
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand compatibility of salacinol-type α-glucosidase inhibitors toward the GH31 family†
Fumihiro Ishikawa a,
Aiko Hiranoa,
Yuuto Yoshimoria,
Kana Nishidaa,
Shinya Nakamurab,
Katsuki Takashimaa,
Shinsuke Marumotoc,
Kiyofumi Ninomiyad,
Isao Nakanishib,
Weijia Xiee,
Toshio Morikawad,
Osamu Muraokad and
Genzoh Tanabe*ad
a,
Aiko Hiranoa,
Yuuto Yoshimoria,
Kana Nishidaa,
Shinya Nakamurab,
Katsuki Takashimaa,
Shinsuke Marumotoc,
Kiyofumi Ninomiyad,
Isao Nakanishib,
Weijia Xiee,
Toshio Morikawad,
Osamu Muraokad and
Genzoh Tanabe*ad
aPharmaceutical Organic Chemistry Lab, Faculty of Pharmacy, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
bComputational Drug Design and Discovery Lab, Faculty of Pharmacy, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
cJoint Research Center, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
dPharmaceutical Research and Technology Institute, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
eState Key Laboratory of Natural Medicines, Department of Medicinal Chemistry, China Pharmaceutical University, Nanjing 2100009, P. R. China. E-mail: g-tanabe@phar.kindai.ac.jp
First published on 15th January 2021
Abstract
We show that salacinol-type α-glucosidase inhibitors are ligand-compatible with the GH 31 family. Salacinol and its 3′-O-benzylated analogs inhibit human lysosomal α-glucosidase at submicromolar levels. Simple structure-activity relationship studies reveal that the salacinol side-chain stereochemistry significantly influences binding to GH31 α-glucosidases.
Introduction
GH31 α-glucosidases are retaining α-glucosidases that catalyze the hydrolysis of α-glycosidic linkages in oligosaccharides and glycoconjugates.1,2 GH31 α-glucosidases are involved in several physiological processes, including the processing of newly biosynthesized glycoproteins in the endoplasmic reticulum, the breakdown of glycogen in the lysosome, and the hydrolysis of disaccharides in the gastrointestinal tract. GH31 α-glucosidases perform essential biological functions that have attracted considerable attention as therapeutic targets, with drugs for lysosomal storage disease,3 diabetes,4 obesity,5 virus infections,6 and tumors7 having been developed. Furthermore, the therapeutic benefits of targeting GH31 α-glucosidases have facilitated the development of new inhibitor classes, including disaccharides,8,9 iminosugars,10 carbasugars,11 peudoaminosugars,12,13 and non-glycosidic derivatives.14–20 