
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInhibitors of pantothenate synthetase of Mycobacterium tuberculosis – a medicinal chemist perspective
Amaroju Suresha,
Singireddi Srinivasaraoa,
Yogesh Mahadu Khetmalisa,
Shashidhar Nizalapurb,
Murugesan Sankaranarayananc and
Kondapalli Venkata Gowri Chandra Sekhar *a
*a
aDepartment of Chemistry, Birla Institute of Technology & Science-Pilani, Hyderabad Campus, Medchal District, Hyderabad-500078, Telangana, India. E-mail: kvgc@hyderabad.bits-pilani.ac.in; kvgcs.bits@gmail.com; Tel: +91 40 66303527
bSchool of Chemistry, UNSW, Australia, Sydney, NSW 2052, Australia
cMedicinal Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology & Science-Pilani, Pilani Campus, Pilani 333031, Rajasthan, India
First published on 7th October 2020
Abstract
Tuberculosis (TB), one of the most prevalent infections, is on the rise today. Although there are drugs available in the market to combat this lethal disorder, there are several shortcomings with the current drug regimen, such as prolonged treatment period, drug resistance, high cost, etc. Hence, it is inevitable for the current researchers across the globe to embark on new strategies for TB drug discovery, which will yield highly active low cost drugs with a shorter treatment period. To achieve this, novel strategies need to be adopted to discover new drugs. Pantothenate Synthetase (PS) is one such striking drug target in Mycobacterium tuberculosis (MTB). It was observed that the pantothenate biosynthetic pathway is crucial for the pathogenicity of MTB. Pantothenate is absent in mammals and needs to be obtained from dietary sources. Hence, the pantothenate biosynthesis pathway is an impending target for emerging new therapeutics to treat TB. Worldwide, several approaches have been implemented by researchers in the quest for these inhibitors such as high-throughput screening, simulating the reaction intermediate pantoyl adenylate, use of vibrant combinatorial chemistry, hybridization approach, virtual screening of databases, inhibitors based on the crystal structure of MTB PS, etc. The present review recapitulates current developments in PS inhibitors, important analogues of numerous metabolic intermediates, and newly established inhibitors with innumerable chemical structures.
Introduction
Tuberculosis (TB), triggered by the pathogenic bacteria, Mycobacterium tuberculosis (MTB), is the most fatal and widespread infectious disease in the world. When MTB attacks the lungs it develops into pulmonary TB and assaults other parts of the body resulting in extrapulmonary TB. Though several researchers are trying to develop new agents to combat this lethal disorder, this prehistoric hazard could not be diminished. It remains one of the main communal challenges, next to the human immunodeficiency virus (HIV). Globally in 2017, 10 million people were infected with TB, including 464![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 633 cases amongst people living with HIV. 1.3 million deaths occurred due to TB, and additionally, 0.3 million HIV-positive people also died.1 0.558 million people developed resistance to rifampicin (RR-TB) – the most effective first-line drug – and amongst these, 82% had multidrug-resistant TB (MDR-TB). Around 0.23 million deaths occurred due to MDR/RR-TB. MDR-TB occurs once the MTB strain becomes resistant to the most active antitubercular drugs, i.e., isoniazid and rifampin. Around 8.5% of MDR-TB cases have extensively drug-resistant TB (XDR-TB).1 XDR-TB arises when the MTB strain is resistant to isoniazid and rifampin as well as being resistant to one of the fluoroquinolones, and also to either amikacin, kanamycin or capreomycin, one of the second-line injectable drugs.
633 cases amongst people living with HIV. 1.3 million deaths occurred due to TB, and additionally, 0.3 million HIV-positive people also died.1 0.558 million people developed resistance to rifampicin (RR-TB) – the most effective first-line drug – and amongst these, 82% had multidrug-resistant TB (MDR-TB). Around 0.23 million deaths occurred due to MDR/RR-TB. MDR-TB occurs once the MTB strain becomes resistant to the most active antitubercular drugs, i.e., isoniazid and rifampin. Around 8.5% of MDR-TB cases have extensively drug-resistant TB (XDR-TB).1 XDR-TB arises when the MTB strain is resistant to isoniazid and rifampin as well as being resistant to one of the fluoroquinolones, and also to either amikacin, kanamycin or capreomycin, one of the second-line injectable drugs.
When MTB strain develops resistance to the entire first and second-line drugs, it transpires to totally drug-resistant TB (TDR-TB) or extremely drug-resistant TB (XDR-TB). After the whole genome sequencing of MTB was completed in 1998,2 TB Structural Genomics Consortium (TBSGC) is putting efforts to identify novel drug targets. TBSGC is a worldwide consortium whose objective is to decide the structures of over 400 probable targets from the MTB genome and scrutinize their structures in the milieu of functional information.3 Of the 185 distinctive targets from several biosynthetic pathways, 16 have protein database structures and 102 are in various phases of advancement at the TBSGC.4 Further, over 200 potential targets have been identified through the understanding of critical growth phase and alternative biosynthesis pathways during non-replicating persistent MTB.5
The prevailing drugs have a number of limitations, the most significant of them being the occurrence of resistance to drugs. No new anti-TB drugs have emerged over the past three decades for treating TB. The drug discovery process for TB has seen remarkable progress only in the last 10 years, with bedaquiline (TMC 207), a diarylquinoline, a novel adenosine triphosphate synthase inhibitor being approved for treatment of MDR TB.6 Currently, moxifloxacin and gatifloxacin (fluoroquinolones) are in phase III clinical development.7 Two novel compounds based on nitroimidazoles, PA-824 and delamanid (OPC-67683) are in phase III and approved in the European Union under the trade names pretomanid and deltyba respectively, for the treatment of both drug-susceptible and drug-resistant TB.8 Several new compounds viz., TBA-354 (nitroimodazole), SQ641 (capuramycin), SQ609 (dipiperidine), DC-159a (fluoroquinolone), BTZ043 (benzothiazinone) and CPZEN-45 (caprazene nucleoside) are in preclinical stage (Fig. 1).9
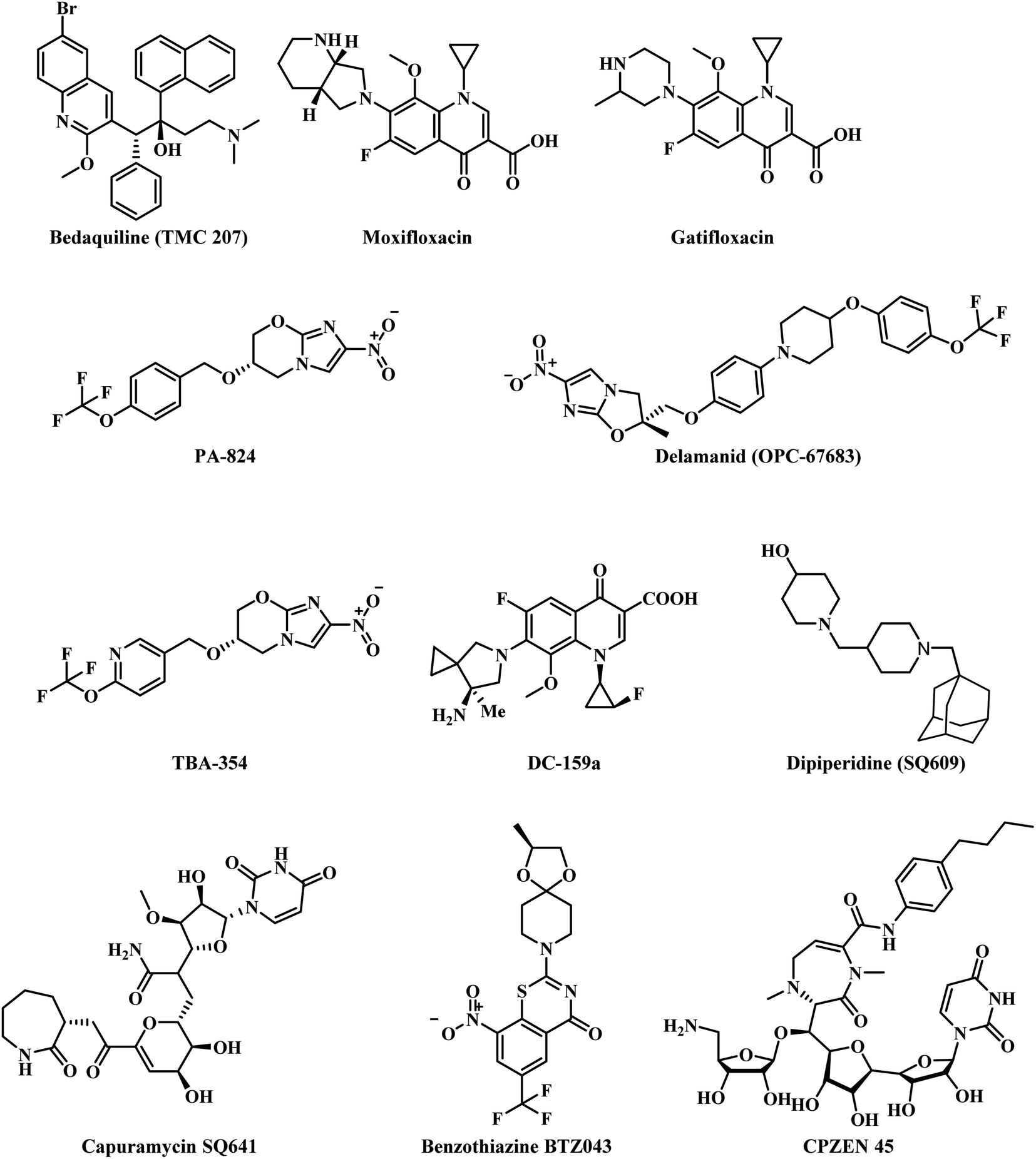 | ||
| Fig. 1 Various antitubercular compounds. | ||
Enzymatic assays often use either spectrophotometric/colorimetric (or) fluorometric (or) calorimetric (or) light scattering method of detection of a signal at a particular wavelength of appropriate electromagnetic radiation. Currently, many clinically used drugs are either inhibiting or antagonize the activity of enzymes involved in mediating the disease processes. Hence, understanding the exact mechanism of action of the target enzyme is very much challenging in the early phases of discovery and development of new chemical entities through extensive Structure–Activity Relationship (SAR) studies. In such a situation, through enzyme-based studies only, types of inhibition such as competitive inhibition, non-competitive inhibition, uncompetitive inhibition, allosteric inhibition, partial inhibition, tight-binding inhibition, time-dependent inhibition exhibited by the test compounds can be determined.10
It is crucial to have new chemical entities forthcoming during the novel drug discovery development. Screening of compound libraries to detect new leads is briskly increasing in TB drug development. Currently, several possible leads are being recognized by the high-throughput screening approach.11 Nevertheless, lack of acquaintance about the specific target(s) hinders lead advancement and further improvement.12 Hence, a target-based screening possibly offers an attractive solution to the constraint of phenotypic screening.
