
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
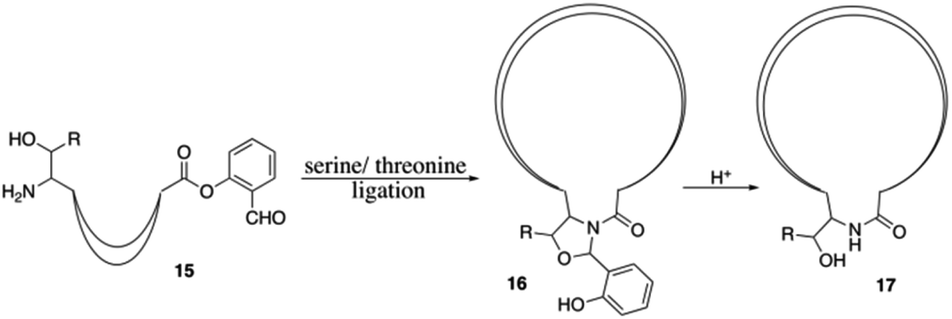
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent applications of solid-phase strategy in total synthesis of antibiotics
Yuxin Zhouc and
Xiao-Wei Liang *ab
*ab
aHunan Key Laboratory of Skin Cancer and Psoriasis, Xiangya Hospital, Central South University, Changsha, 410008, China. E-mail: xiaowei.liang@csu.edu.cn
bNational Clinical Research Center for Geriatric Disorders, Xiangya Hospital, Central South University, Changsha, 410013, China
cJinling High School, 169 Zhongshan Road, Nanjing, Jiangsu 210005, China
First published on 24th November 2021
Abstract
Antibiotics produced by soil microorganisms have been widespread and have cured the most prevalent diseases since 1940s. However, recent bacterial resistance to existing antibacterial drugs is causing a public health crisis. The structure–activity relationship of antibiotics needs to be established to search for existing antibiotics-based next-generation drug candidates that can conquer the challenge of bacterial resistance preparedness, which relies on the development of highly efficient total synthesis strategies. The solid-phase strategy has become important to circumvent tedious intermediate isolation and purification procedures with simple filtrations. This review will give a brief overview of recent applications of solid-phase strategy in the total synthesis of antibiotics.
1. Introduction
The intrinsic complexity of natural products is of great value with regards to physicochemical favorability and have been extensively important to establish their structure–activity relationship for natural products-based clinical drug candidates.1 Natural products have been pre-optimized through natural evolution to bind biomolecules and penetrate cells, with functional romps, stereo-genic centers, and ring motifs with high density.2 Throughout the history of new clinical drug development, natural products have served as leads for drug development, through anecdotal reports of active extracts, traditional medicine, or high-throughput screen. Natural products like Taxol, Artemisinin, Penicillin, Quinine, Morphine, Vancomycin (Fig. 1) have changed the world of medicine and have been a huge benefit to science and society with remarkable achievements for over a century.3 In 2020, Newman and Cragg updated a review and suggested that 32% of approved small molecules for the last 39 years and 38% of approved antitumor drugs for the last 73 years belong to natural products and their derivatives, continuing to emphasize the significance of natural products as sources for new drug development.4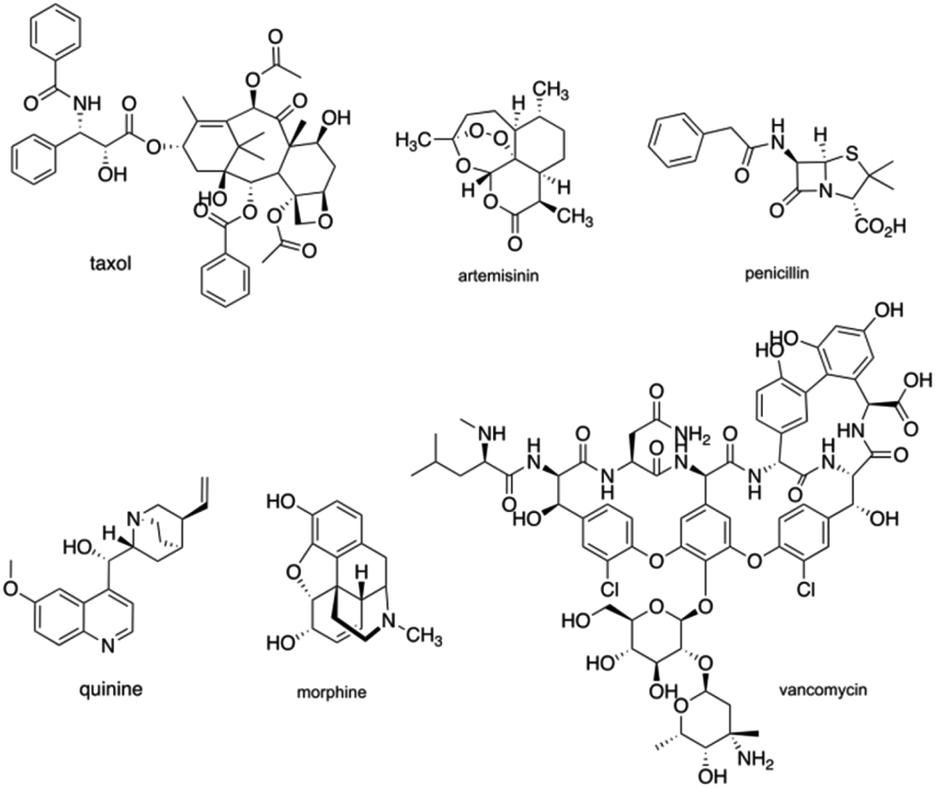 | ||
| Fig. 1 Famous natural products representatives. | ||
However, despite these advantages, natural products suffer from synthetic intractability, which turns out to be a challenge for further research on their structure–activity relationships. One solution is to target a structurally simplified version of a natural product that retains its activity. In this way, the intrinsic complexity of the original natural product could be reduced, which could enable discoveries for function-oriented synthesis. Another solution is to make the best use of the diversity of the structure of natural products in new drug discovery and pursue the essence of nature and natural products in total synthesis.5
Among all kinds of natural products, antibiotics produced by soil microorganisms have been a widespread cure for the most prevalent diseases since 1940s. However, bacterial resistance to existing antibacterial drugs is causing a public health crisis.6 One strategy to address this problem is to study the structure–activity relationship (SAR) of antibiotics to develop new drugs based on existing antibiotics, and efficient synthesis strategies would make it possible to prepare a series of analogues, hence enabling a comprehensive SAR study to search more potent drug candidates.
The solid phase strategy was first proposed by Robert Bruce Merrifield in the 1960s7 and has become an important strategy to circumvent tedious intermediate isolation and purification procedures with simple filtrations.8 Although solid phase synthesis based on total synthesis of natural products has been reported for several years,9 challenges and problems remained particularly in the total synthesis of non-natural amino acids containing cyclic-peptides. In this review, we will take a closer look at the recent applications of solid-phase approaches in synthesizing antibiotics and discuss the studies of their SAR (since 2016).
2. Total synthesis of daptomycin
2.1 Total synthesis of daptomycin by Li group
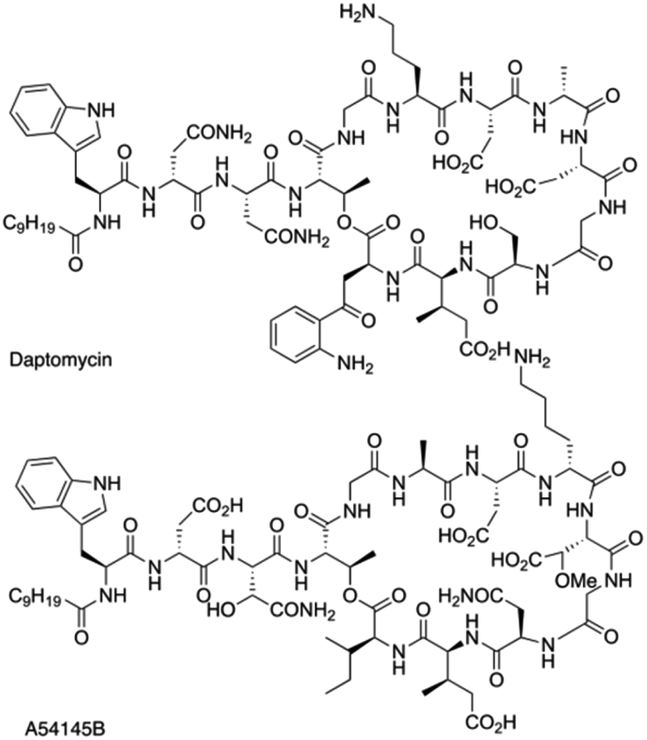
Daptomycin (1) is a Ca2+-dependented lipodepsipeptide isolated from Streptomyces roseoporus by researchers at Eli Lily in the 1980s (Fig. 2) and approved by the FDA in 2003 to treat skin infections caused by antibiotic-resistant Gram-positive pathogens, including Staphylococcus aureus (MRSA), vancomycin-resistant Enterococci (VRE), and vancomycin-resistant S. aureus (VRSA).10 Although the resistance of Gram-positive bacteria is uncommon to daptomycin, increasing reports were found through intrinsic resistance and acquired resistance, such as Staphylococcus, Micrococcus, S. aureus.11 The structure–activity relationship of daptomycin needs to be established to search for daptomycin-based next-generation antibiotics to conquer the challenge of bacterial resistance preparedness, which relies on the development of highly efficient total synthesis strategies, but only a few daptomycin analogues have been produced by semisynthesis, genetic engineering and chemoenzymatic synthesis.12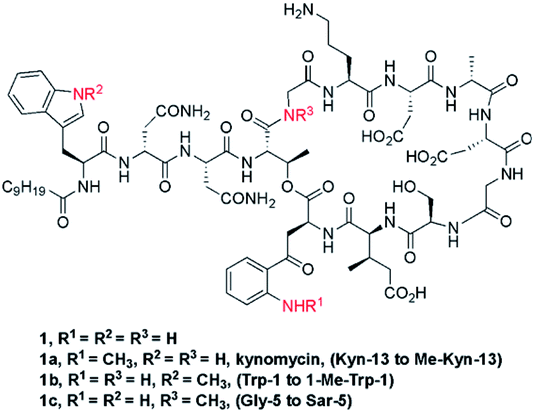 | ||
| Fig. 2 The structures of daptomycin and its derivatives. | ||
The challenge for total synthesis of daptomycin was hindered by the structural complexity of two critical nonproteinogenic amino acids, Kyn (kynurenine) and 3-mGlu (3-methylglutamic acid) and its 13 amino acid-containing cyclic lipodepsipeptide backbone. The first version of total synthesis of daptomycin was reported in a patent literature by Cubist Pharmaceuticals Inc., and a solution-phase strategy was proposed to realize the total synthesis of daptomycin; however, the segment-coupling approach was highly labor intensive.13 In 2013, Li and coworkers reported an elegant approach for the total synthesis of daptomycin using a hybrid synthesis strategy of solid-phase and solution-phase synthesis; they also described this successful story of synthesis in their personal account.14
To start the total synthesis of daptomycin, Li and coworkers first established the concise synthesis of two key fragments, the Kyn containing fragment 4 and 3-mGlu fragment containing 8 (Scheme 1). Fragment 4 was synthesized from glycine 2 with some modification from literatures15 and fragment 8 was prepared by oxidative ring-opening of the compound 7.16 Then, to finish the synthesis of daptomycin, solid-phase synthesis strategy was applied and amino acids residues were assembled via the standard Fmoc-SPPS (Fmoc solid-phase peptide synthesis) strategy. The trityl-resin-linked pentatpeptide 11 was obtained via standard Fmoc-SPPS by coupling with fragment 10 and 8. Then, followed by a reduction of the N-terminal azide group with dithiothreitol (DTT) and a peptide homologation via Fmoc-SPPS, the peptide 13 was successfully constructed with the whole linear sequence (Scheme 2).
