
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA phytochemical-based medication search for the SARS-CoV-2 infection by molecular docking models towards spike glycoproteins and main proteases†
Anju Choorakottayil Pushkarana,
Prajeesh Nath ENb,
Anu R. Melgea,
Rammanohar Puthiyedath b and
C. Gopi Mohan*a
b and
C. Gopi Mohan*a
aComputational Biology and Bioinformatics Lab, Center for Nanosciences and Molecular Medicine, Amrita Vishwa Vidyapeetham, Kochi 682041, Kerala, India. E-mail: cgmohan@aims.amrita.edu; cgopimohan@yahoo.com; anjucp5@gmail.com; anurm19924@aims.amrita.edu; Tel: +91-484-4001234 ext. 8769
bAmrita School of Ayurveda, Amrita Vishwa Vidyapeetham, Kollam 690525, Kerala, India. E-mail: drprajeeshnath@ay.amrita.edu; rammanohar@ay.amrita.edu
First published on 24th March 2021
Abstract
Identifying best bioactive phytochemicals from different medicinal plants using molecular docking techniques demonstrates a potential pre-clinical compound discovery against SARS-CoV-2 viral infection. The in silico screening of bioactive phytochemicals with the two druggable targets of SARS-CoV-2 by simple precision/extra precision molecular docking methods was used to compute binding affinity at its active sites. phyllaemblicin and cinnamtannin class of phytocompounds showed a better binding affinity range (−9.0 to −8.0 kcal mol−1) towards both these SARS-CoV-2 targets; the corresponding active site residues in the spike protein were predicted as: Y453, Q496, Q498, N501, Y449, Q493, G496, T500, Y505, L455, Q493, and K417; and Mpro: Q189, H164, H163, P168, H41, L167, Q192, M165, C145, Y54, M49, and Q189. Molecular dynamics simulation further established the structural and energetic stability of protein–phytocompound complexes and their interactions with their key residues supporting the molecular docking analysis. Protein–protein docking using ZDOCK and Prodigy server predicted the binding pose and affinity (−13.8 kcal mol−1) of the spike glycoprotein towards the human ACE2 enzyme and also showed significant structural variations in the ACE2 recognition site upon the binding of phyllaemblicin C compound at their binding interface. The phyllaemblicin and cinnamtannin class of phytochemicals can be potential inhibitors of both the spike and Mpro proteins of SARS-CoV-2; furthermore, its pharmacology and clinical optimization would lead towards novel COVID-19 small-molecule therapy.
Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection commonly referred to as novel coronavirus disease 2019 (COVID-19) has been declared pandemic by the World Health Organization. This contagious viral infection around the world reminds us the vulnerability of the human race towards this invisible enemy. Different zoonotic viral diseases that emerged earlier including SARS, Ebola, and Middle East respiratory syndrome were highly infectious and resulted in millions of death.1 However, these viruses are successfully contained without causing a global pandemic. In contrast, the present COVID-19 is highly contagious and more than 43 million people were infected globally with a mortality rate of ∼4%. Different neurological symptoms and respiratory manifestations associated with COVID-19 patients were revealed. The most prevalent symptom is the severe acute respiratory complication leading to ventilator support; other neurological signs include headache, fever, nausea, pneumonia, loss of smell, unconsciousness, ataxia, epilepsy, neuralgia, and cerebrovascular and musculoskeletal disease.2,3 In some patients, this viral infection causes acute encephalopathy and acute hemorrhagic necrotizing encephalopathy, which might lead to blood–brain barrier breakdown.4The bioactive phytochemical compounds are well known in pharmaceutical industries for developing treatment towards different health conditions such as inflammation, cancer, and infectious diseases. Phytochemicals isolated from various natural sources have gained great interest in the development of anti-viral treatment for chikungunya, HIV, influenza, dengue, and SARS.5 Since the outbreak of SARS-CoV-1 in 2003, several phytochemicals from flora and fauna were tested for their anti-viral activity, which include alkaloids, flavones, flavonols, fatty acids, tannins and terpenes. Jo et al.6 reported key pharmacophoric chemical features such as hydrophobic groups, electron donors (hydroxyl) and carbohydrate moieties for anti-SARS-CoV-1 infection apart from lipophilic and hydrophilic groups seen in other anti-viral drugs. Furthermore, different computational and in vitro studies have been recently reported, which demonstrated the anti-viral properties of bioactive phytochemicals from medicinally important plant sources such as green tea, turmeric, gooseberry, and basil against SARS-CoV-2.7–9 It is to be noted at this juncture that the development of bioactive natural products is more desirable than specific vaccine design for this new virus despite heavy experimental measurements involving extraction, chemical complexity and diversity of natural compounds.
Different studies are in progress with the aim of developing an effective therapeutic for the treatment of COVID-19 and its spike receptor binding domain (RBD) mutants and other target mutants.10,11 Among them, structure-based drug design is playing an important role to understand the molecular mechanism of interaction at the atomic level of SARS-CoV-2 druggable proteins with the host cell receptor for the discovery of clinically efficacious drugs.12–17 Furthermore, a computational strategy can be employed to identify bioactive compounds from natural sources by a virtual screening technique in order to shorten the time period required for anti-viral drug discovery.6,18,19 Therefore, this technique has become the main strategy for the scientific community for developing novel inhibitors. For example, Ngo et al.,20,21 used a database of natural products to identify putative drug candidates targeting SARS-CoV-2. Moreover, in silico drug repurposing techniques for the identification of SARS-CoV-2 inhibitors have been reported.22,23 This is possible due to the powerful atomic scale view of the three-dimensional (3D) structure of different druggable proteins solved for SARS-CoV-2 by X-ray crystallography, NMR and cryo-EM techniques. At present, ∼330 structures account for SARS-CoV-2 druggable targets in protein data bank (PDB), which includes non-structural and structural proteins,24–29 and all are freely available to the scientific community.
Recently, many types of SARS-CoV-2 vaccine candidates have been reported, some of which are reported to be highly effective for the management of COVID-19.30,31 However, the long-term effect as well as the efficacy of the vaccine in pregnant women and children needs to be evaluated urgently. The present work constitutes the structure-based phytochemical mechanism of inhibition studies on two key pharmacological SARS-CoV-2 targets: (i) the receptor binding domain (RBD) of the spike glycoprotein and (ii) main protease (Mpro). The spike glycoprotein is present on the surface of the SARS-CoV-2, which interacts directly with the peptidase domain of the human ACE-2 receptor found on the surface of the human epithelial cells via membrane fusion and endocytosis and creates the fundamental key functional mechanism through the virus gains entry into the host living cell. Mpro is an essential enzyme required for SARS-CoV-2 viral replication, where it is involved in the proteolytic functional processing of viral polyproteins.24
The main objective of the present work is to identify the best phytochemical from five different medicinal herbs for inhibiting the two key SARS-CoV-2 druggable targets and to further understand its molecular mechanism of inhibition. These include (i) Phyllanthus emblica (Amalaki), (ii) Cinnamomum zeylanica (Tvak), (iii) Embelia ribes (Vidanga), (iv) Curcuma longa (Haridra) and (v) Justicia adhatoda (Vasa) herbs which are well documented in classical Ayurvedic texts. Out of these five herbs, P. emblica is ascribed with immunomodulatory (rasayana) and anti-ageing (vayasthapana) properties,32 C. zeylanica is indicated for the management of oropharyngeal (kantharuk) and respiratory afflictions (kasa).33 E. ribes34 and C. Longa35 are attributed with microbicidal activity (krimighna). J. adhatoda is specifically indicated in the management of respiratory illnesses (kasa and svasa).36 In addition, the present computational modeling revealed for the first time the structure–function relationships of key phytochemicals in blocking the human ACE-2 receptor recognition site towards the binding of the SARS-CoV-2 spike protein.
