
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the molecular mechanisms of proteolysis of SARS-CoV-2 Mpro by QM/MM computational methods†
Katarzyna
Świderek
 * and
Vicent
Moliner
*
* and
Vicent
Moliner
*
Departament de Química Física i Analítica, Universitat Jaume I, 12071 Castelló, Spain. E-mail: swiderek@uji.es; moliner@uji.es
First published on 25th June 2020
Abstract
SARS-CoV-2 Mpro is one of the enzymes essential for the replication process of the virus responsible for the COVID-19 pandemic. This work is focused on exploring its proteolysis reaction by means of QM/MM methods. The resulting free energy landscape of the process provides valuable information on the species appearing along the reaction path and suggests that the mechanism of action of this enzyme, taking place in four steps, slightly differs from that of other cysteine proteases. Our predictions, which are in agreement with some recently published experimental data, can be used to guide the design of COVID-19 antiviral compounds with clinical potential.
Introduction
Novel severe acute respiratory syndrome – coronavirus 2 (SARS-CoV-2) has been identified as the virus responsible for COVID-19, the name designated to the global coronavirus 2019 pandemic disease emerged in late December 2019. At present there are no vaccines or antiviral drugs available for the prevention or treatment of COVID-19 infections. The main coronavirus protease (SARS-CoV-2 Mpro, also called 3CLpro) plays a key role in processing the polyproteins that are translated from the viral RNA in the replication of SARS-CoV-2 virus. Thus, inhibiting the activity of this enzyme would block the viral life cycle. In addition, a conserved feature of SARS-CoV-2 Mpro is its specificity in cleaving proteins after the Gln residue, a characteristic observed in other coronavirus proteases but not in human enzymes.1 Consequently, inhibitors of SARS-CoV-2 Mpro shouldn't present toxic effects on humans, thus becoming an excellent target to synthesize new anti-viral drugs. Within this context, the existence of recently solved X-ray structures of SARS-CoV-2 Mpro,1–3 opens the door for computer simulations focused on understanding how this enzyme works, which can assist in the design of new compounds with higher and more selective inhibitory activity.SARS-CoV-2 Mpro is a cysteine protease (CP) with an active site catalytic dyad (Cys145/His41) similar to other CPs. It is proposed that the imidazole group of His polarizes and activates the SH group of Cys to form a highly nucleophilic CysS−/HisH+ ion pair that would react with the substrate.4 In this regard, according to our recent QM/MM study on the proteolysis reaction catalyzed by the cruzain CP, the proton from the protonated HisH+ is transferred to the N atom of the scissile peptide bond after the Cys attacks the carbonyl carbon atom of the peptide.5 After this acylation step, the recovery of the enzyme in the following deacylation stage would be assisted by a water molecule activated by the His.
The present paper is focused on exploring the proteolysis reaction catalyzed by SARS-CoV-2 Mpro by means of multiscale QM/MM molecular dynamics (MD) simulations. This appears to be the first QM/MM study revealing the mechanism of action of SARS-CoV-2 Mpro at the atomistic level. The polypeptide Ac-Val-Lys-Leu-Gln-ACC (ACC is the 7-amino-4-carbamoylmethylcoumarin fluorescent tag) was used as the substrate because rate constants have been recently reported.6 Then, a valuable comparison between our computational results and experimental data will allow the validation of our predictions. The structure of SARS-CoV-2 Mpro and a schematic representation of the reaction are depicted in Fig. 1. As shown, the full dimer will be considered for the simulations of the proteolysis reaction taking place in one of the active sites.
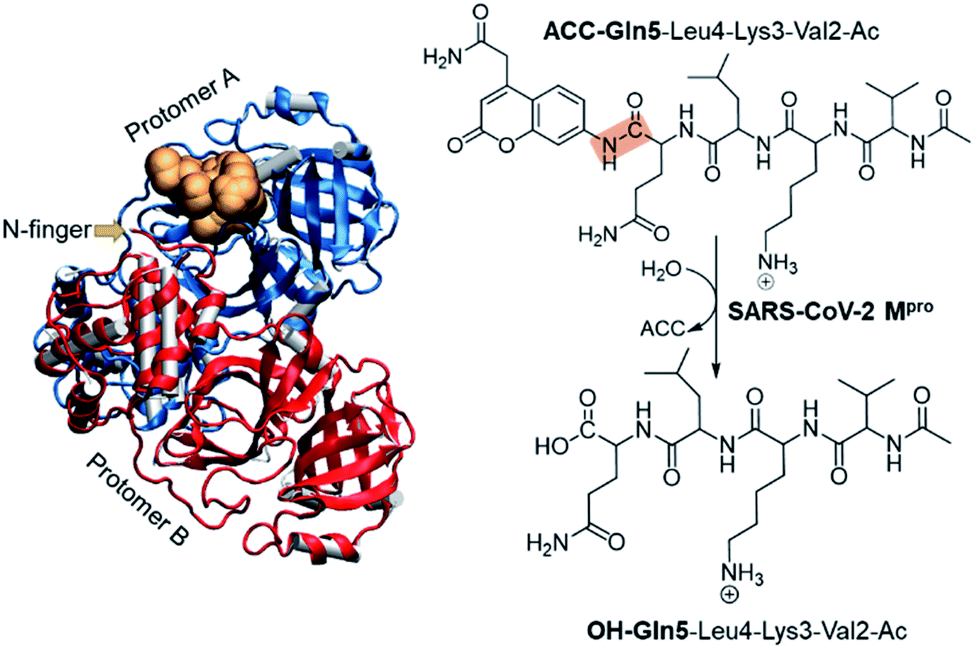 | ||
| Fig. 1 Structure of dimer SARS-CoV-2 Mpro and the substrate in an active site (gold balls), together with the schematic representation of the SARS-CoV-2 Mpro catalyzed reaction. The scissile peptide bond between Gln5 and ACC is highlighted in orange. | ||
Computational methods
The molecular system was prepared based on the crystal structure of wild type Mpro in complex with a peptidyl Michael acceptor inhibitor (PDB ID 6LU7).2 The inhibitor was replaced by the polypeptide Ac-Val-Lys-Leu-Gln-ACC by means of the Discovery Studio Visualizer program.7 In particular, the side-chains of the P1–P4 amino acids of the inhibitor were mutated to the corresponding residues of the selected polypeptide while the ACC group was manually generated.Once the fully solvated SARS-CoV-2 Mpro dimer was set up and equilibrated by means of classical molecular dynamics (MD) simulations (Fig. S1†), QM/MM free energy surfaces (FESs) were obtained, in terms of potentials of mean force (PMFs), for every step of the reaction using the umbrella sampling approach8 combined with the weighted histogram analysis method (WHAM).9 The semiempirical AM1 (ref. 10) method and the density functional M06-2X11 were selected to describe the QM sub-set of atoms (77 atoms, including ACC and Gln5 of the substrate, the side chain of His41, full Cys145 residue and part of the backbone of its preceding and posterior residues, Ser144 and Gly146, respectively. See Fig. S2†), while the protein and solvent water molecules (71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 819 atoms) were treated with the AMBER12 and TIP3P13 force fields, respectively (see the electronic supplementary information† for details). It is important to point out that despite limitations of some standard DFT functionals for modelling sulfur nucleophiles with covalent reactivity have been identified, M06-2X has been revealed to be reasonably accurate.14 This is in good agreement with our previous studies of the catalysis and inhibition of different cysteine proteases carried out with this hybrid functional and available experimental data.5,15,16
819 atoms) were treated with the AMBER12 and TIP3P13 force fields, respectively (see the electronic supplementary information† for details). It is important to point out that despite limitations of some standard DFT functionals for modelling sulfur nucleophiles with covalent reactivity have been identified, M06-2X has been revealed to be reasonably accurate.14 This is in good agreement with our previous studies of the catalysis and inhibition of different cysteine proteases carried out with this hybrid functional and available experimental data.5,15,16
Results and discussion
Analysis of the FESs (Fig. S3 and S4†) shows that both the acylation and the deacylation reactions take place in a stepwise manner (Fig. 2). Thus, according to the most favorable reaction path, as depicted in Scheme 1, first a proton is transferred from Cys145 to His41 concomitant with the nucleophilic attack on the carbonyl carbon atom of the peptide bond by the sulfur atom of Cys145, thus leading to a thiohemiketal (THA) intermediate, E-I1. Then, the cleavage of the peptide bond is assisted by the proton transfer from the protonated His41 to the nitrogen atom of the substrate, forming the acyl–enzyme complex intermediate E-I2. Once the first product of the reaction, ACC, is released from the active site, an activated water molecule attacks the carbonyl carbon atom of the Gln5 of the peptide, concomitant with the proton transfer to His41; E-I3. The covalent bond between Cys145 and the peptide in this protonated THA intermediate is then broken to release the second product species of the reaction in the last step. Details of the active site in the key states of the reaction are depicted in Fig. 3.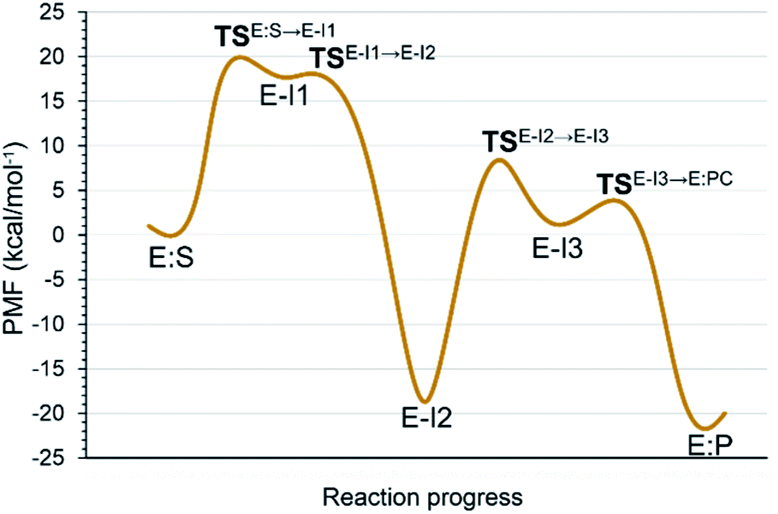 | ||
| Fig. 2 M06-2X:AM1/MM free energy profile of the proteolysis reaction catalyzed by SARS-CoV-2 Mpro. | ||
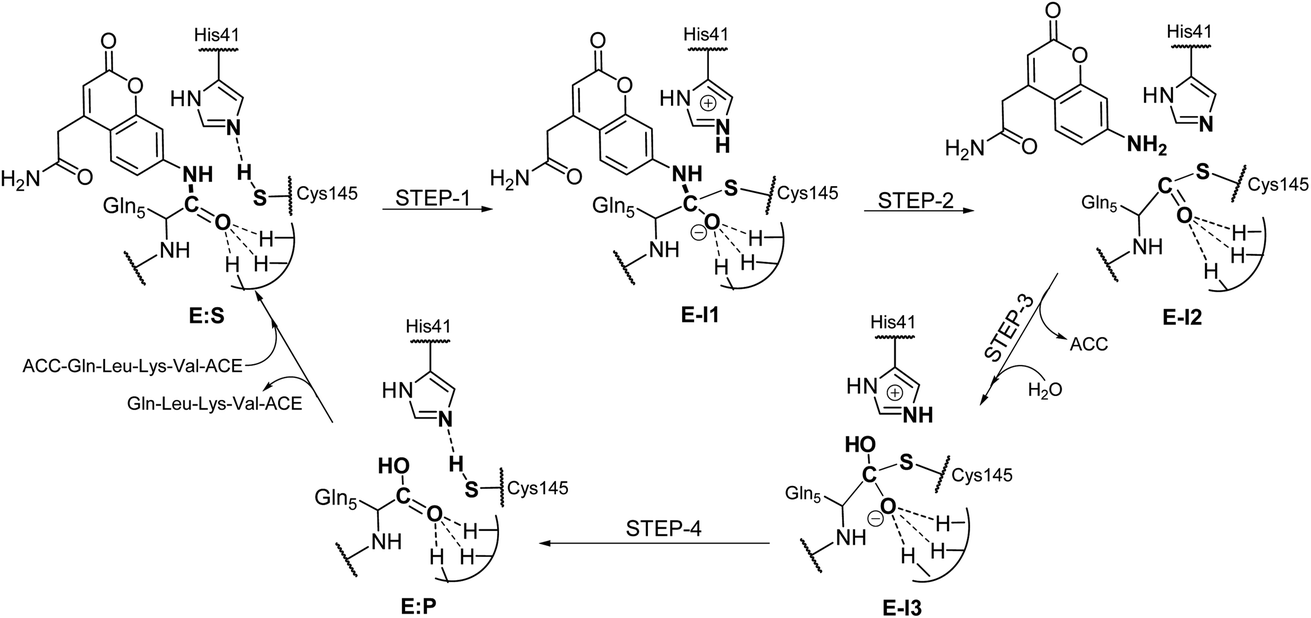 | ||
| Scheme 1 Molecular mechanism of the proteolysis catalyzed by SARS-CoV-2 Mpro as deduced from QM/MM FESs. | ||
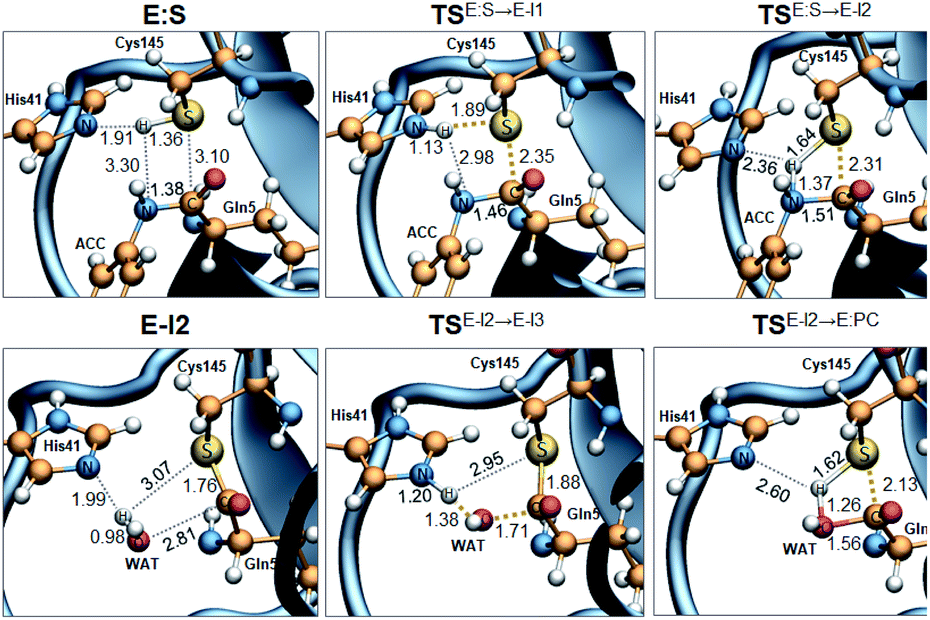 | ||
| Fig. 3 M06-2X/MM optimized structures of the key states of the acylation (top panels) and deacylation (bottom panels): E:S, rate determining TS of the acylation TSE:S→E-I1, TSE:S→E-I2, TSE-I2→E-I3, E-I2, rate determining TS of the deacylation TSE:I2→E-I3, and TSE:S→E-I2. Distances are in Å. | ||
The full free energy landscape of the proteolysis reaction is shown in Fig. 2 and is derived from the M06-2X:AM1/MM free energy surfaces explored for the acylation and the diacylation step (Fig. S3 and S4,† respectively). It is important to point out that the analysis of the full reaction energy profile considered as the sum of two consecutive processes relies on the idea that the product of the acylation step, when ACC species is released, can be considered as the initial state for the following diacylation step. This is a good approximation if the exploration of the energy landscape of multi-step reactions is carried out in terms of free energy surfaces using statistical simulations. According to the results, the rate limiting step of the acylation step is determined by TSE:S→E-I1, with a free energy barrier of 19.9 kcal mol−1 computed at 310 K. This value is in very good agreement with the value that can be derived from the rate constant reported by Rut et al., 19.4 kcal mol−1.6 The kinetics of the deacylation step is governed by the TSE-I2→E-I3, as in the histidine-assisted thiomethanolysis of formamide and the hydrolysis of the resulting thioester in solution.17 The resulting free energy barrier, 22.8 kcal mol−1, is close to the 26.6 kcal mol−1 computed in our previous QM/MM study for the deacylation step of the cruzain CP,5 even though in SARS-CoV-2 Mpro the reaction takes place in two steps through a protonated THA E-I3, and a concerted path was located in cruzain. Anyway, E-I3 and specially E-I1, must be considered as metastable intermediates, considering the low energy barriers required for their decomposition. In fact, as observed in Fig. 3, the peptide bond is already elongated in TSE:S→E-I1 with respect to that of E:S, although the proton transfer from His41 to the scissile peptide bond is not involved in this transition state (H⋯N distance = 2.98 Å). The E-I1 → E-I2 step can be defined as a shoulder in the free energy profile thus suggesting an acylation reaction taking place through an almost concerted but very asynchronous mechanism. Downhill trajectories starting from TSE:S→E-I1 can end in E-I2 at 310 K. The concerted processes for the acylation and the deacylation reactions have been also explored and located (see optimized TSE:S→E-I2 and TSE-I2→E:P structures in Fig. 3 and FESs in Fig. S5†) providing significantly higher activation energies; 25.6 and 40.6 kcal mol−1, respectively. Regarding the thermodynamics, a significantly exergonic process is obtained in both the acylation (−18.7 kcal mol−1) and the deacylation (−21.3 kcal mol−1) steps.
Further insights into our results can be provided by comparison with previous studies on other CPs. First, according to the present results, the most stable protonation state of the Cys145/His41 dyad in E:S (and in E:P) corresponds to that where both residues are neutral (Fig. 3), in contrast with previous computational studies of the proteolysis5 and inhibition15,16,18 of other CPs. The Cys145−/His41+ ion pair is located in high energy regions of the FESs of the first and last steps (Fig. S3†). Analysis of the structures of E:S and those recently derived from X-ray diffraction studies of SARS-CoV-2 Mpro in complex with different inhibitors,1–3 suggests that the absence of a conserved residue (Asp/Glu) interacting with the δN atom of His, thus modulating its pKa, can be the origin of this singular behavior of SARS-CoV-2 Mpro. The presence of a stable THA intermediate during the acylation step in CPs is also a question of debate. While this has been supported by X-ray diffraction studies,19 its presence has been questioned by others,20 including ourselves.5 Moreover, previous attempts to explore the viability of the inhibition of CPs, such as cruzain by peptidyl halomethyl ketones,15 reveal that a stepwise mechanism involving the formation of a THA intermediate was unsuccessful. Nevertheless, despite being a metastable intermediate as discussed above, it appears that the presence of a THA (E:I1) in the active site of SARS-CoV-2 Mpro is favored by strong hydrogen bond interactions with an “oxyanion hole” formed by Gly143, Ser144 and Cys145 (Table S3†). A THA has been also detected in a recent study of the inhibitory reaction of SARS-CoV-2 Mpro by α-ketoamide inhibitors, stabilized by the same triad of residues.1 Regarding the deacylation step in CPs, there is also controversy over its mechanism. Thus, while some authors have proposed that it occurs in a concerted manner,5,21 others support the presence of an intermediate similar to the protonated THA intermediate E-I3.17 As in the case of E-I1, the “oxyanion hole” would stabilize E-I3 and favor a step-wise deacylation process (Fig. S4 and Table S4†).
Finally, the specific protein–substrate interactions have been analyzed, to provide further results to guide the design of COVID-19 antiviral compounds. Fig. 4, together with Fig. S6 and S7,† shows the main interactions between the individual fragments of the substrate and the residues of each sub-site of the binding pocket in the E:S non-covalent reactant complex. First conclusion that can be derived from these results is that Gln is the residue with the largest amount of favorable interactions with the enzyme thus becoming the most important amino acid for substrate recruitment. This conclusion, in agreement with the specificity of SARS-CoV-2 Mpro in cleaving proteins after the Gln residue, can pave the way for future inhibitor design. It appears that these interactions clearly dictate the favorable orientation of the peptide in the active site for the proteolysis to take place, including the interaction with the catalytic Cys145 whose sulfur atom is well oriented towards the carbon atom of the scissile peptide bond (Fig. 3). Interactions with Asn142 and His163, the later through a strong hydrogen bond with the oxygen atom of amide group of Gln5, appear to be relevant (Table S3 and S4†). Asn142 also interacts with the previous residue of the substrate and, to some extent, with the next residue (ACC and Leu4, respectively). Interestingly, another protein amino acid that interacts with these three fragments of the peptide is Ser1, which belongs to the N finger of the other protomer. This terminal group, located on the bottom of the S1 pocket establishes two strong hydrogen bond interactions with the Glu166 of the protomer A. This linkage is possibly responsible for the depth of S1 thus limiting the size of substituents at this position in future inhibitor design. These results, apart from supporting the fact that an individual SARS-CoV-2 Mpro monomer is inactive, are in agreement with previous structural studies on SARS-CoV Mpro in other coronaviruses that also pointed out the relevance of Glu166, Phe140 and the N terminus of the partner protomer in Mpro dimerization.22–24 Residues Phe140, Leu141, Asn142, Glu166, His163, His172 and Ser1-B were identified to be involved in the S1 subsite in the analysis of the X-ray structure of SARS-CoV-2 Mpro covalently bound to the N3 Michael acceptor inhibitor,2 and residues Phe140, His163 and Glu166 in the X-ray structure of SARS-CoV-2 Mpro covalently bound to α-ketoamide inhibitors.1 Other residues of this and the rest of sub-sites identified in previous structural studies do not appear to be involved in significant stabilizing interactions with the rest of amino acids of the peptide used in the present study. In addition, even though it has been demonstrated that the inhibition of CPs is dependent on the interactions between the peptidic framework (the P2) of the inhibitor and the S2 pocket of the enzyme,16,25 this is probably not the case in SARS-CoV-2 Mpro where S2 is a small hydrophobic pocket without possibly strong hydrogen bond interactions (Fig. 4), same as the S4 sub-site, as already pointed out in previous structural studies.2,3 Finally, the S3 sub-site is completely exposed to the solvent and the three significant interactions with the Lys3 of the substrate (Met165, Glu166 and Gln189) are established with the peptide backbone atoms.
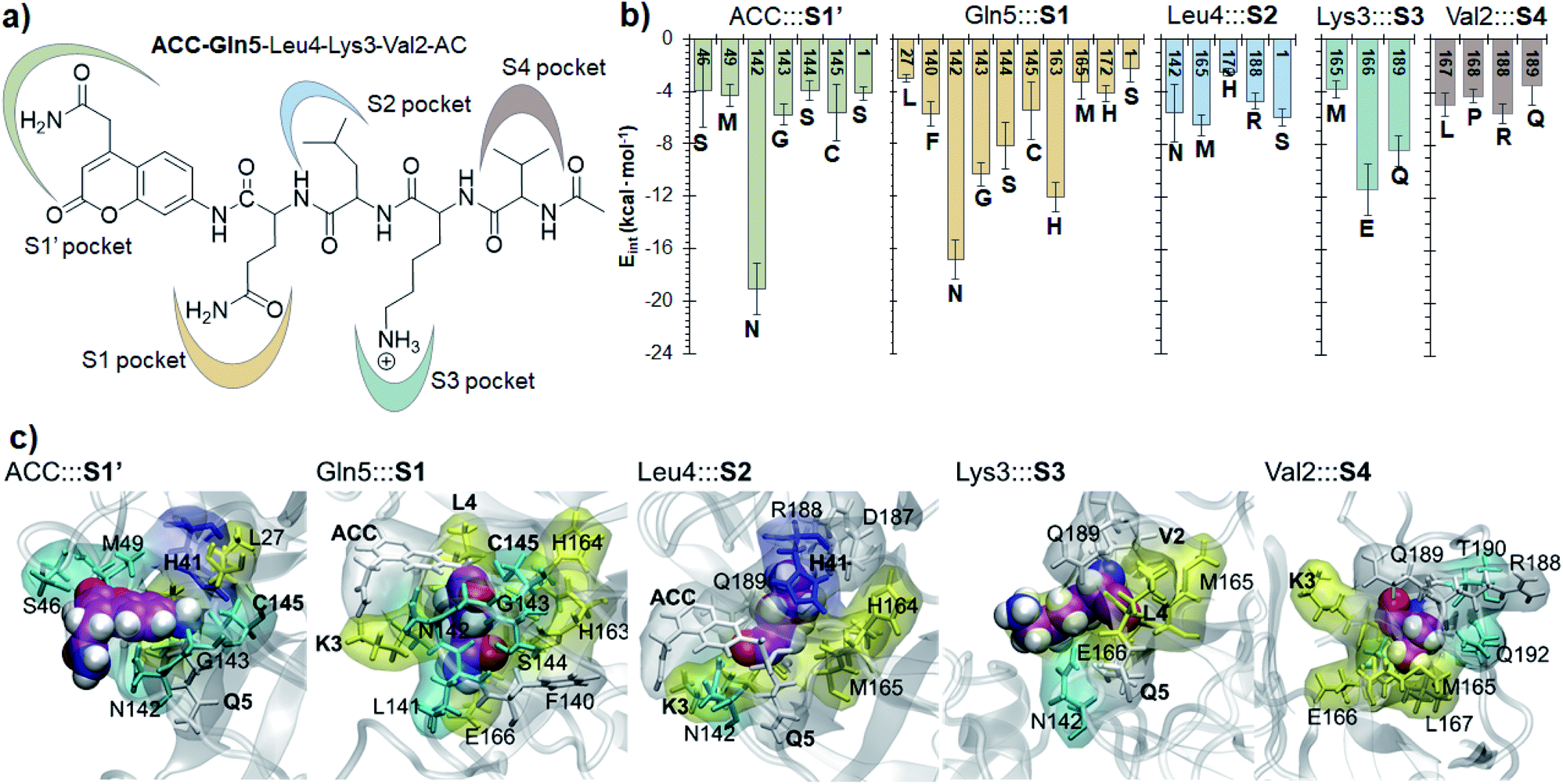 | ||
| Fig. 4 Protein-peptide interactions in E:S. (a) Definition of the protein subsites; (b) main average interaction energies (electrostatic plus Lennard–Jones, larger than 3 kcal mol−1) between the individual fragments of the substrate and its binding pocket obtained as an average over 1000 structures from the AM1/MM MD simulation. (c) Structural representation of each binding pocket for the E:S structure optimized at the M06-2X/MM level. Each fragment of the substrate is presented as spheres. Residues in yellow belong to the extended β-sheet motif, the residues of β-turns are indicated in cyan, residues of the coil are in white and residues belonging to the 310-helix are indicated in blue. | ||
Conclusions
We have described the SARS-CoV-2 Mpro catalytic proteolysis mechanism at the atomistic level with high level DFT/MM methods, including the structures of the different states appearing along the most favorable reaction path. The predicted activation free energy of the rate limiting step is in very good agreement with recently reported kinetic data using the same peptidyl substrate, Ac-Val-Lys-Leu-Gln-ACC. Supported by X-ray diffraction structures, our results suggest noticeable differences between this and other CPs. They include the protonation state of the catalytic His41/Cys145 dyad, the presence of a THA intermediate in the acylation state, and the interactions that are established between the sub-sites of the protein and the different fragments of a natural peptidyl substrate. According to our results, it can be concluded that SARS-CoV-2 Mpro is capable of catalyzing the proteolysis reaction of peptide-like substrates by the combination of short distance and long distance substrate–enzyme interactions, basically in the S1 pocket.Considering that the reaction of inhibition of SARS-CoV-2 Mpro by covalent (peptidyl) irreversible SARS-CoV-2 Mpro inhibitors is equivalent to the acylation step of the proteolysis, the results presented in this study can be relevant for future developments of drugs for COVID-19. Thus, these covalent inhibitors must be designed to form a stable enzyme–inhibitor complex similar to the product of the acylation step (E-I2). Consequently, focus must be shifted not only on obtaining an exergonic process with low activation energy, but also on avoiding its hydrolysis, which would be equivalent to our study of the deacylation step (E-I2 → E:P). Then, apart from a reactive warhead, the interactions between the rest of the inhibitor and the different sub-sites of the binding pocket must be taken into account, which can be guided by the results derived from the present study.
Author contributions
The study was designed by K. Ś. and V. M., who discussed and analyzed the results, and wrote the manuscript. K. Ś. carried out all calculations. Both authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades (Grant PGC2018-094852-B-C21), the Spanish Ministerio de Ciencia e Innovación (Grant PID2019-107098RJ-I00), the Generalitat Valenciana (Grant AICO/2019/195 and SEJI/2020/007) and Universitat Jaume I (project UJI-A2019-04). KŚ thanks the MINECO for the Juan de la Cierva – Incorporación (ref. IJCI-2016-27503) contract. The authors acknowledge computational resources from the Servei d’Informàtica of Universitat Jaume I.References
- L. Zhang, et al. , Science, 2020, 368, 409–412 CAS.
- Z. Jin, et al. , Nature, 2020, 582, 289–293 CrossRef CAS.
- W. Dai, et al. , Science, 2020, 368, 1331–1335 CrossRef CAS.
- J. W. Keillor and R. S. Brown, J. Am. Chem. Soc., 1992, 114, 7983–7989 CrossRef CAS.
- K. Arafet, S. Ferrer and V. Moliner, ACS Catal., 2017, 7, 1207–1215 CrossRef CAS.
- W. Rut, K. Groborz, L. Zhang, X. Sun, M. Zmudzinski, R. Hilgenfeld and M. Drag, bioRxiv, 2020 DOI:10.1101/2020.03.07.981928.
- Dassault Systèmes BIOVIA, Discovery Studio Visualizer v.19, San Diego Search PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swendsen, P. A. Kollman and J. M. Rosenberg, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- W. E. I. Zhang, et al. , J. Comput. Chem., 2003, 24, 1999–2012 CrossRef.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- (a) E. Awoonor-Williams, W. C. Isley III, S. G. Dale, E. R. Johnson, H. Yu, A. D. Becke, B. Roux and C. N. Rowley, J. Comput. Chem., 2020, 41, 427–438 CrossRef CAS; (b) A. Voice, G. Tresadern, H. van Vlijmen and A. Mulholland, J. Chem. Inf. Model., 2019, 59, 4220–4227 CrossRef CAS.
- K. Arafet, S. Ferrer and V. Moliner, Biochemistry, 2015, 54, 3381–3391 CrossRef CAS.
- K. Arafet, F. V. González and V. Moliner, Chem.–Eur. J., 2020, 26, 2002–2012 CrossRef CAS.
- M. Strajbl, J. Florian and A. Warshel, J. Phys. Chem. B, 2001, 105, 4471–4484 CrossRef CAS.
- A. Silva, et al. , J. Chem. Inf. Model., 2020, 60, 1666–1677 CrossRef.
- J. Drenth, K. H. Kalk and H. M. Swen, Biochemistry, 1976, 15, 3731–3738 CrossRef CAS.
- (a) D. H. Wei, X. Q. Huang, J. J. Liu, M. S. Tang and C. G. Zhan, Biochemistry, 2013, 52, 5145–5154 CrossRef CAS; (b) K. Byun and J. L. Gao, J. Mol. Graphics Modell., 2000, 18, 50–55 CrossRef CAS.
- S. Ma, L. S. Devi-Kesavan and J. Gao, J. Am. Chem. Soc., 2007, 129, 13633–13645 CrossRef CAS.
- K. Anand, et al. , EMBO J., 2002, 21, 3213–3224 CrossRef CAS.
- H. Yang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13190–13195 CrossRef CAS.
- T. Hu, et al. , Virology, 2009, 388, 324–334 CrossRef CAS.
- (a) S. A. Gillmor, C. S. Craik and R. J. Fletterick, Protein Sci., 1997, 6, 1603–1611 CrossRef CAS; (b) E. Dunny, W. Doherty, P. Evans, J. P. G. Malthouse, D. Nolan and A. J. S. Knox, J. Med. Chem., 2013, 56, 6638–6650 CrossRef CAS; (c) X. Zhai and T. D. Meek, Biochemistry, 2018, 57, 3176–3190 CrossRef CAS; (d) S. Royo, S. Rodriguez, T. Schirmeister, J. Kesselring, M. Kaiser and and F. V. Gonzalez, ChemMedChem, 2015, 10, 1484–1487 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Computational methods, FF parameters for ACC, computed pKa values of titratable residues, details of active sites and QM-MM partitioning, M06-2X:AM1/MM FESs, list of key inter-atomic distances in key states along the reaction path optimized at M06-2X/MM, protein–substrate non-bonding interaction energies per residue, Cartesian coordinates of the QM sub-set of atoms of the rate limiting TSs and the concerted TS optimized at the M06-2X/6-31+G(d,p)/MM level, and full structures of E:S, E-I2 and E:P and rate limiting TSE:S→E-I1, TSE-I2→E-I3, TSE:S→E-I2 and TSE:I2→E:P optimized at the M06-2X/6-31+G(d,p)/MM level in PDB format. See DOI: 10.1039/d0sc02823a |
| This journal is © The Royal Society of Chemistry 2020 |