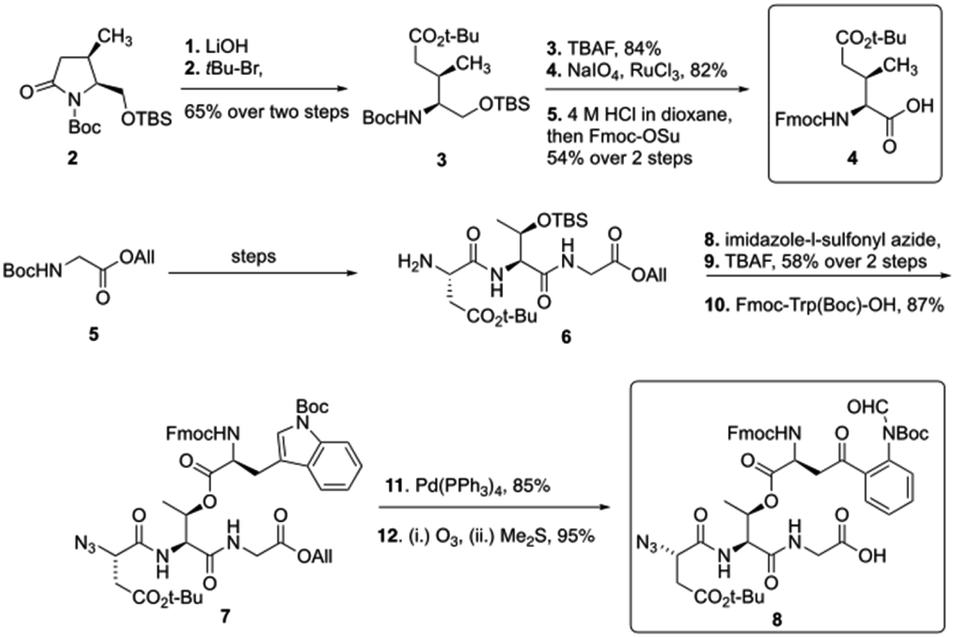 | ||
| Scheme 1 The synthesis of Kyn and 3-mGlu fragment by the Li group. | ||
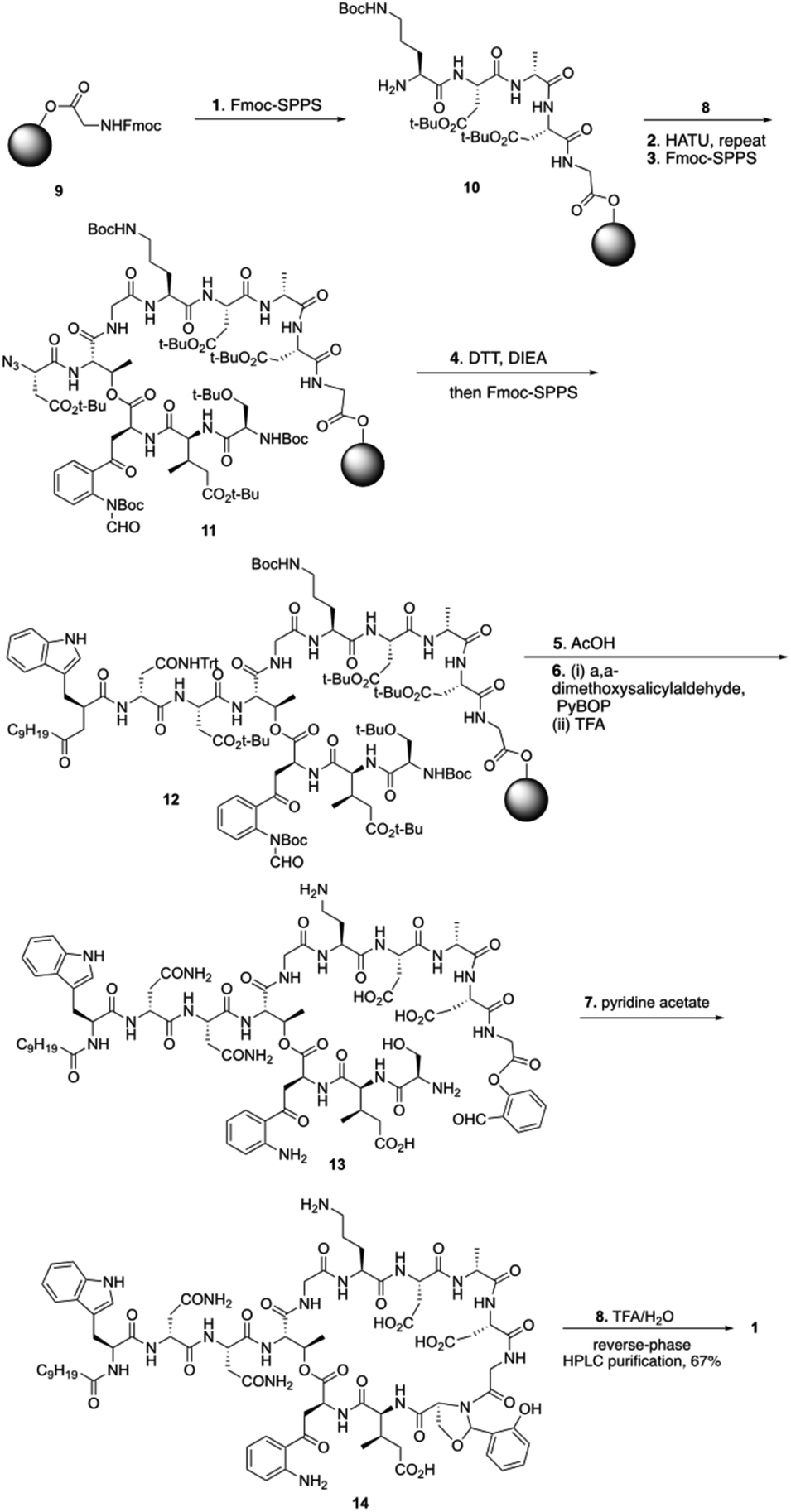 | ||
| Scheme 2 The total synthesis of daptomycin by the Li group. | ||
Finally, the synthesis of daptomycin was completed via a serine ligation-mediated cyclization (Scheme 3). The side-chain-protected linear peptide of 12 was released to give the peptide acid 13 with the protecting groups intact. Global deprotection of all side-chain-protecting groups with TFA afforded peptide salicylaldehyde ester 14 with 17% yield. Herein, to perform cyclization via serine ligation, peptide 14 was dissolved in a pyridine acetate buffer (mol mol−1, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 5 mM concentration for 4 h at room temperature, the N,O-benzylidene acetal-linked cyclic product was obtained without isolation and treated with TFA for 10 min, and daptomycin (1) was obtained at 67% yield after reverse-phase HPLC purification (Scheme 2).
:1) at 5 mM concentration for 4 h at room temperature, the N,O-benzylidene acetal-linked cyclic product was obtained without isolation and treated with TFA for 10 min, and daptomycin (1) was obtained at 67% yield after reverse-phase HPLC purification (Scheme 2).
 | ||
| Scheme 3 The strategy of macrocyclization by the Li group. | ||
This strategy of Li and coworkers is a considerable and practical synthesis of daptomycin. Later, the same team studied the SAR of daptomycin, and a series of daptomycin derivatives were synthesized. Li and coworkers prepared over 80 daptomycin analogs and found out that the methylation on daptomycin (Fig. 2), including N-methylations of Kyn-13 (1a), Trp-1 (1b), and Gly-5 (1c), could maintain or enhance the in vitro antibacterial activities of the original molecule.14 Specifically, kynomycin (1a), the daptomycin analogue with methylated Kyn-13, even showed a 2–4 times greater ability in eradicating both daptomycin-susceptible and daptomycin resistant MRSA and VRE. Moreover, kynomycin exhibits slightly lower toxicity. This analogue is further developed as a potential new antibiotic against multidrug-resistant pathogens.
