
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of indole derivatives as prevalent moieties present in selected alkaloids
Majid M. Heravi
 *,
Zahra Amiri
,
Kosar Kafshdarzadeh
and
Vahideh Zadsirjan
*
*,
Zahra Amiri
,
Kosar Kafshdarzadeh
and
Vahideh Zadsirjan
*
Department of Chemistry, School of Physics and Chemistry, Alzahra University, Vanak, Tehran, Iran. E-mail: mmh1331@yahoo.com; mmheravi@alzahra.ac.ir; z_zadsirjan@yahoo.com; Fax: +98 2188041344; Tel: +98 9121329147
First published on 15th October 2021
Abstract
Indoles are a significant heterocyclic system in natural products and drugs. They are important types of molecules and natural products and play a main role in cell biology. The application of indole derivatives as biologically active compounds for the treatment of cancer cells, microbes, and different types of disorders in the human body has attracted increasing attention in recent years. Indoles, both natural and synthetic, show various biologically vital properties. Owing to the importance of this significant ring system, the investigation of novel methods of synthesis have attracted the attention of the chemical community. In this review, we aim to highlight the construction of indoles as a moiety in selected alkaloids.
 Majid M. Heravi | Majid M. Heravi was born in 1952 in Mashhad, Iran. He received his BSc degree from the National University of Iran in 1975, his MSc and PhD degrees from Salford University, England, in 1977 and 1980. He started his career as a research fellow in Daroupakhsh in 1981 and joined as an assistant professor at Ferdowsi University of Mashhad. In 1999 he moved to Alzahra University of Tehran. He has so far published 880 ISI cited papers including 12 chapters in Advances in heterocyclic chemistry and one chapter in Alkaloids and also three books (published by Elsevier). |
 Zahra Amiri | Zahra Amiri was born in Lorestan, Iran and received her BSc degree in Pure Chemistry from Lorestan University. Currently, she is MSc student in Organic Chemistry at Alzahra University under the supervision of Professor Majid M. Heravi. Her research interests focus on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Kosar Kafshdarzadeh | Kosar Kafshdarzadeh was born in 1995 in Karaj, Iran. She received her BSc degree in Applied Chemistry from University of Tehran in 2018. Since 2018, she is MSc student in Organic Chemistry at Alzahra University under the supervision of Professor Majid M. Heravi. Her research focuses on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. |
 Vahideh Zadsirjan | Vahideh Zadsirjan was born in 1979 in Shiraz. She received her BSc in Pure Chemistry from Kharazmi University in 2002, and her MSc in 2007 and PhD degrees in organic chemistry in 2016 under the supervision of Professor Majid M. Heravi from Alzahra University Tehran, Iran. Her research is focused on heterocyclic chemistry, catalysis, organic methodology and green synthetic organic chemistry. He has so far published 80 ISI cited papers including one chapter in Advances in heterocyclic chemistry book series published by Elsevier) and one chapter in Alkaloids, and also two books (published by Elsevier). |
1 . Introduction
Indoles are a significant type of heterocycle as they are found in proteins in the form of amino acids, such as tryptophan. They are also present in several drugs, such as indomethacin and the notorious LSD, and several plants such as strychnine.1 The incorporation of an indole core, a biologically known pharmacophore in medicinal molecules, means it is a useful heterocyclic that can bear a number of biological properties.2 Compounds containing the indole nucleus exhibit several different biological properties, including anti-cancer, anti-fungal, anti-HIV, anti-inflammatory, anti-viral, anti-tubercular, anti-microbial,3 anti-hypertensive,2 and anti-diabetic activities,4 and also photochemotherapeutic properties.5Natural products have been renowned as the source of several active ingredients of medicines. The currently approved natural-product-based drugs have been reported broadly in previous reviews.6–8 They contain compounds from plants (such as huperzine, elliptinium and galantamine), animals (ziconotide and exenatide), microbes (daptomycin) and also synthetic or semi-synthetic compounds that rely on naturally occurring compounds (for example everolimus, micafungin, telithromycin, caspofungin and tigecycline). They have various therapeutic indications, for example, they have anti-diabetic, anti-infective, and anti-cancer properties and so on.
Alkaloids occur as secondary metabolites in plants. They are recognized as nitrogen-containing natural biologically active compounds.9 Chemically, alkaloids are a class of nitrogen-containing compounds, which may contain one or more nitrogen atoms (in heterocyclic rings). In general, alkaloids are basic in nature and are typically obtained from plant sources. There are numerous commercially available drugs available, that are alkaloid based in nature.10
Indole alkaloids contain indoles that are bicyclic in structure, comprising a six membered benzene ring fused to a five-membered nitrogen bearing pyrrole ring. This pyrrole ring has a nitrogen atom, which results in the basic properties of indole alkaloids, making them pharmacologically active.11
Indole alkaloids are broadly distributed in plants belonging to the families of Loganiaceae, Apocynaceae, Nyssaceae and Rubiaceae. Significant indole alkaloids that have been extracted from plants include the anti-hypertensive drug, reserpine from Rauwolfia serpentine12 and also the potent anti-tumor drugs, vincristine and vinblastine, obtained from Catharanthus roseus.
Various indole alkaloids exert significant pharmacological properties, but quite diverse influences can be attained even from alkaloids of one genus, for example the Strychnos alkaloid strychnine can strongly affect muscle contraction, whereas the toxiferines serve as muscle relaxants.13
The indole unit is a near-ubiquitous component of biologically potent naturally occurring compounds. For instance, melatonin (1) is a hormone known in plants, animals, and microbes, in which the difference in duration of melatonin production each day, is used as a seasonal clock in animals.14 Tryptophan (2), a vital amino acid, partakes in various critical biological processes.15 Serotonin or 5-hydroxytryptamine (5-HT) (3), which is biochemically obtained from tryptophan, is known as a neurotransmitter and is found in all bilateral animals.16
In addition, the indole unit is recognized as one of the most significant moieties for drug discovery, and it has attracted the attention of researchers for generations.17 Reserpine (4), an indole alkaloid, is utilized in the treatment of high blood pressure and also in the treatment of severe agitation in patients that have mental disorders.18 Ajmalicine (5), an indole alkaloid which is present in various plants, is an anti-hypertensive drug that is utilized for the treatment of high blood pressure.19 Vinblastine (6) is applied for the treatment of various kinds of cancer, such as Kaposi's sarcoma, Hodgkin's disease, non-Hodgkin's lymphoma, and testicular or breast cancer (Fig. 1).20
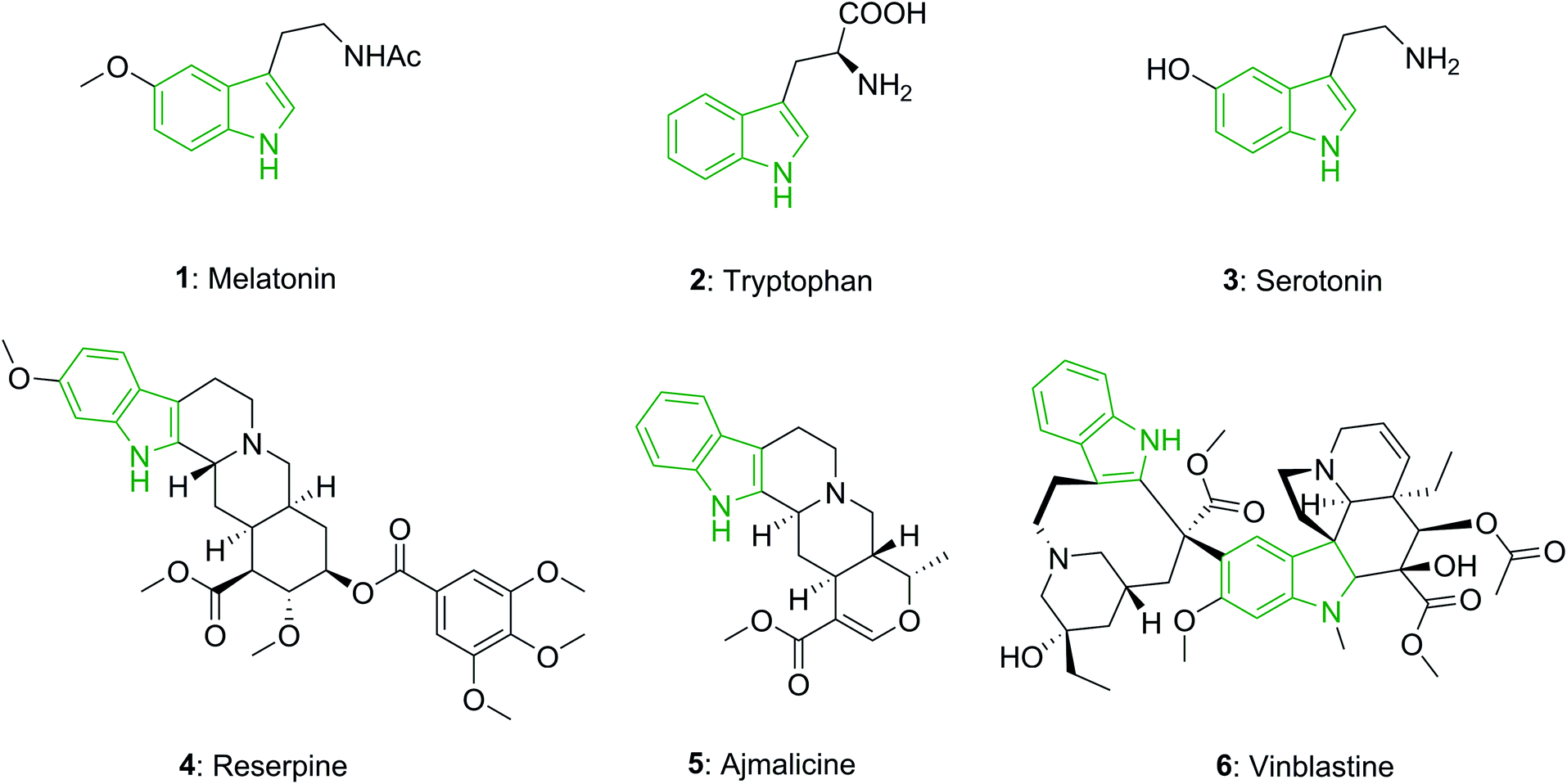 | ||
| Fig. 1 The structures of natural products and drugs that possess indole moieties. | ||
Various methods have been reported for the synthesis of indoles, for example modern versions of classical synthesis methods (named reactions along with indole synthesis) such as: Bartoli indole synthesis,21 Hemetsberger indole synthesis,22 Bischler indole synthesis,23 Julia indole synthesis,24 Larock indole synthesis,25 Nenitzescu indole synthesis,26 Madelung indole synthesis,27 Reissert indole synthesis28 and the most important one, Fischer indole synthesis.29 Other methods involve the transformation of heterocycles,30 the conversion of indolines into indoles,31 the synthesis of o-alkynylanilines,32 the reduction of oxindoles to indoles,33 synthetic methods using arynes,34 the reductive cyclization of nitrobenzene derivatives35 and catalysis using N-heterocyclic carbenes.36
Owing to the importance of the indole as a scaffold in natural products and biologically active compounds, a plethora of reviews and several chapters have been published in this field.37–64 In a continuation of our interest in heterocyclic chemistry65–67 and the applications of named reactions in the total synthesis of natural products,68–75 in this review we aim to highlight the synthesis of indoles as a moiety in selected alkaloids.
2. Synthesis of indole moieties in alkaloids via named reactions
2.1. Fischer indole synthesis
Murrayanine is the main compound extracted from Murraya Koenigii in 1965 by Chakraborty. Murrayanine shows various anti-mycobacterial, anti-oxidant, and anti-fungal activities. Murraya Koenigii Spreng, an aromatic plant belonging to the family Rutaceae, is usually identified as the curry leaf tree. Notably, it has been utilized in sub-tropical and tropical Asia as a folk medicine.76 This tonic plant has been employed for the treatment of different disease conditions77 and has been demonstrated to have a potential role as a remedy for cancer78 and also inflammation. Moreover, the leaf extracts of M. Koenigii are broadly utilized as having anti-fungal,79 anti-diabetic,80 anti-inflammatory81 and anti-oxidant properties.82 In 1968, Chakraborty and co-workers reported83 the total synthesis of murrayanine (12). The total synthesis of murrayanine (12) started with the Japp–Klingemann reaction of phenyldiazonium chloride (7) and 2-hydroxymethylene-5-methylcyclohexanone (8) affording hydrazine 9. Then, the Fischer indole synthesis of hydrazine 9 using HOAc/HCl under reflux gives 1-oxo-3-methyl-1,2,3,4-tetrahydrocarbazole (10). The latter, after three synthetic steps, gives benzylic alcohol 11, which was oxidized using manganese dioxide in carbon tetrachloride to afford murrayanine (12) (Scheme 1).83 In addition, some alternative methods for the synthesis of murrayanine (12), including some with better overall yields, have been reported.84–89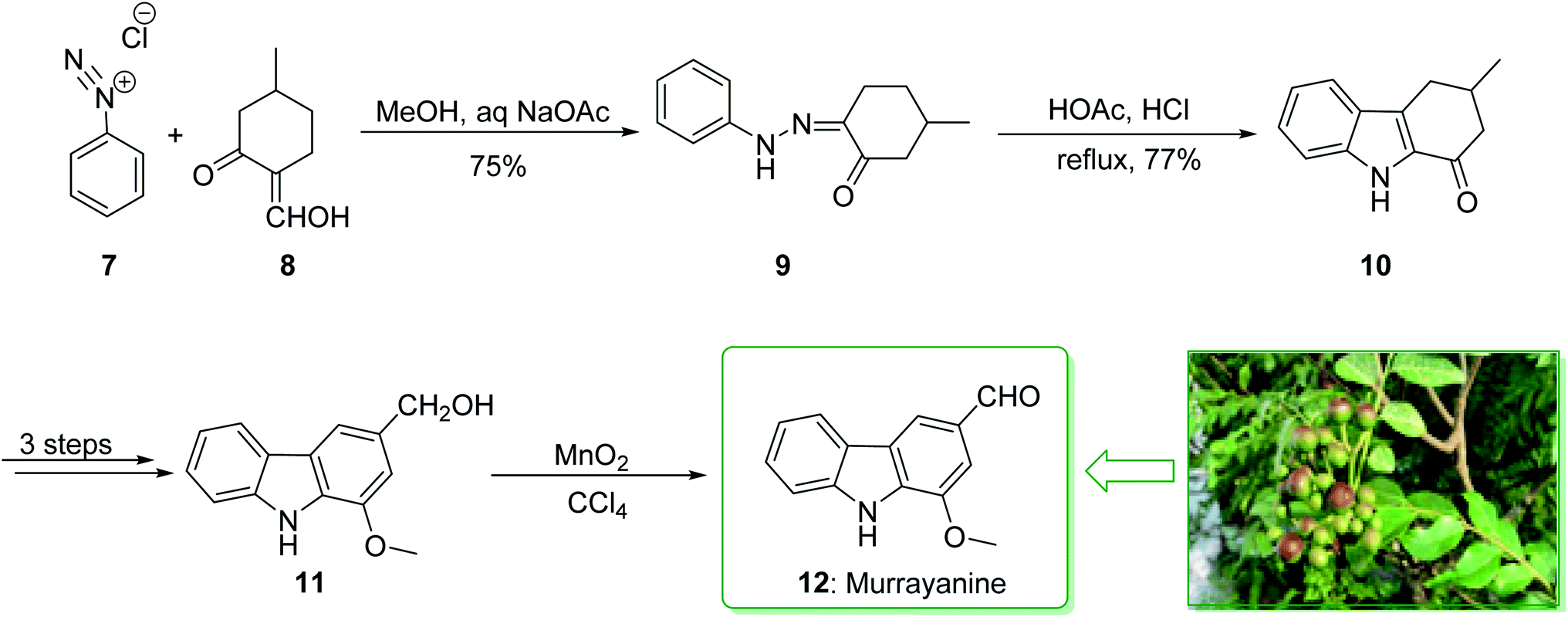 | ||
| Scheme 1 Total synthesis of murrayanine (12). | ||
Pyrano[3,2-a]carbazole alkaloids are very stimulating owing to their structural aspects, and also because of their valuable biological properties.90–94 In 1968, Chakraborty et al. isolated murrayacine (18) from two different natural sources including Clausena heptaphylla95 and Murraya koenigii.96 The total synthesis of murrayacine (18) was achieved and reported by Chakraborty et al. in 1973.97 Using this method, the total synthesis of murrayacine (18) commenced with the reaction between 4-hydroxymethyl-3-hydroxyaniline (13) and formylcyclohexanone (14) via the Japp–Klingemann reaction, which afforded cyclohexane-1,2-dione 4-hydroxymethyl-3-hydroxyphenylhydrazone (15). The latter, through indolization in the presence of a mixture of glacial HOAc and HCl, provided the indole-2-hydroxy-3-hydroxymethyl-8-oxo-5,6,7,8-tetrahydrocarbazole (16). The latter afforded chromenoindole 17 after six steps. Finally, oxidation of the chromenoindole 17 produced the natural product murrayacine (18) (Scheme 2).97 In addition, some alternative synthesis methods for murrayacine (18), some of which have better overall yields, have been reported.98–100
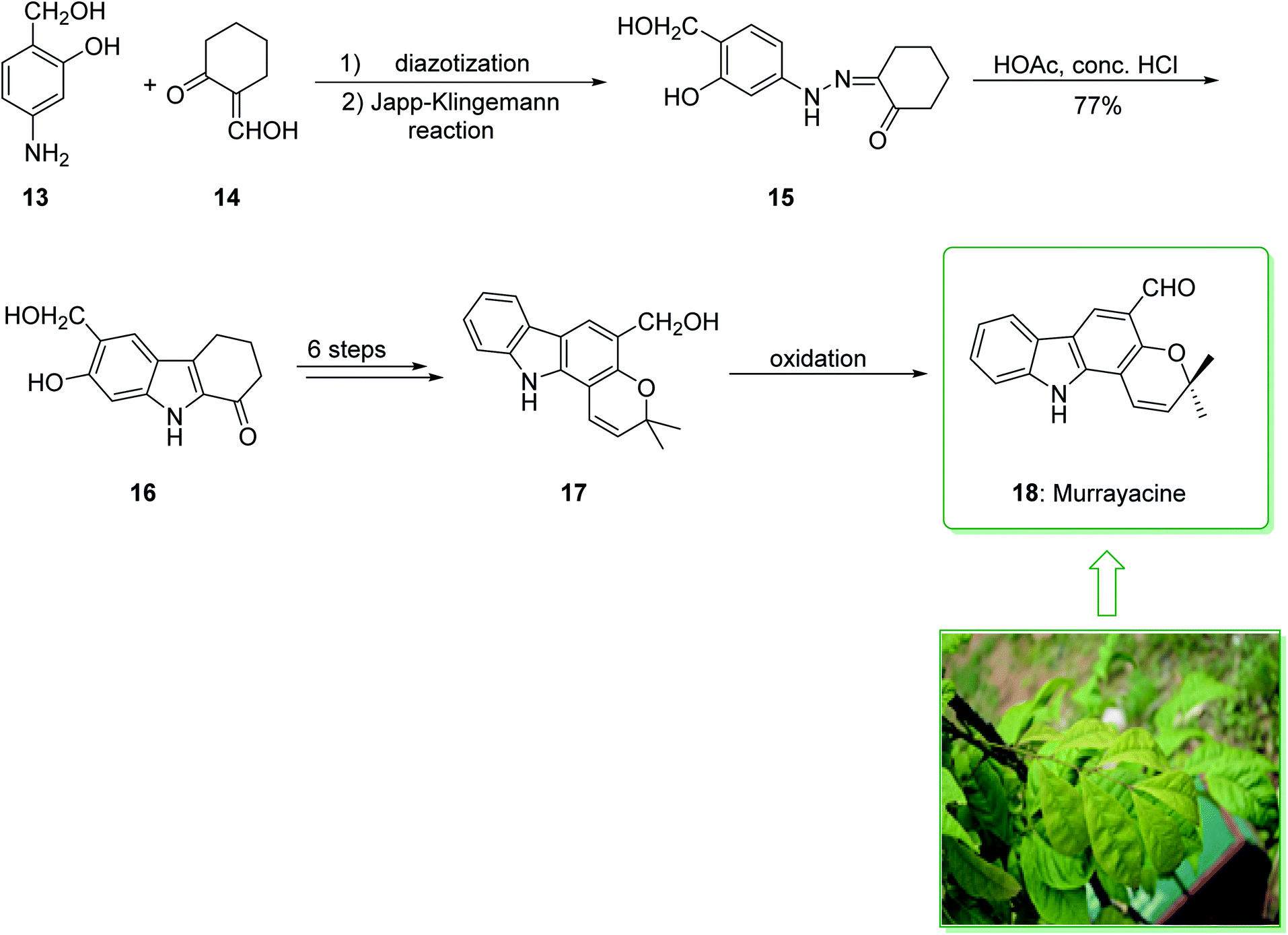 | ||
| Scheme 2 Total synthesis of murrayacine (18). | ||
Evodia rutaecarpa has been used in Chinese medical practice for a long time has been utilized for the treatment of inflammation-related symptoms. Rutaecarpine is an alkaloid extracted from Evodia rutaecarpa.101 Investigations have demonstrated that this anti-inflammatory function is owed to its component rutaecarpine, which shows potent COX-2 inhibited activity. Moreover, rutaecarpine has other functions, for example, it has analgesic, vasorelaxing, anti-anoxic, anti-platelet and cytotoxic properties.102 In 1971, vasicolinone (26), an quinazoline alkaloid, was extracted from the leaves of the Indian plant Adhatoda uasica Nees (Acanthaceae).103 Both kinds of alkaloids have attracted significant attention because of their pharmacological properties.104,105 Rutaecarpine is one of the component parts of traditional ancient Chinese herbal medicine.106 In 1992, Hermecz and co-worker achieved and reported106 the total synthesis of rutaecarpine (24) and vasicolinone (26) through Fischer indolization of 3-(phenylhydrazonomethyl)pyrroloquinazolinone (22) under thermal and acidic reaction conditions, respectively. This research group attempted the total synthesis of rutaecarpine (24) and vasicolinone (26). The total syntheses of these two natural products started with deoxyvasicinone (21). Anthranilic acid (19) and 2-pyrrolidone (20) were reacted together via Kametani's method,107 using thionyl chloride (SOCl2) and gave the deoxyvasicinone alkaloid (21) in a high yield (93% yield),108 after two steps this yielded hydrazone 22. A solution of the hydrazone 22 in Dowtherm A was heated to above 160 °C, and the produced rutaecarpine (24) was slowly precipitated, upon recrystallization from dimethylformamide (DMF), the target natural product rutaecarpine (24) was provided in a moderate yield (49%).
In addition, under acidic reaction conditions, upon the protonation of spiroindoleninpyrroloquinazolinone 23 on the indolenine nitrogen, 23 underwent a retrograde aldol reaction to afford the ring-opened pyrroloquinazolinone 25. In the following, after hydrolysis of the formamide substituent, the natural product vasicolinone (26) was quantitatively provided from pyrroloquinazolinone 25 using dimethylation of the amino group with 37% aqueous formaldehyde (HCHO) using sodium cyanoborohydride (NaBH3CN) in acetonitrile at room temperature109 (Scheme 3).106
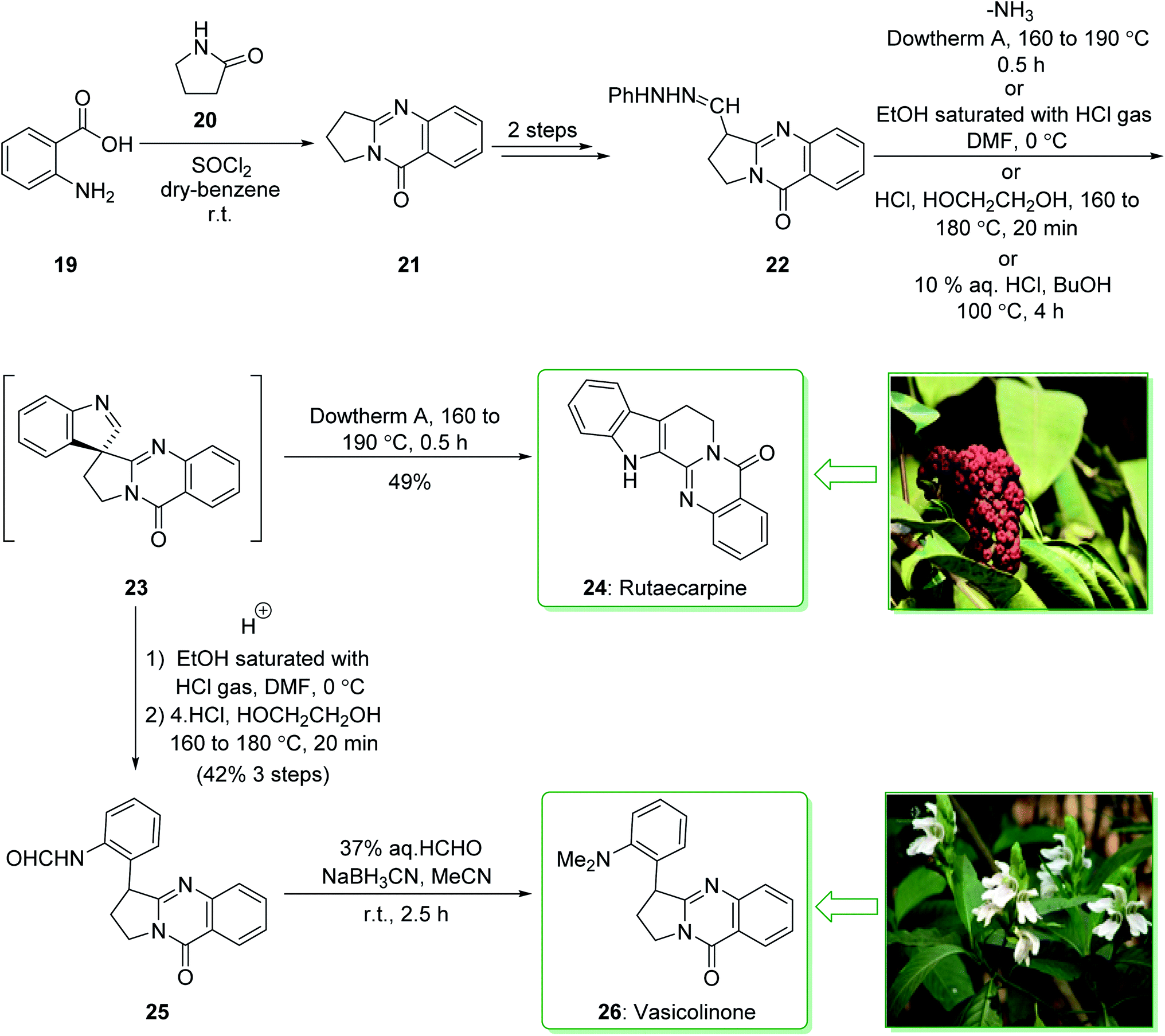 | ||
| Scheme 3 Total synthesis of rutaecarpine (24) and vasicolinone (26). | ||
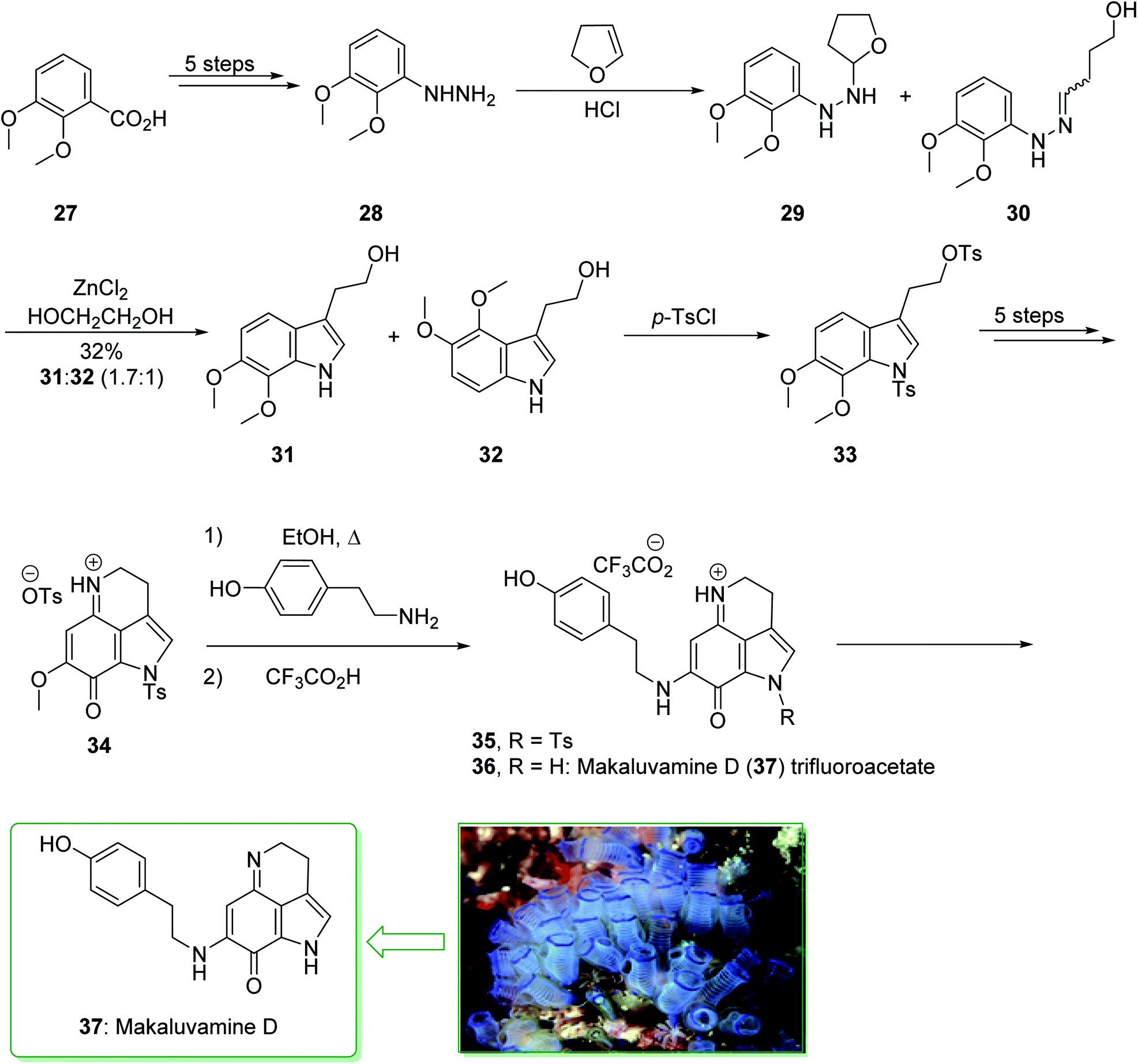
A class of very cytotoxic metabolites that possess the pyrrolo[4,3,2-de]quinoline framework were discovered upon examining the marine sources of anti-neoplastic agents.110 These contain the discorhabdins,111 prianosins,112 isobatzellines, batzellines,113 and damirones,114 all extracted in 1991 from sponges, and also wakayin, isolated from the Fijian ascidian Clauelina sp.115 Other members of the pyrroloiminoquinone family were identified by Ireland in 1993 from the Fijian sponge Zyzzya cf. marsailis.116 These materials, called makaluvamines, have been known to contain outstanding and potentially significant biological properties, for example inhibition of the function of mammalian topoisomerase II. Moreover, they show strong in vitro cytotoxicity towards the human colon tumor cell line HCT 116. The synthesis of the pyrrolo[4,3,2-de]quinoline system, that is typical of a class of marine alkaloids that contains the discorhabdins, prianosins, and also other anti-neoplastic agents, was demonstrated in 1994 by White et al.117 This method is represented in the total synthesis of makaluvamine D (37), a topoisomerase II inhibitor extracted from the sponge Zyzzya cf. marsailis. The total synthesis of makaluvamine D (37) starts from 2,3-dimethoxybenzoic acid (27) and within five steps affords (3,4-dimethoxyphenyl)hydrazine (28). The latter was reacted with dihydrofuran118 and provided a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :l mixture of the tetrahydrofuran (THF) 29 and the hydrazone 30 (as an E/Z mixture). The mixture was exposed to Fischer indolization reaction conditions in the presence of ZnCl2 and gave the desired tryptophol 31, accompanied by the 4,5-dimethoxyindole derivative 32. Next, the elimination of 32 from the corresponding product 31 proved difficult, and to prevent losses during purification, the mixture of 31 and 32 was directly sulfonated using excess p-toluenesulfonyl chloride. The bissulfonyl derivative 33 was provided in a satisfactory yield. The latter afforded tosylate 34 after five steps. In the following, the condensation reaction of 34 with tyramine was correspondingly productive, affording 35. In addition, further prolonged exposure to tyramine under reflux in EtOH eliminated the N-tosyl substituent from 35 and resulted in makaluvamine D (37) (Scheme 4).117
:l mixture of the tetrahydrofuran (THF) 29 and the hydrazone 30 (as an E/Z mixture). The mixture was exposed to Fischer indolization reaction conditions in the presence of ZnCl2 and gave the desired tryptophol 31, accompanied by the 4,5-dimethoxyindole derivative 32. Next, the elimination of 32 from the corresponding product 31 proved difficult, and to prevent losses during purification, the mixture of 31 and 32 was directly sulfonated using excess p-toluenesulfonyl chloride. The bissulfonyl derivative 33 was provided in a satisfactory yield. The latter afforded tosylate 34 after five steps. In the following, the condensation reaction of 34 with tyramine was correspondingly productive, affording 35. In addition, further prolonged exposure to tyramine under reflux in EtOH eliminated the N-tosyl substituent from 35 and resulted in makaluvamine D (37) (Scheme 4).117
 | ||
| Scheme 4 Total synthesis of makaluvamine D (37). | ||
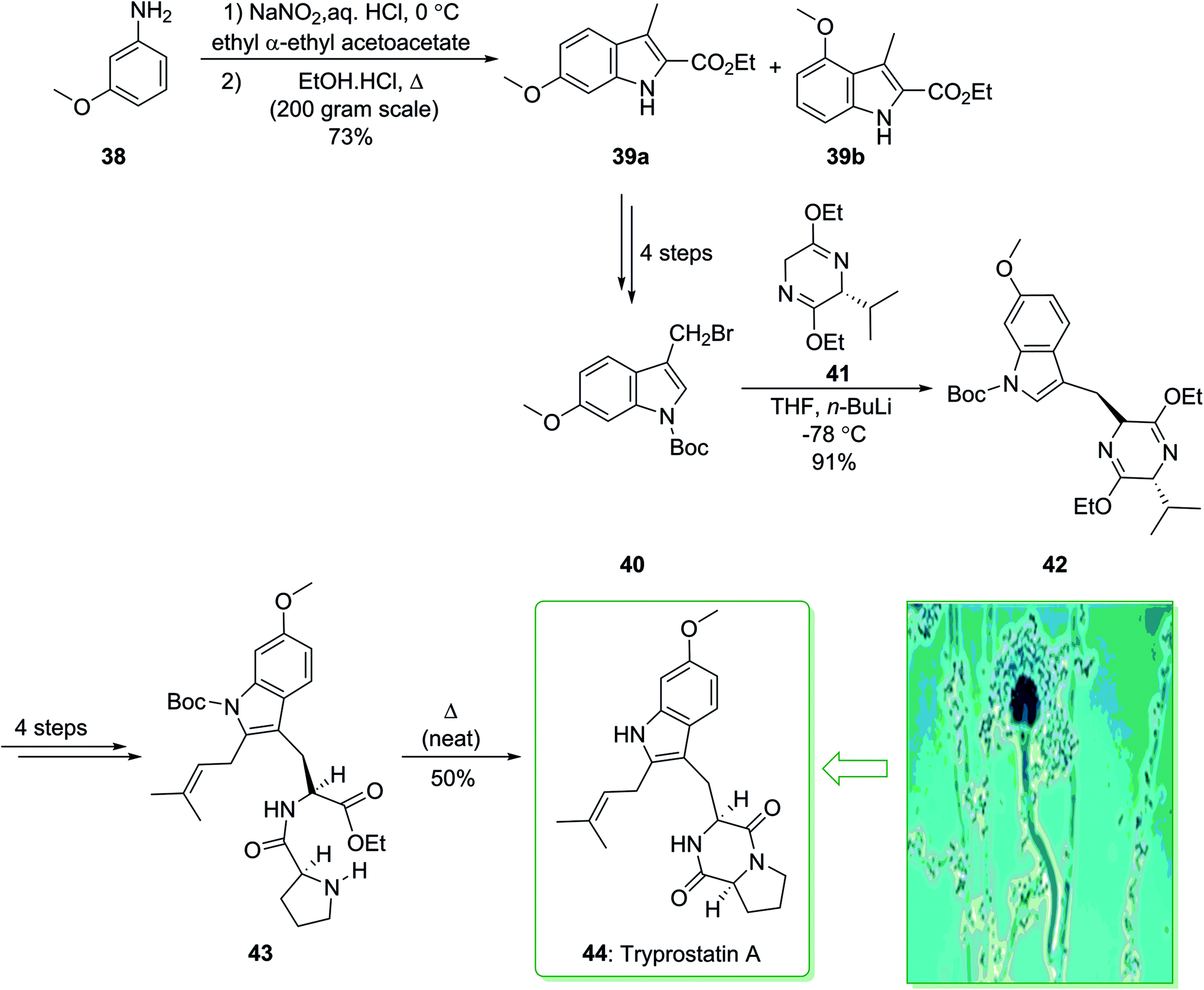