Innumerable approaches were used to overcome the problem of resistance in the past such as antibiotic combination rotation, using new chemical entities for already well-known targets, identification of targets which do not mutate etc.13 Currently, the most urgent goal of TB chemotherapy is to develop highly active, low-cost drugs with a shorter treatment regimen. Pantothenate (vitamin B5) is the precursor to coenzyme A, an essential cofactor that is mandatory in several intracellular processes viz., the metabolism of carbohydrates and fatty acids, cell signaling, synthesis of polyketides and non-ribosomal peptides. As a vitamin, the pathway of its synthesis is confined to plants, fungi, and microorganisms. Animals need to attain it from dietary sources. Consequently, the enzymes of the pathway are potential targets for novel drugs, including herbicides, fungicides and antimicrobial agents. The biosynthetic pathway of pantothenate involves four steps catalyzed by panB, panC, panD, and panE genes.14 PanC encodes the pantothenate synthetase, which catalyzes the ATP-dependent condensation of D-pantoate and β-alanine to form pantothenate.15 The full pantothenate biosynthetic pathway was only determined in the last 40 years. Although a few enzymes in the biosynthetic pathway were previously known, majority of them remained unidentified till 1980.12 The pantothenate biosynthetic pathway is well known in E. coli which comprises of four enzymatic steps16 (Fig. 2). Ketopantoate hydroxymethyl transferase (KPHMT),17 converts α-keto isovalerate into ketopantoate using 5,10-methylene tetrahydrofolate; subsequently, ketopantoate is reduced to pantoate by ketopantoate reductase (KPR)18 using NADPH as the hydrogen donor. In a separate branch, β-alanine is synthesized from L-aspartate by the enzyme L-aspartate-α-decarboxylase (ADC).19 Finally, pantothenate is produced in an ATP consuming condensation reaction between pantoate and β-alanine, catalysed by PS.16
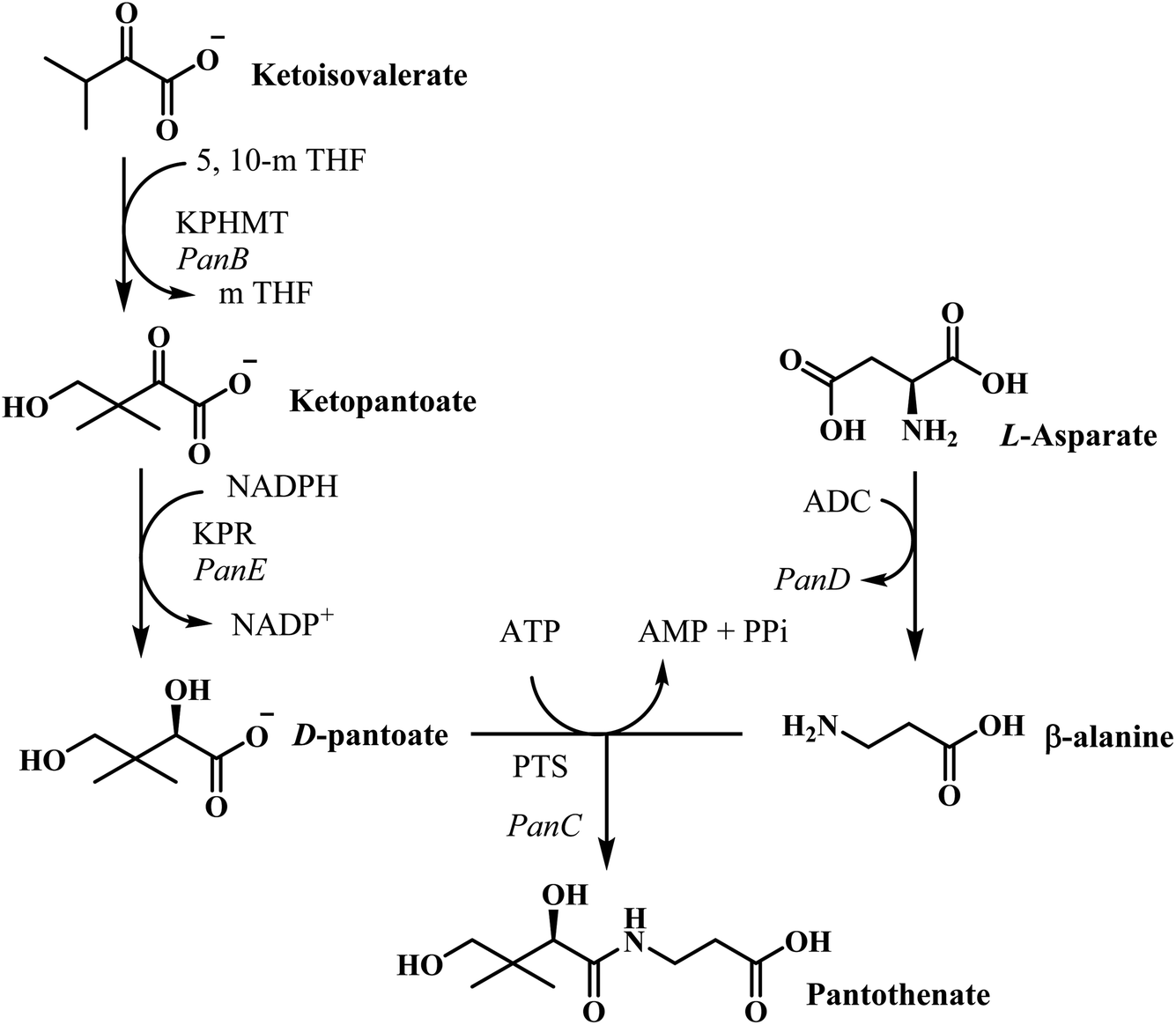 | ||
| Fig. 2 Pantothenate biosynthesis pathway in E. coli.22 The enzymes are KPHMT, ketopantoate hydroxymethyltransferase;20 KPR, ketopantoate reductase; ADC, aspartate decarboxylase;23 PS, pantothenate synthetase; 5,10-mTHF, 5,10-metylene tetrahydrofolate. | ||
Already in the year 1982, using E. coli strains auxotrophic for pantothenate, the genes panB (encoding KPHMT), panC (encoding pantothenate synthetase), and panD (encoding ADC) were physically mapped on the genome,16 which established the gene order panD, panC and panB. The panB and panC genes are next to each other from panD.14 All four enzymes from E. coli pantothenate biosynthetic pathway have been cloned and overexpressed, and their crystal structures have been solved.20,21
PS is the product of the panC gene which catalyzes the synthesis of pantothenate from D-pantoate and β-alanine with the hydrolysis of ATP into AMP and PPi.24 Pantothenate synthetase of Mycobacterium tuberculosis is a homodimer with a subunit molecular mass of 33 kDa and is the last enzyme in the pantothenate biosynthesis pathway. The kinetic mechanism of pantothenate synthetase was found to be Bi Uni Uni Bi Ping Pong, with ATP binding followed by D-pantoate binding, the release of PPi, binding of β-alanine, formation of pantoyl adenylate as a key intermediate followed by the release of pantothenate and AMP.25 Michaelis constants were 0.13, 0.8, and 2.6 mM for D-pantoate, β-alanine, and ATP, respectively and the turnover number, kcat, was 3.4 s−1.15 In the year 2004, Renjian Zheng et al., studied the significance and specific roles of active site conserved residues (His44, His47, Asn69, Gln72, Lys160 and Gln164) of MTB PS in the binding of substrates as well as in the formation and stabilization of pantoyl adenylate intermediate.25 Shuishu Wang and David Eisenberg initially proposed an alternative mechanism for the MTB PS enzyme to open and close the active site cavity, in which a flexible region acts as a gate to the active site cavity.26 They also proposed that non-reactive analogs of the intermediate may act as high affinity and specificity inhibitors of the PS.26 Later, the same authors determined the 3D crystal structure of M. tuberculosis PS enzyme complexed with AMP and showed that the phosphate group of AMP serves as an anchor for the binding of β-alanine. This structure confirms that binding of β-alanine in the active site cavity can occur only after the formation of the pantoyl adenylate intermediate as well as the flexible region (gate to the active site) is locked in an inactive form.27 Atsuko Satoh et al., during the year 2010, reported that the PS apoenzyme from Staphylococcus aureus adopts an open and relatively mobile structure, while the complex (PS and the reaction intermediate pantoyl adenylate) structure is closed and entirely rigid. They also suggested that pantoyl adenylate-based approach, in combination with acetate, would be ideal for novel compounds that selectively inhibit PS activity.28 In the year 2018, Bharati Pandey et al., studied the molecular mechanism of the decreased affinity of the PS enzyme for the substrate ATP caused by alanine mutations using molecular dynamics (MD) simulations and free energy calculations and found that alanine mutations cause distinct conformational changes in the ATP binding region.29
In this report, we described various compounds that were proven to be PS inhibitors and identified through several well-known approaches. PS is very much essential for the survival of MTB in the host.30,31 Apart from this, recently, it has been established that pyrazinamide, one of the widely used first-line antitubercular drug, also acts by inhibition of pantothenate biosynthesis. panD mutations observed among pyrazinamide resistant MTB strains proved that pyrazinamide also acts via PS inhibition.29–32 This discovery led the researchers to strategize this pathway as a potential target for TB drug discovery.33 Initially, PS inhibitors were primarily identified through target-based methods using fragment-based screening,34 rational design based on reaction intermediates,35 and unbiased screening of small compound libraries.36 Currently, due to advancement in technologies and application of conditional knockdown strategy, whole-cell target-based screening against PS was carried out, and the lack of these enzymes in mammals suggest the suitability of this enzyme for inhibitor design or selection.30,37 Based on these facts, PS may be considered as one of the potential antimycobacterial targets that may also be useful for the treatment of the non-replicating persistent forms of MTB.38
We categorise these into various sub-headings based on the way these compounds were discovered.
Pantoyl adenylate intermediate based PS inhibitors
Chris Abell's group was the first to report about the synthesis and identification of potential PS inhibitors.39 They designed, synthesized, and reported ten racemic ester/sulfamoyl analogues of pantoyl adenylate as potential inhibitors of PS. The sulfamoyls were 100 fold more active than the ester counterparts owing to the structural similarity to the pantoyl adenylate intermediate. Compound 1 (Table 1) emerged as the most active compound with Ki of 300 nM.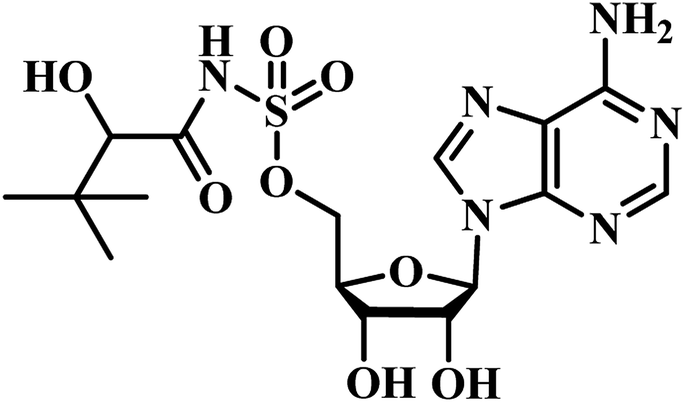
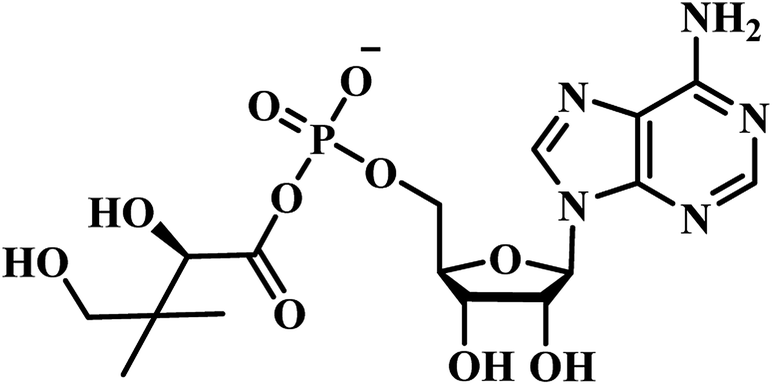
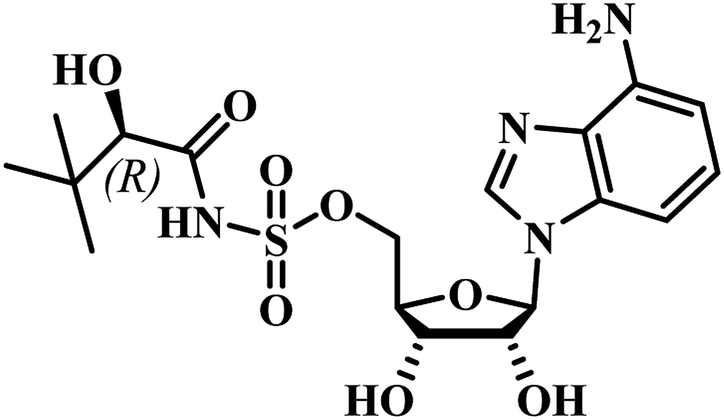
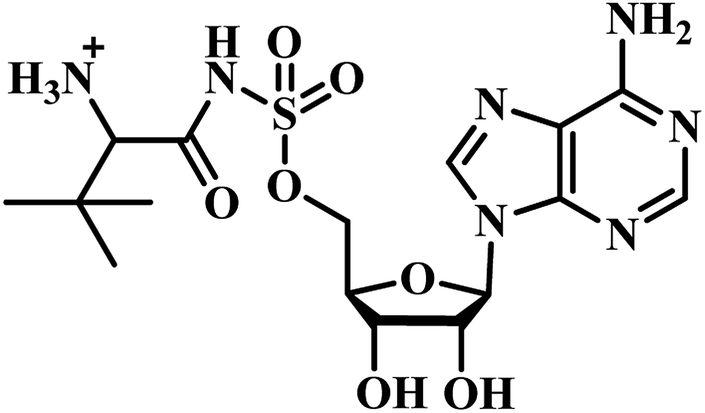
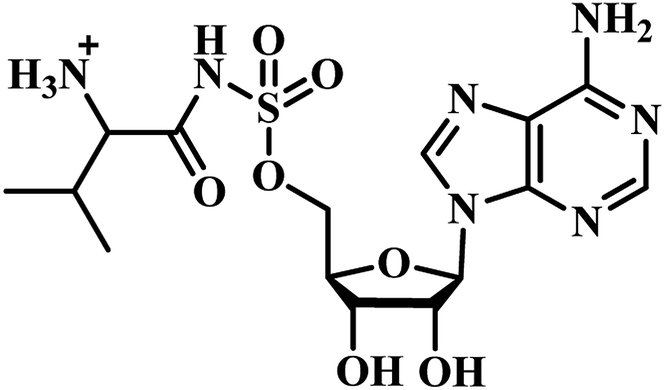
Qiao and researchers from China have synthesized five panC inhibitors that mimic the intermediate pantoyl adenylate.40 They synthesized the analogues with the C-2 stereo center retaining the same configuration (i.e., R) as the reaction intermediate 1. Though, the five compounds closely mimicked the pantoyl-adenylate, none of the compounds were exceptionally potent inhibitors. Ki for these compounds ranged from 0.3 to 3.0 μM with 3 being the most active compound.