Materials and methods
COVID-19 protein target selection
The two important molecular targets of novel SARS-CoV-2 contributing to its virulence are the spike protein and Mpro. The X-ray crystallographic coordinate of the RBD domain of the spike protein in complex with human cell receptor ACE2 with a resolution of 2.45 Å was retrieved from PDB having an accession code ‘6M0J’.26 Initially, the crystal structure was optimized using the Protein Preparation Wizard module of the Schrödinger software. The ACE2 protein and the water molecules were removed from this complex structure. Furthermore, the RBD of the SARS-CoV-2 spike protein structure was pre-processed by adding hydrogen atoms, removing the alternate conformations of the amino acids and also adding the missing atoms. The geometry of the pre-processed structure is optimized followed by energy minimization by employing OPLS2005 force field. Similarly, the X-ray crystal structure of Mpro of SARS-CoV-2 (PDB code – 6LU7)24 was optimized with the same protocol as mentioned above. Finally, these two key druggable targets of SARS-CoV-2 were used for molecular docking studies to understand the molecular mechanism of functional inhibition using these Ayurvedic active ingredients (phytochemical) to suppress the severity of virus infections.Ligand preparation
The following bioactive Ayurvedic ingredients (phytochemical) were studied for their anti-viral activities targeting SARS-CoV-2 viral proteins: procyanidin A2, procyanidine B1, and cinnamtannin B1 from Twak (Cinnamomum zeylanica); phyllaemblicin B and phyllaemblicin C from Amalaki (Phyllanthus emblica Linn.); germacrone from Haridra (Curcuma longa); embelin from Vidanga (Embelia ribes) and Vasicine from Vasa (Justicia adhatoda). The chemical structures of these bioactive phytochemical compounds were retrieved from PubChem database. These ligand structures were inspected and corrected for bond lengths and angles, and missing hydrogen atoms were added. The geometry of the bioactive phytochemical structures was optimized by employing the MMFF94 force field using the LigPrep module of the Schrödinger software. These optimized conformations of the phytochemical structures were used further for our molecular docking studies.Molecular docking of the phytochemicals towards SARS-CoV-2 targets
Initially, the molecular docking grid box was generated using the Glide grid generation module of the Schrödinger software by defining the reported active sites of both the spike protein and the Mpro crystal structure. The amino acid residues N487, K417, Q493, Y505, Y449, T500, N501, G446, Y449, Y489, N487 and G502 present in the RBD of the spike protein are reported to be involved in direct hydrogen bonding interactions with the host receptor ACE2.26 Therefore, the docking grid box for the spike protein was generated by keeping the centroid of these residues as the center of the grid box. The grid box generated for spike protein was enclosing all these interface amino acid residues important for binding to the host cell receptor. Jin et al. has identified a small molecule inhibitor against SARS-CoV-2 Mpro protein and the key residues involved in the ligand binding are Y54, H41, S46, M49, D187, Q189, M49, F185, Q192, T190, A191, P168, H164, C145, M165, H163, H172, G143, and L16724. These Mpro residues were set as the center of the docking grid box. Furthermore, using the GLIDE module of the Schrödinger software,37 docking of the phytochemicals was performed at the active sites of both the spike and the Mpro SARS-CoV-2 protein targets.Glide molecular docking was performed in two sequential steps: (i) standard precision (SP) docking: ligands which are able to bind with protein are screened;38 and (ii) extra precision (XP) docking step: this involves a strict scoring function that will eliminate the false-positive hits.39 The docking protocol using the GLIDE module has been successfully implemented in our previous studies.40–43 The bioactive phytochemicals from different medicinal plants were ranked based on their docking score (or binding affinity) against the SARS-CoV-2 protein targets. The best phytochemicals were further analyzed using the PyMol and Discovery Studio visualizer to understand its molecular mechanism of action for SARS-CoV-2 infections.
Prediction of the pharmacokinetic properties of bioactive phytochemicals
The pharmacokinetic properties of eight phytochemicals selected in this study were predicted using the QikProp module of the Schrödinger software. QikProp predicts the pharmacokinetic parameters that include absorption, distribution, metabolism, excretion and toxicity of the compounds with high accuracy.Molecular dynamics simulation study of key phytochemicals in complex with spike and Mpro proteins
Molecular Dynamics (MD) simulations were carried out for the docked structures of SARS-CoV-2 spike–phyllaemblicin C, spike–cinnamtannin B1 and SARS-CoV-2 Mpro–phyllaemblicin C complexes using the AMBER program.44 Initially, the atoms of the ligands were defined using the GAFF force field in the antechamber module of AMBER. Following ligand preparation, the atom types were assigned for all the atoms in the protein–ligand complexes using the FF99SB force field in the Leap module of AMBER. Total charges of the studied three-complex systems were neutralized by adding two Cl− ions for the spike protein in complex with phyllaemblicin C and cinnamtannin B1 phytochemicals, while four Na+ ions were added to Mpro–phyllaemblicin C complexes. Solvation was performed by immersing the protein-ligand complexes in a 10 Å orthorhombic box of TIP3P water molecules. Furthermore, the energy minimization was done in two steps involving the steepest descent and conjugate gradient methods for 5000 and 1000 MD steps respectively. During the initial minimization