In 1995, Osada and co-workers isolated tryprostatins A and B from the fermentation broth of Aspergillus fumigatus BM939.119 Tryprostatin A (44) possesses promising attributes, as it selectively stops the cell cycle in tsFT210 cells in the mitotic phase.120 Stimulating biological activity, along with a seemingly simple structure, has motivated the synthetic community, and various total syntheses have been demonstrated.121,122 In 1997, Cook and co-workers achieved and reported123 the enantiospecific total synthesis of tryprostatin A (44) through a regiospecific bromination method as a key step. The total synthesis of tryprostatin A (44) started with the reaction of m-anisidine (38) with NaNO2 and concentrated aqueous hydrochloric acid, followed by the addition of the anion of ethyl α-ethylacetoacetate, and the Japp–Klingmann azo-ester intermediate was obtained. Once this intermediate was heated in a solution of ethanolic HCl, a mixture of ethyl 6-methoxy-3-methylindole-2-carboxylate 39a and its 4-methoxy isomer 39b (10:1) was provided. The corresponding 6-methoxyindole isomer 39a was separated from the mixture through facile crystallization and after four synthetic steps gave 3-(bromomethyl)indole (40). The latter was coupled with the anion of the Schollkopf chiral auxiliary 41 (obtained from D-valine),124 and the corresponding trans diastereomer 42 was obtained as the sole product. After four steps the latter yielded dipeptide 43, which was heated at 160 °C (neat) and gave tryprostatin A (44) (Scheme 5).123
 | ||
| Scheme 5 Total synthesis of tryprostatin A (44). | ||
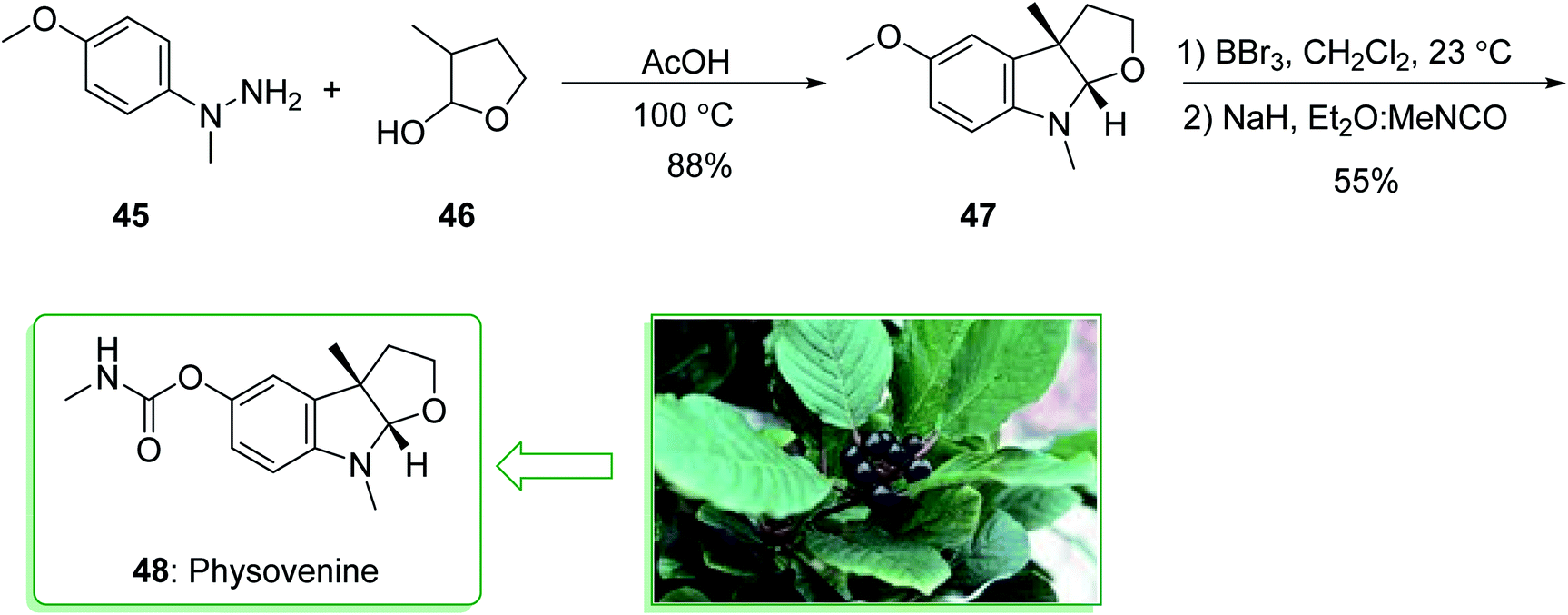
The acetylcholinesterase inhibitors physovenine (48) and physostigmin125,126 contain basic furo- and pyrrolidinoindoline scaffolds, respectively. Pyrrolidinoindoline natural products, which contain the C-3a functionalized hexahydropyrrolo[2,3-b]indole ring scaffold, have been extracted from various natural sources and show diverse biological properties.127 The Physostigma genus (Fabaceae) produces a class of indole alkaloids involving physostigmine as the major basic component. Physovenine, one of the minor alkaloids of a similar plant, and a C-ring oxygenated analogue of physostigmine, was in turn structurally identified in 1964.128 Two of the alkaloids, (−)-physostigmine and (−)-physovenine exhibited strong anti-acetylcholinesterase (AChE) properties upon examination in vitro, and could be used to measure the inhibition of AChE by human erythrocytes.129 In 2010, Garg et al. demonstrated a convergent approach for the construction of the fused indoline ring scaffold found in a multitude of bioactive compounds. This approach includes the condensation reaction of hydrazines with latent aldehydes to eventually provide indoline-comprising products by using an interrupted Fischer indolization sequence.130
This research group tried to demonstrate the formal synthesis of physovenine (48). Based on this approach, the formal synthesis of physovenine (48) was commenced from the Fischer indole synthesis of hydrazine 45 and lactol 46 in HAOc, which afforded furoindoline 47. The latter, after two steps (in a 55% yield), was transformed to physovenine (48)131 (Scheme 6).130
 | ||
| Scheme 6 Formal synthesis of physovenine (48). | ||
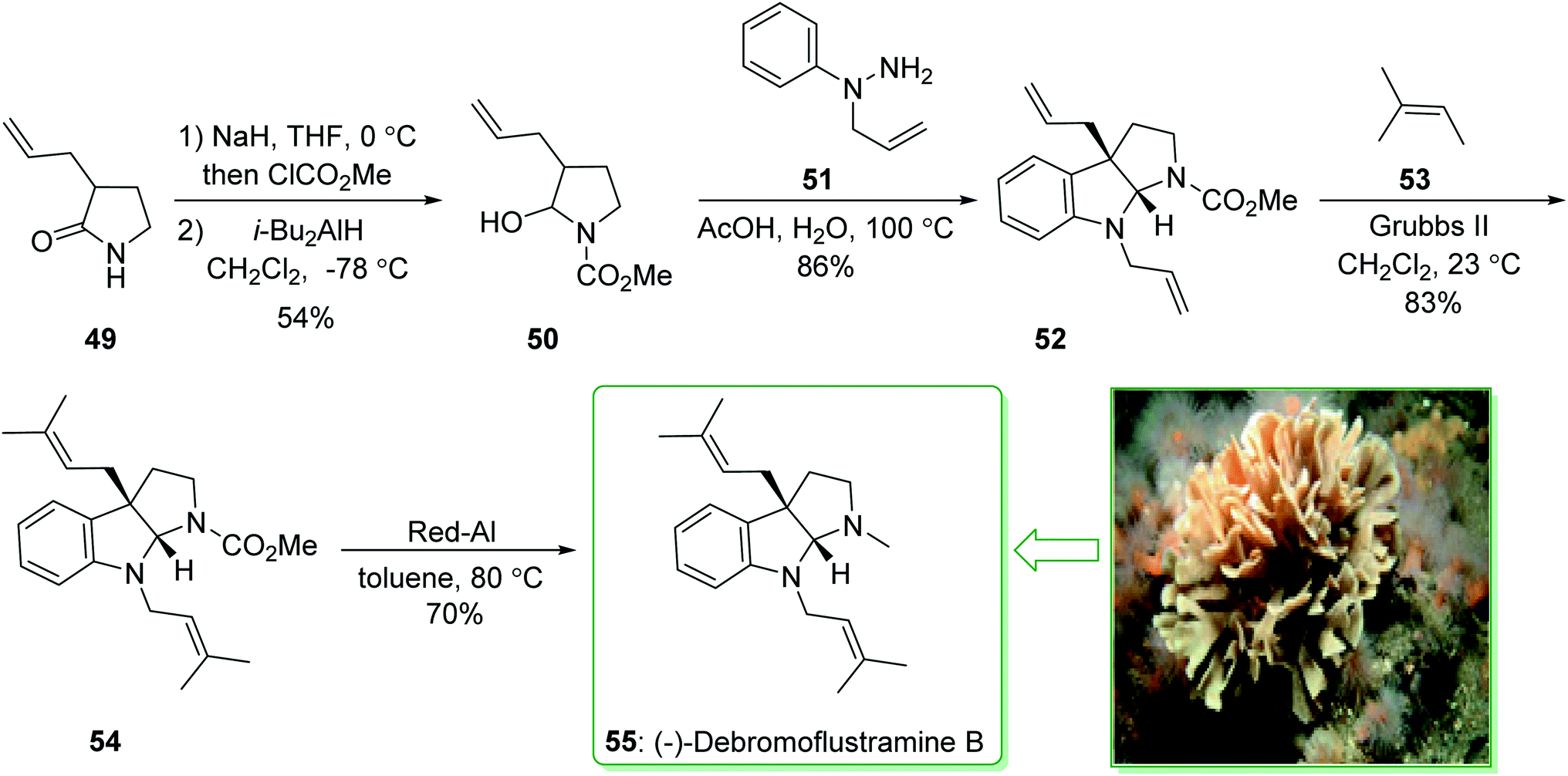
The formal total synthesis of (−)-debromoflustramine B (55) starting from pyrrolidinone 49132 afforded hemiaminal 50 in two steps. The latter with 1-allyl-1-phenylhydrazine (51) in acetic acid/water at 100 °C assisted the key condensation/sigmatropic rearrangement to provide bis(allylated)pyrrolidinoindoline 52. Next, the reaction of compound 52 with 2-methyl-2-butene (53) using Grubbs' second generation catalyst yielded the bis(prenylated) derivative 54, which was transformed into (−)-debromoflustramine B (55) through reduction with Red-Al. It is worth mentioning that compound 54 has formerly been transformed into debromoflustramine B (55) via reduction using lithium aluminum hydride133 (Scheme 7).130
 | ||
| Scheme 7 Formal synthesis of (−)-debromoflustramine B (55). | ||
Colegate and co-workers isolated a furanobisindole alkaloid, called phalarine (63) from Phalaris coerulescens.134 Its structure was identified using a distinctive tricyclic propeller unit building block fused to a piperidine core and a multiply substituted indole scaffold via successive tetrafunctionalized stereogenic centers at C4a and C9a. However, the biological properties of 63 have not been previously reported, and the typical structure attracted the attention of various synthetic chemists,135,136 and their attempts resulted in the seminal enantioselective total synthesis of 63 by Danishefsky137 and Chen.138 In 2010, Danishefsky et al. demonstrated136 the Pictet–Spengler reaction for C2-aryl indoles, and effectively separated the elusive azaspiroindolenine intermediate of the Pictet–Spengler reaction. Total synthesis of phalarine (63) was commenced from β-carboline 56 and after 11 steps afforded the intermediate 57. The latter, using p-toluenesulfonic acid in toluene, gave the corresponding indole product 58 (5% yield), benzofuro[3,2-b]indole 59 (17% yield), benzofuro[3,2-b]indol-8-amine 60 (18% yield), and diindole-2-carboxylate 61 (9% yield). In the following, compound 60 gave diindole 62 after three steps. Upon installation of the gramine side chain on 62 using N,N-dimethylmethylene ammonium chloride in acetic acid, and elimination of the tosyl masking group, phalarine (63) was afforded (Scheme 8).136
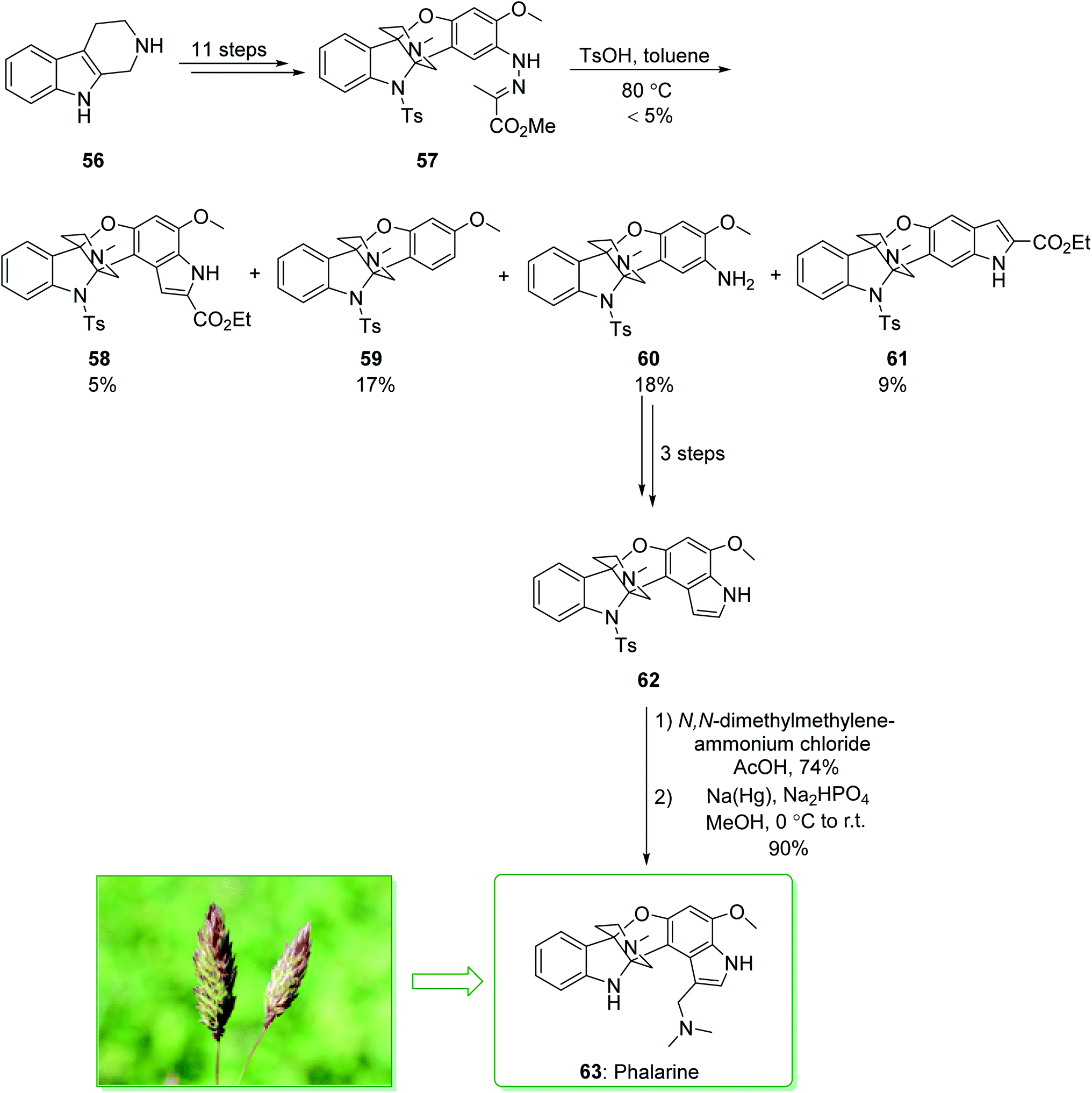 | ||
| Scheme 8 Total synthesis of phalarine (63). | ||
The natural products of lycogarubin C (67) and lycogalic acid (68) were identified in 1994. They were extracted individually by Steglich139 and Akazawa140 from Lycogala epidendrum, a slime mold. Moreover, lycogalic acid, referred to as chromopyrrolic acid (CPA),141–143 has been identified as the usual intermediate in the biosynthesis of the indolo[2,3-a]carbazole alkaloids containing staurosporine and rebeccamycin; these show a broad range of properties as inhibitors of protein kinases and also topoisomerase I.91
Lycogarubin C (67) and lycogalic acid A (68) are naturally occurring key marine compounds that are utilized in investigations into the inhibitor of DNA topoisomerase-I. In 2010, Zhixiang et al. demonstrated that lycogarubin C was synthesized from (but-3-ynyloxy)(tert-butyl)dimethylsilane as a starting precursor in eight steps, through the following key steps: a hetero-/retro-Diels–Alder reaction, the reduction of 1,2-diazine, Swern oxidation, and also Fischer indole synthesis.144
The total synthesis of lycogarubin C (67) and lycogalic acid A (68) was commenced from (but-3-yn-1-yloxy)(tert-butyl)dimethylsilane (64), and after seven synthetic steps, gave 1-(tert-butyl) 2,5-dimethyl 3,4-bis(2-oxoethyl)-1H-pyrrole-1,2,5-tricarboxylate (65). Then, the Fischer indole synthesis of 65 with phenyl hydrazine (66) using PPA in THF under reflux, afforded lycogarubin C (67) in a satisfactory yield (49% yield). Lastly, lycogalic acid A (68) was synthesized from lycogarubin C (67) using potassium hydroxide in THF and H2O under reflux (Scheme 9).144
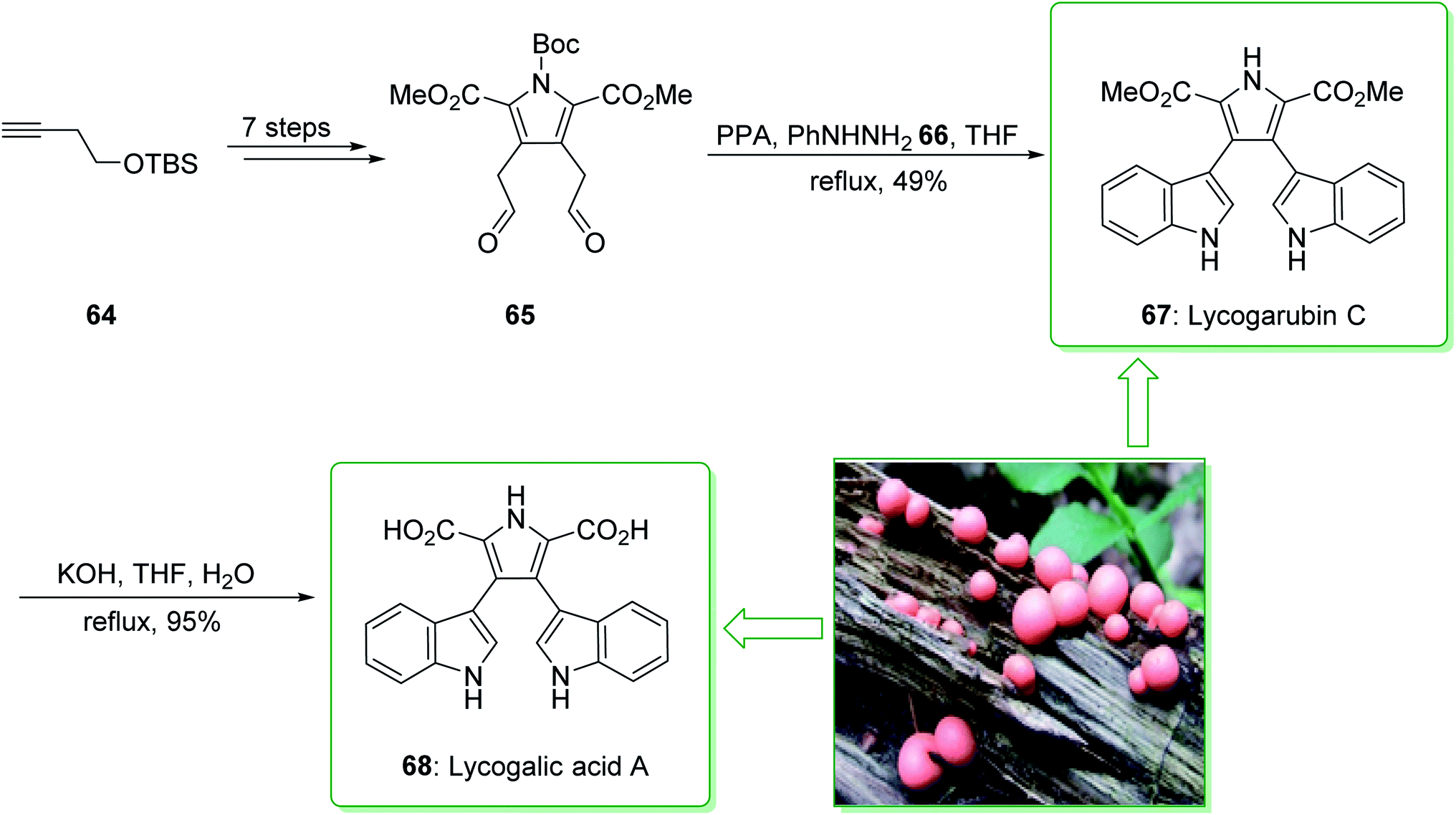 | ||
| Scheme 9 Total synthesis of lycogarubin C (67) and lycogalic acid (68). | ||
Minfiensine (75), an indole alkaloid that possesses important biological properties, such as anti-cancer activities, was initially extracted and isolated by Massiot et al. in 1989 from the African plant Strychnos minfiensis.145,146 In 2011, Zhang et al. demonstrated147 the short total synthesis of (±)-minfiensine. This method involves a Fischer indole synthesis, a Heck alkylation of an intermediate ketone enolate, transformation of a ketone carbonyl into an epoxide, and also conversion of an epoxide into an allylic alcohol. The total synthesis of minfiensine (75) commenced with the Fischer indole synthesis. The condensation reaction of phenylhydrazine (66) and 1,4-cyclohexanedione monoethyleneacetal (69) at ambient temperature followed by heating (at 190 °C for 4.5 h) provided the anticipated indole product 70 in a good yield (89% yield), a further three steps gave the aldehyde 71. Then, the reductive amination of aldehyde 71 using NaBH4 and 2-iodocrotylamine (72) in MeOH at ambient temperature afforded (Z)-2-iodobut-2-en-1-ol (73)148 in a good yield (86%). The latter after five steps afforded the allyl alcohol 74. After hydrolysis of the carbamide group in 74 using NaOH, minfiensine (75) was synthesized in a good yield (82%) (Scheme 10).147
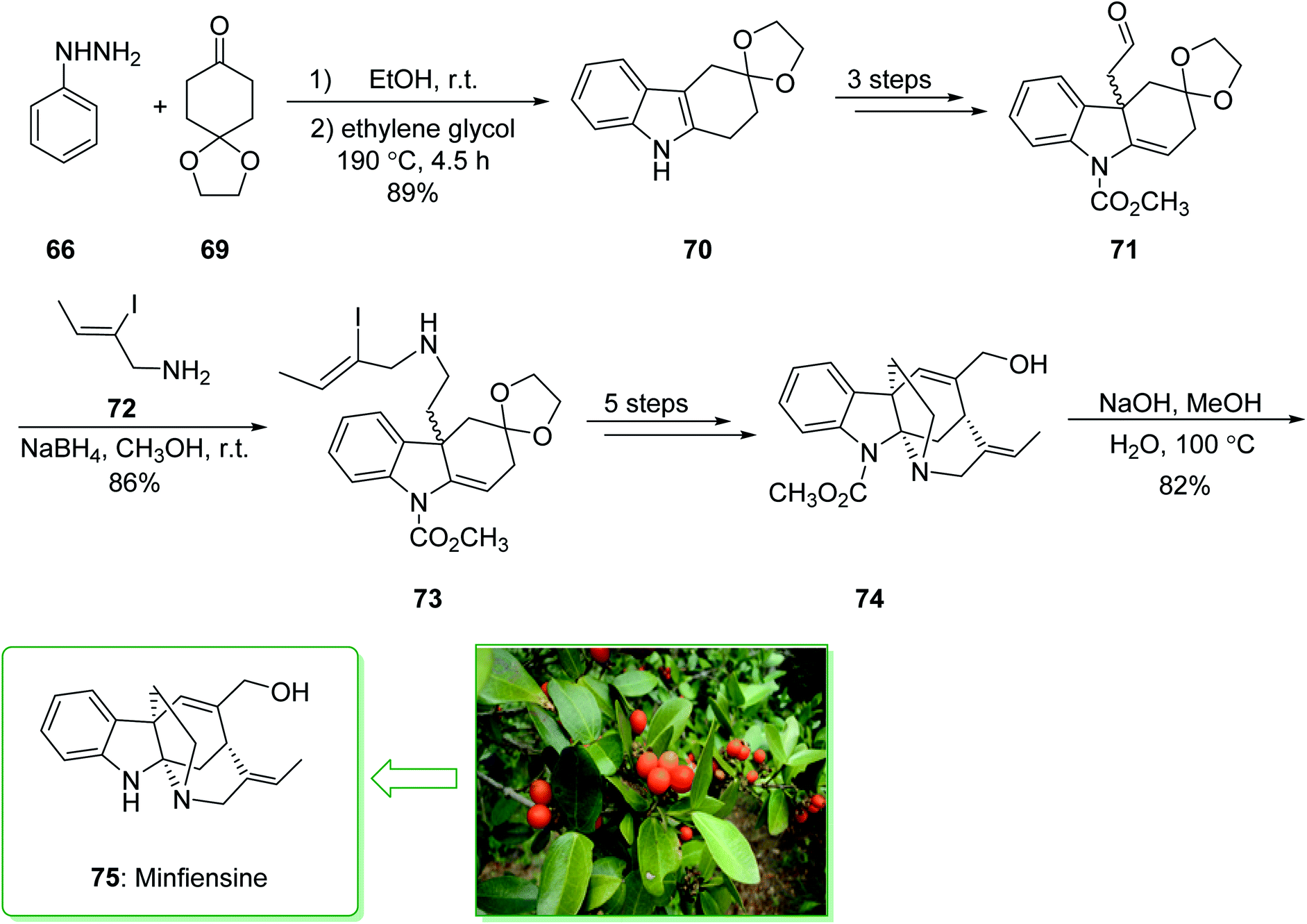 | ||
| Scheme 10 Total synthesis of (±)-minfiensine (75). | ||
(±)-Aspidophylline A (84) was extracted from Malayan Kopsia singapurensis by Kam and co-workers in 2007. It is known to reverse drug resistance in resistant KB cells.149 In 2011, Garg and co-worker reported150 the total synthesis of (±)-aspidophylline A (84), which is one of various complex furoindoline-containing alkaloids. This pathway contains various main conversions, comprising the Heck cyclization to assemble the [3.3.1]-bicyclic motif, as well as a late-stage interrupted Fischer indolization to provide the furoindoline and form the pentacyclic building block natural product. The total synthesis of (±)-aspidophylline A (84) started with the Diels–Alder adduct 78 that was prepared from the thermal Diels–Alder reaction of maleic anhydride (77) and pyridinone 76.151 Bicycle 78 furnished lactone 79 within 15 steps. The Fischer indolization reaction between ketoester 79 and phenylhydrazine using trifluoroacetic acid (TFA) in dichloroethane (DCE) at 40 °C provided the intermediate 81, probably through intermediate 80. In the following, the elimination of the solvent and the addition of potassium carbonate in methanol under heating resulted in lactone methanolysis and cyclization (transition structure 82) and gave pentacycle 83. The elimination of the tosyl masking substituent of 83 and formylation gave (±)-aspidophylline A (84). As a result, the total synthesis of (±)-aspidophylline A (84) was performed in 18 steps from the Diels–Alder adduct 78 (Scheme 11).150
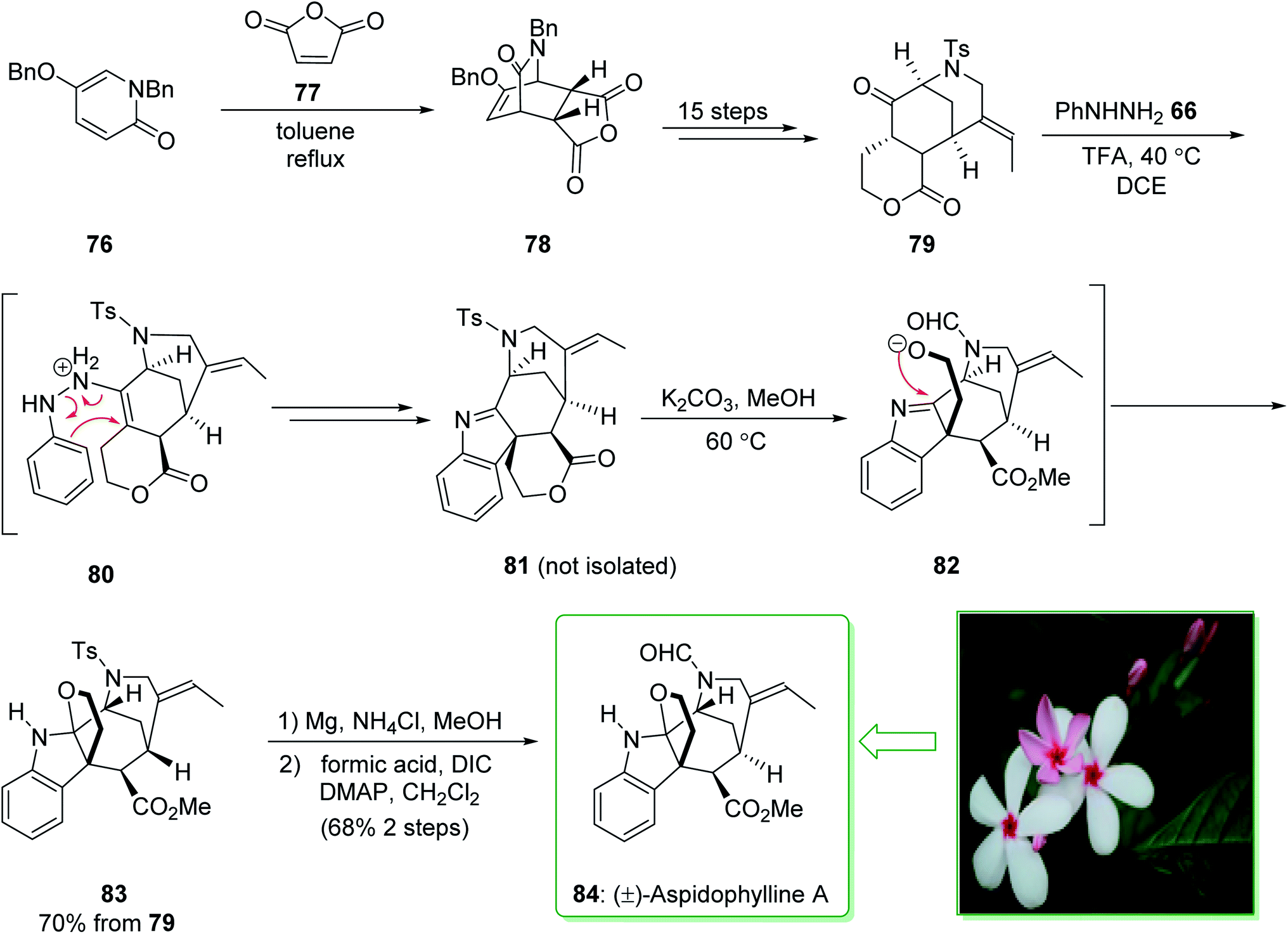 | ||
| Scheme 11 Total synthesis of (±)-aspidophylline A (84). | ||
The scholarisines belong to the family of akuammiline alkaloids.152 Scholarisine A, a monoterpenoid indole alkaloid, having an unprecedented framework with a bridged lactone embedded in a cage like building block, was first extracted in 2008 by Luo et al. from the leaves of Alstonia scholaris.153 The majority of Dengtaiye components are indole alkaloids, in which four major bioactive compounds, comprising vallesamine, scholarisine, 19-epischolarisine, and picrinine show important analgesic, anti-fertility, antibacterial, anti-asthmatic, antitumor and anti-tussive bioactivities.154,155 In 2012, Smith et al. reported156 an effective total synthesis of (+)-scholarisine A (90) through a 20-step reaction. Highlights of the reaction are a reductive cyclization including a nitrile and an epoxide, a modified Fischer indole reaction, a late-stage oxidative lactonization, and an intramolecular cyclization providing the indolenine ring system of (+)-scholarisine A. The total synthesis of (+)-scholarisine A (90) was commenced from bicyclic lactone (−)-85, and after six steps gave the ketone (+)-86. Next, a Fischer synthesis using 1-benzyl-1-phenylhydrazine (87) (pyridine-HCl, 110 °C) gave the masked indole lactone (−)-88. After 12 steps the latter gave indolenine (+)-89. Elimination of the trifluoroacetyl substituent in 89 with a 1:2 mixture of saturated aqueous potassium carbonate and MeOH, followed by aliphatic amine oxidation with iodosobenzene (PHIO) in CH2Cl2, afforded the natural product (+)-scholarisine A (90) (Scheme 12).156
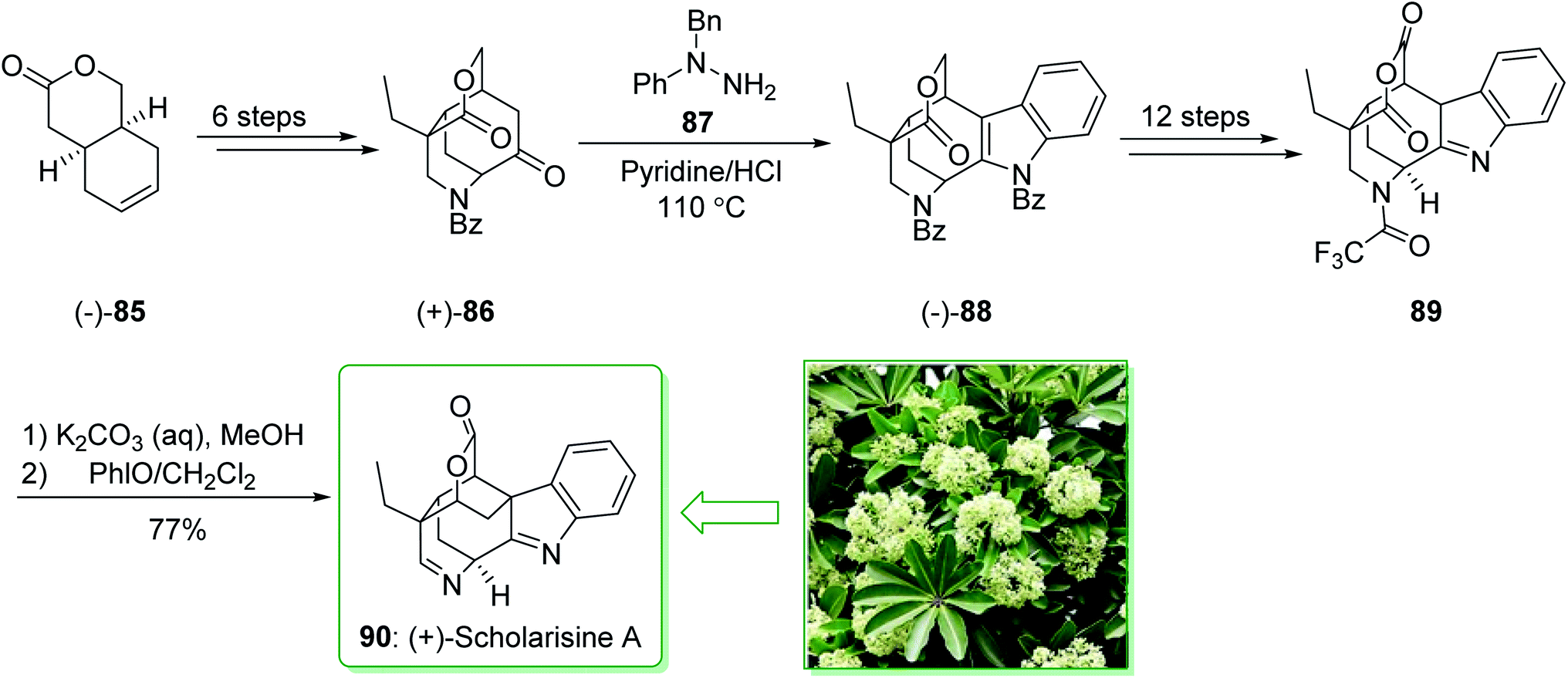 | ||
| Scheme 12 Total synthesis of (+)-scholarisine A (90). | ||
The asymmetric total synthesis of (+)-scholarisine A (90) was achieved and reported in 2013 by Smith et al.157 The key steps involve a novel cyclization, a modified Fischer indolization; an oxidative lactonization and a late-stage cyclization. The total synthesis of (+)-scholarisine A (90) was commenced from the commercially available anhydride 91 and after 12 steps this gave the ketone (+)-86. The latter was reacted with benzyl phenylhydrazine using a slightly acidic medium of pyridine/hydrochloric acid at 110 °C overnight to give the benzyl-protected indole (−)-88 in a good yield (>70%). Next, after 11 steps, the latter gave indolenine 89. In the following, the elimination of the trifluoroacetyl group in 89 was easily accomplished using a mixture of saturated aqueous potassium carbonate and MeOH (1:2) to provide the desired free amine. Finally, oxidation with iodosobenzene (PhIO)158 in CH2Cl2 completed the synthesis of (+)-scholarisine A (90) (Scheme 13).157
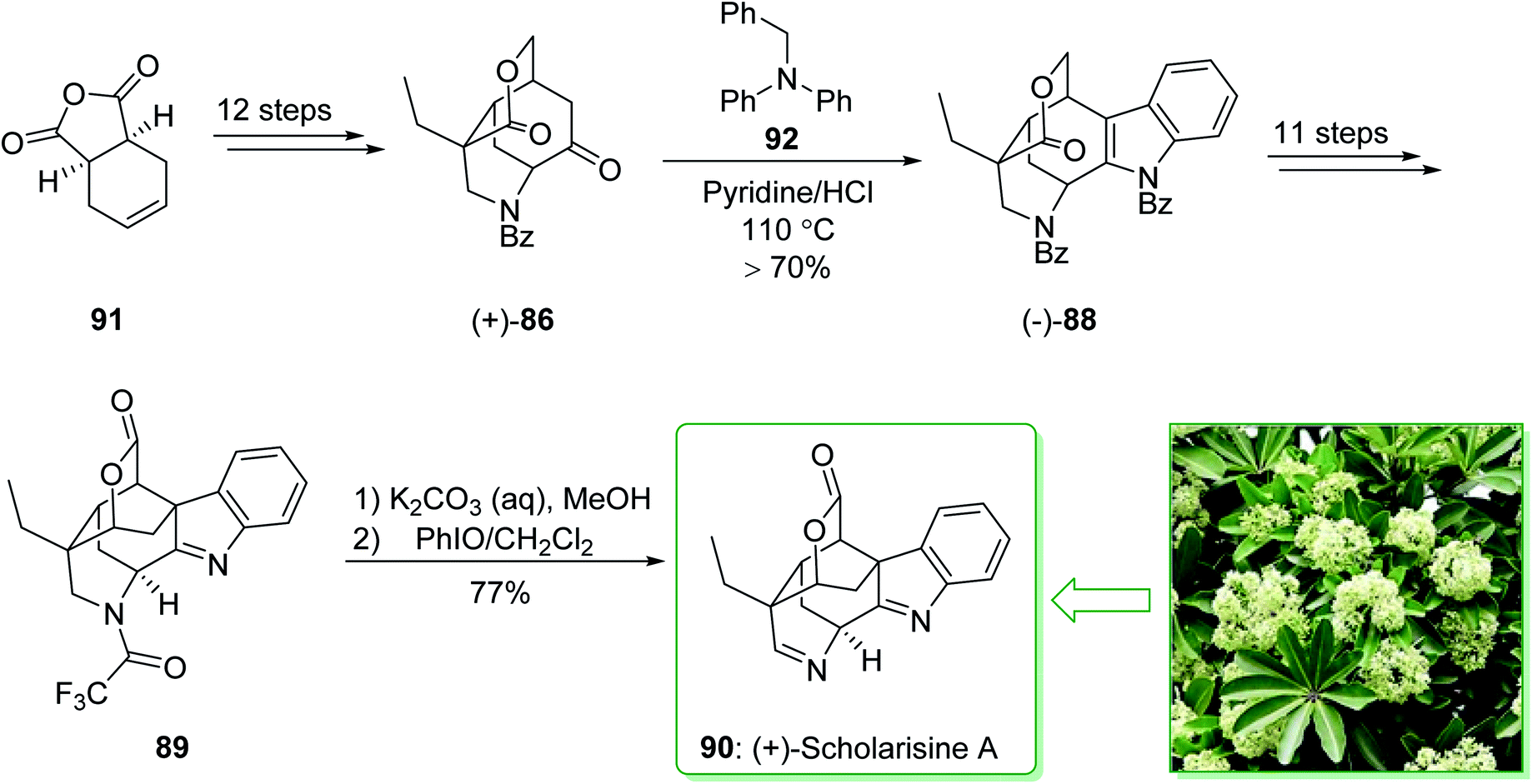 | ||
| Scheme 13 Total synthesis of (+)-scholarisine A (90). | ||
The communesin family of naturally occurring compounds and perophoramidine are prevalent goals for chemical synthesis.159,160 Two indole alkaloids, including perophoramidine161 and communesin,162 that have unique molecular frameworks were extracted in 1993 and 2002 from ascidian Perophora namei and a strain of Penicillium sp., respectively. Both the indole alkaloids contain an analogous polycyclic scaffold comprising two vicinal quaternary centers and having a moderately inverted stereochemistry at the 4-position of the part-saturated quinoline scaffold. It is worth mentioning that perophoramidine 96 is cytotoxic against the HCT116 colon cancer cell line.161 In 2012, Garg et al. demonstrated163 a short strategy for the total synthesis of the communesin alkaloids and perophoramidine. This approach relies on the usage of the interrupted Fischer indolization to construct the tetracyclic indoline unit of the naturally occurring compounds. The synthesis of perophoramidine (96) started with the reaction of hydrazine 45 and N,O-acetal 93 using HOAc in H2O at 75 °C via Fischer indole synthesis to provide the tetracyclic indoline 94. It is worth mentioning that the carbamylated N,O-acetal 93 was used in this approach as it was known to be more easily removed in comparison with the N-Ts group. Next, the tetracyclic indoline 94 afforded imine 95 within two steps as an intermediate for the synthesis of perophoramidine (96) (Scheme 14).163
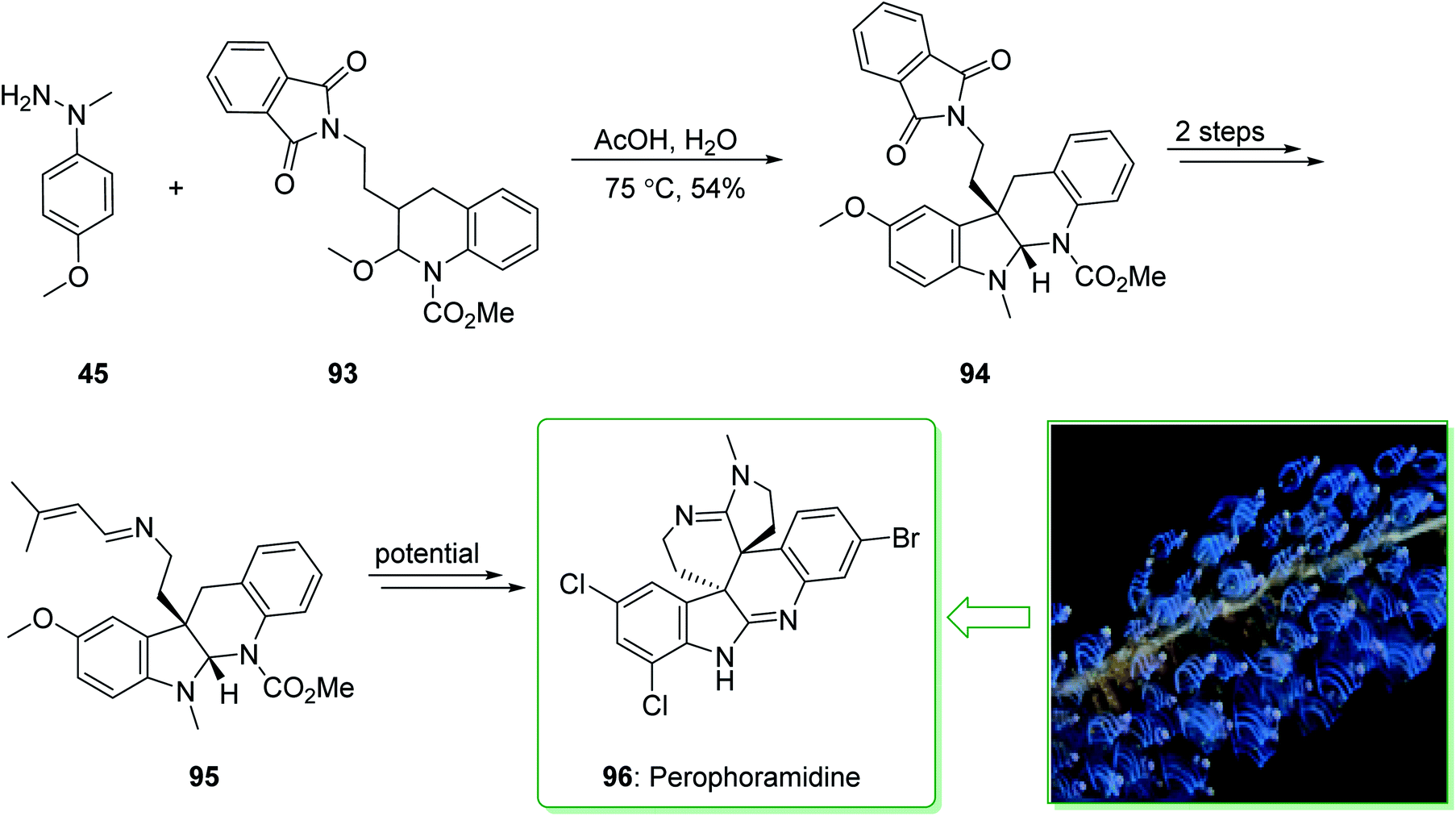 | ||
| Scheme 14 The total synthesis of perophoramidine (96). | ||
Dictyodendrins A–E were extracted from the marine sponge Dictyodendrilla verongiformis collected off the southern Japanese coast in 2003 by Matsunaga and Fusetani.164 These natural products contain an exclusive pyrrolo[2,3-c]carbazole unit, and at least one sulfate substituent in their periphery structure. In addition, these alkaloids display a telomerase inhibitory property. As telomerase is overexpressed in most tumor cell lines,165 telomerase inhibition signifies a unique favorable approach for the establishment of cancer chemotherapy.166 Jia and co-workers in 2014 revealed167 a complete elucidation for the short total synthesis of dictyodendrins E. The total synthesis of dictyodendrins E (105) started from the Ullmann coupling reaction of p-iodoanisole (97) and 1,3-dinitrobenzene (98), and afforded biphenyl 99, after a further three steps the iodide 100 was obtained. Next, the Ullmann coupling reaction of iodide 100 using BocNHNH2 provided the corresponding Boc-masked phenylhydrazine 101 in a satisfactory yield (57%). Then, the Fischer indole synthesis of Boc-masked phenylhydrazine 101 and ketone 102 afforded indole 103. Upon four steps, phenol 104 was provided from indole 103. Finally, dictyodendrins E (105) were successfully synthesized after four synthetic steps ((1) ClSO3, CH2CCl3; (2) BCl3, TBAI; (3) Zn, HCO2NH4; (4) 2,3-dichloro-5,6-dicyano1,4-benzoquinone (DDQ), room temperature) from phenol 104 (Scheme 15).167
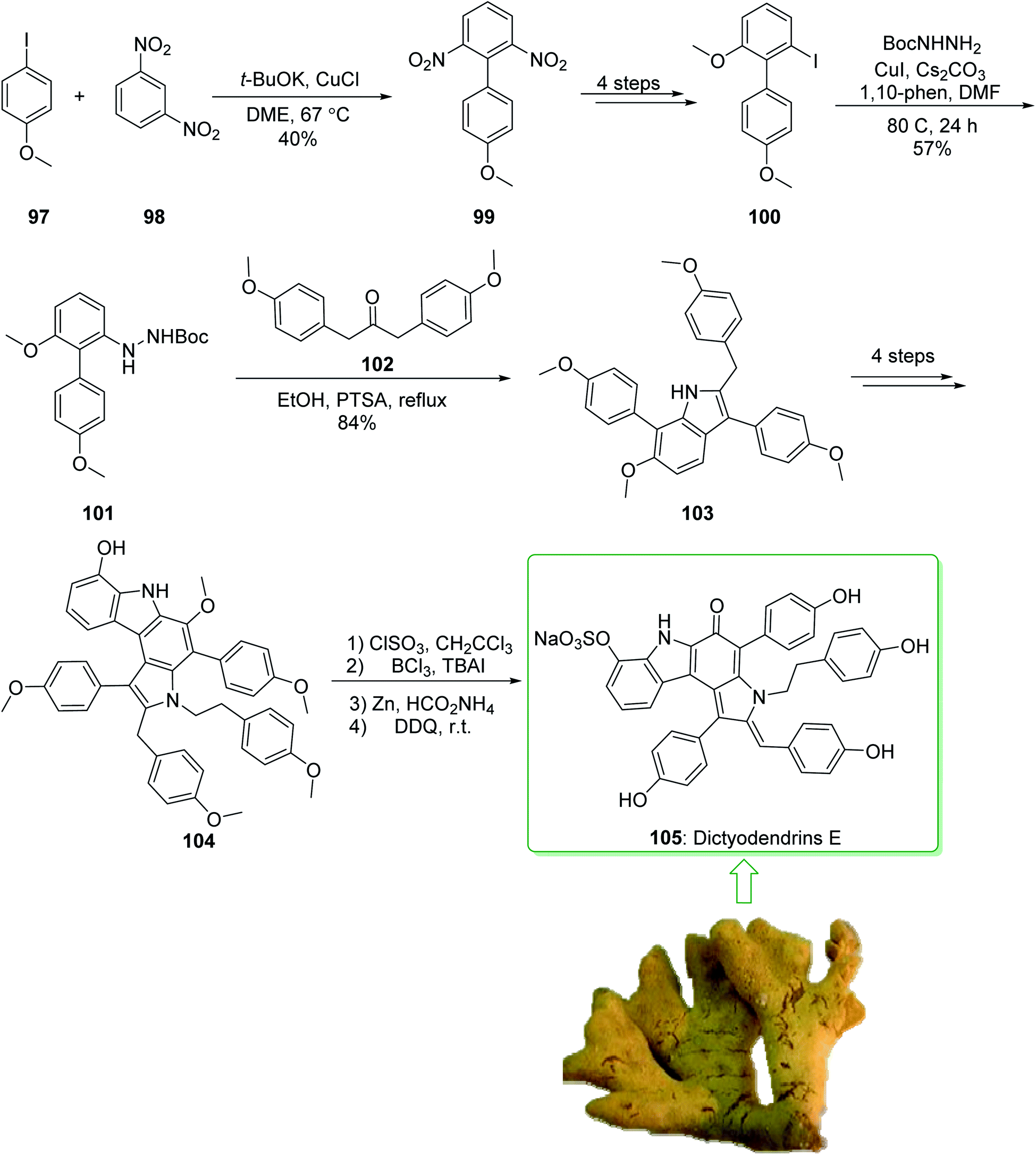 | ||
| Scheme 15 Total synthesis of dictyodendrins E (105). | ||
In 2015, Tokuyama reported168 the total synthesis of the biosynthetically-related monoterpene indole alkaloid (−)-mersicarpine (110). An azepino[3,2-b]indole intermediate was synthesized through d'Angelo's enantioselective Michael addition, Fischer indole synthesis, and DIBALH-catalyzed reductive ring-expansion reaction. The total synthesis of (−)-mersicarpine (110) began from the optically active cyclohexanone 106. The Fischer indole synthesis of the optically active cyclohexanone 106 and phenylhydrazine hydrochloride (107) by using methanesulfonic acid (MsOH) under reflux in MeOH gave the corresponding tricyclic indole (−)-108 in a good yield (84% yield). Indole (−)-108 after six steps, gave azepinoindole 109. The latter, after autoxidation of the resultant 109 and reductive treatment with dimethyl sulfide completed the total synthesis of (−)-mersicarpine (110). As a result, the total synthesis of (−)-mersicarpine (110) was accomplished in a six-pot/nine-step sequence with a 21% overall yield (Scheme 16).168
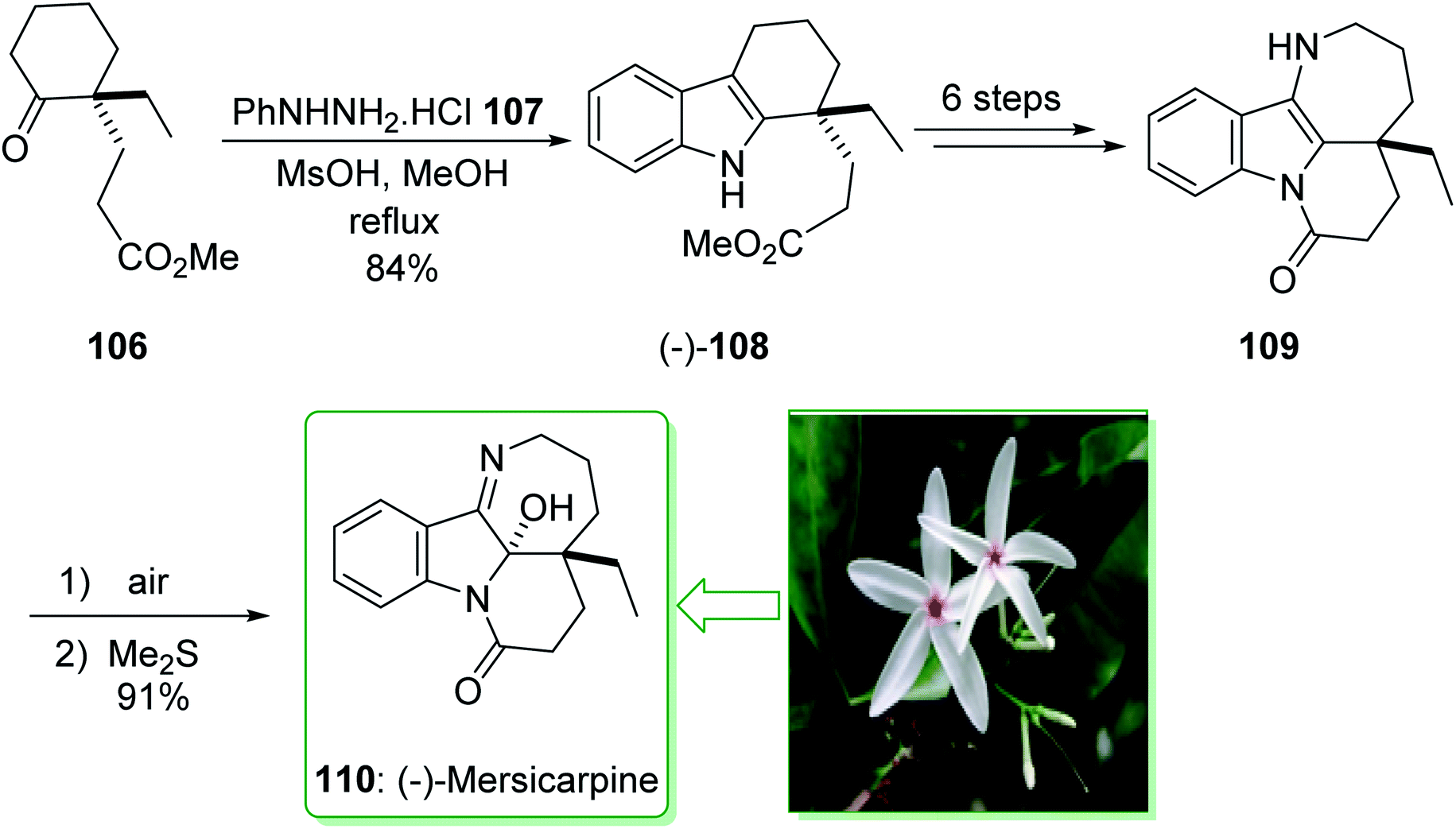 | ||
| Scheme 16 Total synthesis of (−)-mersicarpine (110). | ||
Indolocarbazoles and carbazoles are known to be present in various natural products that are used as significant drugs.91,169,170 Owing to their auspicious anti-microbial, anti-fungal, anti-hypertensive and anti-cancer properties the chemistry of indolo[2,3-a]carbazoles has been broadly investigated.171 Racemosin B (117) was extracted in 2013 by Guo and co-workers from the green alga Caulerpa racemosa, along with the most frequently encountered pigment in the genus Caulerpa, caulerpin.172 The indolo[3,2-a]carbazole framework, one of the isomeric indolocarbazoles, is naturally occurring and was first reported in 2002.173 Three members of this family, including asteropusazole A, B and ancorinazole were extracted from marine sponges by Wright and co-workers in 2013. The first results of the cytotoxicity and anti-microbial assays demonstrated that asteropusazole A is a medicinal candidate that can be used to target psychiatric and neurological disorders.174 In 2016, Kotha et al. reported175 a novel synthetic approach to indolocarbazoles using a two-fold Fischer indolization under eco-friendly reaction conditions using N,N-dimethyl urea and L-(+)-tartaric acid. In this method, atom economical reactions, such as ring-closing metathesis, enyne-metathesis, and also the Diels–Alder reaction have been utilized as key steps.
