oxides (e.g., IrO2, RuO2) with high catalytic activity.5–7 Nonetheless, their high cost and scarcity impedes large-scale implementation, and therefore a pursuit for earth-abundant electrocatalysts with low cost, high stability, and high activity is still in progress.3,8Previous efforts have suggested the use of transition metal carbides,9–11 nitrides,12,13 phosphides,6,7,14 sulfides,2,3,15,16 selenides,3 tellurides,17 oxides and layered double hydroxides18–22 as efficient and cost-effective electrocatalysts for the HER and OER. Among these, amorphous materials have shown exceptional performance compared to their crystalline counterparts, which is attributed to the presence of abundant defects, thus offering more energetic flexibility at the active sites.19,23–31 In addition, heteroatom doping (e.g., N, O, S, P) also improves electrocatalytic activity, conductivity, and even stability for these materials due to advantageous anti-corrosion and electronic properties.24,26
The electrochemical deposition of electrocatalytic films allows for easy creation of materials with plentiful defect sites, as demonstrated by previous reports on metallic phosphides, sulfides, selenides, and oxides/hydroxides.27,31–35 Competitive electrocatalytic activities have been reported, with overpotentials (at 10 mA cm−2) close to 100 mV for the HER and 200 mV for the OER.27,33,36,37 In addition, electrodeposition is a cost-effective and relatively simple technique that enables the fabrication of binder-free electrodes in just one or a few steps.36,37 Furthermore, charge and mass transport properties can be greatly improved when conductive substrates with a large surface area such as nickel foam (NF) are employed.4,38 Yet, several studies in the OER/HER field do not verify the reproducibility of the synthesized materials or the electrochemical testing conditions, and details on the deposition method are usually not fully disclosed. In fact, optimization of the electrodeposition method has not been thoroughly considered in the literature, even though the use of an improved electrodeposition environment is essential to reduce mass transport limitations and guarantee uniform and reproducible films.39–41 Evidently, these aspects hinder the replicability of the materials and impede their transition to large-scale application.
Here, the synthesis of Ni–S–P–O films on NF is addressed by using a mass transport-enhanced electrodeposition approach. In contrast to previous electrodeposition reports in the literature, we highlight the importance of using a reproducible and well-controlled environment to obtain high-quality electrocatalytic films with increased activity for both the HER and the OER. Notably, the deposition is greatly improved using a 3D-printed electrochemical flow cell, which provides superior mass transport and mixing of the plating bath. Critical variables are strategically varied, namely deposition time and electrolyte flow rate to optimize the method. Subsequent evaluation of the as-prepared electrocatalysts for the HER and OER in the same flow cell with alkaline electrolyte demonstrate their superior activity and stability. Finally, the bifunctionality of the best-performing NiSP/NF is validated by performing overall water splitting tests. On the whole, the proposed approach represents an efficient, low-cost and sustainable route to generate high-quality electrocatalysts for large-scale electrochemical water-splitting technologies for a long period of use.
Experimental
Electrode preparation
Nickel foam (NF, 99.99%, 80–110 pores per inch) with a thickness of 1.6 mm was purchased from MTI Co. Pt gauze (99.9%, 52 mesh) and thiourea (CH4N2S) were purchased from Merck KGaA Co. An FAA-3-50 anion exchange membrane (AEM) with a thickness of 50 μm was purchased from FuMATech GmbH. Nickel(II) nitrate [Ni(NO3)2], nickel(II) sulfate hexahydrate (NiSO4·6H2O), sodium hypophosphite monohydrate (NaH2PO2·H2O), acetic acid (CH3COOH), sodium acetate trihydrate (CH3COONa·3H2O), potassium dihydrogen phosphate (KH2PO4), sodium phosphate dibasic (Na2HPO4), and potassium hydroxide (KOH) were purchased from Alfa Aesar. All the aqueous solutions were prepared with deionized water (DIW). Importantly, incidental activity enhancement during OER tests due to Fe impurities was avoided by purifying the KOH electrolyte with a previously reported method.42 Cleaning of NF pieces is detailed in the ESI.†Electrodeposition of nickel sulfide, nickel phosphide and nickel sulfide/phosphide (hereinafter, referred to as NiS, NiP, and NiSP, respectively) films on NF was performed using a 3D-printed electrochemical flow cell. Its features, geometry, hydrodynamics, and mass transport properties are detailed in our previous work.40Fig. 1 displays the overall electrodeposition process. In a typical synthesis, clean NF pieces with a geometric area of 12 cm2 were placed inside the cathodic compartment of the flow cell. Three pieces of Pt gauze were stacked and placed in the anodic compartment. An AEM was located between both sections to avoid incidental Pt incorporation on the NF electrodes. The inter-electrode gap was 5 mm. Fig. S1 in the ESI† details the flow cell configuration. The plating bath (100 mL) was recirculated at a constant flow rate using a peristaltic pump. Compositions were adapted from previous reports in the literature: 10 mM Ni(NO3)2, 0.5 M CH4N2S and 100 mM phosphate buffer (pH 8.0) for Ni + S deposition,33,34 and 10 mM NiSO4, 100 mM NaH2PO2·H2O and 100 mM acetate buffer (pH 3.6) for Ni + P deposition.43,44 For the NiSP/NF electrode, Ni + S deposition was performed initially, followed by Ni + P deposition just by changing the plating bath in situ. Electrodeposition was carried out using chronopotentiometry (CP) runs at −50 mA cm−2 using an Interface 1000 potentiostat/galvanostat (Gamry). A three-electrode configuration with a Ag/AgCl reference electrode was employed. Three different deposition times (15, 30, and 45 min) and flow rates (3, 6, and 9 L h−1) were evaluated at 25 °C. To verify the reproducibility, five replicates for each condition were studied. The flow rate was controlled using calibrated rotameters. After electrodeposition, the electrodes were rinsed with DIW and stored in a vacuum desiccator (25 °C).
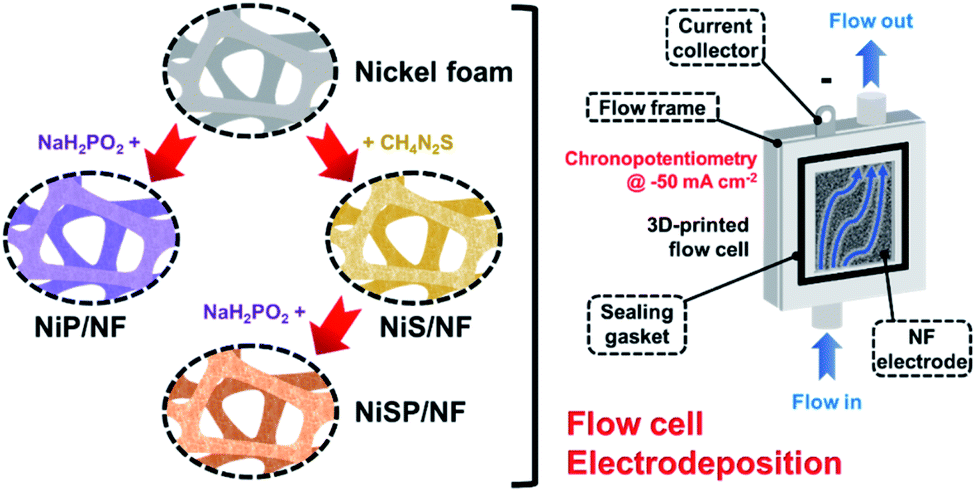 | ||
| Fig. 1 Schematic illustration of the flow electrodeposition approach using a 3D-printed flow cell. | ||
Characterization
Crystallinity of the electrocatalytic films was assessed by X-ray diffraction (XRD) using a Rigaku MiniFlex 600 X-ray diffractometer, in the range from 20 to 80 2θ degree. The morphology and structure of the materials was examined by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) on a FEI Quanta 650 environmental scanning electron microscope and a JEOL 2010f transmission electron microscope at 200 kV accelerating voltage, respectively. Energy-dispersive X-ray spectroscopy (EDX) was performed to obtain elemental mappings. X-ray photoelectron spectroscopy (XPS) was employed to investigate surface chemical composition using a Kratos AXIS Ultra DLD spectrometer. Data was obtained using a monochromatic Al-Kα X-ray source (hv = 1486.5 eV) with a spot size of 300 × 700 μm2. The binding energies of Ni 2p3/2, S 2p, and P 2p were corrected/verified with the C 1s major peak of adventitious carbon at 284.8 eV. XPS data were processed by using CasaXPS software. Total-reflectance X-ray fluorescence (TXRF) was used to perform multi-elemental analysis of the electrolyte with an internal standard (Y, 5 ppm) in a Bruker S2 PICOFOX spectrometer. Finally, the H2 and O2 gas evolution during overall water splitting was measured using the water displacement method in 50 mL burettes.Electrochemical measurements
The as-prepared electrodes were directly used as the working electrode in the electrochemical flow cell to evaluate the OER and HER activity and stability. Tests were performed in Fe-purified 1.0 M KOH electrolyte (pH ∼ 14) using a three-electrode configuration and a Ag/AgCl (saturated KCl) reference electrode. The latter was used due to its robustness and low cost, which allowed us to monitor each type of reaction and synthesis separately with different Ag/AgCl electrodes. The open-circuit potential (OCP) was always measured 15 min prior to testing. Polarization curves were recorded as linear sweep voltammetry (LSV) scans with a scan rate of 10 mV s−1. LSV curves were iR-compensated at 85%. All potentials were referred to the reversible hydrogen electrode (RHE) according to the equation (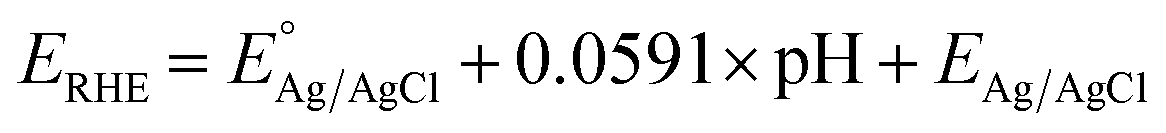 ;