process, the solvent molecules and the ions were allowed to move freely and the protein and ligand were restrained. In the second step of energy minimization, all the atoms in the system were allowed to move freely. The Particle Mesh Ewald (PME) algorithm was applied to handle long-range electrostatic interactions.45 The van der Waals interaction threshold was set to 10 Å and the dielectric constant was fixed at 1.0. Furthermore, each of the protein–ligand complex systems was heated from 0 to 37 °C for 20 ps by applying the NVT ensemble followed by 100 ps of equilibration using the NPT ensemble with a constant pressure of 1 atm. The production MD simulation was carried out for 60 ns time interval, where all the atoms in the three different complex systems were allowed to move and interact without any restraints. The trajectory snapshots and associated energies (bonded and non-bonded) of these three complex systems were saved for each 5000 time step. The high-frequency vibrations in the systems were eliminated by assigning the SHAKE algorithm, wherein all the bonds will be constrained to their equilibrium values. The trajectory files obtained from the MD production simulation were used for analyzing the root mean square deviation (RMSD) using the ptraj module and total energies of the protein–ligand complex systems.SARS-CoV-2 spike–ACE2 complex binding studies in the presence of phytochemicals
The binding mode of the spike protein with the ACE2 receptor was analyzed in the presence of the highest binding affinity phytochemical obtained in our molecular docking studies. The crystal structure of the spike–ACE2 protein complexes solved by protein crystallography technique (PDB id: 6M0J) was used as reference for the present study. To evaluate the accuracy of the docking predictions, we first performed the molecular docking of the spike protein with the ACE2 receptor using the ZDOCK software. ZDOCK is an interactive web-based server for performing the protein–protein docking simulations.46 Different output poses generated by the ZDOCK server were compared with the binding mode of the spike protein and ACE2 in the crystal structure complex. The best mode of binding of ACE2 with the SARS-CoV-2 spike protein was structurally compared by superimposing both structures (6M0J crystal structure with the docked structure) and evaluating its root mean square deviations (RMSD). Furthermore, the complex of SARS-CoV-2 spike protein and the best phytochemical was allowed to dock with the human ACE2 protein, and the modes of binding of ACE2 with the ligand (phytochemical)-free and ligand-bound spike proteins was compared. Furthermore, the binding affinity of these docked protein complexes was calculated using the PRODIGY web server.47Results
Molecular docking of bioactive phytochemicals towards the SARS-CoV-2 spike protein
In the present study, we used the X-ray crystal structure of the spike protein RBD domain of SARS-CoV-2 (6M0J) for the identification of bioactive phytochemical compounds as an entry inhibitor by preventing its direct interaction towards its natural ACE2 host cell receptor binding interface.26 The selected bioactive phytochemical compounds (active ingredients) from different medicinal plants showing anti-viral activities are as follows: (i) procyanidin A2, procyanidine B1 and cinnamtannin B1 from Twak (C. zeylanica); (ii) phyllaemblicin B and phyllaemblicin C from Amalaki (P. emblica); (iii) Germacrone from Haridra (C. longa); (iv) embelin from Vidanga (E. ribes) and (v) vasicine from Vasa (J. adhatoda) (Fig. 1).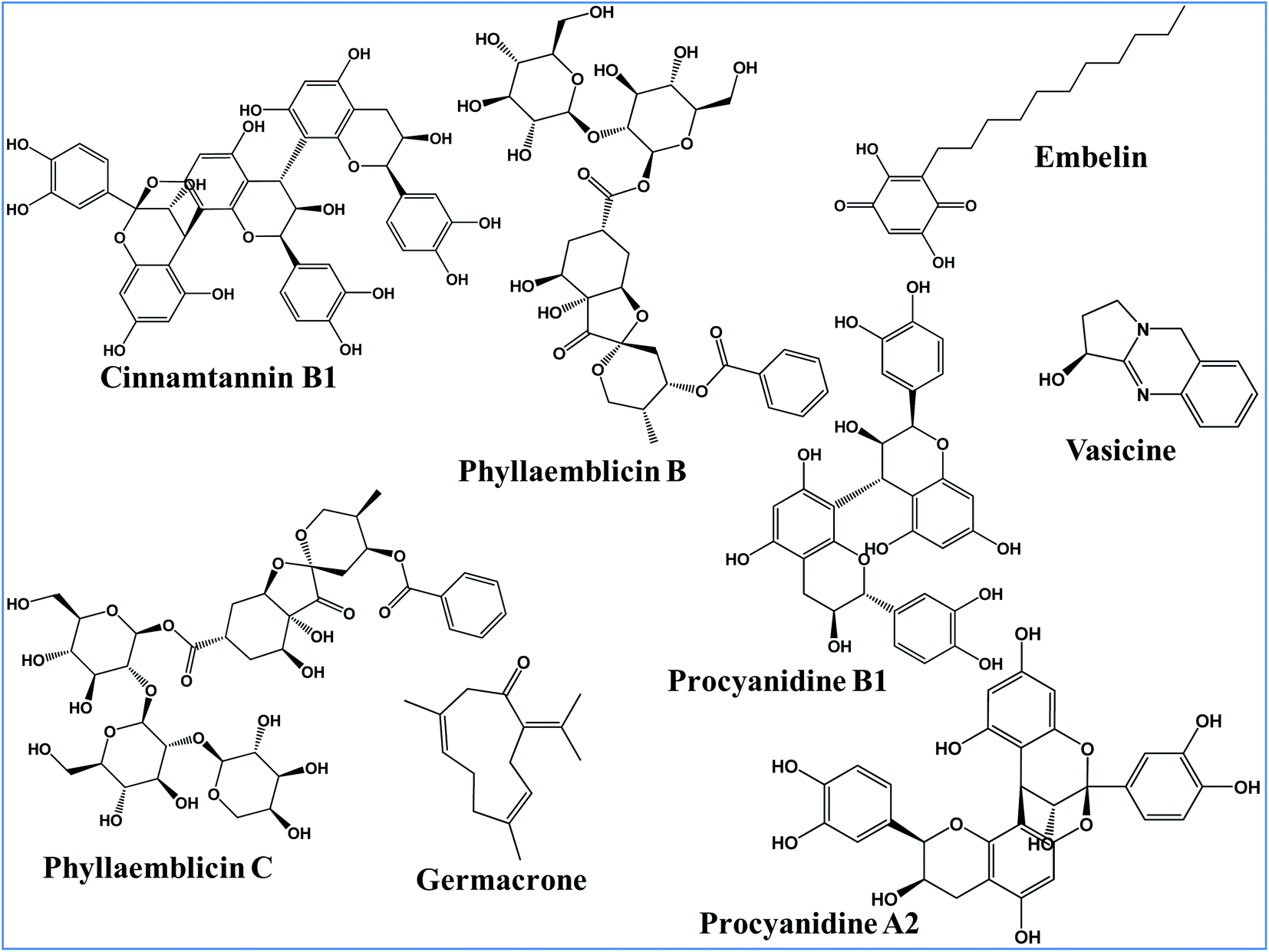 | ||
| Fig. 1 Chemical structure of bioactive phytochemicals from different medicinal plants. | ||
All these eight phytochemical compounds were docked into the active site of the RBD of the SARS-CoV-2 spike glycoprotein. Molecular docking using the Glide software revealed that the key ingredients present in Amalaki and Twak showed the highest binding affinity towards the active sites of the RBD domain of SARS-CoV-2 spike protein by making direct interactions with the key residues involved in ACE2 binding. Phyllaemblicin C, a bioactive phytochemical present in Amalaki, showed the highest binding affinity towards the active site of the RBD of the spike protein with a docking score of −9.131 kcal mol−1 (Table 1). Cinnamtannin B1 and phyllaemblicins B bioactive phytochemicals were obtained as the second and third best compounds with docking scores of −9.008 kcal mol−1 and −7.381 kcal mol−1 respectively, as shown in Table 1. Furthermore, the key ingredients embelin, vasicine and germacrone present in other medicinal plants showed only a weak binding affinity towards the SARS-CoV-2 spike protein in the range of −3.0 to −4.0 kcal mol−1 (Table S1-ESI†).
| SARS-CoV-2 target | Phytochemicals | Binding affinity (kcal mol−1) | Interacting amino acid residues with phytochemical |
|---|---|---|---|
| a The underlined residues are the reported key amino acids of spike protein of SARS-CoV-2 for the direct binding with human ACE2 receptor26,52 and for Mpro protein these residues corresponds to its inhibitor binding.24 | |||
| Spike protein (RBD) | Phyllaemblicin C | −9.131 | 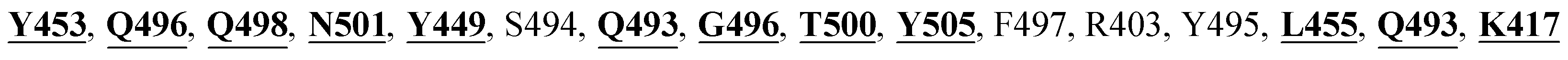 |
| Cinnamtannin B1 | −9.008 | 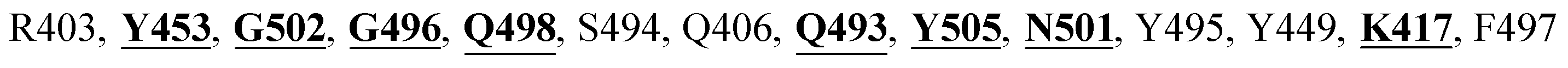 |
|
| Phyllaemblicin B | −7.381 |  |
|
| Procyanidine B1 | −6.275 |  |
|
| Procyanidine A2 | −5.023 |  |
|
| Main protease (Mpro) | Phyllaemblicin C | −9.723 | 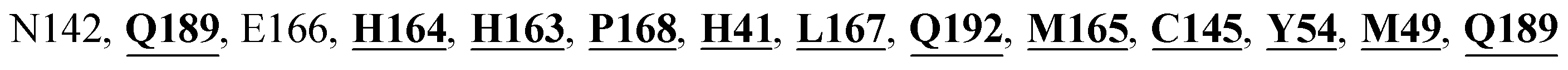 |
| Phyllaemblicin B | −9.151 | 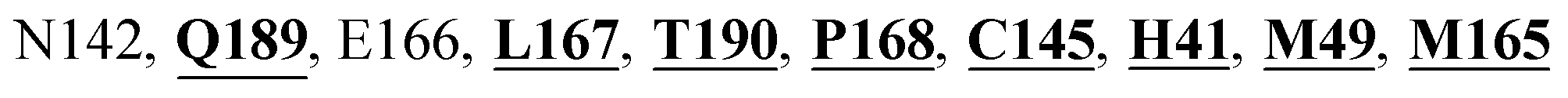 |
|
| Procyanidine B1 | −9.128 | 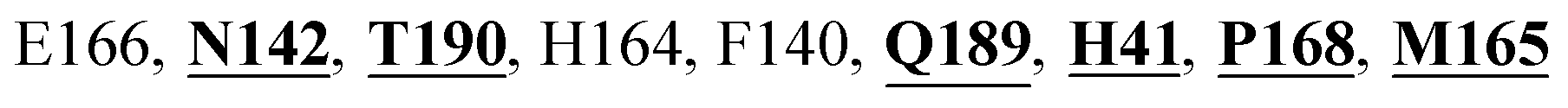 |
|
| Cinnamtannin B1 | −8.385 |  |
|
| Procyanidine A2 | −7.105 | 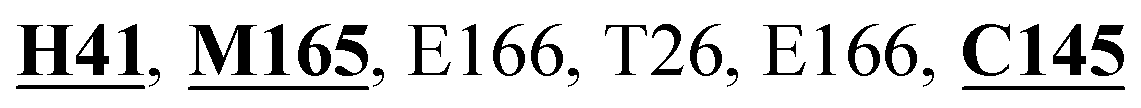 |
|