The unit structure of asteropusazole (118) and racemosin B (117) was synthesized using Fischer indolization of cyclohexanone 113 and N-methylphenylhydrazine (112) (prepared from the commercially available aniline derivative 111)176 that afforded carbazole 114. Using the DDQ oxidation reaction, 111 gave the keto derivative 115. Lastly, the N-masked indole derivative 115 was treated with N-methylphenylhydrazine 112 to afford the highly aromatized product 116 in a moderate yield (51% yield), which is an intermediate for the synthesis of asteropusazole (118) and racemosin B (117) (Scheme 17).175
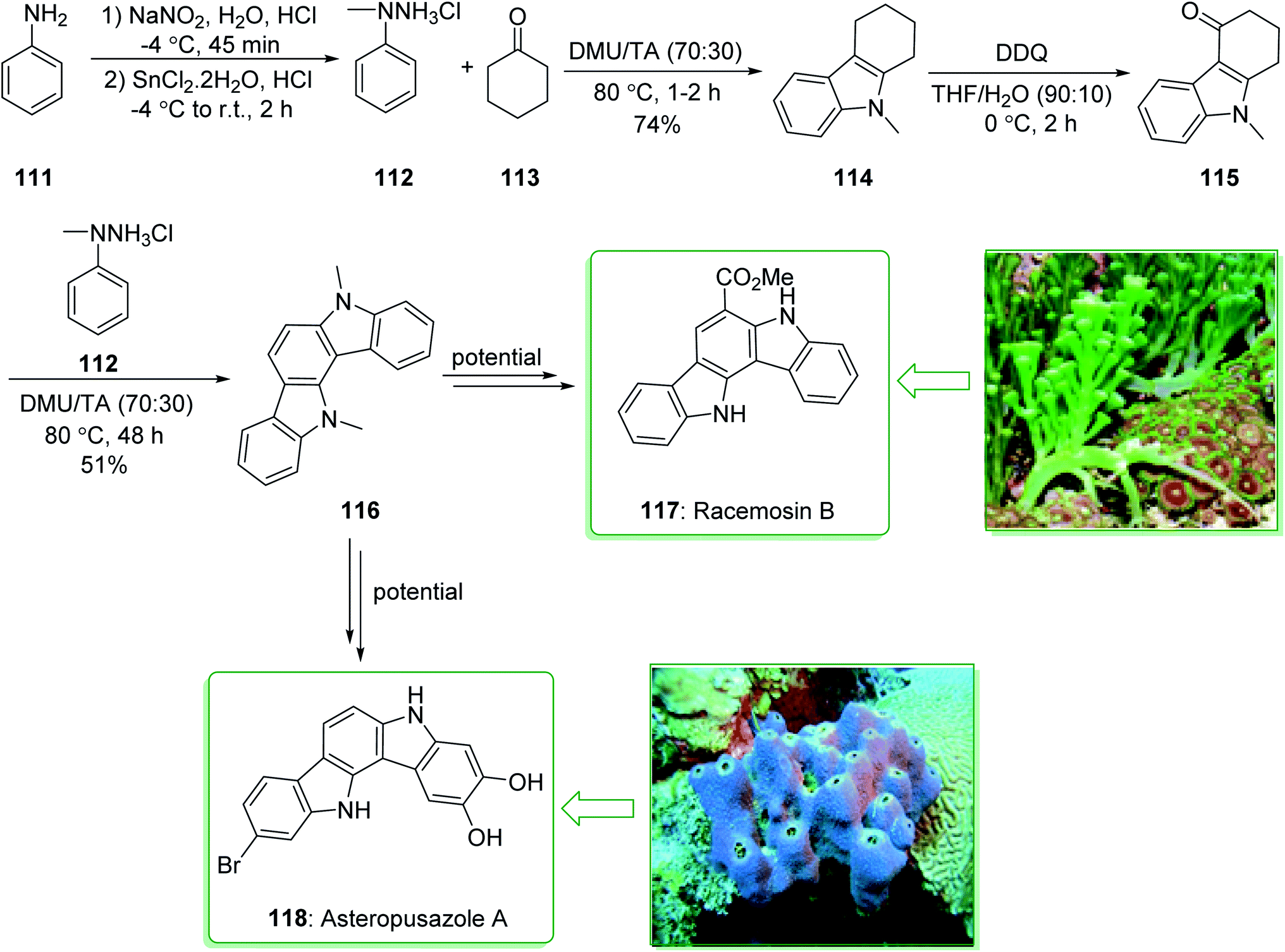 | ||
| Scheme 17 Synthesis of the unit structure of racemosin B (117) and asteropusazole (118). | ||
Scholarisine K and A belong to the family of akuammiline alkaloids.152 Scholarisine K (123) and alstolactine A (125) were extracted by Luo and co-workers in 2015 from Alstonia scholaris.177,178 The first enantioselective total synthesis of scholarisine K (123) and alstolactine A (125) were performed in 2017 by Gao et al.179 The key aspects of these syntheses are the ring closure metathesis and also an intramolecular Heck reaction to make the 1,3-bridged [3,3,1] bicycle (C–D ring), the intramolecular alkylation reaction was followed by the Fischer indolization reaction to make the basic framework of the akuammilines, and also the bioinspired, acid-improved epoxide opening/lactonization to form the second lactone ring of alstolactine A. The total synthesis of scholarisine K (123), and alstolactine A (125) started with aldehyde 119, which after more than 14 steps afforded the corresponding lactone 120. The latter was converted to indolenine 121 through a Fischer indolization. After five steps indolenine 121 gave epoxide 122. The removal of N-Cbz under Pd(OH)2/C and reductive amination in two steps (with 79% yield) provided scholarisine K (123). Moreover, indolenine 121, after more than six steps, gave epoxide 124. Based on similar transformations, the natural product alstolactine A (125) was provided by replacing the Cbz with a methyl group (Scheme 18).179
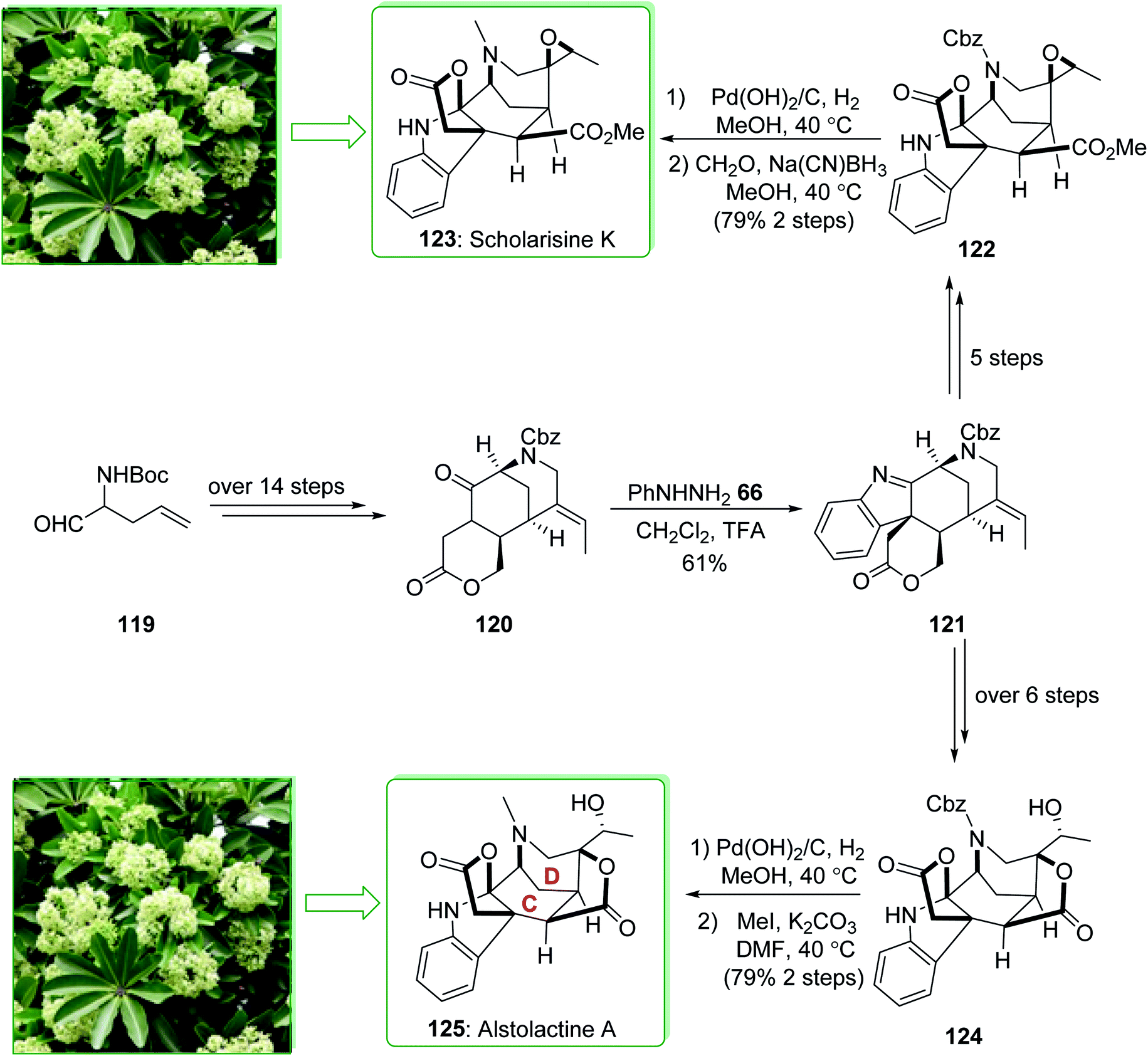 | ||
| Scheme 18 Total synthesis of scholarisine K (123) and alstolactine A (125). | ||
(−)-Pyrifolidine was first extracted by Svoboda et al. from the leaves of Aspidospermu quebrucho blunco (Apocynaceae) in 1973.180 The consistently strategic total syntheses of Aspidosperma alkaloids (+)-vincadifformine (136), (−)-quebrachamine (132), (+)-aspidospermidine (135), (−)-aspidospermine (134), (−)-pyrifolidine (133), and also nine others using the effectively generated tricyclic ketone 129 were demonstrated in 2017 by Jiang et al.181 Key aspects of these synthetic methods are the stereoselective intermolecular [4 + 2] cycloaddition, a palladium/C-mediated hydrogenation/deprotection/amidation cascade method and also the Fischer indolization reaction. The total synthesis of (+)-vincadifformine (136), (+)-aspidospermidine (135), (−)-aspidospermine (134), (−)-pyrifolidine (133), and (−)-quebrachamine (132) were started from the cycloaddition of 3-ethyl-5-bromo-2-pyrone 126 and enecarbamate (S)-127 that afforded the exo-bridged tricycliclactone 128 (54% yield, 77% brsm) with an exo/endo selectivity of 7:1. The latter, after more than nine steps, gave the tricyclic ketone 129. Next, the reaction of ketone 129 with phenylhydrazine (66), 2-methoxylphenyldrazine (130), or 2,3-dimethoxyphenylhydrazine (28) via a Fischer indole cyclization gave (+)-dehydroaspidospermidine (131a) (71% yield),182 (+)-dehydrodeacetylaspidospermine (131b) (66% yield)183 and (+)-dehydrodeacetylpyrifolidine (131c) (46% yield), respectively. Natural products (+)-vincadifformine (136), (+)-aspidospermidine (135), and (−)-quebrachamine (132) were synthesized from 131a (via different routes).181
Moreover, the total synthesis of (−)-aspidospermine (134) was accomplished in two steps from 131b and also the total synthesis of (−)-pyrifolidine (133) was performed in two steps from 131c. As a result, the total synthesis of (+)-vincadifformine (136) (4.4% overall yield), (+)-aspidospermidine (135) (7.1% overall yield), (−)-aspidospermine (134) (3.8% overall yield), (−)-pyrifolidine (133) (3.7% overall yield), and (−)-quebrachamine (132) (4.3% overall yield) were accomplished from the cycloaddition of 3-ethyl-5-bromo-2-pyrone 126 and enecarbamate (S)-127 (Scheme 19).181
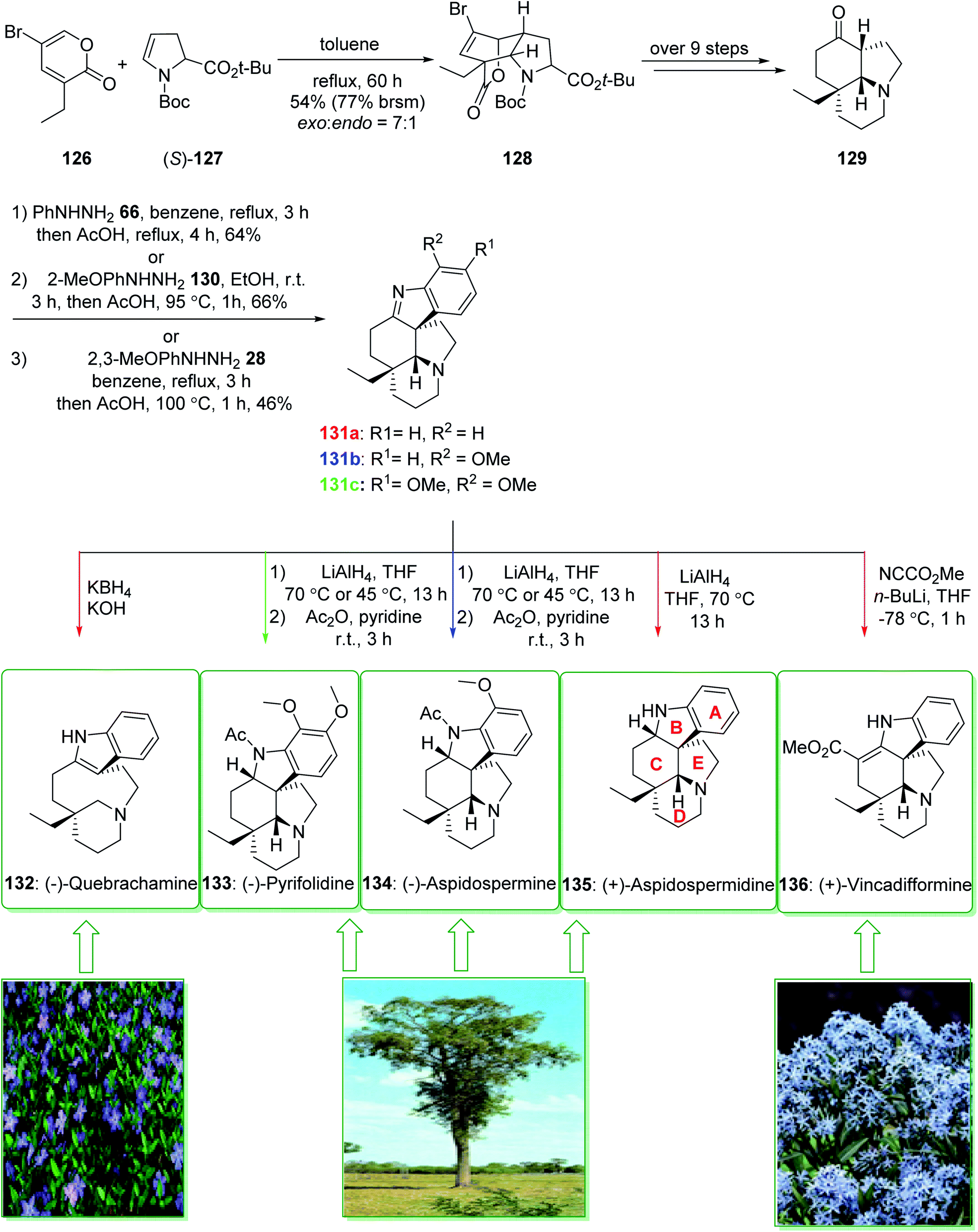 | ||
| Scheme 19 Total synthesis of (−)-quebrachamine (132), (−)-pyrifolidine (133), (−)-aspidospermine (134), (+)-aspidospermidine (135) and (+)-vincadifformine (136). | ||
(−)-Goniomitine, extracted in 1987 by Randriambola et al. from the bark of Gonomia Malagasy, is distinguished from other Aspidosperma alkaloids by its distinctive aminal-comprising tetracyclic unit.184 In 2017, Stoltz et al. demonstrated185 a Fischer indolization method to form a key tricyclic intermediate dihydropyrido[1,2-a]indolone (140) to give (−)-goniomitine (142) in three steps from commercially available precursors. The synthesis of (−)-goniomitine (142) started from 1,3-cyclohexanedione (137), which provided the corresponding C-alkylated product 139 in a satisfactory yield (70% yield) as the enol tautomer. Upon examining various reaction conditions for the Fischer indolization reaction, it was identified that exposing enol-139 to 2N HCl under reflux in toluene gives the key product dihydropyrido[1,2-a]indolone (140), although in an inadequate range of 10–33% yield. After three steps the latter afforded lactam 141 that is an intermediate for the synthesis of (−)-goniomitine (142) (Scheme 20).185
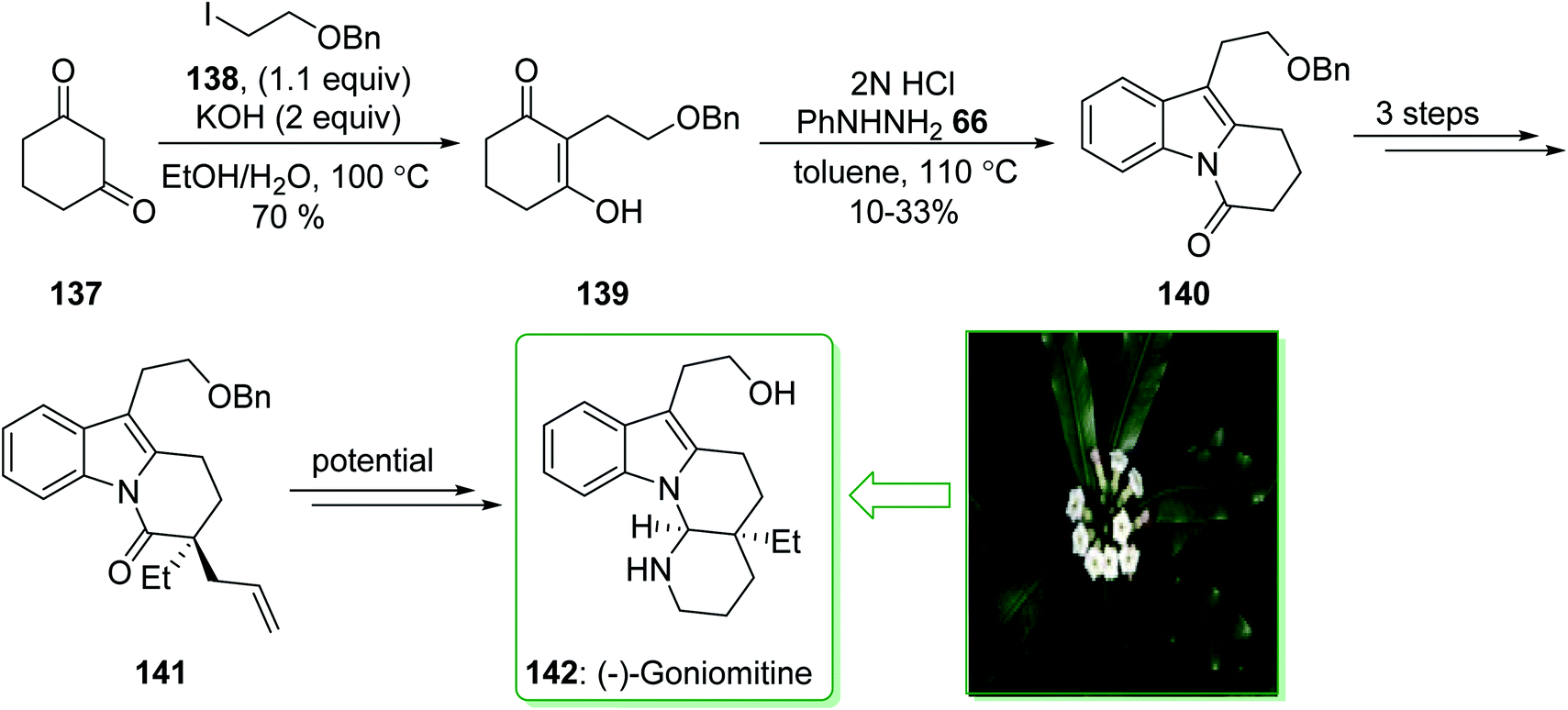 | ||
| Scheme 20 Synthesis of (−)-goniomitine (142). | ||
Carbazole alkaloids exhibit various biological and pharmacological properties. In addition, they have potential uses in electroluminescent materials, especially electrical, thermal, and also optical properties.91,186 Clausine C (153) and clausine M (155) were extracted from Clausena excavate by Huang and co-workers in 1996.187 The siamenol carbazole alkaloid (150) extracted from Murraya siamensis exhibited biological properties, for example anti-HIV properties.173 In addition, clauszoline K (152) and clausine N (154), extracted from the stem bark of Clausena excavate, have been applied in traditional medicine as a detoxification agent and also for the treatment of snake bites.188 In addition, they show powerful anti-cancer, anti-bacterial, and anti-oxidant properties.189 Glycozolicine (156) was isolated for the first time in 1992 from the roots of Glycosmis pentaphylla by Chakraborty and co-workers.190 Furthermore, cytotoxic carbazole alkaloids; clauraila A (148a) and clausenal (148b) were extracted from the roots of Clausena harmandiana (Rutaceae)191 and the leaves of Clausena heptaphylla,192 respectively. Clauraila A (148a) demonstrated an important selective cytotoxicity towards human lung cancer cells (NCIC-H187) and is utilized in Thai folk medicine for the treatment of stomach aches, headaches, and also stomach sickness.193
In 2017, Lokhande et al. reported194 the synthesis of 7-oxygenated carbazole alkaloids through a Fischer–Borsche ring and metal-free reaction conditions. The key step includes the aromatization and a one-pot iodination procedure for functionalization of the Fischer–Borsche ring with molecular iodine. A suitably oxygenated functionality has been introduced to the Fischer–Borsche ring through this approach to produce clauszoline K (152), clausine M (155), clausine N (154), siamenol (150), glycozolicine (156), clauraila A (148a) and clausenal (148b). The total synthesis of clauszoline K (152), glycozolicine (156), clausine C (153), clausine N (154), clausine M (155), clauraila A (148a), clausenal (148b) and also the formal synthesis of siamenol (150) were started from the reaction of (3-methoxy/3-bromo)phenylhydrazine 143a/143b and 4-methylcyclohexanone/4-propionylcyclohexan-1-one 144a/144b under reflux in HOAc via the Fischer–Borsche method, which delivered tetrahydrocarbazoles 145a/146a or 145b/146b. Next, compounds 147a and b were synthesized from tetrahydrocarbazoles 145a and 146a in four steps. Finally, the oxidation of carbazoles 147a and b using DDQ in MeOH/H2O yielded clauraila A (148a) and clausenal (148b). In addition, the total synthesis of glycozolicine (156) was performed in two steps (aromatization and displacement) from tetrahydrocarbazole 146a (Scheme 21).194
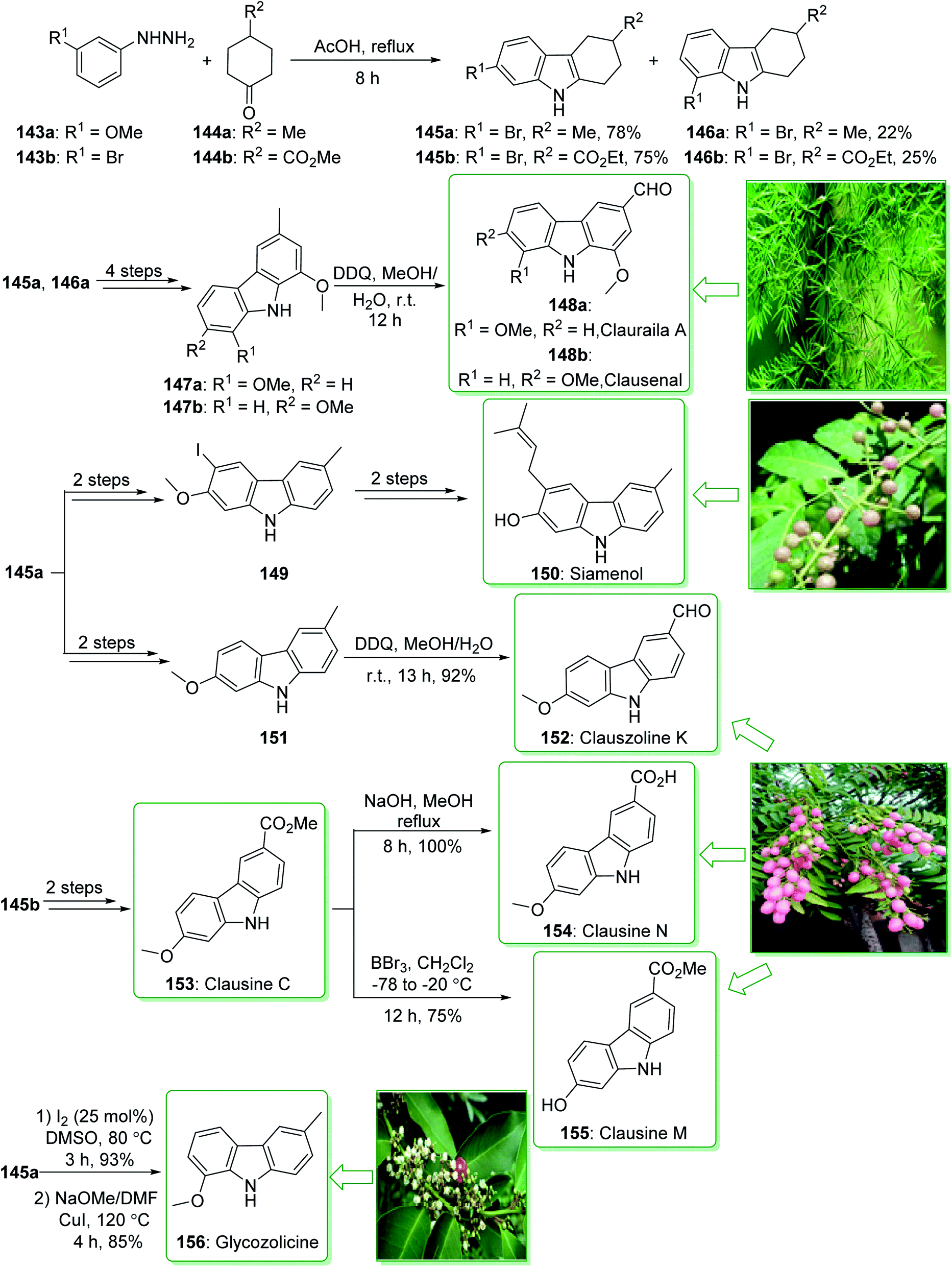 | ||
| Scheme 21 Total synthesis of clauszoline-K (152), glycozolicine (156), clausine C (153), clausine N (154), clausine M (155), clauraila A (148a), clausenal (148b) and the formal synthesis of siamenol (150). | ||
On the other hand, after two steps tetrahydrocarbazole 145a gave the monoiodinated carbazoles 149 or 151 (using different routes). Then, the substrate 151, through an oxidation reaction in the presence of DDQ195 in MeOH/H2O, afforded clauszoline-K (152). Lastly, the formal synthesis of siamenol (150) was accomplished from monoiodinated carbazole 149.194 In the following, clausine C (153) was synthesized from ester 145b, in two steps. Moreover, the reaction of 153 with sodium hydroxide in MeOH gave the clausine N (154) in a 100% yield.
On the other hand, the elimination of the methyl group in clausine C (153) using BBr3 in dichloromethane, gave the natural product clausine M (155) in a satisfactory yield (75% yield) (Scheme 21).194 Some alternative synthesis methods for these carbazole alkaloids, some of which have better overall yields, have been reported.196–199
The akuammiline alkaloids, a significant class of naturally occurring compounds, were extracted from plants found in Africa, India, and Southeast Asia. (+)-Strictamine (166) was extracted in 1966 by Schnoes from the plant Rhazya stricta,200 and (−)-2(S)-cathafoline (167) was isolated in 2014 from Alstonia macrophylla.201 (−)-2(S)-Cathafoline (167) exhibited satisfactory activity in overturning drug resistance in vincristine resistant KB cells.200–202 (+)-Akuammiline (171) was extracted together with (−)-Ψ-akuammigine (169) in 1932 by Henry. (−)-Ψ-Akuammigine (169) was extracted from Picralima klaineana in 1932.203 Biological investigations demonstrated favorable activity for use as an anti-inflammatory agent.204 In 2018, Garg et al. demonstrated205 the initial total synthesis of (+)-strictamine (166), (−)-2(S)-cathafoline (167), (−)-Ψ-akuammigine (169) and (+)-akuammiline (171). This methodology is based on the establishment of the reductive interrupted Fischer indolization reaction to form a pentacyclic intermediate having five contiguous stereocenters, as well as late-stage construction of the methanoquinolizidine building block through a deprotection cyclization cascade reaction. The total synthesis of (−)-Ψ-akuammigine (169) and (+)-akuammiline (171) feature the initial constructions of akuammiline alkaloids comprising both a methanoquinolizidine unit and vicinal quaternary centers. Moreover, this group demonstrated the bioinspired reductive rearrangements of (+)-strictamine (166) and (+)-akuammiline (171) to give (−)-10-demethoxyvincorine and also a novel equivalent thereof.
In this method, the total synthesis of (+)-strictamine (166), (−)-2(S)-cathafoline (167), (−)-Ψ-akuammigine (169) and (+)-akuammiline (171), began with the palladium-mediated Trost desymmetrization of sulfonamide 158 and dibenzoate 157 and providing the alcohol 160. The latter, after 12 steps, gave ketolactone 161, and through the Fischer indolization reaction in the presence of trifluoroacetic acid in 1,2-dichloroethane this afforded the indolenine lactone 162. The latter in the presence of triethylsilane and additional trifluoroacetic acid under stirring at 23 °C afforded the reductive Fischer indolization product 164 in a good yield (83% yield). In the following, indoline 164, after seven steps gave the alkyl chloride 165, which after oxidation, followed by deprotection–cyclization yielded the natural product (+)-strictamine (166). Similarly, compound 165, after N-methylation and deprotection–cyclization afforded (−)-2(S)-cathafoline (167).
Furthermore, indoline 164, after seven steps, gave furoindoline alcohol 168. The latter, after two steps, yielded the natural product (−)-Ψ-akuammigine (169). Moreover, indoline 164, after six steps, afforded aldehyde 170. Finally, aldehyde 170 after four steps yielded (+)-akuammiline (171) (Scheme 22).205
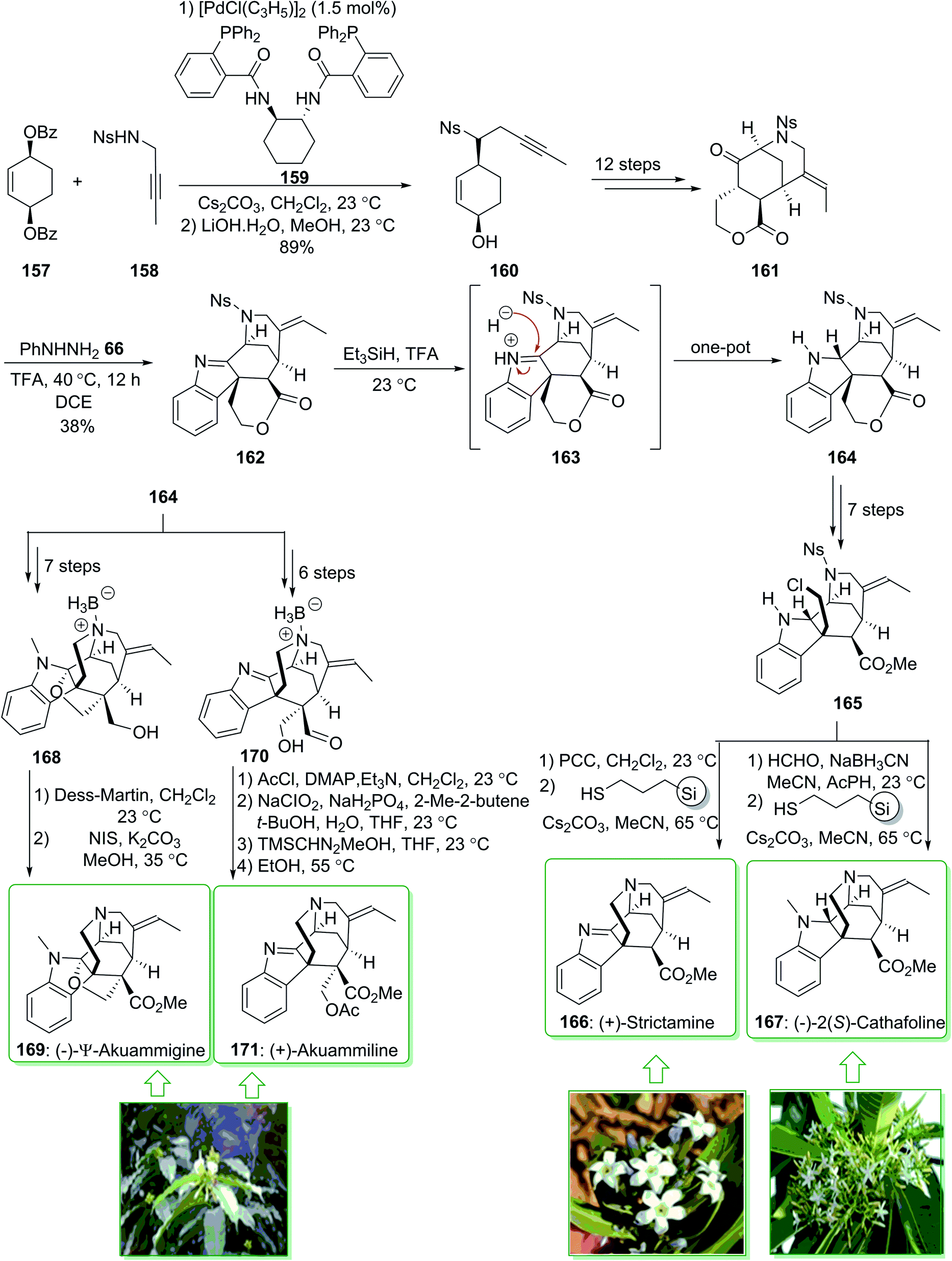 | ||
| Scheme 22 The total synthesis of (+)-strictamine (166), (−)-2(S)-cathafoline (167), (−)-Ψ-akuammigine (169) and (+)-akuammiline (171). | ||
Various carbazole alkaloids have been extracted from terrestrial plants. In addition, numerous carbazole alkaloids have been found in various streptomycillin and algae species.206 In 1979, 6-chlorohyellazole and hyellazole were the first carbazole alkaloids extracted from the blue-green alga Hyella caespitosa.207 In 2018, Chakraborty and co-worker demonstrated208 the total synthesis of marine natural alkaloids chlorohyellazole and hyellazole. In this route, the total synthesis of hyellazole (180) and 4-deoxycarbazomycin B (181) were commenced from 2-methyl-3-nitroaniline (172), which after ten steps afforded 2-bromo-4-methoxy-3-methylaniline hydrochloride (173). The latter, using NaNO2 and diluted HCl in 0–5 °C afforded 2-bromo-4-methoxy-3-methylbenzene diazonium chloride (174), which using the Japp–Klingemann coupling reaction with 2-formylcyclohexanone (175) provided functionalized phenylhydrazonocyclohexanone (176). Fischer indole cyclization of 176 in the presence of glacial AcOH and a concentrated HCl mixture afforded 1-ketotetrahydrocarbazole (177).209 The latter, after two steps including a Wolff–Kishner reduction and aromatization, gave a mixture of 3-methoxy-2-methyl-9H-carbazole (178) with an 11% yield and 1-bromo-3-methoxy-2-methyl-9H-carbazole (179) with a 65% yield. Finally, the Suzuki cross-coupling reaction of carbazole 179, phenylboronic acid and methylboronic acid, respectively, in the presence of a palladium catalyst, afforded the natural products hyellazole (180) and 4-deoxycarbazomycin B (181) (Scheme 23).208 Some alternative synthetic methods for hyellazole (180), chlorohyellazole, and 4-deoxycarbazomycin B (181), some of which have better overall yields, have been reported.210–218
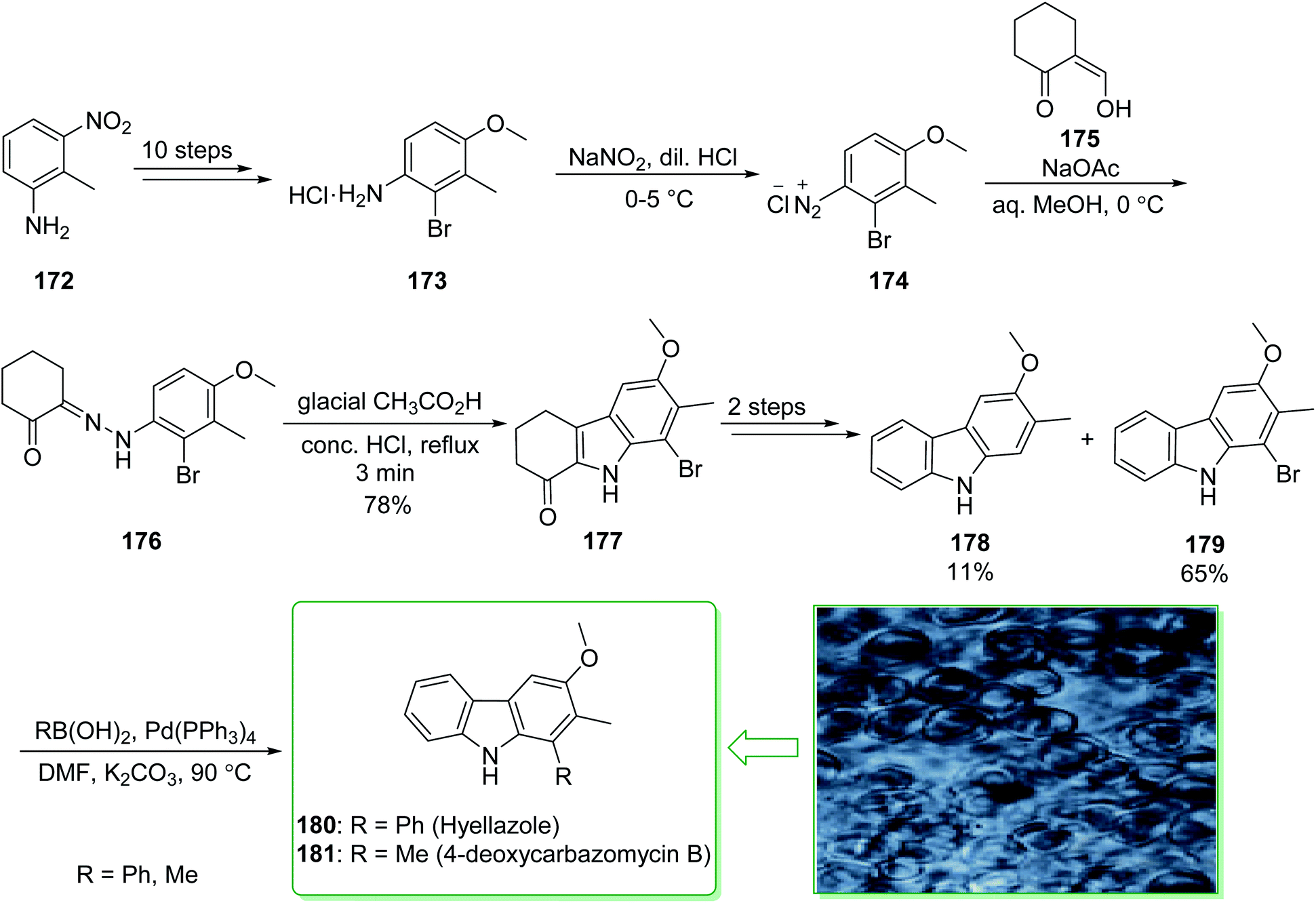 | ||
| Scheme 23 The total synthesis of hyellazole (180) and 4-deoxycarbazomycin B (181). | ||
Fontanesines A–C, bearing both quinazoline and the pyrano[3,2-e] indole framework, were extracted from the leaf fractions and stem bark of Conchocarpus fontanesianus that were gathered in Brazil by Queiroz and co-workers in 2016.219 These alkaloids have not been completely examined biologically, owing to restricted accessibility. Fontanesines are a rare type of pyrano[3,2-e]indoloquinazoline alkaloid. However, although pyranocarbazole and pyranoindole alkaloids have been widely prepared,91,94 pyrano[3,2-e]indoloquinazoline alkaloids have not been reported previously. A previous investigation on the leaf extracts of C. fontanesianus stated their cytotoxic, anti-fungal, and also anti-microbial activities, but the potent principles were not developed.220 In 2019, Abe et al. reported221 a short synthesis of pyrano[3,2-e]indole alkaloid fontanesine B via a Fischer indolization. The isomer of fontanesine B exhibited a greater anti-proliferative property in comparison with the natural product, fontanesine B (187). The total synthesis of fontanesine B (187) was started from 4-acetamidophenyl acetate (182), which after seven steps gave the arylhydrazine 183. The reaction between arylhydrazine 183 and quinazolinone 185 (prepared from isatoic anhydride (184) in five steps) using acetic acid afforded hydrazine 186 in a moderate yield (50% yield). In the following, the extraordinary Fischer indolization of hydrazine 186 was examined. It was found that the pyran ring was injured under the acidic conditions, thus establishment of the Fischer indolization using the hydrazine containing pyran scaffold is fairly stimulating. Hence, examination using different acids was performed, it was found that acetic acid and propionic acid were appropriate for improving the Fisher indolization and the pyran-ring and alkene remained intact. Hydrazine 186 under reflux in acetic acid gave a mixture of 187 and 188 in a satisfactory yield (71% yield, 187/188 = 25:75). Among the various acids, propionic acid was the most effective acid at providing the desired cyclized products (88% yield, 187/188 = 33:67) (Scheme 24).221
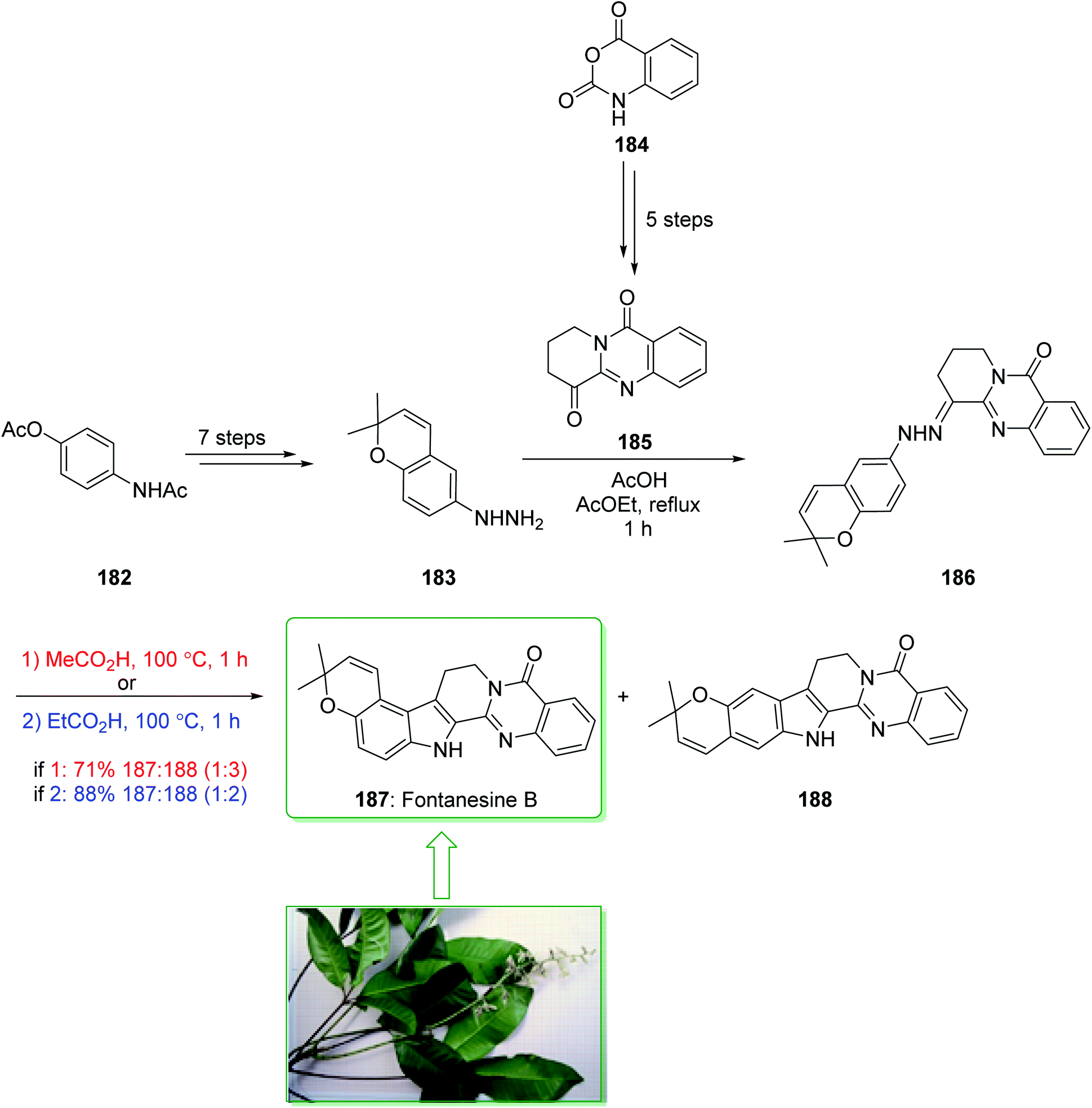 | ||
| Scheme 24 Total synthesis of fontanesine B (187). | ||
The Tabernaemontana genus belongs to the family Apocynaceae containing various species distributed throughout subtropical and tropical regions of the world, including Brazil.222 (±)-Minovincine was extracted from Tabernaemontana riedelii by Szántay in 1997.223 Aspidofractinine-type alkaloids were extracted from the leaf extract of Kopsia teoi by Kam and co-workers in 1997.224 In 2020, Soós and co-workers demonstrated225 the eight-step synthesis of (−)-minovincine (190) and (−)-aspidofractinine (195) using simply accessible reagents and a catalyst. A key aspect of the methodology was the application of the chain of cascade reactions to quickly make the penta- and hexacyclic building blocks. These cascade conversions involved the organocatalytic Michael–Aldol condensation, a multistep anionic Michael-SN2 cascade reaction and also a Mannich reaction interrupted Fischer indolization. The total synthesis of (−)-minovincine (190) began with the formation of tricyclic 186, which was provided using the Michael addition–Aldol condensation organocascade reaction of the Nazarov reagent 184 and ω-chloro-formylpentenoate 185 (ref. 226) in three steps. Then, by using the selective deprotection of the t-Bu-ester, the resultant β-oxo carboxylic acid was decarboxylated in situ and gave ketone 187 in a high yield (95% yield). Then, reaction of product 187 with phenylhydrazine (66) gave aspidospermane-type indolenine 189 (50% yields) and its structural isomer 188 (31%, yields) respectively. Finally, after two steps, pentacyclic 189 gave (−)-minovincine (190). As a result, (−)-minovincine (190) was synthesized in eight steps with an 11% overall yield. On the other hand, after three steps the tricyclic ketone 186 afforded the C-5 acetyl functionalized tricyclic ketone 191. Next, the Fischer indoleMannich cascade reaction occurred efficiently to give the relevant oxoaspidofractinine 194 in a moderate yield (55% yield) (alongside its isomer 192) through a recognized indolenine intermediate 193. Finally, substrate 194 was submitted to hydrazine to provide the (−)-aspidofractinine (195) in a high yield (89% yield). As a result, (−)-aspidofractinine (195) was synthesized in eight steps with a 19% overall yield (Scheme 25).225
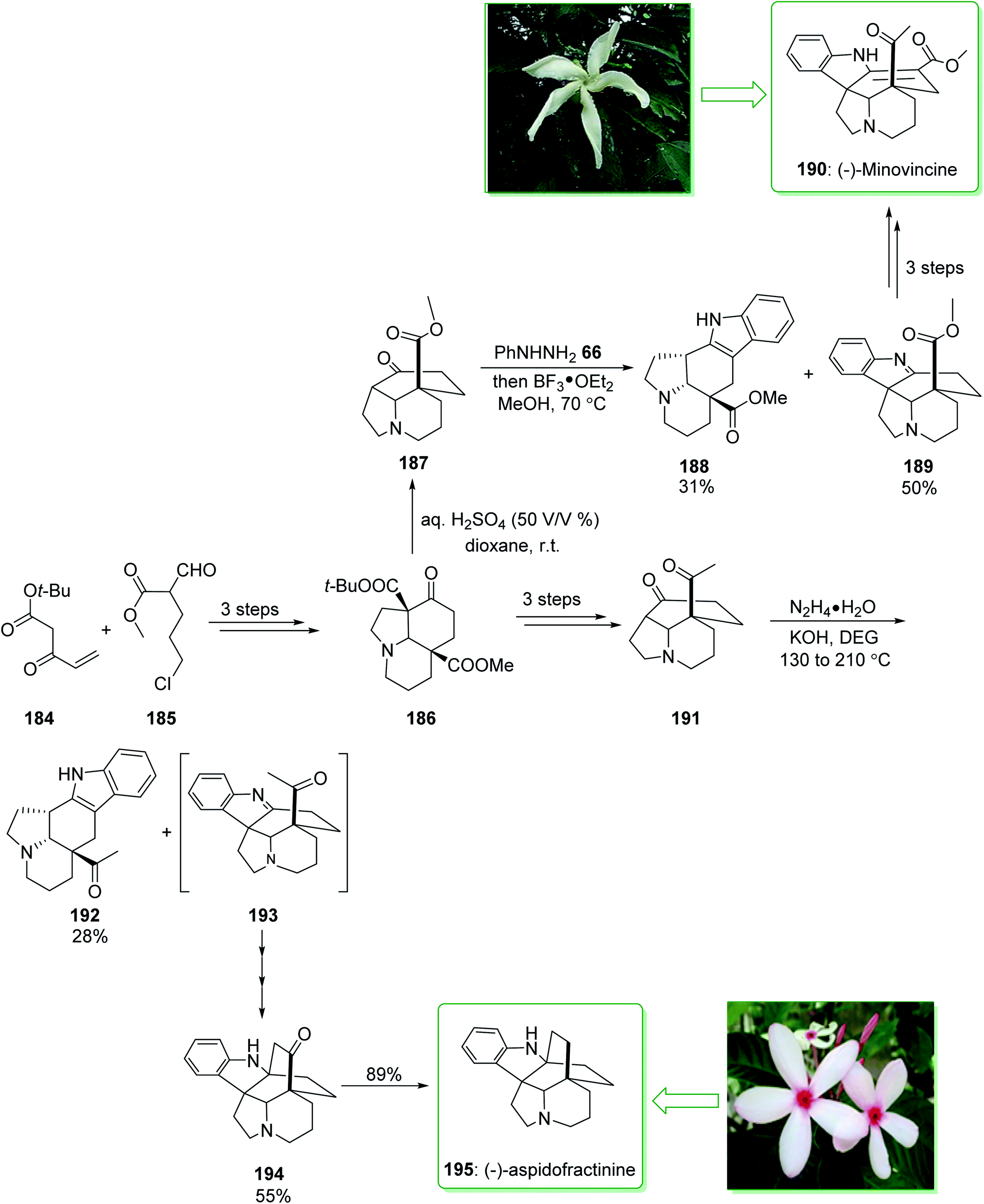 | ||
| Scheme 25 Total synthesis of (−)-minovincine (190) and (−)-aspidofractinine (195). | ||
The genus Strychnos, the biggest genus of the family Loganiaceae, was identified by Linnaeus on the basis of Strychnos nux uomica, the type species, and Strychnos colubrina.227 The uleine-type alkaloids constitute a significant subgroup of the Strychnos alkaloids that are identified by the 1,5-methanoazocino[4,3-b]indole scaffold as having an ethyl substituent at the bridging carbon atom. Uleine was extracted by Schmutz et al. from Aspidosperma ulei Mgf228 and its accurate structure was suggested by Warnhoft and Buchi in the late 1950s.229 (−)-Tubifolidine was extracted from the Strychnos species by Amat and co-workers in 1997.230 In 2020, Cho and co-workers reported231 the enantioselective synthesis of (+)-uleine (204) and (−)-tubifolidine (210). The regioselective construction of enol triflates from 2-azabicyclo[3.3.1]nonane ketones and also indolizations of the resulting ene-hydrazides permitted the effective formation of key indole intermediates, assisting the total synthesis of the desired natural alkaloids. The total synthesis of (+)-uleine (204) was commenced from the key chiral cyclohexenone 197 prepared in a 93% enantiomeric excess (ee) by using modification of the process previously published by Ma et al. from ethyl (E)-4-oxopent-2-enoate (196).232 In the following, compound 197, after five steps, afforded the bicyclic ketone 198. The latter was reacted with potassium tert-butoxide and the Comins' reagent at −78 °C in THF/DMF (1:6) to provide enol triflate 199 as the main product, together with trace quantities of 199′, which was exposed to the two-step indolization reaction for the end-game synthesis of (+)-uleine (204). In this route, the carbon–nitrogen coupling of 199 and 199′ with phenyl hydrazide 200 afforded enehydrazides 201 and 201′ as an inseparable 37:1 mixture in a satisfactory yield (71% total yield). Next, indolization was applied in the presence of zinc chloride by heating to 90 °C in toluene using a molecular sieve, which afforded the corresponding indole 202 (76% yield) and its isomer 202′ (3% yield), after separation. After a further two steps, indol 202 gave (+)-dasycarpidone (203), and after the addition of MeLi and the subsequent dehydration reaction (+)-uleine (204) was yielded. As a result, (+)-uleine (204) was synthesized in 12 steps from cyclohexenone 197 with a 9.2% total yield (Scheme 26).231
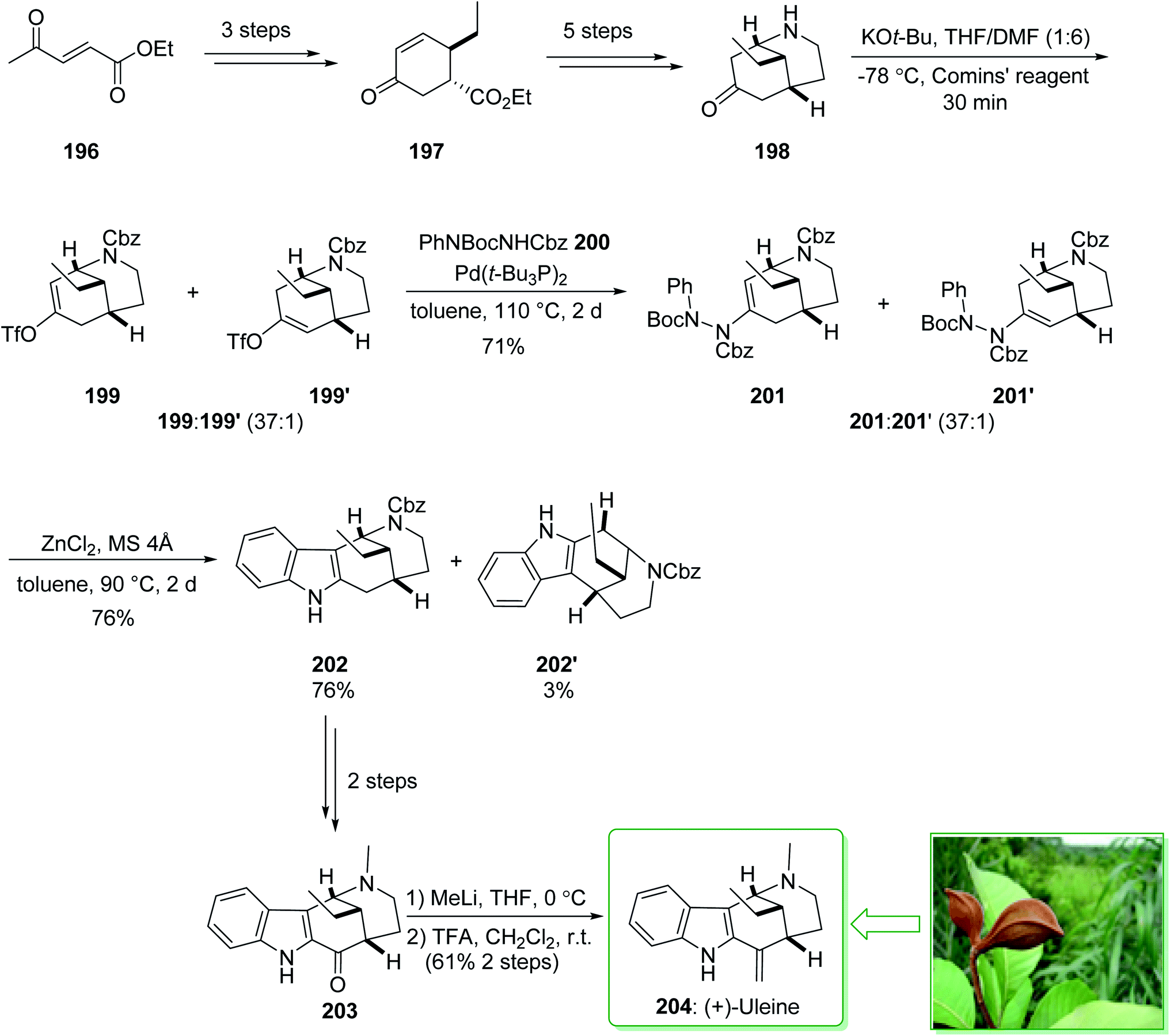 | ||
| Scheme 26 Total synthesis of (+)-uleine (204). | ||
The formal synthesis of (−)-tubifolidine (210) was started from cyclohexenone 205 in an enantiomerically enriched form with a high ee (98% ee).233 After five steps cyclohexenone 205, afforded the corresponding ketone 206. After two steps the latter gave ene-hydrazides 207 and 207′ as an inseparable 21:1 mixture. Submission to the zinc-catalyzed indolization reaction gave indole 208 in a good yield (74% yield).231 Then, the indole intermediate 208, after six steps, including the protection of the indole nitrogen, elimination of the methoxymethyl (MOM) substituent, pyridinium chlorochromate (PCC) oxidation, Wittig olefination, palladium-mediated hydrogenation, and N-alkylation afforded the recognized indole intermediate 209, this was formerly transformed to (−)-tubifolidine (210) (Scheme 27).234
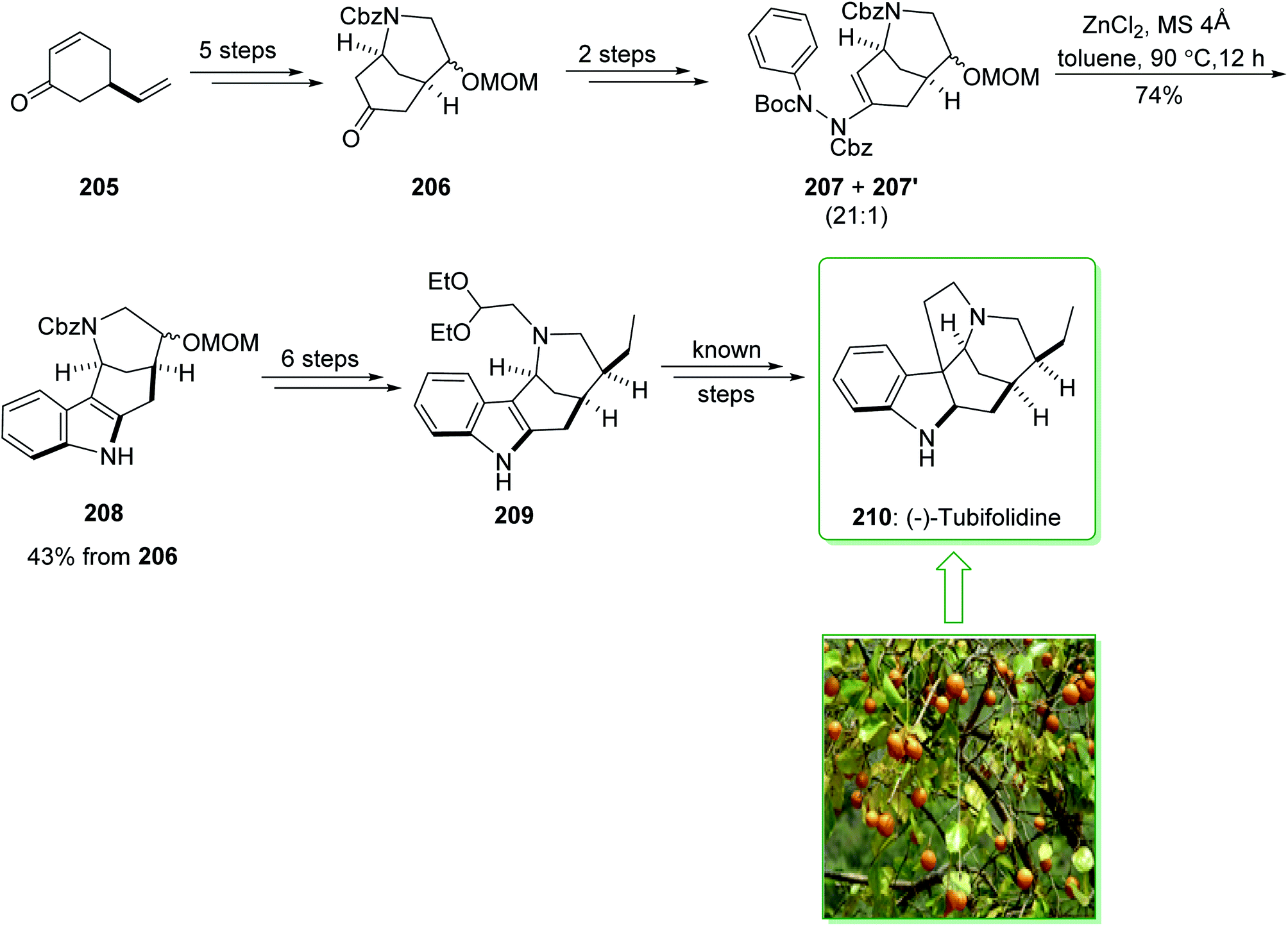 | ||
| Scheme 27 The formal synthesis of (−)-tubifolidine (210). | ||
The paraherquamides are an uncommon family of naturally occurring fungal compounds that include a bicyclo[2.2.2]diazaoctane unit structure, a spiro-oxindole, and a functionalized proline scaffold. Among them, paraherquamide A, was extracted from cultures of Penicillium paraherquei by Yamazaki et al. in 1981.235 VM55599, a minor metabolite from culture extracts of a Penicillium spp. was isolated in 1993 by Everett et al.236 Sarpong et al. in 2020 demonstrated237 a full explanation of the investigations into reverse-prenylated indole alkaloids containing a bicyclo[2.2.2] unit. A different pathway was described that led to the formation of (+)-VM-55599, preparaherquamide and premalbrancheamide. An intramolecular Dieckmann cyclization of an isocyanate and an enolate was utilized to make the bicyclo[2.2.2]diazaoctane unit. The pentacyclic indole framework was formed via a one-pot Hoffman rearrangement and Fischer indole synthesis. The total synthesis of preparaherquamide (219), (+)-VM-55599 (220) and premalbrancheamide (216) were started from enone 211 (prepared in gram-scale amounts from 1-tert-butyl 2-ethyl-3-oxopyrrolidine-1,2-dicarboxylate in nine steps with a 37% overall yield).238 In the following, enone 211, after three steps, gave isocyanate 212. The latter, using H2SO4, was transformed into the relevant ammonium intermediate 213 that was exposed to phenylhydrazine (66) to influence Fischer indolization, affording pentacyclic indole 214 in a single-pot operation. Next, the latter, after three steps, gave ketone 215, which after two steps (Wolff–Kishner reduction and chemoselective reduction of the tertiary amide) gave the natural product premalbrancheamide (216). Moreover, ketone 215, after two steps, afforded a mixture of the exocyclic alkene (217) and endocyclic alkene (218) in a 71% yield (1:2 mixture). Hydrogenation of the mixture of alkenes (i.e., 217 and 218) using Pd/C and chemoselective tertiary amide reduction using DIBAL-H gave the natural products preparaherquamide (219) and (+)-VM-55599 (220) (Scheme 28).237
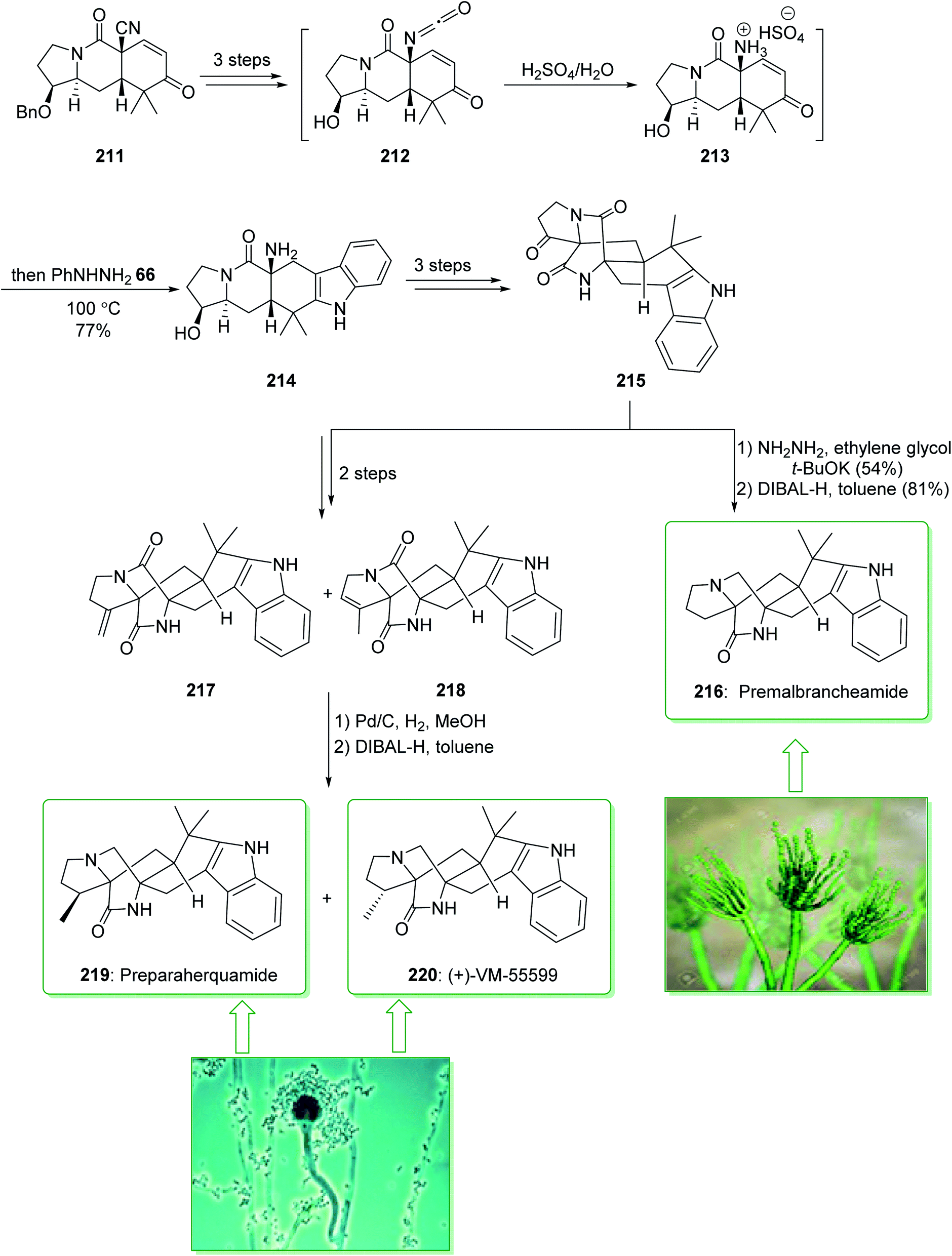 | ||
| Scheme 28 Total synthesis of preparaherquamide (219), (+)-VM-55599 (220) and premalbrancheamide (216). | ||
In 2020, Varga et al. demonstrated239 a unique reductive interrupted Fischer indolization method for the short synthesis of the 20-oxoaspidospermidine scaffold. This fast complexity producing pathway covers the route towards different dihydroindole Aspidosperma alkaloids having various C-5 side chain redox patterns. The end-game redox modulations were performed by using a modified Wolff–Kishner reaction and also a photo-Wolff rearrangement, allowing the total synthesis of (−)-aspidospermidine (229), (−)-limaspermidine (232), and also (+)-17-demethoxy-N-acetylcylindrocarine (231).239 The total synthesis of (−)-aspidospermidine (229), (−)-limaspermidine (232), and (+)-17-demethoxy-N-acetylcylindrocarine (231) were started from the reaction of tert-butyl 3-oxopent-4-enoate (221) and methyl 5-chloro-2-formylpentanoate (222), that afforded 1,3-dicarboxylate 223. The latter, after three steps, gave the stereochemically complex intermediate 225. It was found that the chemoselective reduction of the imine scaffold is similar to a Meerwein–Ponndorf–Verley type reduction,240 in which the hydride source was the sacrificial alcohol solvent. Based on this result, isopropanol was used as a solvent to increase the efficiency of the reductive conversion and elevate the reaction temperature from 85 to 115 °C. Pleasantly, these modifications afforded the corresponding pentacycle 228 as the main product (63% yield) along with the 3H-indole 227 (32% yield), the side-product of Fischer indolization. After three steps, ester 230 was synthesized from the key intermediate 20-oxoaspidospermidine (228). Then, ester 230, after two steps including reduction and removal, yielded (−)-limaspermidine (232). Moreover, ester 230, after treatment using TFA and Ac2O, gave (+)-17-demethoxy-N-acetylcylindrocarine (231).