A54145 is another lipodepsipeptide antibiotic that was isolated from Streptomyces fradiae and exhibits remarkable similarities to daptomycin (Fig. 3).17 In addition to the nonproteinogenic 3-methyl-glutamic acid (3-mGlu) in daptomycin, A54145 contains two other nonproteinogenic amino acids: L-methoxy-aspartic acid (L-HO-Asn) and L-hydroxy-asparagine (L-3R-MeO-Asp). Li and coworkers reported the efficient total synthesis of A54145 with solid phase strategy (Fmoc-SPPS) and revised the proposed structure of A54145 with L-3S-MeO-Asp.18
 | ||
| Fig. 3 Antibiotic A54145 and daptomycin. | ||
2.2 Total synthesis of daptomycin by the Brimble group
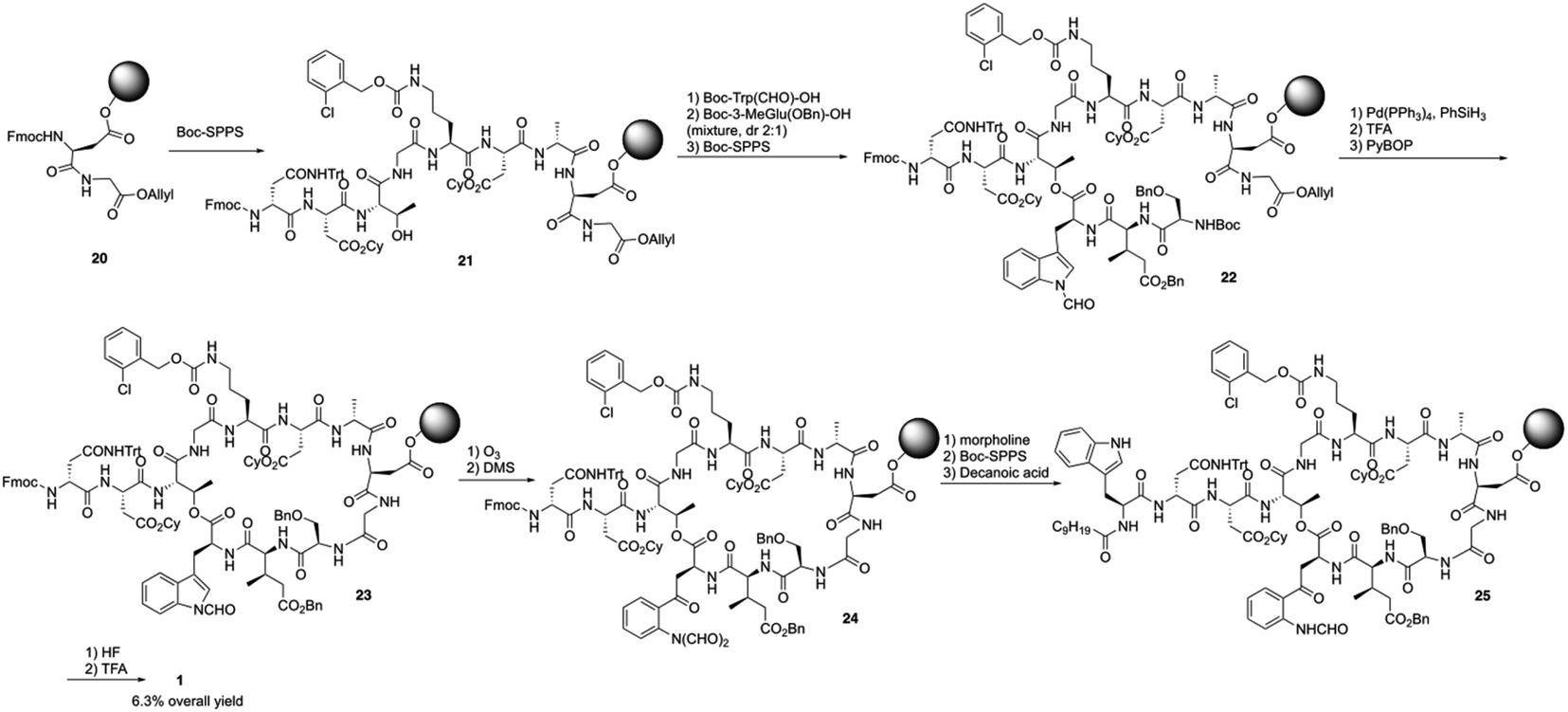
Boc-SPPS could reduce peptide aggregation and suppress aspartimide formation, thus it has several advantages over Fmoc-SPPS, especially when the synthetic target contains base-sensitive moieties. In 2019, Harris, Brimble and coworkers employed a Boc-SPPS approach and realized the total synthesis of daptomycin16 (Scheme 5).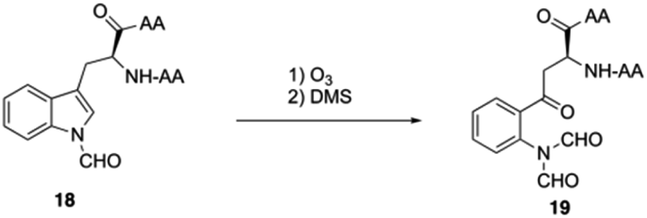 | ||
| Scheme 4 Kyn synthon synthesis via the on-resin ozonolysis of Trp. | ||
In their initial efforts towards the synthesis of daptomycin, the esterification between Thr4-Kyn13 failed to give positive results under various condensation reaction conditions, which were previously studied by the Li and Taylor groups. Harris, Brimble and coworkers tried to use a similar protocol with Taylor's approach to install the Kyn with the Fmoc protected octapeptide, but no desired product was formed. Finally, inspired by a synthesis of the Kyn-containing peptide cyclomontanin B, they successfully developed an alternative route towards daptomycin via an on-resin ozonolysis of Trp residue to Kyn residue (Scheme 4).19 Compound 21 was first synthesized using standard Boc-SPPS conditions with Fmoc-D-Asn-OH to achieve orthogonality to Boc-chemistry, and it was linked with Boc-Trp(CHO)-OH to the side chain of Thr4 residue quantitatively, thus solving the esterification (Scheme 5). Subsequently, through peptide chain elongation and deprotection with Pd(PPh3)4 and phenylsilane to remove the allyl group, along with TFA to remove the Boc group, the on-resin macrolactamisation was delivered quantitatively. Finally, the on-resin ozonolysis of Trp to construct a Kyn-containing peptide 24 was successfully carried out without any notable side-products. Subsequent removal of protecting groups, along with coupling with Boc-Trp-OH and capping with decanoic acid, the final product daptomycin 1 was afforded after deprotecting.
 | ||
| Scheme 5 The total synthesis of daptomycin employed Boc-SPPS approach by the Brimble group. | ||
2.3 Two versions of total synthesis of daptomycin by the Taylor group
Over the last several years, Taylor and coworkers completed two versions of total synthesis of daptomycin.20 In their initial approach, they failed to form the ester bond between Thr4 and Fmoc-Kyn (Boc, CHO)-OH with a variety of coupling reaction conditions.20a By using an orthogonal protecting strategy, they successfully solved this problem and found the decanoyl tail affected on the esterification reaction, pointing out that the decanoyl tail might interact with the hydrophobic resin, making it less accessible to the hydroxyl group of Thr4.Subsequently, they redesigned their approach and achieved the total synthesis of daptomycin smoothly. By combining the α-azido and α-Fmoc-protected amino acids in their solid phase synthesis of daptomycin, the coupling of Fmoc-Kyn(Boc, CHO)-OH with Thr4 was accomplished before the decanoyl tail was installed (Scheme 6). After repeating and treating the peptide with SPPS and side chain deprotection, daptomycin 1 was synthesized with a 9% overall yield. Some analogues of daptomycin were synthesized to study their SAR and they found that R-configuration of the methyl group in the 3-mGlu residue of daptomycin is crucial to its biological activity.
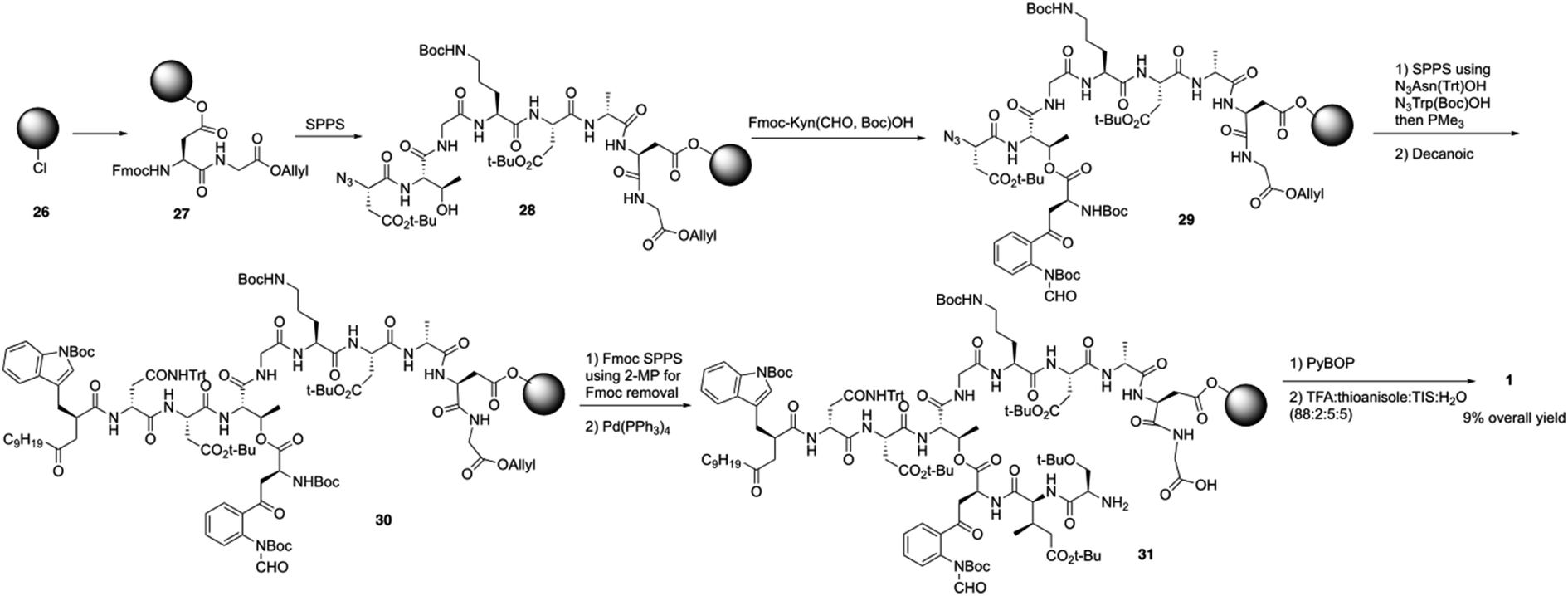 | ||
| Scheme 6 The total synthesis of daptomycin by the Taylor group. | ||
Recently, to overcome the shortcoming of their previous synthesis of daptomycin, especially the commercially unavailablility of 3 azido acids and incompleteness of on-resin cyclization, Taylor and coworkers developed a second version of a high-yielding total synthesis of daptomycin using a stable Fmoc-L-Kyn(Boc)-OH as a suitably protected Kyn synthon.20b An unprecedented side reaction was observed, affording the 4-quinolone containing final peptide when using Fmoc-L-Kyn(Boc, CHO)-OH as Kyn synthons (Scheme 7). They believed that the side product was formed via a Camps cyclization by repeatedly treating the reaction intermediates with 2-methylpiperidine/DMF. Finally, they started to redesign the synthesis approach by using a single azide acid and an off-resin cyclization, successfully realizing the highest yielding synthesis of daptomycin by combining solid-phase synthesis and a Fmoc SPPS stable Kyn synthon (Scheme 8).