; 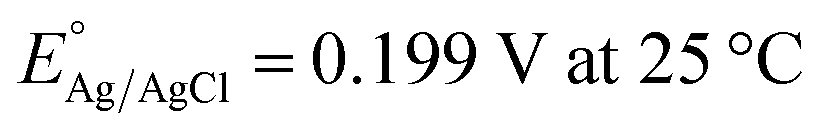 ). The electrochemically-active surface area (ECSA) was estimated through the double-layer capacitance (CDL). The latter was estimated from cyclic voltammetry (CV) scans (at ±50 mV around the OCP) recorded from 200 to 10 mV s−1. Cathodic and anodic currents in the middle of the potential window were averaged and plotted against the scan rate, giving the linear behavior of an ideal capacitor. The slope of the fitted line was associated with the value of CDL, which was compared between electrodes. Tafel slopes were calculated from LSV curves using the Tafel equation: η = a + b
). The electrochemically-active surface area (ECSA) was estimated through the double-layer capacitance (CDL). The latter was estimated from cyclic voltammetry (CV) scans (at ±50 mV around the OCP) recorded from 200 to 10 mV s−1. Cathodic and anodic currents in the middle of the potential window were averaged and plotted against the scan rate, giving the linear behavior of an ideal capacitor. The slope of the fitted line was associated with the value of CDL, which was compared between electrodes. Tafel slopes were calculated from LSV curves using the Tafel equation: η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log|j|, where η represents the overpotential, b is the Tafel slope, a is a constant, and j is the current density. Stability of the electrodes was assessed through CP runs for 20 h at 10 and 20 mA cm−2 for the HER and OER, respectively. Polarization curves and CV scans were registered during and after stability runs to monitor activity and the ECSA, respectively. Finally, in overall water splitting tests, the flow cell was assembled as a two-electrode cell. Polarization curves and galvanostatic runs were recorded to evaluate activity and stability, respectively. For gas evolution tests, an AEM was placed between electrodes to allow quantification. Faradaic efficiency calculation is detailed in the ESI.†
log|j|, where η represents the overpotential, b is the Tafel slope, a is a constant, and j is the current density. Stability of the electrodes was assessed through CP runs for 20 h at 10 and 20 mA cm−2 for the HER and OER, respectively. Polarization curves and CV scans were registered during and after stability runs to monitor activity and the ECSA, respectively. Finally, in overall water splitting tests, the flow cell was assembled as a two-electrode cell. Polarization curves and galvanostatic runs were recorded to evaluate activity and stability, respectively. For gas evolution tests, an AEM was placed between electrodes to allow quantification. Faradaic efficiency calculation is detailed in the ESI.†
Results and discussion
Characterization of electrodes
Electrocatalytic NiP, NiS, and NiSP films were deposited onto clean NF substrates using a forced-flow electrodeposition procedure in which the plating bath was recirculated within a compact 3D-printed flow cell with a tailored design. Fig. S2† shows photos of the as-prepared materials. A uniform deposition is evident, which is mainly attributed to the superior mixing and flow dispersion of the electrochemical flow cell.40 Notice that large substrates with a projected geometric area of 12 cm2 were employed, contrarily to most of the reports in the literature which usually utilize small electrodes (1 cm2). As a comparison, the NiSP film was prepared in a conventional electrochemical cell stirred by a magnetic stir bar (details in the ESI†). Opposite to the flow cell-assisted approach, a non-uniform film was obtained (Fig. S2d†). The uneven distribution of colors on the NF denote a partial deposition in some areas, which is attributed to the irregular vortex flow caused by the magnetic stirrer. Not surprisingly, this reveals that electrolyte mixing is essential during electrodeposition, in particular for large substrates. More details on flow cell-assisted electrodeposition and its optimization are described in the next section.The morphology of the as-obtained films was assessed by SEM. Pristine NF exhibited a smooth surface as shown in Fig. S3.† On the other hand, the morphology of the synthesized films was quite rough, as depicted in the SEM images of Fig. 2. In the case of NiP (Fig. 2a), the surface was covered by numerous grains with sizes ranging from 80 to 250 nm. These particles are covered by abundant nanosheets following a beehive-like pattern, in a similar fashion as described previously.33 These features can be seen in more detail at additional magnifications in Fig. S4.† Furthermore, the NiS film exhibited a comparable morphology in which grains protruded from the NF backbone (Fig. 2b). Additional SEM images confirm the roughened surface (Fig. S5†). However, while grain size seems to be comparable to that observed in the NiP film, the NiS film lacks abundant nanosheet arrays on the surface. Finally, by carrying out an additional electrodeposition step of P to the NiS film, the morphology is modified further as depicted in Fig. 2c. Numerous nanosheet arrays again cover the grains, which is evident especially at higher magnifications (Fig. 2d and S6†). The roughness of the grains appears to be increased, as smaller particles seem to protrude from the initial microparticles. Furthermore, SEM-EDX elemental mappings clearly revealed a uniform distribution of S and P in the synthesized electrodes, as shown in Fig. 2e–g. Interestingly, mappings also revealed a small amount of oxygen in the materials, which suggests that partial oxidation occurred.
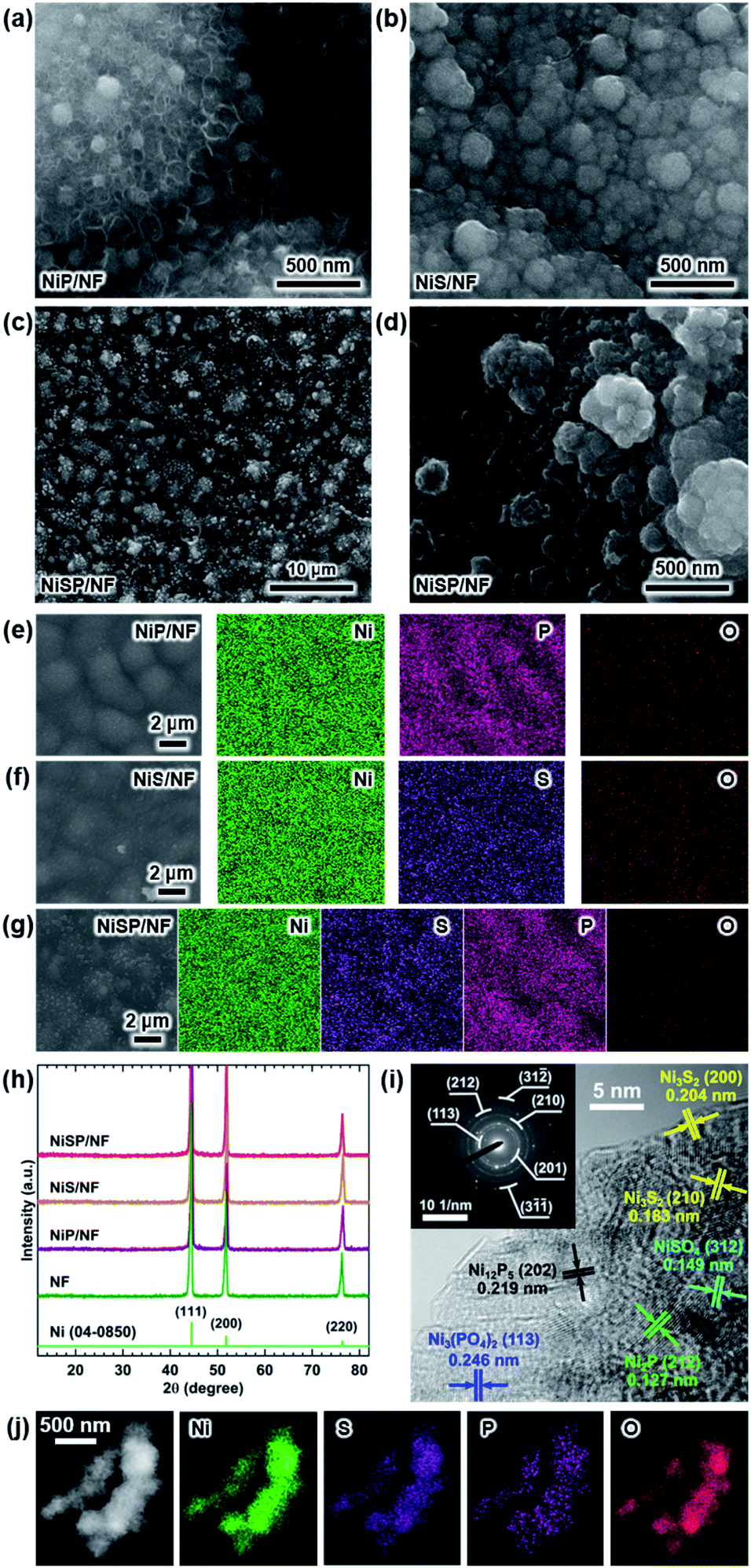 | ||
| Fig. 2 Morphological and structural characterization of the as-prepared electrodes: SEM images for (a) NiP/NF, (b) NiS/NF, and NiSP/NF at (c) low and (d) high magnifications. SEM/EDX elemental mappings for (e) NiP/NF, (f) NiS/NF, and (g) NiSP/NF. (h) XRD results. (i) HRTEM image of NiSP/NF. The inset displays the corresponding SAED pattern. (j) TEM/EDX elemental mappings for NiSP/NF. | ||
The observed morphology in the as-obtained films is attributed to the features of the electrodeposition process. First, the grain structures protruding from the NF surface can be explained by the simultaneous deposition of both Ni metal (Ni2+ → Ni0) and Ni + S/P co-deposition. According to the literature, co-deposition of Ni and P is assumed to occur in a way analogous to the formation of CoP, where elemental Co and P produced during electrodeposition react to yield CoP.45,46 Elemental P is produced from reduction of H2PO2− in acidic conditions as follows:47
| H2PO2− + 2H+ + e− → P + 2H2O | (1) |
On the other hand, it is assumed that co-deposition of Ni and S, using thiourea as S source, proceeds as follows in alkaline media:36,43,48
| NO3− + H2O + 2e− → NO2− + 2OH− | (2) |
| SC(NH2)2 + 2OH− → S2− + OC(NH2)2 + H2O | (3) |
| Ni2+ + S2− → NiS | (4) |
Crystallinity of the as-obtained films was assessed by means of XRD. Results are depicted in Fig. 2h. Only three diffraction peaks at 44.2, 52, and 76.4° can be seen, which are attributed to cubic Ni metal (ICDD PDF# 04-0850).50 No intense peaks neither for NixPy nor NixSy species were found, which suggests that heteroatom sites could be confined to the surface. To gain more structural information of the materials without the interference of the NF support, TEM was performed. The samples were subjected to intense sonication in ethanol for two days to scratch the surface of the electrodes. As depicted in Fig. 2i, the high-resolution TEM (HRTEM) images of NiSP exhibit lattice fringes corresponding to the (202) facet of Ni12P5, the (212) facet of Ni2P, the (210) and (200) facets of Ni3S2, the (312) facet of NiSO4 and the (113) facet of Ni3(PO4)2.33,51–53 This suggests that the as-prepared NiSP was nano-polycrystalline. Corresponding selected area electron diffraction (SAED) pattern (inset of Fig. 2i) displays up to seven distinguishable rings with several discrete spots, which confirms the polycrystalline nature of the NiSP film. Rings also match the planes found in the HRTEM image and add two extra facets of Ni3S2. Furthermore, TEM-EDX elemental mappings demonstrate the presence of Ni, S, P, and O atoms in the material (Fig. 2j). Additional TEM images exhibited the characteristic planes of Ni3S2 and NiSO4 for the NiS film and Ni12P5 for the NiP film (Fig. S7†). Notably, the NiP film exhibited a more amorphous nature among the as-prepared films, with limited crystalline spots where Ni12P5 resides. Evidently, the NiSP film exhibited a higher density of S/P-rich spots, which is attributed to the two-step electrodeposition.