However, there remains a major need to discover and design selective α-glucosidase inhibitors from the perspectives of both cellular tools and therapeutic agents.Salacinol (1), a natural product, was isolated from the stems and roots of Salacia reticulata, which has been used to treat diabetes in Ayurvedic medicine (Fig. 1).21,22 After the discovery of salacinol (1), related sulfonium sulfates, such as kotalanol,23 ponkoranol,24 and salaprinol,24 as well as desulfonated analogs, including neosalacinol (2) (Fig. 1),25 neokotalanol,26 neoponkoranol,27 and neosalaprinol,27 were subsequently isolated from the same plant genus plant as the compounds responsible for antidiabetic activity. In vitro and in vivo activity studies revealed that the antidiabetic activity is due to the inhibition of intestinal α-glucosidases.28 Furthermore, Lineweaver–Burk plots of the inhibition of intestinal α-glucosidases by 1 revealed a competitive type of inhibition on intestinal α-glucosidases.22 On the basis of these results, clinical trials using the extract of S. reticulata on patients with type-2 diabetes showed promising therapeutic effects and minimal side effects.29 Intensive structure-activity-relationship (SAR) studies around 1 have also been conducted around the world; indeed, we revealed the following important structural features of the side-chain structure of 1: cooperativity between 2′S–OH and 4′-OH moieties is essential for the onset of the potent α-glucosidase inhibitory activity,30 while the O-sulfonate anion moiety on the 3′-oxygen is not necessary.31 We subsequently have developed an array of neosalacinols 3–7 bearing 3′-O-(ortho-substituted benzyl) groups through comprehensive SAR studies (Fig. 1).32 The 3′-O-benzylated analogs 3–7 displayed in vitro inhibitory activities toward rat intestinal maltase (IC50 = 0.13–0.66 μM) (Table 1), which highlighted that they are the most potent thiosugar-based inhibitors synthesized to date.32 Furthermore, in vivo activity studies involving 3–7 revealed the effective suppression of blood glucose levels in mice.28 While salacinol (1) and its analogs 3–7 have been studied in detail using intestinal α-glucosidases, thiosugar-sulfonium salts remain an underexplored sector of lysosomal α-glucosidase chemical space. With this in mind, we sought to evaluate the compatibilities of thiosugar-based intestinal α-glucosidase inhibitors with lysosomal α-glucosidase GAA. To complete these SAR studies, we synthesized 8, 10, and 12, and the related de-O-sulfonated versions 9, 11, and 13 (Scheme 1). We analyzed the abilities of these thiosugar sulfonium salts (1–13) to inhibit the enzymatic activities of recombinant human α-glucosidase GAA and rat intestinal disaccharidases, as determined by their inhibitory constants (Ki) or IC50 values, and also demonstrated their inhibitory properties toward β-glucosidase.
 | ||
| Fig. 1 Structures of naturally occurring salacinol (1), neosalacinol (2), and their derivatives 3–13 described in this study. | ||
| r | α-Glucosidase GAAb | A. niger β-glucosidasec |
|---|---|---|
| a Mean ± SEM.b Apparent Ki. Assays conducted at pH 5.2 using α-p-NPG as the substrate.c Apparent IC50. Assays conducted at pH 4.6 using β-p-NPG as the substrate. | ||
| 1 | 0.12 ± 0.02 | >1000 |
| 2 | 3.6 ± 0.3 | >1000 |
| 3 (H) | 0.022 ± 0.007 | >1000 |
| 4 (o-CH3) | 0.034 ± 0.009 | >1000 |
| 5 (o-Cl) | 0.030 ± 0.009 | >1000 |
| 6 (o-CF3) | 0.017 ± 0.010 | >1000 |
| 7 (o-NO2) | 0.17 ± 0.05 | >1000 |
| 8 (3′-epi-1) | 1.0 ± 0.1 | >1000 |
| 9 (3′-epi-2) | 25 ± 2 | >1000 |
| 10 (2′-epi-1) | 2794 ± 294 | >1000 |
| 11 (2′-epi-2) | 2742 ± 230 | >1000 |
| 12 (2′,3′-epi-1) | 3893 ± 262 | >1000 |
| 13 (2′,3′-epi-2) | 463 ± 36 | >1000 |
| Voglibose | 7.6 ± 0.8 | >1000 |
| Acarbose | 40 ± 2 | >1000 |
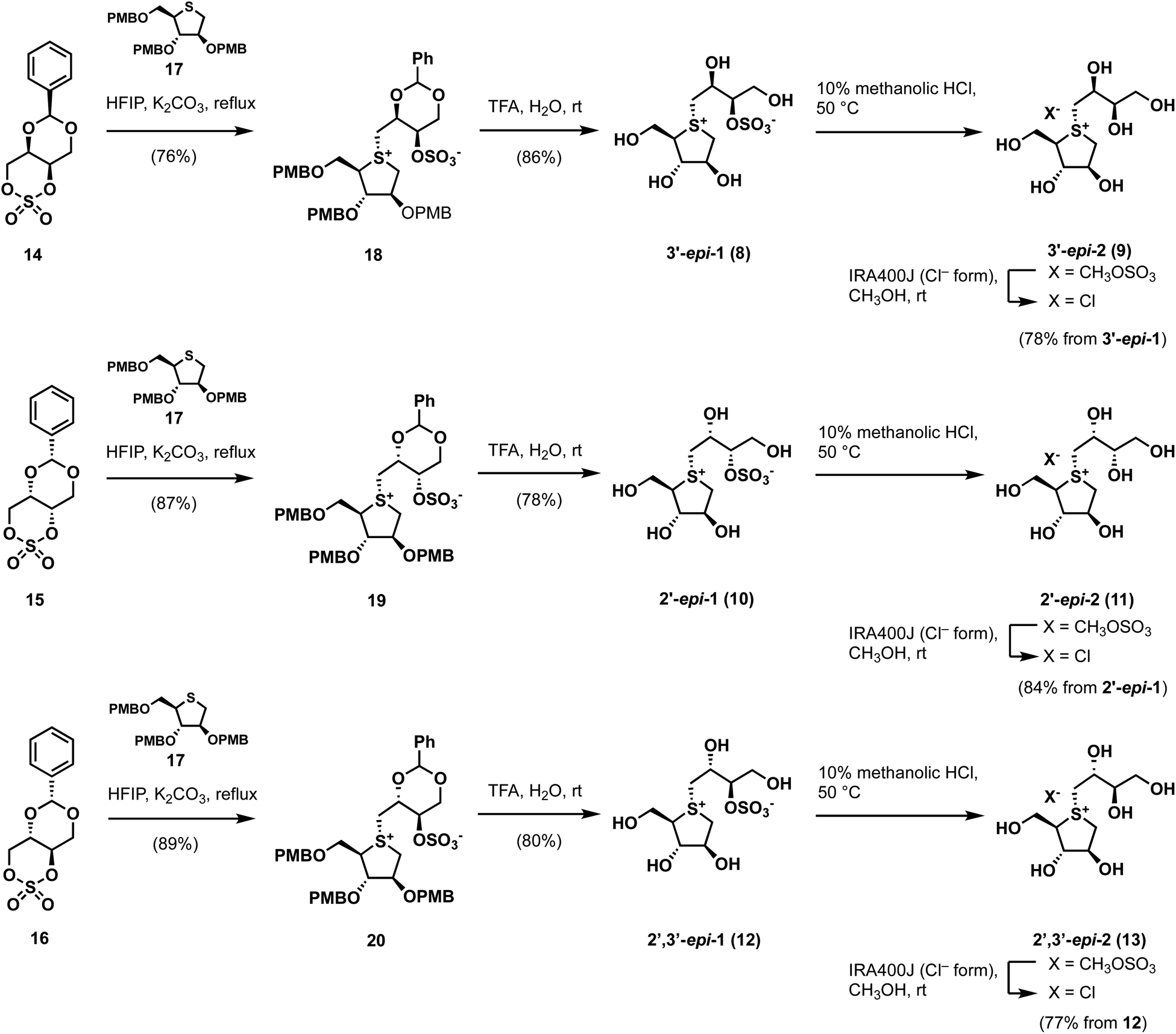 | ||
| Scheme 1 Synthetic routes to side-chain stereo-derivatives of 1. | ||
Results and discussion
By applying Ghavami's conditions for the synthesis of salacinol (1),33 coupling reactions of thiosugar 17 was reacted with cyclic sulfates 14–16 in 1,1,3,3,3-hexafluoroisopropanol (HFIP); α-facial attack of 14–16 at the sulfur atom of 17 preferentially occurred to give coupled products 18–20 in yields of 76%, 87%, and 89%, respectively. Compounds 18–20 were subsequently treated with aqueous TFA to simultaneously remove each p-methoxybenzyl (PMB) group and benzylidene acetal moiety, which gave the desired sulfonium salts 3′-epi-1, 2′-epi-1, and 2′,3′-epi-1 in good yields (Scheme 1). As shown in Table S1† (ESI), 3′-epi-1, 2′-epi-1, and 2′,3′-epi-1 showed 13C NMR spectral data that are similar to those of 1, which confirms the formation of salacinol-type sulfonium inner-salt structures.To remove the sulfo group at the C-3′ oxygen atom, 3′-epi-1, 2′-epi-1, and 2′,3′-epi-1 were subjected to acidic methanolysis, which give the corresponding sulfonium salts, namely 3′-epi-2, 2′-epi-2, and 2′,3′-epi-2 (X = CH3OSO3); their counterions were then exchanged using IRA400J (Cl− form) to give the corresponding chlorides (3′-epi-2, 2′-epi-2, and 2′,3′-epi-2, X = Cl) in good yields. 