The X-ray crystal structure of the SARS-CoV-2 spike glycoprotein in the complex with the human ACE2 receptor has already been solved experimentally.26 The contact residues of the RBD domain of the spike protein interacting with ACE2 are K417, G446, Y449, Y453, L455, F456, A475, F486, N487, Y489, Q493, G496, Q498, T500, N501, G502, and Y505. Among these, K417, G446, Y449, N487, Y489, Q493, T500, N501, G502 and Y505 residues made direct hydrogen bonding interactions with the human ACE2 residues.26
The detailed molecular interaction analyses of these phytochemical compounds at the active site of the SARS-CoV-2 spike protein revealed that most of the interactions are within the experimentally reported active site amino acid residues. Phyllaemblicin C also made hydrogen bonding interactions with the Y453, G496, Q498, N501, Y449, S494, Q493, and G498 RBD residues. Apart from these, Y505, G496, F497, R403, Y495, Y453, L455 and K417 RBD residues also made direct interactions with the phyllaemblicin C, as shown in Fig. 2a and Table 1. The second best binding compound against the RBD domain of spike protein was cinnamtannin B1. The molecular interactions of cinnamtannin B1 with the spike protein–RBD consisted of hydrogen bonding interactions with Y453, G502, G496, Q498, S494, Q406, G493, R403 and Y505 residues, electrostatic interactions with R403 and hydrophobic interactions with Y453 and Y505 residues, as shown in Fig. 2b and Table 1. The 2D-interaction plot of phyllaemblicin C and cinnamtannin B1 in the complex with the spike protein–RBD is presented in Fig. S1a and b. In addition, the atomic level interaction distances of the best three complex systems (phyllaemblicin C, cinnamtannin B1 and phyllaemblicin B) are presented in Table S2.†
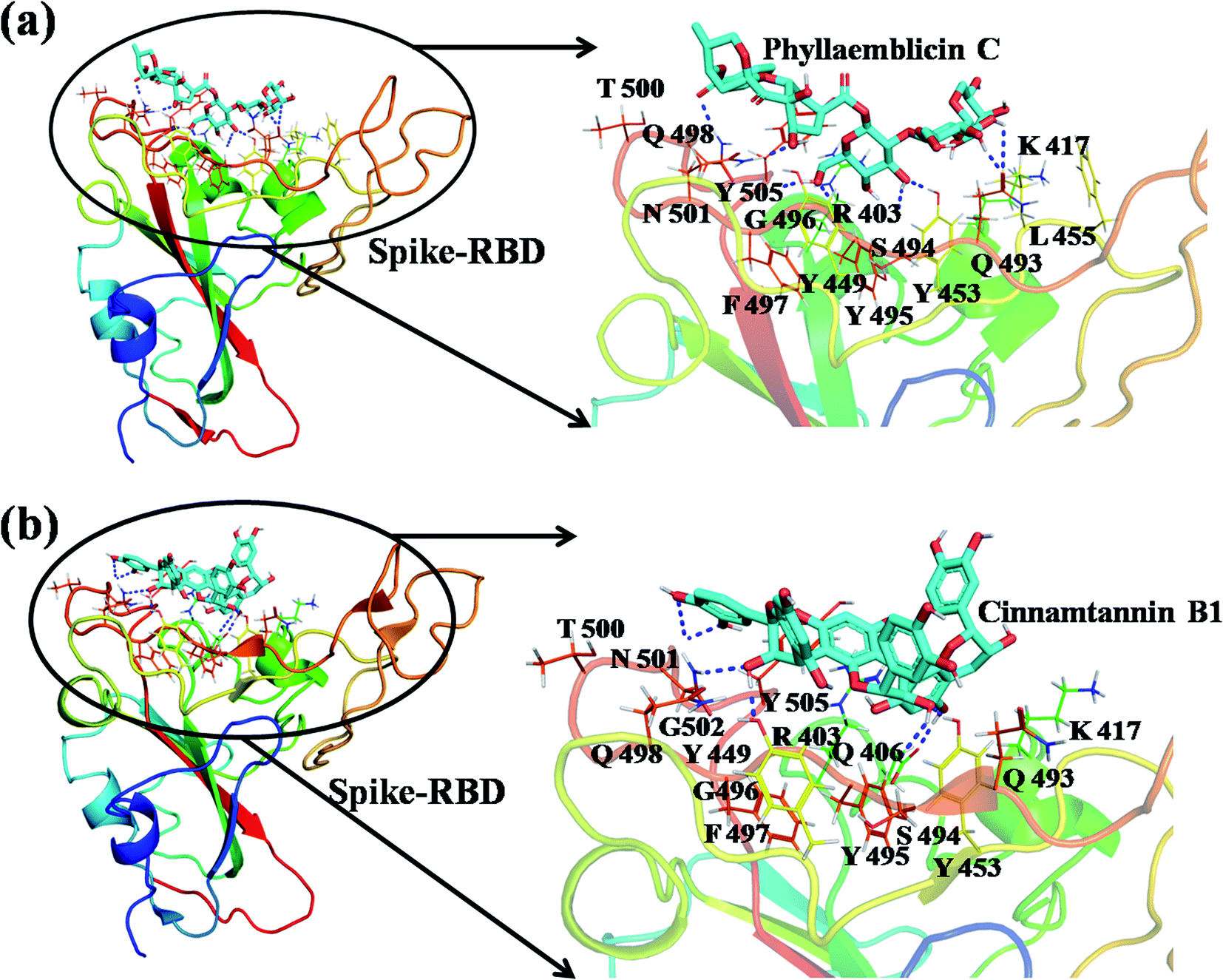 | ||
| Fig. 2 Spike protein RBD domain of SARS-CoV-2 in complex with the phytochemical obtained by molecular docking. (a) Molecular interactions of phyllaemblicin C compound with the RBD domain of the spike protein with the interacting amino acid residues. (b) Molecular interaction of cinnamtannin B1 compound with the RBD of the spike protein along with the interacting residues. The interacting residues of the spike protein are shown as lines, the ligands (phyllaemblicin C and cinnamtannin B1) are represented as cyan sticks and the hydrogen bonding interactions between the molecular complexes are represented as blue dotted lines. | ||
Molecular docking of phytochemical compounds towards Mpro of SARS-CoV-2
Mpro protein of SARS-CoV-2 represents another important druggable target for the anti-viral drug discovery. Many studies have been reported recently regarding the discovery and evaluation of small-molecule inhibitors targeting the Mpro protein. Using X-ray crystallography technique, the key active site residues of Mpro for the ligand binding are reported to be consisting of Y54, H41, S46, M49, D187, Q189, M49, F185, Q192, T190, A191, P168, H164, C145, M165, H163, H172, G143, and L167 (ref. 24) residues. The function of the Mpro enzyme in SARS-CoV-2 is to cleave the polypeptide into its functional proteins, which is carried out by acylation and deacylation steps.24 Mpro is a cysteine protease enzyme having a cysteine–histidine (C145–H41) catalytic dyad.24,48In the present study, different phytochemicals were docked into the active site of the Mpro protein of SARS-CoV-2 and ranked based on the docking score (binding affinity). Phyllaemblicin C was obtained as the best hit with a docking score of (−9.723 kcal mol−1) towards the active site of the Mpro protein of SARS-CoV-2. Phyllaemblicin B and procyanidin B1 showed the second and third best binding affinities towards Mpro with docking scores of −9.151 kcal mol−1 and −9.128 kcal mol−1 respectively, as shown in Table 1. Both phyllaemblicin C and phyllaemblicin B are found in Indian gooseberry and procyanidin B1 is found in cinnamon medicinal plants. Procyanidin B1 is a polyphenolic flavonoid having anti-inflammatory and immune-modulatory activities.49 The molecular interaction of phyllaemblicin C towards the Mpro protein of SARS-CoV-2 was analyzed: it formed hydrogen bonding interactions with N142, Q189, E166, H164, H163, P168 and H41 residues. Moreover, L167, Q192, M165, C145, Y54, and M49 residues were involved in other non-bonded interactions, as shown in Fig. 3a and Table 1.
 | ||
| Fig. 3 SARS-CoV-2 Mpro in complex with phytochemicals: the molecular interactions of phyllaemblicin C with the Mpro protein (a) and phyllaemblicin B with the Mpro protein (b). The protein is represented as cartoon, the interacting amino acid residues are shown as lines and the hydrogen bonding interactions are represented using blue dotted lines. The ligand (phytochemical) is represented as cyan sticks. | ||
Furthermore, the interaction of the next best phytochemical compound, phyllaemblicin B, with Mpro was analyzed. This compound made hydrogen bonding as well as hydrophobic interactions with the Mpro active site residues. The Mpro residues H41, N142, Q189, E166, L167, T190 and P168 are involved in the direct hydrogen bonding with the phytochemicals. The hydrophobic interactions are with C145, H41, M49 and M165 residues of Mpro, as shown in Fig. 3b and Table 1. The 2D-interaction plot of phyllaemblicin C and cinnamtannin B1 in the complex with the spike protein–RBD is presented in Fig. S2a and b.† In addition, the atomic level interaction distances of the three best complex systems (phyllaemblicin C, phyllaemblicin B and procyanidine B1) are presented in Table S2.† Furthermore, a weak binding affinity in the range of −2 to −3 kcal mol−1 with the Mpro protein was observed for key ingredients embelin, vasicine and germacrone present in other medicinal plants. These docking results follow a similar trend on the basis of binding affinity towards two key SARS-CoV-2 targets (Tables 1 and S1†).