To further clarify the surface chemical compositions of the as-obtained samples, XPS analysis was performed. Fig. 3 shows the resultant Ni 2p3/2–1/2, P 2p, and S 2p XPS core-level spectra. The Ni 2p3/2 spectra displayed five peaks located at ∼852.4–852.8 eV for nickel sulfide (Ni–S) or nickel phosphide (Ni–P), ∼855.3 eV for Ni(OH)2, ∼856.6 eV for Ni3(PO4)2, ∼857.0 eV for NiSO4, and ∼861 eV for satellites.54–60 In the P 2p spectra, the two P 2p3/2–1/2 doublets, which correspond to nickel phosphide and nickel phosphate,14,59,61 are confirmed. The S 2p spectra consist of three components which can be assigned to nickel sulfide, S–O species and nickel sulfate.2,62–64 The XPS results show that the NiP, NiS, and NiSP sample surfaces consist of [Ni–P + Ni3(PO4)2 + Ni(OH)2], [Ni–S + NiSO4 + Ni(OH)2], and [Ni–S + NiSO4 + Ni3(PO4)2 + Ni(OH)2], respectively. The presence of sulfate and phosphate species agrees with TEM results. Furthermore, the low intensity of the Ni–P peak in NiP/NF (Fig. 3b) agrees with the low abundance of Ni12P5 from HRTEM images (Fig. S7b†) and suggests that most of the NiP film is composed of amorphous Ni(OH)2 + Ni3(PO4)2. Interestingly, the Ni–S 2p3/2 and 2p1/2 peaks of the NiSP film slightly shifted toward higher binding energies compared to the NiS film, which suggests that charge transfer is modified to some extent possibly by P and O species. A similar effect has been reported on Fe3O4/FeS2 catalysts,65 which indicates the presence of heterogeneous interfaces between Ni–S–P–O species in our deposited films. HRTEM images (Fig. 2i) demonstrate the high abundance of S/P-rich domains next to each other, which provides a complex film with variations on electron density and structural defects. Accordingly, these results confirm that our deposition approach produced films with heterogeneous Ni–P–O, Ni–S–O, and Ni–S–P–O interfaces on NF substrates.
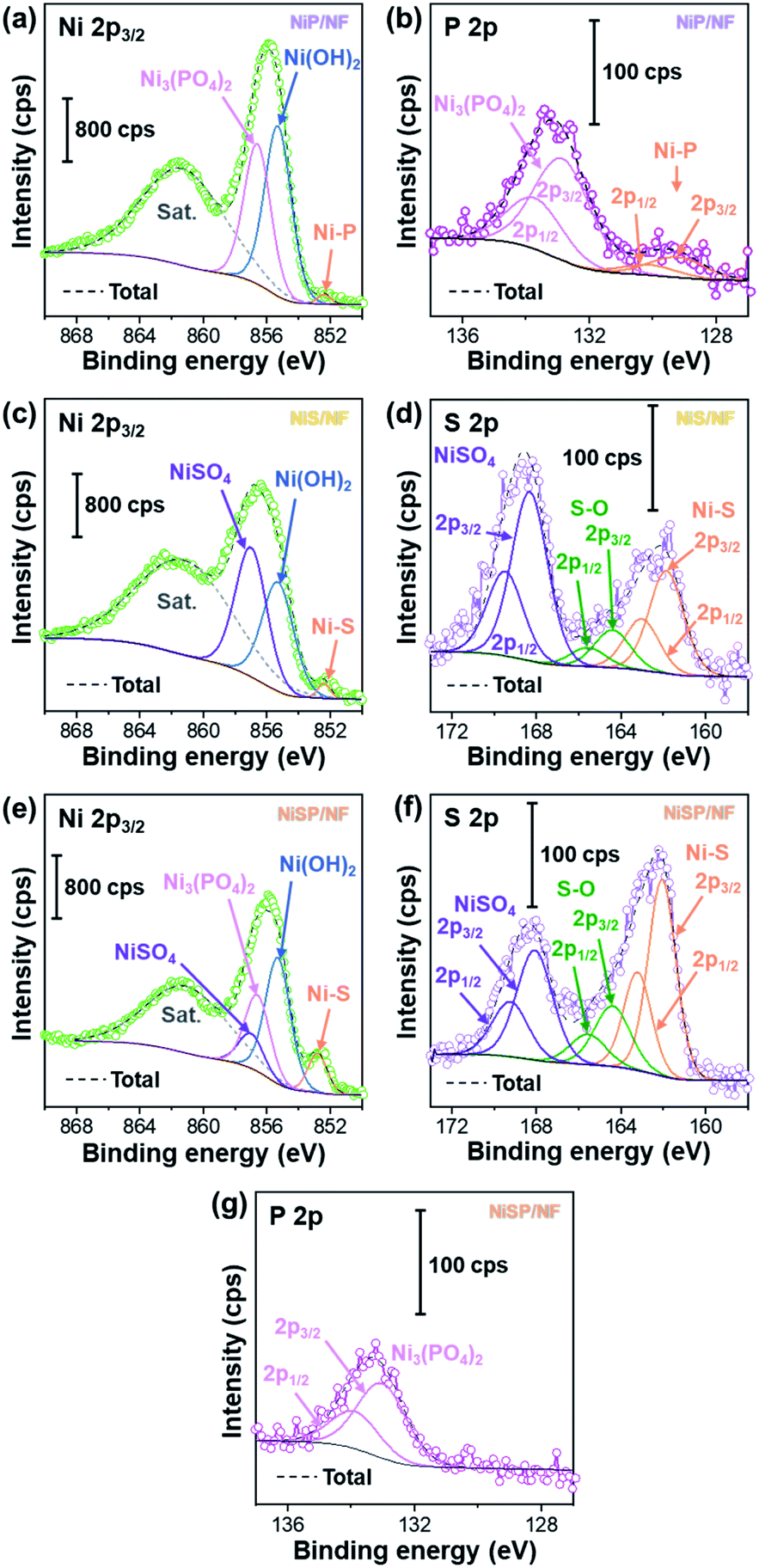 | ||
| Fig. 3 Ni 2p3/2, P 2p, and S 2p XPS core-level spectra of (a and b) NiP/NF, (c and d) NiS/NF, and (e–g) NiSP/NF samples. | ||
Oxygen evolution reaction
The electrocatalytic activity for the OER was evaluated in Fe-purified 1 M KOH electrolyte using the electrochemical flow cell due to its improved mass transport and reproducible environment. Overall results are shown in Fig. 4, where pristine NF is included as comparison. From polarization curves (Fig. 4a), a remarkable oxidation peak emerges between 1.30 and 1.45 V vs. RHE, which can be ascribed to the reversible transformation of β-Ni(OH)2 into β-NiOOH.13,31,33 For the NiSP film the intensity of this oxidation peak is remarkable, compared to the other deposited films, which denotes a high degree of oxidation.31 Moreover, the NiSP film exhibited the smallest overpotentials among the synthesized materials, as depicted in Fig. 4b. In particular, the NiSP/NF electrode required 259 mV to reach a current density of 10 mA cm−2, which is lower than those for NiS (294 mV) and NiP (325 mV) films, as well as pristine NF (441 mV). These values are similar to those reported in previous electrocatalysts prepared via electrodeposition, such as NiCoP–NiCoSe2 (243 mV),34 Ni–S–P (219 mV),33 CoP–MNA (290 mV),46 Ni0.51Co0.49P (239 mV),37 Ni–Fe–Co–S (207 mV),36 Co–S (361 mV),44 and CoS-A (390 mV).43 A more detailed activity comparison of OER electrocatalysts is shown in the ESI (Table S1†). Please note that, in contrast to the studies cited in Table S1,†33–37,43–46,66,67 the overpotentials reported in the current paper were obtained in Fe-free KOH electrolyte, thus neglecting any activity enhancement due to Fe incorporation. This might not be the case for several Ni(OH)2-based OER catalysts in the literature, in which Fe impurities might be responsible for a dramatic increase in activity.42 Finally, the NiSP film requires an overpotential of only 341 mV to reach the 50 mA cm−2 mark, and current densities surpass the 100 mA cm−2 mark for all the deposited films (Fig. S8†), which demonstrates the applicability of large, NF-based electrodes for the OER at high current densities.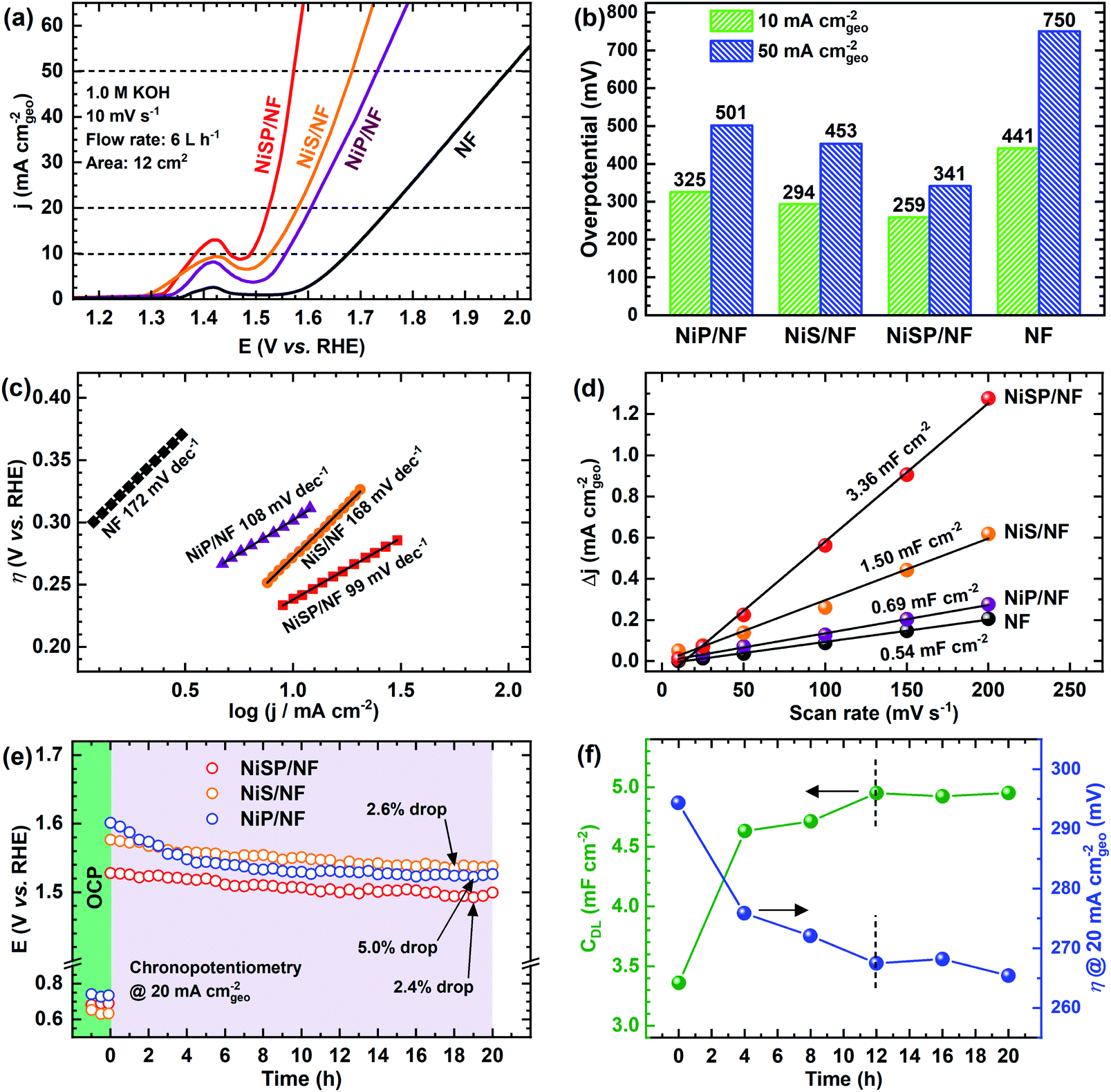 | ||
| Fig. 4 OER electrochemical characterization results: (a) anodic i–V curves, (b) overpotentials at 10 and 50 mA cm−2, (c) Tafel slopes, (d) double-layer capacitances and (e) chronopotentiometric runs at 20 mA cm−2 during a period of 20 h, using the as-prepared electrodes in 1 M KOH electrolyte. (f) Double layer capacitance and overpotential at 20 mA cm−2 of the NiSP/NF electrode with time. | ||
OER kinetics were assessed by means of Tafel plots, which are shown in Fig. 4c. The NiSP film exhibited the smallest Tafel slope (99 mV dec−1), followed closely by NiP (108 mV dec−1). These values are close to the theoretical value of 120 mV dec−1, associated to the first electron transfer reaction in the OER mechanism as the rate-limiting step.68 Furthermore, the ECSAs were estimated from CDL measurements of the as-prepared films using CV scans at different scan rates (Fig. S9†). Results are shown in Fig. 4d. The ECSA follows the trend NiSP (3.36 mF cm−2) > NiS (1.50 mF cm−2) > NiP (0.69 mF cm−2) > pristine NF (0.54 mF cm−2). The superior ECSA for the NiSP film is consistent with the observed morphology from SEM images, where small grains and numerous nanosheets covered the electrode compared to the NiS and NiP films. This provides a large surface area and improves mass transport.40,69
Electrode OER stability was assessed through long-term CP runs as shown in Fig. 4e. After 1 h measuring the OCP during continuous recirculation of the KOH electrolyte, a constant current density of 20 mA cm−2 was applied and the electrode potential was monitored with time. As shown in Fig. 4e, it is evident that overpotential decreases slightly with time, with the NiP film having the most notable potential drop (5.0%). The potential drop for the NiSP (2.4%) and NiS (2.6%) films was comparable. Although there was no drastic change in the overpotential with time, this behavior indicates an improvement in activity. Therefore, LSV curves were recorded after CP runs to evaluate this activity change. Fig. S10† displays the polarization curves for each electrode material after OER stability tests. Notice that intensities for the Ni oxidation peak greatly increased for all the deposited films, surpassing the 20 mA cm−2 mark. This clearly demonstrates that the films underwent a substantial oxidation process during OER stability tests, which might also cause a decrease in overpotentials. This behavior can be clearly seen for the NiSP electrode in Fig. 4f, as overpotentials at 20 mA cm−2 (measured from LSV curves) decrease with time. The overpotential drastically decreases in the first 8 h, and almost stabilizes after 12 h. The ECSA also exhibits a sudden increase in the beginning until a constant value of CDL is achieved in the same timescale (CV scans shown in Fig. S11†). This suggests that a morphology change occurs, which also enhances the electrocatalytic activity of the deposited films during the OER. A similar behavior has been reported previously with Co3C electrocatalysts, where the overpotential decreased due to a gradual formation of porous nanostructures of amorphous CoO, which also caused an increase of the ECSA.9 Therefore, structural and morphological characterizations after OER tests were carried out, and these are detailed later in the manuscript.