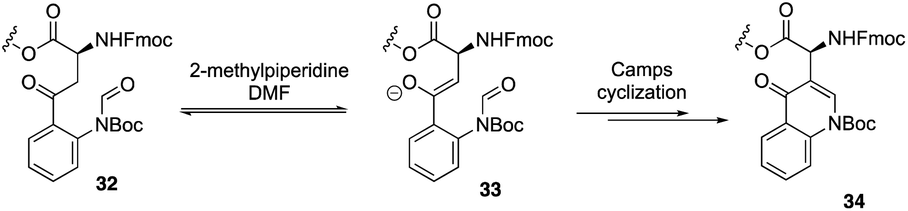 | ||
| Scheme 7 Proposed pathway for the side reaction by the Taylor group. | ||
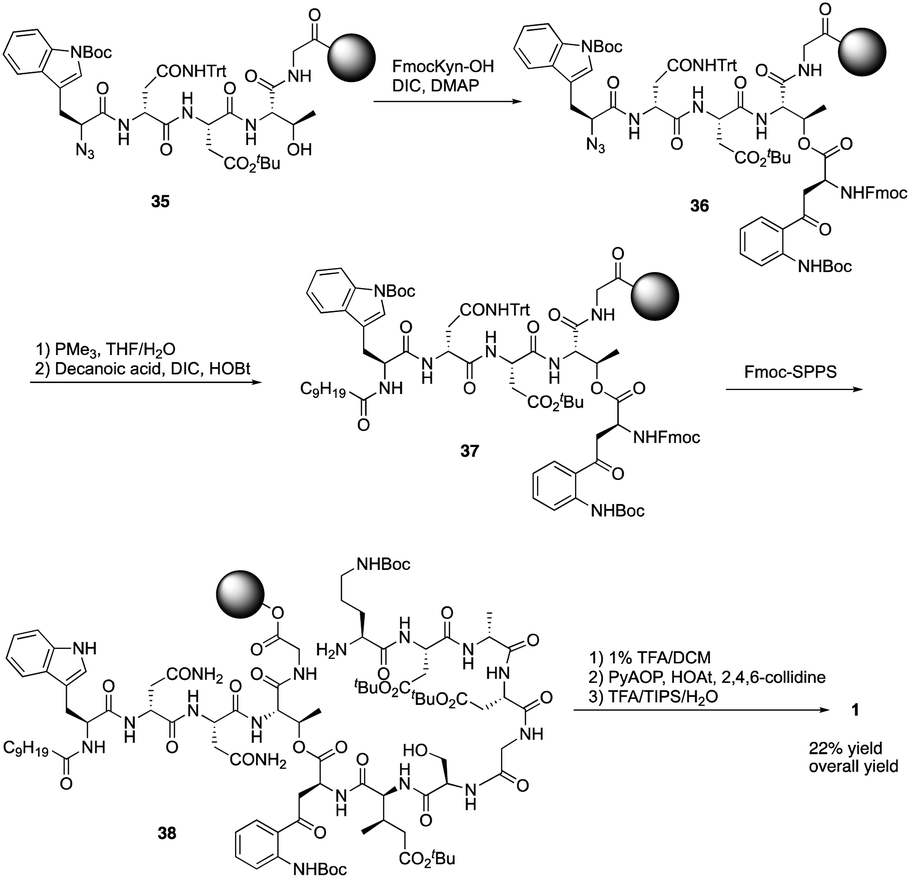 | ||
| Scheme 8 The high-yielding total synthesis of daptomycin by the Taylor group. | ||
3. Total synthesis of AS-48
The circular bacteriocin AS-48 (43) is one of the largest cyclic antimicrobial peptides that is broadly active against bacteria by inducing cell membrane disorganization,21 and it is accessible only by biosynthesis from E. faecalis in low yields. In 2016, Tam and co-workers reported the total synthesis of AS-48 with an efficient chemoenzymatic strategy.22The total synthesis of AS-48 via chemical synthesis has not been reported because of the synthetic challenge of cyclic backbone and hydrophobic resudues (>50%). AS-48 contains no cysteine, thus leading to difficulty in synthesizing AS-48 by the commonly used chemical macrocyclization method like the thia-zip cyclization. Recently, Tam and coworkers combined SPPS and chemoenzymatic strategy and successfully applied them to the efficient total synthesis of AS-48 (Scheme 9). First, they construct the unfolded AS-48 with all the amino acids and creatively added butelase 1 as a highly efficient Asp/Asn (Asx)-specific ligase to catalyze peptide cyclization, which was discovered by their group in their previous study of naturally occurring cyclic peptides.23
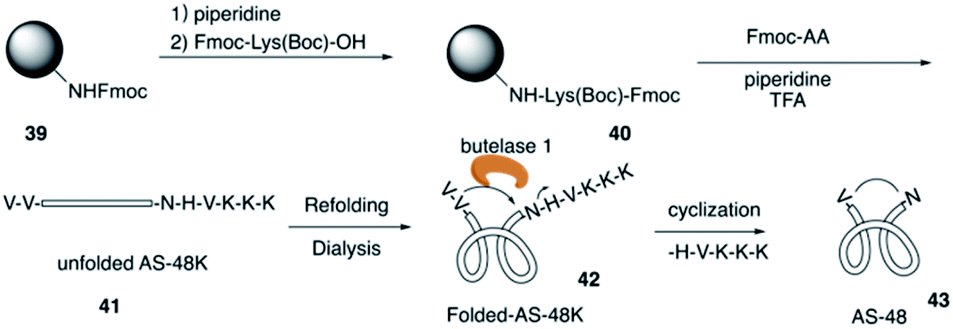 | ||
| Scheme 9 The total synthesis of AS-48 by the Tam group. | ||
After they completed the total synthesis of the largest circular bacterocin by an efficient chemoenzymatic approach, they evaluated their biological activity. Antimicrobial assays suggested that the linear precursor of AS-48 is inactive up to 100 μM while the macrocyclic AS-48 is potently active against all tested bacteria with minimal inhibitory concentrations (MIC) at a sub-μM range.
4. Total synthesis of lysocin E
Lysocin E (50) is a novel antibiotic isolated from Lysobacter sp. RH2180-5 strain and exhibits potent antimicrobial activity aganst MRSA.24 It is a 37-membered macrocycle containing 12 amino acid residues with an unusual N-Me-D-Phe5. In order to well establish the SAR of lysocin E, solid phase synthesis strategy was proposed to efficiently synthesize lysocin E and its derivatives. In 2015 and later, Inoue and co-workers completed the first total synthesis of lysocin E with a fully solid phase strategy.25In 2015, Inoue and coworkers reported the application of SPPS in the total synthesis of lysocin E by first synthesizing the two fragments 46 and 47 (Scheme 10). Then, they repeated the SPPS process and used MW-assisted SPPS to install amino acid residues and piperidine to remove the Fmoc group and successfully construct the intermediates 45 and 49, ultimately completing the total synthesis of lysocin E elegantly with an 8.0% overall yield. They also synthesized enantiomeric, epimeric, N-demethylated analogues and 14 new side-chain analogues of lysocin E using solid phase strategy, and they evaluated the biological activity against Staphylococcus aureus. They found that an unnatural enantiomer has the comparable antibacterial activity and the cationic guanidine of Arg-2/7, and the hydrophobic acyl group of C2 or C11 at Thr-1 and the indole ring at Trp-10 are critical for the potent liposomal and antimicrobial activities. Their elegant total synthesis of lysocin E and analogues highlights the power of the solid phase strategy for systematic SAR study.