Molecular dynamics simulation study of protein–phytochemical complexes
Molecular Dynamics (MD) simulation of the following three different complex systems was studied for 60 ns time interval using the AMBER software and is presented in Fig. 4a–f: (i) spike–phyllaemblicin C, (ii) spike–cinnamtannin B1 and (iii) Mpro–phyllaemblicin C. The RMSD and total energies of the backbone atoms were calculated for every 5000 steps to determine the degree of stability of the complexes. The spike–phyllaemblicin C complex showed slightly greater RMSD than the spike–cinnamtannin B1 complex while Mpro–phyllaemblicin C complex exhibited lower RMSD values after 30 ns showing a higher complex stability than the other two spike complexes (Fig. 4a, c, and e). Furthermore, total energies of the three complex systems were also calculated for 60 ns time interval to understand its energetic stability, as shown in Fig. 4d–f. As expected, the total energy of Mpro–phyllaemblicin C complex was lower than the spike–phytochemical complexes, as shown in Fig. 4 b, d, and f. These results are in good agreement with the molecular docking study in which the binding energy of Mpro–phyllaemblicin C complex was better than that of the spike–phyllaemblicin C and spike–cinnamtannin B1 complexes (Table 1).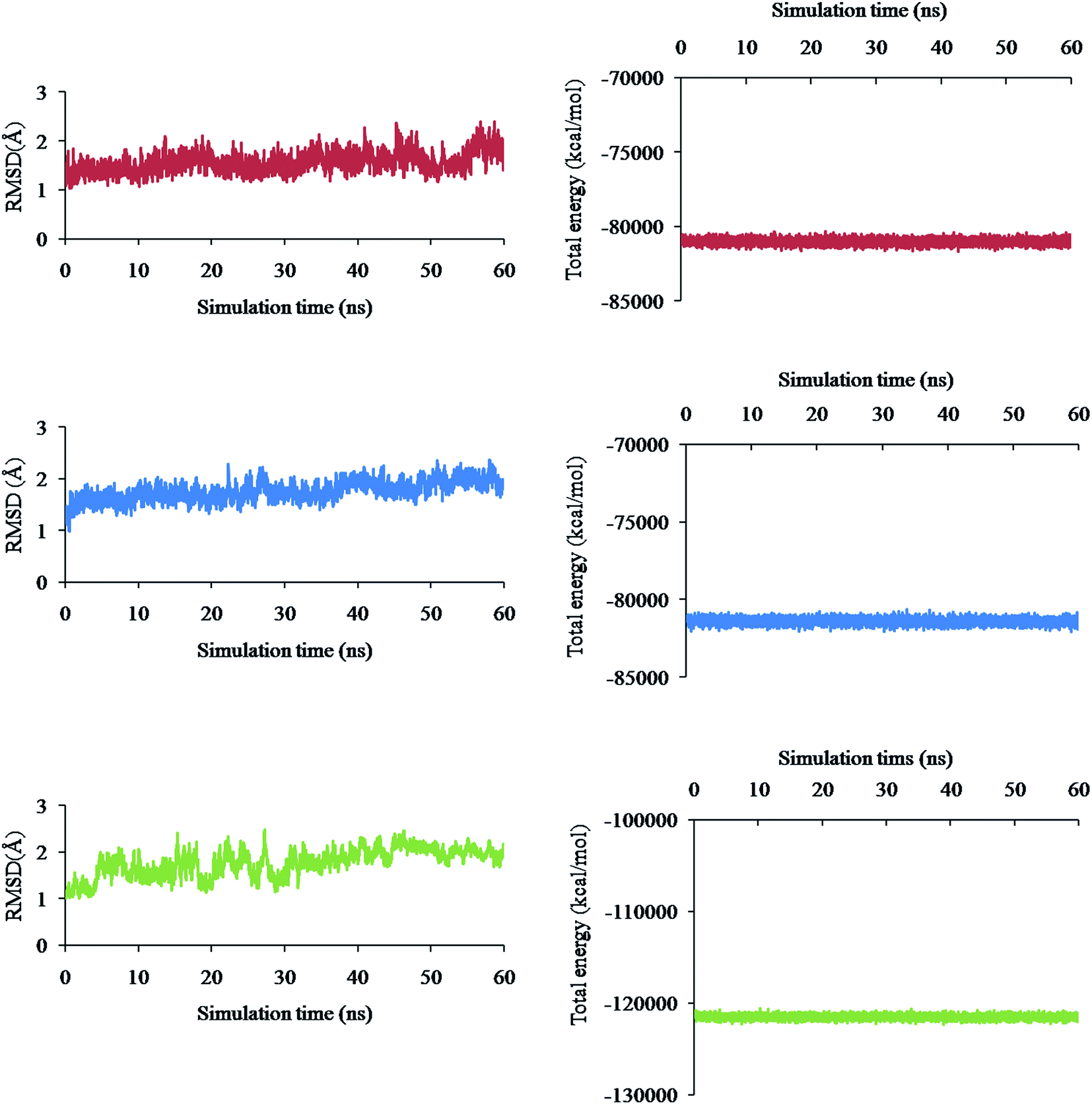 | ||
| Fig. 4 Molecular dynamic simulation studies of RBD domain of spike protein and Mpro of SARS-CoV-2 in complex with phytochemicals: The RMSD plots of spike protein in complex with phyllaemblicin C (red) (a) and cinnamtannin B1 (blue) (c), and Mpro protein in complex with phyllaemblicin C (green) (e). Total energy plots of spike protein in complex with phyllaemblicin C (red) (b) and cinnamtannin B1 (blue) (d) and Mpro protein in complex with phyllaemblicin C (green) (f) were derived from the 60 ns MD simulations of the respective complexes. | ||
Binding effect of phyllaemblicin C at the interface of SARS-CoV-2 spike protein – RBD and the host ACE2 receptor
The viral entry into the host cell is made possible by the atomic level direct interactions of the SARS-CoV-2 spike protein with the host cell receptor ACE2.50 We have investigated how the phytochemical binding at the RBD domain of spike protein has affected its ACE2 binding efficiency by adopting protein–protein docking technique using the ZDOCK software. Initially, the accuracy of docking program and our computational strategy was validated by performing docking of the RBD domain of spike protein with the human ACE2 receptor. Furthermore, the mode of binding was compared with its X-ray crystal structure complex (PDB id: 6M0J). Interestingly, our ZDOCK binding study predicted a highly similar complex association of SARS-CoV-2 spike protein–RBD with the ACE2 host cell receptor, which was highly similar to the 3D folding observed in the corresponding X-ray crystal structure (Fig. 5a). The RMSD between the predicted and corresponding X-ray complex structure showed a perfect 3D alignment of the backbone atoms, as shown in ESI Fig. S3.†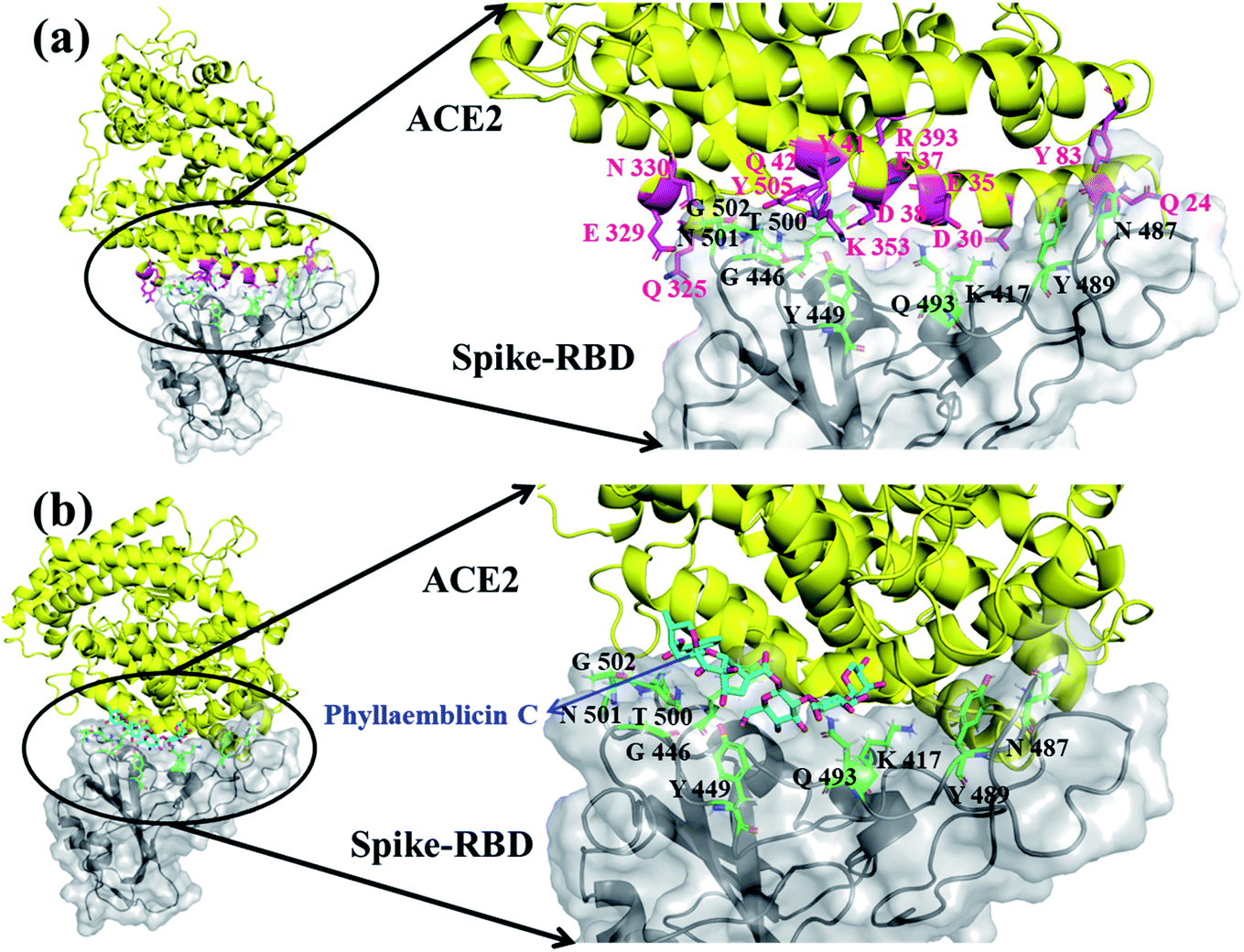 | ||
| Fig. 5 Molecular docking of human ACE2 receptor in complex with RBD spike protein without phytochemical (a) and with phytochemical phyllaemblicin C (b). (a) The docked pose of spike protein RBD interacting residues (black color)with ACE2 interacting residues (magenta color) is shown in magnified view (right zoom out panel) and (b) the docked pose of spike protein RBD interacting residues (black color) with phyllaemblicin C (Cyan sticks) at the binding interface of RBD-spike–ACE2 complexes, which was highly distorted for ACE2 binding. The ACE2 protein is shown as yellow cartoon with the interacting amino acid residues in magenta sticks. The RBD of spike protein is represented by grey cartoon and its key interacting residues are shown as green sticks (right zoom out panel). | ||
The binding affinity between the spike protein–RBD and ACE2 receptor was computed by the PRODIGY web server and the energy was −13.8 kcal mol−1. Furthermore, the amino acid residues involved in molecular interactions between RBD and ACE2 complexes were identified, and they are shown in Fig. 5a and ESI Table S3.†
In order to understand the influence of small molecule binding at this interface, we used the bound structure of spike–RBD and phyllaemblicin C and docked this complex against the human ACE2 protein using the ZDOCK software. The mode and mechanism of binding of the spike protein–phyllaemblicin C complex with the ACE2 receptor was analyzed. Their binding pose revealed that the binding of the spike protein with the ACE2 receptor was highly distorted due to the presence of phyllaemblicin C at its binding interface. In the absence of a small molecule at the interface of spike–RBD and human ACE2 receptor, most of the interactions of RBD residues are directed towards the N-terminal alpha helix of the human ACE2 protein. Here, we observed that the presence of a small molecule (phyllaemblicin C) at its binding interface has resulted in the binding of spike protein to the C-terminal region of the ACE2 instead of the N-terminal helix, as shown in Fig. 5b. The 2D-interacting amino acid residues of the spike–phyllaemblicin C complex and the ACE2 receptor are presented in ESI Fig. S4 and Table S3.†
Pharmacokinetic property prediction of phytochemical compounds
The pharmacokinetic properties of eight phytochemical compounds used in this study were predicted, and they are presented in Table 2. The absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of most of these phytochemical compounds were in the satisfactory range. Generally, phytochemical compounds have a large molecular weight with many stereo centers and do not follow the basic Lipinski rule of five. Some of the present active phytochemicals studied in this work also follow the same category.| Phytochemical compounds | MW | QPlogPo/w | QPlogS | QPlog HERG | QPPCaco | QPlog Khsa | Human oral absorption |
|---|---|---|---|---|---|---|---|
| a Molecular weight (MW) 130–725; predicted octanol/water partition coefficient (QPlogPo/w) −2.0 to 6.5; predicted aqueous solubility (QPlogS) −6.5–0.5; predicted IC50 value for blockage of HERG potassium channel (QPlogHERG) – concern below −5; predicted apparent Caco-2 cell permeability in nm s−1 (QPPCaco) <25 poor, >500 great; prediction of binding to human serum albumin (QPlogKhsa) −1.5 to 1.5; human oral absorption – 1 low, 2 medium, 3 high. | |||||||