Finally, additional CP tests were carried out to evaluate the reproducibility of the measurements in our 3D-printed flow cell. As depicted in Fig. S12,† potential drops were comparable to the ones shown in Fig. 4e, which confirms the reproducibility of the materials and operating conditions. Importantly, although the CP measurements lasted only 20 h, our stability tests were performed under turbulent flow and constant mixing, which also added considerable mechanical stress to the electrocatalysts.40 This means that our testing environment was more realistic compared to most of the typical stability tests in the literature.
Novel systematic optimization of deposition times and recirculation flow rates was employed to evaluate the synthesis of the electrodeposited films. The OER activity at 20 mA cm−2 was used to compare the effect of the evaluated conditions. Significant differences were determined using an ANOVA-Tukey statistical test (95% confidence level). Fig. S13† displays the results when deposition times varied from 15 to 45 min. In general, a significant decrease of the overpotential was observed when the deposition time was increased from 15 to 30 min. A longer deposition time is expected to provide a thicker electrocatalyst film. However, notice that the overpotential increased when a 45 min deposition took place for NiS and NiSP films. Only for the NiP film, the overpotential was slightly improved. This suggests that longer deposition times might excessively cover the active sites, thus decreasing electrocatalytic activity and conductivity. Flow rate optimization is shown in Fig. S14.† Similarly, activity significantly changed when the flow rate was increased from 3 to 6 L h−1, which is attributed to superior mixing that enhances flow distribution and mass transport.40 These properties are critical for electrodeposition, and must be considered seriously in order to obtain uniform and reproducible films.69,70 A further increase of the flow rate up to 9 L h−1 does not produce a significant change in activity, which indicates that the ideal flow rate is 6 L h−1. This is important for electrochemical flow cells, especially when pressure drop, pumping requirements and energy consumption are critical.40 A similar trend can be seen from ECSA measurements varying both variables (Fig. S15†), in which the CDL stabilized above 30 min and a flow rate of 6 L h−1. Notably, the uncertainty bars denote a low variability between replicates (RSD < 5%), which demonstrate the high reproducibility of the electrodeposition approach. Therefore, it can be assumed that an efficient, reproducible and practical deposition can be successfully achieved by applying −50 mA cm−2 for 30 min, at a flow rate of 6 L h−1 using our flow cell geometry and 12 cm2 NF electrodes.
These results demonstrate that the deposition time and flow rate are crucial variables that have a serious impact on the electrocatalytic activity and quality of the synthesized materials. In an unusual strategy in the OER/HER field, we optimized the electrodeposition process by evaluating two variables in an electrochemical flow cell with a controllable and well-defined architecture, which allowed a uniform and reproducible deposition. Compared to traditional lab-scale electrodeposition using conventional cells and magnetic stirring, it is evident that the use of compact flow cells greatly improves the viability of electrodeposition for electrocatalyst synthesis. Electrodeposition of S,P-based films using flow cells also improves the sustainability of electrocatalytic water splitting, because: (i) flow cells provide a higher surface-to-volume ratio, which means that less electrolyte/plating bath volume is necessary, (ii) other synthetic routes, which rely on toxic or dangerous chemicals such as PH3 or H2S, can be avoided, and (iii) the number of synthetic steps can be reduced, since one or two electrodeposition steps can be performed using the same device, as described in this study. 3D-printing also favors this approach, since flow cells with tailored designs can be fabricated depending on specific size requirements, which may vary between laboratories or applications.40 We expect that the use of well-controlled electrodeposition approaches, like the one described in this study, will improve the reproducibility and scale-up of OER/HER electrocatalysts in the future. This is essential to guarantee a fair comparison and replicability of the materials. Thus, we encourage the application of flow electrodeposition due to its simplicity, reproducibility, and sustainability, especially at the laboratory scale.
Hydrogen evolution reaction
To further demonstrate the bifunctionality of the as-prepared films, the HER activity was tested in 1 M KOH electrolyte using the same flow cell configuration. As seen in Fig. 5a, all the electrocatalytic films exhibited improved HER activity compared to pristine NF. Onset potentials for all films is ca. 0.0 V vs. RHE, and current further increases with no evident redox peaks. In addition, Fig. S16† shows the LSV curves at higher current densities, demonstrating the robustness of the large electrodes prepared by electrodeposition. From Fig. 5b, the overpotential at 10 mA cm−2 follows the trend: NiSP (93 mV) < NiP (129 mV) < NiS (143 mV) < pristine NF (194 mV). These values can be contrasted with those available in previous electrodeposition studies: Ni–S (174 mV),36 Ni–Co–S (144 mV),36 NiCoP (218 mV),34 Ni–P@CP (117 mV),66 Ni–S–P (120 mV), Ni–P (134 mV) and Ni–S (315 mV).33 Notice that a poor performance of the Ni–S film for the HER, among the synthesized materials, agrees with a previous trend reported by Xu et al.33 Moreover, the overpotential at 50 mA cm−2 follows the same trend, as at the lower current density, with the NiSP film having the lowest value (174 mV). A detailed activity comparison of HER electrocatalysts is also provided in the ESI (Table S2†).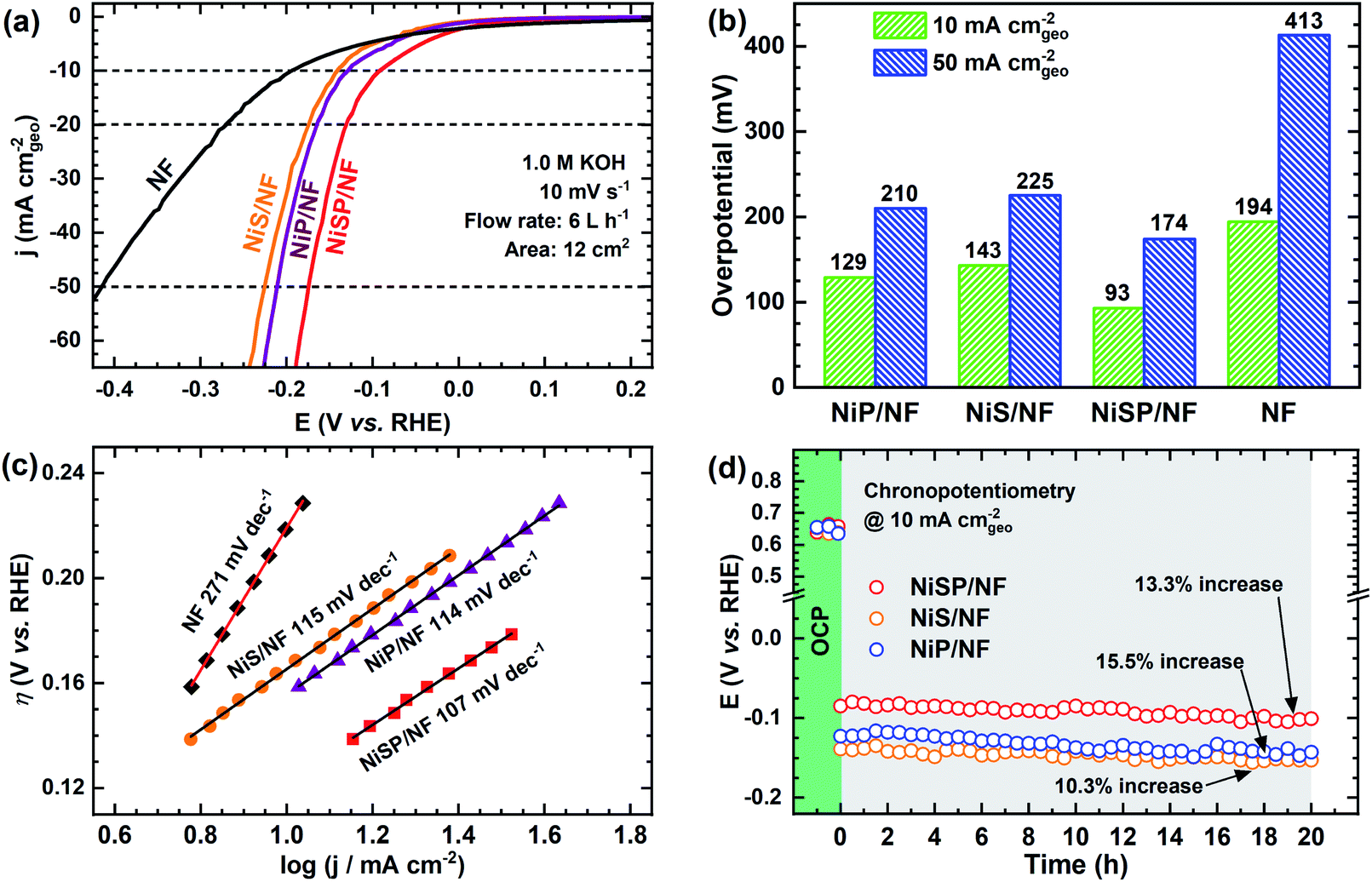 | ||
| Fig. 5 HER electrochemical characterization results: (a) cathodic i–V curves, (b) overpotentials at 10 and 50 mA cm−2, (c) Tafel slopes, and (d) chronopotentiometric runs at 10 mA cm−2 for 20 h using the as-prepared electrodes in 1 M KOH electrolyte. | ||
Tafel plots were estimated to evaluate the HER kinetics and are shown in Fig. 5c. The NiSP film showed a Tafel slope of 107 mV dec−1, which is smaller than those of NiP (114 mV dec−1) and NiS (115 mV dec−1). All Tafel slopes of the deposited films can be associated with the Volmer step of the HER mechanism (i.e., proton adsorption via electron transfer) as the rate-determining step, with a theoretical value of 120 mV dec−1.36 These values are also comparable to those reported by similar studies: Ni–S (109 mV dec−1),33 Ni–P@CP (85.4 mV dec−1),66 Ni–Fe–S (116 mV dec−1) and Ni–S (142 mV dec−1).36 Finally, HER stability results are shown in Fig. 5d. The NiS film exhibited the lowest potential increase (10.3%), followed closely by the NiSP (13.3%) and NiP (15.5%) films. Polarization curves after HER stability tests are shown in Fig. S17.† For the NiSP film, overpotentials were increased only by 7 mV (10 mA cm−2) and 9 mV (50 mA cm−2). The NiP film exhibited the most remarkable increase: 12 mV (10 mA cm−2) and 10 mV (50 mA cm−2). Reproducibility was also verified with additional stability tests (Fig. S12†). These subtle fluctuations demonstrate a reasonable stability for the HER.