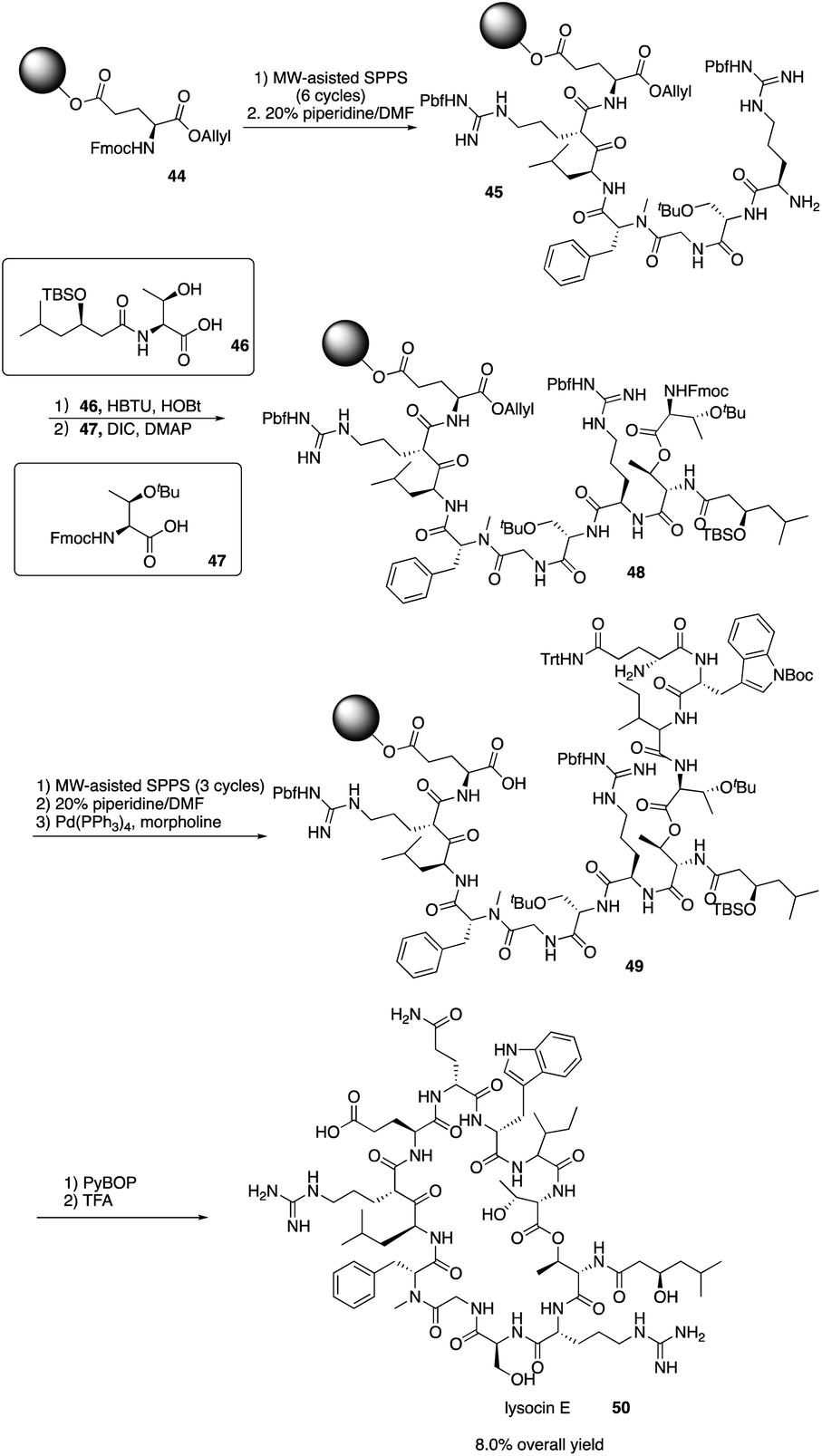 | ||
| Scheme 10 The total synthesis of Lysocin E by the Inoue group. | ||
5. Total synthesis of mannopeptimycin β
Isolated from Streptomyces hygroscopicus LL-AC98 by the Wyeth Research scientists in the 1950s,26,27 Mannopeptimycins have been proved to have antibacteral activity against antibiotic-resistant Gram-positive pathogens like MRSA and VRE, but they have been neglected for almost half a century due to their unidentified complex structures and their modest antibacterial activity.28 Mannopeptimycins (MPPs) didn't get attention from scientists for almost half century until they were found in a retrospective survey of antibiotics against superbug pathogens by inhibiting the transglycosylation step to stop bacterial cell-wall peptidoglycan synthesis via lipid II sequestration.28 Mannopeptimycins are cyclic glycopeptide antibiotics comprising of six amino acids, four of which are nonproteinogenic: L-β-methyl phenylalanine (βmPhe), D-tyrosine and D- and L-β-hydroxyenduracididines (βhEnd). Due to the intriguing structure of mannopeptimycins, few SAR investigations were made to obtain analogues at the L-and D-βhEnd moieties. For the last several years, total synthesis of mannopeptimycins were reported to continue the study of mannopeptimycins such as the novel mode of action and SAR study. In 2004, Fuse and Doi reported the first total synthesis of the mannopeptimycin aglycon by using a solution phase strategy,29 and in 2016, Chen and coworkers delivered the first total synthesis of mannopeptimycin α and β highlighted with a gold catalyzed late-stage N-mannosylation30To reduce the tedious purification process of oligopeptide intermediates, Li and coworkers recently reported the total synthesis of mannopeptimycin β (76) via a hybrid solid-solution phase synthesis and peptide ligation strategy.31 Two key fragments 62 and 69 were first prepared with elegant strategies to control regioselectivity and enantioselectivity from similar starting materials 51 and 63, respectively. Fragment 62 was synthesized via B(OMe)3-epoxide chelation (Scheme 11), and fragment 69 was afforded via a guanidine-epoxide ring-opening strategy (Scheme 12). With well-synthesized building blocks in hand, Li and coworkers started the peptide assembly via solid phase strategy (Scheme 13). Among their unsuccessful attempts, they found that the N-α-Man-D-βhEnd must be installed to the peptide very carefully to avoid the epimerization; therefore, they prepared the tripeptide residue 62 in the solution phase. The 2′-hydroxycinnamoyl AM resin 70 was assembled with amino acid residues via the standard Boc-SPPS and 74 was afforded with the full peptide sequence. Subsequently, to efficiently apply the peptide ligation strategy, the Boc deprotected resin 74 was released into the peptide C-terminal salicylaldehyde ester via ozonolysis to give the cyclized intermediate 75. Finally, mannopeptimycin β (76) was obtained with an 11% overall yield. They could complete a one-round synthesis of mannopeptimycin β in two weeks, which emphasizes the power of a solid phase strategy in the total synthesis of cyclic peptides.