| Phyllaemblicin C | 876.8 | −3.91 | −0.54 | −4.69 | 0.72 | −2.01 | 1 |
| Cinnamtannin B1 | 864.8 | 0.37 | −5.15 | −6.81 | 0.06 | −0.14 | 1 |
| Phyllaemblicin B | 744.7 | −2.44 | −1.91 | −5.02 | 1.69 | −1.52 | 1 |
| Procyanidin B1 | 578.5 | 0.24 | −3.60 | −5.21 | 1.28 | −0.23 | 1 |
| Procyanidin A2 | 576.5 | 0.36 | −4.08 | −5.60 | 1.64 | −0.19 | 1 |
| Embelin | 294.4 | 2.15 | −3.76 | −4.78 | 201.35 | −0.18 | 3 |
| Germacrone | 218.3 | 3.38 | −3.75 | −2.81 | 4641.3 | 0.43 | 3 |
| Vasicine | 188.2 | 1.90 | −2.47 | −4.10 | 2420.9 | −0.19 | 3 |
Discussion
The molecular mechanism of the SARS-CoV-2 entry into the human host epithelial cell via the interaction between the viral spike protein and the ACE2 host cell receptor was studied extensively.26,50,51 It was reported that the spike protein of SARS-CoV-2 is ten times potent than SARS-CoV. This makes spike protein a prime druggable target in the present scenario for preventing the SARS-CoV-2 viral entry into the host cell. Many studies are undergoing in order to identify suitable inhibitors targeting the spike protein of SARS-CoV-2. The spike protein is composed of two subunits, namely, S1 and S2, among which the S1 subunit possesses the RBD. The RBD domain residues directly recognize the ACE2 host cell receptor, and the molecular interactions between them allow the human cell attachment and subsequent viral entry into the host cell.The SARS-CoV-2 spike glycoprotein X-ray crystal structure in the complex with the human ACE2 receptor revealed key residues at its binding interface as explained earlier.26 Yi et al. demonstrated that the single amino acid substitutions at positions R439, K452, E484, T470, Q498, and N501 residues of the SARS-CoV-2 spike protein resulted in a reduced binding affinity with the human ACE2 receptor protein. Moreover, the substitutions at P499, Q493, F486, A475 and L455 residues have enhanced the binding of spike protein towards the human ACE2 receptor.52 Hence, these residues play a critical role in binding with the host cell receptor and subsequent viral entry into the host cell.
The phytochemical compounds have been highly promising since ancient times for treating a wide range of infectious diseases.5–9 In the present study, eight phytochemicals from five different medicinal plants were virtually screened against two important SARS-CoV-2 druggable targets, spike protein and Mpro. Among these eight compounds, phyllaemblicin C showed the highest binding affinity towards both spike and Mpro protein targets. phyllaemblicin B and phyllaemblicin C of Amalaki have been found to have activity against Influenza A virus strain H3N2 causing respiratory infections.53 It was reported earlier that procyanidin A2, procyanidine B1, and cinnamtannin B1 of Twak have been found to have activity against coronavirus (wt SARS-CoV) and SARS-CoV S pseudovirus infections.54 Germacrone of Haridra has been found to have activity against H1N1 and H3N2 viruses causing respiratory illness.55 Embelin from Vidanga has been found to have anti-viral activity against influenza virus H1N1, H5N2 and H3N2 causing respiratory illness.56 Vasicine from Vasa also showed anti-viral activity against Influenza virus causing respiratory disease.57
Our studied phytochemical compounds binding at the active site of SARS-CoV-2 spike protein revealed molecular mechanism of interactions between experimentally known key active site residues. Phyllaemblicin C, a bisabolane-type sesquiterpenoid isolated from gooseberry, was shown to efficiently bind with the RBD of spike protein in comparison to other studied phytochemical compounds. Its key residue interactions include Y453, Q496, Q498, N501, Y449, Q493, G496, T500, Y505, L455, Q493, and K417, which are reported to be important for binding of SARS-CoV-2 spike protein with the ACE2 host cell receptor.26,52 Therefore, phyllaemblicin C would be able to block binding of the spike protein with the host cell receptor ACE2, thereby preventing the virus entry into the host cell (Fig. 2 and 4). This compound was reported to have several medicinal properties including anti-proliferative and anti-viral activities.53
Cinnamtannin B1 is a type of proanthocyanidin that is commonly found in the cortex of cinnamon58 and possesses anti-microbial, anti-platelet and anti-oxidant properties.58,59 Cinnamtannin B1 interacted with Y453, G502, G496, Q498, Q493, Y505, N501, and K417, which are key amino acid residues of the SARS-CoV-2 spike protein-RBD contributing to its direct interactions with the ACE2 host receptor for the viral entry. The structure-based phytochemical study towards the Mpro enzyme, another key druggable target of SARS-CoV-2, showed phyllaemblicin C and B as the best binding affinity compounds. These compounds made direct interactions with the Mpro inhibitor binding site residues, which include Q189, H164, H163, P168, H41, L167, T190, Q192, M165, C145, Y54 and M49, as shown in Table 1 and Fig. 3.24 Furthermore, the procyanidinB1 phytocompound also showed good binding strength towards Mpro, and anti-viral activity of procyanidin B1 from Cinnamomi cortex was already reported. Moreover, procyanidin B1 inhibited the progression of infection of vesicular stomatitis virus and Hepatitis C virus pseudotype by inhibiting the viral replication.60
MD simulation studies of three different complex systems (i) spike–phyllaemblicin C, (ii) spike–cinnamtannin B1 and (iii) Mpro–phyllaemblicin C for 60 ns time interval revealed Mpro–phyllaemblicin C complex to be more stable with lower RMSD values and total energy than those of the spike–phytochemical complexes, as shown in Fig. 4. This MD results are in agreement with our docking studies in which the Mpro–phyllaemblicin C complex showed better binding affinity than that of its spike complexes (Table 1). Modelling and simulation results further revealed that these phytochemicals bind with the key residues of SARS-Cov2 targets with good binding affinities for its molecular mechanism of inhibition and may further prevent increase in viral load and infections.