Post-characterization of electrodes
Several reports in the literature have suggested that metallic carbides, pnictides and chalcogenides endure an in situ transformation into corresponding metal hydroxides or (oxy)hydroxides during the OER,9,51,71,72 or get reduced into metallic sites during the HER.73 Therefore, we investigated the morphological, structural, and chemical changes of the electrocatalytic films after OER and HER stability tests. Fig. 6 shows overall results after the OER. The morphology was retained in the NiP film according to Fig. 6a. Additional SEM images in Fig. S18a† reveal that grain size and distribution was not modified during OER, and nanosheets still cover the surface. Yet, the thickness of the nanosheets after OER testing seems to be reduced, as shown in Fig. 6a. Elemental mappings depict a uniform distribution of P; however, the EDX spectrum shows a decrease in its content after the OER (Fig. S18†). Also, there is a significant increase in the atomic O content from EDX analysis (Table S3†), which indicates that the surface was partially oxidized. No further peaks arise from XRD measurements, which indicate that low crystallinity prevails (Fig. S18d†). XPS results confirm the formation of nickel oxyhydroxide (NiOOH) and phosphate species (PO43−) after the OER (Fig. S18e†), and the Ni 2p and P 2p components corresponding to nickel phosphide disappeared.58 Interestingly, phosphate species remained at the surface. The generation of hydroxides and soluble anions from metallic phosphides has been reported previously.34,74,75 This suggests that the nickel phosphide acted as a “precatalyst”, term used to describe those compounds that experience partial or full transformation into the electrocatalytically active species.2 For the NiS film (Fig. 6b and S19a†) grains appear to be covered by nanosheet arrays after the OER. EDX elemental mappings show a uniform distribution of S (Fig. S19†), and atomic compositions also show a significant increase in the oxygen content (Table S3†). Similar to the NiP sample, the oxidation of nickel sulfide species was observed in XPS results (Fig. S19e†), as NiOxHy/sulfate (SO42−) species were produced.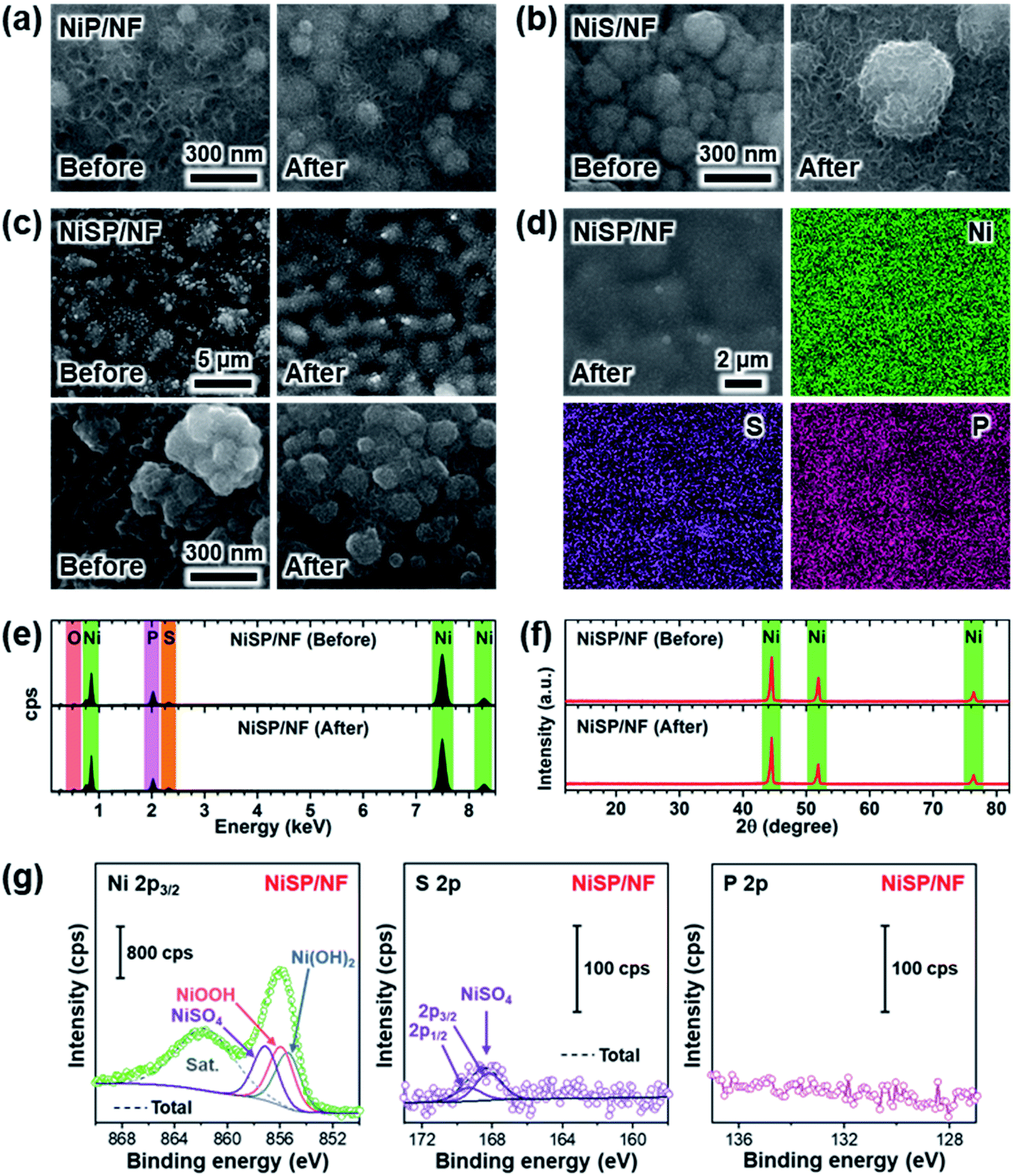 | ||
| Fig. 6 OER electrochemical post analysis results: SEM images before and after the OER for (a) NiP, (b) NiS and (c) NiSP films, (d) elemental mapping after the OER, (e) EDX spectra and (f) XRD comparison for NiSP film, (g) XPS post analysis. | ||
The NiSP film also retained the beehive-like nanosheet arrays after the OER, and the grains appear to be more rounded, as shown in Fig. 6c (additional magnifications in Fig. S20†). Both S and P are uniformly distributed, and EDX spectra and XRD patterns before and after the OER are similar. However, the oxygen content increased (Table S3†). Notably, bright grains in the SEM images coincide with O-rich spots according to EDX elemental mappings, which suggest the presence of oxidized spots on the surface. XPS results of Fig. 6g confirmed the formation of nickel (oxy)hydroxide, which explains the beehive-like patterns. Furthermore, phosphate species disappeared after the OER whereas sulfate species remained. Importantly, the presence of P and S in the electrolyte was confirmed after OER tests using TXRF measurements (Table S4†), which suggests that sulfate/phosphate species were dissolved during the stability tests. Together, similar to the NiS and NiP samples, the in situ generated hydroxides/(oxy)hydroxides can be considered as the true OER active sites, which is consistent with previous reports.6,19,76,77
Morphological and chemical changes of the NiP film after the HER were assessed using the same set of characterization techniques (SEM, EDX, XRD and XPS), as depicted in Fig. S21.† SEM images display an evident loss of roughness and grain morphology, and even the nanosheet arrays are more scattered over the surface. Accordingly, the P content decreased, as suggested by elemental mappings, EDX spectra and atomic compositions (Table S5†). XRD patterns depict a comparable low crystallinity, and XPS analysis show almost no change in the surface chemical composition after the HER (Fig. S21e†), implying that some nickel phosphide remained after the HER. A similar roughness is observed for the NiS film (Fig. S22†). Even though some grains are distributed over the surface, they appear to be smoother, and nanosheets are completely absent on the surface. Elemental mappings suggest that S is uniformly distributed over the surface and the atomic content is similar after the HER (Table S5†). From XPS results, a stronger oxidation of nickel sulfide into nickel hydroxide was observed. This may suggest that Ni–S has a lower oxidation resistance than Ni–P. However, the initial S and P contents (Table S5†) were not equal, which impedes a readily comparison of the oxidation resistance and further studies are necessary in this regard. Finally, the NiSP film retained the nanosheet arrays (Fig. S23†), although the P content reduced as well (Table S5†). Similar to NiS, the NiSP film also experienced the formation of oxidized species after the HER. S and P were also detected in the electrolyte for all the films from TXRF results (Table S4†), which suggests that the reconstruction process also generates soluble ions. Still, XPS results (Fig. S23e†) suggest that a fraction of SO42− and PO43− resided in the NiSP films. Interestingly, oxygen content was significantly reduced for the NiSP film, which can be attributed to the reduction of metallic hydroxides into metals due to the cathodic potentials applied during the HER, as reported for CoSe2 electrocatalysts.73
From these results, the origin behind the superior activity of our electrodeposited films can be debated. Nickel oxyhydroxides have been extensively regarded as the real active OER sites in Ni-based phosphides and sulfides.19,33,34,51,78 Certainly, NiOxHy species were generated during the OER tests, as demonstrated by the increase of the redox peak area after the OER (see Fig. 4a and S10†) and the surface oxidation from EDX and XPS. On the other hand, P and S were transformed into sulfate and phosphate species. Therefore, it is unlikely that the Ni–P and Ni–S species acted as active sites directly. However, even if NiOxHy species are considered as the active sites, it is evident that the generation of these species was not equal for all the synthesized films. Just by looking at the redox peaks in Fig. 4a and S10,† one can readily see that the NiSP film exhibited the largest peak area, followed closely by the NiS film. This suggests that the NiSP film was easily oxidized and more active sites were generated as compared with the NiS and NiP films. Thus, instead of the direct participation of S or P in the OER mechanism, we attribute the superior performance to the structural and electronic effects caused by the incorporation of S and P into the catalytic films, enhancing the subsequent reconstruction of the surface during the OER.