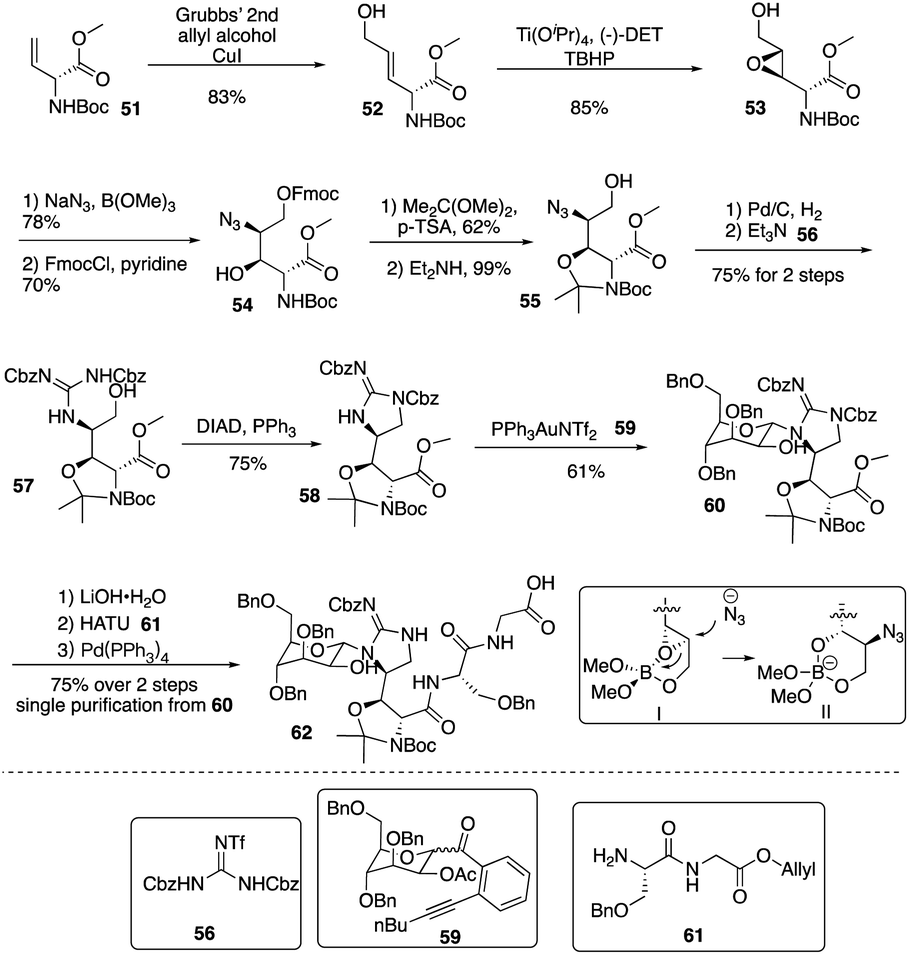 | ||
| Scheme 11 The synthesis of key fragment 62 of mannopeptimycin β by the Li group. | ||
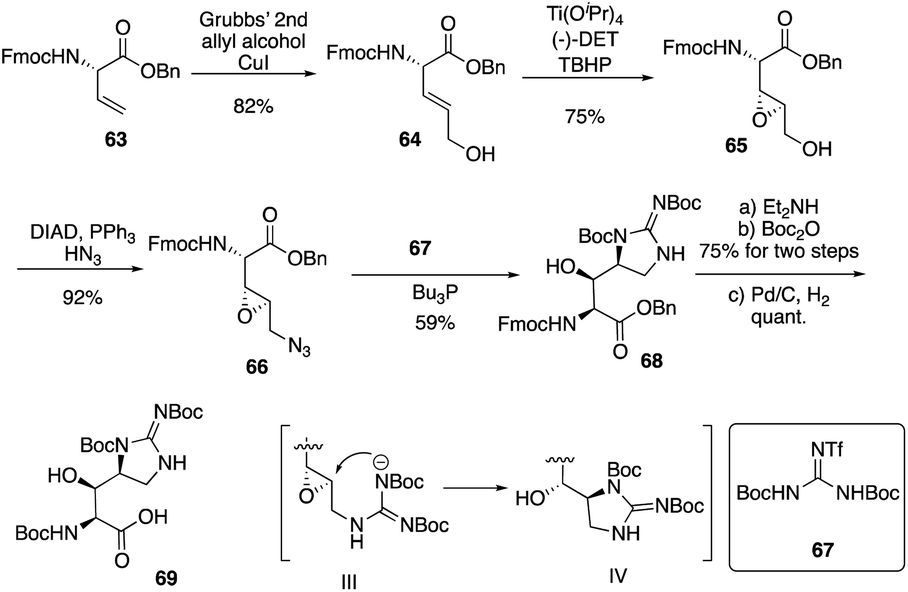 | ||
| Scheme 12 The synthesis of key fragment 69 of mannopeptimycin β by the Li group. | ||
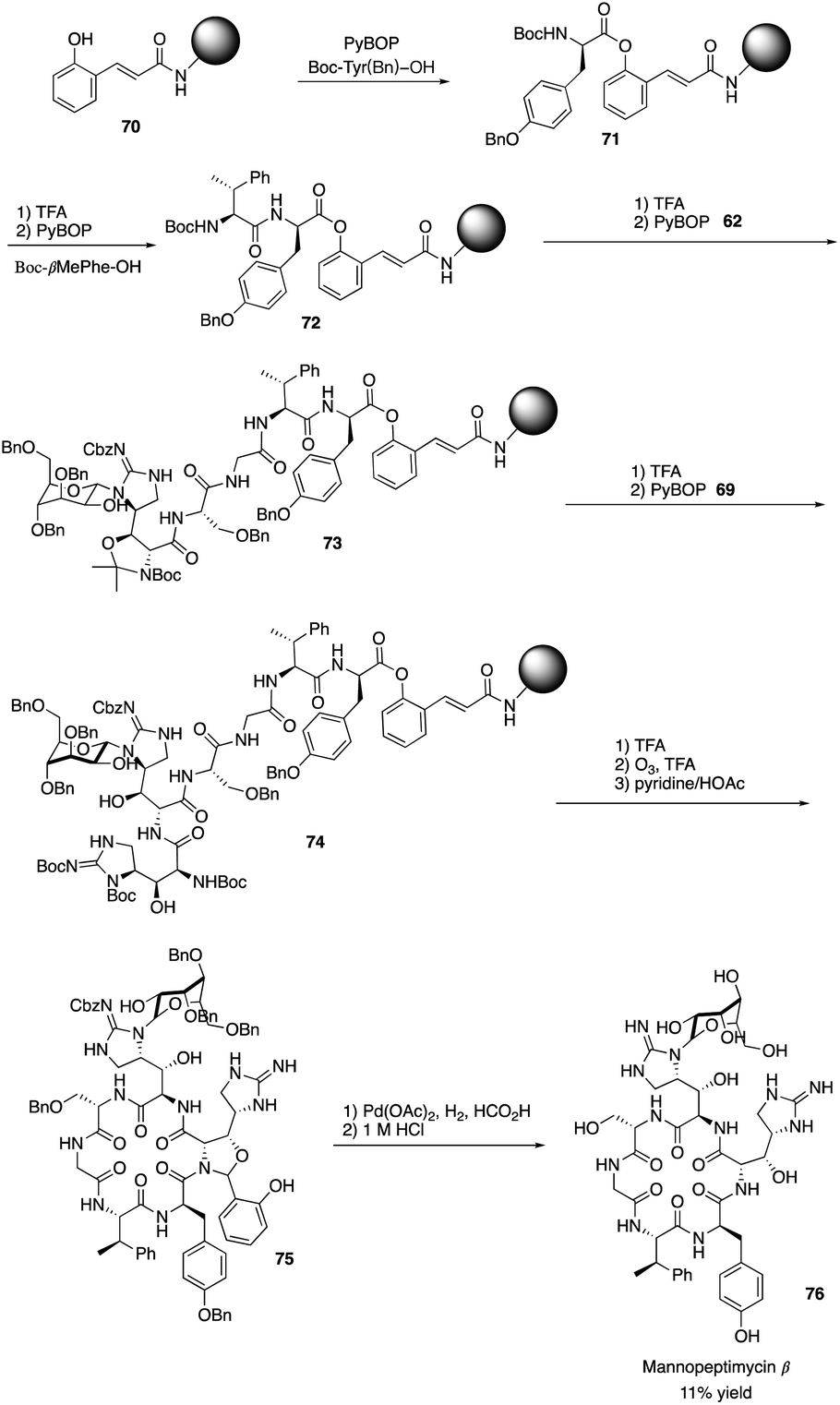 | ||
| Scheme 13 The total synthesis of mannopeptimycin β by the Li group. | ||
6. Total synthesis of CDA3a, and CDA4a
6.1 Total synthesis of CDA3a by the Li group
The calcium-dependent antibiotics (CDAs) are acidic antibacterial cyclic lipopeptides first isolated from Streptomyces coelicolor A3(2), and they display a wide range of antibacterial biological activity.32 Similar to its group members, CDA3a and CDA4a comprise an unusual trans-2,3-epoxyhexanoyl moiety as their exocyclic tail and three nonproteinogenic amino acid residues, including a Z-dehydrotryptophan residue along with a cyclic decadepsipeptide (Fig. 4). Due to widespread bacterial resistance to existing antibacterial drugs, new antibiotics and their derivatives are needed to establish their SAR. For CDAs, the modifications by bioengineering were limited to the specific amino acid residues,33 but chemical synthesis would be an alternate way to overcome the challenge in generating analogues. Last year, Li and coworkers reported the first version of total synthesis of CDAs and analogues via late-stage serine ligation.34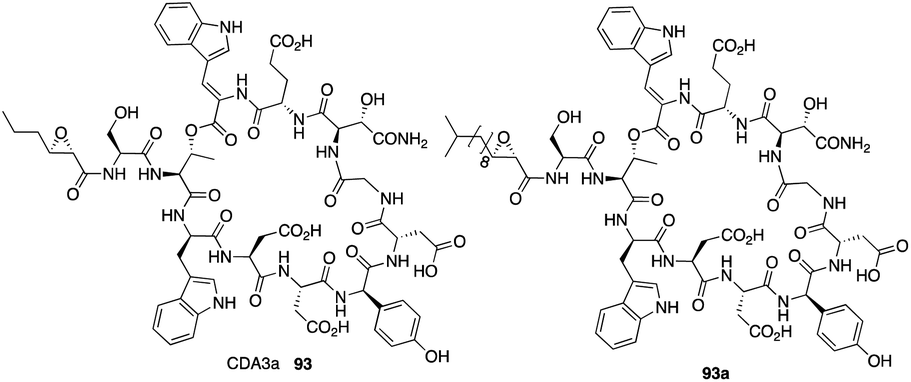 | ||
| Fig. 4 The structures of CDA3a and its analogue. | ||
They first prepared two important fragments, the dehydrotryptophan-containing 81 and the side chain 84, and the unique amino acid residue 81 was synthesized successfully by direct dehydrogenation on the Trp residue (Scheme 14). With all building blocks in hand, they started to synthesize CDA3a by using a solid phase strategy. After incorporating amino acid residue, including fragments 81 and 84 into resin, the macrocyclization proceeded very well to afford the cyclic peptide 91 in the presence of PyBOP. Then the global deprotection was carried out to introduce side chain fragment, and the N,O-benzylidene acetal intermediate 92 was delivered by coupling with 84 (Scheme 15). CDA3a (93) was finally synthesized with a 9.3% overall yield after the HPLC purification. They also synthesized several analogues of CDA3a and found that the length, α,-substitution, and the terminal substitution of the side chain are critical for their bioactivity. Specially, compound 93a had a 256−512-fold greater potency against VRE and MRSA than the parent CDA3a, thus identifying it as the most potent analogue (Fig. 4).