Zhuang et al.54 reported anti-viral activities of the active ingredients of Cinnamomi cortex against other families of coronavirus (SARS-CoV). Three compounds cinnamtannin B1, pprocyanidinA2, and procyanidin B1 exhibited good in vitro anti-viral activities towards SARS-CoV (Table 3). Among them, cinnamtannin B1 showed the most potent inhibitory activity in micromolar concentrations.54 Our structure-based virtual screening study revealed that these phytochemicals from cinnamon can also bind with the spike and Mpro proteins of SARS-CoV-2 with good binding affinity, as shown in Table 3. Though the in vitro activity studies are performed with SARS-CoV, a close homolog of SARS-CoV-2, we can presume that these compounds could have broad anti-viral activity towards this virus family. Furthermore, Liu et al. reported anti-coxsackievirus B3 norsesquiterpenoids which include phyllaemblicin C and phyllaemblicin B from the roots of Phyllanthus emblica and it exhibited potent anti-viral (cytotoxic and inhibitory) activity shown in Table 3.61 Our docking simulations also showed that both these compounds exhibit good binding affinity towards spike and Mpro of SARS-CoV-2 druggable targets. The in vitro anti-viral activity against SARS-CoV and coxsackievirus was identified on the basis of thorough literature survey of the phytochemicals selected in the present computational study. The direct involvement of phytochemicals towards the druggable targets in SARS-CoV or coxsackievirus was not specifically addressed in these in vitro studies; however, its influence on the endocytic pathway of cellular entry was taken into account for anti-viral activity studies.54,61–63 Hence, the phytochemicals selected in the present study need to be validated by different in vitro/in vivo techniques for inhibiting both spike and Mpro viral proteins and could serve as an alternative pre-clinical finding for the treatment of COVID-19 infections.
| Bioactive Phytochemical | SARS-CoV-2 | Anti-viral activity | ||
|---|---|---|---|---|
| Binding affinity (kcal mol−1) | Cytotoxic concentration CC50 (μM) | Inhibitory concentration IC50 (μM) | ||
| Spike (RBD) | Mpro | |||
| a In vitro activity towards coxsackievirus.61b In vitro activity towards SARS-CoV.54 | ||||
| Phyllaemblicin C | −9.131 | −9.723 | 67.7a | 11.0a |
| Cinnamtannin B1 | −9.008 | −8.385 | 242.3b | 32.9b |
| Procyanidin B1 | −6.275 | −9.151 | 656.2b | 161.1b |
| Procyanidin A2 | −5.023 | −7.105 | 796.6b | 120.7b |
| Phyllaemblicin B | −7.381 | −9.151 | 50.2a | 7.8a |
There are different experimental studies reported involving the elucidation of molecular recognition between SARS-CoV-2 spike protein and ACE2 host receptor.26,52 These biomolecular observations are in good agreement with our predicted complex model system obtained using the ZDOCK and Prodigy server. The RBD of spike protein exhibits a concave surface with a ridge on one side, and this molecular surface is where the extracellular part of the host ACE2 receptor will make direct molecular interactions. It was reported that most of the key residues of SARS-CoV-2 RBD spike protein interact with the N-terminal helix of the human ACE2 protein, which include Q24, D30, E35, E37, D38, Y41, Q42, Y83, Q325, E329, N330, K353 and R393 residues.26 Interestingly, in our molecular docking simulations using the ZDOCK software, we also obtained similar binding residue pairs at its complex binding interface between SARS-CoV-2 spike-RBD and ACE2 receptor (binding affinity of −13.8 kcal mol−1), as shown in Fig. 5a and ESI Table S3.† Further, the binding effect of phyllaemblicin C at the interface of SARS-CoV-2 spike protein with the host ACE2 receptor was studied using the ZDOCK and PRODIGY server. Its mode and mechanism of binding clearly indicates that the binding of phyllaemblicin C at the active site of the spike protein–RBD caused large structural variations resulting in the steric hindrance with the host receptor ACE2 (Fig. 5b and Table S3†). This binding recognition has no biological (or pharmacological) significance with respect to SARS-CoV-2 infection. In other words, the spike protein RBD in complex with phyllaemblicin C showed complete failure towards biomolecular recognition of the active site residues in the ACE2 receptor. These docking model further signifies that the phytochemical, phyllaemblicin C, blocks the direct binding of the spike protein RBD with the ACE2 receptor active sites, thereby preventing the viral entry into host cells. Therefore, this important molecular mechanism was revealed in the present modeling work, according to which blocking using a small molecule (phytochemical) causes the direct interaction between spike protein and ACE2 receptor, which should have key influence on the SARS-CoV-2 viral entry and thereby controlling the severity of the infection, spread and morbidity.
Conclusions
The present work forms the basis for identifying the best phytochemical compound towards inhibiting two key SARS-CoV-2 functional druggable targets by structure-based phytochemical modeling techniques. Molecular docking of key ingredients phyllaemblicin C, phyllaemblicin B, procyanidin B1 and cinnamtannin B1 compounds from different medicinal plants revealed its binding affinity towards the active sites of spike and Mpro viral proteins, and was also supported by MD simulations of 60 ns time interval of these complexes. These studies further revealed that the phytochemicals in complex with spike and Mpro proteins are stable and able to retain key interactions with the active site residues to perform its molecular mechanism of anti-viral action. The binding affinity obtained from our docking studies on these phytochemicals is in good agreement with the corresponding in vitro anti-viral activity studies performed in another close homolog of the coronavirus family. Another interesting conclusion that emerged from the present modeling study is that the mode of binding of SARS-CoV-2 spike protein with the human ACE2 receptor in the presence of phyllaemblicin C shows significant structural variations and the receptor recognition of its binding interface is completely obstructed in the presence of phytochemical. Thus, the present computational study forms the basis of further in vitro/in vivo evaluation of the small molecule (phytochemical) for controlling the SARS-CoV-2 infections.Author contributions
ACP, ARM and CGM performed all in silico studies, and writing of the manuscript; PNE and RP contributed in Ayurvedic aspects of the study and reviewing the study.Data availability
The data in this study is available from the corresponding author upon request.Financial support
CGM gratefully acknowledges the Department of Biotechnology (DBT), India, for providing financial support in procuring Schrödinger molecular modeling software license used in the present study (grant no. BT/PR21018/BID/7/776/2016).Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge Computational biology and Bioinformatics Lab, Centre for Nanosciences and Molecular Medicine, Amrita Vishwa Vidyapeetham, Kochi for the computing infrastructure support.References
- L. Wang and G. Crameri, Rev. Sci. Tech., 2014, 33, 569–581 CrossRef.
- C.-C. Lai, W.-C. Ko, P.-I. Lee, S.-S. Jean and P.-R. Hsueh, Int. J. Antimicrob. Agents, 2020, 56, 106024 CrossRef CAS PubMed.
- H.-Y. Wang, X.-L. Li, Z.-R. Yan, X.-P. Sun, J. Han and B.-W. Zhang, Ther. Adv. Neurol. Disord., 2020, 13, 1–2 Search PubMed.
- A. Franceschi, O. Ahmed, L. Giliberto and M. Castillo, Am. J. Neuroradiol., 2020, 41, 1173–1176 CrossRef CAS PubMed.
- I. E. Orhan and F. S. S. Deniz, Nat. Prod. Bioprospect., 2020, 10, 171–186 CrossRef CAS PubMed.
- S. Jo, S. Kim, D. H. Shin and M.-S. Kim, J. Enzyme Inhib. Med. Chem., 2020, 35, 145–151 CrossRef CAS PubMed.
- R. V. Chikhale, S. K. Sinha, R. B. Patil, S. K. Prasad, A. Shakya, N. Gurav, R. Prasad, S. R. Dhaswadikar, M. Wanjari and S. S. Gurav, J. Biomol. Struct. Dyn., 2020, 1–15 Search PubMed.
- M. T. ul Qamar, S. M. Alqahtani, M. A. Alamri and L.-L. Chen, J. Pharm. Anal., 2020, 10, 313–319 CrossRef PubMed.
- D. Shanmugarajan, P. Prabitha, B. P. Kumar and B. Suresh, RSC Adv., 2020, 10, 31385–31399 RSC.
- A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman and A. D. Mesecar, ChemMedChem, 2020, 15, 907–932 CrossRef CAS PubMed.
- S. Shahinshavali, K. A. Hossain, A. V. D. N. Kumar, A. G. Reddy, D. Kolli, A. Nakhi, M. V. B. Rao and M. Pal, Tetrahedron Lett., 2020, 61, 152336 CrossRef PubMed.
- M. O. Olujide, O. Olubiyi, J. L. Monika Keutmann and B. Strodel, Molecules, 2020, 25, 3193 CrossRef PubMed.
- M. T. Khan, A. Ali, Q. Wang, M. Irfan, A. Khan, M. T. Zeb, Y.-J. Zhang, S. Chinnasamy and D.-Q. Wei, J. Biomol. Struct. Dyn., 2020, 1–11 Search PubMed.
- B. Shah, P. Modi and S. R. Sagar, Life Sci., 2020, 252, 117652 CrossRef CAS PubMed.
- R. Yu, L. Chen, R. Lan, R. Shen and P. Li, Int. J. Antimicrob. Agents, 2020, 56, 106012 CrossRef CAS PubMed.