Inspired by the nanomolar potency of sulfamoyl adenylate mimic of the salicyl adenylate intermediate in 2006, Abell's group synthesized three more derivatives (2, 4 and 5) (Table 1) based on the reaction intermediate, pantoyl adenylate 2 (Table 1), which binds firmly into the active site of the enzyme. Out of these three derivatives, compound 3, similar to intermediate 2, was found to be the most active compound with Ki 220 nM (Fig. 3).3
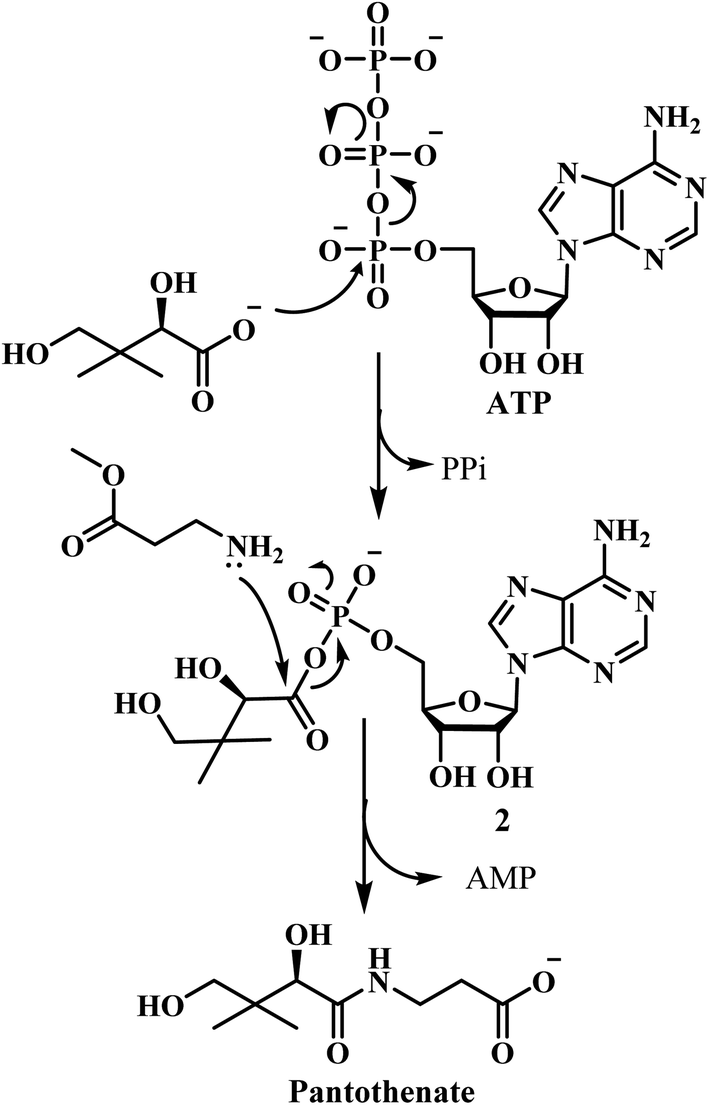 | ||
| Fig. 3 Steps involved in pantothenate synthesis. | ||
PS inhibitors identified through high throughput screening
High-throughput screening is a method for scientific experimentation, mainly utilized in drug discovery and is relevant to biology and chemistry. High-throughput screening facilitates screening a large number of compounds against more targets to generate hits, which in turn will help in identifying new leads, finally producing more excellent products. This technique was widely used in PS drug discovery, which is outlined below.In 2007, White and co-workers36 optimized and developed high throughput method for screening 2880 compounds based on the technique earlier reported by Zheng and Blanchard.14,15 Nafronyl oxalate emerged as the most potent compound with Ki 12 μM. However, the compound did not inhibit MTB growth in vitro but served as a prospective lead in the identification of novel drug targets.
In 2008, Petukhov et al., have reported derivatives of 5-tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamides as novel potent inhibitors of MTB PS.41 High-throughput screening of over ten thousand compounds suggested the importance of a tert-butyl group and absence of functional groups in positions 3 and 5 of the pyrazole ring for PS inhibitory activity of the active ligands. With these inputs handy, they synthesized thirteen derivatives and screened them for PS antagonism. They reported analogues with activity ranging from IC50 90 nM to 7.13 μM.41 They found that groups on the pyrazole ring are essential to fit into the active site of the receptor. Compound 6 (Table 2) emerged to be the most active of the series with IC50 90 nM. They found that hydrophobic substituents on benzene ring led to an increase in activity while polar and nonpolar groups on pyrazole ring resulted in a decrease in activity.41
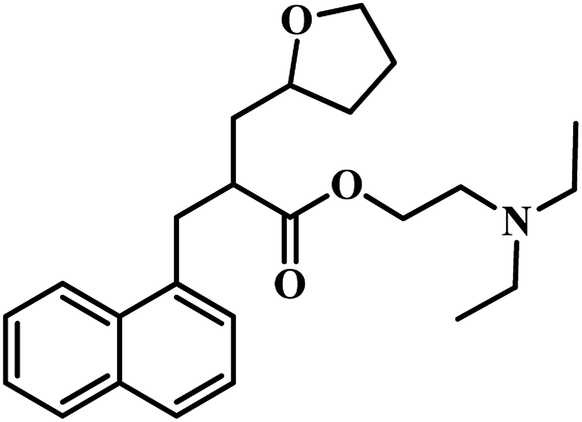
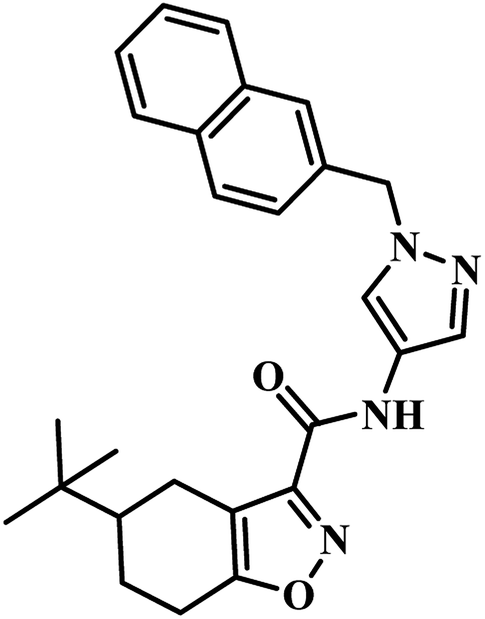
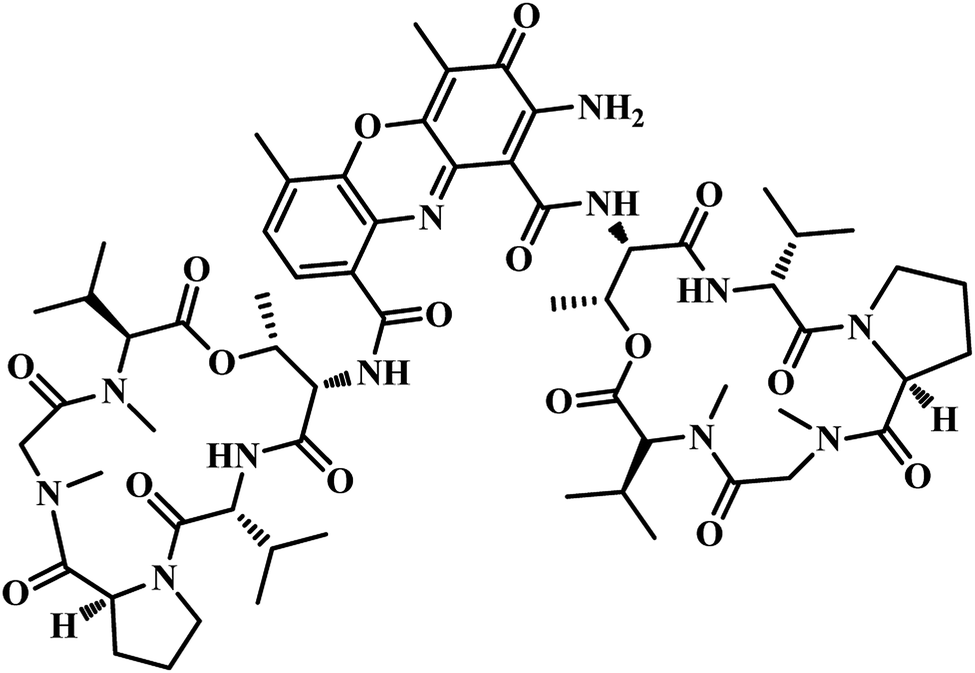
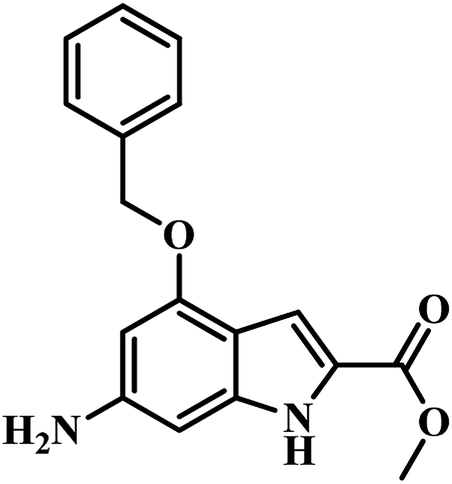
In 2011, Chunling Xiao group from China used a novel method and discovered new inhibitors based on the interaction between a lead inhibitor and ligands as evaluated by circular dichroism spectra and fluorescence methods. They followed the HTS model of MTB PS proposed by White et al., on a small library of 3112 compounds.36,42 Actinomycin D (ActD) emerged as a weak inhibitor of PS from the screening with IC50 of 250.72 μM. Based on the docking results of ActD onto the active site of MTB PS, they discovered the importance of the inner ester of the cyclopeptide for ActD inhibition of MTB PS. They generated a final pharmacophoric query which resulted in the identification of two potential candidates 7 and 8 (Table 2). The IC50 of most active compound 7 against MTB PS was 22.44 μM.42
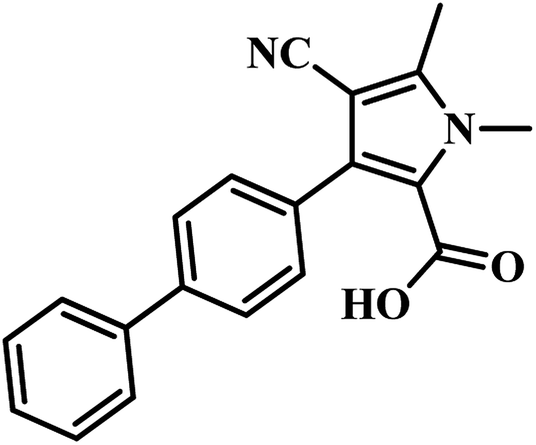
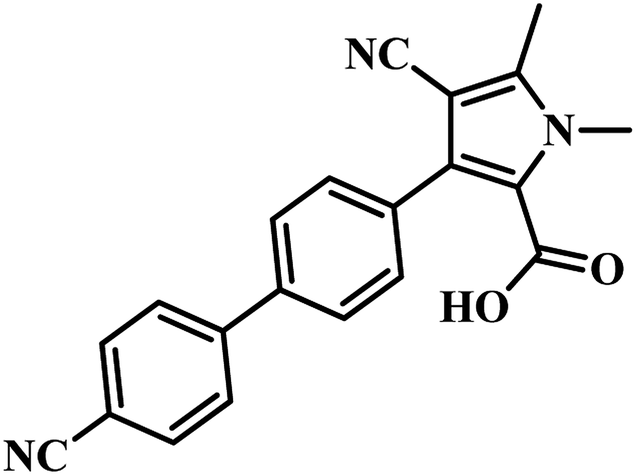
Tanya Parish and the group established an enzyme-based assay to identify inhibitors of panC. They optimized the process through HTS and screened an extensive library of compounds for activity. They screened two molecular libraries that they obtained from Eli Lilly. They screened a totally 27582 compounds from Lilly Strategic Screening Paradigm fourth iteration and a set of another 62651 compounds from generally diverse compounds (diversity fourth iteration). This led to the identification of 222 primary hits from the identified libraries. After further processing and going through various iterations, two compounds belonging to the chemical class of 3-biphenyl-4-cyanopyrrole-2-carboxylic acids emerged as the most potent compounds. Compounds 9 and 10 (Table 2) exhibited Ki of 174.1 and 297.1 nM, respectively. Further, both the compounds also inhibited the growth of live MTB in a manner consistent with panC inhibition with MIC 55 and 118 μM, respectively.31
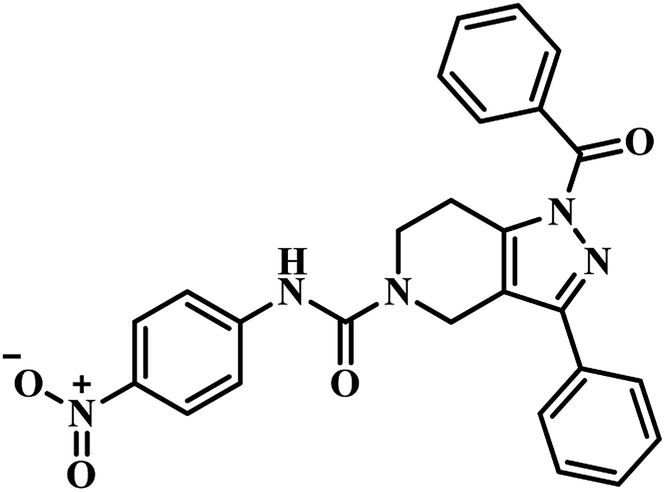
Sriram et al. have designed and synthesized 3-phenyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridine analogues from piperidin-4-one by five-step synthetic process and screened the compounds for MTB PS inhibition assay, in vitro anti-TB activity against MTB and cytotoxicity against RAW 264.7 cell line. They designed the compounds by HTS of their in-house (BITS-Pilani) database using glide extra precision docking and identified compound 11 (Table 2) as prospective lead, which exhibited inhibition of 60.6% at 100 μM against MTB PS. They synthesized a library of forty compounds based on the obtained lead, and among them, compound 12 (Table 2) was found to be the most active one, with IC50 21.8 μM against MTB PS. It inhibited MTB growth with MIC 26.7 μM and further was not cytotoxic at 50 μM against Raw 264.7 cell line.43
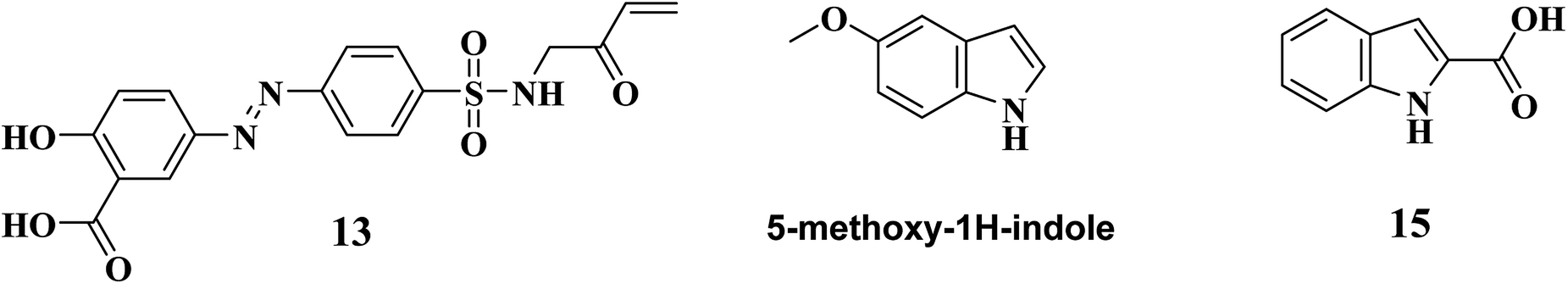
During the year 2018, Sayantan Pradhan and Chittaranjan Sinha reported various sulfonamide derivatives as PS inhibitors. (E)-2-hydroxy-5-((4-(N-(2-oxobut-3-en-1-yl)sulfamoyl)phenyl)diazenyl)benzoic acid, 13 (Fig. 4) emerged as the best PS inhibitor of MTB using high throughput screening technique. In their work, around a hundred and fifty four amide analogues were screened by Discovery Studio molecular docking programme. Pharmacophore generation was also done to recognize the binding method of inhibitors in the receptor active site. To observe the stability and flexibility of inhibitors, molecular dynamics (MD) simulation study has been done; Lipinski's rule of five protocol was also followed to screen drug-likeness, and ADMET (absorption, distribution, metabolism, excretion and toxicity) filtration was also used to value toxicity of the identified hits. DFT computation of optimized geometry and derivation of molecular orbitals was used to correlate the drug-likeness. They also proposed that the recognized hit, 13, may also bind to adenine–thymine region of tuberculosis DNA.44
 | ||
| Fig. 4 Structure of lead compounds that helped in identifying PS inhibitors through high throughput screening, fragment-growing and fragment-linking approach. | ||
Fragment-growing and fragment-linking approach based PS inhibitors
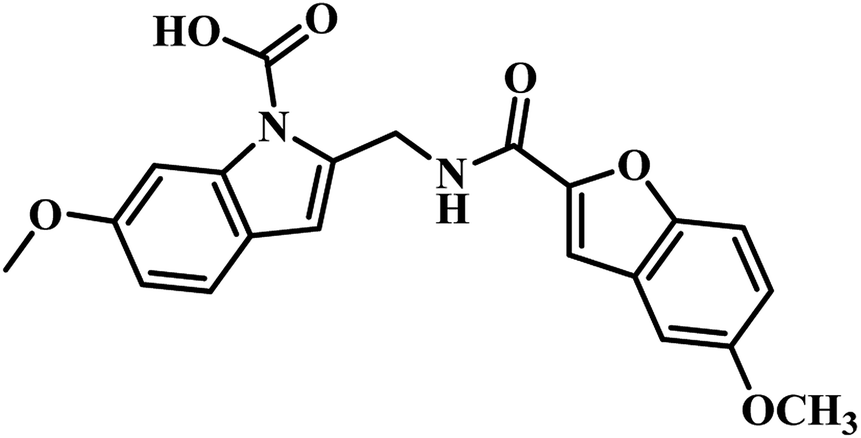
In 2009, Abell's group focused on developing novel PS inhibitors by combining fragment-growing and fragment-linking approach34 rather than developing analogues based on reaction intermediate. Initially, they identified the prominent binding modes of the key fragments, and this was followed by design, synthesis and docking of the compounds in the active site of the enzyme and the iterations were repeated to increase the ligand potency. They identified the compound 5-methoxy-1H-indole (Fig. 4) as the potential hit, and after further fragment growing and iterations, it resulted in compound 14 (Table 3) as the most active compound with Ki of 9 μM. In the subsequent year, Abell's group focused on identifying new leads for PS antagonism via inter-ligand Overhauser effect. Based on 5-methoxy-1H-indole, compound 15 (Fig. 4) was identified by their group from fragment screening against PS, and later on, they came up with compound 16 (Table 3) as the most potent PS inhibitor with Ki 5.4 μM.45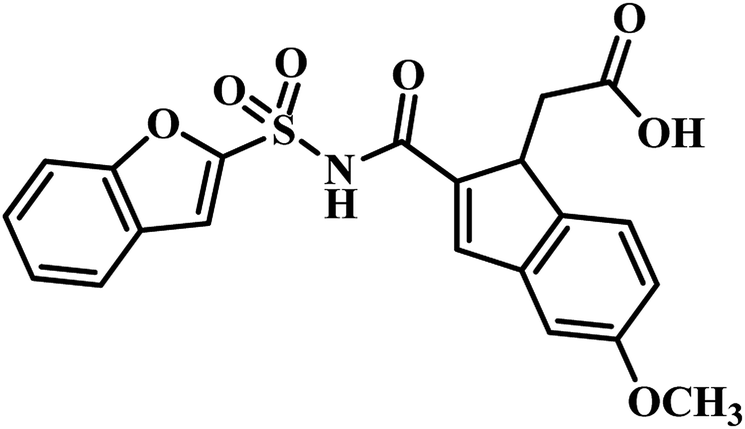

PS inhibitors identified through virtual screening
Virtual screening is a computational technique used to pick chemical systems which are expected to have a precise target. For instance, in the context of drug discovery, virtual screening involves looking for vast libraries of chemical systems and discover those systems which in all likelihood bind to a drug target.Kang and co-workers have used a virtual screening technique for identifying active MTB PS inhibitors. They designed new inhibitors against this target46 and docked the set of pyrazole-based inhibitors, reported by the Petukhov group into the enzyme active site. The obtained docking results were then scrutinized by molecular dynamics to identify the most probable binding mode of the compounds. The docking results were further processed with molecular mechanics energies combined with generalized Born (MM/GBSA) and molecular mechanics energies combined with Poisson Boltzmann surface area (MM/PBSA) continuum solvation methods. The results obtained were scrutinized to verify whether both procedures could clearly distinguish between active and inactive inhibitors. From the analysis, they concluded that the docking method using Gold or Glide correctly predicted the binding modes for the majority of the active inhibitors but the scoring methods (including GoldScore, ChemScore, Standard Precision) did not discriminate the active from the inactive ones.
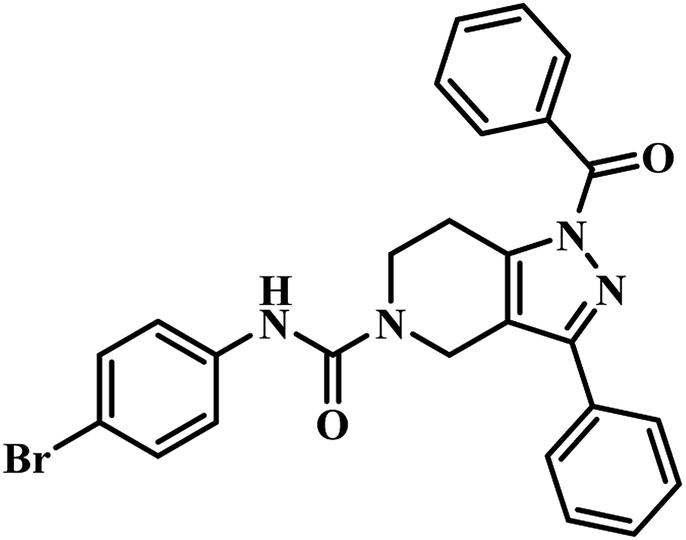
The same year in their subsequent work, they used the crystal structure of mycobacterial PS in complex with 2-(2-(benzofuran-2-yl-sulfonylcarbamoyl)-5-methoxy-1H-indol-1-yl) acetic acid inhibitor as a framework for virtual screening of known anti-TB compound database to search for new leads against this enzyme. The identified prime hit was synthetically tailor-made to obtain thirty novel compounds. The synthesized analogues were screened for MTB PS inhibition study, in vitro anti-TB activity, and cytotoxicity against RAW 264.7 cell line. The compounds showed IC50 in the range of 1.90–9.20 μM. Among the compounds tested, compound 17 (Table 3) was the most active one with IC50 1.90 μM against MTB PS, MIC of 4.53 μM against MTB, and did not exhibit cytotoxicity at 50 μM.47
E-pharmacophore based PS inhibitors
Ligand-based pharmacophore modeling and structure-based protein–ligand docking are both recognized as quite essential parts of drug discovery. Ligand-based technology, which includes 3D-pharmacophore modelling is useful for speedy screening of large compound databases. Shape-based approach, on the other hand, yields more diverse and essential target insights but is time-consuming. The e-pharmacophore technique achieves the advantages of each ligand and shape-based approaches by way of producing energetically optimized, structure-primarily based on pharmacophores that may be used to screen hundreds of thousands of compounds.48Sriram's group took advantage of structure-based e-pharmacophore modelling to identify various inhibitors of the MTB-PS enzyme based on the crystal structure co-crystallized with another inhibitor. Seven active compounds from varied structural classes were primarily identified. From these four molecules exhibited MTB PS activity with IC50 < 10 μM, and compound 18 (Table 4) was selected to further synthesize series of nineteen molecules. The IC50 values for the novel compounds were found to be in the range of 0.35 to 5.86 μM. Compound 19 (Table 4) emerged as the most active compound with IC50 350 nm and an MTB MIC of 1.55 μM.35
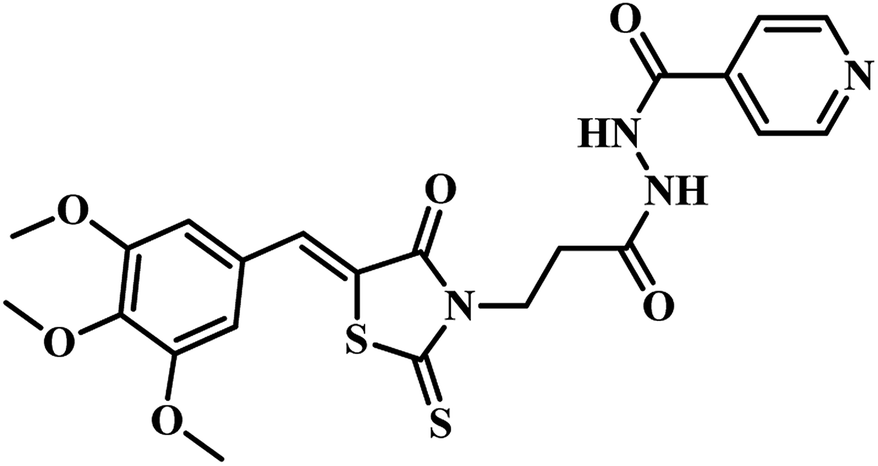
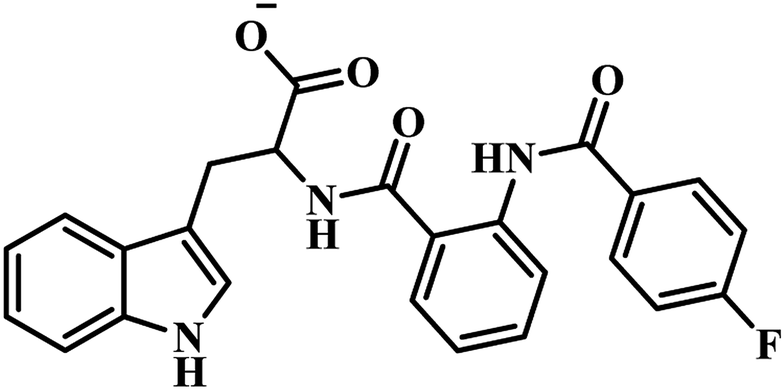
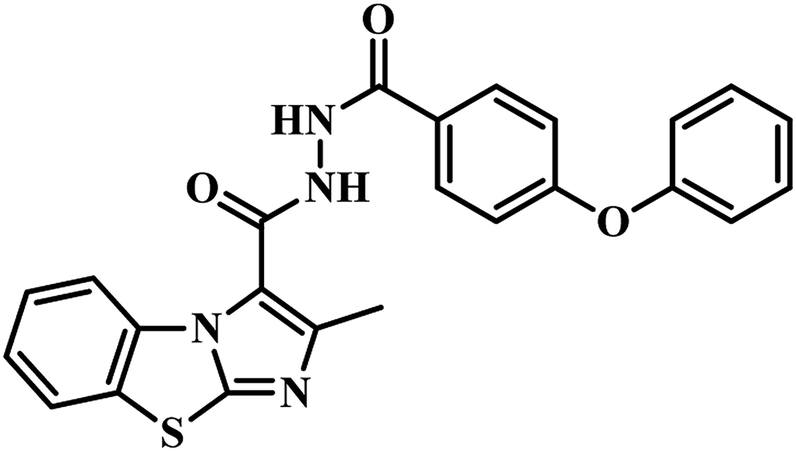
In the subsequent work also Sriram's group used the same strategy of the e-pharmacophore model. They developed the energy-based pharmacophore modeling of the available protein inhibitor complex and virtually screened an extensive commercial library. The e-pharmacophore model is comprised of a ring aromatic, negative ionizable and acceptor sites. Based on the results obtained, fourteen compounds were finally short-listed, and in vitro MTB PS inhibitory assay was carried out. Compounds 20 and 21 (Table 4) emerged as promising hits with IC50 2.18 μM and 6.63 μM, respectively.48 Later, synthetic modification of the potential lead compound 21 (Table 4) was undertaken to derivatize to get novel MTB PS inhibitors. Six compounds exhibited inhibitory activity of <6 μM against MTB PS. The most potent compound 22 (Table 4) showed IC50 2.28 μM against MTB PS.48
PS inhibitors identified through lead optimization strategy
Lead optimization is the technique through which a drug candidate is designed after a preliminary lead compound is recognized. The procedure includes iterative rounds of synthesis and characterization of an ability to become a drug.In continuation of their ongoing research in the year 2016, Sriram's group reported imidazo-[2,1-b]thiazole and benzo[d]imidazo[2,1-b]thiazole derivatives as MTB PS inhibitors.36 They synthesized these compounds based on the lead obtained from their previous work. Thirty-two compounds were developed and were evaluated for MTB PS inhibition, in vitro and in vivo anti-TB activity. Compound 23 (Table 4) emerged as the most active one with IC50 0.53 ± 0.13 μM against MTB PS and MIC 3.53 μM against MTB H37 RV strain. Further, this compound exhibited only 10.4% cytotoxicity at 50 μM against mouse macrophage RAW 264.7 cell line.49
Molecular hybridization-based PS inhibitors
Molecular hybridization is a structural modification strategy in which various bioactive pharmacophores are fused into a single framework anticipating improved activity compared to the parent moieties. This technique is well explored by researchers in the new drug discovery process.Twenty-six 2,6-disubstituted 4,5,6,7-tetrahydrothieno[2,3-c]pyridine-3-carboxamide derivatives were designed by Sriram et al., using a molecular hybridization approach. They developed novel MTB inhibitors by hybridizing their previously reported MTB PS inhibitor 24 (IC50 = 21.4 μM)43 (Table 5) and TAACF inhibitor SID 92097880 (25)47 (Table 5). Compounds were screened for inhibition study against MTB PS, in vitro anti-TB activity, and active analogues for cytotoxicity against RAW 264.7 cell line. Among the compounds, 26 (Table 5) was the most potent compound with IC50 5.87 μM against MTB PS; it inhibited MTB with MIC 9.28 μM and did not exhibit cytotoxicity at 50 μM.50 In the year 2017, our group reported pyrazolo[4,3-c]-piperidine carboxamide coupled triazoles as MTB PS inhibitors. Here, we synthesized twenty-six compounds and evaluated them for their MTB PS inhibition and in vitro anti-TB activity. In this series, compound 27 (Table 5) showed the most promising activity with IC50 1.01 ± 0.32 μM against MTB PS and MIC 24.72 μM against MTB H37 RV strain.51
| S. no. | Compound code/name | Structures | Ki/MIC/IC50 value | Ref. |
|---|---|---|---|---|
| 1 | 24 | 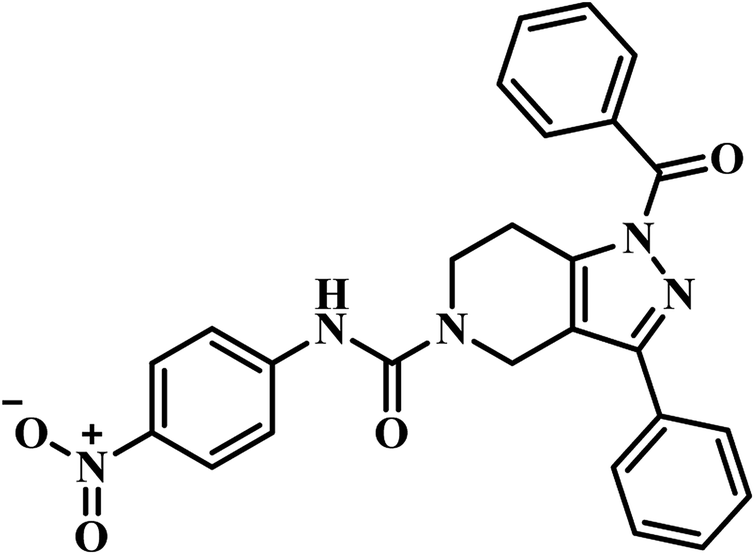 |
IC50 = 21.4 μM | 43 |
| 2 | 25 | 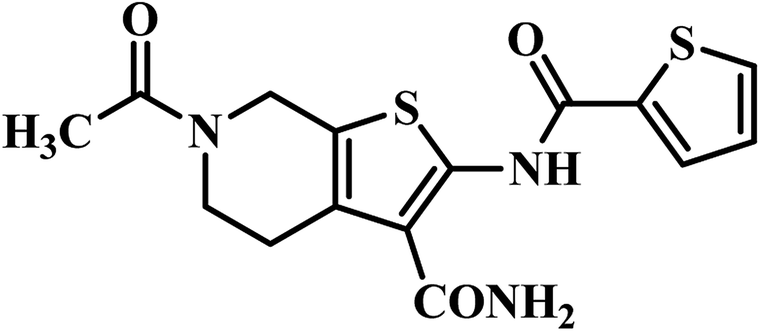 |
MIC = 3.2 μg mL−1 | 47 |
| 3 | 26 | 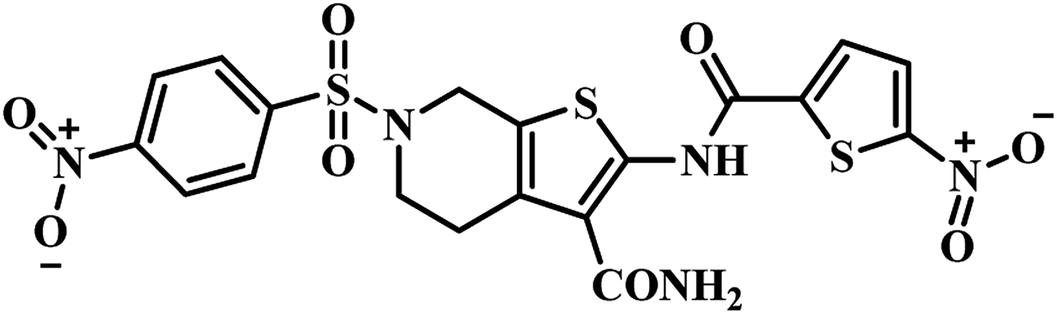 |
IC50 = 5.87 μM | 50 |
| 4 | 27 | 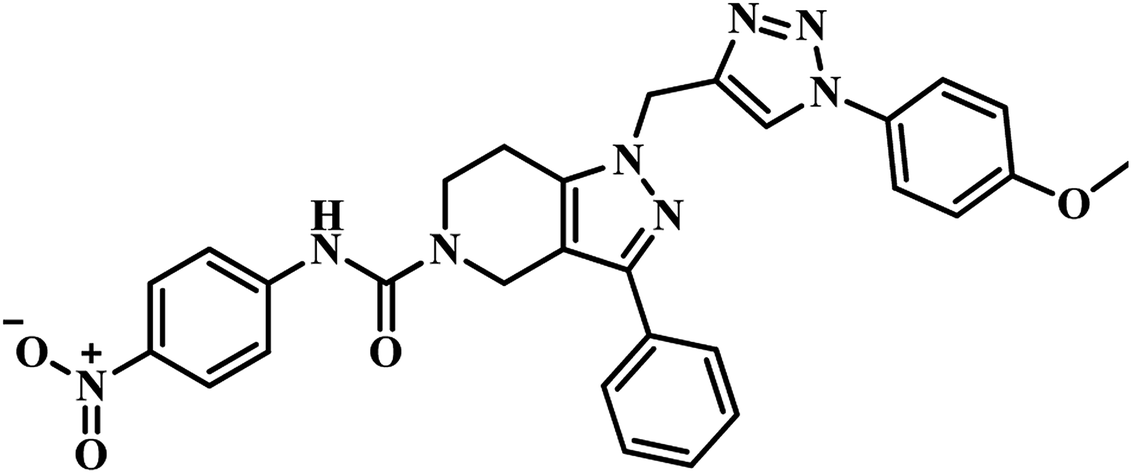 |
IC50 = 1.01 μM | 51 |
| 5 | 28 | 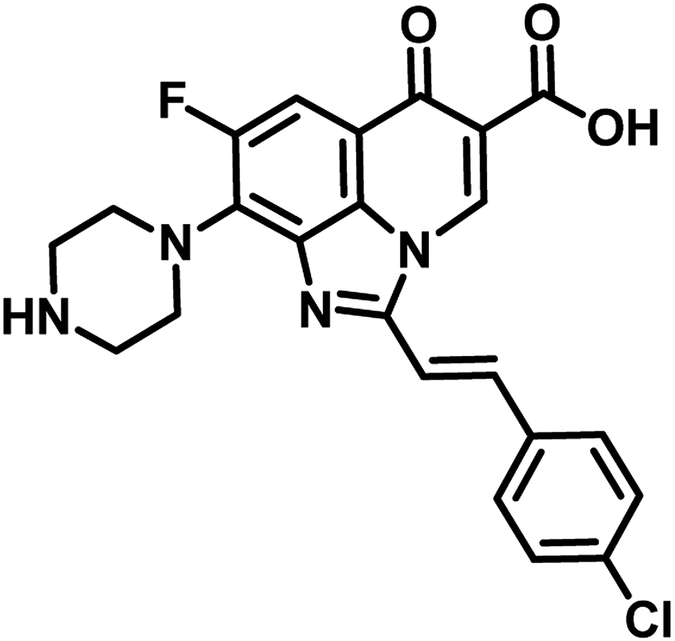 |
MIC = 28.62 μM | 52 |
| 6 | 29 | 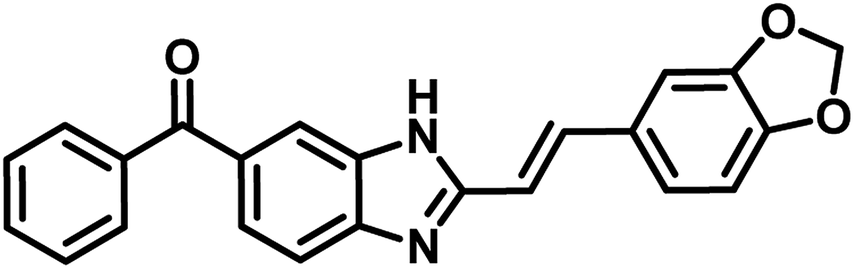 |
Ki = 65.64 μM | 53 |
| 7 | 30 | 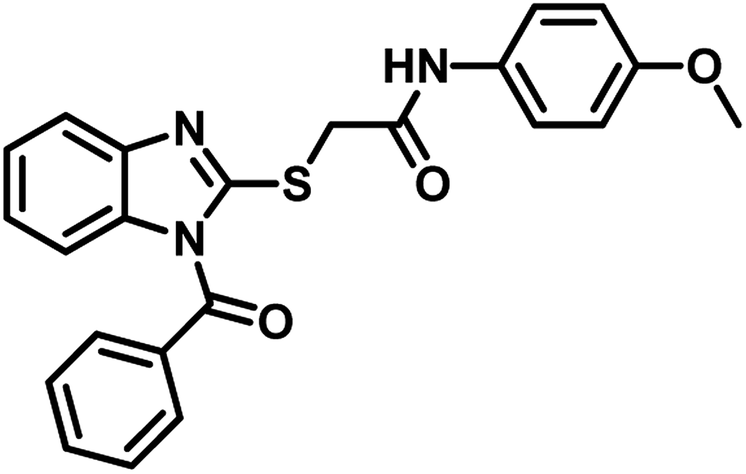 |
MIC = 12.5 μg mL−1 | 54 |
| 8 | 31 | 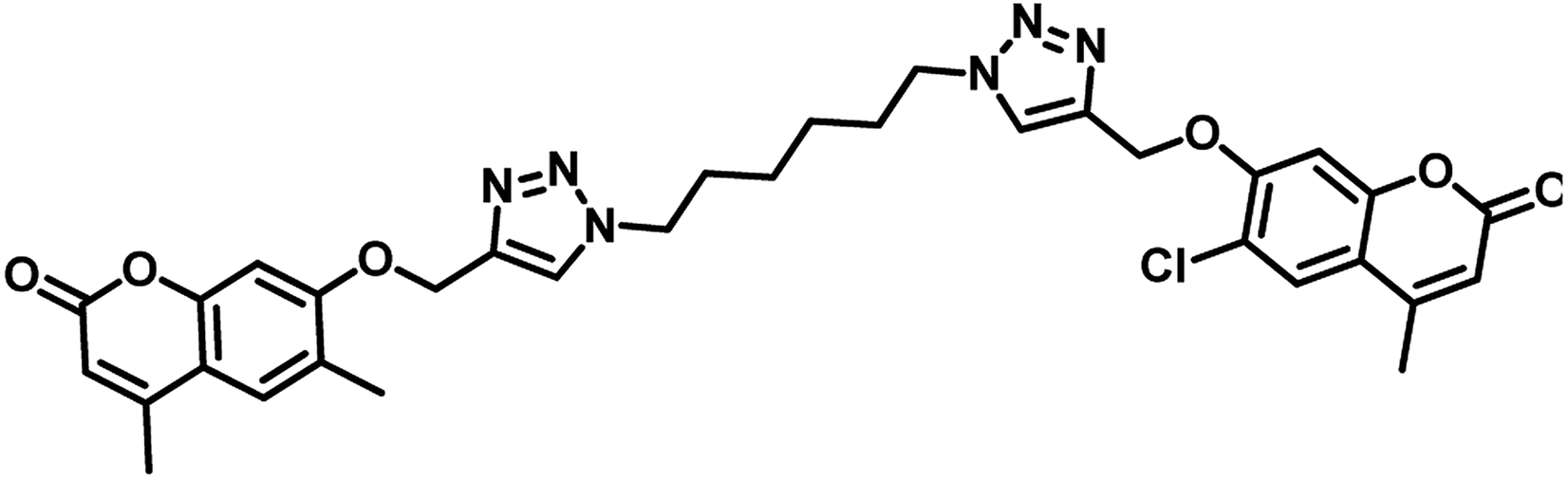 |
MIC = 1.56 μg mL−1 | 55 |
| 9 | 32 | 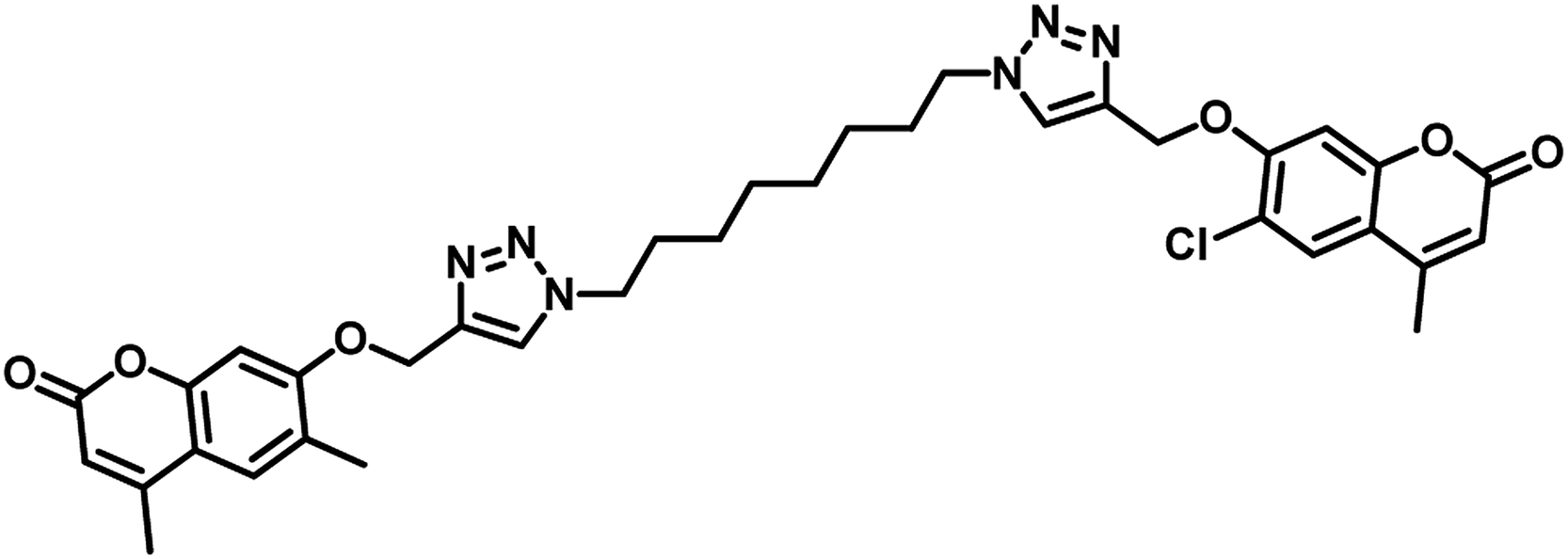 |
MIC = 1.56 μg mL−1 | 55 |
| 10 | 33 | 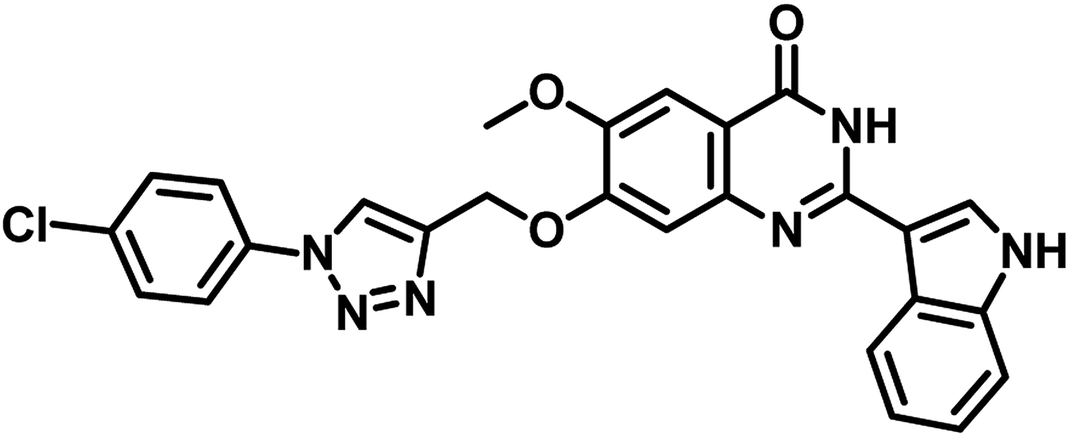 |
MIC = 7 μg mL−1 | 56 |
| 11 | 34 | 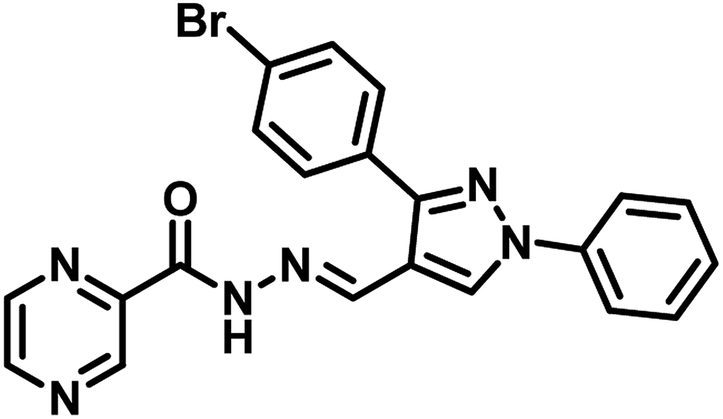 |
MIC = 0.78 μg mL−1 | 57 |
| 12 | 35 | 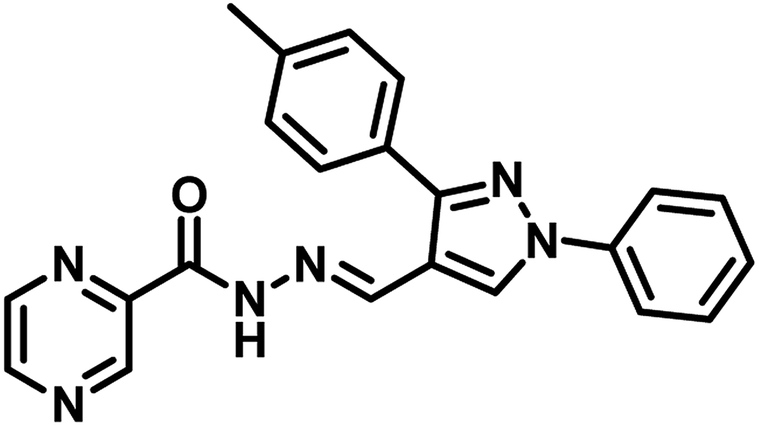 |
MIC = 1.56 μg mL−1 | 57 |
| 13 | 36 |  |
MIC = 1.56 μg mL−1 | 57 |
In the year 2017, Mohana Rao et al. reported the synthesis, molecular docking and antimycobacterial evaluation of nine novel analogues of (E)-8-fluoro-6-oxo-9-(piperazin-1-yl)-2-styryl-2,6-dihydro-1H-imidazo[4,5,1-ij]quinoline-5-carboxylic acid as inhibitors of MTB PS. Among the compounds, analogue 28 exhibited significant activity against the tested strain with MIC 28.62 μM as well as highest binding free energy (−6.92 kcal mol−1 and Ki = 8.45 μM) with interactions of ARG200, THR85 and GLU189 amino acid residues of the active site of the target protein PS during docking study.52
Mohana Rao's group in 2017, reported a series of fourteen novel 2-heterostyryl benzimidazole derivatives as antitubercular agents. Amongst the analogues, compound 29 (Table 5) showed promising inhibition of MTB with MIC 16 μg mL−1 as well as the same compound exhibited highest binding free energy (−9.80 kcal mol−1 and Ki = 65.64 μM) with two amino acid (Lys160, Val187) interactions at the active site residue of MTB PS.53
Snehalata Yadav in the year 2018 reported 2-(1-benzoyl-1H-benzo[d]imidazol-2-ylthio)-N-substituted acetamide analogues. They evaluated these derivatives for antimicrobial, antitubercular, and anticancer activities. Compound 30 (Table 5) was the most active compound against M. tuberculosis H37Rv with MIC 12.5 μg mL−1 and inhibits the MTB PS with percentage inhibition of 60.12 IU per L.54
In the year 2018, Dongamanti Ashok et al. reported synthesis, antimycobacterial, and antimicrobial evaluation of several dimers of novel series of coumarin-1,2,3-triazole hybrids. They found that among the titled analogues, compounds 31 and 32 (Table 5) were the most active with MIC 1.56 μg mL−1 against the tested Mycobacterium tuberculosis H37Rv strain. Further, they also performed a molecular docking study of the least active, moderately active and significantly active anti-TB compounds against PS enzyme of MTB and concluded that the docking results are in well agreement with the in vitro antitubercular evaluation results.55
Narendra Kumar Maddali et al. reported the design, synthesis and molecular docking studies of ten quinazolin-4-ones linked to 1,2,3-triazole hybrids as Mycobacterium tuberculosis H37Rv inhibitors in the year 2019. Among the titled analogues, compound 33 (Table 5) exhibited promising inhibition against MTB H37Rv strain with MIC 7 μg mL−1 as well as profound, strong interaction with three essential amino acids (Gln92, Ser196, Arg200) at the active site of MTB PS with large interaction energy (ΔG = −8.16 kcal mol−1).56
Nayera W. Hassan et al., reported design, synthesis and antitubercular evaluation of thirty one novel analogues of various hybrid molecules comprising pyrazine scaffold in the year 2020. Among the titled compounds, compound 34, ((E)-N′-[(3-(4-bromophenyl)-1-phenyl-1H-pyrazol-4-yl)-methylene]pyrazine-2-carbohydrazide) (Table 5) exhibited anti-TB activity (MIC = 0.78 μg mL−1) higher than pyrazinamide and ethambutol. Compounds 35 ((E)-N′-[(1-phenyl-3-(p-tolyl)-1H-pyrazol-4-yl)methylene]-pyrazine-2-carbohydrazide) and 36 ((E)-N′-[(3-(4-chlorophenyl)-1-(4-nitrophenyl)-1H-pyrazol-4-yl)-methylene]pyrazine-2-carbohydrazide) (Table 5) showed higher potency than pyrazinamide and equal to that of ethambutol (MIC = 1.56 μg mL−1). The molecular docking study of these compounds in the active site of the PS enzyme demonstrated favourable binding modes and interaction pattern.57
Overall, in this manuscript, seven different methods used for the identification of potential PS inhibitors reported by various scientists across the globe have been systematically compiled, and we observe the following SAR pattern:
(a) Pantoyl adenylate intermediate based PS inhibitor – three reports, available in the literature, have been collected and compiled. Among these, sulfamoyl adenylate compound 3, reported by Abell's group, was found to be the most active compound with Ki 220 nM when compared to all other reported compounds. This may be attributed to the firm binding of compound 3 into the active site of the PS enzyme.
(b) PS inhibitor identified through high throughput screening – around six reported reports have been summarized. Among these, compound 6 (analogue of 5-tert-butyl-N-pyrazol-4-yl-4,5,6,7-tetrahydrobenzo-[d]-isoxazole-3-carboxamide) reported by Petukhov et al., emerged to be the most active of the series with IC50 90 nM which was more significant than the 3-phenyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]-pyridine analogues reported by Sriram et al. They also analyzed that hydrophobic substituents on benzene ring led to increase in PS inhibitory activity while polar and nonpolar groups on pyrazole ring resulted in a decrease in activity.
(c) Fragment-growing and fragment-linking approach based PS inhibitors – among the two available reports, compound 16 (5-methoxy-1H-indole carboxamide coupled with 5-methoxy-1H-benzofuran analogue) reported by Abell's group exhibited the most potent PS inhibitory activity with Ki 5.4 μM when compared to compound 15 (indole-2-carboxylic acid analogue) and compound 14 (5-methoxy-1H-indole carboxamide coupled with 1H-benzofuran analogue Ki 5.4 μM). Hence, the benzofuran ring with methoxy substitution at fifth position seems to be essential for PS inhibitory activity.
(d) PS inhibitors identified through virtual screening – two published reports were compiled, and was found that compound 18 (pyrido pyrrole fused naphthyl hydrazide analogue) showed significant activity against MTB PS with IC50 1.90 μM without any cytotoxicity at a tested concentration of 50 μM. This finding signifies the importance of hydrophilic linker hydrazide portion between pyrido pyrrole and naphthyl ring portion of the target molecule against MTB PS activity.
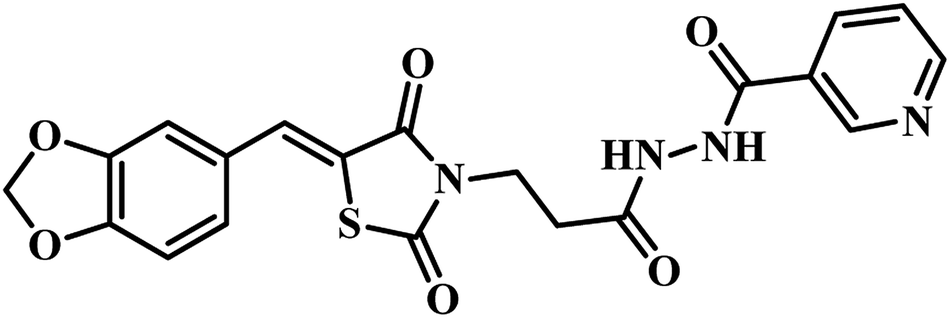
(e) E-pharmacophore based PS inhibitors – out of two published reports by Sriram et al., compound 19 (Z)-N′-(3-(4-oxo-2-thioxo-5-(3,4,5-trimethoxybenzylidene)thiazolidin-3-yl)propanoyl)isonicotinohydrazide , emerged as most potent analogue with an IC50 of 350 nM against the tested MTB PS than compound 18 (Z)-N′-(3-(5-(benzo[d][1,3]dioxol-5-ylmethylene)-2,4-dioxothiazolidin-3-yl)propanoyl)nicotinohydrazide. From these results, it has been observed that, ring opening of 1,3-benzodioxole increases the PS inhibitory activity.
(f) PS inhibitors through lead optimization strategy – compound 23 (benzo[d]imidazo[2,1-b]thiazole hydrazide analogue) emerged as the most active analogue with IC50 0.53 μM against MTB PS without any cytotoxicity at a tested concentration of 50 μM. This indicates the significance of hydrophilic linker hydrazide portion in between benzo[d]imidazo[2,1-b]thiazole and diphenyl oxy portion of the target molecule for MTB PS inhibitory activity.
(g) Molecular hybridization-based PS inhibitors – around eight reports were collected and compiled. Among these, compound 27 (pyrazolo[4,3-c]-piperidine carboxamide coupled triazole) showed potent activity with IC50 1.01 ± 0.32 μM against MTB PS than it's structural analogue compound 24 (pyrazolo[4,3-c]-piperidine carboxamide analogue). These findings also clearly confirm the necessity of the triazole nucleus in enhancing the MTB PS inhibitory profile of the titled analogues.
Conclusion
To minimize prolonged TB treatment or drug resistance, it is essential to identify novel pathways to combat TB. PS is one such attractive target in order to combat both drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. Several researchers across the globe are working in this area and identified reasonably good lead molecules with significant PS inhibition activity through various techniques such as pantoyl adenylate intermediate inhibition, high throughput screening, fragment-growing and fragment-linking approach, virtual screening, e-pharmacophore, lead optimization strategy, molecular hybridization-based inhibition and were used for the further drug development process. From the overview, very promissory groups are tetrahydrobenzo[d]isoxazole-3-carboxamides, tetrahydro pyrazolo pyridine-5-carboxamides, tetrahydro thieno[2,3-c]-pyridine-3-carboxamides, 3-biphenyl-4-cyano pyrrole-2-carboxylic acid, and 5-methoxy-1H-indoles. The greatest activity (IC50 90 nM) was observed in 5-(tert-butyl)-N-(1-(naphthalen-2-ylmethyl)-1H-pyrazol-4-yl)-4,5,6,7-tetrahydrobenzo[d]isoxazole-3-carboxamide (6) which was identified through high throughput screening methodology. Compounds with activity against the whole cell of MTB as a culmination of inhibition of PS might bring great benefits and reduce the spread of TB.Conflicts of interest
The authors declare that there is no conflict of interest.Acknowledgements
The authors are grateful to the Department of Biotechnology (BT/IN/Spain/39/SM/2017-18), Government of India, New Delhi for funding the project.References
- World Health Organization (WHO), Global Tuberculosis factsheet, Geneva, Switzerland, 2018 Search PubMed.
- S. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris and F. Tekaia, Nature, 1998, 393, 537–544 CrossRef CAS.
- A. Ciulli, D. E. Scott, M. Ando, F. Reyes, S. A. Saldanha, K. L. Tuck, D. Y. Chirgadze, T. L. Blundell and C. Abell, ChemBioChem, 2008, 9, 2606–2611 CrossRef CAS.
- S. Anishetty, M. Pulimi and G. Pennathur, Comput. Biol. Chem., 2005, 29, 368–378 CrossRef CAS.
- S. H. Cho, D. Goodlett and S. Franzblau, Tuberculosis, 2006, 86, 445–460 CrossRef CAS.
- S. Tiberi, M. Muñoz-Torrico, R. Duarte, M. Dalcolmo, L. D'Ambrosio and G. B. Migliori, Rev. Port. Pneumol., 2018, 24, 86–98 CAS.
- C. S. Merle, K. Fielding, O. B. Sow, M. Gninafon, M. B. Lo, T. Mthiyane, J. Odhiambo, E. Amukoye, B. Bah, F. Kassa, A. N'Diaye, R. Rustomjee, B. C. De Jong, J. Horton, C. Perronne, C. Sismanidis, O. Lapujade, P. L. Olliaro and C. Lienhardt, N. Engl. J. Med., 2014, 371, 1588–1598 CrossRef.
- J. L. Cabrera-Rivero, D. E. Vargas-Vasquez, M. Gao, D. Ph, M. Awad, B. Ch, S. Park, D. Ph, T. S. Shim, D. Ph and G. Y. Suh, N. Engl. J. Med., 2012, 366, 2151–2160 CrossRef.
- L. G. Dover and G. D. Coxon, J. Med. Chem., 2011, 54, 6157–6165 CrossRef CAS.
- J. Strelow, W. Dewe, P. W. Iversen, H. B. Brooks, J. A. Radding, J. McGee and J. Weidner, The Assay Guidance Manual, Mechanism of Action Assays for Enzymes, 2012, https://www.ncbi.nlm.nih.gov/books/NBK92001/ Search PubMed.
- P. Badrinarayan and G. Narahari Sastry, Comb. Chem. High Throughput Screening, 2011, 14, 840–860 CrossRef CAS.
- S. L. Miller and G. Schlesinger, J. Mol. Evol., 1993, 36, 308–314 CAS.
- Y. T. Tan, D. J. Tillett and I. A. McKay, Mol. Med. Today, 2000, 6, 309–314 CrossRef CAS.
- W. K. Merkel and B. P. Nichols, FEMS Microbiol. Lett., 1996, 143, 247–252 CrossRef CAS.
- R. Zheng and J. S. Blanchard, Biochemistry, 2001, 40, 12904–12912 CrossRef CAS.
- J. E. Cronan, K. J. Littel and S. Jackowski, J. Bacteriol., 1982, 149, 916–922 CrossRef CAS.
- S. U. E. G. Powers and E. E. Snell, J. Biol. Chem., 1976, 251, 3786–3793 CAS.
- M. E. Frodyma and D. Downs, J. Bacteriol., 1998, 180, 4757–4759 CrossRef CAS.
- R. C. Reynolds, S. Ananthan, E. Faaleolea, J. V. Hobrath, C. D. Kwong, C. Maddox, L. Rasmussen, M. I. Sosa, E. Thammasuvimol, E. L. White, W. Zhang and J. A. Secrist, Tuberculosis, 2012, 92, 72–83 CrossRef CAS.
- F. Von Delft, T. Inoue, S. A. Saldanha, H. H. Ottenhof, F. Schmitzberger, L. M. Birch, V. Dhanaraj, M. Witty, A. G. Smith, T. L. Blundell and C. Abell, Structure, 2003, 11, 985–996 CrossRef CAS.
- R. Zheng and J. S. Blanchard, Biochemistry, 2000, 39, 16244–16251 CrossRef CAS.
- C. M. C. Lobley, F. Schmitzberger, M. L. Kilkenny, H. Whitney, H. H. Ottenhof, E. Chakauya, M. E. Webb, L. M. Birch, K. L. Tuck, C. Abell, A. G. Smith and T. L. Blundell, Biochem. Soc. Trans., 2003, 31, 563–571 CrossRef CAS.
- C. Albert, A. Dhanaraj, V. Genschel, U. Khan, G. Ramjee, M. K. Pulido and R. Abell, Nat. Struct. Biol., 1998, 5, 289–293 CrossRef.
- G. D. Novelli, A. Kreiling, T. Wieland, K. Miyatake, Y. Nakano, S. Kitaoka, Y. Nakano and S. Kitaoka, Methods Enzymol., 1979, 62, 215–219 Search PubMed.
- R. Zheng, T. K. Dam, C. F. Brewer and J. S. Blanchard, Biochemistry, 2004, 43, 7171–7178 CrossRef CAS.
- S. Wang and D. Eisenberg, Protein Sci., 2003, 12, 1097–1108 CrossRef CAS.
- S. Wang and D. Eisenberg, Biochemistry, 2006, 45, 1554–1561 CrossRef CAS.
- A. Satoh, S. Konishi, H. Tamura, H. G. Stickland, H. M. Whitney, A. G. Smith, H. Matsumura and T. Inoue, Biochemistry, 2010, 49, 6400–6410 CrossRef CAS.
- B. Pandey, S. Grover, S. Goyal, A. Kumari, A. Singh, S. Jamal, J. Kaur and A. Grover, Sci. Rep., 2018, 8, 1–13 CrossRef CAS.
- G. L. Abrahams, A. Kumar, S. Savvi, A. W. Hung, S. Wen, C. Abell, C. E. Barry, D. R. Sherman, H. I. M. Boshoff and V. Mizrahi, Chem. Biol., 2012, 19, 844–854 CrossRef CAS.
- A. Kumar, A. Casey, J. Odingo, E. A. Kesicki, G. Abrahams, M. Vieth, T. Masquelin, V. Mizrahi, P. A. Hipskind, D. R. Sherman and T. Parish, PLoS One, 2013, 8, 72786 CrossRef.
- K. Patil, S. Bagade, S. Bonde, S. Sharma and G. Saraogi, Biomed. Pharmacother., 2018, 99, 735–745 CrossRef CAS.
- T. J. De Wet, D. F. Warner and V. Mizrahi, Acc. Chem. Res., 2019, 52, 2340–2348 CrossRef CAS.
- A. W. Hung, H. L. Silvestre, S. Wen, A. Ciulli, T. L. Blundell and C. Abell, Angew. Chem., Int. Ed., 2009, 48, 8452–8456 CrossRef CAS.
- P. B. Devi, G. Samala, J. P. Sridevi, S. Saxena, M. Alvala, E. G. Salina, D. Sriram and P. Yogeeswari, ChemMedChem, 2014, 9, 2538–2547 CrossRef CAS.
- E. L. White, K. Southworth, L. Ross, S. Cooley, R. B. Gill, M. I. Sosa, A. Manouvakhova, L. Rasmussen, C. Goulding, D. Eisenberg and T. M. Fletcher, J. Biomol. Screening, 2007, 12, 100–105 CrossRef CAS.
- S. Wellington and D. T. Hung, ACS Infect. Dis., 2018, 4, 696–714 CrossRef CAS.
- K. P. Rakesh, C. S. Shantharam, M. B. Sridhara, H. M. Manukumar and H. L. Qin, MedChemComm, 2017, 8, 2023–2039 RSC.
- K. L. Tuck, S. A. Saldanha, L. M. Birch, A. G. Smith and C. Abell, Org. Biomol. Chem., 2006, 4, 3598–3610 RSC.
- Z. Xu, W. Yin, L. K. Martinelli, J. Evans, J. Chen, Y. Yu, D. J. Wilson, V. Mizrahi, C. Qiao and C. C. Aldrich, Bioorg. Med. Chem., 2014, 22, 1726–1735 CrossRef CAS.
- S. Velaparthi, M. Brunsteiner, R. Uddin, B. Wan, S. G. Franzblau and P. A. Petukhov, J. Med. Chem., 2008, 51, 1999–2002 CrossRef CAS.
- Y. Yang, P. Gao, Y. Liu, X. Ji, M. Gan, Y. Guan, X. Hao, Z. Li and C. Xiao, Bioorg. Med. Chem. Lett., 2011, 21, 3943–3946 CrossRef CAS.
- G. Samala, P. B. Devi, R. Nallangi, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2013, 69, 356–364 CrossRef CAS.
- S. Pradhan and C. Sinha, In Silico Pharmacol., 2018, 6, 9 CrossRef.
- P. Sledz, H. L. Silvestre, A. W. Hung, A. Ciulli, T. L. Blundell and C. Abell, J. Am. Chem. Soc., 2010, 132, 4544–4545 CrossRef CAS.
- F. Ntie-Kang, S. Kannan, K. Wichapong, L. C. Owono Owono, W. Sippl and E. Megnassan, Mol. BioSyst., 2014, 10, 223–239 RSC.
- G. Samala, P. B. Devi, R. Nallangi, J. P. Sridevi, S. Saxena, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 4223–4232 CrossRef CAS.
- G. Samala, P. B. Devi, S. Saxena, N. Meda, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2016, 24, 1298–1307 CrossRef CAS.
- P. B. Devi, S. Jogula, A. P. Reddy, S. Saxena, J. P. Sridevi, D. Sriram and P. Yogeeswari, Mol. Inf., 2015, 34, 147–159 CrossRef CAS.
- G. Samala, P. B. Devi, R. Nallangi, J. P. Sridevi, S. Saxena, P. Yogeeswari and D. Sriram, Bioorg. Med. Chem., 2014, 22, 1938–1947 CrossRef CAS.
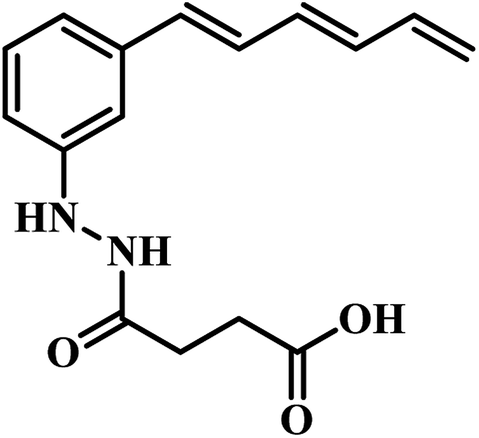
- S. Amaroju, M. N. Kalaga, S. Srinivasarao, A. Napiórkowska, E. Augustynowicz-Kopeć, S. Murugesan, S. Chander, R. Krishnan and K. V. G. Chandra Sekhar, New J. Chem., 2017, 41, 347–357 RSC.
- M. R. Anguru, A. K. Taduri and R. D. Bhoomireddy, Med. Chem. Res., 2017, 9, 299–306 CAS.
- M. R. Anguru, A. K. Taduri, R. D. Bhoomireddy, M. Jojula and S. K. Gunda, Chem. Cent. J., 2017, 11, 1–11 CrossRef.
- S. Yadav, S. M. Lim, K. Ramasamy, M. Vasudevan, S. A. A. Shah, A. Mathur and B. Narasimhan, Chem. Cent. J., 2018, 12, 1–14 CrossRef.
- D. Ashok, S. Gundu, V. K. Aamate, M. G. Devulapally, R. Bathini and V. Manga, J. Mol. Struct., 2018, 1157, 312–321 CrossRef CAS.
- N. K. Maddali, I. K. Viswanath, Y. L. N. Murthy, R. Bera, M. Takhi, N. S. Rao and V. Gudla, Med. Chem. Res., 2019, 28, 559–570 CrossRef CAS.
- N. W. Hassan, M. N. Saudi, Y. S. Abdel-Ghany, A. Ismail, P. A. Elzahhar, D. Sriram, R. Nassra, M. M. Abdel-Aziz and S. A. El-Hawash, Bioorg. Chem., 2020, 96, 103610 CrossRef.
| This journal is © The Royal Society of Chemistry 2020 |