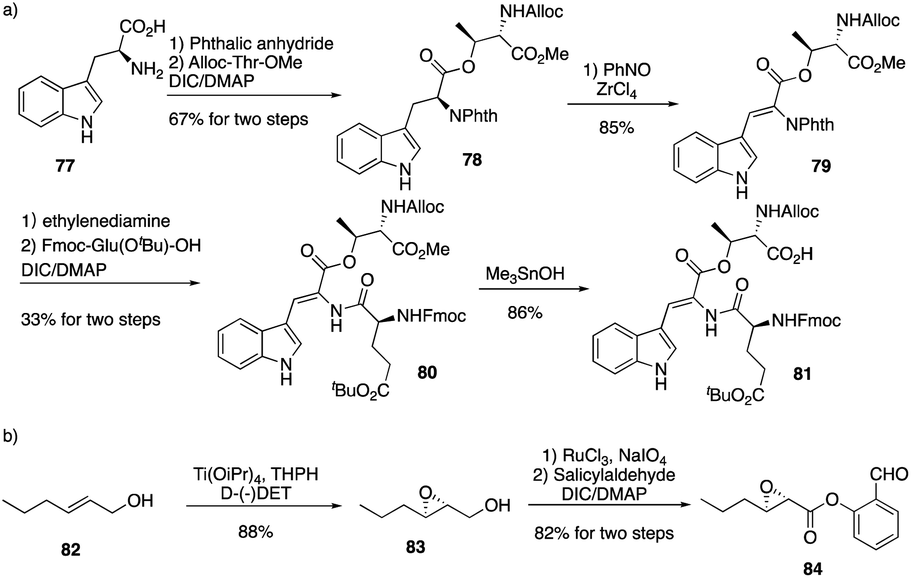 | ||
| Scheme 14 The synthesis of key fragments of CDA3a by the Li group. | ||
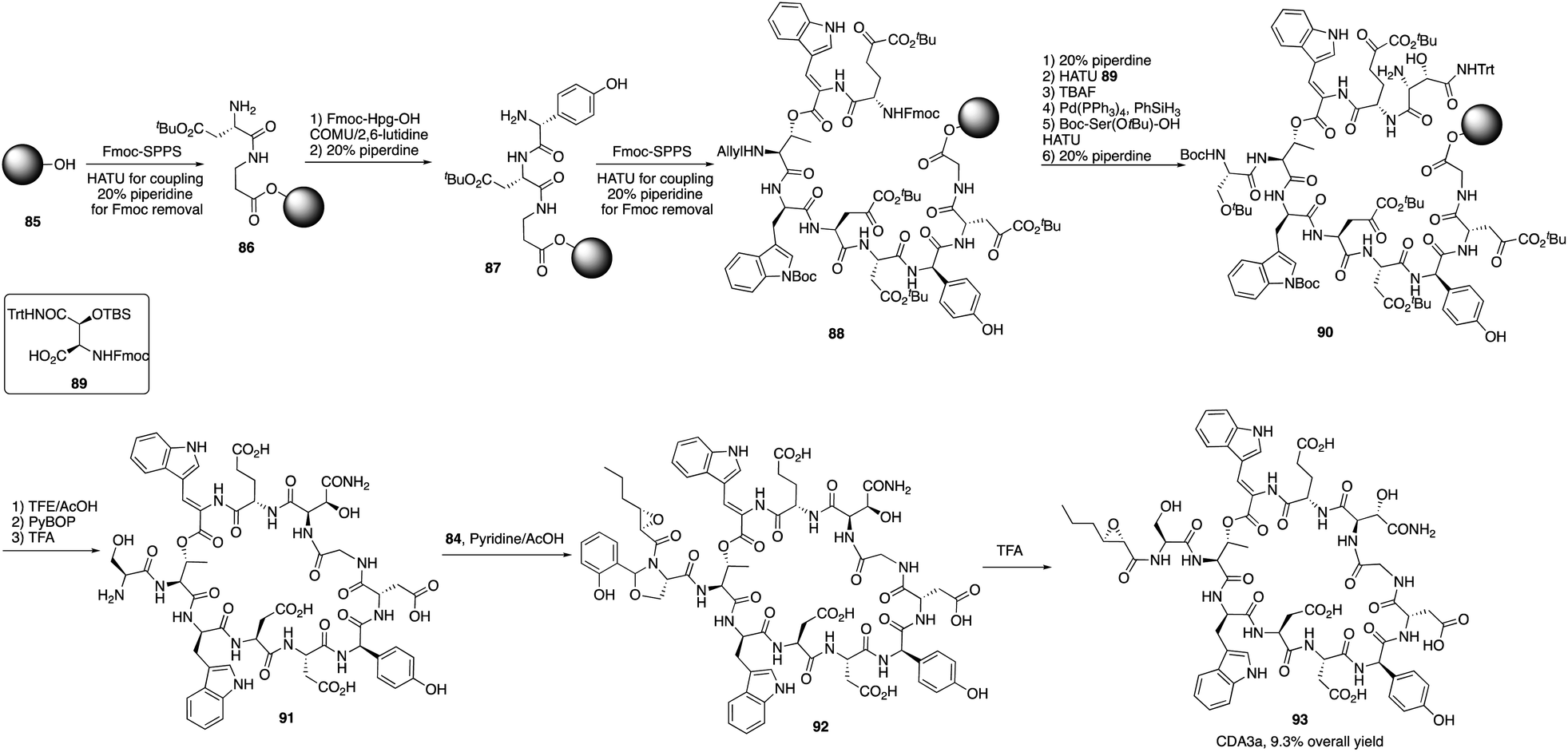 | ||
| Scheme 15 The total synthesis of CDA3a by the Li group. | ||
6.2 Total synthesis of CDA3a, and CDA4a by the Taylor group
Recently, Taylor and coworkers developed a new approach to synthesize the Z-dehydrotryptophan residue, and they completed the total synthesis of CDA3a and CDA4a with a solid phase synthesis strategy.35 CDA3a and CDA4a share the same uncommon amino acids like dehydrotryptophan, D-hydroxyphenylglycine and D-erythro-hydroxyasparagine. The only difference between CDA3a and CDA4a is at position 10 where CDA4a has another uncommon amino acid 3-mGlu.Taylor and coworkers started to synthesize CDA3a and CDA4a by the coupling of Fmoc-D-HOPhGly(OTBS)-OH with the resin-bound peptide 94 (Scheme 16).36 It's worth point out that COMU/2,6-lutidine was used to coupling with HOPhGly to minimize the racemization-prone problem. After repeating this process using Fmoc SPPS to elongate the peptide and coupling with unusual amino acid residues 99 and others, peptide 101 was afforded and treated with Pd(PPh3)4 and 2-MP to remove allyl group and Fmoc group, successfully delivering the macrocyclization product 102 with PyAOP/HOBt. Finally, with a mild deprotection from TFA, CDA3a (93) was obtained with a 6% overall yield, and CDA4a (103) had an 8.5% overall yield after purification by HPLC.
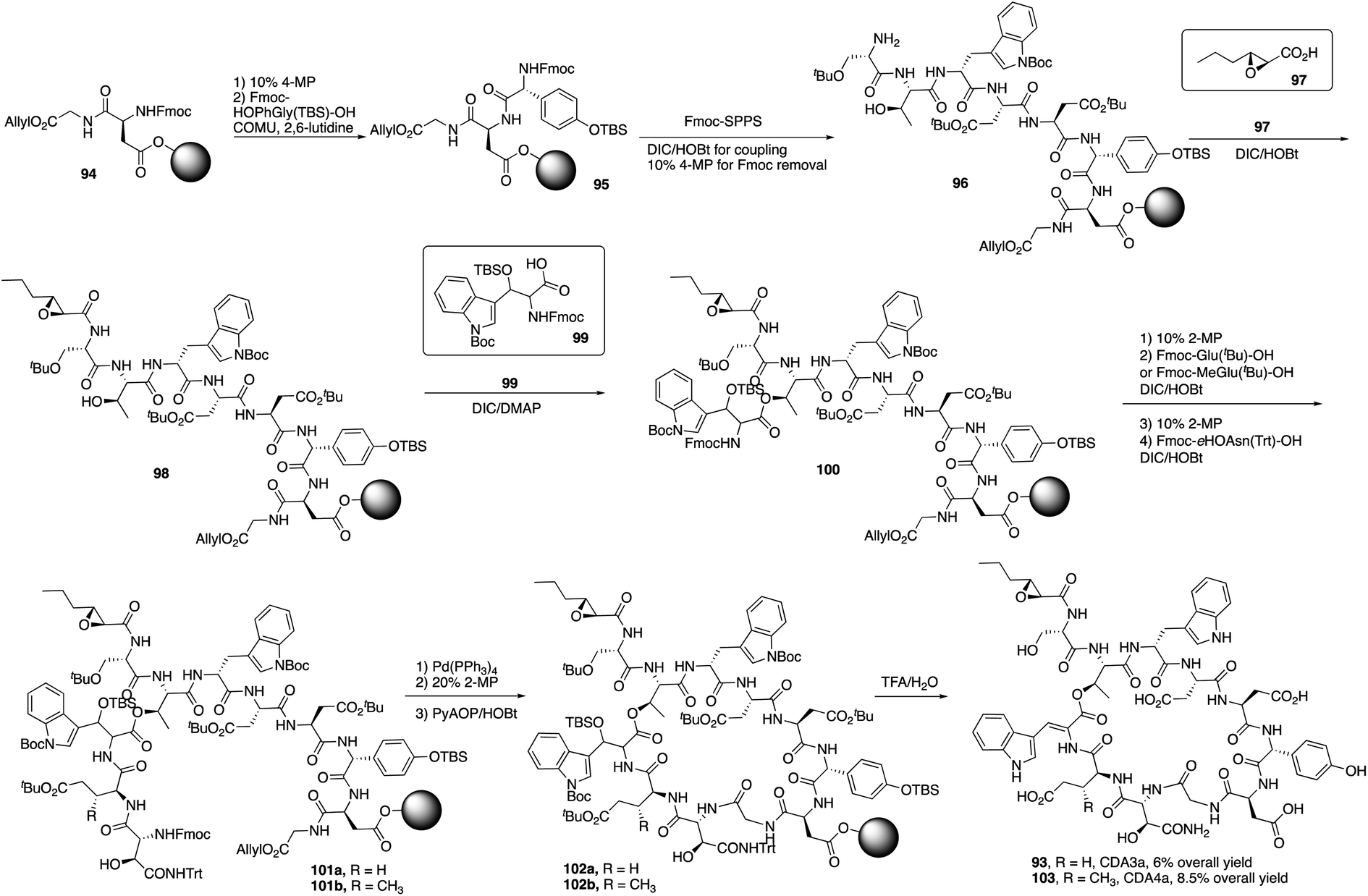 | ||
| Scheme 16 The total synthesis of CDA3a and CDA4a by the Taylor group. | ||
Taylor and coworkers also studied the biological activity of CDAs against Bacillus subtilis 1046. They found the CDA4a was active only at 16 nM Ca+ and at 16 nM Ca+, and the MIC values of CDA3a and CDA4a were 16 and 8 μg mL−1, respectively.
7. Summary and outlook
Throughout the past decades, researchers have devoted their efforts to overcome the challenges of efficient, economical, and benign synthetic plans for natural products, specifically for those used as medicines which suffer from low isolation yields and nonrenewable natural sources. By simplifying the structures and breaking down their complex components, novel synthetic processes have emerged and are still being updated. Scientists are chasing the perfect synthesis in a meaningful way. The applications of a solid phase synthesis strategy in complex natural products provide new ways of investigating natural product leads, and these applications offer new possibilities of natural product analogues development in new drug developments.Natural products, like antibiotics with their unique architectures and stereochemical challenges, are the best targets for synthetic chemists, and the concise synthesis of them means an elegant creativity of organic chemistry and also a tremendous success of new drug development. In this review, we have summarized the recent application of solid phase strategy in total synthesis of antibiotics. Some of complex antibiotics could be completed a one-round synthesis in a very short time, which emphasizes the power of a solid phase strategy in the total synthesis of cyclic peptides. With the help of AI, we think the applications of solid phase synthesis strategy in total synthesis of antibiotics could be completed autonomously by a mobile robotic chemist, which enhance the development of SAR study.37
We believe the currently developed concise synthetic routes of antibiotics provide a practical and economical approach for further SAR study of their analogues. However, the antibiotic resistance is spreading faster than the discovery of new treatments, which suggests the need for new strategies to enhance the development of new drugs.
Abbreviations
| (SAR) | Structure–activity relationship |
| (SPPS) | Solid-phase peptide synthesis |
| (MRSA) | Staphylococcus aureus |
| (VRE) | Vancomycin-resistant enterococci |
| (VRSA) | Vancomycin-resistant S. aureus |
| (DTT) | Dithiothreitol |
| (Kyn) | Kynurenine |
| (3-mGlu) | 3-Methylglutamic acid |
| (L-HO-Asn) | L-Methoxy-aspartic acid |
| (L-3R-MeO-Asp) | L-Hydroxy-asparagine |
| (MIC) | Minimal inhibitory concentrations |
| (MPPs) | Mannopeptimycins |
| (βmPhe) | L-β-Methyl phenylalanine |
| (βhEnd) | D-Tyrosine and D- and L-β-hydroxyenduracididines |
| (CDAs) | Calcium-dependent antibiotics |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China for their financial support. Specially, we thank ViaX for useful comments and language editing during preparation of this manuscript.References
- E. Patridge, P. Gareiss, M. S. Kinch and D. Hoyer, Drug Discovery Today, 2016, 21, 204–207 CrossRef CAS PubMed.
- C. Jimenez, ACS Med. Chem. Lett., 2018, 9, 959–996 CrossRef CAS PubMed.
- K. C. Nicolaou and T. Montagnon in Molecules That Changed the World, Wiley-VCH, 2008 Search PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, S. Rigol and R. Yu, CCS Chem., 2019, 1, 3–37 CAS; (b) R. A. Shenvi and B. Huffman, J. Am. Chem. Soc., 2019, 141, 3332–3346 CrossRef PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- (a) R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS; (b) B. Merrifeld, Science, 1986, 232, 341–347 CrossRef PubMed.
- F. Guillier, D. Orain and M. Bradley, Chem. Rev., 2000, 100, 2091–2158 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, N. Winssinger, J. Pastor, S. Ninkovic, F. Sarabia, Y. He, D. Vourloumis, Z. Yang, T. Li, P. Giannakakou and E. Hamel, Nature, 1997, 387, 268–272 CrossRef CAS PubMed; (b) J. P. Nandy, M. Prakesch, S. Khadem, P. T. Reddy, U. Sharma and P. Arya, Chem. Rev., 2009, 109, 1999–2060 CrossRef CAS PubMed; (c) O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523–1527 CrossRef CAS PubMed.
- R. H. Baltz, Curr. Opin. Chem. Biol., 2009, 13, 144–151 CrossRef CAS PubMed.
- M. Heidary, A. D. Khosravi, S. Khoshnood, M. J. Nasiri, S. Soleimani and M. Goudarzi, J. Antimicrob. Chemother., 2018, 73, 1–11 CrossRef CAS PubMed.
- (a) K. T. Nguyen, D. Ritz, J.-Q. Gu, D. Alexander, M. Chu, V. Miao, P. Brian and R. H. Baltz, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 17462–17467 CrossRef CAS PubMed; (b) J. Grünewald, S. A. Sieber, C. Mahlert, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2004, 126, 17025–17031 CrossRef PubMed; (c) J. Hill, J. Siedlecki, I. Parr, M. Morytko, X. Yu, Y. Z. Zhang, J. Silverman, N. Controneo, V. Laganas, T. C. Li, J. J. Lai, D. Keith, G. Shimer and J. Finn, Bioorg. Med. Chem. Lett., 2003, 13, 4187–4191 CrossRef CAS PubMed.
- The first reported in the patent literature by Cubist Pharmaceuticals Inc., WO Patent, WO2006110185, 2007 Search PubMed.
- (a) H. Y. Lam, Y. Zhang, H. Liu, J. Xu, C. T. T. Wong, C. Xu and X. Li, J. Am. Chem. Soc., 2013, 135, 6272–6279 CrossRef CAS PubMed; (b) H. Y. Lam, R. I. Gaarden and X. Li, Chem. Rec., 2014, 14, 1086–1099 CrossRef CAS PubMed; (c) S. Chen, H. Y. Chow, K. H. L. Po, K. Jin, G. Qiao, Z. Sun, W. Ma, X. Ye, N. Zhou and X. Li, ACS Med. Chem. Lett., 2020, 11, 1442–1449 CrossRef PubMed; (d) S. Chen, H. Y. Chow, K. H. L. Po, P. Gao, P. Blasco, X. Wang, C. Li, L. Ye, K. Jin, K. Chen, E. W. C. Chan, X. You, R. Y. Kao and X. Li, J. Med. Chem., 2020, 63, 3161–3171 CrossRef PubMed.
- (a) F. C. Ross, N. P. Botting and P. D. Leeson, Bioorg. Med. Chem. Lett., 1996, 6, 875–878 CrossRef CAS; (b) C. Maitrani, D. J. Heyes, S. Hay, S. Arumugam, V. V. Popik and R. S. Phillips, Bioorg. Med. Chem. Lett., 2012, 22, 2734–2737 CrossRef CAS PubMed; (c) L. H. J. Kleijn, F. M. Müskens, S. F. Oppedijk, G. de Bruin and N. I. Martin, Tetrahedron Lett., 2012, 47, 6430–6432 CrossRef.
- B. Xu, Y. Hermant, S. H. Yang, P. W. R. Harris and M. A. Brimble, Chem.–Eur. J., 2019, 25, 14101–14107 CrossRef CAS PubMed.
- D. S. Fukuda, R. H. D. Bus, P. J. Baker, D. M. Berry and J. S. Mynderse, J. Antibiot., 1990, 43, 594–600 CrossRef CAS PubMed.
- D. Chen, H. Y. Chow, K. H. L. Po, W. Ma, E. L. Y. Leung, Z. Sun, M. Liu, S. Chen and X. Li, Org. Lett., 2019, 21, 5639–5644 CrossRef CAS PubMed.
- C. T. Wong, H. Y. Lam and X. Li, Org. Biomol. Chem., 2013, 11, 7616–7620 RSC.
- (a) C. R. Lohani, R. Taylor, M. Palmer and S. D. Taylor, Org. Lett., 2015, 17, 748–751 CrossRef CAS PubMed; (b) R. Moreira, J. Wolfe and S. D. Taylor, Org. Biomol. Chem., 2021, 19, 3144 RSC.
- A. Galvez, M. Maqueda, M. Martinez-Bueno and E. Valdivia, J. Bacteriol., 1991, 173, 886–892 CrossRef CAS PubMed.
- X. Hemu, Y. Qiu, G. K. T. Nguyen and J. P. Tam, J. Am. Chem. Soc., 2016, 138, 6968–6971 CrossRef CAS PubMed.
- (a) G. K. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732–738 CrossRef CAS PubMed; (b) Y. Cao, G. K. Nguyen, J. P. Tam and C. F. Liu, Chem. Commun., 2015, 51, 17289–17292 RSC; (c) G. K. Nguyen, Y. Cao, W. Wang, C. F. Liu and J. P. Tam, Angew. Chem., Int. Ed., 2015, 54, 15694–15698 CrossRef CAS PubMed.
- H. Hamamoto, M. Urai, K. Ishii, J. Yasukawa, A. Paudell, M. Murai, T. Kaji, T. Kuranaga, K. Hamase, T. Katsu, J. Su, T. Adachi, R. Uchida, H. Tomoda, M. Yamada, M. Souma, H. Kurihara, M. Inoue and K. Sekimizu, Nat. Chem. Biol., 2015, 11, 127–133 CrossRef CAS PubMed.
- (a) M. Murai, T. Kaji, T. Kuranaga, H. Hamamoto, K. Sekimizu and M. Inoue, Angew. Chem., Int. Ed., 2015, 54, 1556–1560 CrossRef CAS PubMed; (b) T. Kaji, M. Murai, H. Itoh, J. Yasukawa, H. Hamamoto, K. Sekimizu and M. Inoue, Chem.–Eur. J., 2016, 22, 16912–16919 CrossRef CAS PubMed.
- S. E. De Voe and M. P. Kunstmann, US Pat., 3495004, 1970 Search PubMed.
- The structure of mannopeptimycins was elucidated in 2002: H. He, R. T. Williamson, B. Shen, E. I. Graziani, H. Y. Yang, S. M. Sakya, P. J. Petersen and G. T. Carter, J. Am. Chem. Soc., 2002, 124, 9729–9736 CrossRef CAS PubMed.
- M. P. Singh, P. J. Petersen, W. J. Weiss, J. E. Janso, S. W. Luckman, E. B. Lenoy, P. A. Bradford, R. T. Testa and M. Greenstein, Antimicrob. Agents Chemother., 2003, 47, 62–69 CrossRef CAS PubMed.
- S. Fuse, H. Koinuma, A. Kimbara, M. Izumikawa, Y. Mifune, H. He, K. Shin-ya, T. Takahashi and T. Doi, J. Am. Chem. Soc., 2014, 136, 12011–12017 CrossRef CAS PubMed.
- B. Wang, Y. Liu, R. Jiao, Y. Feng, Q. Li, C. Chen, L. Liu, G. He and G. Chen, J. Am. Chem. Soc., 2016, 138, 3926–3932 CrossRef CAS PubMed.
- J. Wang, D. Lin, M. Liu, H. Liu, P. Blasco, Z. Sun, Y. C. Cheung, S. Chen and X. Li, J. Am. Chem. Soc., 2021, 143, 12784–12790 CrossRef CAS PubMed.
- (a) J. H. Lakey, E. J. A. Lea, B. A. M. Rudd, H. M. Wright and D. A. A. Hopwood, Microbiology, 1983, 129, 3565–3573 CrossRef CAS PubMed; (b) C. Kempter, D. Kaiser, S. Haag, G. Nicholson, V. Gnau, T. Walk, K. H. Gierling, H. Decker, H. Zahner, G. Jung and J. W. Metzger, Angew. Chem., Int. Ed., 1997, 36, 498–501 CrossRef CAS; (c) Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. M. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175–1187 CrossRef CAS PubMed.
- C. Milne, A. Powell, J. Jim, M. A. Nakeeb, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2006, 128, 11250–11259 CrossRef CAS PubMed.
- D. Chen, K. H. L. Po, P. Blasco, S. Chen and X. Li, Org. Lett., 2020, 22, 4749–4753 CrossRef CAS PubMed.
- M. Diamandas, R. Moreira and S. D. Taylor, Org. Lett., 2021, 23, 3048–3052 CrossRef CAS PubMed.
- C. Liang, M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Tetrahedron Lett., 2017, 58, 2325–2329 CrossRef CAS.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |