
Shining a new light on the structure of polyurea/polyurethane materials†
Piangtawan
Phoungtawee
and
Daniel
Crespy
 *
*
Department of Materials Science and Engineering, School of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), Rayong 21210, Thailand. E-mail: daniel.crespy@vistec.ac.th
First published on 17th June 2021
Abstract
Polyurea and polyurethane containing urea bonds are widely used in the preparation of coatings, foams, and micro- and nanocapsules. The molecular structures of such materials are typically difficult to characterize quantitatively due to their poor solubility and because mixtures with strong acids tend to hydrolyze the polymers. In a mixture of trifluoroacetic acid (TFA) and acetone, a very strong hydrogen bonding occurs between the carbonyl oxygen of acetone and the proton of TFA, which leads to its deshielding. This peculiar mixture of solvent was used for the quantitative characterization of polyurethane/urea coatings, microcapsules, and nanocapsules by NMR spectroscopy. By using aromatic and aliphatic diisocyanates, diamines, and diols, the extent of urea groups, and hence hydrolyzed isocyanates, in the polymer can be determined. This method is shining light on materials that were not yet quantitatively characterized such as polyurea nanocapsules prepared by interfacial polymerization in miniemulsion.
Introduction
Polyurethanes and polyureas are polymers which are typically prepared by the reaction between isocyanates and molecules containing hydroxyl or amine groups.1 While polyurethanes are ubiquitous in our daily life,2 polyureas have more limited applications due to their difficult processibility. Polyureas were used to prepare coatings for antifouling,3 antibacterial4 and anti-icing applications.5 Moreover, due to the hydrophobic nature of isocyanates and the hydrophilicity of many diols and diamines, interfacial polyaddition in emulsion were performed to fabricate nanocapsules or nanoparticles for the encapsulation of catalysts,6–9 fragrances,10 phase change materials,11 pesticides,12 dyes,13 corrosion inhibitors,14 contrast agents for magnetic resonance imaging,15,16 self-healing agents,17–20 and drugs.21The presence of urea bonds in polyurethanes can be beneficial for the fabrication of intrinsic self-healing materials which can be remended due to the presence of strong hydrogen bonds.22–24 Moreover, the addition of controlled and low amounts of water to isocyanates was found to generate aerogels with high porosity.25 However, the concurrent reaction between water and isocyanates in the synthesis of polyurethanes and polyureas is often not controlled, leading to the hydrolysis of isocyanates to carbamic acids. The latter are unstable and yields CO2 and an amine, which can react with isocyanates to form unwanted urea groups in the structure of the main polymer. This fact has lead various research groups to use anhydrous oil-in-oil emulsions for the fabrication of polyurethane particles without urea bonds.26,27 Indeed, polymers containing many urea groups tend to become intractable and are difficult to process. This is due to the formation of abundant and strong intermolecular hydrogen bonding,28 which can form readily ordered structures at room temperature.29 For example, the presence of urea segments in polyurethane foams can lead to phase separation and the creation of fibril structures.30
Urea groups are typically detected by Fourier-transform infrared spectroscopy (FTIR) because the vibration of the carbonyl bond in urea is significantly different from the vibration of the carbonyl in urethane groups.31–39 Nuclear magnetic resonance (NMR) spectroscopy can be performed but usually lead to weak signals due to the limited solubility of polyureas in conventional deuterated solvents such as DMSO-d6,40,41 or require a high-resolution magic angle spinning (HR-MAS) setup and a large quantity of material to be measured.28,42
Herein, we aim to determine quantitatively the extent of formation of urea groups in polyurea and polyurethane, with a focus on the groups formed after hydrolysis of isocyanates. Recently, the mixture acetone/TFA was used for dissolving polyamides for the preparation of transparent and homogenous films.43 Inspired by this finding, we dissolved polyureas and polyurethanes in this mixture and studied their structure by 1H NMR spectroscopy. This investigation is particularly relevant for nanoparticles and nanocapsules prepared by interfacial polymerization and for coatings.
Experimental section
Materials
Polyglycerol polyricinoleate (PGPR, Danisco) was purified by washing with hexane before use. 1,6-Hexanediamine (HMDA, 99.5%, Acros Organics), 1,6-hexanediol (HDO, >97%, TCI chemicals), 1,3-bis(aminoethyl)-cyclohexane (BAC, >98%, TCI chemicals), m-xylylenediamine (XDA, >99%, TCI chemicals), 2,4-toluene diisocyanate (TDI, 80%, Acros Organics), hexamethylene diisocyanate (HMDI, ≥98%, Sigma-Aldrich), cyclohexane (≥99.8%, Carlo Erba), sodium chloride (NaCl, 99%, Carlo Erba), calcium chloride dried granular (CaCl2, 90%, Carlo Erba), acetone (99.8%, Carlo Erba), hexane (99.97%, Honeywell), resorcinol (>99%, TCI), sodium dodecyl sulfate (SDS, 99%, Acros), tetrahydrofuran (THF, 99.99%, Honeywell), xylene (mixture of isomers, 98.5%, RCI Labscan), isophorone diisocyanate (IPDI, 98%, Acros Organics)dimethyl sulfoxide (DMSO, 99%, Loba Chemie), trifluoroacetic acid (TFA, 99%, Acros Organics), and acetone-d6 (99.8%, Eurisotop) were used as received. Deionized water was used throughout this work.Preparation of polyurea/polyurethane nanocapsules
HMDA or BAC or HDO (0.1 g) and 30 mg NaCl or CaCl2 were mixed in 1.3 g of water or DMSO. These solutions were mixed at 1000 rpm for 1 h at 27 °C with a solution of 75 mg of PGPR in 7.5 g cyclohexane. Afterwards, the obtained emulsion was further emulsified with an ultrasonicator (Hielscher UP200St, diameter 7 mm) for 6 min in a pulse mode (1 s on, 1 s off) at 50% amplitude in an ice bath. Then, known amounts of TDI and 35 mg PGPR in 5 g cyclohexane were slowly dropped in the miniemulsion for 5 min, which were stirred at 500 rpm at 27 °C for polyurea or 60 °C for polyurethane nanocapsules. The reactions were further stirred for 2 h to complete the reaction. The dispersions (2 g) were then added into 20 g water and the mixture was centrifuged (2-16KL model from Sigma) at 2655 g and 25 °C for 5 min. Phase separation occurred to facilitate the purification of the polymers. The pellets were re-dispersed in 2 mL cyclohexane. The process was repeated 2 more times by changing the precipitation solvent to acetone and then hexane. Finally, the pellets were dried in a vacuum desiccator.For the determination of the completion of reaction, dispersions of polyurea and polyurethane nanocapsules (2 g) (P2, P5–7) were added to 20 g hexane. The dispersions were then centrifuged (2-16KL model from Sigma) at 2655 g and 25 °C for 5 min. The supernatants were collected and filtered through a filter paper and the filtrate were evaporated with a rotary evaporator at 40 °C. Around 10 mg of the resulting product was dissolved in 700 μL DMSO-d6 containing 2 mg of ethylene carbonate as internal standard.
Preparation of polyurea microcapsules
Polyurea microcapsules were synthesized following a slightly modified procedure.20 A 1.0 wt% aqueous solution (50 mL) of SDS was added into a 100 mL beaker. Then, IPDI (1.63 g) was dissolved into 10 mL of xylene and added to the beaker. The mixture was homogenized with a high-speed disperser (Ultraturrax T18) at 5000 rpm at 28 °C for 5 min to obtain a stable emulsion. Then, XDA (1 g) in 10 mL of a 1.0 wt% aqueous solution of SDS was added dropwise to the emulsion for 5 min, stirred at 300 rpm for 30 min at 28 °C, and further stirred 1.5 h at 50 °C. The obtained suspension (2 g) was added in 20 g water and then centrifuged (2-16KL, Sigma) at 2655 g at 25 °C for 5 min. The pellets were re-dispersed in 2 mL cyclohexane. The process was repeated 2 more times by changing the precipitation solvent to acetone and then hexane. Finally, the pellets were dried in a vacuum desiccator.Preparation of the polyurethane coating
Resorcinol (500 mg) was dissolved in a mixture of 400 μL THF and 4 μL water. HMDI (750.5 mg) was added in the aforementioned solution and stirred at 500 rpm for 5 min. Then, the mixture was drop-cast on a glass slide and cured 24 h at 28 °C, followed by 24 h at 60 °C. A transparent coating with a thickness of 170 μm was obtained. The coating was peeled off and dissolved in 1 mL DMSO. The solution was precipitated in 10 mL methanol and then centrifuged (2-16KL, Sigma) at 2655 g for 10 min at 25 °C. The pellet was dissolved and reprecipitated two more times and dried in a vacuum oven at 60 °C 24 h.Analytical tools
1H NMR spectra of polyurea/polyurethane nanocapsules were recorded on a Bruker 600 MHz NMR spectrometer. Dried polyurea/polyurethane (4–6 mg) of nanocapsules were dissolved in a mixture of 300 μL acetone-d6 (40% mol) and 460 μL TFA (60% mol) for NMR experiments, which were acquired with a 5 mm double resonance broad band probe equipped with a z-gradient on a 600 MHz console (Bruker AVANCE III HD System). For the calculation of the molar ratios of monomer units in the polymers, the integral associated with the signals given by the 4 protons of the 2 methylene groups neighbouring the urea or urethane groups was compared with the aromatic protons. Attenuated total reflectance Fourier transform infrared (ATR-FTIR) measurements were performed on a Frontier FTIR spectrometer (PerkinElmer) between 400 to 4000 cm−1 with a resolution of 4 cm−1. Dispersions of polyurea or polyurethane nanocapsules (50 μL) were diluted with 3 mL cyclohexane for measuring the hydrodynamic diameter by dynamic light scattering (DLS, NanoPlus, Particulate systems). The morphologies of nanocapsules were observed with a transmission electron microscope (TEM, JEM-ARM200F, JEOL). Dispersions of polyurea microcapsules (5 μL) were diluted with 5 mL cyclohexane and dropped on silicon wafers and left for drying. The morphology of polyurea microcapsules were observed with a scanning electron microscope (SEM, JSM-7610F, JEOL).Results and discussion
Polyureas, because of the high density of hydrogen bonding inside their structures, are difficult to dissolve. Even the presence of alkali halide salts such as LiCl, LiBr2, or CaCl2 in organic solvents, typically used for dissolving polyamides, are often not sufficient to dissolve them. Solvents such sulfuric acid or formic acid at high temperatures are required for protonating the carbonyl bond and breaking hydrogen bonding between polymer molecules, which limit their characterization in the liquid state. Recently, the mixture of trifluoroacetic acid (TFA) and acetone was used to dissolve polyamide 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 44,45 and polyamide 11 at room temperature to prepare composites and ferroelectric coatings.43 The TFA/acetone mixture allowed the formation of smooth homogeneous films with polyamide 11 crystallizing in the δ′ phase. 1H NMR spectroscopy investigations have shown that the acidic proton of TFA was shared with acetone.43 Whereas no intermolecular hydrogen bonding was detected in pure TFA or pure acetone, strong hydrogen bonding occurred between the oxygen of acetone and the proton in TFA, which is one the hydrogen bond with the largest enthalpy (−10 kcal mol−1).
44,45 and polyamide 11 at room temperature to prepare composites and ferroelectric coatings.43 The TFA/acetone mixture allowed the formation of smooth homogeneous films with polyamide 11 crystallizing in the δ′ phase. 1H NMR spectroscopy investigations have shown that the acidic proton of TFA was shared with acetone.43 Whereas no intermolecular hydrogen bonding was detected in pure TFA or pure acetone, strong hydrogen bonding occurred between the oxygen of acetone and the proton in TFA, which is one the hydrogen bond with the largest enthalpy (−10 kcal mol−1).
We investigated herein the structure of polyurea and polyurethane containing urea groups by 1H NMR spectroscopy in a TFA/acetone-d6 mixture. First of all, we prepared polyurea and polyurethane nanocapsules by interfacial polymerization in inverse miniemulsions. The process involves the reaction between diamines or diols that are dissolved in water or a polar solvent and subsequently emulsified in an apolar solvent (here cyclohexane).14,46–48 After addition of diisocyanates through the continuous phase of the miniemulsions, nanocapsules are formed (see Fig. 1). Water and DMSO were used as dispersed phase for the dissolution of diols and diamines so that polyurethanes, polyureas, and polyurethane/ureas materials were obtained (Table 1). After the interfacial polymerization, colloids with a hydrodynamic diameter of 150–500 nm were obtained. The morphology of the colloids varied from nanocapsules (P1–4, 6) to nanoparticles (P5), and multi-hollow nanoparticles (P7), as observed by transmission electron microscopy (see Fig. 1). Derivatives of this procedure have been widely used and reported by several groups in the literature.49–55 We collected polyurea/polyurethane nanocapsules prepared in water-in-oil or DMSO-in-oil miniemulsions just after (start of the reaction) and 2 h (end of the reaction) after ultrasonication. Whereas signals for the isocyanate groups at 2265 cm−1 were observed by Fourier transform infrared (FTIR) spectroscopy directly after ultrasonication, no signals were observed after 2 h of the reaction (Fig. S1, ESI†). Furthermore, the dispersion of nanocapsules after 2 h reaction was added to hexane, centrifuged, and the supernatant was concentrated by evaporation of hexane. The remaining liquid was dissolved in a solution of ethylene carbonate (internal standard) in DMSO-d6. As shown in Fig. S2, ESI,† no signals that could be assigned to TDI were found, confirming that the isocyanates were not present in significant amounts after reaction.
 | ||
| Fig. 1 Scheme for the preparation of polyurea nanocapsules with either DMSO or water as core and transmission electron micrographs of polyurea (P1–5) and polyurethane (P6–7) nanocapsules and nanoparticles. | ||
| Entry | Dispersed phase | Polymer | Polar monomer | M:TDI ratio |
D h | ||
|---|---|---|---|---|---|---|---|
| Feed | Polymer | nm | PDI | ||||
| P1 | Water | Polyurea | HMDA | 1:0.9 |
1:1.3 |
243 | 0.300 |
| P2 | Water | Polyurea | HMDA | 1:1 |
1:1.3 |
276 | 0.194 |
| P3 | Water | Polyurea | HMDA | 1:1.1 |
1:1.5 |
245 | 0.307 |
| P4 | Water | Polyurea | 1,3-BAC | 1:1 |
1:1.9 |
173 | 0.187 |
| P5 | DMSO | Polyurea | HMDA | 1:1 |
1:1.2 |
319 | 0.264 |
| P5′ | Anh. DMSO | Polyurea | HMDA | 1:1 |
1:1.1 |
301 | 0.216 |
| P6 | Water | Polyurethane/urea | HDO | 1:1 |
1:2.8 |
490 | 0.308 |
| P7 | DMSO | Polyurethane | HDO | 1:1 |
1:2.1 |
386 | 0.084 |
The qualitative analysis of the chemical structure of these compounds was usually reported by measuring them by Fourier transform infrared spectroscopy.14,56 As expected, vibrations from carbonyl groups of urea at 1634 cm−1 and for –NH bending at 1548 cm−1 were observed for polyurea nanocapsules (P1–5), as shown in Fig. 2a and Fig. S3, ESI.† For the polyurethane specimen (P6–7), vibrations associated to the carbonyl groups of urethane groups at 1710 cm−1, 1600 cm−1 for the –NH bending, ∼1510 cm−1 for amide II band57 and 1066 cm−1 for C–O bonds were detected (Fig. 2a). Furthermore, a small signal for the polyurethane P6 at 1634 cm−1 indicated the presence of urea groups.
 | ||
| Fig. 2 (a) FT-IR spectra of the polyurea (P2–3) and polyurethane/urea (P6–7) nanocapsules. (b) Scheme for the reactions between the diamines monomers and from hydrolyzed diisocyanates, water, and diols with the toluene-2,4-diisocyanate (TDI) (c) Mechanism of the hydrolysis of isocyanate and decarboxylation of the carbamic acid in the presence of water. | ||
The urea groups produced during the reactions in the presence of isocyanates, amines, and water (P1–4) originated from the reaction between diisocyanates and diamines (i and ii in Fig. 2b) or from the reaction between diisocyanates and hydrolyzed diisocyanates. Indeed, isocyanates react with water to form unstable carbamic acid compounds, which are subsequently decomposed in amines and carbon dioxide (iii) (Fig. 2c). The rate of the reaction between isocyanates with water is typically around ∼10−3 less than for the reaction between isocyanates and primary amines,58 meaning that diamines are primarily consumed, even in the presence of water. The hydrolyzed diisocyanate can react with non-hydrolyzed diisocyanates to yield new urea segments in the polymer (v). Finally, diisocayanates react with diols (iv), with a kinetic constant in the absence of catalyst typically estimated as similar as for the reaction between water and isocyanates.58
In order to assess the proportion of urea groups in the synthesized nanocapsules, 1H-NMR spectroscopy of polyurea and polyurethane nanocapsules after purification and dissolved in TFA–acetone-d6 was performed. Solutions of polyurea and polyurethane in acetone-d6/TFA (40:60 mol%) were transparent until a concentration of ∼42 and 13 mg ml−1, respectively (Fig. S4, ESI†). Then, we verified that the mixture TFA–acetone-d6 was not degrading the polymers during at least the time of measurements by using Diffusion Ordered Spectroscopy (DOSY) technique. DOSY is a 2D NMR spectroscopy technique displaying chemical shifts and self-diffusion coefficients of dissolved molecules. The loss of magnetization/resonance intensity of molecules during with time yields information about their diffusion coefficient. The diffusion coefficient D is related to molecular weight M by the following equation:59D = AMa where A and a are constants specific to the polymer. High polydispersity in molecular weight hence leads to widening of the values of diffusion coefficient. As examples, poly(ethylene oxide) with molecular weights of 85000 g mol−1 and 90000 g mol−1 displayed coefficients of diffusion measured by 1H DOSY NMR spectroscopy in D2O of 2.4 × 10−11 m2 s−1 and 2.2 × 10−11 m2 s−1, respectively.60 Besides, coefficients of diffusion of 6.5 × 10−11 m2 s−1 and 5.4 × 10−11 m2 s−1 were measured for poly(ethylene terephthalate) with weight-average molecular weights of 18000 g mol−1 and 25000 g mol−1.611H DOSY NMR was employed for monitoring the hydrolysis and degradation of nylon 11.431H-NMR DOSY spectra of polyurethane and polyurea from dissolved nanocapsules showed that the coefficient of diffusion of the polymer was constant (Fig. S5, ESI†), hereby indicating that no significant decomposition occurred. In the 1H-NMR spectra, the signal from the proton of TFA was indeed detected at around 12.5 ppm in every 1H NMR spectra (Fig. S6, ESI†). The signal appears at very high chemical shift (downfield) because of the strong electron withdrawing group in TFA, which decreases the electron density around the proton nucleus. The integral of the signal associated to the methylene proton near the nitrogen of the urea group at 3.1 ppm or near the oxygen of the urethane group at 4.0 ppm was compared with the integral of the signal associated to the protons of the aromatic rings (7.0–7.2 ppm). Because the latter solely originated from the TDI, this method allows to estimate the amount of TDI that was hydrolyzed and reacted with itself after hydrolysis.
For the preparation of polyurea nanocapsules from hexamethylene diamine (HMDA) and toluene-2,4-diisocyante (TDI), the feed molar ratio HMDA:TDI was varied to 1:0.9, 1:1, 1:1.1 (entries P1–3 in Table 1). Integration of the 1H-NMR spectra (Fig. S7, ESI†) showed that the ratio between HMDA and TDI monomer units in the polyurea were 1:1.3, 1:1.3, 1:1.5, respectively. This increase of TDI units in the polymer compared to the feed ratio indicates that TDI was partially hydrolyzed and reacted with non-hydrolyzed, participating in the formation of polyurea nanocapsules. Replacing HMDA with 1,3-bis(aminoethyl)cyclohexane (BAC) led to nanocapsules with more TDI units inside the polymer (Table 1, Fig. S7, ESI†), implying that HMDA displayed a higher reactivity than BAC.2
Published literature mention the replacement of water by aprotic polar solvents such as DMSO for water-sensitive reactions or when the encapsulated payload is not water-soluble.26,62,63 As expected, the polymerization with DMSO (P5) (Fig. 3a) as dispersed phase instead of water (P2) yielded a polyurea with less aromatic units. Using anhydrous DMSO (P5′) further decreased the content of TDI monomer units in the polyurea shell, yielding a HMDA:TDI composition in the polymer close to the feed molar ratio. The very small discrepancy was attributed to the fact that moisture can be absorbed by DMSO during the ultrasonication of the samples. The polymerization between diols and diisocyanates is slower than the polymerization between diamines and diisocyanates, especially in the absence of catalyst. It is then inferred that the extent of side-reactions, such as hydrolysis of isocyanates, in the former reaction increases. Indeed, larger amounts of TDI was incorporated in the polyurethane/urea (P6 and P7) than their comparable polyurea counterparts (P2 and P5), both with water (P2 and P6) or DMSO as dispersed phase (P5 and P7). In the light of this study, we can conclude that polyurethane and polyurea nanocapsules prepared with a similar procedure contain substantial amounts of TDI units in their structure, with amounts superior to their feed ratios. For biomedical applications, the presence of TDI units in the polymer is not suitable as its degradation products can be toxic. The concept of the study was then generalized to other systems such as microcapsules and coatings. Instead of preparing nanocapsules in water-in-oil miniemulsions, we prepared polyurea microcapsules in an oil-in-water process by suspension polymerization. Polyurea was produced by the reaction between equimolar amounts of IPDI and XDA. In principle, similar reactions are possible also in oil-in-water miniemulsions.64–66 Microcapsules with an average diameter of 12 ± 2 μm were obtained (see Fig. 3b). In this system, the diamine was aromatic while the isocyanate was aliphatic. A ratio of IPDI:XDA units in the polyurea microcapsules of 1.6:1 was detected by 1H NMR spectroscopy (Fig. S8a, ESI†). The larger amounts of aliphatic moieties compared with the feed ratio was attributed to the hydrolysis of IPDI, yielding a product that reacted with non-hydrolyzed IPDI concurrently to its reaction with XDA. The relative content of water in this experiment was much larger than for the nanocapsules because water was used as continuous phase. Besides, a polyurethane coating was prepared from the reaction between resorcinol and HMDI on a glass substrate, followed by thermal curing (Fig. 3c). The reaction took place in the presence of a small amount of water to simulate situations of high humidity or long storage in imperfectly sealed container. However, the content of water being however too low to allow foaming. The ratio between aliphatic (from the diisocyanate) and aromatic units (from the diol) was 1.1:1, hence suggesting that HMDI reacted partially with water (Fig. S8b, ESI†).
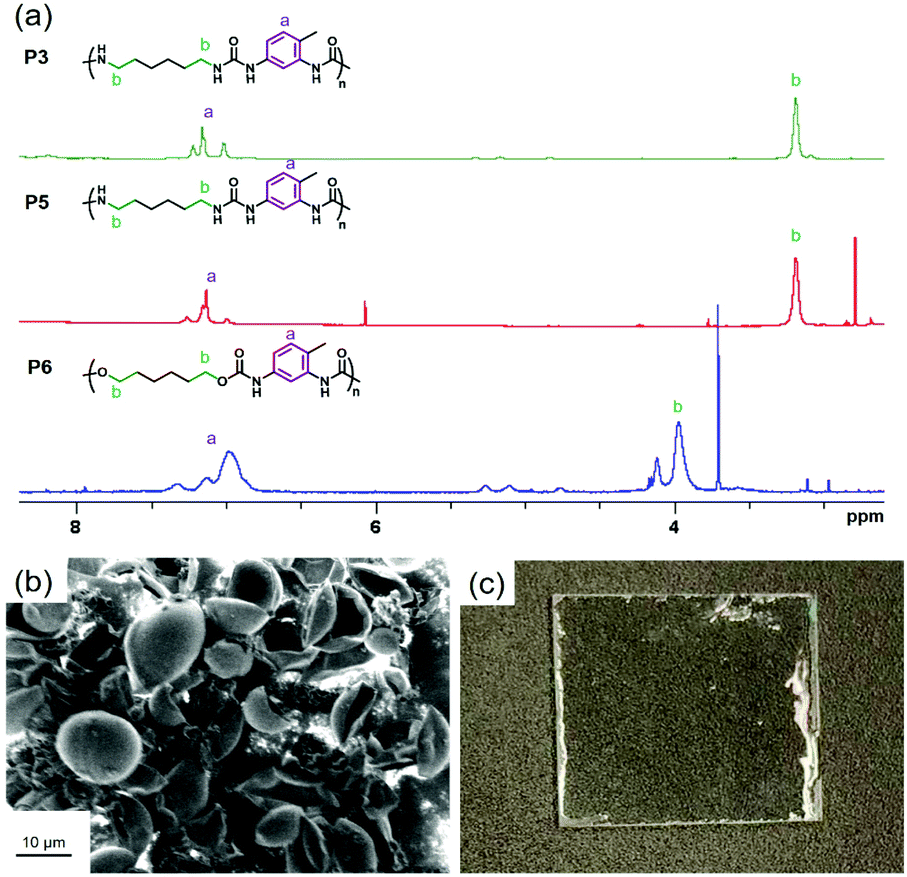 | ||
| Fig. 3 (a) 1H-NMR spectra of polyurea/urethane nanocapsules (entries P3, 5 and 6) in acetone-d6:TFA (40:60). (b) SEM micrograph of polyurea microcapsules prepared by interfacial polymerization in an oil-in-water emulsion. (c) Photograph of a polyurethane coating prepared by the reaction between resorcinol and HMDI. | ||
Conclusions
Polyurethane/urea nanocapsules and coatings were characterized by nuclear magnetic resonance spectroscopy in a mixture of deuterated acetone and trifluoroacetic acid. The extent of monomeric units in the polymers was determined by comparing and quantifying aliphatic and aromatic protons. In water-in-oil miniemulsions, the hydrolysis of isocyanates and its subsequent reaction with non-hydrolyzed isocyanates was found to be very significant and led to the incorporation of large extent of urea units in the polymers. The concept was further extended to the analysis of microcapsules prepared in oil-in-water emulsions and to coatings. These findings show that assumption of composition based on starting monomers is not suitable and that the polymeric materials should be accurately characterized. This work has hence important implications for the design of formulations of polyurea and polyurethanes.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Vidyasirimedhi Institute of Science and Technology (VISTEC), the National Research Council of Thailand (NRCT5-RSA63025-01), and the Office of the Higher Education Commission of Thailand (OHEC).Notes and references
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- J. Fang, A. Kelarakis, D. Wang, E. P. Giannelis, J. A. Finlay, M. E. Callow and J. A. Callow, Polymer, 2010, 51, 2636–2642 CrossRef CAS.
- L. A. T. W. Asri, M. Crismaru, S. Roest, Y. Chen, O. Ivashenko, P. Rudolf, J. C. Tiller, H. C. van der Mei, T. J. A. Loontjens and H. J. Busscher, Adv. Funct. Mater., 2014, 24, 346–355 CrossRef CAS.
- H. Yu, Z. He, G. Qian, X. Gong and X. Qu, Constr. Build. Mater., 2020, 242, 117793 CrossRef CAS.
- S. V. Ley, C. Mitchell, D. Pears, C. Ramarao, J.-Q. Yu and W. Zhou, Org. Lett., 2003, 5, 4665–4668 CrossRef CAS PubMed.
- H. Han, Y. Zhou, S. Li, Y. Wang and X. Z. Kong, ACS Appl. Mater. Interfaces, 2016, 8, 25714–25724 CrossRef CAS PubMed.
- H. Sun, Y. Wei, X. Z. Kong and X. Jiang, Polymer, 2021, 216, 123432 CrossRef CAS.
- Y. Sui, Y. Cui, G. Xia, X. Peng, G. Yuan and G. Sun, Process Biochem., 2019, 87, 73–82 CrossRef CAS.
- M. Jacquemond, N. Jeckelmann, L. Ouali and O. P. Haefliger, J. Appl. Polym. Sci., 2009, 114, 3074–3080 CrossRef CAS.
- C. Liang, X. Lingling, S. Hongbo and Z. Zhibin, Energy Convers. Manage., 2009, 50, 723–729 CrossRef.
- L. Ren, B. Huang, W. Fang, D. Zhang, H. Cheng, Z. Song, D. Yan, Y. Li, Q. Wang, Z. Zhou and A. Cao, ACS Appl. Mater. Interfaces, 2021, 13, 1333–1344 CrossRef CAS PubMed.
- S. Behzadi, C. Rosenauer, M. Kappl, K. Mohr, K. Landfester and D. Crespy, Nanoscale, 2016, 8, 12998–13005 RSC.
- K. Thongchaivetcharat, R. Jenjob, F. Seidi and D. Crespy, Carbohydr. Polym., 2019, 217, 217–223 CrossRef CAS PubMed.
- K. Malzahn, S. Ebert, I. Schlegel, O. Neudert, M. Wagner, G. Schütz, A. Ide, F. Roohi, K. Münnemann, D. Crespy and K. Landfester, Adv. Healthcare Mater., 2016, 5, 567–574 CrossRef CAS PubMed.
- I. Schlegel, R. Muñoz-Espí, P. Renz, I. Lieberwirth, G. Floudas, Y. Suzuki, D. Crespy and K. Landfester, Macromolecules, 2017, 50, 4725–4732 CrossRef CAS.
- M. M. Caruso, B. J. Blaiszik, H. Jin, S. R. Schelkopf, D. S. Stradley, N. R. Sottos, S. R. White and J. S. Moore, ACS Appl. Mater. Interfaces, 2010, 2, 1195–1199 CrossRef CAS PubMed.
- G. Wu, J. An, D. Sun, X. Tang, Y. Xiang and J. Yang, J. Mater. Chem. A, 2014, 2, 11614–11620 RSC.
- D. Sun, J. An, G. Wu and J. Yang, J. Mater. Chem. A, 2015, 3, 4435–4444 RSC.
- P. D. Tatiya, R. K. Hedaoo, P. P. Mahulikar and V. V. Gite, Ind. Eng. Chem. Res., 2013, 52, 1562–1570 CrossRef CAS.
- W. He, X. Gu and S. Liu, Adv. Funct. Mater., 2012, 22, 4023–4031 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- C. Liu, C. Ma, Q. Xie and G. Zhang, J. Mater. Chem. A, 2017, 5, 15855–15861 RSC.
- J. Xu, W. Chen, C. Wang, M. Zheng, C. Ding, W. Jiang, L. Tan and J. Fu, Chem. Mater., 2018, 30, 6026–6039 CrossRef CAS.
- N. Leventis, C. Sotiriou-Leventis, N. Chandrasekaran, S. Mulik, Z. J. Larimore, H. Lu, G. Churu and J. T. Mang, Chem. Mater., 2010, 22, 6692–6710 CrossRef CAS.
- C. Herrmann, D. Crespy and K. Landfester, Colloid Polym. Sci., 2011, 289, 1111–1117 CrossRef CAS.
- K. Müller, M. Klapper and K. Müllen, Colloid Polym. Sci., 2007, 285, 1157–1161 CrossRef.
- W.-G. Qiu, F.-L. Zhang, X.-B. Jiang and X.-Z. Kong, Chin. J. Polym. Sci., 2018, 36, 1150–1156 CrossRef CAS.
- J. Mattia and P. Painter, Macromolecules, 2007, 40, 1546–1554 CrossRef CAS.
- R. Neff, A. Adedeji, C. W. Macosko and A. J. Ryan, J. Polym. Sci., Part B: Polym. Phys., 1998, 36, 573–581 CrossRef CAS.
- B. Di Credico, G. Griffini, M. Levi and S. Turri, ACS Appl. Mater. Interfaces, 2013, 5, 6628–6634 CrossRef CAS PubMed.
- P. Scarfato, E. Avallone, P. Iannelli, V. De Feo and D. Acierno, J. Appl. Polym. Sci., 2007, 105, 3568–3577 CrossRef CAS.
- W. Chen, X. Liu and D. W. Lee, J. Mater. Sci., 2012, 47, 2040–2044 CrossRef CAS.
- J.-S. Cho, A. Kwon and C.-G. Cho, Colloid Polym. Sci., 2002, 280, 260–266 CrossRef CAS.
- G. Li, Y. Q. Feng, X. G. Li, P. Gao, J. Wang and J. Y. Xie, J. Mater. Sci., 2007, 42, 4838–4844 CrossRef CAS.
- S.-K. Wang and C. S. P. Sung, Macromolecules, 2002, 35, 883–888 CrossRef CAS.
- P. W. Loscutoff, H. Zhou, S. B. Clendenning and S. F. Bent, ACS Nano, 2010, 4, 331–341 CrossRef CAS PubMed.
- S. Lu, T. Shen, J. Xing, Q. Song, J. Shao, J. Zhang and C. Xin, Mater. Lett., 2018, 211, 36–39 CrossRef CAS.
- S. Park, Y. Lee, Y. S. Kim, H. M. Lee, J. H. Kim, I. W. Cheong and W.-G. Koh, Colloids Surf., A, 2014, 450, 46–51 CrossRef CAS.
- J. Xu, H. Han, L. Zhang, X. Zhu, X. Jiang and X. Z. Kong, RSC Adv., 2014, 4, 32134–32141 RSC.
- H. Han, S. Li, X. Zhu, X. Jiang and X. Z. Kong, RSC Adv., 2014, 4, 33520–33529 RSC.
- X. Jiang, X. Zhu, A. A. Arnold, X. Z. Kong and J. P. Claverie, Ind. Eng. Chem. Res., 2017, 56, 2993–2998 CrossRef CAS.
- S. Anwar, D. Pinkal, W. Zajaczkowski, P. von Tiedemann, H. Sharifi Dehsari, M. Kumar, T. Lenz, U. Kemmer-Jonas, W. Pisula, M. Wagner, R. Graf, H. Frey and K. Asadi, Sci. Adv., 2019, 5, eaav3489 CrossRef CAS PubMed.
- E. L. Papadopoulou, J. A. Heredia-Guerrero, M. I. Vázquez, J. Benavente, A. Athanassiou and I. S. Bayer, Polymer, 2017, 120, 255–263 CrossRef CAS.
- E. L. Papadopoulou, F. Pignatelli, S. Marras, L. Marini, A. Davis, A. Athanassiou and I. S. Bayer, RSC Adv., 2016, 6, 6823–6831 RSC.
- D. Crespy, M. Stark, C. Hoffmann-Richter, U. Ziener and K. Landfester, Macromolecules, 2007, 40, 3122–3135 CrossRef CAS.
- W. He, M. Parowatkin, V. Mailänder, M. Flechtner-Mors, R. Graf, A. Best, K. Koynov, K. Mohr, U. Ziener, K. Landfester and D. Crespy, Biomacromolecules, 2015, 16, 2282–2287 CrossRef CAS PubMed.
- Q. Luo, Y. Wang, Z. Chen, P. Wei, E. Yoo and E. Pentzer, ACS Appl. Mater. Interfaces, 2019, 11, 9612–9620 CrossRef CAS PubMed.
- D. Crespy and K. Landfester, Beilstein J. Org. Chem., 2010, 6, 1132–1148 CrossRef CAS PubMed.
- A. Elzayat, I. Adam-Cervera, O. Álvarez-Bermúdez and R. Muñoz-Espí, Colloids Surf., B, 2021, 203, 111764 CrossRef CAS PubMed.
- S. K. Pramanik, U. Pal, P. Choudhary, H. Singh, R. J. Reiter, A. Ethirajan, S. Swarnakar and A. Das, ACS Appl. Bio Mater., 2019, 2, 5218–5226 CrossRef CAS.
- O. Álvarez-Bermúdez, I. Adam-Cervera, A. Aguado-Hernándiz, K. Landfester and R. Muñoz-Espí, ACS Sustainable Chem. Eng., 2020, 8, 17956–17966 CrossRef.
- S. Seneca, S. K. Pramanik, L. D'Olieslaeger, G. Reekmans, D. Vanderzande, P. Adriaensens and A. Ethirajan, Mater. Chem. Front., 2020, 4, 2103–2112 RSC.
- E. P. Barros Silva Soares de Souza, G. Trindade, M. V. Lins Dantas Gomes, L. A. Santos Silva, R. Grespan, L. J. Quintans Junior, R. L. Cavalcanti de Albuquerque Júnior, S. Shanmugan and A. Antunes de Souza Araújo, Food Chem. Toxicol., 2020, 135, 110958 CrossRef CAS PubMed.
- D. Bresolin, V. Mazurek, A. Valério, C. Sayer, P. H. H. de Araújo and D. de Oliveira, Eur. Polym. J., 2018, 108, 529–535 CrossRef CAS.
- F. Wang and C. E. Diesendruck, Polym. Chem., 2017, 8, 3712–3720 RSC.
- P. J. Peruzzo, P. S. Anbinder, O. R. Pardini, J. R. Vega and J. I. Amalvy, Polym. J., 2012, 44, 232–239 CrossRef CAS.
- W. D. Vilar, Chemistry and Technology of Polyurethanes, Rio deJaneiro Vilar Consultoria Tecnica Ltd, 2002 Search PubMed.
- A. Chen, D. Wu and C. S. Johnson, J. Am. Chem. Soc., 1995, 117, 7965–7970 CrossRef CAS.
- M. Urbańczyk, D. Bernin, A. Czuroń and K. Kazimierczuk, Analyst, 2016, 141, 1745–1752 RSC.
- J.-G. Rosenboom, J. De Roo, G. Storti and M. Morbidelli, Macromol. Chem. Phys., 2017, 218, 1600436 CrossRef.
- D. Crespy and K. Landfester, Soft Matter, 2011, 7, 11054–11064 RSC.
- C. Bohlender, K. Landfester, D. Crespy and A. Schiller, Part. Part. Syst. Charact., 2013, 30, 138–142 CrossRef CAS.
- M. Takasu and H. Kawaguchi, Colloid Polym. Sci., 2005, 283, 805–811 CrossRef CAS.
- Q. Zhang, Y. Shi, X. Zhan and F. Chen, Colloids Surf., A, 2012, 393, 17–26 CrossRef CAS.
- L. Torini, J. F. Argillier and N. Zydowicz, Macromolecules, 2005, 38, 3225–3236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information of polyurea/polyurethane nanocapsules (FTIR, 1H-NMR, 1H-NMR DOSY spectra). See DOI: 10.1039/d1py00649e |
| This journal is © The Royal Society of Chemistry 2021 |