We support this idea using three important observations from previous studies. First, the surface of the catalyst should be considered as a “dynamic” layer. Deng et al. demonstrated that redox activation processes induce changes on the morphology of Ni(OH)2 nanosheets, increasing the porosity and permeability to ions.79 Further work by Dette et al. proved that Ni(OH)2 to NiOOH transitions induce mechanical stress due to the change in the Ni–Ni distance, leading to the cracking of NiOxHy nanosheets into nanoparticle assembles.80 Importantly, the addition of metals such as Co or Fe leads to different structural behaviors due to changes in the bond strength.79–81 Second, the inclusion of non-metals induces important structural effects. Zhu et al. showed that the lattice distortion caused by P substitution boosted the oxidation of Co into CoOOH in CoSe1.26P1.42 electrodes compared to unsubstituted CoSe2. Experimentally, in situ X-ray absorption (XAS) measurements confirmed that the presence of P induced more vacancies, which facilitated the oxidation of the metal cation.73 This has crucial implications for Ni active sites, as theoretical studies suggest that OH* species are preferentially adsorbed on Ni atoms with low coordination numbers at edge sites, which results in superior OER activities.82,83 Third, the inclusion of non-metals also induce important electronic effects at the heterointerfaces. Previous density functional theory (DFT) studies reported that oxygen induces charge transfer at the interface with adjacent metal and sulfur sites, decreasing the activation energy for oxygen-containing intermediates.65 Moreover, XPS and XAS studies suggest that S and P promote the generation of high-valent Ni sites due to their vacant 3d orbitals.19,51 Notably, these observations can be also applied to the HER. The vacancies caused by P substitution have been also linked to the enhanced reduction of Co into metallic Co, which was suggested to be the active site for the HER in Co–Se precatalysts.73 Furthermore, electronegative species such as P also stabilize adjacent metal sites and increase the conductivity.19,72
Based on these findings, we hypothesize that our catalytic films behaved like a dynamic reconstruction layer as depicted in Fig. 7. During the OER redox activation, the surface layer in contact with the electrolyte was oxidized and fragmented into an amorphous, nano-porous layer with abundant NiOxHy “flakes”. Notably, the incorporation of S and P atoms into the Ni lattice during the electrodeposition process favored the fragmentation and reconstruction of this layer due to the abundance of vacancies and defects. The enhanced OER activity could be explained by the increase of Ni active sites with low coordination numbers, edge sites and the formation of high-valent Ni at the heterogeneous interfaces. Furthermore, the presence of S and P atoms provides sites of higher electronegativity, thereby affecting the electronic nature of the films. Most of the S and P atoms were oxidized by the penetrating electrolyte and diffusing oxygen, which produced a multilayer structure with intercalated K+, HO−, SO42−, and PO43− ions.19,80 Notice that a fraction of SO42− and PO43− was initially intercalated in the as-prepared films (Fig. 3), which suggests that these ions might be involved in the activation process as well. For instance, these ions can improve the ionic conductivity inside the porous layer and stabilize the high-valent Ni sites, as these anions cannot be further oxidized.19 Evidently, the NiSP film exhibited the best performance due to the increased number of vacancies, defects, and species in the nano-polycrystalline domains (S + P + SO42− + PO43−). This is supported by TEM/SAED (Fig. 2i) and XPS (Fig. 3) results, and electrochemically by the redox peak area (Fig. S10†) and ECSA (Fig. 4d) changes.
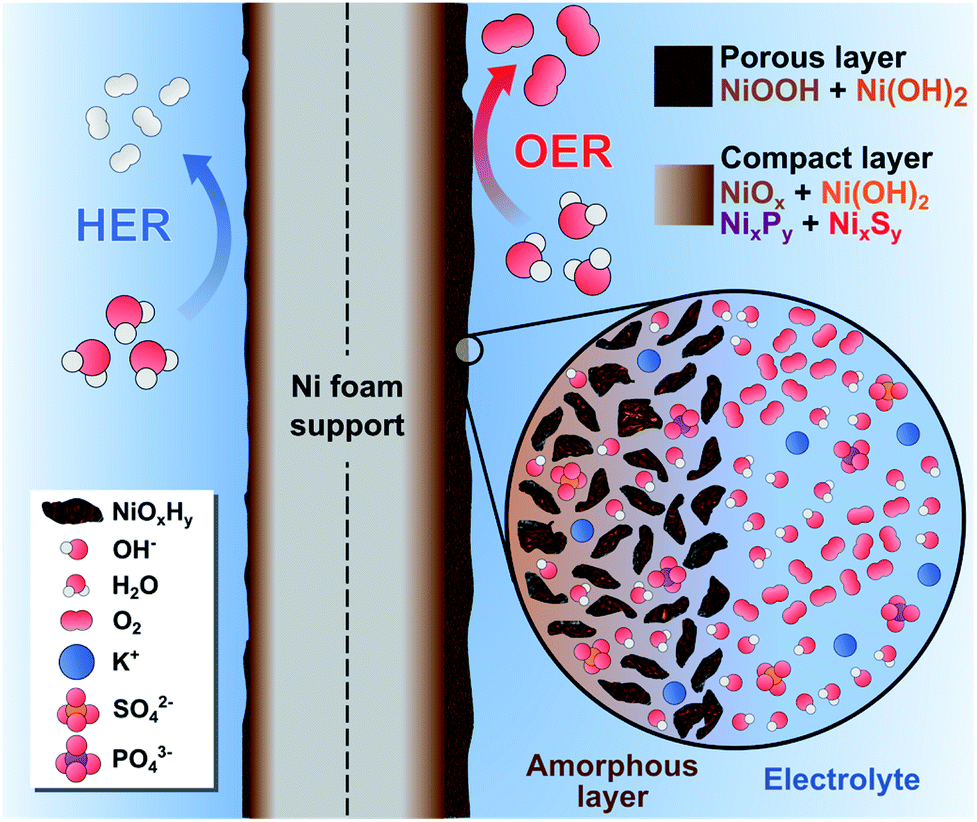 | ||
| Fig. 7 Schematic illustration of the dynamic reconstruction process occurred during the HER and the OER on the NiSP electrocatalytic film. | ||
We hypothesize that the thickness of the nanoporous layer constantly changed until it was stabilized, according to the activity and ECSA trends in Fig. 4f. As suggested by previous studies, this layer might be around 200 nm thick, as severe morphological changes were not observed in the OER post-analysis.19 A more compact internal layer could also contain disperse NixPy and NixSy species with more metallic character, which might increase the conductivity of the electrode.72 The presence of a dual layer could also explain why SO42− ions were retained after the OER, whereas PO43− ions disappeared from the NiSP film (Fig. 6g). As depicted in Fig. 1, phosphorous was deposited in the second step, which means that more S atoms occupied the inner, compact layer while P atoms occupied the outermost, nanoporous layer. As PO43− ions were rapidly removed from the outermost layer, SO42− ions migrated more slowly from the compact layer to the nanoporous layer. This migration and removal of anions suggests that the reconstruction is highly dynamic, and ions are not fixed in the multilayer structure, especially in the nanoporous layer. For the HER, it is possible that the thickness of these layers was reduced, as XPS and EDX results suggest a decrease of the oxygen content, while SEM/EDX revealed a loss of roughness, grains and the disappearance of nanosheets. If metallic Ni is considered as the HER active site, it could explain the lower activity of the highly-oxidized NiS film. This behavior is supported by previous studies, and it is likely to occur due to the reductive potentials.73,84
Although this model explains the observed trends of the OER, describing the true role of P and S species for the HER is difficult. Although XPS results suggest that the oxidized layer was reduced, some hydroxides remained. Presence of hydroxides could be explained by the exposure to the alkaline environment.78 However, according to a recent study about nickel sulfide in alkaline media, a similar oxidation process induced the amorphization and enhanced the HER activity.16 Still, this idea does not explain why the NiS film exhibited a lower HER activity in our study. Furthermore, since some Ni–P remained at the surface (Fig. S21†), P might also increase the HER activity as proposed previously.53,78 This could explain why the Ni–P film exhibited a better performance only for the HER compared to the Ni–S film. Yet, it is difficult to test this hypothesis in our approach, as a low abundance of NixPy species was observed. Similarly, the influence of sulfate and phosphate ions in the resultant OER activity is not completely understood. Evidently, S and P atoms were oxidized and dissolved into the electrolyte, which is consistent with recent works.31,34,66,73,85 Future studies should carefully address this aspect, as the number of publications concerning metal sulfides and phosphides has considerably increased. More fundamental studies are necessary to unravel the role of S and P species, particularly using catalytic materials with well-defined crystal structures and in situ/operando characterization techniques such as XAS or Raman spectroscopy.
Overall water splitting
Due to the satisfactory performance for both the OER and HER using the NiSP/NF electrode, a two-electrode flow cell was assembled in which the NiSP films were used as cathode and anode for overall water splitting as shown in Fig. 8. The polarization curves (Fig. 8a) show that the NiSP/NF couple requires a low cell voltage of only 1.70 V to reach a current density of 10 mA cm−2. This value is close to other cell voltages reported in the literature: Ni–S–P‖Ni–S–P (1.58 V),33 NiCoP–NiCoSe2 (1.70 V),34 CoNiP‖CoNiP (1.61 V),35 CoP–MNA‖CoP–MNA (1.62 V),46 Ni–P‖@Ni–P (1.63 V),66 NiPx‖NiPx (1.61 V)67 and Ni–Fe–Co–S‖Ni–Fe–Co–S (1.54 V).36 A broader comparison of different overall water splitting systems is shown in the ESI (Table S6†). Moreover, cell potentials of 1.73, 1.78, and 1.85 V were required to afford current densities of 20, 50 and 100 mA cm−2, respectively.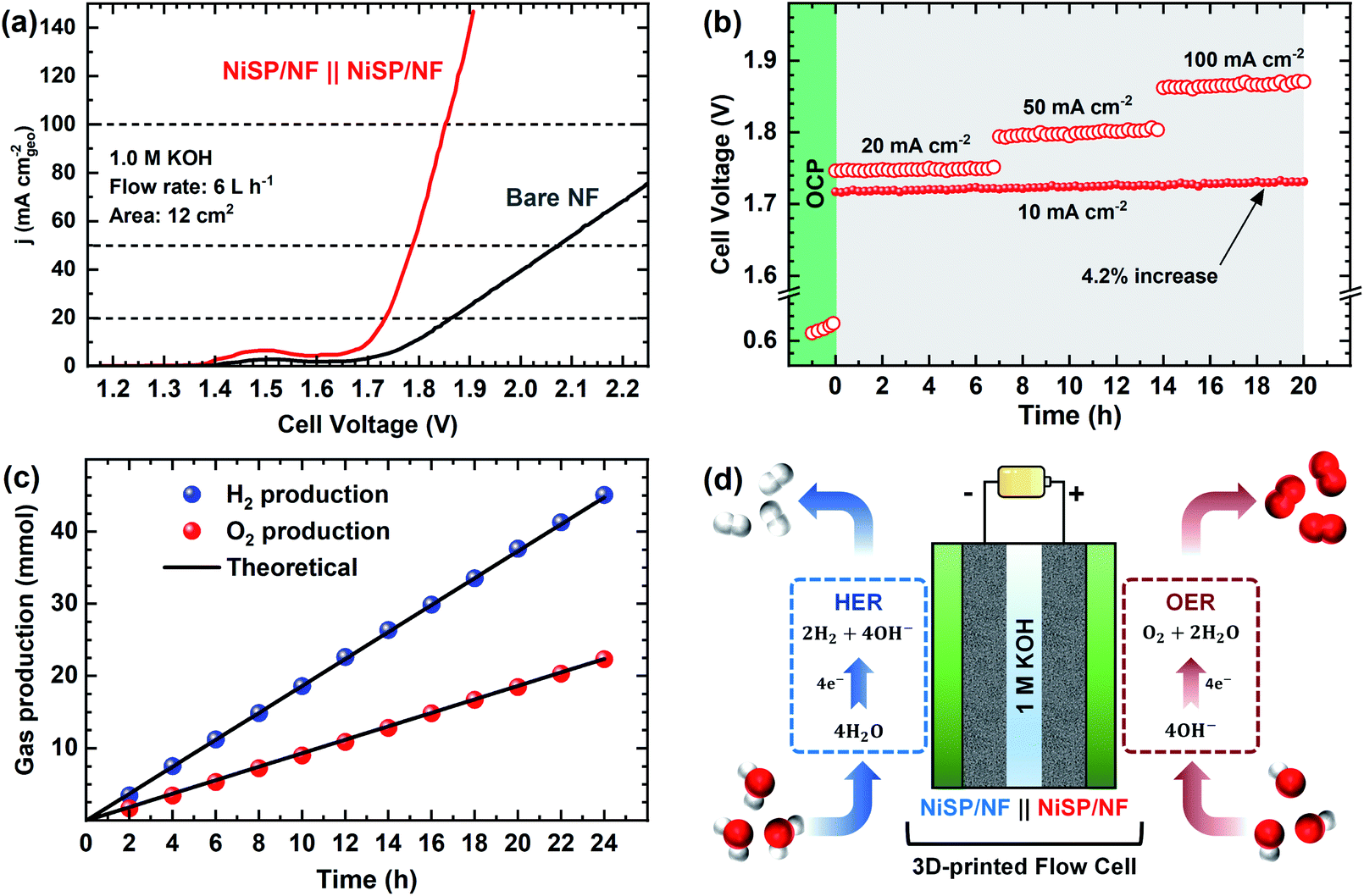 | ||
| Fig. 8 Overall water splitting results: (a) i–V curves, (b) chronopotentiometric run at different current densities for 20 h, (c) gas evolution measurements at 10 mA cm−2, and (d) schematic illustration of the assembled flow cell with NiSP/NF electrodes using 1 M KOH electrolyte. | ||
Stability during galvanostatic tests was also evaluated, as shown in Fig. 8b. After applying a constant current density of 10 mA cm−2, only a subtle increase of the voltage (4.2%) was seen and was further verified by comparing polarization curves before and after electrolysis (Fig. S24†). These results demonstrate a solid performance for the two-electrode flow cell. Durability was also assessed by multi-step galvanostatic runs, where current density was increased every 7 h from 20 to 100 mA cm−2. Cell voltage was constant, and only slight fluctuations were observed during the final stage, which is attributed to the pronounced evolution of H2 and O2 bubbles. To validate the applicability at high current densities, long-term electrolysis at 100 mA cm−2 was sustained for 48 h with a power supply (Fig. S25†). A cell voltage increase of only 3.6% was observed. This demonstrates that the NiSP/NF easily achieves high current densities, especially when compared with the behavior of pristine NF. Finally, gas production at each electrode was measured using a simple water displacement method, which allowed the estimation of H2/O2 production rates and faradaic efficiencies. A molar ratio of 2.01:1 was calculated from the slopes of H2 and O2 production trends in Fig. 8c, which is close to the theoretical value of 2:1.34 Faradaic efficiencies were 99.5 ± 3.5 and 99.8 ± 5.6% for H2 and O2 production, respectively. Furthermore, even though cell voltage was close to 2.3 V when an AEM was placed between the electrodes (Fig. S26†), gas evolution was efficiently measured in a more realistic approach. This further demonstrates the applicability of the electrochemical flow cell to test electrocatalytic materials in a more reproducible and practical way.40
Conclusions
In summary, a novel mass transport-enhanced electrodeposition approach was utilized to optimize the synthesis of high-quality electrocatalytic films. Remarkably, the NiP, NiS, and NiSP films prepared in this work exhibited noteworthy activity and durability in alkaline electrolyte, which is attributed to: (i) improved morphology with high surface area, (ii) polycrystalline nature with numerous defect sites and variable electronic densities on the surface, (iii) fast charge transfer according to their Tafel slopes, and (iv) a dynamic reconstruction process enhanced by the inclusion of S/P species which provided numerous active sites for the OER and HER. Post-analysis of the electrodes using complementary techniques suggest that the as-prepared NiP, NiS and NiSP films acted as precatalysts, and the OER and HER active sites were generated in situ. Particularly, we encourage future strategies to unravel the role of sulfate and phosphate species in the HER/OER, as our results suggest that these species intercalate the nanoporous reconstruction layer, but they are also dissolved from the catalytic films into the electrolyte.Notably, the use of a novel 3D-printed electrochemical flow cell played an important role by effectively optimizing the electrodeposition process on large electrodes (12 cm2) and providing a realistic testing environment. These features should be considered by future studies as an effective way to validate the reproducibility of the materials. Superior features of the approach include: (i) improved architecture, specifically designed for the application, (ii) compact design which provides a small and constant distance between electrodes, (iii) forced flow which improves mixing of the electrolytes and bubble removal during electrolysis, and (iv) the use of an AEM to allow separation of electrode compartments during electrodeposition and collection of gases during electrolysis tests. In addition, the approach proposed herein also involves important sustainability and practical improvements, such as: (i) a higher surface-to-volume ratio which reduces the consumption of the plating bath/electrolyte and allows the use of larger substrates, (ii) the use of simple electrodeposition steps without employing toxic solvents, dangerous gases or complex thermal steps during synthesis, and (iii) the use of a robust 3D-printed device made from low-cost materials, which demonstrates the applicability of 3D-printing technologies in the laboratory and allows research groups fast-prototyping capabilities as appropriate for their specific requirements.
Author contributions
The manuscript was written through contributions by all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
R. A. A. M., K. K., Y. J. S., J. A. W., H. H. S., and C. B. M. gratefully acknowledge funding from the National Science Foundation (NSF) via Grant CHE-1664941 and the Welch Foundation through Grant F-1436 for their generous support. V. H. R. S. and R. A. A. M. acknowledge the 3D printing services of the UACH's Innovation and Technological Entrepreneurship Center, particularly the help of Fernando Ledezma Millán.References
- A. Li, Y. Sun, T. Yao and H. Han, Chem.–Eur. J., 2018, 24, 18334–18355 CrossRef CAS PubMed.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef CAS.
- N. K. Chaudhari, H. Jin, B. Kim and K. Lee, Nanoscale, 2017, 9, 12231–12247 RSC.
- Z. Chen, X. Duan, W. Wei, S. Wang and B.-J. Ni, J. Mater. Chem. A, 2019, 7, 14971–15005 RSC.
- A. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2017, 8, 144–152 CrossRef CAS PubMed.
- Y. Pei, Y. Cheng, J. Chen, W. Smith, P. Dong, P. M. Ajayan, M. Ye and J. Shen, J. Mater. Chem. A, 2018, 6, 23220–23243 RSC.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J.-H. Kim, K. Kawashima, B. R. Wygant, O. Mabayoje, Y. Liu, J. H. Wang and C. B. Mullins, ACS Appl. Energy Mater., 2018, 1, 5145–5150 CAS.
- K. Kawashima, C. L. Cao, H. Li, R. A. Márquez-Montes, B. R. Wygant, Y. J. Son, J. V. Guerrera, G. Henkelman and C. B. Mullins, ACS Sustainable Chem. Eng., 2020, 8, 14101–14108 CrossRef CAS.
- K. Kawashima, K. Shin, B. R. Wygant, J.-H. Kim, C. L. Cao, J. Lin, Y. J. Son, Y. Liu, G. Henkelman and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 3909–3918 CrossRef CAS.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- M. Shalom, D. Ressnig, X. Yang, G. Clavel, T. P. Fellinger and M. Antonietti, J. Mater. Chem. A, 2015, 3, 8171–8177 RSC.
- O. Mabayoje, S. G. Dunning, K. Kawashima, B. R. Wygant, R. A. Ciufo, S. M. Humphrey and C. B. Mullins, ACS Appl. Energy Mater., 2020, 3, 176–183 CrossRef CAS.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- S. Anantharaj, H. Sugime and S. Noda, Chem. Eng. J., 2020, 127275 Search PubMed.
- K. S. Bhat, H. C. Barshilia and H. S. Nagaraja, Int. J. Hydrogen Energy, 2017, 42, 24645–24655 CrossRef CAS.
- D. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. Li, H. Wang, Q. Zhang and S. W. Boettcher, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- X. Xu, C. Li, J. G. Lim, Y. Wang, A. Ong, X. Li, E. Peng and J. Ding, ACS Appl. Mater. Interfaces, 2018, 10, 30273–30282 CrossRef CAS PubMed.
- J.-H. Kim, D. H. Youn, K. Kawashima, J. Lin, H. Lim and C. B. Mullins, Appl. Catal., B, 2018, 225, 1–7 CrossRef CAS.
- B. R. Wygant, K. A. Jarvis, W. D. Chemelewski, O. Mabayoje, H. Celio and C. B. Mullins, ACS Catal., 2016, 6, 1122–1133 CrossRef CAS.
- J.-H. Kim, K. Shin, K. Kawashima, D. H. Youn, J. Lin, T. E. Hong, Y. Liu, B. R. Wygant, J. Wang, G. Henkelman and C. B. Mullins, ACS Catal., 2018, 8, 4257–4265 CrossRef CAS.
- U. P. Suryawanshi, M. P. Suryawanshi, U. V. Ghorpade, M. He, D. Lee, S. W. Shin and J. H. Kim, ACS Appl. Energy Mater., 2020, 3, 4338–4347 CrossRef CAS.
- P. Cai, J. Huang, J. Chen and Z. Wen, Angew. Chem., Int. Ed., 2017, 56, 4858–4861 CrossRef CAS PubMed.
- H. Ren, X. Sun, C. Du, J. Zhao, D. Liu, W. Fang, S. Kumar, R. Chua, S. Meng, P. Kidkhunthod, L. Song, S. Li, S. Madhavi and Q. Yan, ACS Nano, 2019, 13, 12969–12979 CrossRef CAS PubMed.
- T. Wang, C. Wang, Y. Jin, A. Sviripa, J. Liang, J. Han, Y. Huang, Q. Li and G. Wu, J. Mater. Chem. A, 2017, 5, 25378–25384 RSC.
- L. Yu, H. Zhou, J. Sun, I. K. Mishra, D. Luo, F. Yu, Y. Yu, S. Chen and Z. Ren, J. Mater. Chem. A, 2018, 6, 13619–13623 RSC.
- F. Hu, Y. Zhang, X. Shen, J. Tao, X. Yang, Y. Xiong and Z. Peng, J. Power Sources, 2019, 428, 76–81 CrossRef CAS.
- L. Yang, Z. Guo, J. Huang, Y. Xi, R. Gao, G. Su, W. Wang, L. Cao and B. Dong, Adv. Mater., 2017, 29, 1704574 CrossRef PubMed.
- Z. Wu, D. Nie, M. Song, T. Jiao, G. Fu and X. Liu, Nanoscale, 2019, 11, 7506–7512 RSC.
- U. K. Sultana and A. P. O'Mullane, ChemElectroChem, 2019, 6, 2630–2637 CrossRef CAS.
- U. K. Sultana, T. He, A. Du and A. P. O'Mullane, RSC Adv., 2017, 7, 54995–55004 RSC.
- Q. Xu, W. Gao, M. Wang, G. Yuan, X. Ren, R. Zhao, S. Zhao and Q. Wang, Int. J. Hydrogen Energy, 2020, 45, 2546–2556 CrossRef CAS.
- T. Wang, X. Liu, Z. Yan, Y. Teng, R. Li, J. Zhang and T. Peng, ACS Sustainable Chem. Eng., 2020, 8, 1240–1251 CrossRef CAS.
- L. Zhou, S. Jiang, Y. Liu, M. Shao, M. Wei and X. Duan, ACS Appl. Energy Mater., 2018, 1, 623–631 CrossRef CAS.
- G. B. Darband, M. Aliofkhazraei, S. Hyun, A. S. Rouhaghdam and S. Shanmugam, Nanoscale, 2019, 11, 16621–16634 RSC.
- J. Yu, Q. Li, Y. Li, C.-Y. Xu, L. Zhen, V. P. Dravid and J. Wu, Adv. Funct. Mater., 2016, 26, 7644–7651 CrossRef CAS.
- L. Wei, K. Goh, Ö. Birer, H. E. Karahan, J. Chang, S. Zhai, X. Chen and Y. Chen, Nanoscale, 2017, 9, 4401–4408 RSC.
- L. F. Arenas, C. P. de León, R. P. Boardman and F. C. Walsh, J. Electrochem. Soc., 2017, 164, D57–D66 CrossRef CAS.
- R. A. Márquez-Montes, V. H. Collins-Martínez, I. Pérez-Reyes, D. Chávez-Flores, O. A. Graeve and V. H. Ramos-Sánchez, ACS Sustainable Chem. Eng., 2020, 8, 3896–3905 CrossRef.
- R. A. Márquez-Montes, K. Kawashima, K. M. Vo, D. Chávez-Flores, V. H. Collins-Martínez, C. B. Mullins and V. H. Ramos-Sánchez, Environ. Sci. Technol., 2020, 54, 12511–12520 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- B. Liu, S. Qu, Y. Kou, Z. Liu, X. Chen, Y. Wu, X. Han, Y. Deng, W. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 30433–30440 CrossRef CAS PubMed.
- T. Liu, Y. Liang, Q. Liu, X. Sun, Y. He and A. M. Asiri, Electrochem. Commun., 2015, 60, 92–96 CrossRef CAS.
- C.-W. Chiu, I.-W. Sun and P.-Y. Chen, J. Electrochem. Soc., 2017, 164, H5018 CrossRef CAS.
- Y.-P. Zhu, Y.-P. Liu, T.-Z. Ren and Z.-Y. Yuan, Adv. Funct. Mater., 2015, 25, 7337–7347 CrossRef CAS.
- N. Kovalska, N. Tsyntsaru, H. Cesiulis, A. Gebert, J. Fornell, E. Pellicer, J. Sort, W. Hansal and W. Kautek, Coatings, 2019, 9, 189 CrossRef.
- J. Shi, X. Li, G. He, L. Zhang and M. Li, J. Mater. Chem. A, 2015, 3, 20619–20626 RSC.
- P. Ganesan, A. Sivanantham and S. Shanmugam, J. Mater. Chem. A, 2016, 4, 16394–16402 RSC.
- N. Wang, B. Tao, F. Miao and Y. Zang, RSC Adv., 2019, 9, 33814–33822 RSC.
- H. Q. Fu, L. Zhang, C. W. Wang, L. R. Zheng, P. F. Liu and H. G. Yang, ACS Energy Lett., 2018, 3, 2021–2029 CrossRef CAS.
- H. Li, Y. Du, Y. Fu, C. Wu, Z. Xiao, Y. Liu, X. Sun and L. Wang, Int. J. Hydrogen Energy, 2020, 45, 565–573 CrossRef CAS.
- H. Zhou, F. Yu, J. Sun, R. He, S. Chen, C.-W. Chu and Z. Ren, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5607 CrossRef CAS PubMed.
- G. Wang, C. Li and H. Shan, ACS Catal., 2014, 4, 1139–1143 CrossRef CAS.
- R. Karthikeyan, D. Thangaraju, N. Prakash and Y. Hayakawa, CrystEngComm, 2015, 17, 5431–5439 RSC.
- J. Li, J. Li, X. Zhou, Z. Xia, W. Gao, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2016, 8, 10826–10834 CrossRef CAS PubMed.
- W. Zhou, H. Xin, H. Yang, X. Du, R. Yang, D. Li and C. Hu, Catalysts, 2018, 8, 153 CrossRef.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- D. Fa, B. Yu and Y. Miao, Proc. Natl. Acad. Sci. U. S. A., 2019, 564, 31–38 CAS.
- R. B. Shalvoy and P. J. Reucroft, J. Vac. Sci. Technol., 1979, 16, 567–569 CrossRef CAS.
- N. Jiang, B. Jiang, J. Wang and H. Song, New J. Chem., 2020, 44, 8379–8385 RSC.
- X. Ou and Z. Luo, RSC Adv., 2016, 6, 10280–10284 RSC.
- G. Zhou, L.-C. Yin, D.-W. Wang, L. Li, S. Pei, I. R. Gentle, F. Li and H.-M. Cheng, ACS Nano, 2013, 7, 5367–5375 CrossRef CAS PubMed.
- L. Zhang, L. Ji, P.-A. Glans, Y. Zhang, J. Zhu and J. Guo, Phys. Chem. Chem. Phys., 2012, 14, 13670–13675 RSC.
- M. J. Wang, X. Zheng, L. Song, X. Feng, Q. Liao, J. Li, L. Li and Z. Wei, J. Mater. Chem. A, 2020, 8, 14145–14151 RSC.
- X. Wang, W. Li, D. Xiong, D. Y. Petrovykh and L. Liu, Adv. Funct. Mater., 2016, 26, 4067–4077 CrossRef CAS.
- Z. Zhang, S. Liu, J. Xiao and S. Wang, J. Mater. Chem. A, 2016, 4, 9691–9699 RSC.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed.
- L. F. Arenas, C. Ponce de León and F. C. Walsh, Electrochem. Commun., 2017, 77, 133–137 CrossRef CAS.
- L. F. Arenas, N. Kaishubayeva, C. P. de León and F. C. Walsh, Trans. IMF, 2020, 98, 48–52 CrossRef CAS.
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- Y. Zhu, H.-C. Chen, C.-S. Hsu, T.-S. Lin, C.-J. Chang, S.-C. Chang, L.-D. Tsai and H. M. Chen, ACS Energy Lett., 2019, 4, 987–994 CrossRef CAS.
- K. N. Dinh, X. Sun, Z. Dai, Y. Zheng, P. Zheng, J. Yang, J. Xu, Z. Wang and Q. Yan, Nano Energy, 2018, 54, 82–90 CrossRef CAS.
- Y. Lian, H. Sun, X. Wang, P. Qi, Q. Mu, Y. Chen, J. Ye, X. Zhao, Z. Deng and Y. Peng, Chem. Sci., 2019, 10, 464–474 RSC.
- W. Li, X. Gao, X. Wang, D. Xiong, P.-P. Huang, W.-G. Song, X. Bao and L. Liu, J. Power Sources, 2016, 330, 156–166 CrossRef CAS.
- K. Liu, F. Wang, P. He, T. A. Shifa, Z. Wang, Z. Cheng, X. Zhan and J. He, Adv. Energy Mater., 2018, 8, 1703290 CrossRef.
- P. W. Menezes, A. Indra, C. Das, C. Walter, C. Göbel, V. Gutkin, D. Schmeiβer and M. Driess, ACS Catal., 2017, 7, 103–109 CrossRef CAS.
- J. Deng, M. R. Nellist, M. B. Stevens, C. Dette, Y. Wang and S. W. Boettcher, Nano Lett., 2017, 17, 6922–6926 CrossRef CAS PubMed.
- C. Dette, M. R. Hurst, J. Deng, M. R. Nellist and S. W. Boettcher, ACS Appl. Mater. Interfaces, 2019, 11, 5590–5594 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- X. P. Wang, H. J. Wu, S. B. Xi, W. S. V. Lee, J. Zhang, Z. H. Wu, J. O. Wang, T. D. Hu, L. M. Liu, Y. Han, S. W. Chee, S. C. Ning, U. Mirsaidov, Z. B. Wang, Y. W. Zhang, A. Borgna, J. Wang, Y. H. Du, Z. G. Yu, S. J. Pennycook and J. M. Xue, Energy Environ. Sci., 2020, 13, 229–237 RSC.
- J. Huang, J. Chen, T. Yao, J. He, S. Jiang, Z. Sun, Q. Liu, W. Cheng, F. Hu, Y. Jiang, Z. Pan and S. Wei, Angew. Chem., Int. Ed., 2015, 54, 8722–8727 CrossRef CAS PubMed.
- C. Hu, Q. Ma, S.-F. Hung, Z.-N. Chen, D. Ou, B. Ren, H. M. Chen, G. Fu and N. Zheng, Chem, 2017, 3, 122–133 CrossRef CAS.
- D. Li, H. Baydoun, B. Kulikowski and S. L. Brock, Chem. Mater., 2017, 29, 3048–3054 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental section, electrochemical testing set-up, additional electrochemical and physicochemical characterization before and after OER and HER, additional post-analysis results. See DOI: 10.1039/d0ta12097a |
| This journal is © The Royal Society of Chemistry 2021 |