13C NMR spectroscopic data for 3′-epi-2, 2′-epi-2, and 2′,3′-epi-2 are similar, with the exception of the C-3′ methine carbon signals (δC3′ 74.2–75.1), which are significantly upfield shifted compared to those of 3′-epi-1, 2′-epi-1, and 2′,3′-epi-1 (δC3′ 82.0–82.8), confirming their de-O-sulfonated structures (Table S1†). The syntheses of cyclic sulfates 14–16 are described in Scheme S1.†
Our biochemical studies began by examining of the inhibitory properties of salacinol (1) and neosalacinol (2) toward α-glucosidase GAA (recombinant myozyme from genzyme, family GH31) at pH 5.2 using 4-nitrophenyl-α-D-glucopyranoside (α-p-NPG) as the substrate (Fig. S1† and Table 1). Prototypal 1 exhibited tight binding toward GAA, with a calculated Kappi values of 0.12 ± 0.02 μM (Fig. S1† and Table 1). In contrast, the Kappi value of the de-O-sulfonated analog 2 toward GAA was calculated to be 3.6 ± 0.3 μM, which is clearly inferior to that of 1 (Table 1 and Fig. S1†). In addition, voglibose and acarbose, which are widely used clinical intestinal α-glucosidase inhibitors, are interestingly less potent toward GAA than 1 and 2, with Kappi values of 7.6 ± 0.8 μM and 40 ± 2 μM, respectively (Table 1 and Fig. S2†). Based on these results, we conducted a comparative SAR study against GAA using potent intestinal α-glucosidase inhibitors 3–7. Of the five, four analogs 3–6 showed strong inhibitory activities toward GAA, with calculated Kappi values of 0.022 ± 0.007 μM, 0.034 ± 0.009 μM, 0.030 ± 0.009 μM, and 0.017 ± 0.010 μM, respectively (Fig. S3† and Table 1). Interestingly, the most potent intestinal α-glucosidase inhibitor (7)32 was less potent inhibition (Kappi = 0.17 ± 0.05 μM) (Table 1 and Fig. S3†) than the other 3′-O-benzylated analogs 3–6. We conclude that ortho-substitution of the benzene ring and/or the electronic effects of the benzene-ring substituents appear to have no effect on GAA-inhibition characteristics, albeit with one exception. We therefore suggest that 3′-O-benzylation is an effective protocol for increasing binding affinity to GAA. Furthermore, in silico docking studies of salacinol (1), neosalacinol (2), and the 3′-O-benzylated analogs 3–7 with GAA strongly support the observed Kappi trend (Fig. S4 and S5†). Although GAA and intestinal α-glucosidases are classified in family GH31, these results may highlight microenvironmental active-site differences between them.
We next evaluated the role of the stereochemistry of the salacinol side chain on inhibitory activity toward GH31 α-glucosidases (rat intestinal disaccharidases vs. GAA). As shown in Table 2, the 3′-epi-1 (8) exhibited weak binding affinity toward rat intestinal sucrase and isomaltase (IC50: 19 and 6.4 μM for sucrase and isomaltase, respectively); however, it completely lacked activity toward rat intestinal maltase (IC50 > 100 μM). In contrast, the de-O-sulfonated version of 8, 3′-epi-2 (9) exhibited stronger inhibitory activities toward three rat intestinal disaccharidases than 8 (9: IC50 = 0.69 μM for sucrase, IC50 0.58 μM for isomaltase, and IC50 4.3 μM for maltase). It was especially interesting that de-O-sulfonation significantly enhanced inhibitory activity toward rat intestinal maltase. Furthermore, by benchmarking against the inhibitory potency of 2 (IC50 25 μM for maltase), we conducted that a combination of the 3′R-OH and 2′S-OH moieties is more suitable than 3′S-OH and 2′S-OH for the onset of potent maltase inhibitory activity. On the other hand, four stereo-derivatives 10–13, each of which contains the 2′R-OH unit, exhibited virtually no binding to the rat intestinal disaccharidases, which highlights the importance of the cooperative roles of the 2′S-OH and 4′-OH moieties.
| Compound | Sucrase | Isomaltase | Maltase |
|---|---|---|---|
| a Apparent IC50 in (μM).b Ref. 24.c Ref. 32.d Ref. 35.e Ref. 36. | |||
| 1 | 1.6b | 5.2b | 5.2b |
| 2 | 3.6 | 0.45 | 25 |
| 3 (H) | 0.44c | 0.14c | 0.32c |
| 4 (o-CH3) | 0.41c | 0.48c | 0.66c |
| 5 (o-Cl) | 0.090c | 0.26c | 0.31c |
| 6 (o-CF3) | 0.15c | 0.19c | 0.33c |
| 7 (o-NO2) | 0.042c | 0.21c | 0.13c |
| 8 (3′-epi-1) | 19 | 6.4 | >100 |
| 9 (3′-epi-2) | 0.69 | 0.58 | 4.3 |
| 10 (2′-epi-1) | >100 | >100 | >100 |
| 11 (2′-epi-2) | 85 | >100 | >100 |
| 12 (2′,3′-epi-1) | >100 | >100 | >100 |
| 13 (2′,3′-epi-2) | 84 | 34 | >100 |
| Voglibose | 0.20d | 2.1d | 1.2d |
| Acarbose | 1.5e | 646e | 1.7e |
We next assessed the binding properties of 8–13 toward GAA (Table 1 and Fig. S6†); 3′-epi-1 (8) and 3′-epi-2 (9) are GAA-binding compounds, with Kappi values determined to be 1.0 ± 0.1 μM and 25 ± 2 μM, respectively, which are nine- and seven-times higher than those of 1 and 2 (Table 1 and Fig. S6†). On the other hand, stereo-derivatives 10–13 exhibited no inhibitory activities was observed (Table 1 and Fig. S6†). As a result, the molecular recognizing abilities of GH31 α-glucosidases appear to be relatively tolerant of the stereochemistry at the 3′-position, but can strictly discriminate the stereochemistry at the 2′-position.
We next assessed the ability of 1–9 to specifically inhibit GH31 α-glucosidases. Hence, we examined their abilities to inhibit β-glucosidase from Aspergillus niger34 at pH 4.6 using 4-nitrophenyl-β-D-glucopyranoside (β-p-NPG) as the substrate. None of these compounds displayed any inhibitory activity toward β-glucosidase from A. niger under our assay conditions, with apparent IC50 values > 1000 μM (Table 1 and Fig. S7†). These results demonstrate that thiosugar-based sulfonium salts 1–9 appear to be highly selective for GH31 α-glucosidases over A. niger β-glucosidase.
Conclusions
In summary, we demonstrated that salacinol-type α-glucosidase inhibitors exhibit ligand compatibility for the GH 31 family. Salacinol (1) and its 3′-O-benzylated analogs 3–7 displayed submicromolar-inhibitory activities toward human lysosomal α-glucosidase. Simple SAR studies demonstrated that the side-chain stereochemistry has a large effect on binding to GH31 α-glucosidases. We expect that the thiosugar skeleton may be valuable for the design of selective inhibitors that target glycosidases that recognize and process differently configured and substituted carbohydrates.Author contributions
G. T. designed this project. A. H., Y. Y., K. Nishida, W. X., and G. T. synthesized compounds. F. I, K. Ninomiya, and T. M. conducted biochemical studies. S. N and I. N carried out in silico calculation. F. I., G. T., and O. M. evaluated the data. F. I. and G. T. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by a Grant-in-Aid for Research (C) (17K08377 to G. T.) from the Japan Society of the Promotion of Science and the Hoansha Foundation (G. T.). This work was partly supported by a Grants-in-Aid for Scientific Research on Innovative Areas (17H05438 and 19H04664 to F. I.), a Grant-in-Aid Research (C) (19K05722 to F. I.), and grants from the Takeda Science Foundation (F. I.) and the Noda Institute for Scientific Research (F. I.). We are also grateful for the financial support extended by the Antiaging Project for Private Universities.Notes and references
- S. A. K. Jongkees and S. G. Withers, Unusual enzymatic glycoside cleavage mechanisms, Acc. Chem. Res., 2014, 47(1), 226–235 CrossRef CAS.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, The carbohydrate-active enzymes database (CAZy) in 2013, Nucleic Acids Res., 2014, 42(D1), D490–D495 CrossRef CAS.
- T. D. Butters, R. A. Dwek and F. M. Platt, Imino sugar inhibitors for treating the lysosomal glycosphingolipidoses, Glycobiology, 2005, 15(10), 43R–52R CrossRef CAS.
- A. Y. Y. Cheng and R. G. Josse, Intestinal absorption inhibitors for type 2 diabetes mellitus: prevention and treatment, Drug Discov. Today, 2004, 1(2), 201–206 CAS.
- C. P. Kordik and A. B. Reitz, Pharmacological treatment of obesity: therapeutic strategies, J. Med. Chem., 1999, 42(2), 181–201 CrossRef CAS.
- M. J. Papandreou, R. Barbouche, R. Guieu, M. P. Kieny and E. Fenouillet, The α-glucosidase inhibitor 1-deoxynojirimycin blocks human immunodeficiency virus envelope glycoprotein-mediated membrane fusion at the CXCR4 binding step, Mol. Pharmacol., 2002, 61(1), 186–193 CrossRef CAS.
- N. Asano, Glycosidase inhibitors: update and perspectives on practical use, Glycobiology, 2003, 13(10), 93R–104R CrossRef CAS.
- R. A. Ugalde, R. J. Staneloni and L. F. Leloir, Microsomal glucosidases of rat liver. Partial purification and inhibition by disaccharides, Eur. J. Biochem., 1980, 113(1), 97–103 CrossRef CAS.
- M. H. D. Postema, J. L. Piper, L. Liu, J. Shen, M. Foust and P. Andreama, Synthesis and partial biological evaluation of a small library of differentially-linked β-C-disaccharides, J. Org. Chem., 2003, 68(12), 4748–4754 CrossRef CAS.
- K. Afarinkia and A. Bahar, Recent advances in the chemistry of azapyranose sugars, Tetrahedron: Asymmetr, 2005, 16(7), 1239–1287 CrossRef CAS.
- O. Arjona, A. M. Gómez, J. C. López and J. Plumet, Synthesis and conformational and biological aspects of carbasugars, Chem. Rev., 2007, 107(5), 1919–2036 CrossRef CAS.
- V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Recent developments of transition-state analogue glycosidase inhibitors of non-natural product origin, Chem. Rev., 2002, 102(2), 515–554 CrossRef CAS.
- T. Mahmud, The C7N aminocyclitol family of natural products, Nat. Prod. Rep., 2003, 20(1), 137–166 RSC.
- S. Sou, S. Mayumi, H. Takahashi, R. Yamasaki, S. Kadoya, M. Sodeoka and Y. Hashimoto, Novel α-glucosidase inhibitors with a tetrachlorophthalimide skeleton, Bioorg. Med. Chem. Lett., 2000, 10(10), 1081–1084 CrossRef CAS.
- T. Niwa, U. Doi and T. Ogasawara, Inhibitory activity of corn-derived bisamide compounds against α-glucosidase, J. Agric. Food Chem., 2003, 51(1), 90–94 CrossRef CAS.
- K. Takada, T. Uehara, Y. Nakao, S. Matsunaga, R. W. M. van Soest and N. Fusetani, Schulzeines A-C, new α-glucosidase inhibitors from the marine sponge Penares schulzei, J. Am. Chem. Soc., 2004, 126(1), 187–193 CrossRef CAS.
- Y. Nakao, T. Maki, S. Matsunaga, R. W. M. van Soest and N. Fusetani, Penarolide sulfates A1 and A2, new α-glucosidase inhibitors from a marine sponge Penares sp., Tetrahedron, 2000, 56(46), 8977–8987 CrossRef CAS.
- Y. Nakao, T. Maki, S. Matsunaga, R. W. M. van Soest and N. Fusetani, Penasulfate A, a new α-glucosidase inhibitor from a marine sponge Penares sp., J. Nat. Prod., 2004, 67(8), 1346–1350 CrossRef CAS.
- Y. Nakao, T. Uehara, S. Matsunaga, N. Fusetani and R. W. M. Soest, Callyspongynic Acid, a Polyacetylenic Acid Which Inhibits α-Glucosidase, from the Marine Sponge Callyspongia truncate, J. Nat. Prod., 2002, 65(6), 922–924 CrossRef CAS.
- J. Kawabata, K. Mizuhata, E. Sato, T. Nishioka, Y. Aoyama and T. Kasai, 6-Hydroxyflavonoids as α-glucosidase inhibitors from Marjoram (Origanum majorana) leaves, Biosci., Biotechnol., Biochem., 2003, 67(2), 445–447 CrossRef CAS.
- M. Yoshikawa, T. Murakami, H. Shimada, H. Matsuda, J. Yamahara, G. Tanabe and O. Muraoka, Salacinol, potent antidiabetic principle with unique thiosugar sulfonium sulfate structure from the Ayurvedic traditional medicine Salacia reticulata in Sri Lanka and India, Tetrahedron Lett., 1997, 38(48), 8367–8370 CrossRef CAS.
- M. Yoshikawa, T. Morikawa, H. Matsuda, G. Tanabe and O. Muraoka, Absolute stereostructure of potent α-glucosidase inhibitor, salacinol, with unique thiosugar sulfonium sulfate inner salt structure from Salacia reticulata, Bioorg. Med. Chem., 2002, 10(5), 1547–1554 CrossRef CAS.
- M. Yoshikawa, T. Murakami, K. Tashiro and H. Matsuda, Kotalanol, a potent α-glucosidase inhibitor with thiosugar sulfonium sulfate structure, from antidiabetic Ayurvedic Medicine Salacia reticulata, Chem. Pharm. Bull., 1998, 46(8), 1339–1340 CrossRef CAS.
- M. Yoshikawa, F. Xu, S. Nakamura, T. Wang, H. Matsuda, G. Tanabe and O. Muraoka, Salaprionol and ponkoranol with thiosugar sulfonium sulfate structure from Salacia prinoides and α-glucosidase inhibitory activity of ponkoranol and kotalanol desulfate, Heterocycles, 2008, 75(6), 1397–1405 CrossRef CAS.
- Y. Minami, C. Kuriyama, K. Ikeda, A. Kato, K. Takebayashi, I. Adachi, W. J. G. Fleet, A. Kettawan, T. Okamoto and N. Asano, Effect of five-membered sugar mimics on mammalian glycogen-degrading enzymes and various glucosidases, Bioorg. Med. Chem., 2008, 16(6), 2734–2740 CrossRef CAS.
- O. Muraoka, W. Xie, G. Tanabe, F. A. M. Amer, T. Minematsu and M. Yoshikawa, On the structure of the bioactive constituent from ayurvedic medicine Salacia reticulata: revision of the literature, Tetrahedron Lett., 2008, 49(51), 7315–7317 CrossRef CAS.
- W. Xie, G. Tanabe, J. Akaki, T. Morikawa, K. Ninomiya, T. Minematsu, M. Yoshikawa, X. Wu and O. Muraoka, Bioorg. Med. Chem., 2011, 19(6), 2015–2022 CrossRef CAS.
- F. Ishikawa, K. Jinno, E. Kinouchi, K. Ninomiya, S. Marumoto, W. Xie, O. Muraoka, T. Morikawa and G. Tanabe, Diastereoselective synthesis of salacinol-type α-glucosidase inhibitors, J. Org. Chem., 2018, 83(1), 185–193 CrossRef CAS.
- M. H. S. Jayawardena, N. M. W. de Alwis, V. Hettigoda and D. J. S. Fernando, A double blind randomised placebo controlled cross over study of a herbal preparation containing Salacia reticulata in the treatment of type 2 diabetes, J. Ethnopharmacol., 2005, 97(2), 215–218 CrossRef CAS.
- O. Muraoka, K. Yoshikawa, T. Hatanaka, T. Minematsu, G. Lu, G. Tanabe, T. Wang, H. Matsuda and M. Yoshikawa, Synthesis and biological evaluation of deoxy salacinols, the role of polar substituents in the side chain on the α-glucosidase inhibitory activity, Bioorg. Med. Chem., 2006, 14(2), 500–509 CrossRef CAS.
- G. Tanabe, K. Yoshikai, T. Hatanaka, M. Yamamoto, Y. Shao, T. Minematsu, O. Muraoka, T. Wang, H. Matsuda and M. Yoshikawa, Biological evaluation of de-O-sulfonated analogs of salacinol, the role of sulfate anion in the side chain on the α-glucosidase inhibitory activity, Bioorg. Med. Chem., 2007, 15(11), 3926–3937 CrossRef CAS.
- G. Tanabe, S. Nakamura, N. Tsutsui, G. Balakishan, W. Xie, S. Tsuchiya, J. Akaki, T. Morikawa, K. Ninomiya, I. Nakanishi, M. Yoshikawa and O. Muraoka, In silico design, synthesis and evaluation of 3′-O-benzylated analogs of salacinol, a potent α-glucosidase inhibitor isolated from an Ayurvedic traditional medicine “Salacia”, Chem. Commun., 2012, 48(69), 8646–8648 RSC.
- A. Ghavami, K. S. Sadalapure, B. D. Johnston, M. Lobera, B. B. Snider and B. M. Pinto, Improved syntheses of the naturally occurring glycosidase inhibitor salacinol, Synlett, 2003, 2003(9), 1259–1262 Search PubMed.
- T. Watanabe, T. Sato, S. Yoshioka, T. Koshijima and M. Kuwahara, Purification and properties of Aspergillus niger β-glucosidase, Eur. J. Biochem., 1992, 209(2), 651–659 CrossRef CAS.
- G. Tanabe., T. Otani, W. Cong, T. Minematsu, K. Ninomiya, M. Yoshikawa and O. Muraoka, Biological evaluation of 3′-O-alkylated analogs of salacinol, the role of hydrophobic alkyl group at 3′ position in the side chain on the α-glucosidase inhibitory activity, Bioorg. Med. Chem. Lett., 2011, 21(10), 3159–3162 CrossRef CAS.
- O. Muraoka, T. Morikawa, S. Miyake, J. Akaki, K. Ninomiya, Y. Pongpiriyadacha and M. Yoshikawa, Quantitative analysis of neosalacinol and neokotalanol, another two potent α-glucosidase inhibitors from Salacia species, by LC-MS with ion pair chromatography, J. Nat. Med., 2011, 65(1), 142–148 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10038b |
| This journal is © The Royal Society of Chemistry 2021 |