In the following, the intermediate 228, after two steps (modification of the original Stork–Dolfini synthesis) afforded the natural product (−)-aspidospermidine (229). Notably, the total synthesis of (−)-aspidospermidine (229) was performed in eight reaction vessels with an overall yield of 14%. In addition, the total synthesis of (−)-limaspermidine (232) was performed in 12 steps and gave an overall yield of 7%. Moreover, the total synthesis of (+)-17-demethoxy-N-acetylcylindrocarine (231) was accomplished in 12 steps with a 5% overall yield (Scheme 29).239
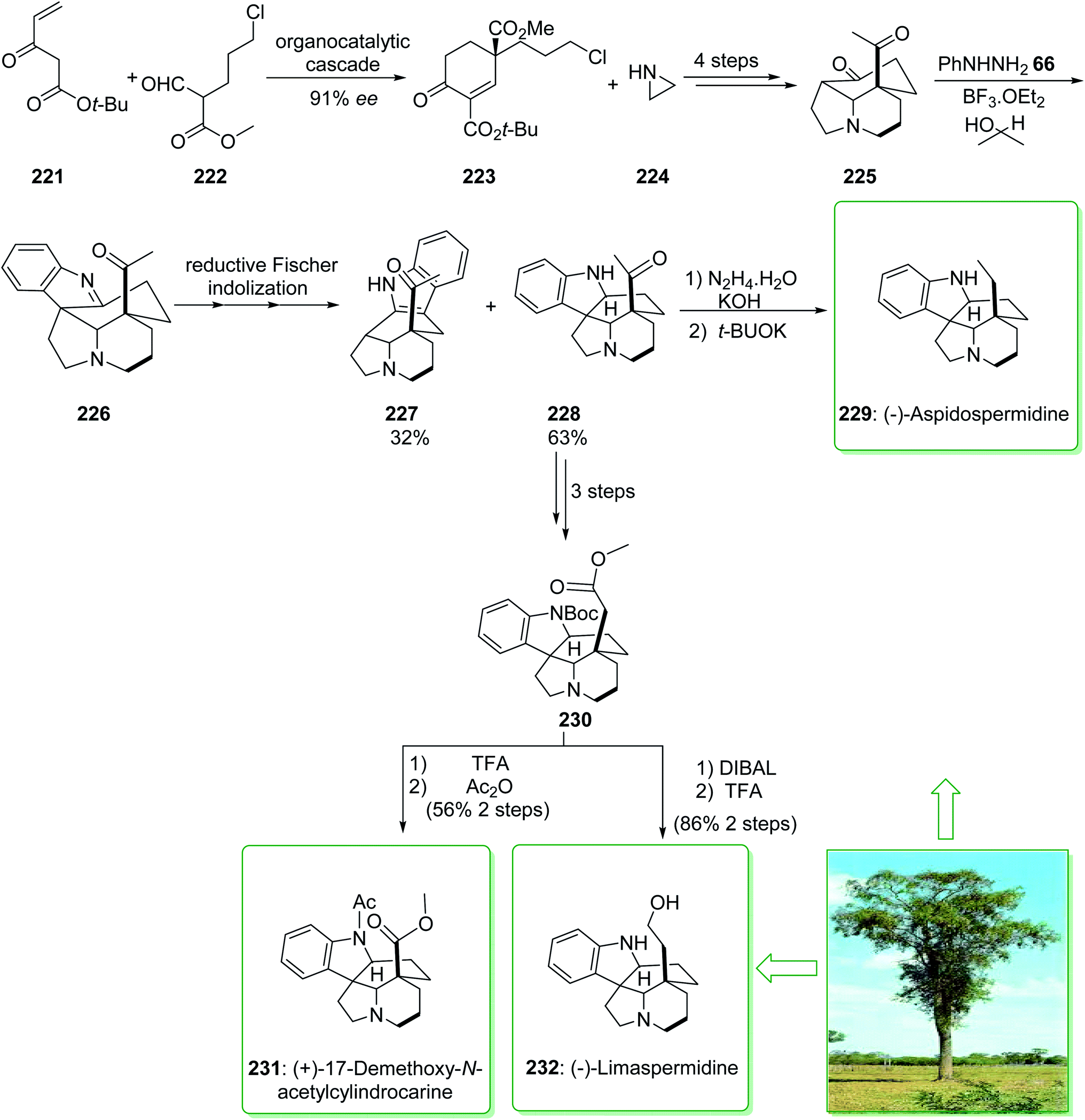 | ||
| Scheme 29 Total synthesis of (−)-aspidospermidine (229), (+)-17-demethoxy-N-acetylcylindrocarine (231) and (−)-limaspermidine (232). | ||
2.2. Larock indole synthesis
Sarpagine and Ajmaline are biogenetically correlated alkaloids that have been extracted from different species of Rauwolfia that are generally dispersed throughout Africa and Asia.241,242 These plants are broadly utilized in folk Chinese medicine for the treatment of neuralgia, hypertension,243 and migraines.244 (+)-12-Methoxy-Na-methylvellosimine (239) and (+)-12-methoxyaffinisine (240) were extracted by Kato and co-workers in 2002 from the bark of Rauwolfia bahiensis.245 In addition, (−)-fuchsiaefoline (242) was extracted from Peschiera fuchsiaefolia in 1987 by Reis and co-worker.246 Cook and co-workers in 2004 demonstrated247 that the asymmetric synthesis of 7-methoxy-D-tryptophan ethyl ester (237) was accomplished by a combination of the Larock heteroannulation reaction with a Schollkopf-based chiral auxiliary in a satisfactory yield. Next, this ester 237 was used in the initial total synthesis of (+)-12-methoxy-Na-methylvellosimine (239), (+)-12-methoxyaffinisine (240), and (−)-fuchsiaefoline (242) using the regiospecific and stereospecific approach with very high overall yield. The enantioselective Pictet–Spengler reaction, and also the enolate-driven Pd-mediated cross coupling reactions, acted as key steps. Based on this method, the total synthesis of (+)-12-methoxy-Na-methylvellosimine (239), (+)-12-methoxyaffinisine (240) and (−)-fuchsiaefoline (242) were started from the Larock heteroannulation of 2-iodo-6-methoxyaniline (233) and the propargyl-functionalized Schollkopf chiral auxiliary 234 using palladium(II) acetate, potassium carbonate, and lithium chloride in DMF at 100 °C. The ratio of the corresponding indole 235 to the byproduct 236 was identified on the basis of the integration of the proton at C3 in the 1H-NMR spectrum of the crude reaction mixture. The ratio was increased to 15:1 (235:236) when a 2% catalyst was used rather than a 5% catalyst (palladium(II) acetate). The corresponding indole 235 was isolated from the side-product 236 by using flash chromatography. In the following, after seven steps, indole 235 gave the pentacyclic ketone 238. The latter was transformed into 12-methoxy-Na-methylvellosimine 239 through a Wittig reaction and hydrolysis. Next, the aldehyde 239 was reduced using sodium borohydride and gave 12-methoxyaffinisine 240 in a high yield (95% yield). On the other hand, the aldehyde group for the intermediate 239, using iodine (I2) and potassium hydroxide in ethanol, was oxidized to the ethyl ester 241.248 Finally, quaternization of the Nb nitrogen function in ester 241 using methyl iodide afforded the Nb-methiodide salt that was transformed into the chloride 242 after reacting with silver chloride in ethanol (Scheme 30).247 Moreover, this alkaloid was synthesized by this research group in 2006.249
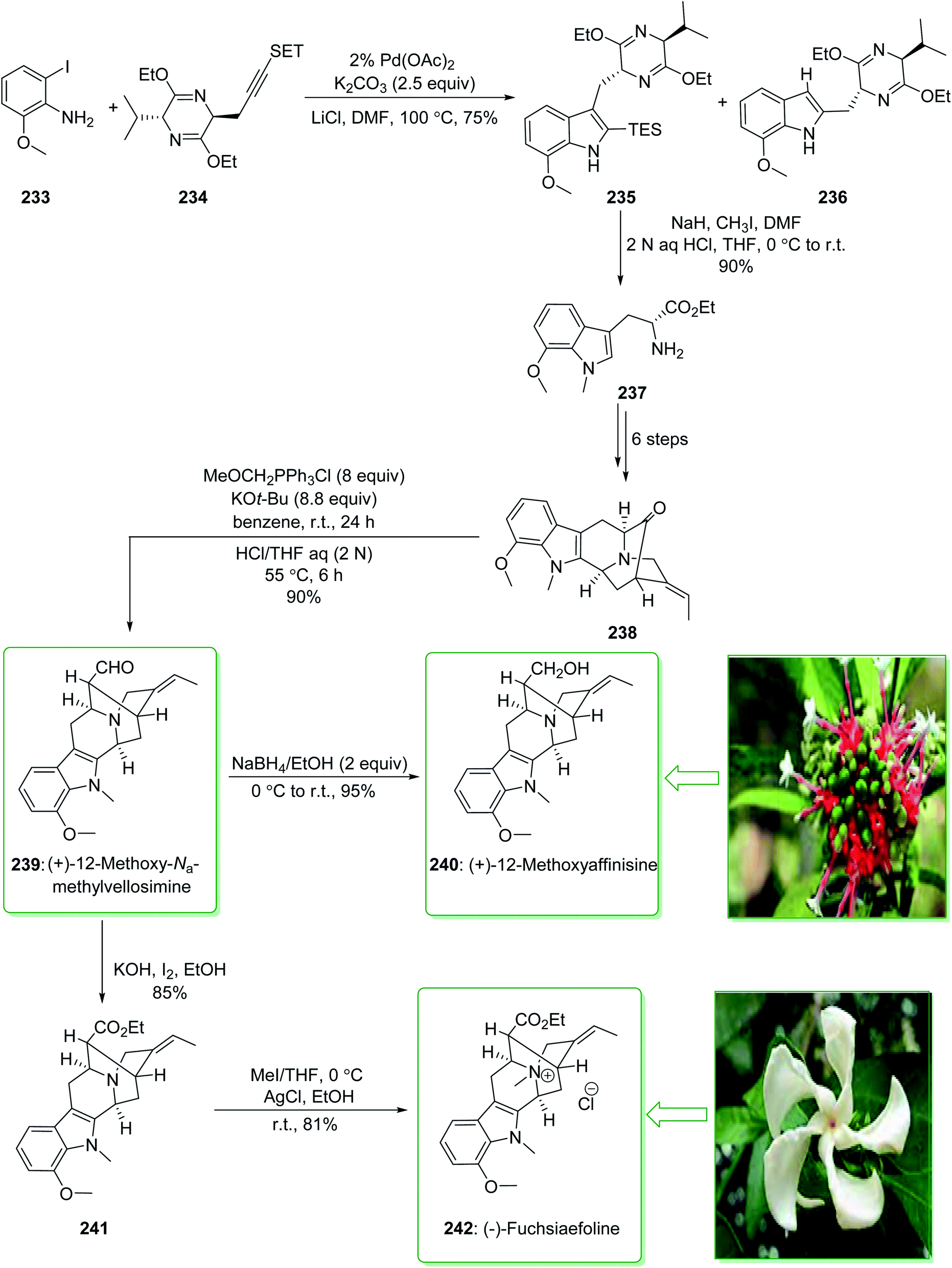 | ||
| Scheme 30 Total synthesis of (+)-12-methoxy-Na-methylvellosimine (239), (+)-12-methoxyaffinisine (240) and (−)-fuchsiaefoline (242). | ||
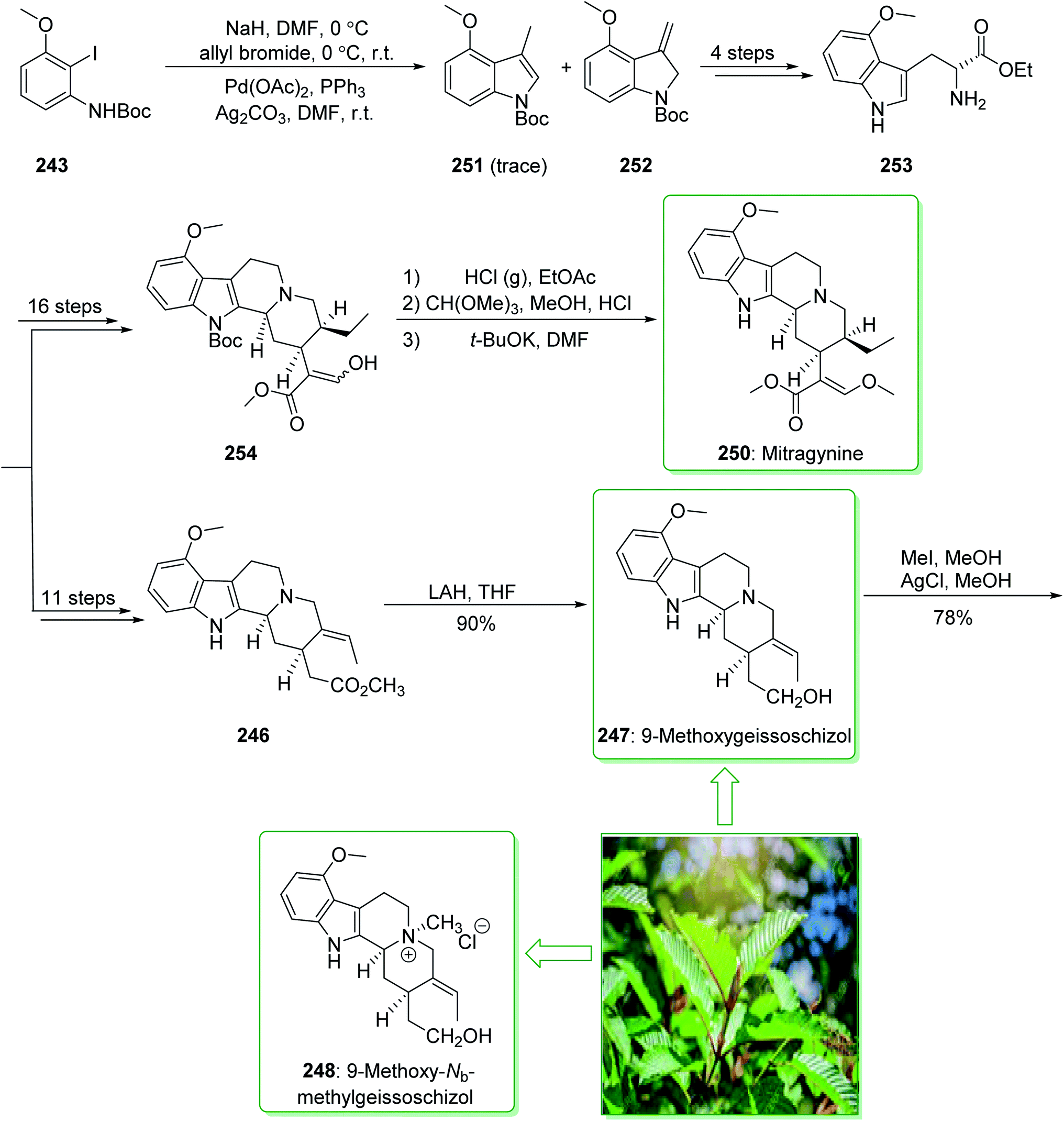
Kratom is the usual name of Mitragyne speciosa Korth, found in Thailand, which can be utilized as an opium substitute by chewing, smoking, or drinking a broth form of the kratom leaves. Noticeably, the alkaloid content of the leaves of the Mitragyne speciosa is about 0.5%, about half of which contains mitragynine (250). However, the structural identification of 250 contains a rich history,250,251 the actual structure of 250 was definitely determined by Zacharias in 1964.252 The formal investigation of the pharmacology of mitragynine (250) demonstrated that it was a central nervous system (CNS) stimulant.253 Then, in vivo and in vitro investigations showed mitragynine mostly acted on the μ-opioid receptors.254 Cook and co-workers in 2007 demonstrated255 an asymmetric approach for the formation of 4-methoxytryptophan through a regiospecific Larock heteroannulation. This method was used for the first total synthesis of 9-methoxygeissoschizol (247), 9-methoxy-Nb-methylgeissoschizol (248), and also the total synthesis of mitragynine (250). The total synthesis of mitragynine (250), 9-methoxygeissoschizol (247), and 9-methoxy-Nb-methylgeissoschizol (248) began from the Larock heteroannulation of Boc-masked 2-iodo-3-methoxyaniline (243)256 and the TES alkyne 244 (ref. 257) using palladium(II) acetate, potassium carbonate, and lithium chloride in DMF at 100 °C, which afforded the corresponding indole 245 in an 82% yield. The latter, after 12 steps, gave the 9-methoxy-substituted tetracyclic intermediate 246. In the following, ester 246 was reduced using lithium aluminum hydride and afforded 9-methoxygeissoschizol (247) in a high yield (90% yield). Next, 9-methoxy-Nb-methylgeissoschizol (248) was provided through the Nb methylation of compound 247 with MeI and followed by exchange of the iodide to the chloride in the presence of silver chloride (Scheme 31).255 On the other hand, after two steps, ester 246 gave the corresponding ester 249. Lastly, ester 249 was exposed to formylation, and the Boc substituent was removed in ethyl acetate that had been saturated with HCl gas. Lastly, acetal construction and potassium tert-butoxide catalyzed the removal of methanol and afforded the natural product mitragynine (250) (Scheme 31).
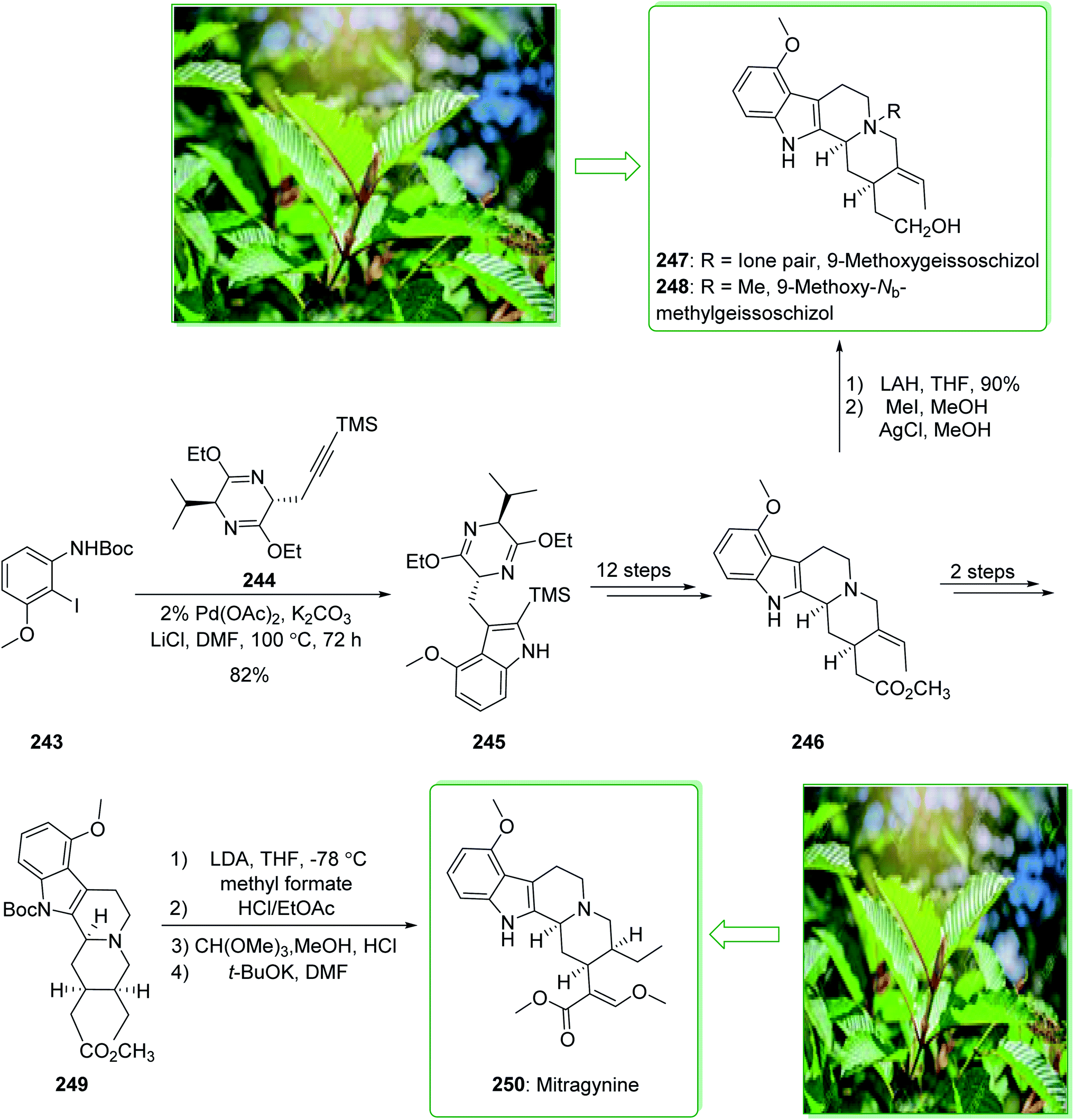 | ||
| Scheme 31 Total synthesis of 9-methoxygeissoschizol (247), 9-methoxy-Nb-methylgeissoschizol (248), and mitragynine (250). | ||
In the following, this method was used in 2009 for the total synthesis of mitragynine (250), 9-methoxygeissoschizol (247), and 9-methoxy-Nb-methylgeissoschizol (248) by Cook and co-workers.258 This research group demonstrated the total synthesis of these alkaloids through the Mori–Ban–Hegedus indole synthesis. The total synthesis of mitragynine (250), 9-methoxygeissoschizol (247), and 9-methoxy-Nb-methylgeissoschizol (248) were started from easily accessible aryl iodide 243 that was exposed to the allylic alkylation and the Mori–Ban–Hegedus indole synthesis and afforded a 1:1 mixture of 3-methylindoline (252) and 3-methylindole 251. To diminish the isomerization of the 3-methyleneindoline (252) to the 3-methylindole 251 in the Mori–Ban–Hegedus indole synthesis, it was essential to change the base from K2CO3 to Ag2CO3.259 Pleasantly, upon using Ag2CO3 in the Heck reaction, this approach was much quicker, and the reaction could be performed at ambient temperature. The 3-methyleneindoline (252) was provided, along with a trace quantity of the 3-methylindole 251. In the following, 3-methyleneindoline 252, upon four steps, gave the optically active 4-methoxytryptophan ethyl ester (253). The latter, after 11 steps, gave the chiral tetracyclic ester 246. Next, the reduction of the ester 246 in the presence of LiAlH4 in THF at room temperature provided 9-methoxygeissoschizol (247) in a high yield (90% yield). Then, methylation of compound 247 using MeI and the exchange of the iodide anion to the chloride anion using silver chloride afforded 9-methoxy-Nb-methylgeissoschizol (248).258
On the other hand, after 16 steps the optically active 4-methoxytryptophan ethyl ester (253) gave enol 254. The latter was reacted with HCl (g) in ethyl acetate to eliminate the Boc substituent, and this was followed by reaction with anhydrous methanolic HCl solution using trimethyl orthoformate to give the desired acetal intermediate. The acetal was dissolved in DMF and treated with potassium tert-butoxide and provided mitragynine (250) (Scheme 32).
 | ||
| Scheme 32 Total synthesis of 9-methoxygeissoschizol (247), 9-methoxy-Nb-methylgeissoschizol (248), and mitragynine (250). | ||
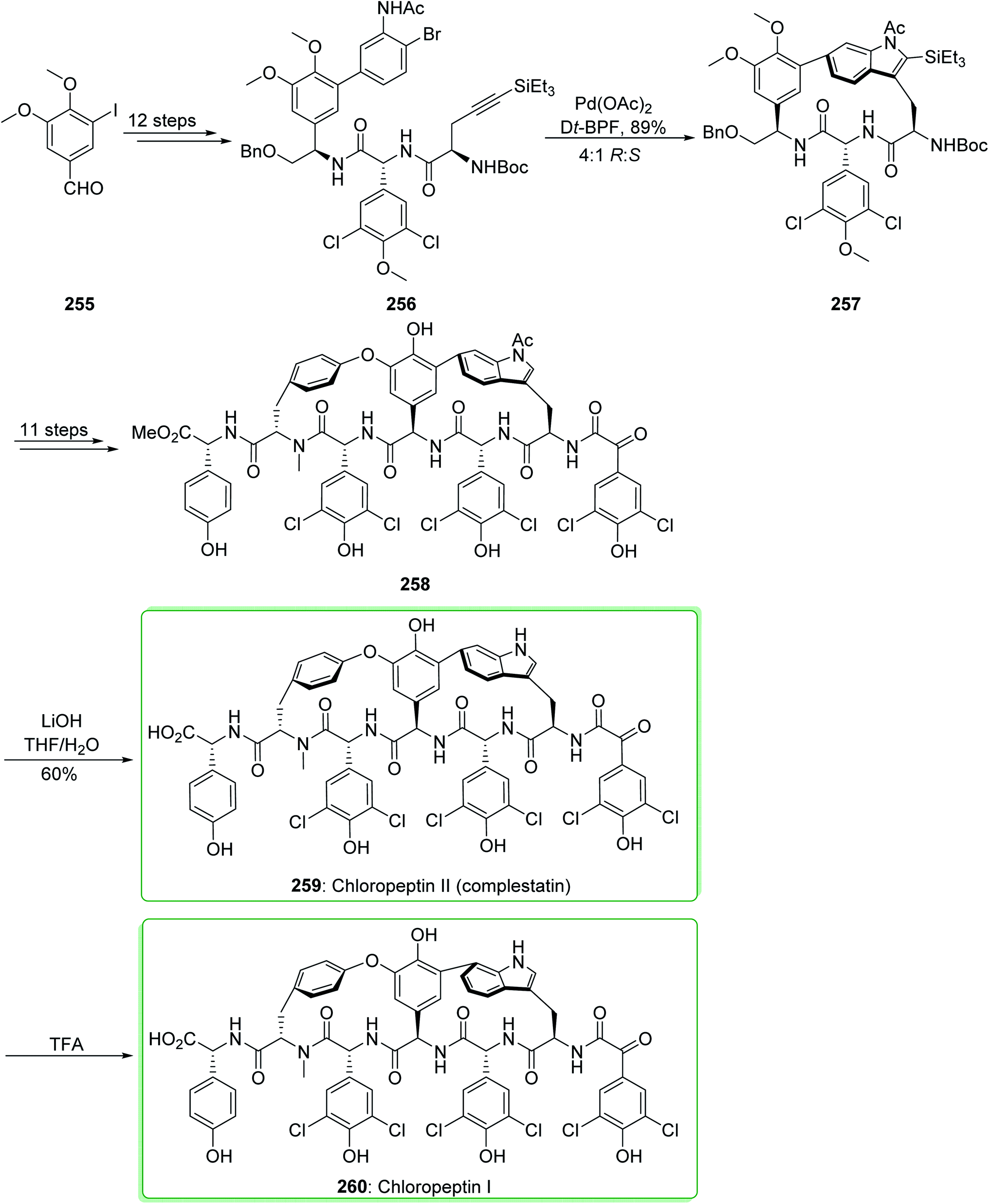
Complestatin (259, chloropeptin II) was first revealed in 1980 as an inhibitor of the alternate pathway of human complement.260 Then, the first results of its activity against HIV infectivity and its cytopathic effects were revealed.261–263 In the following, Omura demonstrated the separation of both chloropeptin I (260) and chloropeptin II (259) from Streptomyces sp. In 2009, Boger and co-workers reported264 the first total synthesis of chloropeptin II (259, complestatin). Key to this method is the usage of an intramolecular Larock indole synthesis. The first macrocyclization, using conditions that allow application of 2-bromoaniline and incorporating a removable terminal alkyne group (-SiEt3), sterically dictates the indole cyclization regioselectivity.265 The total synthesis of chloropeptin II (complestatin) (259) and chloropeptin I (260) commenced from 3-iodo-4,5-dimethoxybenzaldehyde (255), which after 12 steps afforded the cyclization substrate 256. The reaction of 256 with palladium(II) acetate in the presence of the bidentate ligand Dt-BPF and the soluble base triethylamine under reflux in toluene/acetonitrile (1:1, 1 mM at 110 °C for 1 h) efficiently afforded indole 257 (71% yield) and its (S)-atropisomer (not shown) in a superb, combined yield (89% yield) using a reaction which enables whole cyclization regioselectivity and a good atropdiastereoselectivity (4:1 R:S) to select the natural isomer. After 11 steps, indole 257 provided the penultimate precursor 258. Next, deprotection of 258 to afford 259 was performed with LiOH (THF/H2O at 0 °C for 3 h, 60% yield) in a reaction in which the indole N-acetyl substituent was eliminated quicker (<30 min) than the methyl ester hydrolysis. However, this group did not perform the reaction on a preparative scale affording an isolated yield, the clean acid-mediated transformation of 259 to 260 was performed on a small scale with both synthetic and authentic 259 and checked using liquid chromatography mass spectrometry. The two examples acted in a similar way giving 260 as the sole product, and the optimum results were obtained when it was performed with trifluoroacetic acid (50%)/H2O at 50 °C proceeding at a rate that could be easily observed (5 hours, vs. <5–15 min with neat trifluoroacetic acid at 50 °C (ref. 266)) (Scheme 33).264
 | ||
| Scheme 33 Total synthesis of chloropeptin II (259) and chloropeptin I (260). | ||
This method was also used for the total synthesis of chloropeptin II (259) and chloropeptin I (260) by Boger et al. in 2010. This research group used 3-iodo-4,5-dimethoxybenzaldehyde (255) as a starting material thus, chloropeptin II (259) and chloropeptin I (260) were synthesized in 22 and 23 synthetic steps, respectively.267
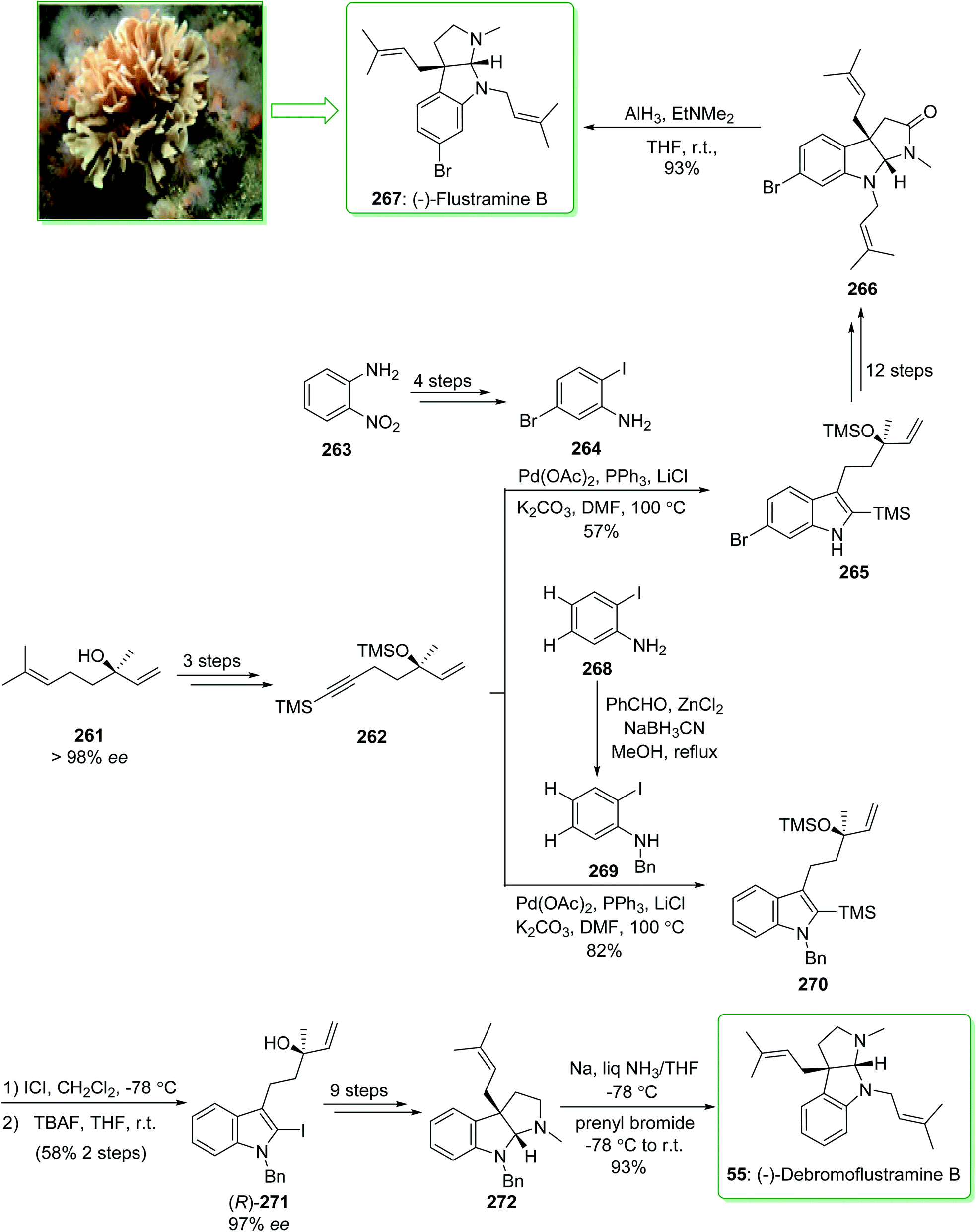
The simplest members of the hexahydropyrrolo[2,3-b]indole alkaloids are flustramines, which exhibit noteworthy biological properties.268 (−)-Flustramine B and (−)-debromoflustramine B were extracted from the marine byrozoan Flustra foliacea in 1979 by Carle and co-worker.269 (−)-Flustramine B has been known to show muscle relaxant activity, influencing both smooth and skeletal muscles.270 The hexahydropyrrolo[2,3-b]indole, bearing a quaternary carbon prenylated at C-3a, is the defining structural aspect of these alkaloids and thus various effective approaches have been reported for their synthesis.271 In 2009, Kobayashi and co-workers reported272 the total synthesis of (−)-flustramine B (267) through a one-pot intramolecular Ullmann coupling reaction and Claisen rearrangement. The enantioselective total synthesis of (−)-flustramine B (267) was started from the reaction of the iodoaniline derivative 264 (brominated iodoaniline 264 was synthesized from o-nitroaniline (263) in four steps) and silyl acetylene 262 having a chiral center (synthesized from (−)-linalool (261) in three steps). The Larock indole synthesis, using the iodoaniline derivative 264 and silyl acetylene 262, was accomplished in the presence of lithium chloride, potassium carbonate (K2CO3), triphenylphosphine, and palladium(II) acetate in DMF at 100 °C and gave the desired silyl indole 265. The latter, after 12 steps, afforded (−)-flustramide B (266). Lastly, the latter was reacted with AlH3·EtNMe2 (1.5 equiv.) at ambient temperature to decrease the unreacted lactum carbonyl substituent. As a result, (−)-flustramine B (267) was synthesized in a high yield (93% yield) (Scheme 34).272
 | ||
| Scheme 34 Enantioselective total synthesis of (−)-flustramine B (267) and (−)-debromoflustramine B (55). | ||
The enantioselective total synthesis of (−)-debromoflustramine B (55) was performed by Kobayashi and co-workers in 2013.273 In this method, the enantioselective total synthesis of (−)-debromoflustramine B was started from (R)-(−)-linalool (261), which after three steps gave the non-racemic silylalkyne 262. The Pd-mediated Larock indole synthesis was performed using Walsh's reaction conditions.274 Next, the reaction of 262 with N-benzyl-ortho-iodoaniline (269) in DMF at 100 °C in the presence of palladium(II) acetate and triphenylphosphine using lithium chloride and potassium carbonate gave the 2-silylindole 270 in a good yield (82% yield). It should be mentioned that the N-benzyl-ortho-iodoanilines 269 were synthesized through the reductive amination reaction of benzaldehyde and iodoaniline using sodium cyanoborohydride (NaBH3CN) and zinc chloride in methanol.275 In the following, the iododesilylation of 2-silylindole 270 using ICl and also the elimination of the TMS substituent with tetra-n-butylammonium fluoride afforded the tertiary allylic alcohol (R)-271 with a high ee (97% ee) and a satisfactory yield (58%). The latter, after nine steps, gave the hexahydropyrrolo[2,3-b]indole derivative 272. Lastly, reductive debenzylation of 272 using Na in liquid NH3 and quenching of the resultant amide anion with prenyl bromide led to the total synthesis of (−)-debromoflustramine B (55) in a high yield (93%) (Scheme 34).273
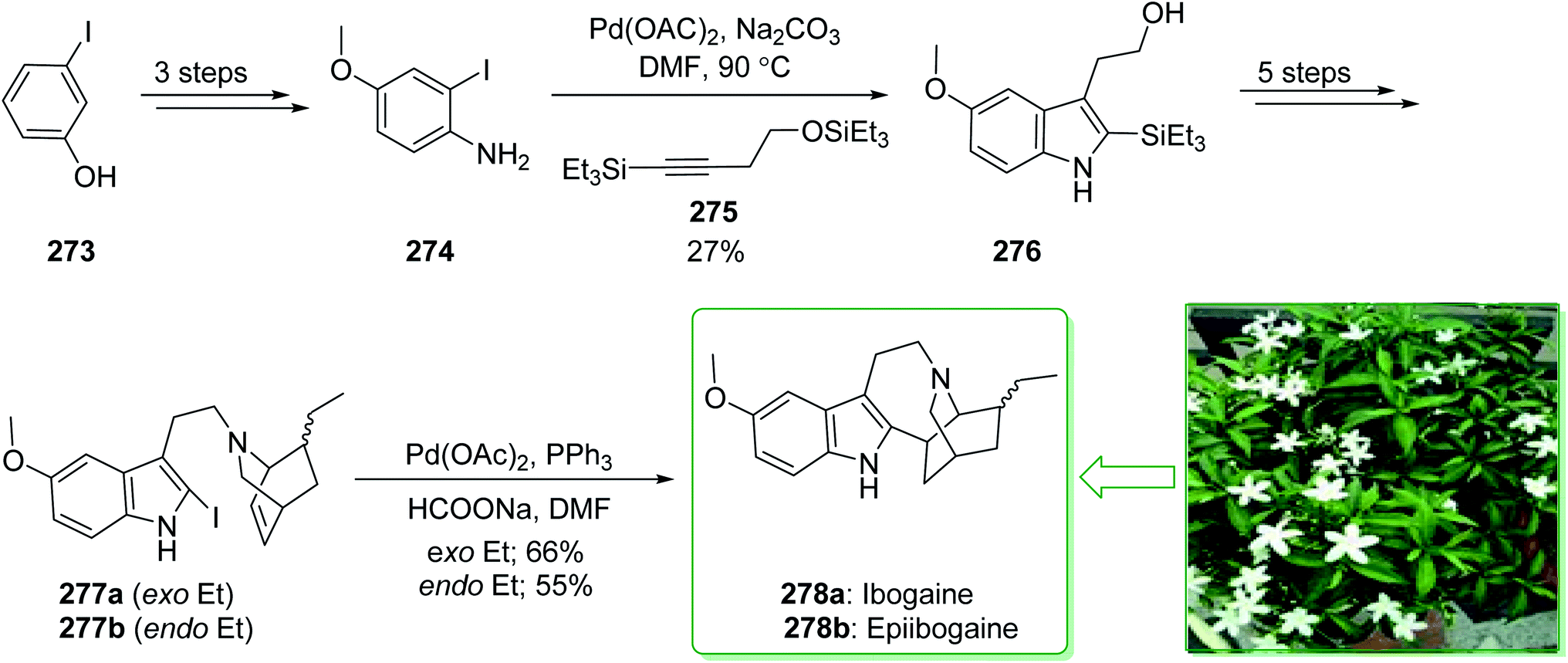
Most of the iboga alkaloids were extracted by Sundberg and co-workers in 2002 from the Tabernaemontana or Tabernanthe species of plants from the Apocynaceae family. Ibogaine (278a) is known as a naturally occurring plant indole alkaloid of the iboga family.276 Members of this group of alkaloids contain a typical bridgehead nitrogen comprising a tricyclic building block by the fusion of a seven-membered indoloazepine ring, along with a rigid isoquinuclidine ring. Ibogaine shows various pharmacological activities and it has been under active examination as an anti-addictive agent.277 In 2012, Sinha and Jana demonstrated278 the effective total synthesis of ibogaine (278a), epiibogaine (278b) and their analogues. An intramolecular reductive-Heck type cyclization reaction was applied for the formation of a seven-membered indoloazepine ring to provide the iboga-skeleton. Larock's heteroannulation reaction was used for the formation of an appropriately functionalized indole and also the Diels–Alder reaction was used for the formation of the isoquinuclidine ring. The total synthesis of ibogaine (278a) and epiibogaine (278b) were commenced from Larock's heteroannulation reaction of 4-methoxy-2-iodoaniline (274) (prepared in three steps from m-iodophenol (273)) and disilylated alkyne 275 that gave the 5-methoxy-2,3-disubstituted indole 276.279 The latter, after five steps, gave compounds 277a and 277b. Lastly, the reductive Heck coupling reaction of 277a and 277b was performed individually in DMF and afforded ibogaine (278a) in a moderate yield (66% yields) and also epiibogaine (278b) in a 55% yield. As a result, ibogaine (278a) (9.8%) and epiibogaine (278b) (9.7%) were synthesized from 4-methoxy-2-iodoaniline (274) in an overall yield of 19.5% (Scheme 35).278
 | ||
| Scheme 35 Total synthesis of ibogaine (278a) and epiibogaine (278b). | ||
The natural product, terreusinone was extracted in 2003 by Son and co-workers from the algicolous marine fungus Aspergillus terreus.280 Terreusinone includes a pyrrolo[2,3-f]indole-4,8-dione ring scaffold. Terreusinone exhibits significant UV-A masking properties, indicating terreusinone may act to shield the host organism from the destructive influences of solar UV radiation.281,282 The initial synthesis of (+)-terreusinone (286) was reported in 2011 by Sperry and Wang.283 Key steps involve a one-pot Larock indolization-Sonogashira coupling and the hydroamination of an unfunctionalized ortho-alkynylaniline mediated by a cationic Au(I) complex. The total synthesis of (+)-terreusinone (286) was started from 2-methoxy-4-nitroaniline 279, which after three steps gave bromide 280 and 1,2-dibromide 281 in a 2:1 ratio. Then, dibromide 280 was exposed to an excess of (R)-283 (prepared from racemic propargylic alcohol (±)-282 in two steps)284 through Senanayake's modified Larock indolization reaction.285 Both the Larock indolization and also the Sonogashira coupling reaction afforded indole 284 in a low yield (26% yields). The latter, after three steps, gave pyrroloindole 285. Upon oxidation of 285 with Frémy's salt under buffered conditions, (+)-terreusinone (286) was provided in a good yield. This synthetic method was performed in eight steps from commercially available starting precursors (Scheme 36).283
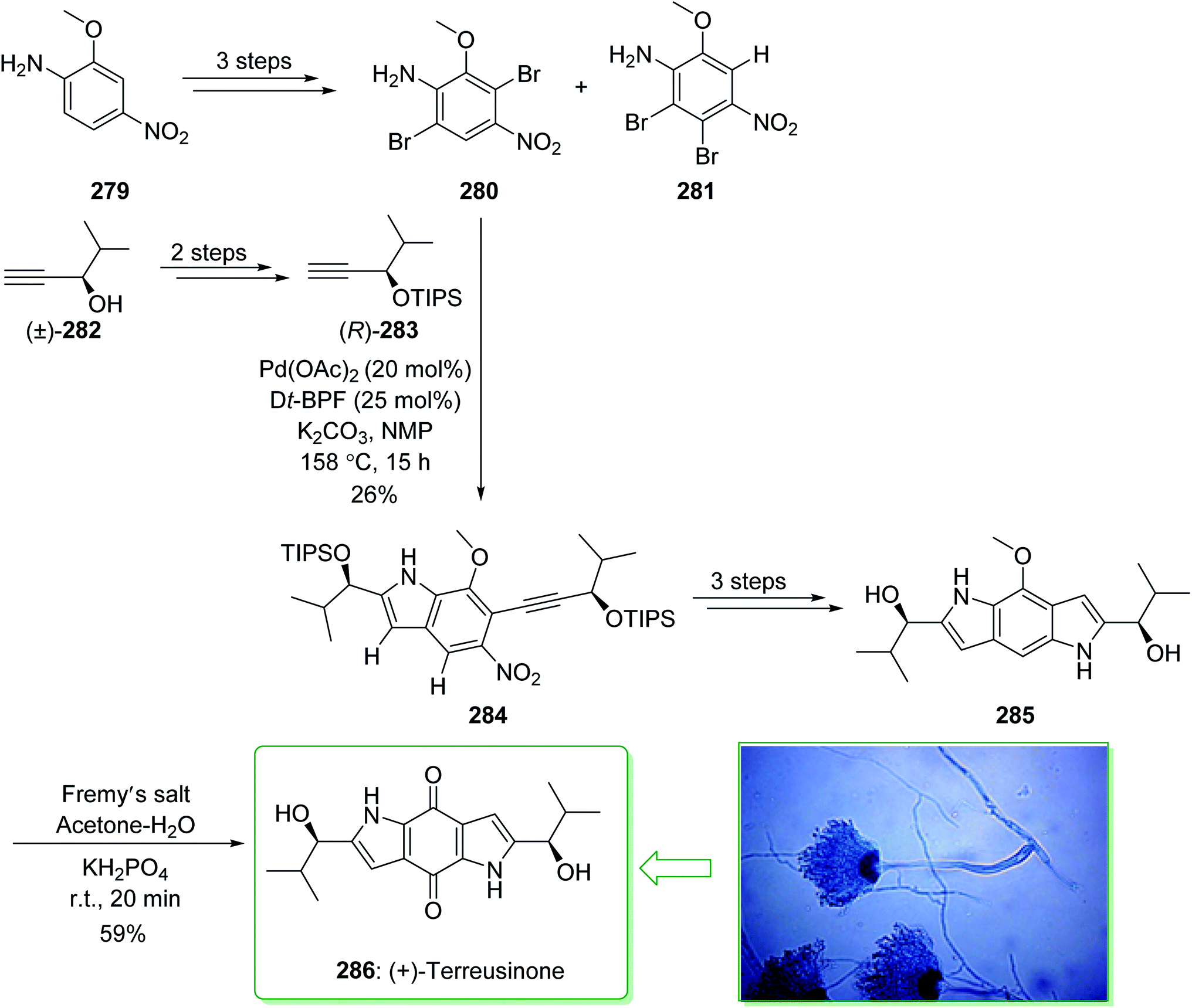 | ||
| Scheme 36 Total synthesis of (+)-terreusinone (286). | ||
An effective, bidirectional synthesis of the photoprotecting dipyrrolobenzoquinone (+)-terreusinone (286) was performed by Sperry and Wang in 2012.286 Key steps involve a Cu- and amine-free double Sonogashira reaction between an electron-rich 1,4-dibromide and a masked propargylic alcohol and also pyrrolo[2,3-f]indole construction through double hydroamination mediated by Echavarren's cationic Au(I) complex. Based on this method, the bidirectional synthesis of (+)-terreusinone (286) was commenced from the double Sonogashira reaction of the newly synthesized electron-rich 2,5-dibromo-3-methoxy-1,4-dianiline (287) and an excess of propargylic alcohol (R)-283 with Pd(OAc)2, 1,1′-bis(di-tert-butylphosphino)ferrocene and K2CO3 in N-methyl-2-pyrrolidone at 138 °C (for 2 h),287,288 which afforded the double Sonogashira product 288. (It should be mentioned that 2,5-dibromo-3-methoxy1,4-dianiline (287) was easily provided by using the simple reduction of p-nitroaniline 280).283 Next, the double hydroamination reaction of compound 288 using Echavarren's cationic Au(I) complex (acetonitrile)[(2-biphenyl)di-tert-butylphosphine]gold(I) hexafluoroantimonate (289) gave the pyrrolo[2,3-f]indole 290. It is worth mentioning that the desilylation of 290 did not perform as well as expected, returning only satisfactory yields of the desired pyrrolo[2,3-f]indole 285 with a yield of 43% over two steps from 288 (path A, Scheme 37).
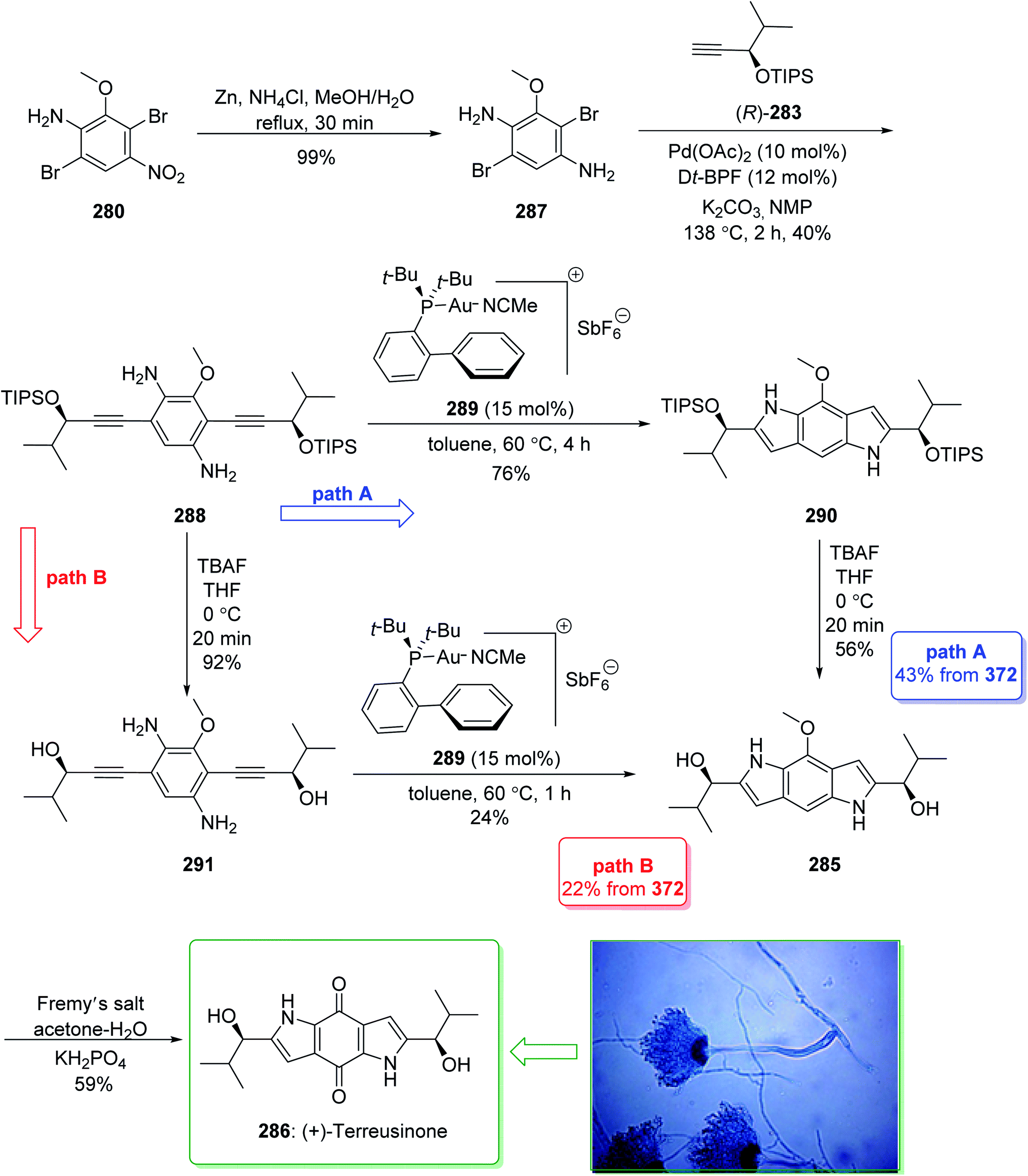 | ||
| Scheme 37 Bidirectional synthesis of (+)-terreusinone (286). | ||
In the following, in an effort to increase the above mentioned yield, initially the order of synthetic steps (from 288 → 285) was reversed and the desilylation was accomplished. However, the deprotection of 288 into 291 was performed and gave a very high yield, the double hydroamination of 287 to afford 285 was low yielding, meaning this alternate route afforded 285 in a 22% yield from 288 (path B, Scheme 37). Nevertheless, the two distinct routes depicted in Scheme 37 demonstrate the formal synthesis of the natural product (+)-286.286 The pyrrolo[2,3-f]indole 285 was oxidized and afforded terreusinone [(+)-286] (Scheme 37).283
Moreover, this method was applied by Sperry and Wang in 2013 for the total synthesis of (+)-terreusinone (286), that commenced from a one-pot Larock–Sonogashira coupling reaction.289
The short synthesis of dictyodendrins B (297) and E (105) was accomplished in just 9 and 11 steps by Jia and co-workers in 2013.290 The very convergent methodology used a Pd-mediated Larock indole synthesis and a Pd-catalyzed one-pot consecutive Buchwald–Hartwig amination/C–H activation reaction as key steps. The total synthesis of dictyodendrin B (297) and the formal synthesis of dictyodendrin E (105) were commenced from the iodination of aniline 292. In this route, aniline 292, in the presence of iodine monochloride (ICl), afforded o-iodoaniline (293) in a good yield (83% yield). The o-iodoaniline (293) and alkynyl ketone 294 were reacted under Larock reaction conditions (palladium(II) acetate, triphenylphosphine, sodium carbonate, lithium chloride, and DMF at 100 °C). Pleasantly, the reaction was very regioselective and afforded the corresponding indole 295 as the only regioisomer, although in a poorer yield (44% yield). The latter, after four steps comprising N-alkylation, bromination, a Buchwald–Hartwig amination/C–H activation reaction, and elimination by using hydrogenolysis, yielded compound 296. Next, the latter was easily transformed into dictyodendrin B (297), after a three-step sequence. Therefore, the total synthesis of dictyodendrin B (297) was accomplished in only nine steps and a 27% overall yield from aniline 292.290 On the other hand, indole 295, after nine steps, gave dictyodendrin E (105).291 Thus, the total synthesis of dictyodendrin E (105) was accomplished in only 11 steps and with a 29% overall yield from aniline 292 (Scheme 38).
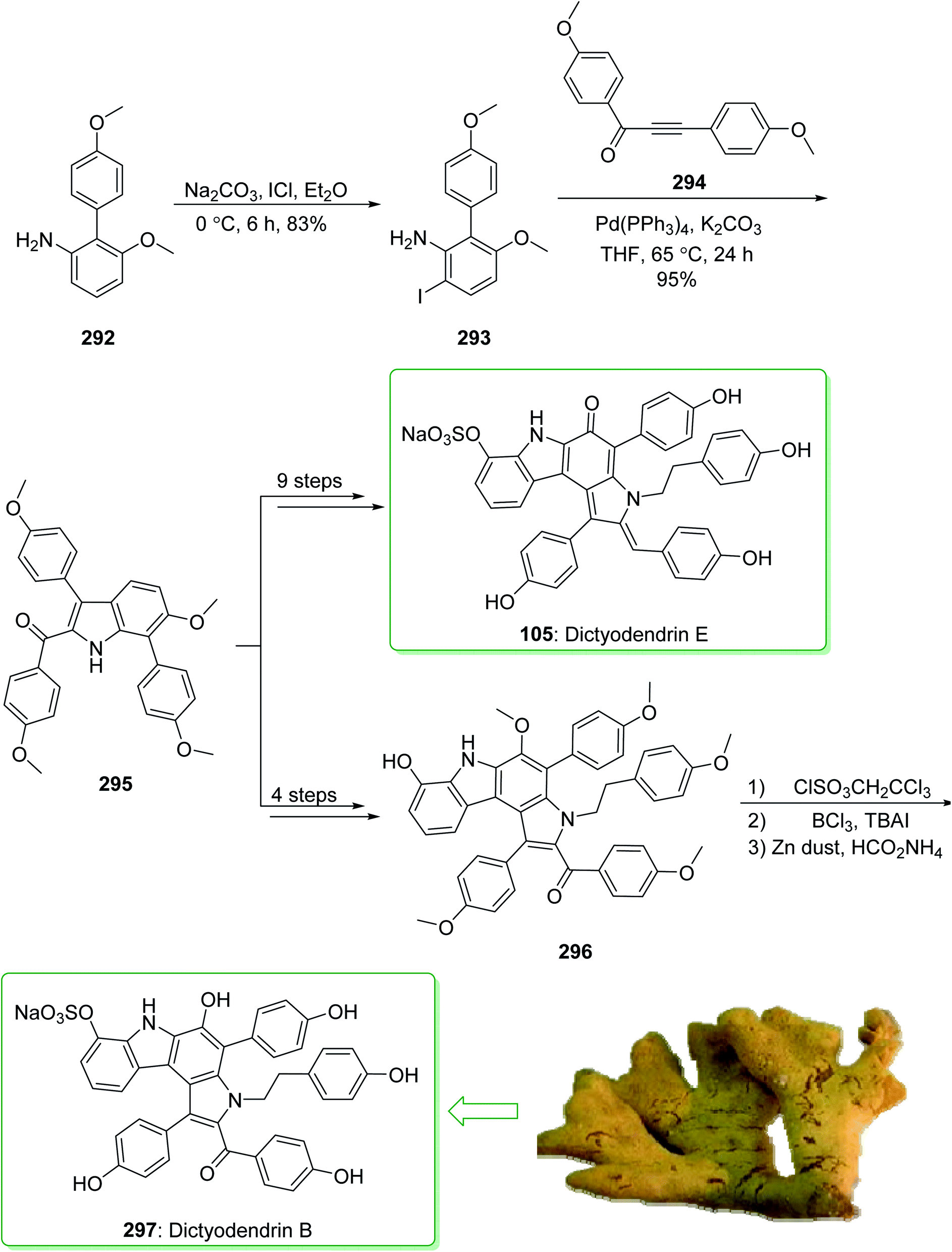 | ||
| Scheme 38 Total synthesis of dictyodendrin B (297) and the formal synthesis of dictyodendrin E (105). | ||
In 2014, Jia et al. published167 a complete explanation of the short total synthesis of dictyodendrins B and also the formal synthesis of dictyodendrin C. In this method, a palladium-catalyzed Larock indole synthesis was utilized to make the very functionalized indole unit, and a Pd-catalyzed one-pot sequential Buchwald–Hartwig amination/C–H activation reaction was utilized to form the key pyrrolo[2,3-c]carbazole unit. The total synthesis of dictyodendrins B (297) began with the Ullmann coupling reaction of p-iodoanisole (97) and 1,3-dinitrobenzene (98), that afforded biphenyl 99. The latter, after three steps, gave o-iodoaniline 298. In the following, the reaction of o-iodoaniline 298 and alkyne 299 through the Larock reaction afforded the corresponding indole 300 in a good yield (87% yield). After six steps, phenol 301 was provided from indole 300. Finally, after three steps ((1) ClSO3, CH2CCl3; (2) BCl3, TBAI; (3) Zn, HCO2NH4), the total synthesis of dictyodendrins B (297) was performed successfully (Scheme 39).167
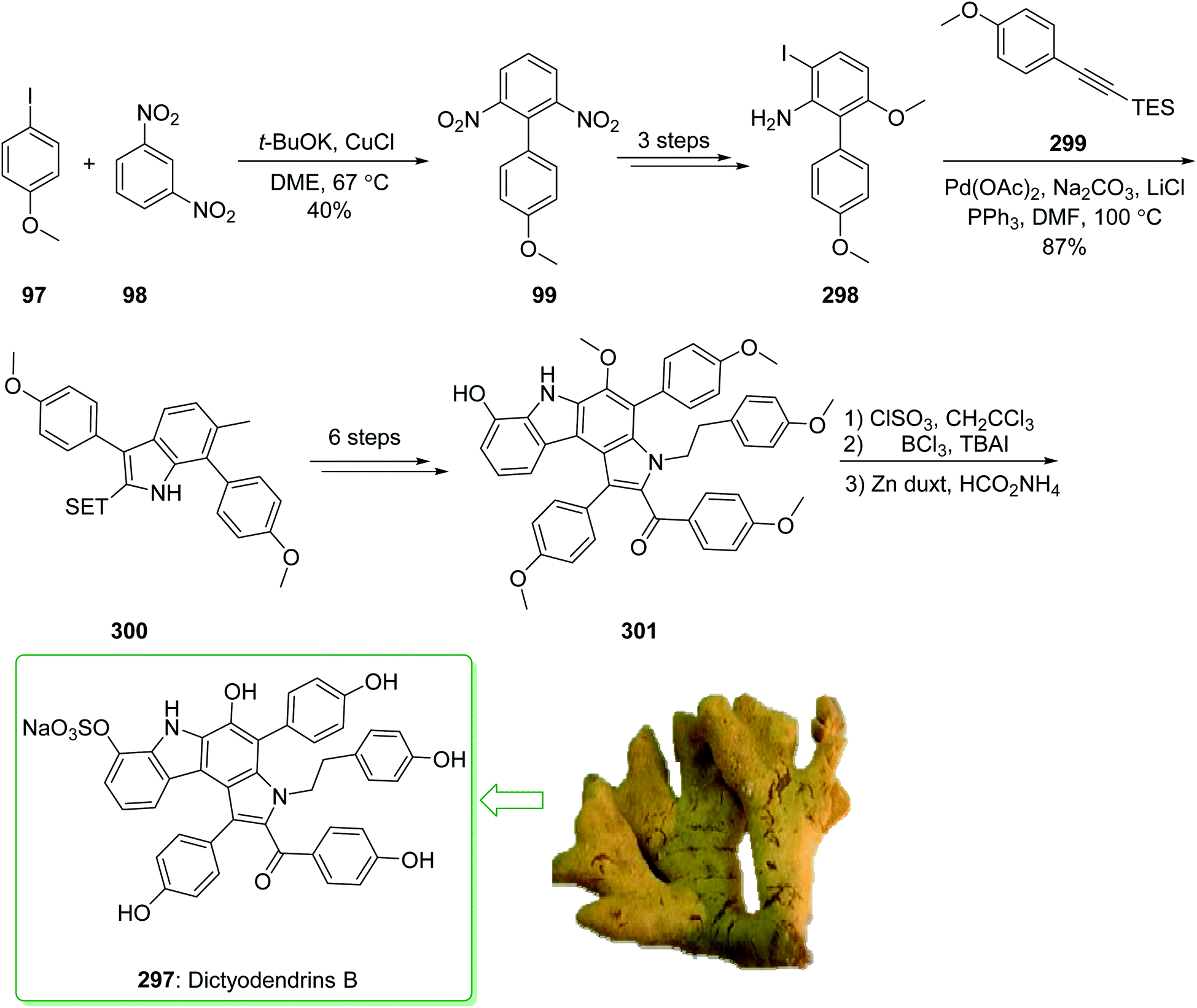 | ||
| Scheme 39 Total synthesis of dictyodendrins B (297). | ||
Moreover, the formal synthesis of dictyodendrins C (305) was commenced from the Ullmann coupling reaction of p-iodoanisole (97) and 1,3-dinitrobenzene (98), that afforded aniline 99. After four steps aniline 99 gave o-iodoaniline 302. Then, the reaction of o-iodoaniline 302 with alkyne 299 using the Larock indole reaction afforded the corresponding indole 303. The latter, after four steps, gave phenol 304, which after several steps afforded dictyodendrins C (305). As a result, the formal synthesis of dictyodendrins C (305) was accomplished in a 14% overall yield (Scheme 40).292
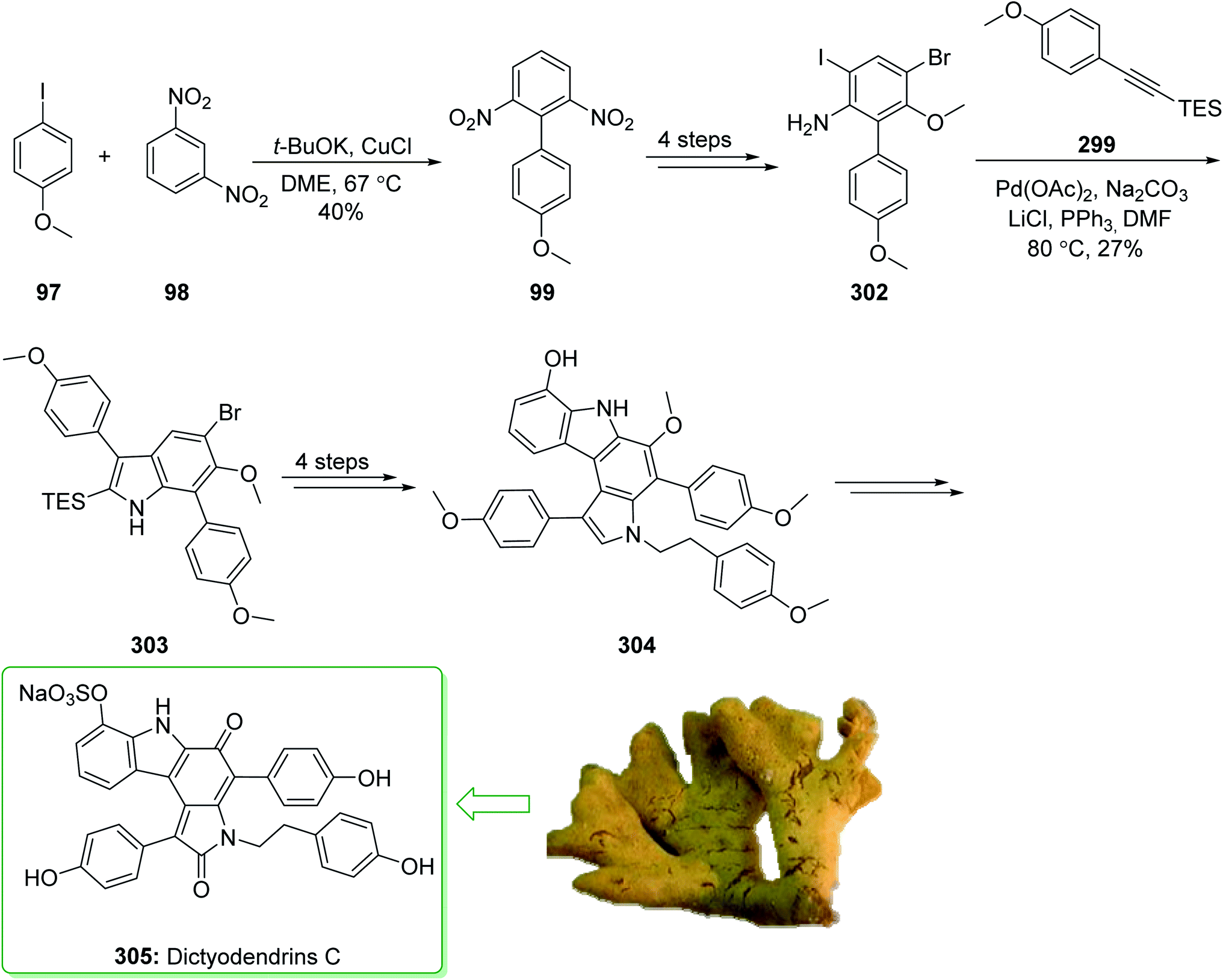 | ||
| Scheme 40 Formal synthesis of dictyodendrins C (305). | ||
Fargesine, an N-oxide alkaloid, was extracted by Zhu and co-workers from the stem and root of Evodia fargesii Dode, in which fruits are used in folk medicine for pain relief from bellyache.293 In 2013, Jia et al. developed294 the commonly used methodology for the formation of 3,4-fused tricyclic indoles through an intramolecular Larock indolization reaction. The total synthesis of fargesine (312) began with the reductive coupling reaction of aldehyde 306 and the primary amine 307 that afforded the secondary amine 308. The latter, after two steps, gave 2-iodoaniline (309). The intramolecular Larock indolization reaction of 309 gave the corresponding tricyclic product 310 in an almost quantitative yield. The latter, after three steps including selective elimination of the Boc substituent on N, reductive amination and oxidation, yielded the desired N-oxide 311. Finally, removal of the Boc group on the oxygen of 311 under basic conditions afforded fargesine (312). As a result, the total synthesis of fargesine (312) was performed in eight steps with a 15% overall yield (Scheme 41).294
 | ||
| Scheme 41 Total synthesis of fargesine (312). | ||
β-Carbolines, a large group of natural indole alkaloids, contain a tricyclic pyrido-3,4-indole ring, analogous to the tryptamine structure.295 β-Carbolines were initially found in Peganum harmala that grows in Central Africa, Asia, and also South America. Indolopyridocoline (319), was extracted in 1965 by Kaschnitz from the bark of Gonioma kamassi E. Mey.296 The commonly used synthetic method for β-carboline-containing alkaloids was reported in 2014 by Bannister and Pan.297 Two sequential Pd-catalyzed methods, a Sonagashira coupling reaction and a Larock indole annulation reaction, are the key steps. This research group tried to synthesize indolopyridocoline (319), norketoyobyrine (323), demethoxycarbonyl dihydrogambirtannine (324), rutaecarpine (329), isonauclefidine triflate (335), norepiisogeissoschizoate (336) and mitragynine (250). The total synthesis of indolopyridocoline (319) began with the Sonogashira coupling reaction of butyne-1-ol (313) and 2-bromopyridine (314), that afforded the corresponding alkyne 315. Then, the Larock indole annulation reaction of alkyne 315 and 2-bromoaniline (316) using palladium(II) acetate, 1,1′bis(diphenylphosphino) ferrocene (dppf), and potassium bicarbonate (KHCO3) in DMF at 110 °C gave the desired indole 317 with a 95% yield and a high regioselectivity. The latter, after two steps, involving the conversion of the alcohol 317 to a triflate 318 and the oxidation of tetracycle 318, gave indolopyridocoline (319) (Scheme 42).297
 | ||
| Scheme 42 Total synthesis of indolopyridocoline (319). | ||
(−)-Demethoxycarbonyldihydrogambirtannine (324) was extracted in 1973 by Peube–Locou from the leaves of Ochrosia lifuana and also Ochrosia miana (Apocynaceae).298 Norketoyobyrine (323) and isonauclefidine (335) were extracted in 2011 by Xu and co-workers from the bark of Anthocephalus chinensis.299 The genus Anthocephalus, a member of the tribe Naucleaceae in the family Rubiaceae, is known in southern China and southern Asia. Their barks have been utilized for treating blood diseases, uterine complaints, dysentery and leprosy in ‘Ayurvedo’, an ancient Indian form of medicine.300 The total syntheses of norketoyobyrine (323) and demethoxycarbonyl dihydrogambirtannine (324) were started from the Sonogashira coupling reaction of isoquinolin-3-yl trifiate (320) and butyne-1-ol (313) that afforded alkyne 321.297 The Larock indolization of alkyne 321 and 2-bromoaniline (316) afforded the corresponding indole isoquinoline 322. In the following, indole isoquinoline 322, after two synthetic steps containing cyclization and oxidation, gave norketoyobyrine (323). In addition, indole isoquinoline 322, after two steps involving cyclization and reduction, yielded demethoxycarbonyl dihydrogambirtannine (324) (Scheme 43). In addition, the alternative synthesis of demethoxycarbonyl-dihydrogambirtannine (324) and norketoyobyrine (323) have been reported.301,302
 | ||
| Scheme 43 Total synthesis of norketoyobyrine (323) and demethoxycarbonyl dihydrogambirtannine (324). | ||
Furthermore, the total synthesis of rutaecarpine (329) was started from the Sonogashira reaction of methyl ether 325 which contained a chloro group and butyne-1-ol (313) that gave alkyne 326. The Larock indolization afforded a regioisomeric mixture that exhibited a preference for the desired 2-heteroaryl indole product 328. Potassium bicarbonate as a base afforded an 8:1 preference for 328 in a very good yield (82%). Next, the cyclization of 328 using HCl/n-butanol showed a preference for rutaecarpine (329) (16:1, 81% yield) (Scheme 44).297
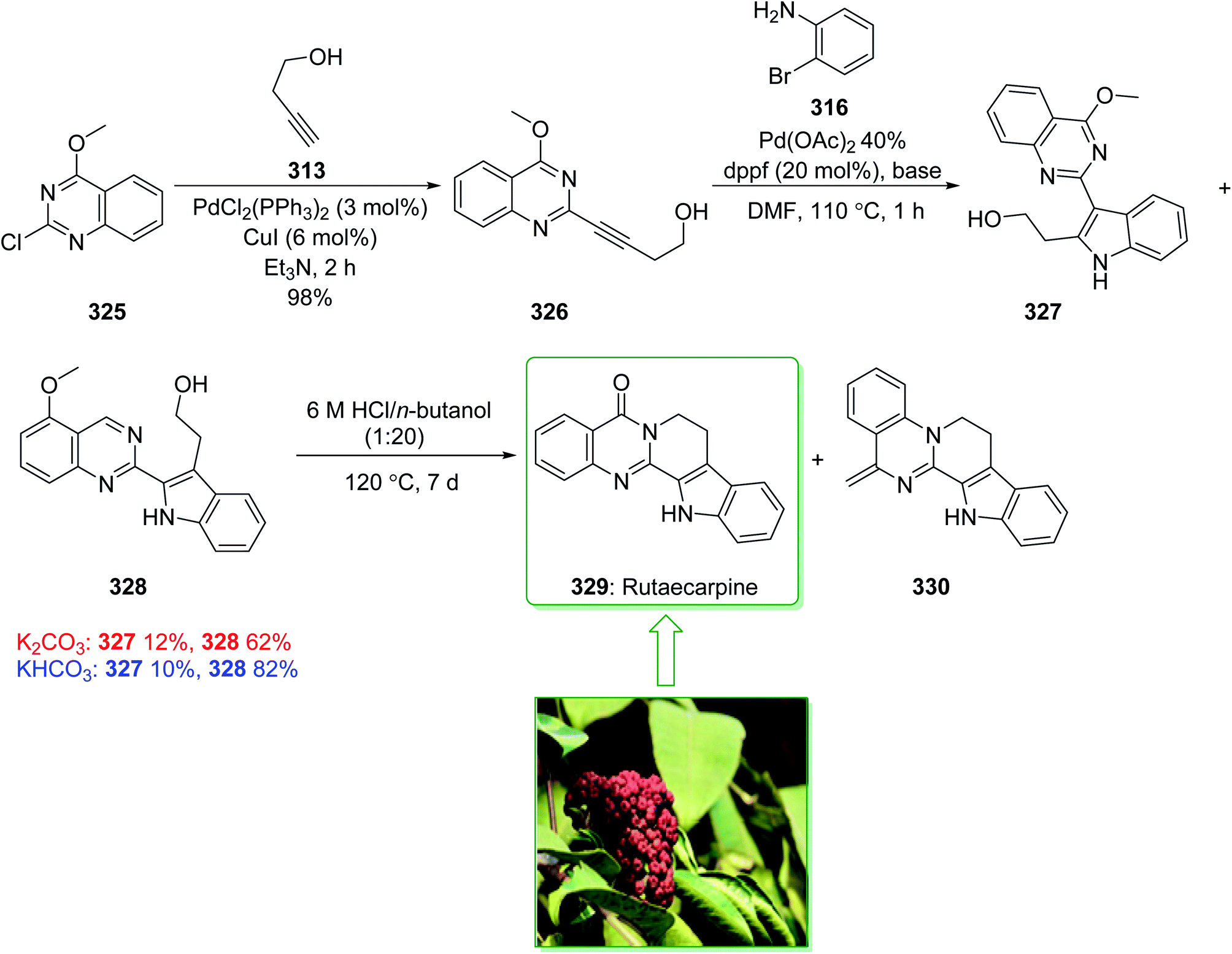 | ||
| Scheme 44 Total synthesis of rutaecarpine (329). | ||
The total synthesis of isonauclefidine triflate (335) and the formal synthesis of norepiisogeissoschizoate (336) began with the Sonogashira coupling of the tert-butyl ester functionalized chloropyridine 331 and butyne-1-ol (313) that afforded the corresponding alkyne 332. Then, the Larock reaction of alkyne 332 and 2-bromoaniline (316) provided indole 333, which after the cyclization reaction gave tetracycle 334. Finally, isonauclefidine triflate (335) was provided from the reaction of tetracycle 334 in the presence of TFA. Moreover, the formal synthesis of norepiisogeissoschizoate (336) was accomplished after three steps from tetracycle 334 (ref. 303) (Scheme 45).297
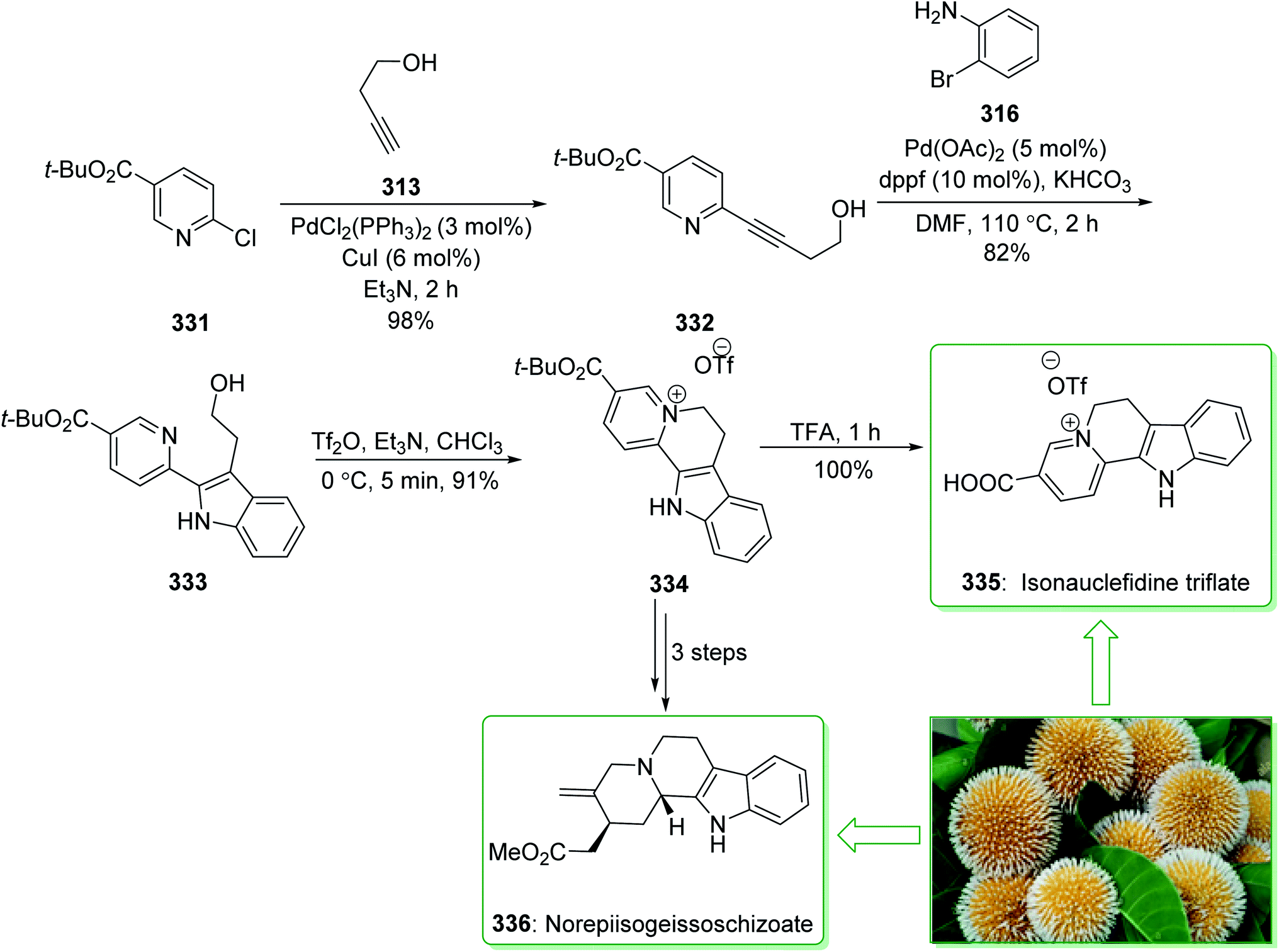 | ||
| Scheme 45 Total synthesis of isonauclefidine triflate (335) and the formal synthesis of norepiisogeissoschizoate (336). | ||
Furthermore, the formal synthesis of mitragynine (250) started with the Larock indolization of alcohol 337 and electron-rich aniline 338, which yielded the corresponding tetracycle 340. The latter after four steps gave mitragynine (250)304 (Scheme 46).297
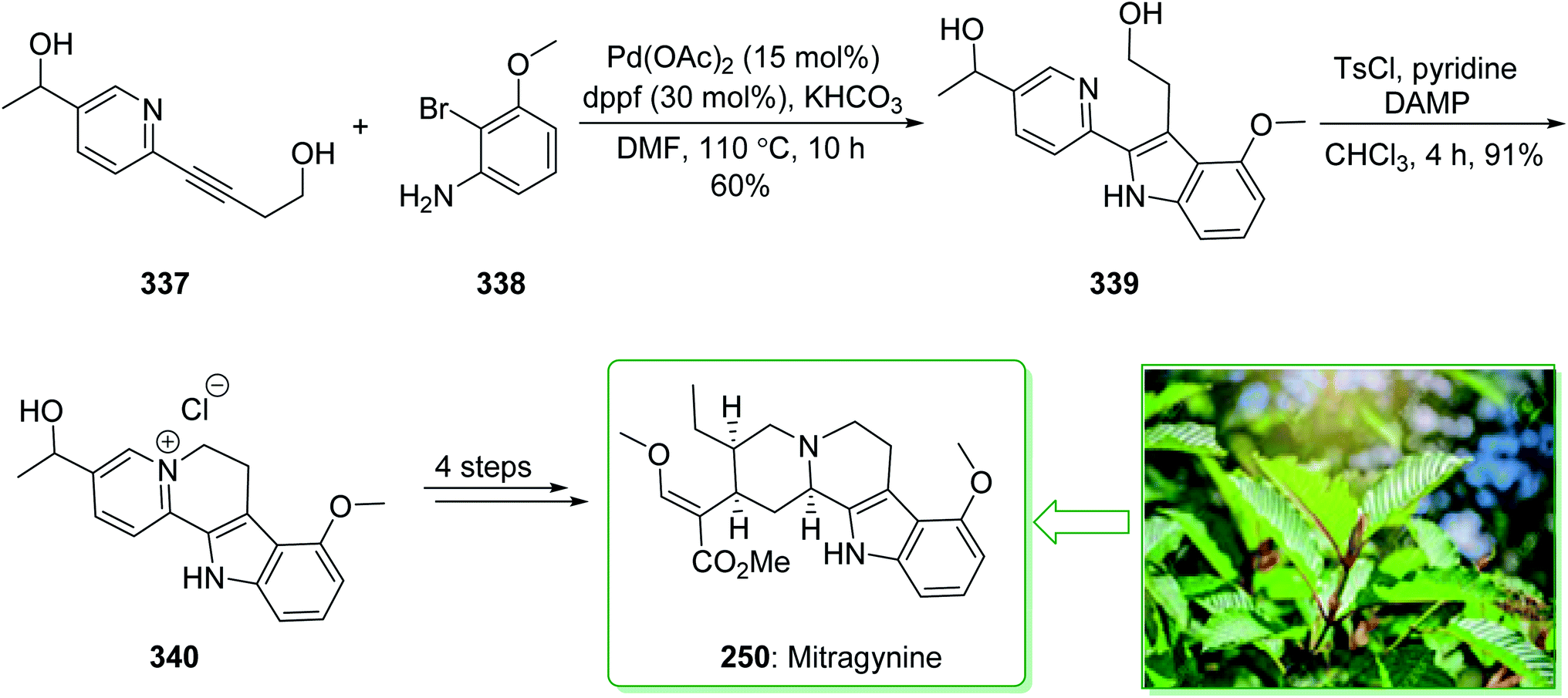 | ||
| Scheme 46 Formal synthesis of mitragynine (250). | ||
Dragmacidin D (349), a secondary metabolite, was extracted by Wright and co-workers in 1992 from a deep-water marine sponge of the genus Spongosorites.305 Dragmacidin D was known to be an active inhibitor of the serine/threonine phosphatases PP2A and PP1. Additional biological properties that have been reported for the dragmacidins involve anti-bacterial, anti-viral, and anti-fungal properties, and also in vitro cytotoxicity against A549 human lung, HCT-8 human colon and P388 murine leukemia.306 In 2015, Zakarian et al. reported the enantioselective synthesis of dragmacidin D (349) in 10 steps.307 The asymmetric total synthesis of dragmacidin D (349) was achieved from the central heteroannulation of propanoate 343 (prepared in four steps from 4-methoxy-2-bromoacetic acid 341) and pyrazine 344 (prepared in four steps from 2,6-dichloropyrazine (342)) in the presence of the [1,1′-bis(di-tert-butylphosphino)ferrocene] PdCl2 catalyst 345 (central Larock indole synthesis) and afforded the 2,3,4,7-tetrasubstituted indole 346. The latter, after three steps, gave the thioester 347. Finally, the total synthesis of dragmacidin D (349) was completed after two additional steps, including the CuOAc-mediated acyl cross-coupling reaction of the thioester 347 with stannane 348 (bearing a guanidinyl) and the cyclocondensation of the resulting guanidinylmethyl ketone using acidic conditions in the presence of trifluoroacetic acid in dichloromethane. Upon purification by using reverse-phase preparative high performance liquid chromatography (HPLC), 15 mg of the trifluoroacetic acid salt of synthetic dragmacidin D (349) was isolated as a brownish red foam (Scheme 47).307
 | ||
| Scheme 47 Asymmetric total synthesis of dragmacidin D (349). | ||
Indole alkaloids bearing a 3α-amino-hexahydropyrrolo-[2,3-b]indole motif were found in various natural products.308 Among the alkaloids in this family, psychotriasine (355) was extracted from Psychotria calocarpa in 2010 by Hao et al.309 In 2015, Deng et al. demonstrated310 the total synthesis of (−)-psychotriasine (355), which relied on the advanced intermediates of 3α-amino-hexahydropyrrolo[2,3-b]indole. To make these structural scaffolds, a cascade reaction containing a BINOL-derived phosphoric anion-paired catalyst for asymmetric or diastereoselective azo-coupling/iminium-cyclizations was established. Other key steps of the synthesis include a sterically hindered amination using hypervalent iodine reagents and the Larock annulation. The asymmetric total synthesis of (−)-psychotriasine (355) was started from 3α-amino-hexahydropyrrolo[2,3-b]indole 350. The latter, after two steps, gave tetrahydropyrrolo[2,3-b]indole 351. Then, the Larock cyclization of 351 and an identified alkyne framework (N-(methoxycarbonyl)-4-(trimethylsilyl)-3-butynylamine) (352), using a palladium(II) acetate catalyst, Dt-BPF ligand, potassium carbonate additive and N-methyl-2-pyrrolidone, provided the tryptamine dimer compound 353 in a good yield (83% yield). Then, N-deprotection was used to remove the Bz group using DIBAL-H and carbamation of the amine using ClCO2Me gave 354 in a 64% yield (over two steps). Finally, N-deprotection of 354 to remove the Boc group using trifluoroacetic acid and further reduction using Red-Al provided the desired natural product (−)-psychotriasine (355) at >96% ee (Scheme 48).310
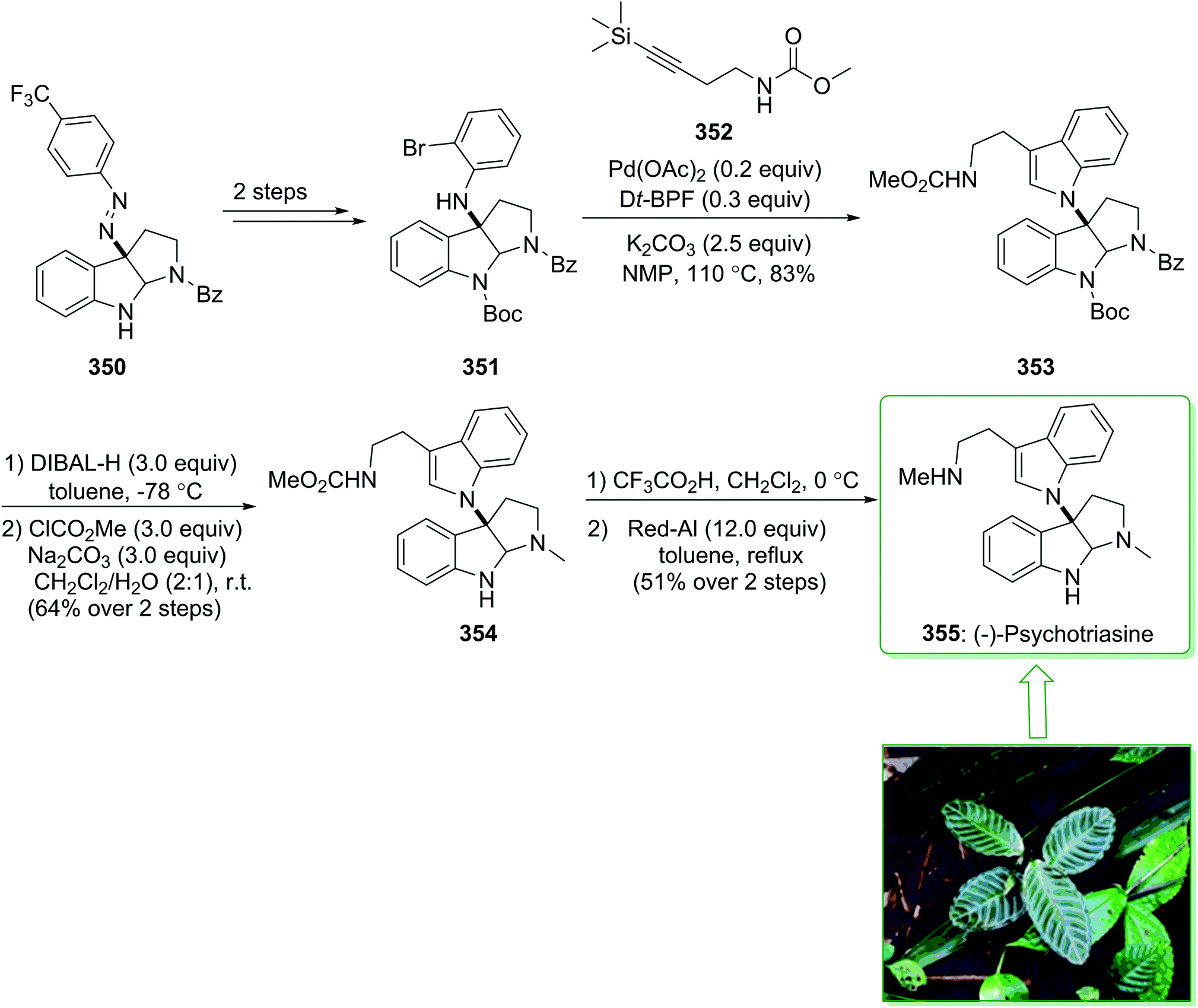 | ||
| Scheme 48 Asymmetric total synthesis of (−)-psychotriasine (355). | ||
(+)-Pestalazine B (358) containing a C3(sp3)–N1 bridge was initially extracted from the plant pathogenic fungus Pestalotiopsis theae in 2008 by Ding et al.311 It is worth mentioning that its structure was revised in 2010 by de Lera and co-worker upon total synthesis.312 The total synthesis of (+)-pestalazine B (358) was commenced from the easily accessible compound 356, and after three steps afforded compound 357. Lastly, the Larock annulation of 357, with the sterically bulky ligand Dt-BPF, afforded (+)-pestalazine B (358) in a 16.2% overall yield over this four-step total synthesis (from the known compound 356) (Scheme 49).
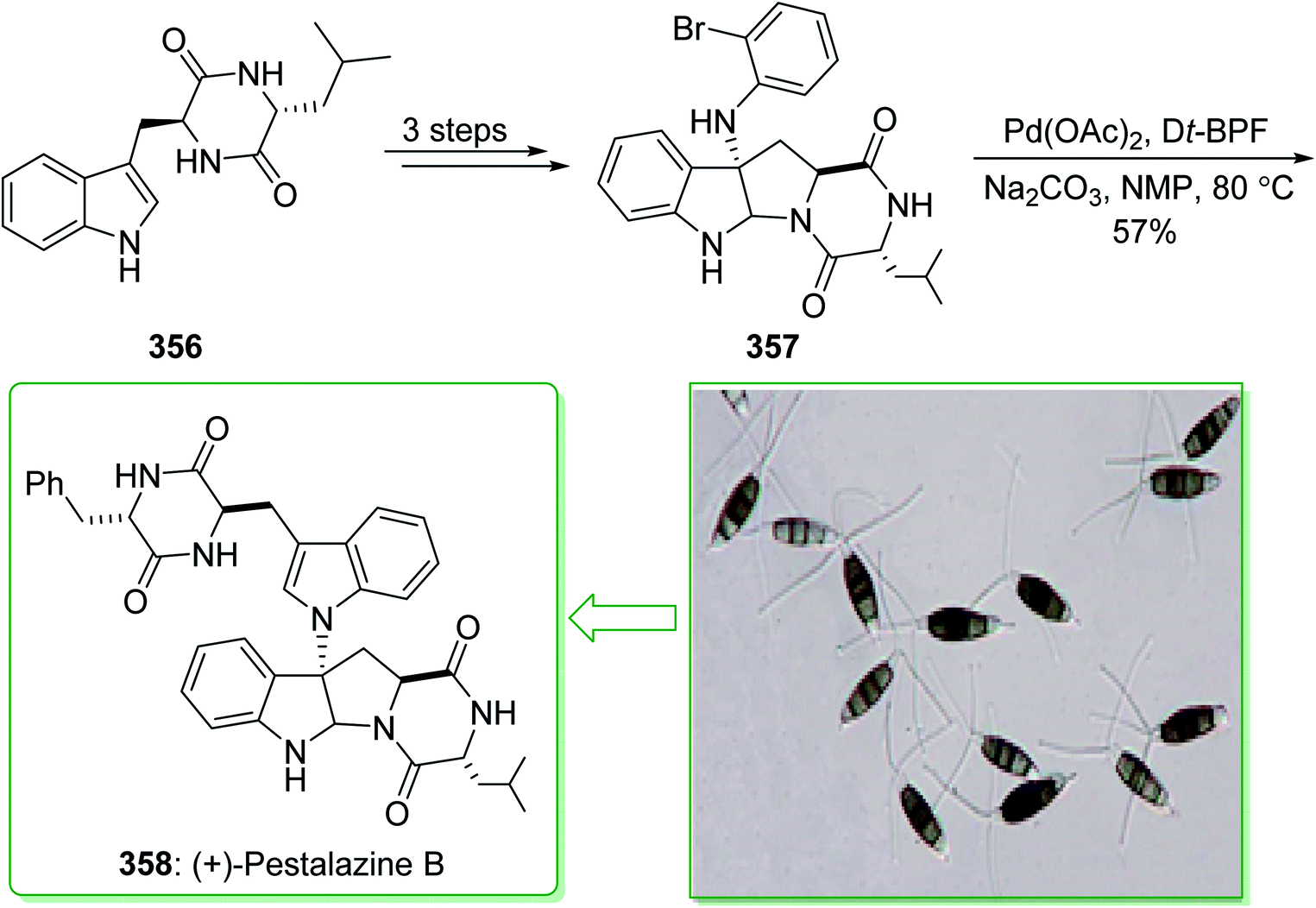 | ||
| Scheme 49 Total synthesis of (+)-pestalazine B (358). | ||
The ergot alkaloids manufactured by the fungus Claviceps purpurea are a different group of naturally occurring indole compounds that show a wide range of powerful pharmacological properties.313,314 Among the ergot alkaloids (EAs) family of naturally occurring compounds, lysergic acid315 is the most broadly identified member and various semisynthetic derivatives have been clinically utilized for treating various neurological diseases. In 2015, Boger et al. demonstrated316 that the total synthesis of dihydrolysergic acid (363) and dihydrolysergol (364) relied on a palladium (0)-mediated intramolecular Larock indole cyclization for the formation of the tricyclic indole and also a consequently inverse electron demand Diels–Alder reaction. The total synthesis of dihydrolysergic acid (363) and dihydrolysergol (364) were commenced from 2-bromoaniline (359). The latter, after five steps, gave homopropargylic alcohol 360, that through an intramolecular palladium(0)-mediated Larock indole annulation gave the tricyclic indole 361. The latter, after eight steps yielded the intermediate 362, which after hydrolysis of the methyl ester (1 N NaOH/MeOH at 40 °C, 3 h) afforded dihydrolysergic acid (363). Moreover, reduction of the intermediate 362 (using LiAlH4, THF at 0 °C) yielded dihydrolysergol (364) (Scheme 50).316
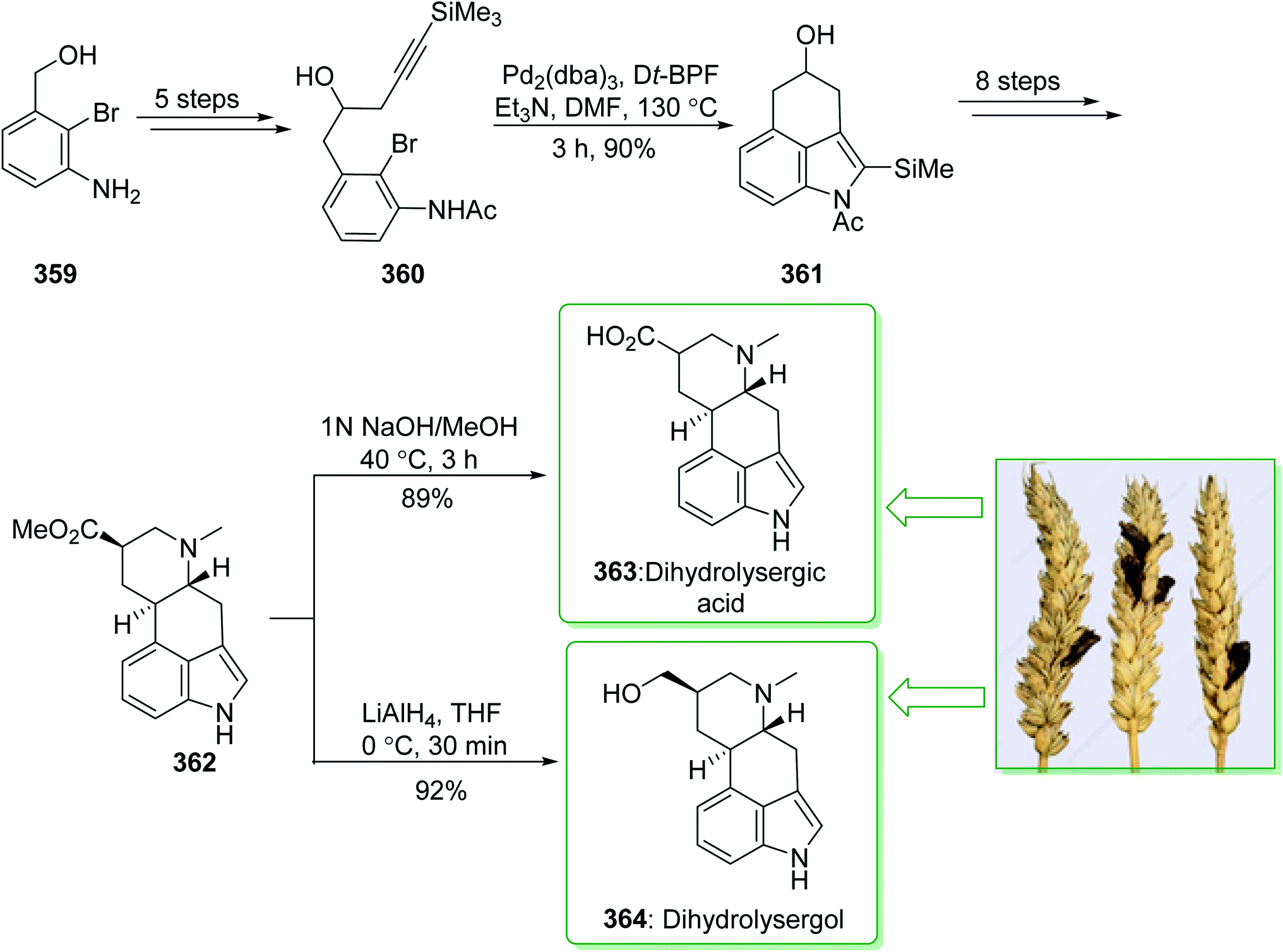 | ||
| Scheme 50 Total synthesis of dihydrolysergic acid (363) and dihydrolysergol (364). | ||
An efficient method for the total synthesis of eight ergot alkaloids was reported in 2017 by Jia et al.317 This method permits the first total synthesis of pyroclavine (371), pibocin (373), 9-deacetoxyfumigaclavine C (372), fumigaclavine G (374), festuclavine (370), and dihydrosetoclavine (369). The key aspect of the synthesis is the usage of a palladium-mediated intramolecular Larock indole annulation/Tsuji–Trost allylation cascade to make the tetracyclic unit in one step. The total synthesis of festuclavine (370), pyroclavine (371), pibocin A (373), 9-deacetoxyfumigaclavine C (372), fumigaclavine G (374) and dihydrosetoclavine (369) began with bromide 365. After six steps, 365 gave homopropargylic amines 366a and b. In the following, the intramolecular palladium-mediated Larock indole annulation of 366a was accomplished using palladium(II) acetate and Me-phos at 100 °C and provided the corresponding tricyclic indole 367 in a high yield (92% yield). Astonishingly, when the reaction was performed on a gram scale, an undesired tetracyclic compound 368 was obtained in a small quantity. It should be mentioned that compound 368 most probably occurred as a result of using the Tsuji–Trost allylation of the tricyclic indole 367. Meanwhile, compound 368 could be utilized as an advanced intermediate to simplify the synthesis and reduce the synthetic pathway, additional optimization of the cascade reaction of 366a to increase the yield of 368 was examined. When the reaction was performed using palladium(II) acetate (0.3 equiv.) and Me-phos (0.9 equiv.), the desired compound 368 was provided in a good yield (65% yield) along with 25% of 367. Following this, the tetracyclic basic framework 368, after four steps, gave dihydrosetoclavine (369).317
In addition, the total synthesis of festuclavine (370) and pyroclavine (371) were started from bromide 365, and after six steps gave 366a and b. After reacting Pd(OAc)2 (20 mol%) and Me-phos (40 mol%), K2CO3, and LiCl in DMF at 100 °C, 366a and b afforded 367, and after a further five steps these gave compounds 370 and 371. In the following, this research group examined the formation of pibocin A (373), 9-deacetoxyfumigaclavine C (372) and fumigaclavine G (374) using the direct functionalization of 370 and 371. The chemoselective bromination reaction of the 2-position of festuclavine (370) using N-bromosuccinimide yielded pibocin A (373) in a good yield (80% yield). Moreover, the chlorination of 370 with tert-butyl hypochlorite and reaction with prenyl-9-BBN using triethylamine gave 9-deacetoxyfumigaclavine C (372) in a good yield (80% yield). Similarly, pyroclavine (371) was converted to fumigaclavine G (374) in a satisfactory yield (50% yield) (Scheme 51).317
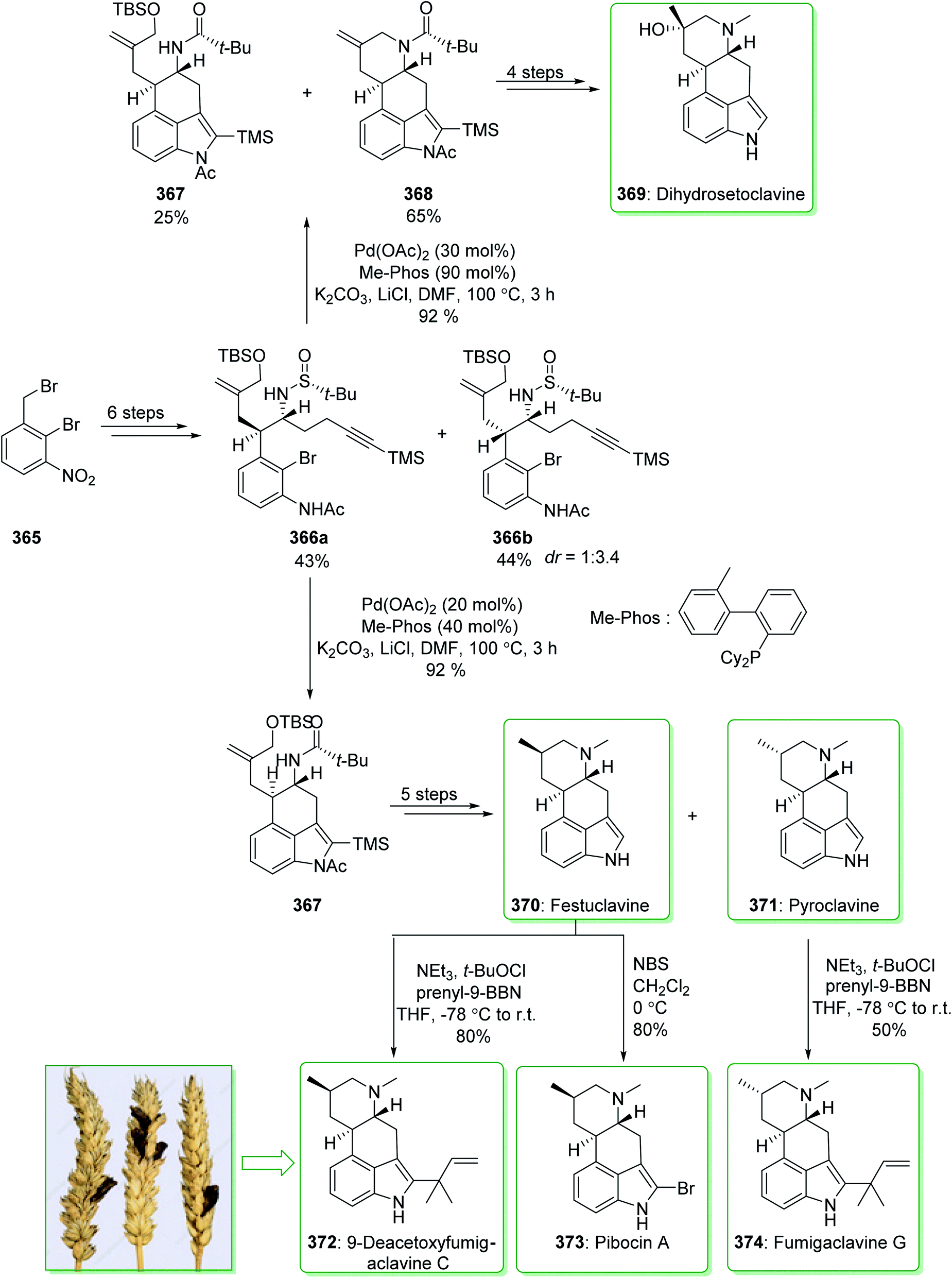 | ||
| Scheme 51 Total synthesis of festuclavine (370), pyroclavine (371), pibocin A (373), 9-deacetoxyfumigaclavine C (372), fumigaclavine G (374) and dihydrosetoclavine (369). | ||
Aspergilazine A, dimerized by two diketopiperazines cores through an unusual N-1 to C-6 linkage, was extracted in 2012 by Gu and co-workers from the marine-derived fungus Aspergillus taichungensis ZHN-7-07.318 The fungal strain Aspergillus taichungensis ZHN-7-07 along with the mangrove plant Acrostichum aureum, are members of the Aspergillus Candidi family.319,320 In 2016, Reisman et al. demonstrated the total synthesis of the bisindole natural product (−)-aspergilazine A (379).321 The total synthesis of the dimeric diketopiperazine natural product (−)-aspergilazine A (379) began with a Buchwald–Hartwig coupling reaction between 1-bromo-2-iodobenzene (375) and diamine 376, which afforded dibromide (377). Next, exposure of a mixture of dibromide 377 and alkyne 378 to Pd[P(t-Bu)3]2 and Cy2NMe in 1,4-dioxane at 80 °C provided bis(triethylsilyl)-(−)-aspergilazine A in a satisfactory yield (62% isolated yield), demonstrating an average reaction proficiency of 79% per indolization. Finally, HCl-catalyzed desilylation efficiently provided the natural product (−)-aspergilazine A (379) (Scheme 52).321
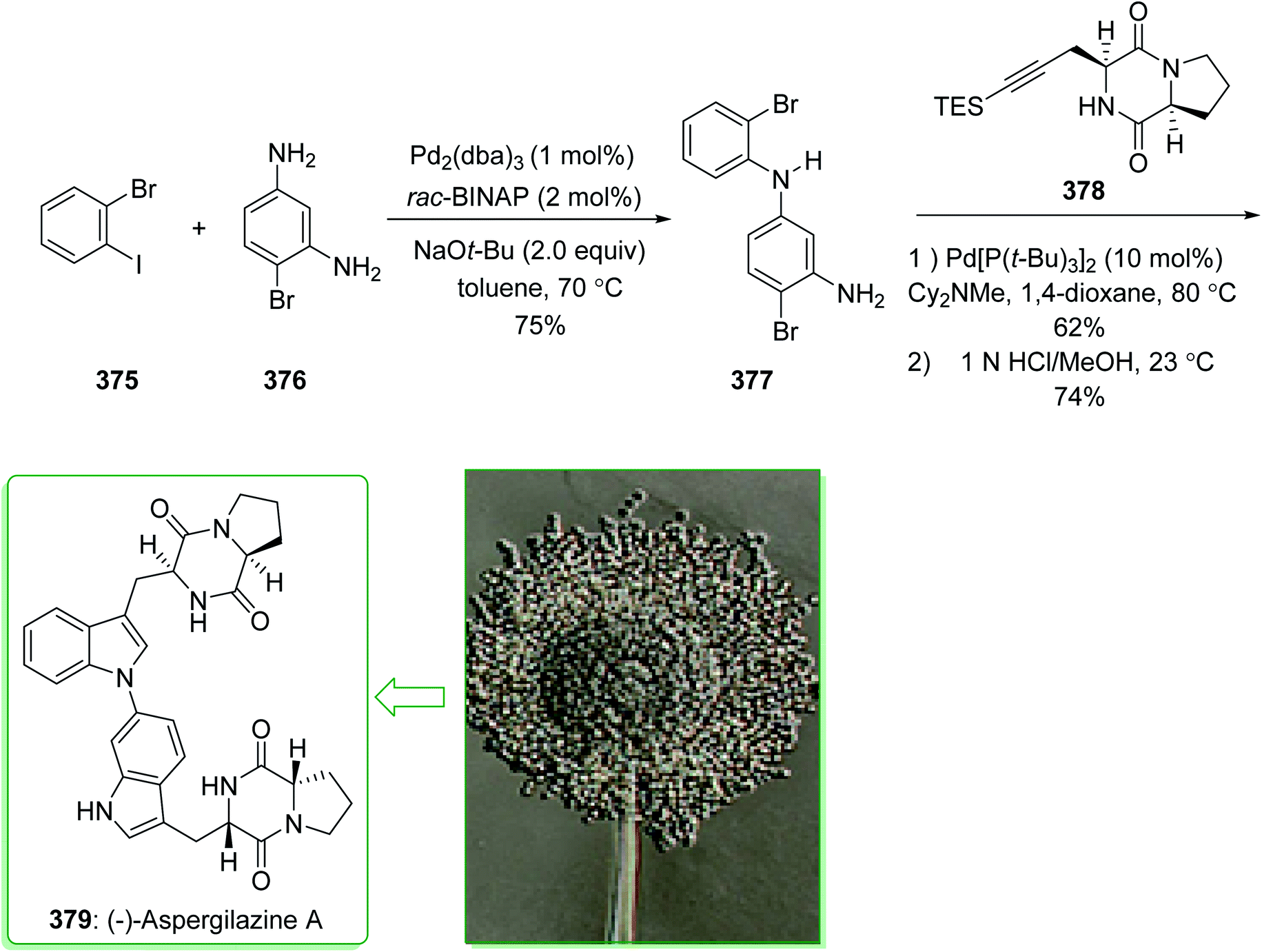 | ||
| Scheme 52 Total synthesis of (−)-aspergilazine A (379). | ||
2.3. Bartoli indole synthesis
In 2001, Dobbs demonstrated322 a concise and effective method for the synthesis of 2,4-dimethylindole (382), 4-(hydroxymethyl)-2-methylindole (386) and 4-(methoxymethyl)-2-methylindole (388). These alkaloids were previously extracted and reported in 1994 by Sterner and co-workers from two species of European Basidomycetes (Tricholoma sciodes and Tricholoma virgatum).323 Tricholoma is one of the well-known genera of the Basidiomycota division. However, several species of this genus have been utilized as culinary mushrooms, only a few insignificant investigations have been performed exploring the phenolic contents of the Tricholoma genus and also their biological properties.324 The total synthesis of 2,4-dimethylindole (382), 4-(hydroxymethyl)-2-methylindole (386), and 4-(methoxymethyl)-2-methylindole (388) started from 4-bromo-3-nitrotoluene (380). The straight reaction between isopropenylmagnesium bromide and 4-bromo-3-nitrotoluene (380) via Bartoli reaction conditions in THF at −40 °C gave 2,4-dimethyl-7-bromoindole (381) in a satisfactory yield (67% yield).322 As with the examination reactions, the radical reduction was performed almost quantitatively (94% yield) to afford 2,4-dimethylindole (382) in moderate yields (62% overall yield).Furthermore, the radical benzylic bromination reaction of 4-bromo-3-nitrotoluene (380) yielded 4-bromo-3-nitro-1-bromomethylbenzene (383) in very high yields (>85% yield). The latter was exposed to the Bartoli reaction again using isopropenylmagnesium bromide, to give 7-bromo-4-(bromomethyl)-2-methylindole (384) in a good yield (61% yield). In the following, the reaction of compound 384 with H2O (under acidic reaction conditions) afforded 7-bromo-4-(hydroxymethyl)-2-methylindole (385) in very good yields (88% yields) and this was transformed into the alkaloid 4-(hydroxymethyl)-2-methylindole (386) by using radical reduction (91% yield) (41% overall yields). In addition, the reaction of 7-bromo-4-(bromomethyl)-2-methylindole (384) with NaOMe provided 7-bromo-4-(methoxymethyl)-2-methylindole (387) in good yields (76% yields) and this was again reduced quantitatively with tributyltin hydride (Bu3SnH) (94% yield) to afford 4-(methoxymethyl)-2-methylindole (388) in a moderate overall yield (37%) (Scheme 53).322
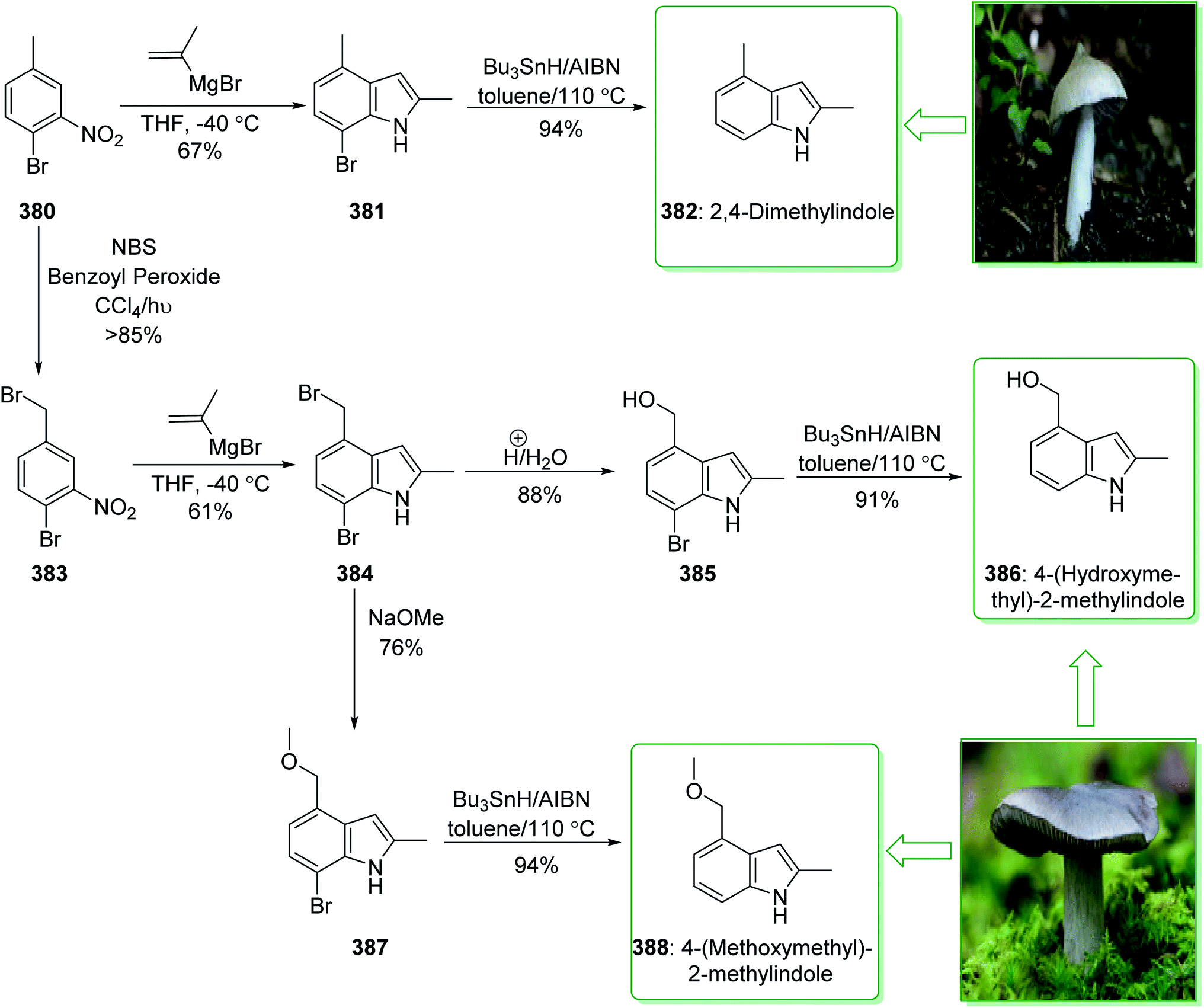 | ||
| Scheme 53 Total synthesis of 2,4-dimethylindole (382), 4-(hydroxymethyl)-2-methylindole (386) and 4-(methoxymethyl)-2-methylindole (388). | ||
The family of trikentrins,325 and herbindoles326 have the most noticeable features of an unusual class of naturally occurring indole alkaloid compounds, in which annulation takes place around the benzene core. These biologically potent compounds are attractive structures.327 The trikentrins were extracted from the marine sponge Trikentrion flabelliforme by Capon et al. in 1986.328 The herbindoles were discovered by Scheuer from the Australian sponge Axinella sp. and contain both anti-feedant and cytotoxic activities.329 Silva and Craveiro in 2008 reported330 an efficient method to synthesize the polyalkylated indole (±)-trans-trikentrin A (393). The synthesis of this alkaloid involves a Tl(III)-catalyzed ring contraction reaction to provide the trans-1,3-difunctionalized five-membered ring through a diastereoselective method. Other key steps are Bartoli's reaction and a Heck coupling reaction. The total synthesis of (±)-trans-trikentrin A (393) started from acetophenone 389 and after three steps gave 1-bromo-4-ethyl-2-nitrobenzene (390) in good yields (79% overall yield). The latter was reacted with CH2CHMgBr in THF to afford the bromo-indole 391 via a Bartoli indole synthesis. In the following, after 14 synthetic steps, indole 391 yielded the corresponding dihydrocyclopenta[g]indole 392, containing the trans-1,3 five-membered ring.331 Then, compound 392, after three steps, including tosylation, and the reduction and removal of the Boc substituent, gave (±)-trans-trikentrin A (393) (Scheme 54).330
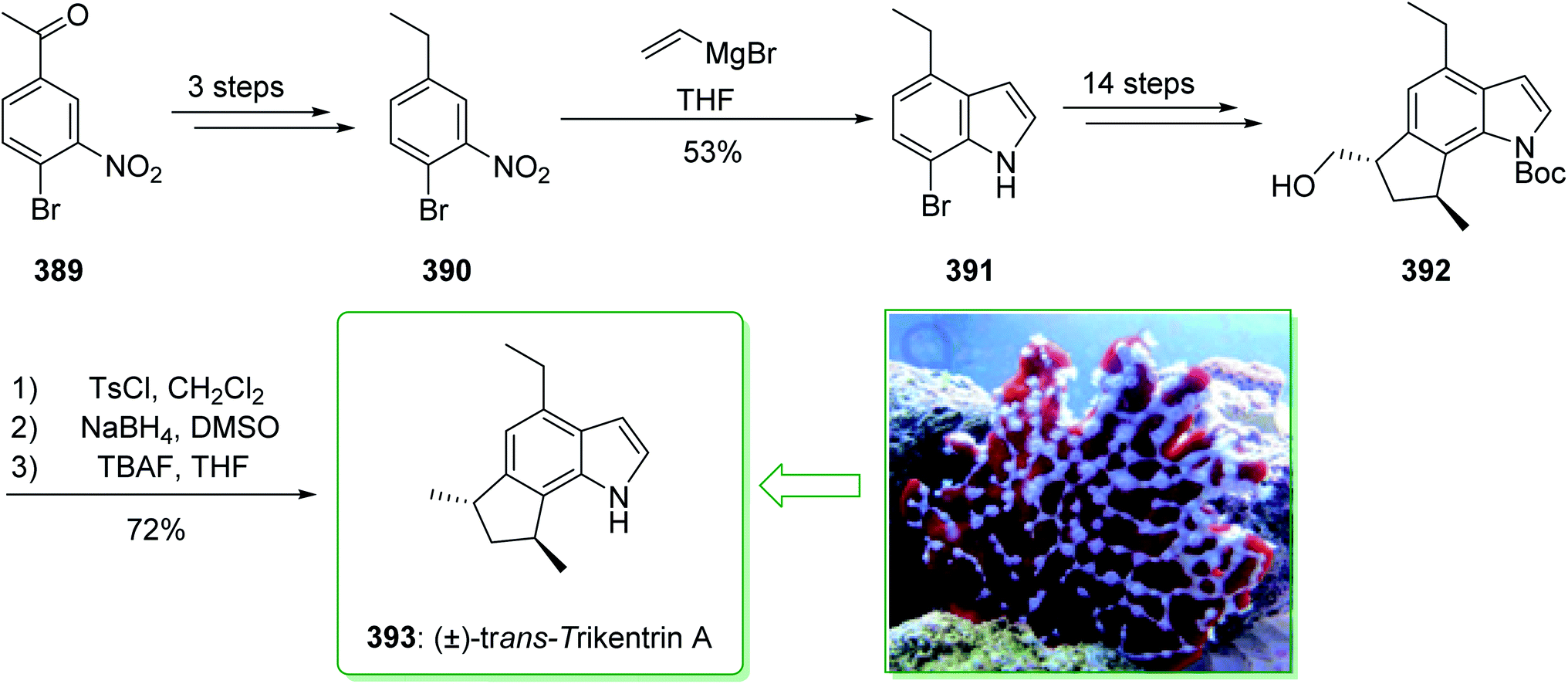 | ||
| Scheme 54 Total synthesis of (±)-trans-trikentrin A (393). | ||
In 2009, Buszek and co-workers demonstrated332 an effective nine-step total synthesis of the annulated indole natural products (±)-cis-trikentrin A (398) and (±)-herbindole A (403) through an intermolecular Diels–Alder cycloaddition using the established indole aryne (indolyne) approach as the key step. This approach enables the trikentrins and the related herbindoles to be quickly admitted. The essential 6,7-indolyne material was easily generated through a Bartoli indole synthesis using functionalized vinyl magnesium bromide and nitrobenzenes. This research group tried to provide (±)-cis-trikentrin A (398) and (±)-herbindole A (403). The total synthesis of (±)-cis-trikentrin A (398) was started from 4-ethylaniline (394) and after two steps, including nitration diazotization and bromination, afforded o-dibromide 395. The Bartoli indole synthesis using vinyl magnesium bromide and THF at −40 °C was progressed uneventfully and afforded the corresponding indole 396 in a moderate yield (52% yield). The latter, after five steps comprising protection, cycloaddition, osmylation, oxidative removal, and concomitant desilylation, provided the desired dithioacetal 397. Lastly, upon Raney Ni reduction of 397, the natural product (±)-cis-trikentrin A (398) was synthesized successfully (Scheme 55).332
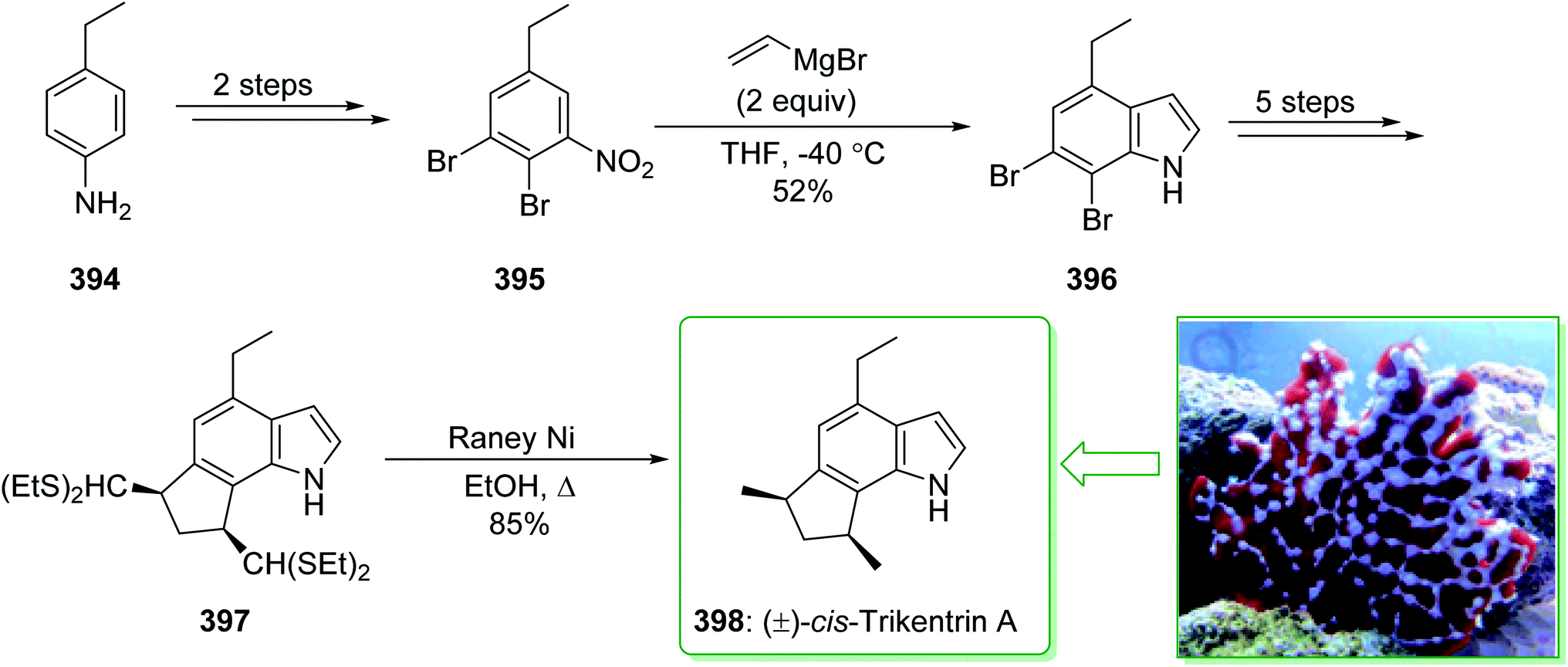 | ||
| Scheme 55 Total synthesis of (±)-cis-trikentrin A (398). | ||
In addition, the total synthesis of (±)-herbindole A (403) began from 3,4-dimethylaniline (399) and after two steps, including nitration, diazotization and bromination, gave o-dibromide (400). Pleasantly, the Bartoli approach provided the corresponding indole 401, but in a moderate yield (36% yield). The latter, after five steps including N-silylation, a Diels–Alder reaction, osmylation, oxidative removal, and a thioacetalization reaction was converted into etrahydrocyclopenta[g]indole 402. Lastly, Raney Ni reduction of 402 gave the racemic herbindole A (403). As a result, (±)-herbindole A (403) was synthesized efficiently in nine steps from 3,4-dimethylaniline (399) (Scheme 56).332
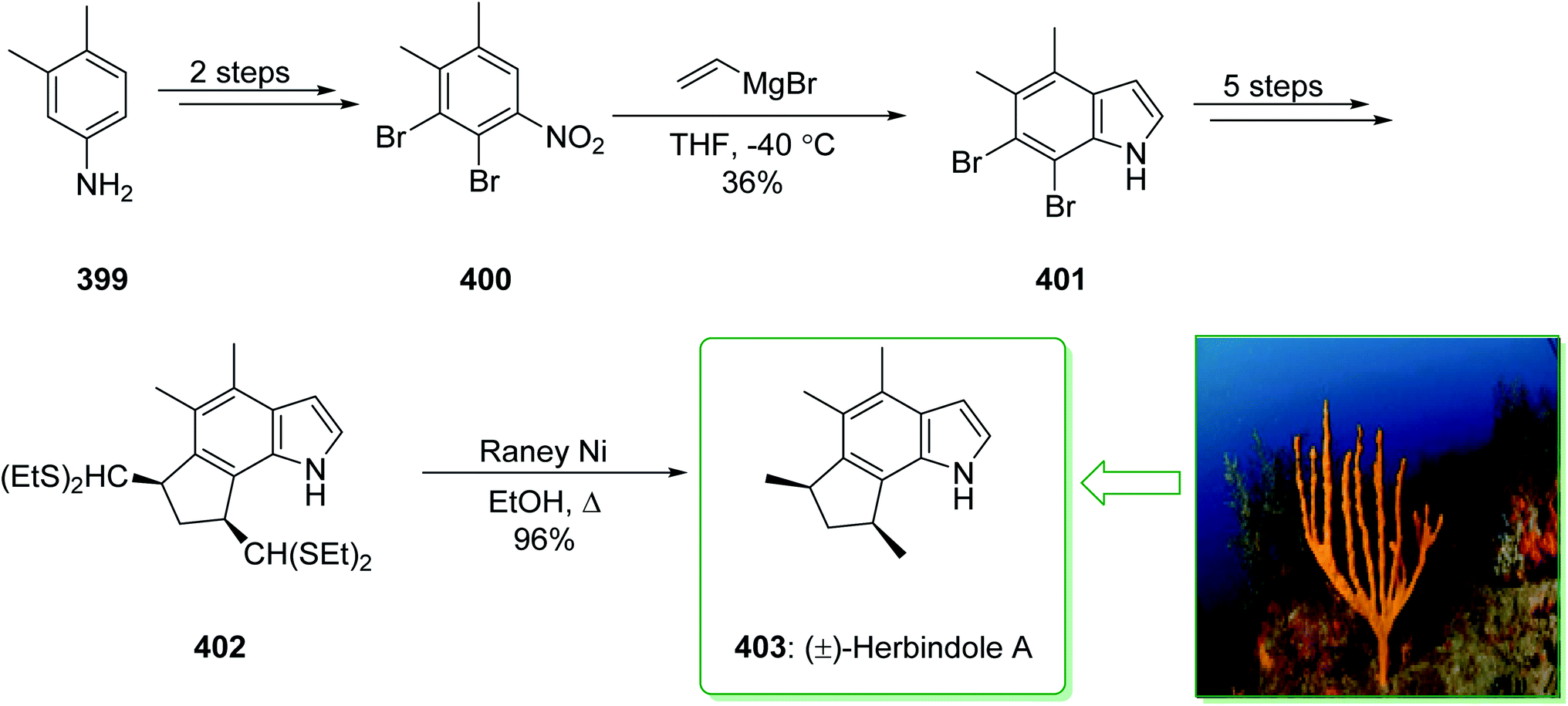 | ||
| Scheme 56 Total synthesis of (±)-herbindole A (403). | ||
2.4. Madelung indole synthesis
Mersicarpine was isolated from the stem-bark extracts of the Kopsia fruticosa and Kopsia arborea by Kam et al. in 2004.333 Mersicarpine contains a typical seven-membered cyclic imine fused along with indoline and δ-lactam.334,335 The monoterpene indole alkaloids represent a significant family of naturally occurring compounds with a rich structural diversity. Different compounds in this class show strong biological activities.336 Various methods for the total synthesis have been reported, involving Kerr's first total synthesis of (±)-mersicarpine337 and Fukuyama's first total synthesis of (−)-Mersicarpine (110).338The mersicarpine and leuconoxine are structurally complex and biologically fascinating aspido-sperma-derived monoterpene indole alkaloids.55 (−)-Scholarisine G (415),339 (+)-melodinine E (416)340 and (−)-leuconoxine (417)341 are pentacyclic alkaloids containing a stimulating [5.5.6.6]diazafenestrane unit342 that possesses two or three contiguous quaternary stereogenic centers. (−)-Mersicarpine (110),333 however, contains a fused tetracyclic 6/5/6/7 ring scaffold, identified by an unusual tetrahydro-2H-azepine ring and also a hemiaminal scaffold.343–346 In 2019, Wang and co-worker reported347 a unified approach for the asymmetric synthesis of (−)-scholarisine G (415), (+)-melodinine E (416), (−)-leuconoxine (417) and (−)-mersicarpine (110) from the 2-alkylated indole intermediate. The total synthesis of (−)-scholarisine G (415), (+)-melodinine E (416), (−)-leuconoxine (417) and (−)-mersicarpine (110) began with o-toluidine (404). The key Smith-modified Madelung indole synthesis was commenced with the formation of N-silylated o-toluidine (405) by treating o-toluidine (404) in the presence of a stoichiometric quantity of n-BuLi and was quenched with chlorotrimethylsilane ((Me)3SiCl). Without separation, this intermediate was subjected to sec-butyllithium solution at a low temperature to make a reactive lithium dianion (406). After slow addition of lactone (+)-409, the cascade acylation/heteroatom Peterson olefination/isomerization reactions progressed effortlessly to yield the 2-quantenary carbon functionalized indole (−)-412 in an overall yield of 85%. It should be mentioned that lactone (+)-409 was synthesized in a good yield (72% yield) and with a good ee (89% ee) from the reaction of 3-ethyltetrahydro-2H-pyran-2-one (407) and allyl methyl carbonate (408) using a Pd catalyst and the (R)-DM-BINAP ligand.348 Next, after four steps indole (−)-412 gave the keto hemiaminal 413. In the following, after a Staudinger-aza-Wittig cyclization reaction, the keto hemiaminal 413 yielded (−)-mersicarpine (110). Moreover, Zhu's intermediate 414, after six steps, was obtained from indole (−)-412. Zhu's intermediate 414 provided the natural product (−)-scholarisine G (415) after an LDA-promoted intramolecular aldol cyclization. Then, the reaction of (−)-scholarisine G (415) with the Burgess reagent in acetonitrile at 70 °C yielded (+)-melodinine E (416) in a high yield (99%). In the following, the hydrogenation of (+)-melodinine E (416) provided another member, (−)-leuconoxine (417), in a high yield (94%). In addition, (−)-mersicarpine (110) was synthesized using five steps from indole (−)-412 (Scheme 57).347
 | ||
| Scheme 57 Total synthesis of (−)-scholarisine G (415), (+)-melodinine E (416), (−)-leuconoxine (417) and (−)-mersicarpine (110). | ||
2.5. Buchwald-modification of the classical Fischer indolization
Alkaloids containing β-carboline have been extracted both from marine and terrestrial sources.349 Haploscleridamine is a unique tryptamine-derived alkaloid350 extracted from a marine sponge of the order Haplisclerida gathered in Palau and was reported in 2002 by Faulkner et al.351 This unique β-tetrahydrocarboline has been shown to prevent cathepsin K, a cysteine protease that results in osteoporosis, and therefore this natural product can act as the main compound in the establishment of treatment methods for this disease. In 2019, Lovely et al. reported352 the enantioselective total synthesis of the imidazole comprising a β-carboline natural product, and (−)-haploscleridamine (421) from the histidine methyl ester (418). Key to the effective assembly of this alkaloid is a ring-closing metathesis reaction of an imidazole resulting in an allylic alcohol to give 3-piperidinone. In addition, the usage of the Buchwald-modification of the classical Fischer indolization and deprotection of the N-tosyl scaffold supplied haploscleridamine. Based on this method, the total synthesis of (−)-haploscleridamine (421) started with the histidine methyl ester (418) and after seven steps this gave piperidinone 419. The latter, through Buchwald modification of the Fischer indole reaction, afforded the indole product 420. Then, after subjecting tosylamide 420 to reductive detosylation with Mg in MeOH,353 (−)-haploscleridamine (421) was obtained in a satisfactory yield (Scheme 58).352 | ||
| Scheme 58 Total synthesis of (−)-haploscleridamine (421). | ||
2.6. Batcho–Leimgruber indole formation
In 2013, Buszek and co-workers demonstrated354 an effective total synthesis of the annulated indole natural product (±)-cis-trikentrin B (427) by using a regioselectively constructed 6,7-indole aryne cycloaddition from 5,6,7-tribromoindole. The unaffected C-5 bromine was successively utilized for a Stille cross-coupling reaction to make the butenyl side chain and complete the synthesis. This methodology gives enables the trikentrins and the relevant herbindoles to be quickly admitted. The requisite 5,6,7-indole aryne precursor was synthesized through the Leimgruber–Batcho indole synthesis. The total synthesis of (±)-cis-trikentrin B (427) started with p-toluidine (422) and after four steps yielded the enamine intermediate 423, which was instantaneously utilized without separation for the next step. An iron(III) chloride-mediated reaction with hydrazine hydrate in MeOH at 60 °C consistently gave the corresponding 5,6,7-tribromoindole (424) (Leimgruber–Batcho indole synthesis). The latter, after seven steps, gave the corresponding indole 425. Lastly, Stille cross-coupling of indole 425 and the vinyl tin reagent 426 in the presence of triphenylarsine and Pd2(dba)3 using MWI easily gave racemic cis-trikentrin B (427) in a satisfactory yield (73% yield) (Scheme 59).354 | ||
| Scheme 59 Total synthesis of (±)-cis-trikentrin B (427). | ||
Indiacen A and B355 are prenyl indoles and were the initially identified secondary metabolites extracted from the bacterium Sandaracinus amylolyticus in 2012 by Müller et al. belonging to a novel species of myxobacteria. These secondary metabolites, indiacen A and its chloro analogue indiacen B, exhibit anti-microbial properties. These are known to be potent against Gram-positive and Gram-negative bacteria and also the fungus Mucor hiemalis. Indiacen B (433) is a 3-formylindole derivative having a dienyl chloride side chain.356 The natural product indiacen B from the myxobacterium Sandaracinus amylolyticus was produced for the first time, revealing its anti-microbial properties. The E-configuration of the chloroalkene scaffold of indiacen B was proved using X-ray analysis.357 In 2015, Lindel et al. demonstrated357 that the total synthesis of indiacen B (433) began from 2-methyl-3-nitroaniline (428), and using sodium nitrite (NaNO2) and potassium iodide (KI) afforded the 1-iodo-2-methyl-3-nitrobenzene 429. The latter, through the Batcho–Leimgruber method (using dimethylformamide dimethyl acetal (DMFDMA) and titanium(III) chloride), gave 4-iodoindole (430), and after two steps this afforded the indole 431. Finally, indiacen B (433) (29%) and its Z-isomer 432 (14%) were synthesized successfully from indole 431 (using POCl3 in DMF at 0 °C) (Scheme 60).357
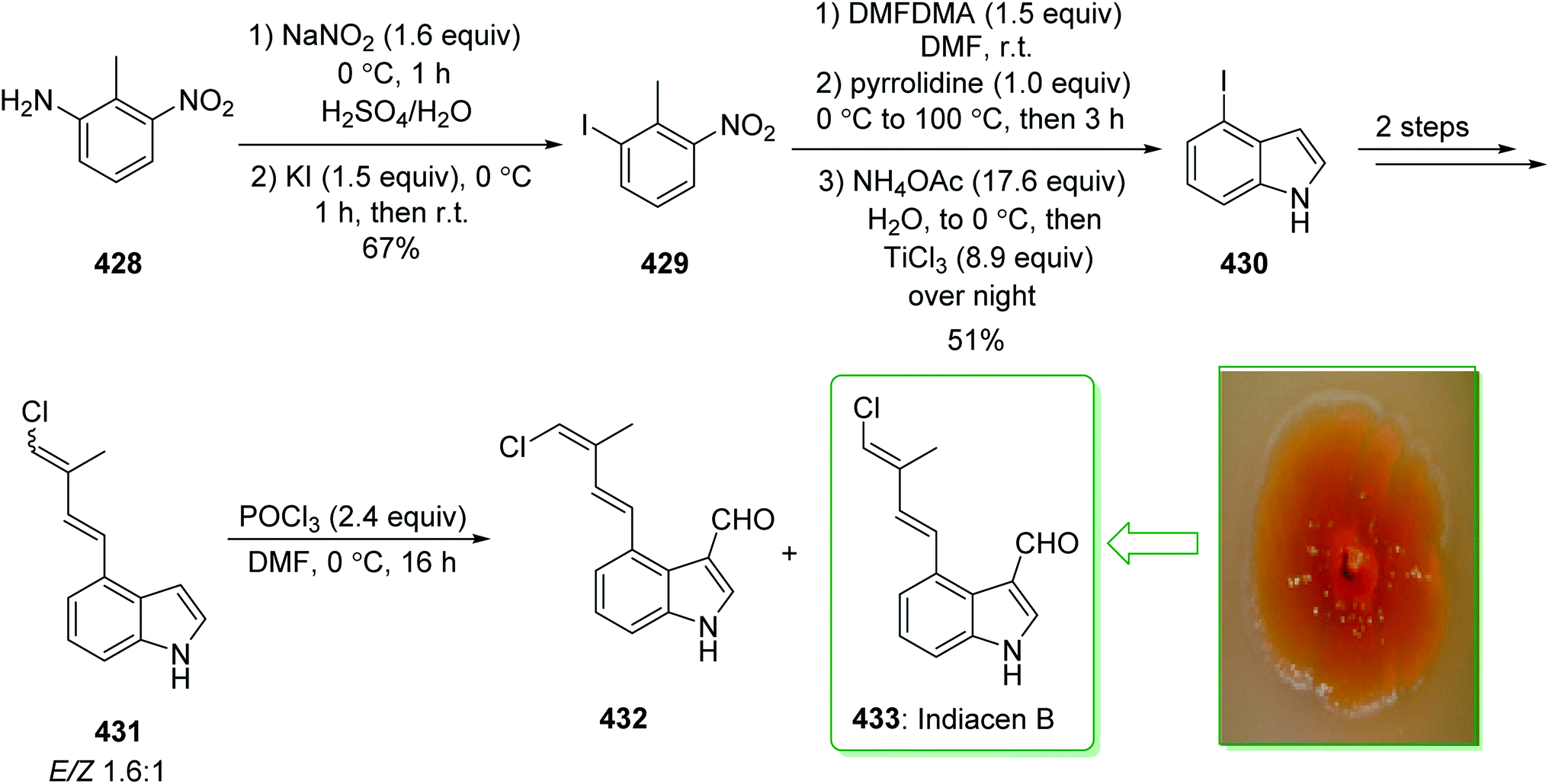 | ||
| Scheme 60 Total synthesis of indiacen B (433). | ||
2.7. Cadogan–Sundberg and SnCl2-mediated reductive cyclization
The phytoalexins are a different group of naturally occurring compounds, which are produced in plants in response to a pathogenic challenge. The indole-comprising phytoalexins are denoted as the indole phytoalexins358 or the crucifer wasabi phytoalexins.359 These phytoalexins have been extracted from cruciferous plants, comprising cabbage, broccoli, mustard, cauliflower and wasabi (Wasabia japonica, syn. Eutrema wasabi).360,361 The initial wasabi phytoalexin, methyl 1-methoxyindole-3-carboxylate (437), was extracted, identified, and manufactured by Soledade et al.362 In 2013, Peet et al. reported the total synthesis of phytoalexin (437). Two synthetic methods have been utilized for the formation of the wasabi indole phytoalexin (437).363 The total synthesis of phytoalexin (437) began with 2-nitrophenylacetic acid (434). The reaction of methyl 2-nitrophenylacetic acid (434) with formaldehyde (HCHO), tetrabutylammonium iodide (n-Bu4NI) and K2CO3 in toluene provided 2-(2-nitrophenyl)acrylate (435) in a good yield (85% yield). Then, phytoalexin (437) was synthesized in a good yield (85%) from 435 using modified Cadogan–Sundberg conditions,364 for instance, using trimethylphosphite (P(OCH3)3) instead of triethylphosphite (P(OEt)3).In the second route for the synthesis of 437, acrylate 435 was used. The reaction of 435 in the presence of SnCl2 and NaOAc in THF provided methyl 1-hydroxyindole-3-carboxylate (436) in a good yield (88%). Finally, the methylation reaction of compound 436 using MeI afforded phytoalexin (437) in a high yield (97%). Based on these methods, phytoalexin (437) was synthesized in yields of 72% and 73%, respectively, that are the maximum yields obtained for all the different synthetic pathways (Scheme 61).363
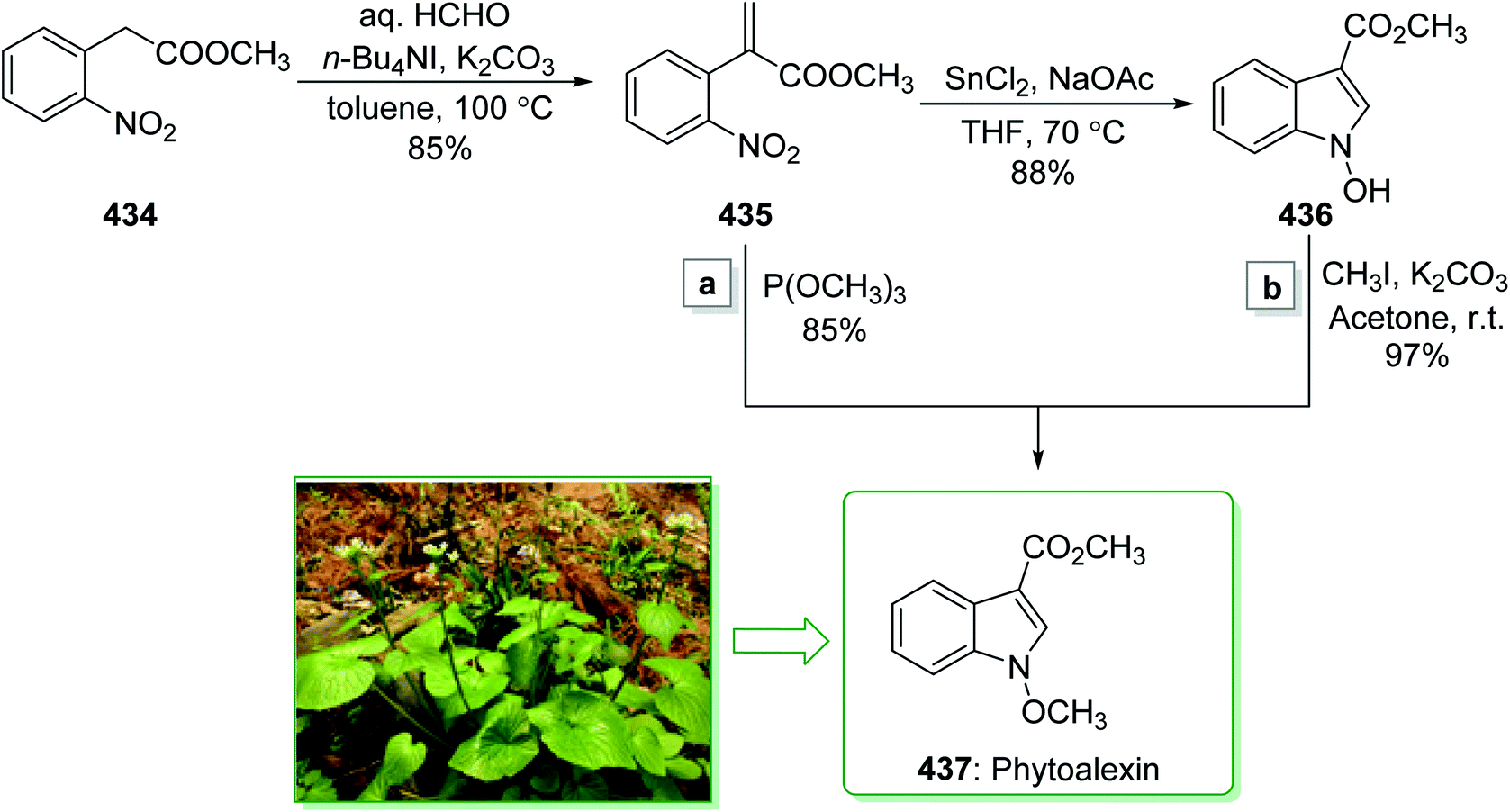 | ||
| Scheme 61 Synthesis of phytoalexin (437). | ||
2.8. Hemetsberger–Knittel reaction
Lyngbyatoxin A (442) was extracted in 1979 by Moore et al. from the lipid extract of sea weed.365 Lyngbyatoxin A (442) is an effective tumor promoter366 and similar to other indolactam alkaloids, applies its biological properties via the activation of the protein kinase C (PKC). In 2006, Tanner and Vital demonstrated367 that indole 441, an advanced intermediate for the asymmetric total synthesis of lyngbyatoxin A (442), was synthesized from allylic alcohol 439 in nine steps and with a high ee (>95% ee). The effective and very asymmetric construction of the all-carbon quaternary stereocentre of lyngbyatoxin A (442) started with the allylic alcohol 439 (prepared from p-aminoacetophenone 438 in four steps), which after eight steps gave azide 440.368 This was performed by heating compound 440 in xylene, that efficiently gave indole 441 in a satisfactory yield (69% yield). The latter is appropriately functionalized so as to permit its transformation into lyngbyatoxin A: the C-3 indolic position is essentially nucleophilic, and the bromine at C-4 gives an appropriate handle for insertion of the amino group; moreover, the TBDMS-masked alcohol scaffold permits the formation of the linalyl appendage (Scheme 62).367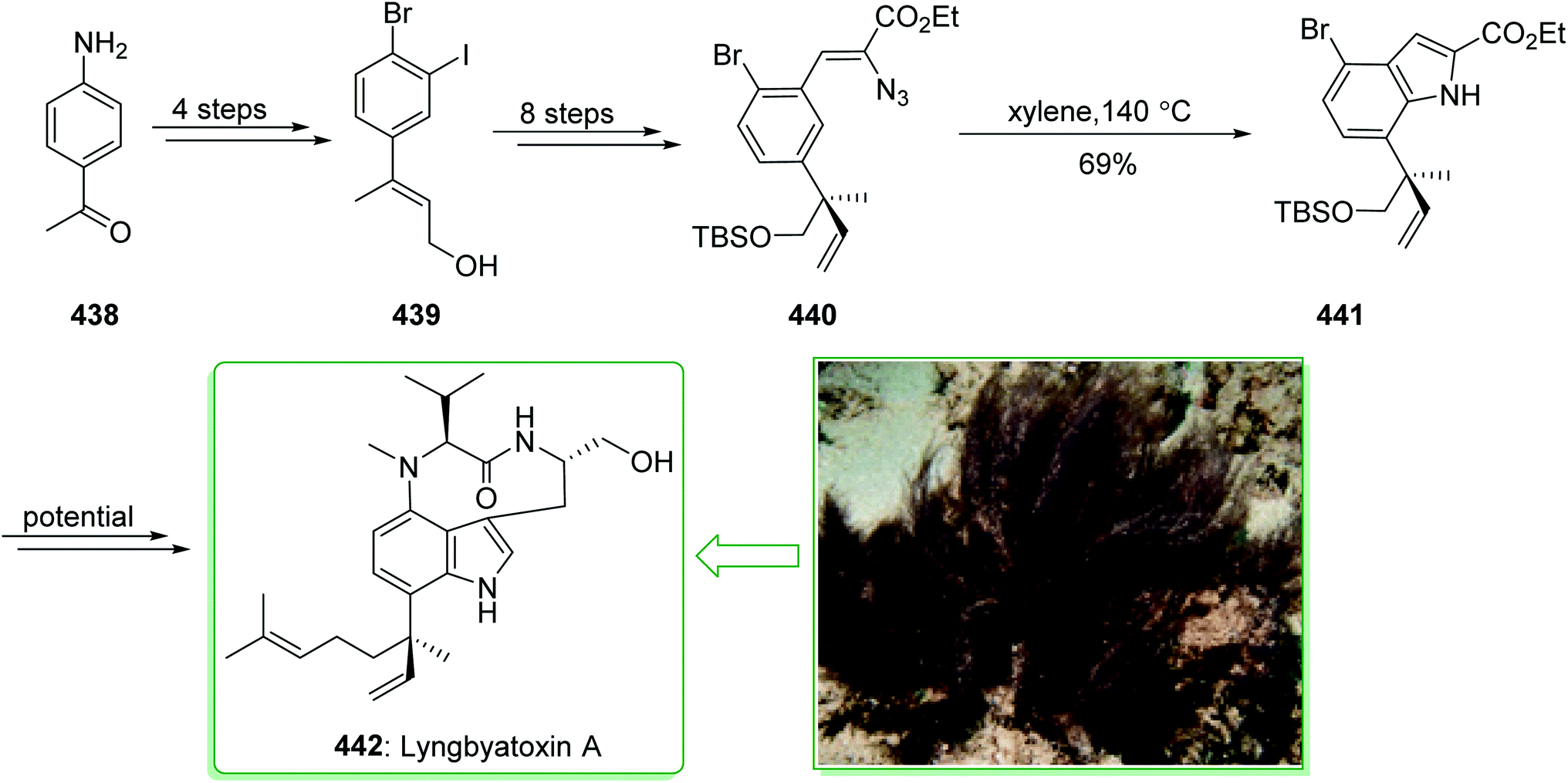 | ||
| Scheme 62 Asymmetric construction of the all-carbon quaternary stereocentre of lyngbyatoxin A (442). | ||
3. Miscellaneous
Rizatriptan, an active selective 5-HT 1B/1D receptor agonist, has been proven to be clinically valuable for the treatment of migraines.369 Rizatriptan selectively limits isolated human middle meningeal arteries and prevents neurogenic dural extravasation and also vasodilatation.370 In 2015, Zhu et al. achieved and reported371 the total synthesis of rizatriptan (447). The reaction of the o-nitrostyrenes using aqueous titanium(III) chloride solution at ambient temperature gives indoles via a formal reductive C(sp2)–H amination reaction. β,β-Difunctionalized o-nitrostyrenes, 2,3-difunctionalized indoles were provided through a domino sequential reaction involving a reduction/cyclization/migration reaction. This approach was utilized as a key step in the short synthesis of rizatriptan and also the formal total synthesis of aspidospermidine. In this route, the synthesis of rizatriptan (447) began with the regioselective Heck reaction of aryl triflate 443 and but-3-en-1-ol (444), which after three steps afforded the triazole 445. The titanium(III) chloride-improved reductive cyclization of 445 gave the corresponding indole 446 (51% yield), which was transformed to rizatriptan (447) (Scheme 63).371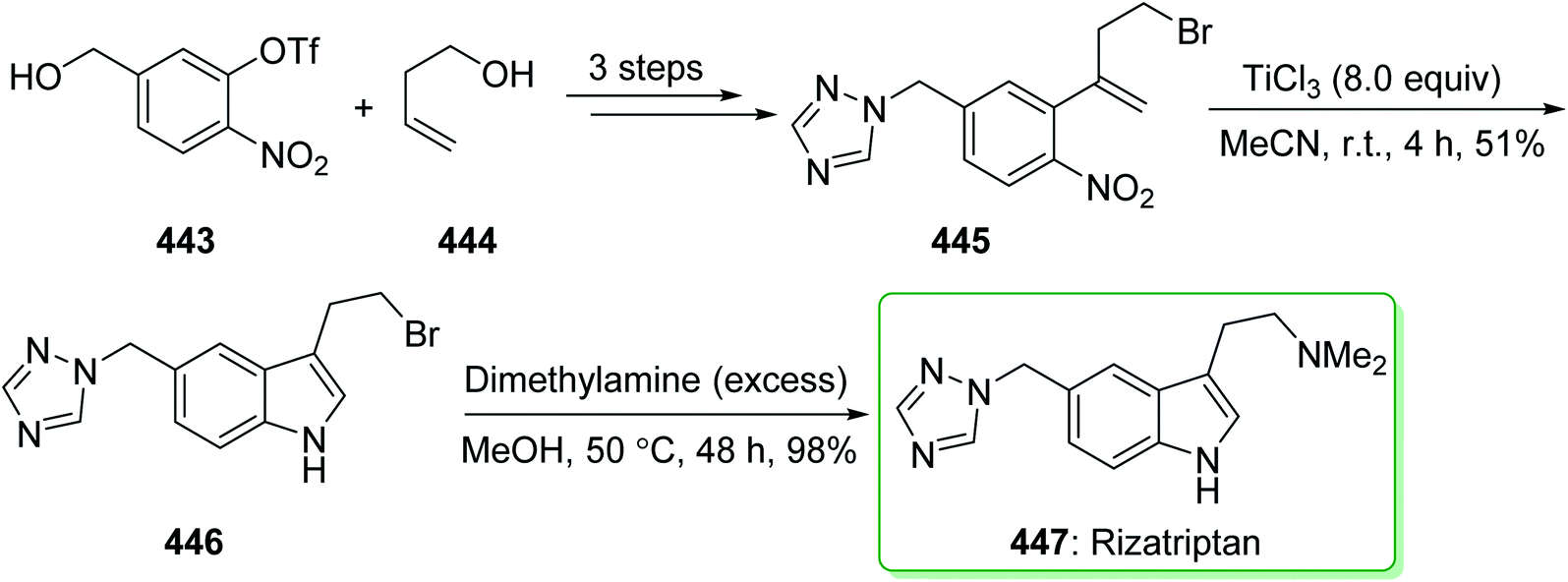 | ||
| Scheme 63 Total synthesis of rizatriptan (447). | ||
The formal total synthesis of (+)-aspidospermidine (135) began with ketone 448. A solution of compound 448 in acetone-ammonium acetate with titanium(III) chloride was stirred at room temperature to give tetrahydrocarbazolone 449 in a satisfactory yield (70% yield). The latter was then transformed into (+)-aspidospermidine (135)372 (Scheme 64).371
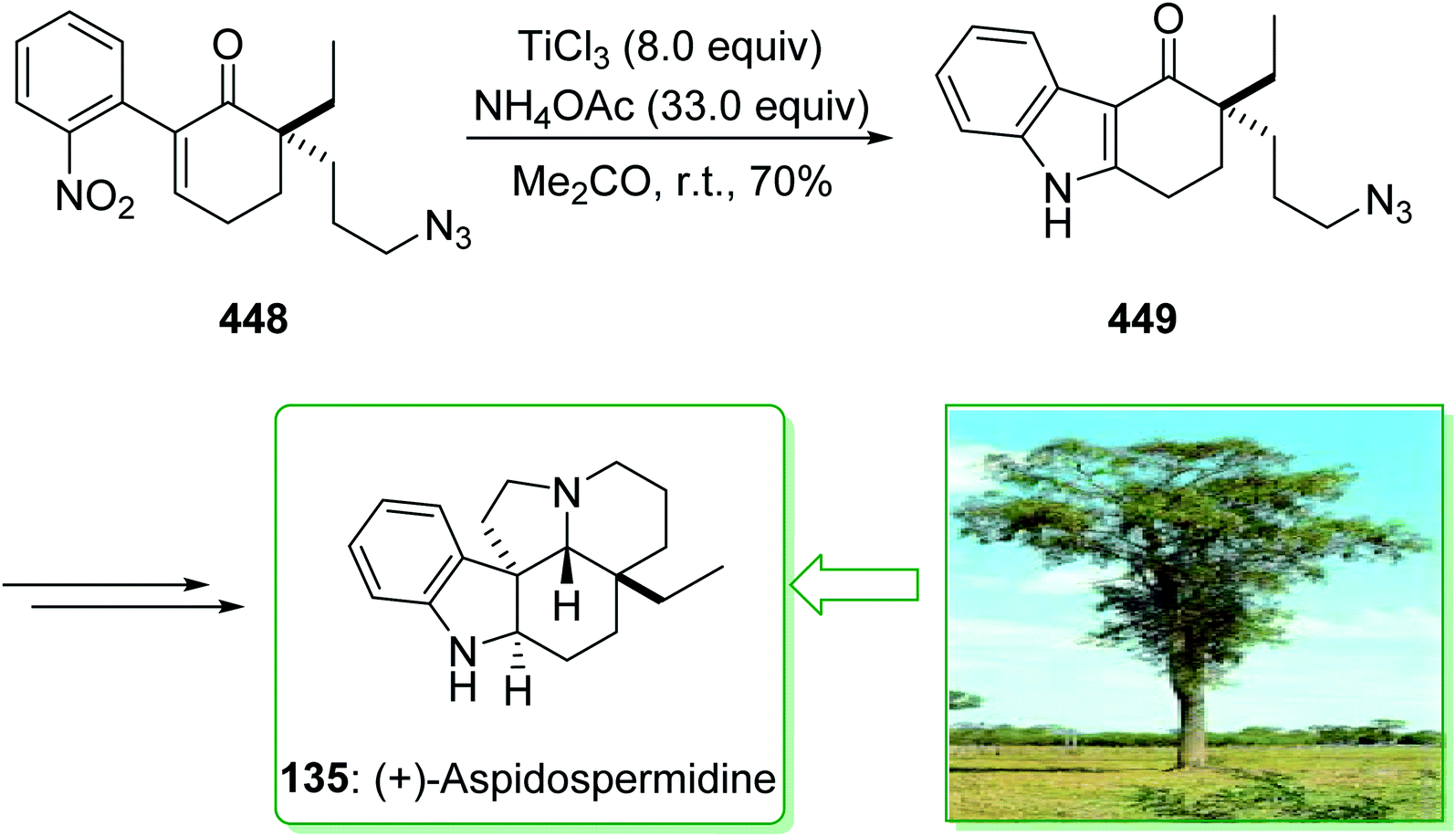 | ||
| Scheme 64 Formal synthesis of (+)-aspidospermidine (135). | ||
Tetracyclic indole alkaloids were extracted from a genus of the bacteria Streptomyces sp. (PA-48561) by Yasui and Kamiguchi in 2000.373 In 2016, Söderberg. et al. reported374 the total synthesis of the tetracyclic indole alkaloid ht-13-A (455) from 3(R)-t-butyldimethylsilyloxypyrrolidin-2-one. The main steps in this method are a Lewis acid catalyzed acyliminium ion allylation reaction, a Mitsunobu reaction, a Pd-mediated Stille–Kelly intramolecular cross coupling, and also a CO catalyzed Pd-mediated reductive N-heterocyclization. The total synthesis of ht-13-A (455) was commenced from the Mitsunobu reaction between the pyrrolidines 451 (prepared from 3(R)-t-butyldimethylsilyloxypyrrolidin-2-one (450) (in four steps) and 2-bromo-3-nitrophenol (452), which after two steps afforded the tricyclic compound 453. The Pd-mediated reductive N-heterocyclization of 453 using CO afforded the desired tetracyclic indole 454. Lastly, the reduction of the methoxycarbonyl substituent to a methyl substituent with sodium bis(2-methoxyethoxy)aluminum hydride (Red-Al) in toluene afforded ht-13-A (455) in a very good yield (Scheme 65).374
 | ||
| Scheme 65 Total synthesis of the tetracyclic indole alkaloid ht-13-A (455). | ||
Kam et al. in 2008 reported375 the extraction of seven indole alkaloids, jerantinines A–G of the Aspidosperma type from an examination of the leaf extract of a similar species.376 The jerantinines exhibited a noteworthy cytotoxicity against human KB cells.377 In 2017, Magauer et al. demonstrated378 the establishment of an enantioselective and very convergent three-component synthesis of the functionalized ABC ring scaffold of the Aspidosperma alkaloid jerantinine E. This synthetic methodology was achieved for fast formation of the tricyclic tetrahydrocarbazolone unit through a Pd-mediated amination and oxidative indole construction. In addition, a secondary amine framework, which includes all carbon atoms of the D and E ring of the natural product can be formed in three additional steps. Synthetic investigations for jerantinine E (459) began with 1,4-cyclohexanedione monoethyleneacetal (69), which after six steps gave cyclohex-2-en-1-one 456. The latter, through oxidative indole construction379 in the presence of Pd(OAc)2 and Cu(OAc)2, provided tetrahydrocarbazolone 457. In the following, benzyl protection of the tetrahydrocarbazolone 457 afforded tetrahydrocarbazolone 458, that is an intermediate for the formation of jerantinine E (459) (Scheme 66).378
 | ||
| Scheme 66 Synthetic studies towards jerantinine E (459). | ||
Furostifoline (463), the furo[3,2-a]carbazole alkaloid, was extracted by Furukawa et al. in 1990 (ref. 380) from the root bark of Murraya euchrestifolia Hayata and is utilized in Chinese traditional medicine.381 The first total synthesis of furostifoline (463) was reported in 1996 (ref. 382) and the optimum synthetic route to furostifoline (463) was reported in 2000 by Knölker and Fröhner.383 Furostifoline is a carbazole alkaloid having a furo[3,2-a]carbazole building block.384,385 In 2002, Yasuhara et al. demonstrated386 that furostifoline was synthesized in four steps with a 10% overall yield from 2-acetyl-3-bromofuran (460). The total synthesis of furostifoline (463) began from 2-acetyl-3-bromofuran (460) and after two steps, including the Wittig reaction and the Sonogashira reaction afforded 2-(isopropenyl)-3-[(2-ethoxycarbonylamino)phenylethynyl]furan (461). Next, the tetra-n-butylammonium fluoride-improved cyclization of 461 in THF under reflux conditions afforded a mixture of 2-[(isopropenyl)furanyl]indole (462). Thus, it was proposed that the photocyclization triggered the polymerization of 462 and afforded the natural product furostifoline (463) in a low yield (24%) (Scheme 67).386
 | ||
| Scheme 67 Total synthesis of furostifoline (463). | ||
The inhibitors of 3α-hydroxysteroid dehydrogenase, 0231A and 0231B, were extracted in 2001 by Gräafe from a fermentation broth of Streptomyces sp. HKI0231. Meanwhile 3α-hydroxysteroid dehydrogenase is an enzyme related to inflammatory processes, these compounds are favorable as main structures for anti-inflammatory agents.387 An advanced intermediate in the Nakatsuka synthesis of 0231B was synthesized through a fluoride-catalyzed indole construction in the key step. It is worth mentioning that both Pd-based methods and hydride-based methods were unsuccessful in forming the indole.388 The formal synthesis of 0231B (468) was commenced from 2-hydroxy-5-methyl-3-nitrobenzaldehyde (464) and after three steps afforded acetamide 465. Auspiciously, the reaction of acetylene 465 with tetrabutylammonium fluoride (TBAF) in THF under refluxing conditions provided indole 466 in a satisfactory yield (yield). After two steps, enone 467 was furnished, and gave 0231B (468) (after six steps)389 (Scheme 68).388
 | ||
| Scheme 68 Formal synthesis of 0231B (468). | ||
In addition, the formal synthesis of 0231B (468) was commenced from the Sonogashira coupling reaction of triflate 469 and trimethylsilylacetylene (470) and afforded acetylene 471. Fluoride-catalyzed cyclization, desilylation and also deacetylation occurred in a one-pot reaction and resulted in the transformation of 471 into indole 472 in a moderate yield (34%). After two steps, enone 473 was provided, and then gave 0231B (468) (after six steps) (Scheme 69).389
 | ||
| Scheme 69 Formal synthesis of 0231B (468). | ||
Pyrido[3,4-b]indoles, frequently identified as β-carbolines, are the key structural scaffolds of different biologically significant alkaloids of synthetic and natural origins.390,391 Naturally occurring compounds bearing a β-carboline core have been extracted from terrestrial plants and several marine invertebrates and their biological activities change from interactions with the benzodiazepine receptor to powerful anti-viral, anti-tumor, and anti-microbial properties.392–394 Eudistomin U (478) was extracted in 1994 by Badre and co-workers from the Caribbean ascidian Lissoclinum fragile and exhibited anti-microbial and DNA binding properties.395–397 A flexible method for the synthesis of functionalized β- and γ-carbolines relied on the transition metal mediated [2 + 2 + 2] cycloaddition reactions of substituted yne-ynamides and methylcyanoformate was achieved and reported by Witulski et al. in 2011.398 The flexibility of this unique reaction sequence is shown by its use in the total synthesis of the marine natural product eudistomin U (478). The total synthesis of eudistomin U (478) was commenced from 2-iodoaniline (474) that after three steps yielded yne-ynamide 475. Next, the Cp*RuCl(cod)-mediated [2 + 2 + 2] cycloaddition of 475 using methylcyanoformate led to the β-carboline ester 476 (94% yield), which was saponificated using in situ elimination of the N-carboline and N-indolyl tosyl substituents to afford the β-carboline carboxylic acid 477 in a high yield (96% yield). Lastly, decarboxylation of 477 using Cu powder under microwave irradiation (MWI) gave eudistomin U (478) in a high yield (88%) (Scheme 70).398
 | ||
| Scheme 70 Total synthesis of eudistomin U (478). | ||
C-aryl glycosides, a class of naturally occurring compounds, show a variety of significant biological activities.399 A number of members of this family exhibit strong anti-viral, anti-biotic, and anti-tumor activities,400 and there is also sufficient experimental proof that C-aryl glycosides bind duplex DNA.401 Two alkaloids extracted from the roots of the plant Isatis indigotica contain an indole-C-glycoside unit.402 Indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (487) exhibits a cytotoxic property against human liver cancer HepG2 cells and also human myeloid leukemia HL60 cells. In 2012, Minehan and Yapremyan demonstrated403 that indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (487), a unique C-glycoside from Isatis indigotica that has significant cytotoxic properties, can be synthesized in ten steps from ethynyl-β-C-glycoside (481) and 2-iodo-3-nitrophenyl acetate (482). Noticeably, key steps in the synthesis involve a Sonogashira coupling reaction and a copper(I) iodide-catalyzed indole construction. The total synthesis of indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (487) was commenced from the Sonogashira reaction between acetoxyaryl iodide 482 (obtained from 2-amino-3-nitrophenol (480) in two steps), and alkyne 481 (obtained from dextrose 479 in seven steps), and gave the corresponding alkyne 483. The latter, after three steps including aminolysis, hydroxyl methylation, and reduction, provided aniline 484. Next, a base-catalyzed indolization reaction was investigated using Knochel's t-BuOK-NMP system.404 The reaction of compound 484 in the presence of t-BuOK in N-methyl-2-pyrrolidone gave variable amounts of indole C-glycoside 485 with yields of 33–55%. In the following, indole C-glycoside 485 after four steps gave nitrile 486. The elimination of the silyl ether masking groups was performed using TBAF in THF and provided indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (487) in satisfactory yields (72%) (Scheme 71).403
 | ||
| Scheme 71 Total synthesis of indole-3-acetonitrile-4-methoxy-2-C-β-D-glucopyranoside (487). | ||
Dueber et al. 2016 demonstrated405 the formal synthesis of the indole alkaloids (±)-cis-trikentrin A (398) and (±)-herbindole B (491) from the usual meso-hydroquinone intermediate synthesized by a Ru-mediated [2 + 2 + 1 + 1] cycloaddition. Key steps involve a sterically demanding Buchwald–Hartwig amination and also a distinctive C(sp3)-H amination/indole construction. The formal synthesis of (±)-herbindole B (491) and (±)-cis-trikentrin A (398) began with the bicyclic alkene 488, which after six steps afforded aniline triflamide 489 and triflamide (492) through two different routes. In the following, aniline triflamide 489, through straight indole construction by using the C–H activation method, gave indole 490 in a satisfactory yield (66%), in which one carbon–nitrogen bond and one carbon–carbon double bond were formed. Next, after more than eight steps, indole 490 gave (±)-herbindole B406 (491).405
On the other hand, triflamide 492 was exposed to the indolization reaction and the desired indole 493 was obtained in a satisfactory yield (61%), in which one carbon–nitrogen bond and one carbon–carbon double bond were constructed (average yield of 78% per event). Finally, after more than eight steps, (±)-cis-trikentrin A (398) was synthesized successfully from indole 493 (Scheme 72).
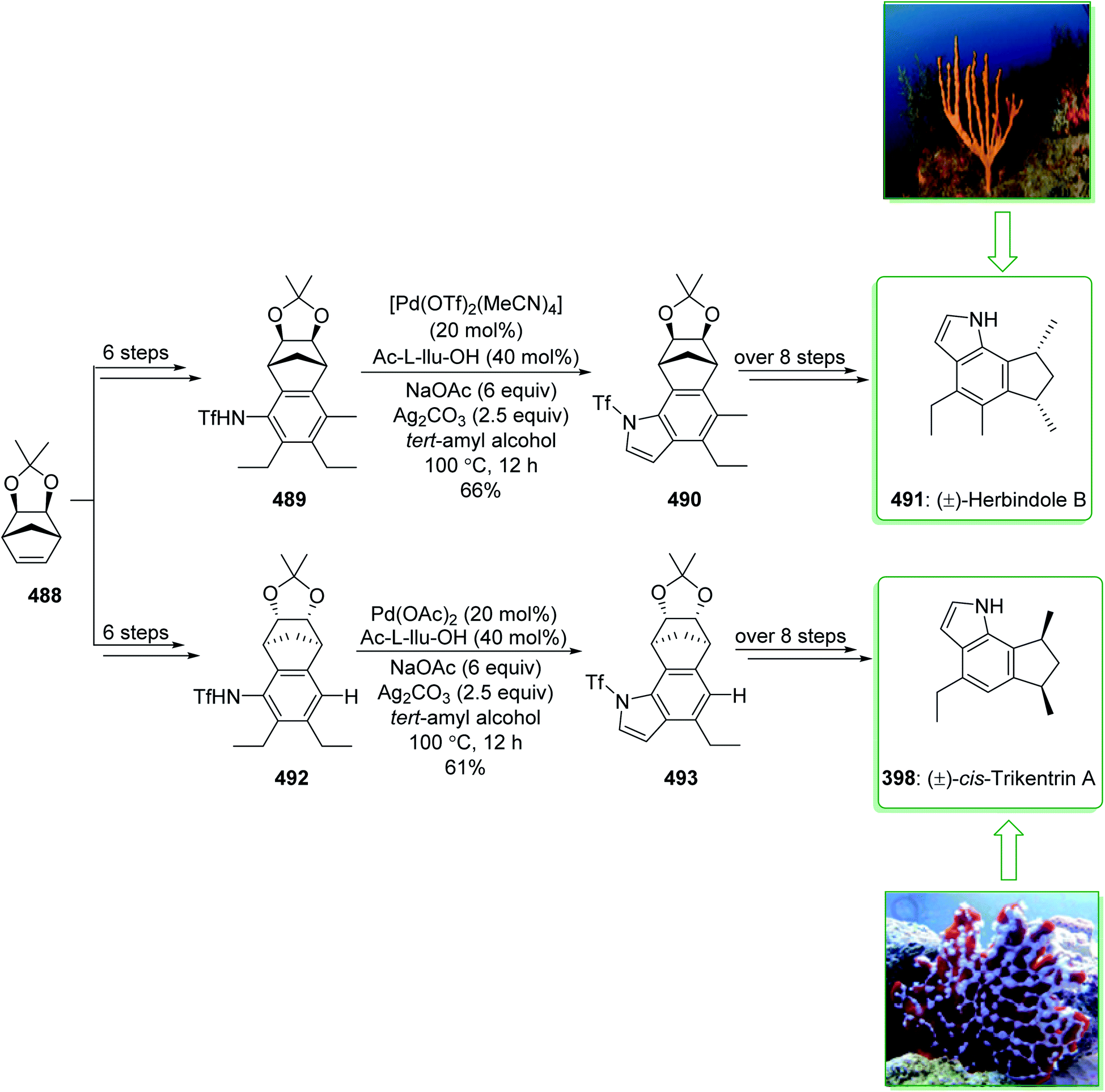 | ||
| Scheme 72 Formal synthesis of (±)-herbindole B (491) and (±)-cis-trikentrin A (398). | ||
Catharanthine is a significant member of the Iboga class of alkaloids.407,408 Catharanthine was extracted in 1985 by Raucher and Bray409 from the leaves of Catharanthus roseous.410,411 Catharanthus roseus includes more than 400 valuable alkaloids, for example vinblastine, vincristine, catharanthine, yohimbine, tabersonine, lochnericine, ajmalicine, vindosine, vindoline and vindolicine.412 Catharanthine has noteworthy biological functions as it has anti-cancer properties.413 A stereocontrolled total synthesis of (±)-catharanthine (502) was accomplished in 1999 by Fukuyama and Reding.414 The key step includes the radical-catalyzed cyclization of a variety of substituted intermediates to provide the desired indole. The cyclization reaction employs a facile phosphorus-based radical-reducing agent. The total synthesis of (±)-catharanthine (502) begins with the carbodiimide coupling of cis-2-alkenyl aniline 497 (prepared as a single diastereomer from quinoline 495 in three synthetic steps with a 42% overall yield) and endo-lactone 496 (prepared from diethyl ethoxymethylenemalonate 494 in four steps). This reaction yielded anilide iodolactone 498, which after three steps gave 2-alkenylthioanilide 499. The cyclization reaction using stoichiometric azobisisobutyronitrile (AIBN), excess aqueous hypophosphorous acid (H3PO2) and Et3N under reflux in 1-propanol consistently gave the corresponding indole 500 in a 40–50% yield. After two further steps, 500 gave intermediate 501. Next, the benzyl carbamate was eliminated under mild conditions and very selective reaction conditions415 to directly give (±)-catharanthine (502) (Scheme 73).414
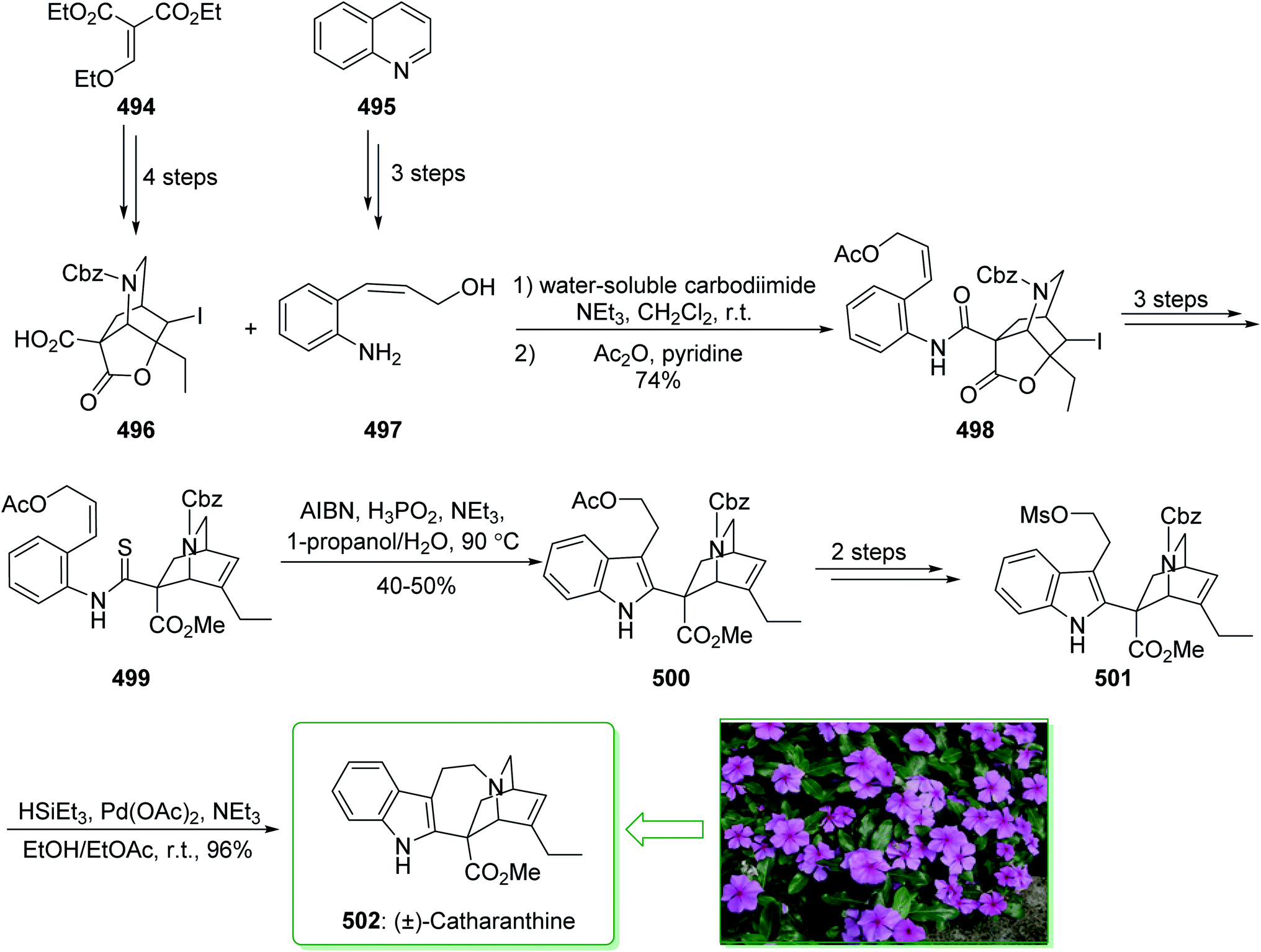 | ||
| Scheme 73 Total synthesis of (±)-catharanthine (502). | ||
Vinblastine416 is a well known chemotherapeutic agent utilized for the treatment of cancer. This natural product was recognized for the first time as a myelosuppressive agent by Noble's group in 1958 during the investigation of anti-diabetic agents in Catharanthus roseus.417 Individually, researchers at Eli Lilly demonstrated that extracts of Catharanthus roseus influenced activity against P-1534 leukemia in mice, and also extracted vinblastine as a potent unit in 1959.418 In 2010, Yokoshima et al. reported419 a stereocontrolled total synthesis of (+)-vinblastine (514). The synthesis of the upper half specifies the stereoselective formation of the tertiary alcohol via a 1,3-dipolar cycloaddition of nitrile oxide and a Baeyer–Villiger oxidation, a simple indole construction using the radical cyclization of o-alkenylthioanilide, and also the macrocyclization of 2-nitrobenzenesulfonamide. The essential coupling reaction of the upper half with synthetic vindoline was effectively accomplished to provide the coupling product in an approximately quantitative yield, and subsequent conversions afforded (+)-vinblastine (514). The total synthesis of (+)-vinblastine (514) started with the construction of the lower half, vindoline (507). The synthesis of the lower half was achieved using a Mitsunobu coupling reaction of indole 505 (prepared from 7-mesyloxyquinoline (503) in seven steps) and 2,4-dinitrobenzenesulfonamide (chiral amine) 506 (prepared from 2-pentenal (504) in six steps), which after over three steps afforded vindoline (507).419 On the other hand, the upper unit 511 was synthesized from n-butyraldehyde (508) and after 16 steps gave the alkenylthioanilides 509. Then, numerous conditions were examined, and it was found that using THF as a solvent inhibited the isomerization to provide indole 510 in a satisfactory yield (67% yield). The latter, after nine steps, gave the upper unit 511. Then, the chlorination of 511 using tert-butyl hypochlorite gave chloroindolenine, which was concisely proven using a neutral silica gel column to eliminate the excess quantity of the reagent. The chloroindolenine and synthetic vindoline (0.9 equivalents) were dissolved in CH2Cl2 and reacted with trifluoroacetic acid (CF3CO2H) to give the corresponding coupled product 512 in a high yield (97%) as a single isomer, which after two synthetic steps provided the secondary amine 513. Lastly, the latter using NaHCO3 in isopropyl alcohol (i-PrOH) and H2O gave (+)-vinblastine (514) in a moderate yield (66% yield) (Scheme 74).419
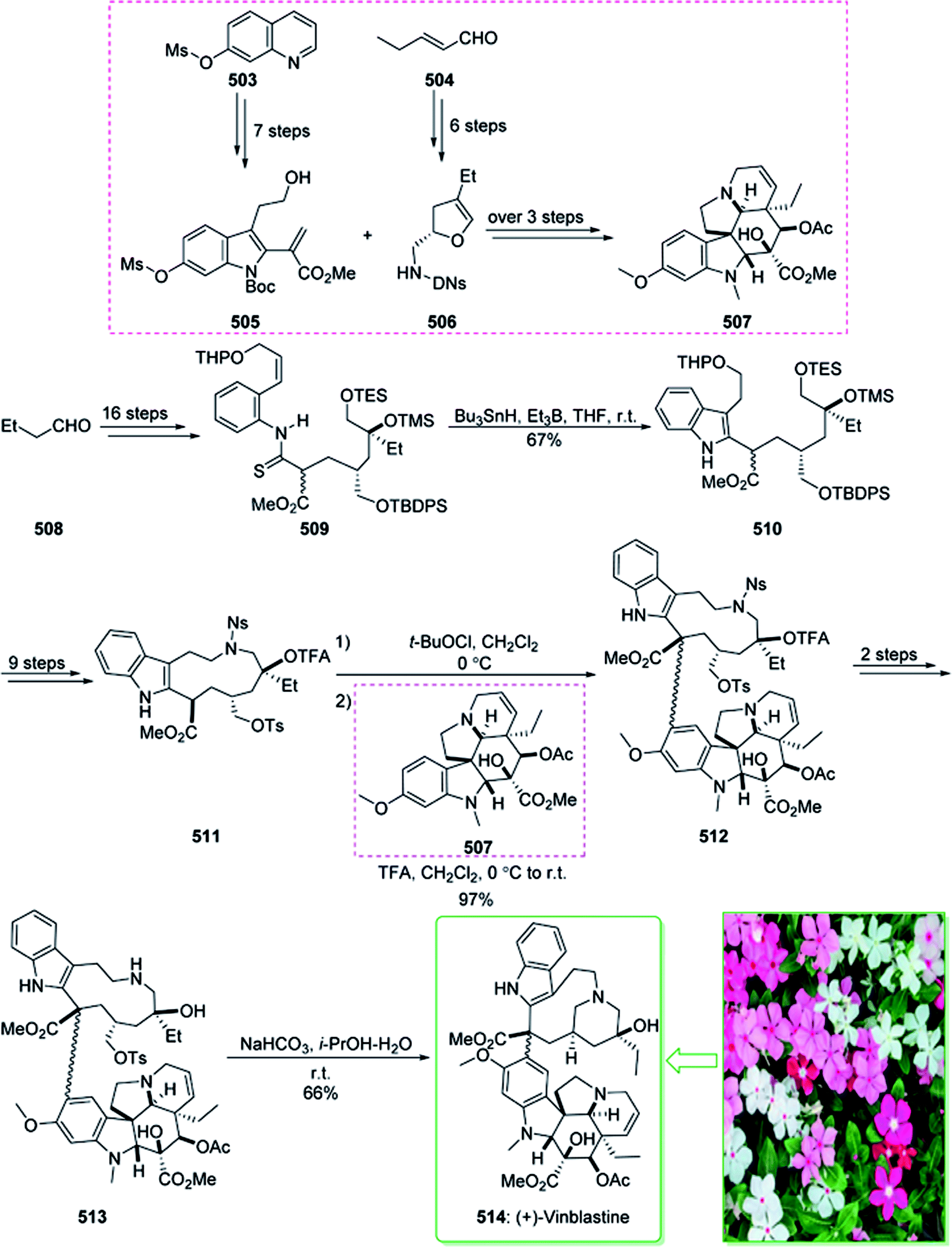 | ||
| Scheme 74 Total synthesis of (+)-vinblastine (514). | ||
4. Conclusion
In summary, indoles represent one of the most significant privileged motifs in drug discovery. Indoles and their derivatives have the exclusive property of mimicking the structure of peptides and can bind reversibly to enzymes, giving incredible opportunities to identify unique drugs that possess various modes of action. In addition, there are a remarkable number of approved indole-comprised drugs on the market. With the improvement in synthetic approaches, the separation of unique compounds from natural sources bearing indole frameworks is another ongoing and increasing area of investigation. The investigation of these novel molecules and the study of their properties and potential applications in the reaction of various diseases is another synergistic feature of the significance of the organic synthesis of indoles. Fischer indole synthesis is an essential reaction used in many natural product syntheses. This important named reaction is broadly used for installing the indole ring. In this review, we aim to demonstrate various methods used for synthesizing indoles as a moiety in selected alkaloids.Conflicts of interest
There are no conflicts to declare.References
- V. Sharma, P. Kumar and D. Pathak, J. Heterocycl. Chem., 2010, 47, 491–502 CAS.
- E. Abele, R. Abele, O. Dzenitis and E. Lukevics, Chem. Heterocycl. Compd., 2003, 39, 3–35 CrossRef CAS.
- H. Panwar, R. Verma, V. Srivastava and A. Kumar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2006, 45, 2099 Search PubMed.
- Y.-Y. Li, H.-S. Wu, L. Tang, C.-R. Feng, J.-H. Yu, Y. Li, Y.-S. Yang, B. Yang and Q.-J. He, J. Pharmacol. Res., 2007, 56, 335–343 CrossRef CAS PubMed.
- P. Barraja, L. Sciabica, P. Diana, A. Lauria, A. Montalbano, A. M. Almerico, G. Dattolo, G. Cirrincione, S. Disarò and G. Basso, Bioorg. Med. Chem. Lett., 2005, 15, 2291–2294 CrossRef CAS PubMed.
- K. S. Lam, Trends Microbiol., 2007, 15, 279–289 CrossRef CAS PubMed.
- M. S. Butler, Nat. Prod. Rep., 2008, 25, 475–516 RSC.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS PubMed.
- B. Debnath, W. S. Singh, M. Das, S. Goswami, M. K. Singh, D. Maiti and K. Manna, Mater. Today Chem., 2018, 9, 56–72 CrossRef CAS.
- B. Debnath, M. Uddin, P. Patari, M. Das, D. Maiti and K. Manna, Int. J. Pharm. Pharm. Sci., 2015, 7, 223–227 CAS.
- M. El-Sayed and R. Verpoorte, Phytochem. Rev., 2007, 6, 277–305 CrossRef CAS.
- S. Sagi, B. Avula, Y.-H. Wang and I. A. Khan, Anal. Bioanal. Chem., 2016, 408, 177–190 CrossRef CAS PubMed.
- N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed.
- L. B. Diss, S. D. Robinson, Y. Wu, S. Fidalgo, M. S. Yeoman and B. A. Patel, ACS Chem. Neurosci., 2013, 4, 879–887 CrossRef CAS PubMed.
- M.-Z. Zhang, N. Mulholland, D. Beattie, D. Irwin, Y.-C. Gu, Q. Chen, G.-F. Yang and J. Clough, Eur. J. Med. Chem., 2013, 63, 22–32 CrossRef CAS PubMed.
- S. N. Young, J. Neuropsychiatry Clin. Neurosci., 2007, 32, 394 Search PubMed.
- S. A. Patil, R. Patil and D. D. Miller, Future Med. Chem., 2012, 4, 2085–2115 CrossRef CAS PubMed.
- F.-E. Chen and J. Huang, Chem. Rev., 2005, 105, 4671–4706 CrossRef CAS PubMed.
- W. Kurz, K. Chatson, F. Constabel, J. Kutney, L. Choi, P. Kolodziejczyk, S. Sleigh, K. Stuart and B. Worth, Planta Med., 1981, 42, 22–31 CrossRef CAS PubMed.
- H. Ishikawa, D. A. Colby and D. L. Boger, J. Am. Chem. Soc., 2008, 130, 420–421 CrossRef CAS PubMed.
- G. Bartoli, R. Leardini, A. Medici and G. Rosini, J. Chem. Soc., Perkin Trans. 1, 1978, 692–696 RSC.
- H. Hemetsberger and D. Knittel, Monatsh. Chem., 1972, 103, 194–204 CrossRef CAS.
- A. Bischler and P. Fireman, Ber. Dtsch. Chem. Ges., 1893, 26, 1336–1349 CrossRef.
- J.-B. Baudin and S. A. Julia, Tetrahedron Lett., 1986, 27, 837–840 CrossRef CAS.
- R. C. Larock and S. Babu, Tetrahedron Lett., 1987, 28, 5291–5294 CrossRef CAS.
- C. Nenitzescu, Bull. Assoc. Chim., 1929, 11, 37–43 Search PubMed.
- W. Madelung, Ber. Dtsch. Chem. Ges., 1912, 45, 1128–1134 CrossRef.
- W. Noland and F. Baude, Org. Synth., 1973, 5, 567–571 Search PubMed.
- E. Fischer and F. Jourdan, Ber. Dtsch. Chem. Ges., 1883, 16, 2241–2245 CrossRef.
- A. Kuznetsov, A. Makarov, A. E. Rubtsov, A. V. Butin and V. Gevorgyan, J. Org. Chem., 2013, 78, 12144–12153 CrossRef CAS PubMed.
- T. Chandra, S. Zou and K. L. Brown, Tetrahedron Lett., 2004, 45, 7783–7786 CrossRef CAS.
- A. Carpita and A. Ribecai, Tetrahedron Lett., 2009, 50, 6877–6881 CrossRef CAS.
- W. Wierenga, J. Griffin and M. A. Warpehoski, Tetrahedron Lett., 1983, 24, 2437–2440 CrossRef CAS.
- D. Hong, Z. Chen, X. Lin and Y. Wang, Org. Lett., 2010, 12, 4608–4611 CrossRef CAS PubMed.
- R. Umeda, Y. Nishimoto and T. Mashino, Heterocycles, 2013, 87, 1241–1247 CrossRef CAS.
- M. T. Hovey, C. T. Check, A. F. Sipher and K. A. Scheidt, Angew. Chem., Int. Ed., 2014, 126, 9757–9761 CrossRef.
- J. J. Li, Name reactions in heterocyclic chemistry, John Wiley & Sons, 2004 Search PubMed.
- J. P. Kutney, in The Total Synthesis of Natural Products, 2007, vol. 3, pp. 273–438 Search PubMed.
- S. M. Bronner, G. Y. J. Im and N. K. Garg, in Heterocycles in Natural Product Synthesis, 2011, pp. 221–265, DOI:10.1002/9783527634880.ch7.
- D. L. Hughes, Org. Prep. Proced. Int., 1993, 25, 607–632 CrossRef CAS.
- R. McDanell, A. E. M. McLean, A. B. Hanley, R. K. Heaney and G. R. Fenwick, Food Chem. Toxicol., 1988, 26, 59–70 CrossRef CAS PubMed.
- R. Dalpozzo and G. Bartoli, Curr. Org. Chem., 2005, 9, 163–178 CrossRef CAS.
- S. Cacchi and G. Fabrizi, Chem. Rev., 2011, 111, PR215–PR283 CrossRef PubMed.
- M. Shiri, Chem. Rev., 2012, 112, 3508–3549 CrossRef CAS PubMed.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195 CrossRef CAS PubMed.
- M.-Z. Zhang, Q. Chen and G.-F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed.
- H. A. Hamid, A. N. Ramli and M. M. Yusoff, Front. Pharmacol., 2017, 8, 96 Search PubMed.
- K. Hemalatha, G. Madhumitha and S. M. Roopan, Chem. Sci. Rev. Lett., 2013, 2, 287–292 Search PubMed.
- B. M. Trost and M. K. Brennan, Synthesis, 2009, 2009, 3003–3025 CrossRef.
- G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045–1075, 10.1039/A909834H.
- M. Inman and C. Moody, Chem. Sci., 2013, 4, 29–41 RSC.
- F. Wu, P. Kerčmar, C. Zhang and J. Stöckigt, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2016, vol. 76, pp. 1–61 Search PubMed.
- G. L. Adams and A. B. Smith III, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2016, vol. 76, pp. 171–257 Search PubMed.
- M. Kitajima and H. Takayama, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2016, vol. 76, pp. 259–310 Search PubMed.
- M. Pfaffenbach and T. Gaich, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2017, vol. 77, pp. 1–84 Search PubMed.
- S. Arai, M. Nakajima and A. Nishida, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2017, vol. 78, pp. 167–204 Search PubMed.
- C. Xia and X. Tong, in Alkaloids Chem. Biol., ed. H.-J. Knölker, Academic Press, London, 2018, vol. 79, pp. 139–189 Search PubMed.
- J. D. Mason and S. M. Weinreb, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2019, vol. 81, pp. 115–150 Search PubMed.
- M. G. Kalshetti and N. P. Argade, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2020, vol. 83, pp. 187–223 Search PubMed.
- S. C. Taylor and S. M. Weinreb, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2021, vol. 85, pp. 177–222 Search PubMed.
- N. R. Tasker, P. Wipf, S. C. Taylor and S. M. Weinreb, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2021, vol. 85, pp. 1–112 Search PubMed.
- G. W. Gribble, Pure Appl. Chem., 2003, 75, 1417–1432 CAS.
- S. Agarwal, S. Cammerer, S. Filali, W. Frohner, J. Knoll, M. P. Krahl, K. R. Reddy and H.-J. Knolker, Curr. Org. Chem., 2005, 9, 1601–1614 CrossRef CAS.
- G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875–2911 CrossRef CAS PubMed.
- S. Khaghaninejad and M. M. Heravi, Adv. Heterocycl. Chem., 2014, 111, 95–146 CrossRef CAS.
- M. M. Heravi and T. Alishiri, Adv. Heterocycl. Chem., 2014, 113, 1–66 CrossRef CAS.
- M. M. Heravi and V. F. Vavsari, Adv. Heterocycl. Chem., 2015, 114, 77–145 CrossRef CAS.
- M. M. Heravi and V. F. Vavsari, RSC Adv., 2015, 5, 50890–50912 RSC.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- M. M. Heravi and L. Mohammadkhani, J. Organomet. Chem., 2018, 869, 106–200 CrossRef CAS.
- M. M. Heravi, T. B. Lashaki, B. Fattahi and V. Zadsirjan, RSC Adv., 2018, 8, 6634–6659 RSC.
- M. M. Heravi, V. Zadsirjan and M. Malmir, Molecules, 2018, 23, 943 CrossRef PubMed.
- M. M. Heravi, V. Zadsirjan, K. Kafshdarzadeh and Z. Amiri, Asian J. Org. Chem., 2020, 9, 1999–2034 CrossRef CAS.
- M. M. Heravi, S. Rohani, V. Zadsirjan and N. Zahedi, RSC Adv., 2017, 7, 52852–52887 RSC.
- M. M. Heravi, V. Zadsirjan, H. Hamidi, M. Daraie and T. Momeni, in The Alkaloids, ed. H.-J. Knölker, Elsevier, Academic Press, London, 2020, vol. 84, pp. 201–334 Search PubMed.
- L. Wei, Y. Z. Wang and Z. Y. Li, BMC Plant Biol., 2012, 14, 190–197 CAS.
- S. Syam, A. B. Abdul, M. Sukari, S. Mohan, S. I. Abdelwahab and T. S. Wah, Molecules, 2011, 16, 7155–7170 CrossRef CAS PubMed.
- S. Mohan, S. I. Abdelwahab, S.-C. Cheah, M. A. Sukari, S. Syam, N. Shamsuddin and M. Rais Mustafa, Evid Based Complement Alternat Med., 2013, vol. 2013 Search PubMed.
- K. Das, D. Chakraborty and P. Bose, Experientia, 1965, 21, 340 CrossRef CAS PubMed.
- A. C. Adebajo, G. Olayiwola, J. Eugen Verspohl, E. O. Iwalewa, N. Omisore, D. Bergenthal, V. Kumar and S. Kolawole Adesina, Pharm. Biol., 2005, 42, 610–620 CrossRef.
- S. Gupta, M. George, M. Singhal, G. N. Sharma and V. Garg, J. Adv. Pharm. Technol. Res., 2010, 1, 68 Search PubMed.
- R. S. Ramsewak, M. G. Nair, G. M. Strasburg, D. L. DeWitt and J. L. Nitiss, J. Agric. Food Chem., 1999, 47, 444–447 CrossRef CAS PubMed.
- D. P. Chakraborty and B. Chowdhury, J. Org. Chem., 1968, 33, 1265–1268 CrossRef CAS.
- T. Martin and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1988, 235–240, 10.1039/P19880000235.
- H.-J. Knölker and M. Bauermeister, J. Chem. Soc., Chem. Commun., 1990, 664–665, 10.1039/C39900000664.
- H.-J. Knölker and M. Bauermeister, Tetrahedron, 1993, 49, 11221–11236 CrossRef.
- H.-J. Knölker and M. Wolpert, Tetrahedron, 2003, 59, 5317–5322 CrossRef.
- P. Bernal and J. Tamariz, Helv. Chim. Acta, 2007, 90, 1449–1454 CrossRef CAS.
- C. Börger, O. Kataeva and H.-J. Knölker, Org. Biomol. Chem., 2012, 10, 7269–7273 RSC.