- A. K. Srivastav, S. K. Gupta and U. Kumar, ChemRxiv, 2020, 18, 100385 Search PubMed.
- C. Wu, Y. Liu, Y. Yang, P. Zhang, W. Zhong, Y. Wang, Q. Wang, Y. Xu, M. Li and X. Li, Acta Pharm. Sin. B, 2020, 10, 766–788 CrossRef CAS PubMed.
- A. da Silva Antonio, L. S. M. Wiedemann and V. F. Veiga-Junior, RSC Adv., 2020, 10, 23379–23393 RSC.
- C. G. Mohan, Structural Bioinformatics: Applications in Preclinical Drug Discovery Process, Springer Nature, ISBN 978-3-030-05282-9, 2019 Search PubMed.
- S. T. Ngo, N. Quynh Anh Pham, L. Thi Le, D. H. Pham and V. V. Vu, J. Chem. Inf. Model., 2020, 60, 5771–5780 CrossRef CAS PubMed.
- D. Gentile, V. Patamia, A. Scala, M. T. Sciortino, A. Piperno and A. Rescifina, Mar. Drugs, 2020, 18, 225 CrossRef CAS.
- R. Alexpandi, J. F. De Mesquita, S. K. Pandian and A. V. Ravi, Front. Microbiol., 2020, 11, 1796 CrossRef PubMed.
- W. R. Ferraz, R. A. Gomes, A. L. S. Novaes and G. H. G. Trossini, Future Med. Chem., 2020, 12, 1815–1828 CrossRef CAS.
- Z. Jin, X. Du, Y. Xu, Y. Deng, M. Liu, Y. Zhao, B. Zhang, X. Li, L. Zhang and C. Peng, Nature, 2020, 582, 289–293 CrossRef CAS PubMed.
- W. Rut, Z. Lv, M. Zmudzinski, S. Patchett, D. Nayak, S. J. Snipas, F. El Oualid, T. T. Huang, M. Bekes, M. Drag and S. K. Olsen, Sci. Adv., 2020, 6, eabd4596 CrossRef CAS PubMed.
- J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang and L. Zhang, Nature, 2020, 581, 215–220 CrossRef CAS PubMed.
- J. Chen, B. Malone, E. Llewellyn, M. Grasso, P. M. Shelton, P. D. B. Olinares, K. Maruthi, E. T. Eng, H. Vatandaslar and B. T. Chait, Cell, 2020, 183, 1560–1573 CrossRef PubMed.
- Y. Gao, L. Yan, Y. Huang, F. Liu, Y. Zhao, L. Cao, T. Wang, Q. Sun, Z. Ming and L. Zhang, Science, 2020, 368, 779–782 CrossRef CAS PubMed.
- Q. Ye, A. M. West, S. Silletti and K. D. Corbett, Protein Sci., 2020, 29, 1890–1901 CrossRef CAS PubMed.
- M. D. Knoll and C. Wonodi, Lancet Infect. Dis., 2021, 397, 72–74 CAS.
- E. E. Walsh, A. R. W. Frenck Jr, A. R. Falsey, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, K. Neuzil, M. J. Mulligan, R. Bailey and K. A. Swanson, N. Engl. J. Med., 2020, 383, 2439–2450 CrossRef CAS PubMed.
- S Murthy, ed. Chowkhambha Orientalia, 2005, Ch. 12, Verse 39–40, p. 268 Search PubMed.
- Anandasrama Samskrita Grandhavali, ed. G. A. Vinayaka, 2nd edn, 1925, Verse 51, p. 79 Search PubMed.
- Anandasrama Samskrita Grandhavali, ed. G. A. Vinayaka, 2nd edn, 1925, Verse 12, pp. 55–56 Search PubMed.
- Anandasrama Samskrita Grandhavali, ed. V. G. Apatte, 2nd edn, Verse 55–56 Search PubMed.
- S. Murthy, ed. Chowkhambha Orientalia, 2005, Ch. 7, Verse p. 119 Search PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley and J. K. Perry, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- A. C. Pushkaran, N. Nataraj, N. Nair, F. Götz, R. Biswas and C. G. Mohan, J. Chem. Inf. Model., 2015, 55, 760–770 CrossRef CAS PubMed.
- A. C. Pushkaran, V. Vinod, M. Vanuopadath, S. S. Nair, S. V. Nair, A. K. Vasudevan, R. Biswas and C. G. Mohan, Sci. Rep., 2019, 9, 1–14 CAS.
- S. Vijayrajratnam, A. C. Pushkaran, A. Balakrishnan, A. K. Vasudevan, R. Biswas and C. G. Mohan, Biochem. J., 2016, 473, 4573–4592 CrossRef CAS PubMed.
- P. S. Panicker, A. R. Melge, L. Biswas, P. Keechilat and C. G. Mohan, Chem. Biol. Drug Des., 2017, 90, 629–636 CrossRef CAS PubMed.
- D. Case, T. Darden, T. Cheatham, C. Simmerling, J. Wang, R. Duke, R. Luo, R. Walker, W. Zhang and K. Merz, Amber 12 reference manual, Universty of California San Francisco, San Francisco, CA, 2012 Search PubMed.
- T. Darden, D. York and L. Pedersen, Int. J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- B. G. Pierce, K. Wiehe, H. Hwang, B.-H. Kim, T. Vreven and Z. Weng, Bioinformatics, 2014, 30, 1771–1773 CrossRef CAS PubMed.
- L. C. Xue, J. P. Rodrigues, P. L. Kastritis, A. M. Bonvin and A. Vangone, Bioinformatics, 2016, 32, 3676–3678 CAS.
- W. Dai, B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng and F. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS.
- J. Xing, R. Li, N. Li, J. Zhang, Y. Li, P. Gong, D. Gao, H. Liu and Y. Zhang, Mol. Cell. Biochem., 2015, 407, 89–95 CrossRef CAS.
- M. Hoffmann, H. Kleine-Weber, S. Schroeder, N. Krüger, T. Herrler, S. Erichsen, T. S. Schiergens, G. Herrler, N.-H. Wu and A. Nitsche, Cell, 2020, 181, 271–280 CrossRef CAS PubMed.
- W. Sungnak, N. Huang, C. Bécavin, M. Berg, R. Queen, M. Litvinukova, C. Talavera-López, H. Maatz, D. Reichart and F. Sampaziotis, Nat. Med., 2020, 26, 681–687 CrossRef CAS PubMed.
- C. Yi, X. Sun, J. Ye, L. Ding, M. Liu, Z. Yang, X. Lu, Y. Zhang, L. Ma and W. Gu, Cell. Mol. Immunol., 2020, 17, 621–630 CrossRef CAS PubMed.
- J.-J. Lv, S. Yu, Y. Xin, R.-R. Cheng, H.-T. Zhu, D. Wang, C.-R. Yang, M. Xu and Y.-J. Zhang, Phytochemistry, 2015, 117, 123–134 CrossRef CAS PubMed.
- M. Zhuang, H. Jiang, Y. Suzuki, X. Li, P. Xiao, T. Tanaka, H. Ling, B. Yang, H. Saitoh and L. Zhang, Antiviral Res., 2009, 82, 73–81 CrossRef CAS PubMed.
- Q. Liao, Z. Qian, R. Liu, L. An and X. Chen, Antiviral Res., 2013, 100, 578–588 CrossRef CAS PubMed.
- M. S. Hossan, A. Fatima, M. Rahmatullah, T. J. Khoo, V. Nissapatorn, A. V. Galochkina, A. V. Slita, A. A. Shtro, Y. Nikolaeva and V. V. Zarubaev, Arch. Virol., 2018, 163, 2121–2131 CrossRef CAS PubMed.
- R. Amber, M. Adnan, A. Tariq and S. Mussarat, J. Pharm. Pharmacol., 2017, 69, 109–122 CrossRef CAS PubMed.
- R. A. Anderson, C. L. Broadhurst, M. M. Polansky, W. F. Schmidt, A. Khan, V. P. Flanagan, N. W. Schoene and D. J. Graves, J. Agric. Food Chem., 2004, 52, 65–70 CrossRef CAS PubMed.
- J. J. López, I. Jardín, G. M. Salido and J. A. Rosado, Life Sci., 2008, 82, 977–982 CrossRef PubMed.
- S. Li, E. N. Kodama, Y. Inoue, H. Tani, Y. Matsuura, J. Zhang, T. Tanaka and T. Hattori, Antiviral Chem. Chemother., 2010, 20, 239–248 CrossRef CAS PubMed.
- Q. Liu, Y.-F. Wang, R.-J. Chen, M.-Y. Zhang, Y.-F. Wang, C.-R. Yang and Y.-J. Zhang, J. Nat. Prod., 2009, 72, 969–972 CrossRef CAS PubMed.
- P. Merilahti, S. Koskinen, O. Heikkilä, E. Karelehto and P. Susi, Adv. Virol., 2012, 2012, DOI:10.1155/2012/547530.
- O. O. Glebov, FEBS J., 2020, 287, 3664–3671 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10458b |
| This journal is © The Royal Society of Chemistry 2021 |