- H. Knölker and K. R. Reddy, in The Alkaloids, ed. G. A. Cordell, Academic Press, Amsterdam, 2008, vol. 65, pp. 1–430 Search PubMed.
- H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef PubMed.
- H.-J. Knolker, Curr. Org. Synth., 2004, 1, 309–331 CrossRef.
- I. Bauer and H.-J. Knölker, Top. Curr. Chem., 2012, 309, 203–253 CrossRef CAS PubMed.
- A. W. Schmidt, K. R. Reddy and H.-J. Knölker, Chem. Rev., 2012, 112, 3193–3328 CrossRef CAS PubMed.
- S. Ray and D. Chakraborty, Phytochemistry, 1976, 15, 356 CrossRef CAS.
- D. Chakraborty and K. Das, Chem. Commun., 1968, 967 RSC.
- D. P. Chakraborty, A. Islam and P. Bhattacharyya, J. Org. Chem., 1973, 38, 2728–2729 CrossRef CAS.
- H.-J. Knölker and C. Hofmann, Tetrahedron Lett., 1996, 37, 7947–7950 CrossRef.
- K. K. Gruner, T. Hopfmann, K. Matsumoto, A. Jäger, T. Katsuki and H.-J. Knölker, Org. Biomol. Chem., 2011, 9, 2057–2061 RSC.
- R. Hesse, K. K. Gruner, O. Kataeva, A. W. Schmidt and H. J. Knölker, Chem.–Eur. J., 2013, 19, 14098–14111 CrossRef CAS PubMed.
- W.-F. Chiou, C.-J. Chou, J.-F. Liao, A. Y.-C. Sham and C.-F. Chen, Eur. J. Pharmacol., 1994, 257, 59–66 CrossRef CAS PubMed.
- J.-F. Liao, W.-F. Chiou, Y.-C. Shen, G.-J. Wang and C.-F. Chen, Chin. Med., 2011, 6, 1–8 CrossRef PubMed.
- S. Johne, D. Gröger and M. Hesse, Helv. Chim. Acta, 1971, 54, 826–834 CrossRef CAS.
- A. Amin, D. Mehta and S. Samarth, Prog. Drug Res., 1970, 218–268 CAS.
- N. Shoji, A. Umeyama, T. Takemoto, A. Kajiwara and Y. Ohizumi, J. Pharm. Sci., 1986, 75, 612–613 CrossRef CAS PubMed.
- J. Kökösi, G. Szász and I. Hermecz, Tetrahedron Lett., 1992, 33, 2995–2998 CrossRef.
- T. Kametani, T. Higa, C. Van Loc, M. Ihara, M. Koizumi and K. Fukumoto, J. Am. Chem. Soc., 1976, 98, 6186–6188 CrossRef CAS.
- T. Kametani, C. Van Loc, T. Higa, M. Koizumi, M. Ihara and K. Fukumoto, J. Am. Chem. Soc., 1977, 99, 2306–2309 CrossRef CAS.
- C. Kaneko, T. Chiba, K. Kasai and C. Miwa, Heterocycles, 1985, 23, 1385–1390 CrossRef CAS.
- T. F. Molinski, Chem. Rev., 1993, 93, 1825–1838 CrossRef CAS.
- N. B. Perry, J. W. Blunt, M. H. G. Munro, T. Higa and R. Sakai, J. Org. Chem., 1988, 53, 4127–4128 CrossRef CAS.
- J. F. Cheng, Y. Ohizumi, M. R. Walchli, H. Nakamura, Y. Hirata, T. Sasaki and J. Kobayashi, J. Org. Chem., 1988, 53, 4621–4624 CrossRef CAS.
- S. Sakemi, H. H. Sun, C. W. Jefford and G. Bernardinelli, Tetrahedron Lett., 1989, 30, 2517–2520 CrossRef CAS.
- D. B. Stierle and D. J. Faulkner, J. Nat. Prod., 1991, 54, 1131–1133 CrossRef CAS.
- B. R. Copp, C. M. Ireland and L. R. Barrows, J. Org. Chem., 1991, 56, 4596–4597 CrossRef CAS.
- D. C. Radisky, E. S. Radisky, L. R. Barrows, B. R. Copp, R. A. Kramer and C. M. Ireland, J. Am. Chem. Soc., 1993, 115, 1632–1638 CrossRef CAS.
- J. D. White, K. M. Yager and T. Yakura, J. Am. Chem. Soc., 1994, 116, 1831–1838 CrossRef CAS.
- B. Mckittrick, A. Failli, R. J. Steffan, R. M. Soll, P. Hughes, J. Schmid, A. A. Asselin, C. Shaw, R. Noureldin and G. Gavin, J. Heterocycl. Chem., 1990, 27, 2151–2163 CrossRef CAS.
- C.-B. Cui, H. Kakeya, G. Okada, R. Onose, M. Ubukata, I. Takahashi, K. Isono and H. Osada, J. Antibiot., 1995, 48, 1382–1384 CrossRef CAS PubMed.
- T. Usui, M. Kondoh, C.-B. Cui, T. Mayumi and H. Osada, Biochem. J., 1998, 333, 543–548 CrossRef CAS PubMed.
- A. S. P. Cardoso, M. M. B. Marques, N. Srinivasan, S. Prabhakar, A. M. Lobo and H. S. Rzepa, Org. Biomol. Chem., 2006, 4, 3966–3972 RSC.
- S. Zhao, K. S. Smith, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, T. L. Macdonald and J. M. Cook, J. Med. Chem., 2002, 45, 1559–1562 CrossRef CAS PubMed.
- T. Gan, R. Liu, P. Yu, S. Zhao and J. M. Cook, J. Org. Chem., 1997, 62, 9298–9304 CrossRef CAS.
- M. S. Allen, L. K. Hamaker, A. J. La Loggia and J. M. Cook, Synth. Commun., 1992, 22, 2077–2102 CrossRef CAS.
- S. Takano and K. Ogasawara, Alkaloids, 1990, 36, 225–251 Search PubMed.
- D. J. Triggle and R. Filler, CNS Drug Rev., 1998, 4, 87–136 CrossRef CAS.
- U. Anthoni, C. Christophersen and P. Nielsen, Alkaloids: Chemical and Biological Perspectives, Wiley, New York, 1999 Search PubMed.
- B. Robinson, J. Chem. Soc., 1964, 1503–1506, 10.1039/JR9640001503.
- F. J. Dale and B. Robinson, J. Pharm. Pharmacol., 1970, 22, 889–896 CrossRef CAS PubMed.
- A. W. Schammel, B. W. Boal, L. Zu, T. Mesganaw and N. K. Garg, Tetrahedron, 2010, 66, 4687–4695 CrossRef CAS PubMed.
- T. Matsuura, L. E. Overman and D. J. Poon, J. Am. Chem. Soc., 1998, 120, 6500–6503 CrossRef CAS.
- N. Khoukhi, M. Vaultier and R. Carrie, Tetrahedron, 1987, 43, 1811–1822 CrossRef CAS.
- A. V. Dix, C. M. Meseck, A. J. Lowe and M. O. Mitchell, Bioorg. Med. Chem. Lett., 2006, 16, 2522–2524 CrossRef CAS PubMed.
- P. A. Cockrum, S. M. Colegate, J. A. Edgar, K. Flower, D. Gardner and R. I. Willing, Phytochemistry, 1999, 51, 153–157 CrossRef.
- C. Li, C. Chan, A. C. Heimann and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 1444–1447 CrossRef CAS PubMed.
- J. D. Trzupek, C. Li, C. Chan, B. M. Crowley, A. C. Heimann and S. J. Danishefsky, Pure Appl. Chem., 2010, 82, 1735–1748 CAS.
- J. D. Trzupek, D. Lee, B. M. Crowley, V. M. Marathias and S. J. Danishefsky, J. Am. Chem. Soc., 2010, 132, 8506–8512 CrossRef CAS PubMed.
- H. Ding and D. Y. K. Chen, Angew. Chem., Int. Ed., 2011, 123, 702–705 CrossRef.
- R. Fröde, C. Hinze, I. Josten, B. Schmidt, B. Steffan and W. Steglich, Tetrahedron Lett., 1994, 35, 1689–1690 CrossRef.
- T. Hashimoto, Y. Akiyo, K. Akazawa, S. Takaoka, M. Tori and Y. Asakawa, Tetrahedron Lett., 1994, 35, 2559–2560 CrossRef CAS.
- C. Sánchez, A. F. Braña, C. Méndez and J. A. Salas, ChemBioChem, 2006, 7, 1231–1240 CrossRef PubMed.
- T. Nishizawa, S. Grüschow, D.-H. E. Jayamaha, C. Nishizawa-Harada and D. H. Sherman, J. Am. Chem. Soc., 2006, 128, 724–725 CrossRef CAS PubMed.
- A. R. Howard-Jones and C. T. Walsh, J. Am. Chem. Soc., 2006, 128, 12289–12298 CrossRef CAS PubMed.
- N. Zhou, Q. Shi and Z. Xie, Chin. J. Org. Chem., 2014, 34, 1104–1109 CrossRef CAS.
- G. Massiot, P. Thépenier, M.-J. Jacquier, L. Le Men-Olivier and C. Delaude, Heterocycles, 1989, 29, 1435–1438 CrossRef CAS.
- A. Ramírez and S. García-Rubio, Curr. Med. Chem., 2003, 10, 1891–1915 CrossRef PubMed.
- P. Liu, J. Wang, J. Zhang and F. G. Qiu, Org. Lett., 2011, 13, 6426–6428 CrossRef CAS PubMed.
- M. E. Krafft and J. W. Cran, Synlett, 2005, 2005, 1263–1266 CrossRef.
- G. Subramaniam, O. Hiraku, M. Hayashi, T. Koyano, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2007, 70, 1783–1789 CrossRef CAS PubMed.
- L. Zu, B. W. Boal and N. K. Garg, J. Am. Chem. Soc., 2011, 133, 8877–8879 CrossRef CAS PubMed.
- C. Herdeis and C. Hartke, Heterocycles, 1989, 29, 287–296 CrossRef CAS.
- G. L. Adams and A. B. Smith III, The chemistry of the akuammiline alkaloids, Academic Press, London, 2016 Search PubMed.
- X.-H. Cai, Q.-G. Tan, Y.-P. Liu, T. Feng, Z.-Z. Du, W.-Q. Li and X.-D. Luo, Org. Lett., 2008, 10, 577–580 CrossRef CAS PubMed.
- P. Kamarajan, N. Sekar, V. Mathuram and S. Govindasamy, Biochem. Int., 1991, 25, 491–498 CAS.
- X.-H. Cai, Z.-Z. Du and X.-D. Luo, Org. Lett., 2007, 9, 1817–1820 CrossRef CAS PubMed.
- G. L. Adams, P. J. Carroll and A. B. Smith III, J. Am. Chem. Soc., 2012, 134, 4037–4040 CrossRef CAS PubMed.
- G. L. Adams, P. J. Carroll and A. B. Smith III, J. Am. Chem. Soc., 2013, 135, 519–528 CrossRef CAS PubMed.
- P. Müller and D. M. Gilabert, Tetrahedron, 1988, 44, 7171–7175 CrossRef.
- Z. Zuo and D. Ma, Isr. J. Chem., 2011, 51, 434–441 CrossRef CAS.
- D. Zhang, H. Song and Y. Qin, Acc. Chem. Res., 2011, 44, 447–457 CrossRef CAS PubMed.
- S. M. Verbitski, C. L. Mayne, R. A. Davis, G. P. Concepcion and C. M. Ireland, J. Org. Chem., 2002, 67, 7124–7126 CrossRef CAS PubMed.
- A. Numata, C. Takahashi, Y. Ito, T. Takada, K. Kawai, Y. Usami, E. Matsumura, M. Imachi, T. Ito and T. Hasegawa, Tetrahedron Lett., 1993, 34, 2355–2358 CrossRef CAS.
- A. W. Schammel, G. Chiou and N. K. Garg, Org. Lett., 2012, 14, 4556–4559 CrossRef CAS PubMed.
- K. Warabi, S. Matsunaga, R. W. van Soest and N. Fusetani, J. Org. Chem., 2003, 68, 2765–2770 CrossRef CAS PubMed.
- F. Lavelle, J.-F. Riou, A. Laoui and P. Mailliet, Crit. Rev. Oncol. Hematol., 2000, 34, 111–126 CrossRef CAS PubMed.
- S. M. Gowan, J. R. Harrison, L. Patterson, M. Valenti, S. Neidle and L. R. Kelland, Mol. Pharmacol., 2002, 61, 1154–1162 CrossRef CAS PubMed.
- P. Tao, J. Liang and Y. Jia, Eur. J. Org. Chem., 2014, 2014, 5735–5748 CrossRef CAS.
- H. Tokuyama, J. Synth. Org. Chem., Jpn., 2015, 73, 1120–1129 CrossRef CAS.
- T. J. Hagen, K. Narayanan, J. Names and J. M. Cook, J. Org. Chem., 1989, 54, 2170–2178 CrossRef CAS.
- T. Janosik, N. Wahlström and J. Bergman, Tetrahedron, 2008, 39, 9159–9180 CrossRef.
- M. Prudhomme, Eur. J. Med. Chem., 2003, 38, 123–140 CrossRef CAS PubMed.
- D.-Q. Liu, S.-C. Mao, H.-Y. Zhang, X.-Q. Yu, M.-T. Feng, B. Wang, L.-H. Feng and Y.-W. Guo, Fitoterapia, 2013, 91, 15–20 CrossRef CAS PubMed.
- K. M. Meragelman, L. M. West, P. T. Northcote, L. K. Pannell, T. C. McKee and M. R. Boyd, J. Org. Chem., 2002, 67, 6671–6677 CrossRef CAS PubMed.
- F. Russell, D. Harmody, P. J. McCarthy, S. A. Pomponi and A. E. Wright, J. Nat. Prod., 2013, 76, 1989–1992 CrossRef CAS PubMed.
- S. Kotha, M. Saifuddin and V. R. Aswar, Org. Biomol. Chem., 2016, 14, 9868–9873 RSC.
- D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426–12429 CrossRef CAS PubMed.
- X.-W. Yang, X.-D. Luo, P. K. Lunga, Y.-L. Zhao, X.-J. Qin, Y.-Y. Chen, L. Liu, X.-N. Li and Y.-P. Liu, Tetrahedron, 2015, 71, 3694–3698 CrossRef CAS.
- X.-W. Yang, X.-J. Qin, Y.-L. Zhao, P. K. Lunga, X.-N. Li, S.-Z. Jiang, G.-G. Cheng, Y.-P. Liu and X.-D. Luo, Tetrahedron Lett., 2014, 55, 4593–4596 CrossRef CAS.
- D. Wang, M. Hou, Y. Ji and S. Gao, Org. Lett., 2017, 19, 1922–1925 CrossRef CAS PubMed.
- R. Lyon, H. Fong, N. Farnsworth and G. Svoboda, J. Pharm. Sci., 1973, 62, 218–221 CrossRef CAS PubMed.
- N. Wang, S. Du, D. Li and X. Jiang, Org. Lett., 2017, 19, 3167–3170 CrossRef CAS PubMed.
- G. Stork and J. E. Dolfini, J. Am. Chem. Soc., 1963, 85, 2872–2873 CrossRef CAS.
- I. Coldham, A. J. Burrell, L. E. White, H. Adams and N. Oram, Angew. Chem., Int. Ed., 2007, 46, 6159–6162 CrossRef CAS PubMed.
- L. Randriambola, J.-C. Quirion, C. Kan-Fan and H.-P. Husson, Tetrahedron Lett., 1987, 28, 2123–2126 CrossRef CAS.
- B. P. Pritchett, J. Kikuchi, Y. Numajiri and B. M. Stoltz, Heterocycles, 2017, 95, 1245 CrossRef CAS PubMed.
- H.-J. Knolker, Curr. Org. Chem., 2004, 1, 309–331 Search PubMed.
- T.-S. Wu, S.-C. Huang and P.-L. Wu, Phytochemistry, 1996, 43, 1427–1429 CrossRef CAS.
- S.-C. Huang, P.-L. Wu and T.-S. Wu, Phytochemistry, 1997, 44, 179–181 CrossRef CAS.
- I. A. Arbab, A. B. Abdul, M. Aspollah, S. I. Abdelwahab, M. Y. Ibrahim and Z. Ali, J. Med. Plants Res., 2012, 6, 5107–5118 CrossRef.
- A. Chakraborty, B. Chowdhury, S. Jash, G. Biswas, S. Bhattacharyya and P. Bhattacharyya, Phytochemistry, 1992, 31, 2503–2505 CrossRef.
- U. Songsiang, T. Thongthoom, C. Boonyarat and C. Yenjai, J. Nat. Prod., 2011, 74, 208–212 CrossRef CAS PubMed.
- A. Chakraborty, C. Saha, G. Podder, B. Chowdhury and P. Bhattacharyya, Phytochemistry, 1995, 38, 787–789 CrossRef CAS PubMed.
- R. Bautista, A. Benavides, H. A. Jiménez-Vázquez and J. Tamariz, Nat. Prod. Res., 2013, 27, 1749–1756 CrossRef CAS PubMed.
- M. S. Naykode, S. D. Tambe and P. D. Lokhande, ChemistrySelect, 2017, 2, 567–570 CrossRef CAS.
- Y. Yang, T. J. L. Mustard, P. H.-Y. Cheong and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 14098–14102 CrossRef CAS PubMed.
- M. P. Krahl, A. Jäger, T. Krause and H.-J. Knölker, Org. Biomol. Chem., 2006, 4, 3215–3219 RSC.
- M. Fuchsenberger, R. Forke and H.-J. Knölker, Synlett, 2011, 22, 2056–2058 Search PubMed.
- C. Schuster, C. Börger, K. K. Julich-Gruner, R. Hesse, A. Jäger, G. Kaufmann, A. W. Schmidt and H. J. Knölker, Eur. J. Org. Chem., 2014, 2014, 4741–4752 CrossRef CAS.
- A. Kishonti, A. Jäger and H. J. Knölker, Eur. J. Org. Chem., 2020, 2020, 5572–5579 CrossRef CAS.
- H. Schnoes, K. Biemann, J. Mokry, I. Kompis, A. Chatterjee and G. Ganguli, J. Org. Chem., 1966, 31, 1641–1642 CrossRef CAS.
- S.-H. Lim, Y.-Y. Low, S. K. Sinniah, K.-T. Yong, K.-S. Sim and T.-S. Kam, Phytochemistry, 2014, 98, 204–215 CrossRef CAS PubMed.
- Y. Ahmad, K. Fatima, J. L. Occolowitz, B. A. Solheim, J. Clardy, R. L. Garnick and P. W. Le Quesne, J. Am. Chem. Soc., 1977, 99, 1943–1946 CrossRef CAS.
- T. A. Henry, J. Chem. Soc., 1932, 2759–2768 RSC.
- M. Duwiejua, E. Woode and D. Obiri, J. Ethnopharmacol., 2002, 81, 73–79 CrossRef CAS PubMed.
- E. Picazo, L. A. Morrill, R. B. Susick, J. Moreno, J. M. Smith and N. K. Garg, J. Am. Chem. Soc., 2018, 140, 6483–6492 CrossRef CAS PubMed.
- D. Chakraborty and S. Roy, Progress in the chemistry of organic natural products, Springer, Wien, 1991 Search PubMed.
- J. Cardellina, Tetrahedron Lett., 1979, 20, 4915–4916 CrossRef.
- S. Chakraborty and C. Saha, Eur. J. Org. Chem., 2018, 2018, 2013–2021 CrossRef CAS.
- S. Chakraborty and C. Saha, Eur. J. Org. Chem., 2013, 2013, 5731–5736 CrossRef CAS.
- H. J. Knölker, M. Bauermeister, D. Bläser, R. Boese and J. B. Pannek, Angew. Chem., Int. Ed. Engl., 1989, 28, 223–225 CrossRef.
- C. J. Moody and P. Shah, J. Chem. Soc., Perkin Trans. 1, 1989, 2463–2471 RSC.
- H.-J. Knölker, M. Bauermeister, P. Jörn-Bernd, D. Bläser and R. Boese, Tetrahedron, 1993, 49, 841–862 CrossRef.
- S. Hibino, A. Tonari, T. Choshi and E. Sugino, Heterocycles, 1993, 35, 441–444 CrossRef CAS.
- E. M. Beccalli, A. Marchesini and T. Pilati, J. Chem. Soc., Perkin Trans. 1, 1994, 579–587 RSC.
- H.-J. Knölker, E. Baum and T. Hopfmann, Tetrahedron Lett., 1995, 36, 5339–5342 Search PubMed.
- T. Choshi, T. Sada, H. Fujimoto, C. Nagayama, E. Sugino and S. Hibino, J. Org. Chem., 1997, 62, 2535–2543 CrossRef CAS PubMed.
- H.-J. Knölker, E. Baum and T. Hopfmann, Tetrahedron, 1999, 55, 10391–10412 CrossRef.
- H.-J. Knoelker, W. Froehner and R. Heinrich, Synlett, 2004, 2004, 2705–2708 CrossRef.
- R. S. A. Cabral, P.-M. Allard, L. Marcourt, M. C. u. M. Young, E. F. Queiroz and J.-L. Wolfender, J. Nat. Prod., 2016, 79, 2270–2278 CrossRef CAS PubMed.
- D. G. Agripino, M. E. L. Lima, M. R. d. Silva, C. I. Meda, V. d. S. Bolzani, I. Cordeiro, M. C. M. Young and P. R. H. Moreno, Biota Neotrop., 2004, 4, 1–15 CrossRef.
- T. Abe, T. Itoh and M. Terasaki, Helv. Chim. Acta, 2019, 102, e1900116 CrossRef.
- F. F. Marinho, A. O. Simões, T. Barcellos and S. Moura, Fitoterapia, 2016, 114, 127–137 CrossRef CAS PubMed.
- G. Kalaus, I. Juhász, I. Greiner, M. Kajtár-Peredy, J. Brlik, L. Szabó and C. Szántay, J. Org. Chem., 1997, 62, 9188–9191 CrossRef CAS.
- T.-S. Kam, K. Yoganathan and S.-L. Mok, Phytochemistry, 1997, 46, 789–792 CrossRef CAS.
- T. Soós, S. Varga, P. Angyal, G. Martin, O. Egyed and T. Holczbauer, Angew. Chem., Int. Ed., 2020, 132, 13649–13653 CrossRef.
- B. Berkes, K. Ozsváth, L. Molnár, T. Gáti, T. Holczbauer, G. Kardos and T. Soós, Chem.–Eur. J., 2016, 22, 18101–18106 CrossRef CAS PubMed.
- F. Ohiri, R. Verpoorte and A. B. Svendsen, J. Ethnopharmacol., 1983, 9, 167–223 CrossRef CAS PubMed.
- J. Schmutz, F. Hunziker and R. Hirt, Helv. Chim. Acta, 1957, 40, 1189–1200 CrossRef CAS.
- G. Büchi and E. Warnhoff, J. Am. Chem. Soc., 1959, 81, 4433–4434 CrossRef.
- M. Amat, M.-D. Coll, J. Bosch, E. Espinosa and E. Molins, Tetrahedron: Asymmetry, 1997, 8, 935–948 CrossRef CAS.
- D.-H. Kim, J.-H. Kim, T.-H. Jeon and C.-G. Cho, Org. Lett., 2020, 22, 3464–3468 CrossRef CAS PubMed.
- J. Wang, A. Ma and D. Ma, Org. Lett., 2008, 10, 5425–5428 CrossRef CAS PubMed.
- E. E. Robinson and R. J. Thomson, J. Am. Chem. Soc., 2018, 140, 1956–1965 CrossRef CAS PubMed.
- M. Amat, A. Linares and J. Bosch, J. Org. Chem., 1990, 55, 6299–6312 CrossRef CAS.
- M. Yamazaki, E. Okuyama, M. Kobayashi and H. Inoue, Tetrahedron Lett., 1981, 22, 135–136 CrossRef CAS.
- S. E. Blanchflower, R. M. Banks, J. R. Everett and C. Reading, J. Antibiot., 1993, 46, 1355–1363 CrossRef CAS PubMed.
- J. B. Roque, E. V. Mercado-Marin, S. C. Richter, D. P. de Sant'Ana, K. Mukai, Y. Ye and R. Sarpong, Chem. Sci., 2020, 11, 5929–5934 RSC.
- E. V. Mercado-Marin and R. Sarpong, Chem. Sci., 2015, 6, 5048–5052 RSC.
- G. B. Martin, P. T. Angyal, O. Egyed, S. R. Varga and T. Soós, Org. Lett., 2020, 22, 4675–4679 CrossRef CAS PubMed.
- J. S. Cha, Org. Process Res. Dev., 2006, 5, 1032–1053 CrossRef.
- A. Chatterjee and S. Bandyopadhyay, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1979, 18, 87–88 Search PubMed.
- H. Arthur, S. Johns, J. Lamberton and S. Loo, Aust. J. Chem., 1968, 21, 1399–1401 CrossRef CAS.
- M. Lin, B.-Q. Yang and D.-Q. Yu, Acta Pharm. Sin., 1986, 21, 114–118 CAS.
- D. Ponglux, S. Wongseripipatana, S. Subhadhirasakul, H. Takayama, M. Yokota, K. Ogata, C. Phisalaphong, N. Aimi and S.-I. Sakai, Tetrahedron, 1988, 44, 5075–5094 CrossRef CAS.
- L. Kato, R. M. Braga, I. Koch and L. S. Kinoshita, Phytochemistry, 2002, 60, 315–320 CrossRef CAS.
- R. M. Braga and F. D. A. Reis, Phytochemistry, 1987, 26, 833–836 CrossRef CAS.
- H. Zhou, X. Liao and J. M. Cook, Org. Lett., 2004, 6, 249–252 CrossRef CAS PubMed.
- S. Yamada, D. Morizono and K. Yamamoto, Tetrahedron Lett., 1992, 33, 4329–4332 CrossRef CAS.
- H. Zhou, X. Liao, W. Yin, J. Ma and J. M. Cook, J. Org. Chem., 2006, 71, 251–259 CrossRef CAS PubMed.
- D. Hooper, Pharm. J., 1907, 78, 453 CAS.
- E. Field, J. Chem. Soc., Trans., 1921, 119, 887–891 RSC.
- D. Zacharias, R. Rosenstein and G. Jeffrey, Acta Crystallogr., 1965, 18, 1039–1043 CrossRef CAS.
- K. L. Jansen and C. J. Prast, J. Ethnopharmacol., 1988, 23, 115–119 CrossRef CAS.
- K. Matsumoto, M. Mizowaki, T. Suchitra, H. Takayama, S.-i. Sakai, N. Aimi and H. Watanabe, Life Sci., 1996, 59, 1149–1155 CrossRef CAS PubMed.
- J. Ma, W. Yin, H. Zhou and J. M. Cook, Org. Lett., 2007, 9, 3491–3494 CrossRef CAS PubMed.
- Y. Kondo, S. Kojima and T. Sakamoto, J. Org. Chem., 1997, 62, 6507–6511 CrossRef CAS.
- C. Ma, X. Liu, X. Li, J. Flippen-Anderson, S. Yu and J. M. Cook, J. Org. Chem., 2001, 66, 4525–4542 CrossRef CAS PubMed.
- J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2009, 74, 264–273 CrossRef CAS PubMed.
- T. Sakamoto, Y. Kondo, M. Uchiyama and H. Yamanaka, J. Chem. Soc., Perkin Trans. 1, 1993, 1941–1942 RSC.
- I. Kaneko, D. T. Fearon and K. Austen, J. Immunol., 1980, 124, 1194–1198 CAS.
- K. Momota, I. Kaneko, S. Kimura, K. Mitamura and K. Shimada, Biochem. Biophys. Res. Commun., 1991, 179, 243–250 CrossRef CAS PubMed.
- I. Kaneko, K. Kamoshida and S. Takahashi, J. Antibiot., 1989, 42, 236–241 CrossRef CAS PubMed.
- H. Seto, T. Fujioka, K. Furihata, I. Kaneko and S. Takahashi, Tetrahedron Lett., 1989, 30, 4987–4990 CrossRef CAS.
- J. Garfunkle, F. S. Kimball, J. D. Trzupek, S. Takizawa, H. Shimamura, M. Tomishima and D. L. Boger, J. Am. Chem. Soc., 2009, 131, 16036–16038 CrossRef CAS PubMed.
- T. Jeschke, D. Wensbo, U. Annby, S. Gronowitz and L. A. Cohen, Tetrahedron Lett., 1993, 34, 6471–6474 CrossRef CAS.
- V. R. Hegde, P. Dai, M. Patel and V. P. Gullo, Tetrahedron Lett., 1998, 39, 5683–5684 CrossRef CAS.
- H. Shimamura, S. P. Breazzano, J. Garfunkle, F. S. Kimball, J. D. Trzupek and D. L. Boger, J. Am. Chem. Soc., 2010, 132, 7776–7783 CrossRef CAS PubMed.
- L. Peters, G. M. König, H. Terlau and A. D. Wright, J. Nat. Prod., 2002, 65, 1633–1637 CrossRef CAS PubMed.
- J. S. Carle and C. Christophersen, J. Am. Chem. Soc., 1979, 101, 4012–4013 CrossRef CAS.
- T. Sjöblom, L. Bohlin and C. Christophersen, Acta Pharm. Suec., 1983, 20, 415–418 Search PubMed.
- P. López-Alvarado, E. Caballero, C. Avendaño and J. C. Menéndez, Org. Lett., 2006, 8, 4303–4306 CrossRef PubMed.
- T. Hirano, K. Iwakiri and H. Miyamoto, Heterocycles, 2009, 79, 805–820 CrossRef CAS.
- H. Miyamoto, T. Hirano, Y. Okawa, A. Nakazaki and S. Kobayashi, Tetrahedron, 2013, 69, 9481–9493 CrossRef CAS.
- T. F. Walsh, R. B. Toupence, F. Ujjainwalla, J. R. Young and M. T. Goulet, Tetrahedron, 2001, 57, 5233–5241 CrossRef CAS.
- D. E. Lizos and J. A. Murphy, Org. Biomol. Chem., 2003, 1, 117–122 RSC.
- R. J. Sundberg and S. Q. Smith, Alkaloids, 2002, 59, 281 CAS.
- C. Gallo, P. Renzi, S. Loizzo, A. Loizzo and A. Capasso, Pharmacology, 2009, 3, 906–920 Search PubMed.
- G. K. Jana and S. Sinha, Tetrahedron, 2012, 68, 7155–7165 CrossRef CAS.
- D. L. Moody, M. Dyba, T. Kosakowska-Cholody, N. I. Tarasova and C. J. Michejda, Bioorg. Med. Chem. Lett., 2007, 17, 2380–2384 CrossRef CAS PubMed.
- S. M. Lee, X. F. Li, H. Jiang, J. G. Cheng, S. Seong, H. D. Choi and B. W. Son, Tetrahedron Lett., 2003, 44, 7707–7710 CrossRef CAS.
- Q. Gao and F. Garcia-Pichel, Nat. Rev. Microbiol., 2011, 9, 791–802 CrossRef CAS PubMed.
- E. P. Balskus, R. J. Case and C. T. Walsh, FEMS Microbiol. Ecol., 2011, 77, 322–332 CrossRef CAS PubMed.
- C. Wang and J. Sperry, Org. Lett., 2011, 13, 6444–6447 CrossRef CAS PubMed.
- C. Raminelli, J. V. Comasseto, L. H. Andrade and A. L. Porto, Tetrahedron: Asymmetry, 2004, 15, 3117–3122 CrossRef CAS.
- M. Shen, G. Li, B. Z. Lu, A. Hossain, F. Roschangar, V. Farina and C. H. Senanayake, Org. Lett., 2004, 6, 4129–4132 CrossRef CAS PubMed.
- C. Wang and J. Sperry, Synlett, 2012, 23, 1824–1828 CrossRef CAS.
- T. Kunisue, Z. Chen, G. M. Buck Louis, R. Sundaram, M. L. Hediger, L. Sun and K. Kannan, Environ. Sci. Technol., 2012, 46, 4624–4632 CrossRef CAS PubMed.
- S. E. Cross, M. S. Roberts, R. Jiang and H. A. E. Benson, J. Invest. Dermatol., 2001, 117, 147–150 CrossRef CAS.
- C. Wang and J. Sperry, Tetrahedron, 2013, 69, 4563–4577 CrossRef CAS.
- J. Liang, W. Hu, P. Tao and Y. Jia, J. Org. Chem., 2013, 78, 5810–5815 CrossRef CAS PubMed.
- S. Hirao, Y. Yoshinaga, M. Iwao and F. Ishibashi, Tetrahedron Lett., 2010, 51, 533–536 CrossRef CAS.
- P. Buchgraber, M. M. Domostoj, B. Scheiper, C. Wirtz, R. Mynott, J. Rust and A. Fürstner, Tetrahedron, 2009, 65, 6519–6534 CrossRef CAS.
- S.-J. Qu, Q.-W. Liu, C.-H. Tan, S.-H. Jiang and D.-Y. Zhu, Planta Med., 2006, 72, 264–266 CrossRef CAS PubMed.
- D. Shan, Y. Gao and Y. Jia, Angew. Chem., Int. Ed., 2013, 52, 4902–4905 CrossRef CAS PubMed.
- P. Heshmati, M. Nasehi and M. R. Zarrindast, J. Paramed. Sci., 2014, 5, 99–104 Search PubMed.
- R. Kaschnitz and G. Spiteller, Monatsh. Chem., 1965, 96, 909–921 CrossRef CAS.
- X. Pan and T. D. Bannister, Org. Lett., 2014, 16, 6124–6127 CrossRef CAS PubMed.
- N. Peube-Locou, M. Plat and M. Koch, Phytochemistry, 1973, 12, 199 CrossRef CAS.
- X. Y. Xu, X. H. Yang, S. Z. Li and Q. S. Song, Helv. Chim. Acta, 2011, 94, 1470–1476 CrossRef CAS.
- I. Kitagawa, H. Wei, S. Nagao, T. Mahmud, K. Hori, M. Kobayashi, T. Uji and H. Shibuya, Chem. Pharm. Bull., 1996, 44, 1162–1167 CrossRef CAS PubMed.
- H. J. Knölker, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 702–705 CrossRef.
- H.-J. Knölker and S. Cämmerer, Tetrahedron Lett., 2000, 41, 5035–5038 CrossRef.
- A. Stoit and U. Pandit, Heterocycles, 1987, 25, 7–10 CrossRef CAS.
- I. P. Kerschgens, E. Claveau, M. J. Wanner, S. Ingemann, J. H. van Maarseveen and H. Hiemstra, Chem. Commun., 2012, 48, 12243–12245 RSC.
- A. E. Wright, S. A. Pomponi, S. S. Cross and P. McCarthy, J. Org. Chem., 1992, 57, 4772–4775 CrossRef CAS.
- N. K. Garg and B. M. Stoltz, Chem. Commun., 2006, 3769–3779 RSC.
- J. J. Jackson, H. Kobayashi, S. D. Steffens and A. Zakarian, Angew. Chem., Int. Ed., 2015, 54, 9971–9975 CrossRef CAS PubMed.
- P. Ruiz-Sanchis, S. A. Savina, F. Albericio and M. Álvarez, Chem.–Eur. J., 2011, 17, 1388–1408 CrossRef CAS PubMed.
- H. Zhou, H. P. He, Y. H. Wang and X. J. Hao, Helv. Chim. Acta, 2010, 93, 1650–1652 CrossRef CAS.
- Q. Li, T. Xia, L. Yao, H. Deng and X. Liao, Chem. Sci., 2015, 6, 3599–3605 RSC.
- G. Ding, L. Jiang, L. Guo, X. Chen, H. Zhang and Y. Che, J. Nat. Prod., 2008, 71, 1861–1865 CrossRef CAS PubMed.
- C. Pérez-Balado and Á. R. de Lera, Org. Biomol. Chem., 2010, 8, 5179–5186 RSC.
- A. N. de Groot, P. W. van Dongen, T. B. Vree, Y. A. Hekster and J. van Roosmalen, Drugs, 1998, 56, 523–535 CrossRef CAS PubMed.
- N. R. Tasker and P. Wipf, in The Alkaloids, ed. H.-J. Knölker, Academic Press, London, 2021, vol. 85, pp. 1–112 Search PubMed.
- A. Stoll, Helv. Chim. Acta, 1949, 32, 506 CrossRef CAS PubMed.
- K. Lee, Y. B. Poudel, C. M. Glinkerman and D. L. Boger, Tetrahedron, 2015, 71, 5897–5905 CrossRef CAS PubMed.
- H. Liu, X. Zhang, D. Shan, M. Pitchakuntla, Y. Ma and Y. Jia, Org. Lett., 2017, 19, 3323–3326 CrossRef CAS PubMed.
- S. Cai, X. Kong, W. Wang, H. Zhou, T. Zhu, D. Li and Q. Gu, Tetrahedron Lett., 2012, 53, 2615–2617 CrossRef CAS.
- T. Yaguchi, A. Someya and S.-i. Udagawa, Mycoscience, 1995, 36, 421–424 CrossRef.
- J. Varga, J. C. Frisvad and R. A. Samson, Stud. Mycol., 2007, 59, 75–88 CrossRef CAS PubMed.
- K. V. Chuang, M. E. Kieffer and S. E. Reisman, Org. Lett., 2016, 18, 4750–4753 CrossRef CAS PubMed.
- A. Dobbs, J. Org. Chem., 2001, 66, 638–641 CrossRef CAS PubMed.
- L. Garlaschelli, Z. Pang, O. Sterner and G. Vidari, Tetrahedron, 1994, 50, 3571–3574 CrossRef CAS.
- G. Tel, M. Apaydın, M. E. Duru and M. Öztürk, Food Anal. Methods, 2012, 5, 495–504 CrossRef.
- T. Yasukouchi and K. Kanematsu, Tetrahedron Lett., 1989, 30, 6559–6562 CrossRef CAS.
- N. Saito, T. Ichimaru and Y. Sato, Org. Lett., 2012, 14, 1914–1917 CrossRef CAS PubMed.
- R. J. Huntley and R. L. Funk, Org. Lett., 2006, 8, 3403–3406 CrossRef CAS PubMed.
- R. J. Capon, J. K. Macleod and P. J. Scammells, Tetrahedron, 1986, 42, 6545–6550 CrossRef CAS.
- R. Herb, A. R. Carroll, W. Y. Yoshida, P. J. Scheuer and V. J. Paul, Tetrahedron, 1990, 46, 3089–3092 CrossRef CAS.
- L. F. Silva Jr and M. V. Craveiro, Org. Lett., 2008, 10, 5417–5420 CrossRef PubMed.
- L. F. Silva Jr, M. V. Craveiro and M. T. Gambardella, Synthesis, 2007, 2007, 3851–3857 CrossRef.
- K. R. Buszek, N. Brown and D. Luo, Org. Lett., 2009, 11, 201–204 CrossRef CAS PubMed.
- T.-S. Kam, G. Subramaniam, K.-H. Lim and Y.-M. Choo, Tetrahedron Lett., 2004, 45, 5995–5998 CrossRef CAS.
- Z. Li and G. Liang, Tetrahedron Lett., 2013, 54, 242–244 CrossRef CAS.
- D. V. Patil, M. A. Cavitt, P. Grzybowski and S. France, Chem. Commun., 2011, 47, 10278–10280 RSC.
- M. Ishikura, K. Yamada and T. Abe, Nat. Prod. Rep., 2010, 27, 1630–1680 RSC.
- J. Magolan, C. A. Carson and M. A. Kerr, Org. Lett., 2008, 10, 1437–1440 CrossRef CAS PubMed.
- R. Nakajima, T. Ogino, S. Yokoshima and T. Fukuyama, J. Am. Chem. Soc., 2010, 132, 1236–1237 CrossRef CAS PubMed.
- T. Feng, X.-H. Cai, P.-J. Zhao, Z.-Z. Du, W.-Q. Li and X.-D. Luo, Planta Med., 2009, 75, 1537–1541 CrossRef CAS PubMed.
- T. Feng, X.-H. Cai, Y.-P. Liu, Y. Li, Y.-Y. Wang and X.-D. Luo, J. Nat. Prod., 2010, 73, 22–26 CrossRef CAS PubMed.
- S.-H. Lim, K.-M. Sim, Z. Abdullah, O. Hiraku, M. Hayashi, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2007, 70, 1380–1383 CrossRef CAS PubMed.
- R. Keese, Chem. Rev., 2006, 106, 4787–4808 CrossRef CAS PubMed.
- Z. Xu, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2013, 135, 19127–19130 CrossRef CAS PubMed.
- Y. Iwama, K. Okano, K. Sugimoto and H. Tokuyama, Chem.–Eur. J., 2013, 19, 9325–9334 CrossRef CAS PubMed.
- A. Biechy and S. Z. Zard, Org. Lett., 2009, 11, 2800–2803 CrossRef CAS PubMed.
- M. Pfaffenbach and T. Gaich, Chem.–Eur. J., 2016, 22, 3600–3610 CrossRef CAS PubMed.
- Y. Liu and H. Wang, Chem. Commun., 2019, 55, 3544–3547 RSC.
- X.-H. Li, S.-L. Wan, D. Chen, Q. R. Liu, C. H. Ding, P. Fang and X. L. Hou, Synthesis, 2016, 48, 1568–1572 CrossRef CAS.
- J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC.
- R. G. Berlinck, A. F. Bertonha, M. Takaki and J. P. Rodriguez, Nat. Prod. Rep., 2017, 34, 1264–1301 RSC.
- A. D. Patil, A. J. Freyer, B. Carte, P. B. Taylor, R. K. Johnson and D. J. Faulkner, J. Nat. Prod., 2002, 65, 628–629 CrossRef CAS PubMed.
- M. S. Roy, X. Meng, K. Koda, S. Rasapalli, D. Gout and C. J. Lovely, Tetrahedron Lett., 2019, 60, 979–982 CrossRef.
- H. L. Maia, L. S. Monteiro and J. Sebastião, Eur. J. Org. Chem., 2001, 2001, 1967–1970 CrossRef.
- N. Chandrasoma, N. Brown, A. Brassfield, A. Nerurkar, S. Suarez and K. R. Buszek, Tetrahedron Lett., 2013, 54, 913–917 CrossRef CAS PubMed.
- H. Steinmetz, K. I. Mohr, W. Zander, R. Jansen, K. Gerth and R. Müller, J. Nat. Prod., 2012, 75, 1803–1805 CrossRef CAS PubMed.
- K. J. Weissman and R. Müller, Bioorg. Med. Chem., 2009, 17, 2121–2136 CrossRef CAS PubMed.
- N. Marsch, P. G. Jones and T. Lindel, Beilstein J. Org. Chem., 2015, 11, 1700–1706 CrossRef CAS PubMed.
- R. Mezencev, M. Galizzi, P. Kutschy and R. Docampo, Exp. Parasitol., 2009, 122, 66–69 CrossRef CAS PubMed.
- M. S. C. Pedras and S. Montaut, Chem. Commun., 2004, 452–453 RSC.
- M. S. C. Pedras, M. G. Sarwar, M. Suchy and A. M. Adio, Phytochemistry, 2006, 67, 1503–1509 CrossRef CAS PubMed.
- M. S. C. Pedras, J. L. Sorensen, F. I. Okanga and I. L. Zaharia, Bioorg. Med. Chem. Lett., 1999, 9, 3015–3020 CrossRef CAS.
- M. S. C. Pedras and J. L. Sorensen, Phytochemistry, 1998, 49, 1959–1965 CrossRef CAS.
- B. Li, J. D. Williams and N. P. Peet, Tetrahedron Lett., 2013, 54, 3124–3126 CrossRef CAS.
- M. Kaur and R. Kumar, ChemistrySelect, 2018, 3, 5330–5340 CrossRef CAS.
- J. H. Cardellina, F.-J. Marner and R. E. Moore, Science, 1979, 204, 193–195 CrossRef CAS PubMed.
- H. Fujiki and T. Sugimura, Cancer Surv., 1983, 2, 539–556 Search PubMed.
- P. Vital and D. Tanner, Org. Biomol. Chem., 2006, 4, 4292–4298 RSC.
- H. Hemetsberger, D. Knittel and H. Weidmann, Monatsh. Chem., 1970, 101, 161–165 CrossRef CAS.
- W. H. Visser, G. M. Terwindt, S. A. Reines, K. Jiang, C. R. Lines and M. D. Ferrari, Arch. Neurol., 1996, 53, 1132–1137 CrossRef CAS PubMed.
- M. J. Cumberbatch, R. G. Hill and R. J. Hargreaves, Eur. J. Pharmacol., 1997, 328, 37–40 CrossRef CAS PubMed.
- S. Tong, Z. Xu, M. Mamboury, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2015, 127, 11975–11978 CrossRef.
- Y. Yang, Y. Bai, S. Sun and M. Dai, Org. Lett., 2014, 16, 6216–6219 CrossRef CAS PubMed.
- T. Kamigauchi and M. Yasui, PCT Int. Appl., WO 2000059909, [CAN 133:267009], 2000.
- Y. Zhang, I. W. McArdle, J. W. Hubbard, N. G. Akhmedov and B. C. Söderberg, Tetrahedron Lett., 2016, 57, 2865–2867 CrossRef CAS.
- T.-S. Kam, K.-M. Sim, H.-S. Pang, T. Koyano, M. Hayashi and K. Komiyama, Bioorg. Med. Chem. Lett., 2004, 14, 4487–4489 CrossRef CAS PubMed.
- K.-H. Lim, O. Hiraku, K. Komiyama and T.-S. Kam, J. Nat. Prod., 2008, 71, 1591–1594 CrossRef CAS PubMed.
- R. Frei, D. Staedler, A. Raja, R. Franke, F. Sasse, S. Gerber-Lemaire and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 13373–13376 CrossRef CAS PubMed.
- T. Huber, T. A. Preuhs, C. K. Gerlinger and T. Magauer, J. Org. Chem., 2017, 82, 7410–7419 CrossRef CAS PubMed.
- J. J. Neumann, S. Rakshit, T. Dröge, S. Würtz and F. Glorius, Chem.–Eur. J., 2011, 17, 7298–7303 CrossRef CAS PubMed.
- C. Ito and h. Furukawa, Chem. Pharm. Bull., 1990, 38, 1548–1550 CrossRef CAS.
- W. Frohner, M. P. Krahl, K. R. Reddy and H.-J. Knölker, Heterocycles, 2004, 63, 2393–2407 CrossRef.
- H.-J. Knölker and W. Fröhner, Tetrahedron Lett., 1996, 37, 9183–9186 CrossRef.
- H.-J. Knölker and W. Froehner, Synthesis, 2000, 2000, 2131–2136 CrossRef.
- T. Soós, G. Timári and G. Hajós, Tetrahedron Lett., 1999, 40, 8607–8609 CrossRef.
- G. Timári, T. Soós, G. Hajós, A. Messmer, J. Nacsa and J. Molnár, Bioorg. Med. Chem. Lett., 1996, 6, 2831–2836 CrossRef.
- A. Yasuhara, N. Suzuki and T. Sakamoto, Chem. Pharm. Bull., 2002, 50, 143–145 CrossRef CAS PubMed.
- P. Kleinwaechter, B. Schlegel, I. Groth, A. Haertl and U. Graefe, J. Antibiot., 2001, 54, 510–512 CrossRef PubMed.
- G. A. Kraus and T. Wu, Tetrahedron, 2005, 61, 9502–9505 CrossRef CAS.
- T. Komoda and S.-i. Nakatsuka, Heterocycl. Commun., 2003, 9, 119–122 CAS.
- E. Magnier and Y. Langlois, Tetrahedron, 1998, 54, IN6201–IN6202 CrossRef.
- R. Cao, W. Peng, Z. Wang and A. Xu, Curr. Med. Chem., 2007, 14, 479–500 CrossRef CAS PubMed.
- R. Sakai, T. Higa, C. W. Jefford and G. Bernardinelli, J. Am. Chem. Soc., 1986, 108, 6404–6405 CrossRef CAS.
- M. Cain, R. W. Weber, F. Guzman, J. M. Cook, S. A. Barker, K. C. Rice, J. N. Crawley, S. M. Paul and P. Skolnick, J. Med. Chem., 1982, 25, 1081–1091 CrossRef CAS PubMed.
- W. D. Inman, W. M. Bray, N. C. Gassner, R. S. Lokey, K. Tenney, Y. Y. Shen, K. TenDyke, T. Suh and P. Crews, J. Nat. Prod., 2010, 73, 255–257 CrossRef CAS PubMed.
- A. Badre, A. Boulanger, E. Abou-Mansour, B. Banaigs, G. Combaut and C. Francisco, J. Nat. Prod., 1994, 57, 528–533 CrossRef CAS PubMed.
- P. Molina, P. M. Fresneda and S. García-Zafra, Tetrahedron Lett., 1995, 36, 3581–3582 CrossRef CAS.
- P. Rocca, F. Marsais, A. Godard, G. Quéguiner, L. Adams and B. Alo, Tetrahedron Lett., 1995, 36, 7085–7088 CrossRef CAS.
- F. Nissen, V. Richard, C. Alayrac and B. Witulski, Chem. Commun., 2011, 47, 6656–6658 RSC.
- M. R. Hansen and L. H. Hurley, Acc. Chem. Res., 1996, 29, 249–258 CrossRef CAS.
- U. Hacksell and G. D. Daves, Prog. Med. Chem., 1985, 22, 1–65 CrossRef CAS PubMed.
- V. Murray, A. G. Moore, C. Matias and G. Wickham, Biochim. Biophys. Acta, Gene Struct. Expression, 1995, 1261, 195–200 CrossRef.
- Y. Wu, Z.-X. Zhang, H. Hu, D. Li, G. Qiu, X. Hu and X. He, Fitoterapia, 2011, 82, 288–292 CrossRef CAS PubMed.
- A. Yepremyan and T. G. Minehan, Org. Biomol. Chem., 2012, 10, 5194–5196 RSC.
- A. L. Rodriguez, C. Koradin, W. Dohle and P. Knochel, Angew. Chem., Int. Ed., 2000, 39, 2488–2490 CrossRef CAS PubMed.
- R. A. Leal, C. Bischof, Y. V. Lee, S. Sawano, C. C. McAtee, L. N. Latimer, Z. N. Russ, J. E. Dueber, J. Q. Yu and R. Sarpong, Angew. Chem., Int. Ed., 2016, 55, 11824–11828 CrossRef CAS PubMed.
- S. K. Jackson and M. A. Kerr, J. Org. Chem., 2007, 72, 1405–1411 CrossRef CAS PubMed.
- G. Büchi, D. Coffen, K. Kocsis, P. Sonnet and F. E. Ziegler, J. Am. Chem. Soc., 1966, 88, 3099–3109 CrossRef.
- M. Gorman, N. Neuss, G. H. Svoboda, A. J. Barnes and N. J. Cone, J. Am. Pharm. Assoc., Sci. Ed., 1959, 48, 256–257 CrossRef CAS PubMed.
- S. Raucher and B. L. Bray, J. Org. Chem., 1985, 50, 3236–3237 CrossRef CAS.
- R. Noble, 1964.
- G. R. Dwivedi, R. Tyagi, S. Gupta, S. Tripathi, S. Pati, S. K. Srivastava, M. P. Darokar and A. Sharma, J. Biomol. Struct. Dyn., 2018, 36, 4270–4284 CrossRef CAS PubMed.
- S. Das and A. B. Sharangi, J. Pharmacogn. Phytochem., 2017, 6, 1695–1701 CAS.
- M. Sottomayor and A. R. Barceló, in Stud. Nat. Prod. Chem., Elsevier, 2006, vol. 33, pp. 813–857 Search PubMed.
- M. T. Reding and T. Fukuyama, Org. Lett., 1999, 1, 973–976 CrossRef CAS.
- M. Sakaitani, K. Hori and Y. Ohfune, Tetrahedron Lett., 1988, 29, 2983–2984 CrossRef CAS.
- A. Brossi and M. Suffness, The Alkaloids: Antitumor Bisindole Alkaloids from Catharanthus roseus, Academic Press, New York, 1990 Search PubMed.
- R. Noble, C. T. Beer and J. H. Cutts, Ann. N. Y. Acad. Sci., 1958, 76, 882 CrossRef CAS PubMed.
- G. H. Svoboda, N. Neuss and M. Gorman, J. Am. Pharm. Assoc., Sci. Ed., 1959, 48, 659–666 CrossRef CAS PubMed.
- S. Yokoshima, H. Tokuyama and T. Fukuyama, Chem. Rec., 2010, 10, 101–118 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |