
Reaction Cascades in Polymer Mechanochemistry
Huan
Hu
a,
Zhiyong
Ma
 *a and
Xinru
Jia
b
*a and
Xinru
Jia
b
aBeijing Advanced Innovation Center for Soft Matter Science and Engineering, State Key Laboratory of Organic–Inorganic Composites, College of Chemical Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: mazhy@mail.buct.edu.cn
bDepartment of Polymer Science and Engineering, Chemistry College, Peking University, Beijing, 100871, China
First published on 27th July 2020
Abstract
This review summarizes the development of polymer mechanochemistry-enabled reaction cascades. According to different functions or purposes, we categorize the reported reaction cascades in polymer mechanochemistry into five parts, including mechanocatalysis, cargo-releasing, self-strengthening, gating, and mechanochromism. Mechanocatalysis refers to the force-induced latent catalyst activation and the following catalysis reaction. All reported mechanocatalysts are organometallic coordinated complexes with activated metallic atoms from ruptured coordinate bonds upon force action. Cargo-releasing represents the small molecules released from the designed mechanophores in polymer chains by mechanical disturbance. Commonly, the generation of cargo involves two steps, production of an unstable intermediate and then spontaneous decomposition into small molecules. Self-strengthening usually involves the crosslinking of polymers or formation of new polymer networks triggered by the force-generated active species, and then the mechanical properties of polymeric materials are reinforced. Gating means regulating the desired chemical reactions by specific stimuli. Mechanophores embedded in polymers can be used to control other stimuli-responsive reactions, for example, mechanochemical ring-opening, photoswitching, or degradable reactions. Cascade-enabled mechanochromism is quite different from one-step mechanochromism and has advantages such as the acquisition of more marvellous functions and avoiding unnecessarily complicated design of mechanophores. Finally, some outlook for reaction cascades in polymer mechanochemistry is provided.
1. Introduction
Polymer mechanochemistry refers to the chemical transformation activated by external mechanical forces through the polymer chains.1 Generally, thermal activation is the most common pathway to accelerate desired chemical reactions. However, thermal energy is unquantized and undirected, leading to unfortunate consequences such as side reactions.2 In principle, mechanochemistry can direct chemical reactions down precise reaction pathways by virtue of the specificity to covalent bonds exerted by external forces.3 However, it is very challenging to macroscopically apply mechanical force to the molecule-level reactive sites precisely. Because of the characteristic chain-like structure, polymers are well-suited to conduct mechanical force along the backbone.4 Therefore, polymer mechanochemistry has been attracting considerable attention both in fundamental research, including exploration of the force-activation mechanism,5–15 and in practical applications such as the construction of smart materials.16–24The mechanophore is the key functional unit in the mechanoresponsive polymers. At the early stage, the greatest difficulty was detecting and monitoring the progress of force-induced chemical transformation. Two effective strategies have been developed in recent years. Firstly, mechanophores with optical signal changes have drawn tremendous interest. Such mechanophores include spiropyran,16 rhodamine,20,25 1,2-dioxetane,17etc. The second strategy is the design of mechanophores with given chemical signals, e.g., easily detected small molecules.26,27
In 2015, Moore announced that the force-action in polymer mechanochemistry has been developed from being destructive to productive.28 By the way of polymer mechanochemistry, a lot of fascinating phenomena and functionalities have been found, for example, generation of radicals,6 monomers,29 acids26 and cations,27 colour change16,20,25 or biasing reaction pathways.5 If these functions can be made good use of, it is possible to obtain smart polymers with attractive properties, such as self-sensing, self-reporting, self-healing or self-strengthening, etc. Thus, the concept of a reaction cascade in polymer mechanochemistry was born at the right moment. The reaction cascade in polymer mechanochemistry means that the active species activated by mechanical force are utilized to trigger subsequent chemical reactions. In other words, a reaction cascade involves at least two chemical reactions with the first one induced by mechanical force. In such a reaction cascade, the product from the first step can be used as a reactant, an initiator or a catalyst in the following reaction. Some reviews referring to polymer mechanochemistry have been published,30–33 but there is still no systematic review on polymer mechanochemistry-based reaction cascades.
In this review, we summarize the recent progress of reaction cascades in polymer mechanochemistry from the perspective of different functions or purposes and mainly focus on mechanocatalysis, cargo-releasing, self-strengthening, gating and mechanochromism.
2. Polymer mechanochemistry-enabled reaction cascades
2.1. Mechanocatalysis
Latent catalysts (LCs) are inactive and more stable in the “locked” state, and they can be activated by appropriate external triggers such as heat, light, chemical agents, and redox species.34 The “locked” state is achieved by relatively strong coordination bonds of which the dissociation energy is lower than that of covalent bonds, making mechanical activation possible. Mechanical forces activate LCs and then induce the catalytic reactions with the ability to control a chemical process, which is named as “Mechanocatalysis”. All described mechanocatalysts/LCs in this section are organometallic coordinated complexes embedded in the centre of polymer chains due to the mechanochemical nature and containing a metal core and two ligands. Different mechanocatalysts and the corresponding catalytic reactions are introduced as follows.Sijbesma et al. pioneered the investigation of mechanocatalysis in 2009 by using silver–N-heterocyclic carbene (NHC) and ruthenium–NHC complexes.35 Mechanocatalysts 1 and 2 with different molecular weight were synthesized (Fig. 1a). Each NHC was functionalized with poly(tetrahydrofuran) (pTHF), which provided efficient mechanical energy transduction along the polymer chain towards the weak metal–ligand bond. They firstly sonicated the mixture of benzyl alcohol (3.86 M) and vinyl acetate (6.5 M) with 1. The silver–NHC generated the active ligands to catalyse transesterification reaction upon sonication (Fig. 1a). It achieved 65% conversion after sonication for 60 min in the presence of 1 (22 kDa, 3 mM). Furthermore, the conversions reached 1% and 10% in 10 min when the same concentration (0.6 mM) of 1 with different molecular weights (5 kDa and 22 kDa) was used, respectively. The evident dependence of conversion on Mn over sonication time indicated the mechanochemical features. The mechanically exposed ruthenium–alkylidene complexes from ruthenium–NHC could catalyse metathesis reactions effectively (Fig. 1a). Then both the ring-closing metathesis (RCM) reaction and ring-opening metathesis polymerization (ROMP) reaction were studied. For RCM, and DEDAM (0.1 M) was catalysed by 2 in toluene to obtain diethylcyclopent-3-ene-1,1-dicarboxylate; the mixtures were sonicated for two 1 hour periods, interrupted for 30 min. The conversion reached 36% for 2 (18 kDa) and 14% for 2 (9 kDa) eventually. It is worth mentioning that the conversion did not increase in the off-state sonication, suggesting the limited lifetime of the species with catalytic activity. This observation was further supported by capturing active species with PCy3 during sonication (Fig. 1b): in the absence of DEDAM, 50% of 2 (18 kDa) were bound; in the presence of DEDAM, all active species were bound so that no diethylcyclopent-3-ene-1,1-dicarboxylate was generated. For ROMP, 2 (21 kDa, 1 mM) and cyclooctene (0.5 M) were employed in the same sonication mode of RCM, the conversion to polyoctenamer achieved about 90%, and 1H NMR spectroscopy and size-exclusion chromatography (SEC) confirmed the corresponding polymers. The occurrence of ROMP along with its high efficiency holds potential applications in self-healing materials.
 | ||
| Fig. 1 (a) Ultrasound-induced mechanochemical scission of polymeric silver–NHC complexes (1) and ruthenium–NHC complexes (2), resulting in the activation of a catalyst. For (1), the active ligand catalyses transesterification and for (2) the metal centre catalyses metathesis reactions. (b) Reaction scheme showing the fate of the ruthenium–benzylidene catalyst in the presence of PCy3 and DEDAM following mechanically induced ligand dissociation. Cl ligands have been omitted for clarity. Cy, cyclohexyl. Reproduced with permission.35 Copyright 2009, Springer Nature. | ||
The effects of impurities,36 molecular weight and concentration37 on catalytic reactivity were systematically discussed by Sijbesma's group in 2012. It was found that the active species in ROMP could survive for several hours,38 offering opportunities for catalysis in bulk polymers. They subsequently studied the ROMP of norbornene in solids and the previously discussed Ru–NHC (2) was used as a mechanocatalyst (Fig. 2a).39 The polymer-functionalized Ru–NHC (2) was blended with norbornene monomers and pTHF to obtain a semi-crystalline polymer matrix, and the mechanical force was transmitted by compressing (Fig. 2b). Conversion towards polynorbornene determined by 1H NMR spectroscopy increased linearly to 25% in five repeated compressions, while the control experiment with n-butyl armed Ru–NHC showed no alteration although moderate reproducibility, as expected. The polynorbornene could be measured by gel permeation chromatography (GPC) coupled with a UV detector as well. They further conducted cross-linking ROMP for detecting self-healing property. In the polymer matrix, they used bisnorbornene connected with an alkyl chain as the monomer instead of mononorbornene (Fig. 2a) and bis(2-ethylhexyl)terephthalate as the internal standard; and capturer butylated hydroxytoluene (BTH) was used to exclude the disturbance of radicals produced in mechanical activation. The existence of residue in dissolution and signals in high-resolution magic angle spinning (HR-MAS) 1H NMR spectroscopy confirmed the formation of cross-linked polynorbornene. These experiments forwarded a step to self-healing materials despite the low efficiency.
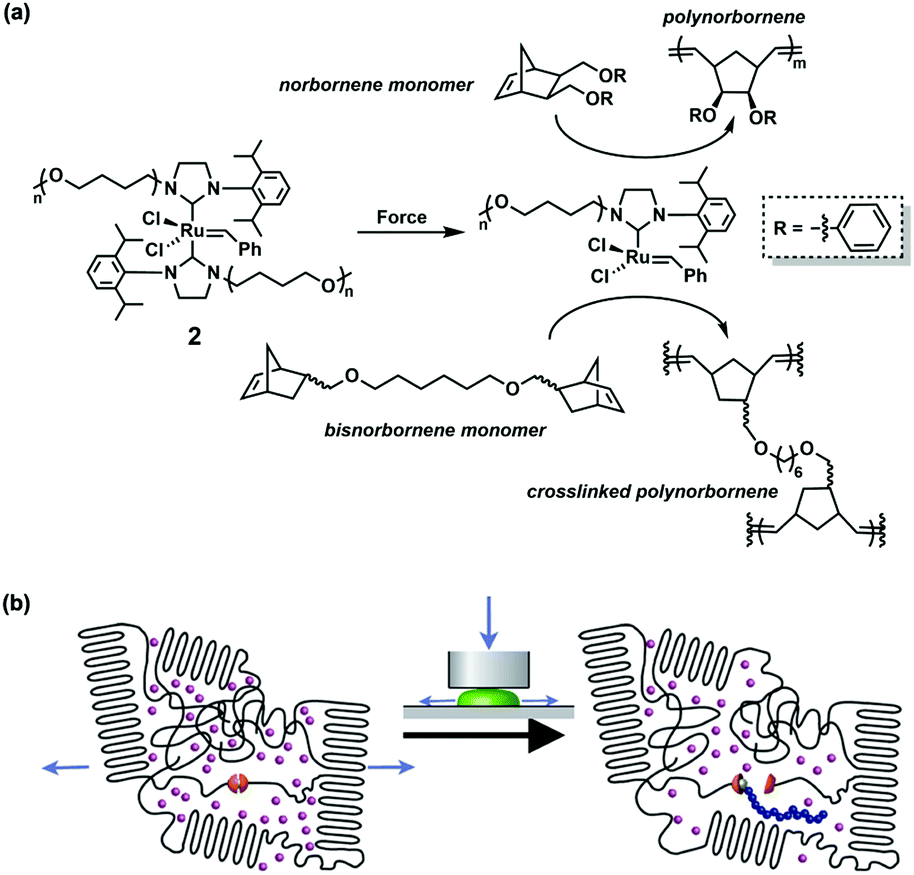 | ||
Fig. 2 (a) Chemical structure of catalyst 2 (![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) ≈ 240) and its mechanical activation to catalyse ring-opening polymerization of the norbornene monomer and bisnorbornene monomer. (b) Schematic representation of activation of catalyst 2 in the semi-crystalline matrix by compressive force and subsequent polymerization of the norbornene monomer. Reproduced with permission.39 Copyright 2013, American Chemical Society. ≈ 240) and its mechanical activation to catalyse ring-opening polymerization of the norbornene monomer and bisnorbornene monomer. (b) Schematic representation of activation of catalyst 2 in the semi-crystalline matrix by compressive force and subsequent polymerization of the norbornene monomer. Reproduced with permission.39 Copyright 2013, American Chemical Society. | ||
Another famous contributor in this field is Binder's group. He and co-workers mechanically catalysed “click” reactions with Cu(I)–NHC,40–43 which had never been studied before. In 2015, Binder et al. conducted mechano-activation of Cu(I)–NHC and subsequent copper-catalysed “click” reaction (CuAAC) both in solution and in a solid polymer matrix (Fig. 3).40 Cu–NHC was introduced into flexible poly(isobutylene) (PIB) and rigid poly(styrene) (PS) to obtain 3a and 3b, respectively. In the solution, phenylacetylene and benzylazide could not react under ultrasound without CuI complexes. The conversion for 3a and 3b was negligible at 60 °C without ultrasound, which means greater stability of Cu–NHC than the Ag–NHC complex (1) and Ru–NHC complex (2). It should be noted that the conversion of 3b was higher than that of 3a (44% vs. 27%) despite its lower Mn (13.1 kDa vs. 17.2 kDa), reflecting the influence of structural difference. To easily detect the CuAAC in the polymer matrix, the authors fabricated a fluorescent “off–on” switch from nonfluorescent 3-azido-7-hydroxy-coumarin and phenylacetylene to a highly fluorescent dye, which enabled visualization and quantitative analysis of reaction conversion inside initiated by compression. After compressing the crystalline pTHF blended with 3a, 3-azido-7-hydroxy-coumarin and phenylacetylene, blue fluorescence was observed by the naked-eye under 254 nm and 366 nm UV light, and the conversion could be calculated through the working curve of concentration towards fluorescence intensity. Such a study provides an opportunity to achieve the “two-in-one” function of self-healing and stress-sensing. The latter is discussed in the next case.
 | ||
| Fig. 3 Mechanochemical activation of latent copper(I)–NHC catalysts (3a, 3b) both in solution and in the solid polymer matrix; the activated catalysts then catalyse “click” reactions. Reproduced with permission.40 Copyright 2015, John Wiley and Son. | ||
Based on the previous study, Binder et al. further applied a fluorescent mechanocatalytic click reaction between 3-azido-7-hydroxy-coumarin and phenylacetylene to monitor stress progress in 2018.41 In other words, they studied the impact of stress recorded by fluorescence intensity due to irreversibility. Four architectures were designed to discuss the effect of the structure on catalytic activity, including low molecular weight, linear (3a, 3b), chain-extended and network-based Cu–NHC complexes (Fig. 4a), among which the polymer skeleton of both the chain-extended and network approach was PS. The designed and synthesized four compounds were respectively blended with pTHF to catalyse the reaction between 3-azido-7-hydroxycoumarin and phenylacetylene. The concentration of mechanocatalyst in the polymer blends was 5.4 × 10−6 mmol mgsample−1. The directly observed increase in fluorescence intensity showed the accumulation of stress during several compression circles (Fig. 4b). After 20 compressions, conversions for linear mechanocatalysts (3a, 3b) reached about 7.2–7.9%, and the low molecular weight mechanocatalyst had negligible conversion (<1%) by calculation due to its inefficient force delivering. Both the chain-extended mechanocatalyst and network-based mechanocatalyst offered ∼44% conversion, but the latter was a better candidate to sense stress for its relatively higher stability.
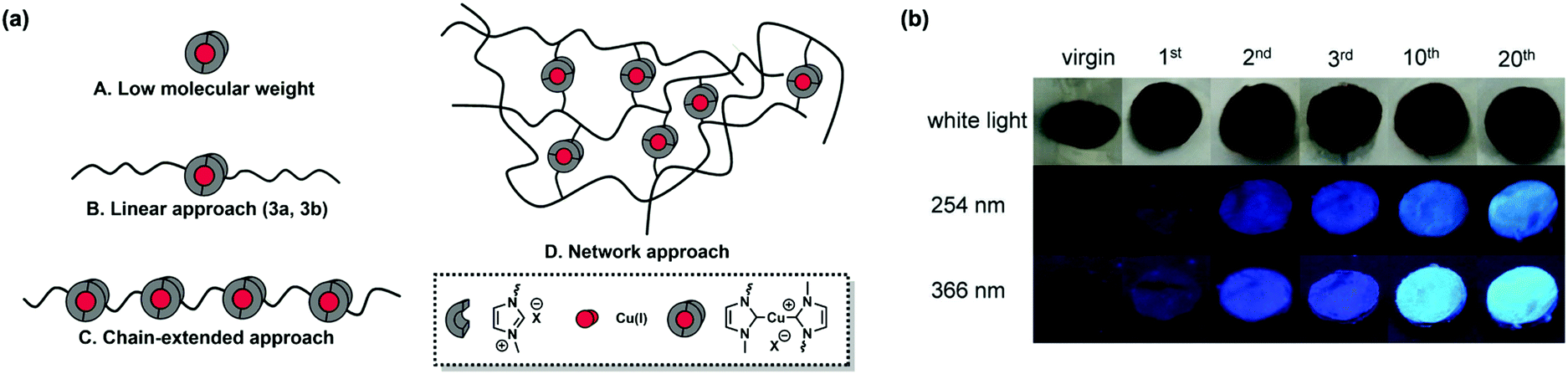 | ||
| Fig. 4 (a) Different architectures of mechanocatalysts: (A) low molecular weight, (B) linear, (C) chain-extended, and (D) network-based mechanocatalysts. (b) Photos of investigated samples (linear mechanocatalyst 3b, 3-azido-7-hydroxycoumarin, and phenylacetylene embedded into a high molecular weight pTHF matrix) before and after the 1st, 2nd, 3rd, 10th, and 20th compression cycle with a hydraulic press (0.74 GPa) visualized under white or UV light (254 or 366 nm), respectively. Reproduced with permission.41 Copyright 2018, John Wiley and Son. | ||
Besides the aforementioned silver, ruthenium and copper latent catalysts, Xu et al. reported a platinum catalyst in 2017.44 They synthesized the platinum–acetylide complex (4) as a mechanocatalyst and embedded it into poly(methyl acrylate) (PMA) (Fig. 5a). In the sonication experiment, the active platinum sites were exposed to performed olefin hydrosilylation, which was extensively used in the laboratory and industry (Fig. 5b). Mechano-activation of the mechanocatalyst was confirmed by GPC coupling with refractive index (RI) and UV detectors, UV-vis absorption spectra and photoluminescence spectra. The mechanochemical reaction is classified into the first-order kinetics according to GPC data. The comparison of kinetics between this mechanocatalyst and PMA homopolymer implied that the breakage of the fragile platinum–alkyne bond is preferred instead of the C–C bond nearby the chain centre. Then the catalytic activity for hydrosilylation was estimated by using 1-octene and (Me3SiO)2MeSiH as the reagents in the presence of 4. GC-MS and 1H NMR measurements found the target product with the conversion of 24% after on–off sonication cycles. The control experiments revealed slight thermal instability of the platinum–acetylide complex under applied conditions. This was similar to the organometallic LCs mentioned above.
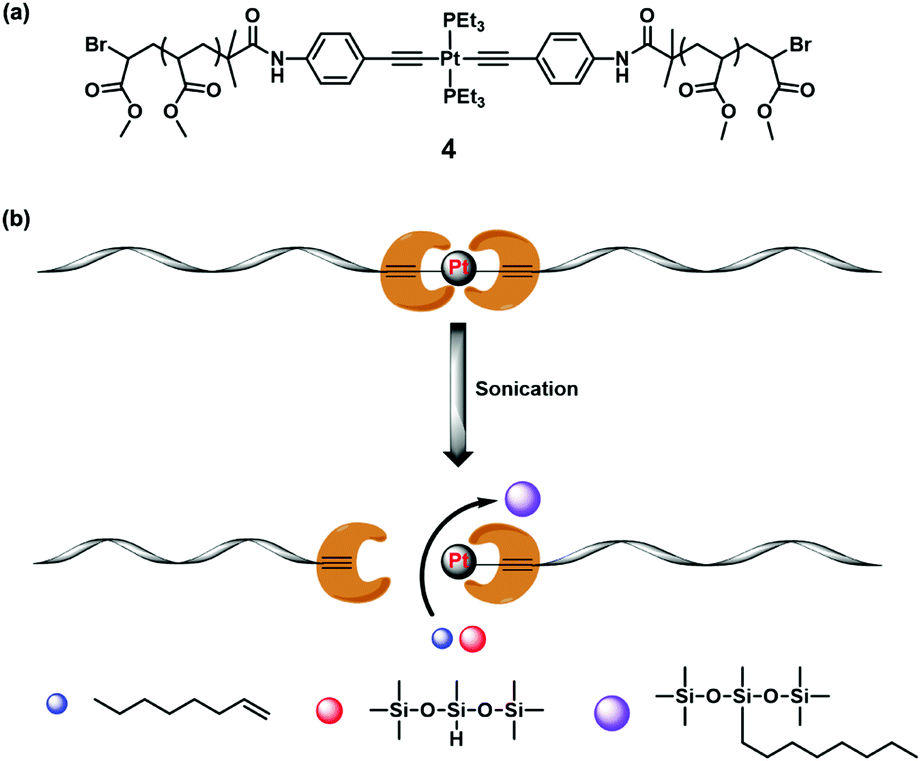 | ||
| Fig. 5 (a) Structure of the PMA containing platinum–acetylide complex. (b) The mechanically induced chain scission was demonstrated to be able to release catalytically active platinum species which could catalyse the olefin hydrosilylation process. Reproduced with permission.44 Copyright 2017, American Chemical Society. | ||
2.2. Cargo-releasing
Small molecules, graphically represented as cargos, can be mechanically released from elaborately designed mechanophores in polymers. In such a process, the mechanophore firstly turns to an unstable intermediate under force, and then decomposes to give small molecules, such as protons, alcohols and iron ions, which shows a promising prospect in catalysis, self-healing and drug delivery etc. Besides, monomer-releasing based on cascade unzipping of the polymer is also mentioned in this section.The most-reported cargo is protons. Moore, as one of the pioneers in the field of polymer mechanochemistry, reported mechanically induced proton releasing for the first time in 2012.26 Based on previously reported thermally induced elimination of gem-dihalocyclopropane systems, Moore et al. attached gem-dichlorocyclopropane (gDCC) onto indene for stabilizing intermediates in the elimination process. Mechanochemical rearrangement occurred at the beginning and aromatization elimination followed to emit HCl (Fig. 6a). The designed mechanophore 5b was covalently incorporated into PMA as the crosslinker, while mechanophore 5a was introduced into PMA in which ethyleneglycol dimethacrylate (EGDMA) served as the crosslinker instead of 5b to obtain the unbound analogue (Fig. 6b). These PMAs were compressed by pellet press under different pressures. The activation of mechanophores 5a and 5b could be analysed qualitatively and quantitatively. Both the compressed and uncompressed samples had an exothermic peak at the same location in differential scanning calorimetry (DSC) profiles corresponding to thermal activation (Fig. 6c). The DSC trace of uncompressed PMA gave a standard curve, and the reduced exothermic peak area correlated with mechanical activation to calculate activation rates. The activation of 5b increased with applied pressure, while the activation ratio of 5a only reached 6% under maximum pressure (352 MPa), whose slight activation could be attributed to thermal instability. The acid generation is further indicated by new aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch peaks in confocal Raman spectra and colour change of a pH indicator (methyl red) form orange to pink (Fig. 6d). The released protons inside the polymers could participate in the next reaction as a catalyst or a reactant to harvest various functions, such as self-healing.
C stretch peaks in confocal Raman spectra and colour change of a pH indicator (methyl red) form orange to pink (Fig. 6d). The released protons inside the polymers could participate in the next reaction as a catalyst or a reactant to harvest various functions, such as self-healing.
 | ||
| Fig. 6 (a) Acid-releasing pathway based on gem-dichlorocyclopropanated indene. (b) Structures of mechanophore-containing polymers. (c) Exotherm in the DSC analyses of PMA containing 5 mol% of EGDMA and 5 mol% of 5a (red); 5 mol% of 5b (blue); 5 mol% of 5b after compression with 352 MPa load (green); and 5 mol% of EGDMA (black). Integration of the peak gives energy density, which is correlated with the quantity of 5a and 5b in the material. DSC conditions: 25–200 °C, 5 °C min−1. (d) Methyl red in acetonitrile added to compressed polymers containing unbound 5a (left) and covalently bound 5b (right). Reproduced with permission.26 Copyright 2012, American Chemical Society. | ||
The application of gem-dichlorocyclopropanated indene is limited owing to its thermal sensitivity (degrades above 40 °C). To overcome this drawback, in 2016, Moore's group discovered thermally stable photoacid generator-oxime sulfonate with mechanical activity.45 The PMA containing oxime sulfonate moiety could break at the S–O bond to release sulfonyl (RSO2⋅) radicals under ultrasound in accordance with CoGEF (Constrained Geometries simulate External Force) simulations, forming aryl sulfonic acid in the presence of H2O (Fig. 7a). They synthesized polymer 6 with two oxime sulfonates; other polymers and pure PMA were also prepared for contrast (Fig. 7b). GPC measurement evidenced the first-order reaction kinetics of polymer 6 and PMA. The chain scission rate of polymer 6 almost doubled as compared to the PMA control (13.6 × 10−5 min−1 kDa−1versus 7.1 × 10−5 min−1 kDa−1), presenting specific activation of oxime sulfonate at the chain centre, which was also supported by growing signal peaks at −66.60 ppm and −71.53 ppm in the 19F NMR spectra during sonication (Fig. 7c). The main products, ketone and sulfonic acid were characterized by the new peak (−71.53 ppm) in the 19F NMR spectra and the pH decreased (Fig. 7d) after sonication. Nevertheless, the supposed by-products NOx were not observed, probably due to limitation of the test method and its physicochemical properties.
 | ||
Fig. 7 (a) Acid-releasing pathway based on oxime sulfonate. (b) Structures of polymers in experiments. (c) 19F NMR spectra of the polymers. CFCl3 (δ = 0 ppm) was used as an internal standard. (d) Change in pH with sonication time. The ultrasonicated polymer solution (1 mg mL−1 in CH3CN) was concentrated, dried under vacuum, and CH3CN/H2O (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was added to obtain a 3 mg mL−1 polymer solution for pH measurements. Reproduced with permission.45 Copyright 2016, American Chemical Society. :1 v/v) was added to obtain a 3 mg mL−1 polymer solution for pH measurements. Reproduced with permission.45 Copyright 2016, American Chemical Society. | ||
Although oxime sulfonate could effectively release protons, one proton was only obtained per chain due to its scissile character. Very recently, Craig et al. summarized shortcomings of the above two cases and replaced indene with methoxy in the gDCC system.46 Methoxy could not only ensure elimination like indene but also provided stability (no decomposition in 110 °C). Furthermore, one polymer chain with multiple methoxy-substituted gDCCs (MeO-gDCCs) could release more protons. The opened cyclopropane in the mechanophore formed an intermediate that was eliminated to deliver HCl and three subchains distinguished in 1H NMR spectra, as expected (Fig. 8a). 67 protons were estimated to release per chain on average when scission occurred. They also introduced MeO-gDCC and rhodamine into poly(dimethylsiloxane) (PDMS). It was noted that the rhodamine was monosubstituted, which could avoid mechano-activation and was synthesized easily. The mechanochemically released HCl induced the ring-opening of rhodamine, leading to a planarized structure and colour-switch under ambient light and UV light (Fig. 8b). The overall mechanochromism could be regarded as an application of proton-releasing, as mentioned before. Interestingly, the colour did not change immediately under force by virtue of slow proton diffusion in the bulk polymers. The reproducible and well-established two-phase exponential fitted UV absorbance kinetics could realize “time-stamped” mechanochromism, i.e., tracing the initial time of mechanical activation according to current absorbance.
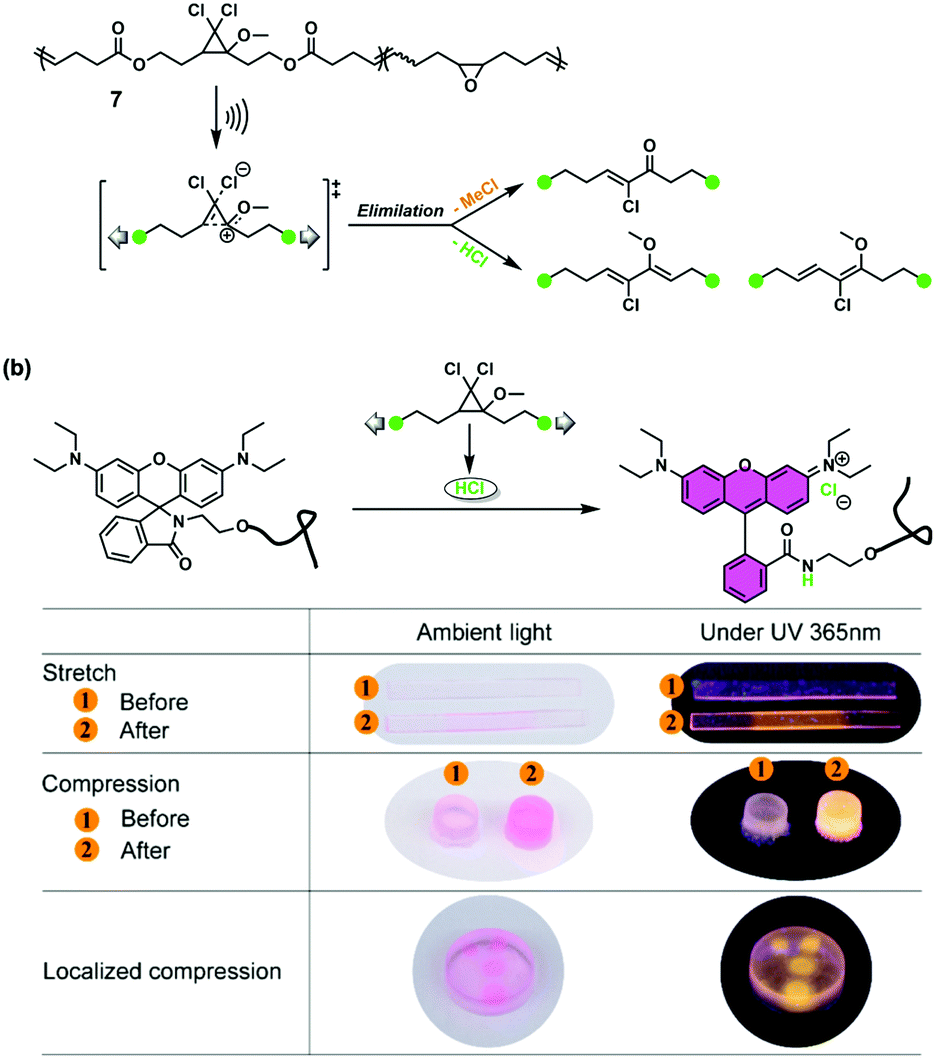 | ||
| Fig. 8 (a) Acid-releasing pathway based on MeO-gDCC. (b) Demonstration of using a mechano-acid to indicate mechanical load under stretching, compression, and localized compression in PDMS elastomer. Protonated rhodamine shows a pink colour under ambient light and yellow fluorescence under UV 365 nm light. Reproduced with permission.46 Copyright 2020, American Chemical Society. | ||
Besides released protons, alcohols also could be obtained in a similar way.47 Recently, Robb et al. designed a furan–maleimide Diels–Alder (DA) adduct with proved mechanochemical activity. The DA adduct was mechanically transformed into metastable furfuryl carbonate, and this unstable intermediate could be decomposed to liberate alcohols by proton transfer at room temperature in a polar protic solvent, such as methanol or water (Fig. 9a). In addition, the α-methyl-substituted DA adduct showed relatively low reaction energy barrier of the corresponding furfuryl carbonate revealed by density functional theory (DFT) calculations. To confirm its reactivity, fluorogenic furfuryl carbonate in the solution of methanol and acetonitrile was monitored (Fig. 9b). As expected, peaks of hydroxycoumarin and furfuryl methyl ether grew in 1H NMR spectra and the fluorescence intensity of hydroxycoumarin in the photoluminescence spectra increased over time, finally determining 98% conversion. To discuss the feasibility of this cascade reaction, polymer 8 was sonicated in methanol and acetonitrile at 0 °C (Fig. 9c). The mechanically triggered reaction cascade did occur with dramatic photoluminescence intensity increase after 150 min, which could be observed by the naked eye (Fig. 9d). In particular, this approach could release a series of alcohols according to different terminal substituents (R in Fig. 9a), which might be more applicable than the specific process of releasing acid.
 | ||
| Fig. 9 (a) Mechanically triggered reaction cascade resulting in small molecule release. (b) Decomposition of furfuryl carbonate in 3:1 MeCN:MeOH generates fluorescent hydroxycoumarin and furfuryl methyl ether. (c) Structures of PMA containing a chain-centred mechanophore equipped with a fluorogenic coumarin probe and a chain-end functional control polymer. (d) Fluorescence spectra of 8 and PMA-control before and after ultrasonication for 150 min. The inset shows photographs of the sonicated solutions excited with 365 nm UV light after 6× dilution and addition of 5% water. Reproduced with permission.47 Copyright 2019, American Chemical Society. | ||
In general, both proton and alcohol release were initiated by homolytic mechanochemical scission in polymer mechanochemistry. However, there were some heterolytic mechanism-controlled cascade reactions coupling with small-molecule release. Moore and co-workers reported the mechanochemically heterolytic cascade unzipping of low-ceiling-temperature poly(o-phthalaldehyde) (PPA) in 2014.29 They synthesized cyclic PPA 9 (Fig. 10a), dissolved it in dilute THF solution, and conducted sonication at −15 °C. The evident decrease of RI signal in GPC over time indicated unzipping from polymers to monomers compared with linear PMA under the same conditions (Fig. 10b and c). They used ab initio steered molecular dynamics calculations to investigate the mechanism inside the unzipping. The cyclic PPA 9 broke to form linear PAA with hemiacetalate and oxocarbenium end groups under force, and then the monomer was quickly released through rearrangement until the molecular weight of PAA reduced to its mechanochemical threshold (Fig. 10a). This simulation revealed the heterolytic mechanism instead of homolytic dissociation, and the authors supposed head-to-tail depolymerization after the formation of the cation and anion. The mechanism was further confirmed by obvious suppression of RI decrease after trapping hemiacetalate and oxocarbenium respectively during sonication of linear PAA. Impressively, the released monomers could recombine to form PPA under anionic polymerization, which was similar to remodelling of biomaterials, expected to extend the lifetime of PAA by recycling.
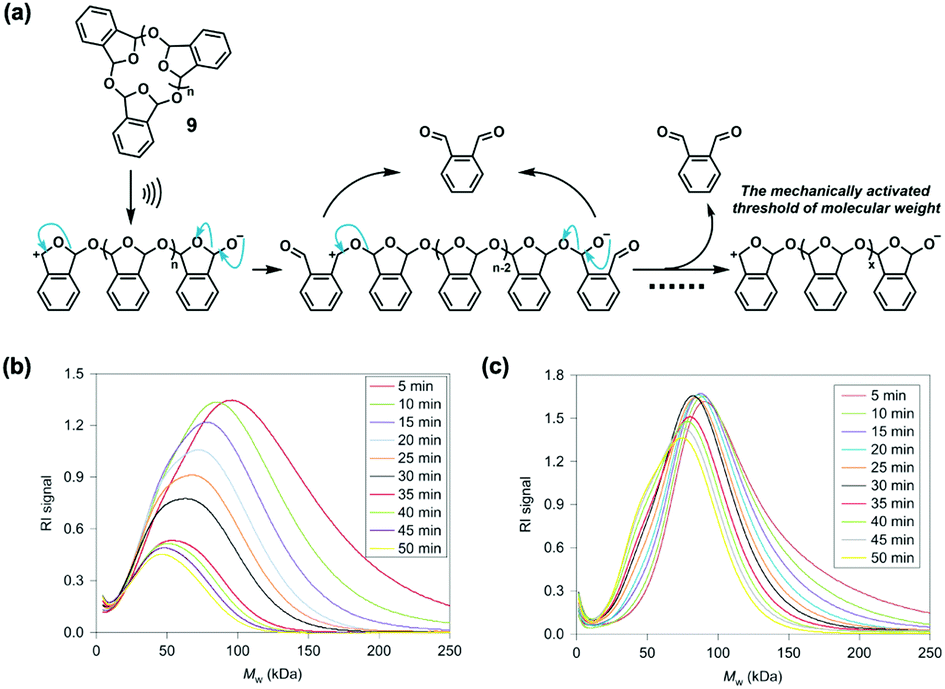 | ||
| Fig. 10 (a) Supposed cascade unzipping based on ab initio steered molecular dynamics calculations. (b) GPC study of the sonicated polymer of 9 (90 kDa). (c) GPC study of the sonicated polymer of linear PMA (103 kDa). Reproduced with permission.29 Copyright 2014, Springer Nature. | ||
Another type of force-activated and heterolytic released cargo was metal ions from metallocene despite its high resistance to heat, UV light, acid and base. In 2018, Fromm and her co-workers firstly confirmed the mechanochemical activity of ferrocene.27 Ferrocene-cantered PMA (10a) was sonicated in dilute THF solution at 0 °C, monitored by SEC. SEC trace revealed that the Mn roughly halved from 133 kDa to 52 kDa after 90 min (elution time: from 12 min to 12.7 min), indicating the specific activation on ferrocene (Fig. 11b). The colourless solution turned red after ultrasound by adding KSCN, which further proved the release and oxidation of iron ions. However, they did not study the mechanism in detail. Soon after, Tang et al. confirmed heterolytic dissociation through experiments and simulation,48 and ferrocene preliminarily turned into zwitterion form under sonication and decomposed to produce ferrous ion immediately (Fig. 11c). They synthesized copolymer 10b with gDCC as an internal standard. The ring-opening ratio of gDCC versus scission cycle could be used to compare mechanochemical activity due to competition between ferrocene and gDCC. After adding tetrabutylammonium bromide (TBAB) salt, the slope reduced from 0.22 to 0.12 (Fig. 11d), implying higher mechanochemical activity of ferrocene, which was achieved by stabilizing the zwitterion with TBAB in heterolytic dissociation. They further captured ferrous ions with phenanthroline directly. When polymer 10c was sonicated in DMF with phenanthroline, two new peaks at 510 nm and 620 nm arose in the UV-vis absorption spectra after sonication (Fig. 11e). The former corresponded to the characteristic peak of Fe(phenanthroline)32+ and the latter might be the complex of half-sandwich ferrocenium and phenanthroline. The heterolytic mechanism was also supported by COGEF calculations. Similarly, they also discussed mechanochemical dissociation of the ruthenocene.49
 | ||
| Fig. 11 (a) Structures of polymer containing ferrocene. (b) Size-exclusion chromatography (SEC) traces revealing an increase of the elution time (concomitant with a decrease of the molecular weight) upon ultrasonication of a 10a solution for the time indicated (in min). (c) Proposed heterolytic dissociation mechanism of a ferrocene-containing polymer by ultrasound-induced chain scission. (d) Ring-opening ratio of gDCC versus scission cycle for 10b and organic salt-stabilized 10b. (e) UV-vis absorption spectra of 10c mixed with phenanthroline before and after sonication. (a and b) Reproduced with permission.27 Copyright 2018, John Wiley and Son. (c–e) Reproduced with permission.48 Copyright 2018, American Chemical Society. | ||
2.3. Self-strengthening
Some active species, including reactive groups and radicals, are commonly generated in the mechanochemical process. Crosslinking of polymers or formation of new polymer networks in the presence of crosslinkers or/and monomers can improve the strength of the initial polymers by self-strengthening. This strategy autonomously reinforces polymeric materials when destructive force is applied.Craig's group reported the mechanochemical strengthening both in solution and in bulk polymers in 2013.50 They proposed the concept of ARM (Activated Remodelling via Mechanochemistry). Extrusion and sonication were conducted on polymers containing gem-dibromocyclopropanes (gDBCs, 11closed); gDBCs underwent 2-electron electrocyclic ring-opening reaction to turn into 2,3-dibromoalkene (11open); then 11open reacted with di-tetrabutylammonium salt of sebacic acid (TBA SA) through nucleophilic displacement, resulting in crosslinking networks (Fig. 12a and b). In their case, crosslinking was favoured over chain scission, and the self-strengthening from being destructive to productive was realized ultimately. They first extruded bulk poly(butadiene) (PB) containing gDBCs mixed with TBA SA; the insoluble part of the extruded polymers indicated crosslinking, which was supported by the disappearance of the TBA SA carboxylate stretch at 1571 cm−1 and the appearance of a new carbonyl stretch at 1721 cm−1 in Fourier transform infrared spectroscopy (FTIR). The polymers showed two orders of magnitude increase of elastic modulus measured by nanoindentation before (2 ± 0.5 MPa) and after (150 ± 85 MPa) extruding, indicating notable promotion of polymer strength. The elastic modulus of the extruded sample even reached up to 280 ± 61 MPa after one week. In a solution state, crosslinking could be detected by observed gelation, and also exhibited in the GPC trace. They further simplified the two-component system by introducing negatively charged ester groups into gDBCs-PB to obtain the single-component system with a more dramatic response. Interestingly, the well-studied gDCC could not achieve crosslinking due to its low reaction activity.
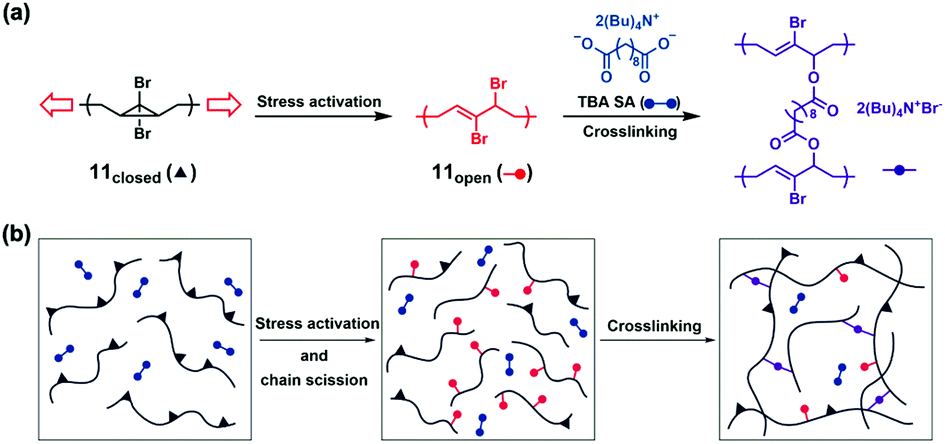 | ||
| Fig. 12 (a) A gDBC mechanophore within a polymer chain under tension undergoes a ring-opening reaction from 11closed to 11open. This increases the contour length and provides an allylic bromide that is capable of self-strengthening through nucleophilic displacement reactions. (b) System-wide force causes chain scission, but also activates the mechanophore (black triangle to red dot), which subsequently reacts with a crosslinker (blue) to form an active crosslink (purple) that overcomes the damage. Reproduced with permission.50 Copyright 2013, Springer Nature. | ||
In 2015, Craig's group presented the second self-strengthening system based on mechano-sensitive benzocyclobutene (BCB) (Fig. 13a).51 One BCB in the polymer skeleton was proved to undergo mechanical activation and achieved interchain crosslinking without a cross-linker. Therefore, they synthesized polymer 12 with multiple BCB, whose Mn increased over sonication time in THF solution (25 mg mL−1) but no gel was formed. When the bismaleimide cross-linker was added at the same concentration, faster gelation than for the gDBC/TBA SA system was observed (Fig. 13b), potentially attributed to the higher reaction activity between ortho-quinodimethide (oQDM) and maleimide than self-crosslinking of oQDMs and chain scission. The bismaleimide cross-linkers were indeed incorporated into the insoluble gel according to the appearance of CO stretch and succinimide C–N–C stretch along with the disappearance of maleimide C–N–C stretch in FTIR. The storage modulus increased by three orders of magnitude during gelation (undetected versus 500–600 Pa). Compared to the gDBC system, this system is more effective and unreactive oQDMs will return to BCB. Moreover, replacing ionic SA with a bismaleimide cross-linker is in favour of practical applications.
 | ||
| Fig. 13 (a) Two crosslinking pathways of oQDM intermediates generated through ring-opening of BCBs in polymer 12 under pulsed ultrasonication. (b) Solution of 25 mg mL−112 + bismaleimide cross-linker before (left) and after (middle) sonication and following removal from the Suslick vessel (right). Reproduced with permission.51 Copyright 2015, American Chemical Society. | ||
Weng et al. continued to expand the library of self-strengthening systems with spirothiopyran (STP) in 2016.52 Colourless STP can convert to green thiomerocyanine (TMC) under mechanical stimuli, similar to spiropyran (SP). Both in solution and in the solid-state, colour switch from yellow to green was observed upon sonication and stretching, respectively. The metastable TMC could be further trapped by CC bonds, such as N-ethylmaleimide (NEM), through thiol Michael addition (Fig. 14a). For crosslinking purposes, 1,6-bismaleimidohexane (BMH) was chosen as the cross-linker, sonicated with linear polyester 13 containing multiple STPs. At the initial stage of sonication, the solution turned green because of mechanochemical isomerization. After 20 min, the solution recovered to yellow and insoluble precipitate began to attach to the walls of the sonication cell (Fig. 14b). The evolution of colour (yellow-green-yellow) indicated mechanochromism of STP, and subsequent capture of TMC resulted in a crosslinked polymer precipitate, which was characterized by FTIR. No direct evidence of crosslinking was found in the bulk polymers. Similar to the BCB system, reversibility of STP enables multiple crosslinking. The combination of mechanochromism and crosslinking may apply in stress-sensing and subsequent remodelling within polymers.
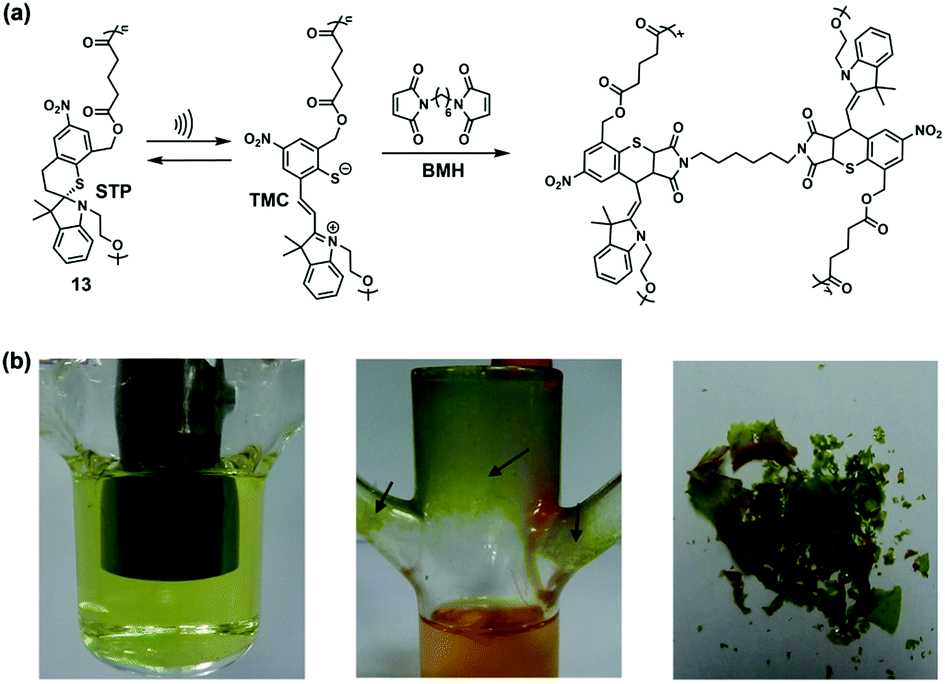 | ||
| Fig. 14 (a) Crosslinking pathway based on the STP system. (b) The sonication apparatus before (left) and after (middle) 20 min of sonication, and the material peeled from the walls of the sonication cell (right). Reproduced with permission.52 Copyright 2016, John Wiley and Son. | ||
Later, Sijbesma et al. explored the second mechanochromism-crosslinking system using colourless hexaarylbiimidazole (HABI).53 HABI can cleave to coloured triphenylimidazolyl (TPI) radicals under force, and TPI initiates radical polymerizations mediated by thiol chain transfer agents. Although they only obtained crosslinked oligomers, the method of machanoradicals provided a novel platform to achieve self-strengthening. In 2017, Kloxin et al. embedded trithiocarbonate (TTC) moieties into linear poly(n-butyl acrylate) (PBA) via addition–fragmentation chain-transfer (RAFT) polymerization.54 The S–C bond of the obtained polymer 14 could preferentially break to form radicals triggered by light and force. The radicals then initiated radical-based polymerization to produce polymer networks (Fig. 15a). They used tetraethylene glycol diacrylate (TEGDA) as a monomer and crosslinker, sonicated polymer 14 in THF, and observed gelation within 10 min (Fig. 15b). To prove the generality of this method, they used triethylene glycol divinyl ether (TEGDVE) and pentaerythritol tetra(3-mercaptopropionate) (PETMP) to conduct thiol–ene polymerization initiated by mechanoradicals, and the gel was observed as well (Fig. 15c). It should be mentioned that the random chain scission can also generate radicals to induce polymerization despite lower efficiency than specific S–C cleavage, which offers an idea to implement the mechanoradical-induced crosslinking or strengthening without using elaborate mechanophores.
 | ||
| Fig. 15 (a) Crosslinking pathways based on the TTC system. (b) Vials are inverted to indicate the formation of the gel after sonicating the liquid solution containing 14 and TEGDA. (c) Vials are inverted to indicate the formation of the gel after sonicating the liquid solution containing TEGDVE, PETMP and 14. Reproduced with permission.54 Copyright 2017, The Royal Society of Chemistry. | ||
Recently, Gong et al. designed an open system based on double-network (DN) hydrogel to achieve mechanochemical self-strengthening, even self-growing, analogous to hypertrophy and strengthening of muscles after training mechanically.55 The DN hydrogel consisted of a rigid and brittle first network, a soft and stretchable second network and dominant water. The pre-stretched first network could give sufficient mechanoradicals through covalent bond scission in polymer chains under mechanical load, while the second network retained the integrity of the hydrogel. Due to great permeability of the hydrogel, the mechanoradicals then induced the polymerization of monomers coming from the surroundings to form new networks, whose properties depended on different types of monomers (Fig. 16a). They chose poly(2-acrylamido-2-methylpropanesulfonic acid) sodium salt (PNaAMPS) and poly(acrylamide) (PAAm) as the first and second networks respectively (Fig. 16b). As a prerequisite, the formed mechanoradicals of the determined DN hydrogel were 1–2 orders of magnitude more than for the single network (SN) hydrogel, guaranteeing the subsequent crosslinking. NaAMPS and N,N′-methylenebisacrylamide (MBA) were used as monomers (Fig. 16b), the same as the first network, enabling repeated formation of mechanoradicals during loading–unloading cycles. The DN hydrogel connected to a weight was placed in the solution of monomers and was lifted to the same height in cycles. The enhancement of size and strength were qualitatively visualized by more obvious appearance of the hydrogel in the load-free state and higher altitude of weight in a lifting state, respectively (Fig. 16c). It was noted that the DN hydrogel failed after 5–6 cycles on account of more brittle networks inside, and the monomers were depleted as well, limiting its applications.
 | ||
| Fig. 16 (a) Crosslinking pathways based on an open DN hydrogel system. (b) Chemical structure and schematic representation of the polymeric network structure of the DN gel and used monomers. (c) Optical images showing the growth in strength and length of a DN gel caused by repetitive mechanical training. An aqueous solution of 0.08 M NaAMPS and 0.08 M MBA (cross-linker) was used in the experiment depicted here. Reproduced with permission.55 Copyright 2019, American Association for the Advancement of Science. | ||
2.4. Gating
The gating concept, i.e., desired chemical reactions only happen after exerting specific stimuli, is commonly used in photochemistry to regulate some chemical processes, even mechanochemical scission.56 In this section, we introduce three exploratory examples in polymer mechanochemistry, which use mechanophores to gate other stimuli-responsive reactions including mechanochemical ring-opening, photoswitching and degradable reaction. The mechanochemical gating is expected to provide a novel method to control the reactivity of chemical reactions.Craig et al. proposed the gating concept in polymer mechanochemistry firstly in 2016.57 The molecular gate can protect the mechanophore substrate from mechanical activation until the gate is unlocked mechanically (Fig. 17a). In that report, they chose cyclobutane and cis-gDCC as the gate and substrate, respectively, to obtain 5,5-dichlorotricyclo(7.2.0.0)undecane (DCTCU) embedded in polymer 15a and 15b (Fig. 17b). According to DFT calculations, cyclobutane and cis-gDCC experience ring-opening processes in turn accompanied by dissociation of three C–C bonds as expected. In single-molecule force spectroscopy (SMFS), the experimental results were in good agreement with the theoretical predictions. Extra >0.8 nN force was needed to activate cis-gDCC in 15a compared to poly(cis-gDCC) (2.2 ± 0.15 nN vs. 1.3 nN), as a result of cyclobutene-gating. In sonication experiments, the ratio of activated cyclobutene and activated cis-gDCC in 15a was 1:1 determined from 1H NMR spectra, whereas 1:3.5 for 15b. The activation of ungated cis-gDCC was 2.5-fold excess than DCTCU, indicating the protection of gating. It is noted that the activation free-energy barrier of the loaded-gate must surpass the loaded substrate but is less than the unloaded substrate, which enables orderly gated mechanochemical reactions. Interestingly, this gated system not only tuned the mechanochemical reactivity of cis-gDCC but also controlled the configurations of the product despite no complete experimental data.
 | ||
| Fig. 17 Schematic representation and molecular design of mechanical reaction gating. (a) A molecular gate (red block) prevents a protected mechanophore substrate (green) from feeling force. Applying sufficiently high force to the gate unlocks it mechanically, allowing the force to be transmitted to the substrate. (b) A cyclobutane mechanophore functions as the gate, and its mechanical cycloreversion unlocks the system so that initially protected cis-gDCC is activated. Structures of the gated system (15a, 15b) are shown below. Reproduced with permission.57 Copyright 2016, Springer Nature. | ||
The method of mechanochemical gating can also regulate photochemical reaction in addition to mechanochemical reactivity.58 In 2018, Robb and co-workers used thermally stable cyclopentadiene–maleimide DA adduct to achieve mechanochemically gated photoswitching. The mechanoresponsive DA adduct in PMA underwent retro [4+2] cycloaddition to give diarylethene (DAEopen) and maleimide under force. Colourless DAEopen turned to coloured DAEclosed through 6π electrocyclic ring-closing reaction under UV light, and this photochromic process could be reversibly triggered by visible light as well, while the initial polymer 16 is non-photochromic due to lacking a 6π electronic structure (Fig. 18a). In sonication experiments of 16, Mn of 16 approximately halved from 90 kg mol−1 to 48 kg mol−1, revealing its mechanochemical activity. The average rate of the selective scission was 11.2 × 10−5 kDa−1 min−1. Moreover, a new peak at 460–550 nm emerged in the trace of GPC equipped with a UV-vis detector after irradiating the sonicated solution with UV light, corresponding to the daughter polymers, which indicated the mechanochemical formation of photochromic species (Fig. 18b). Under UV light, the UV-vis absorption spectra of the initial polymer 16 remained unchanged, while a broad peak centred at 505 nm (characteristic absorption peak of DAE) appeared along with the visible colour change from colourless to red after 90 min sonication (Fig. 18c), directly reflecting mechanochemically gated photoswitching.
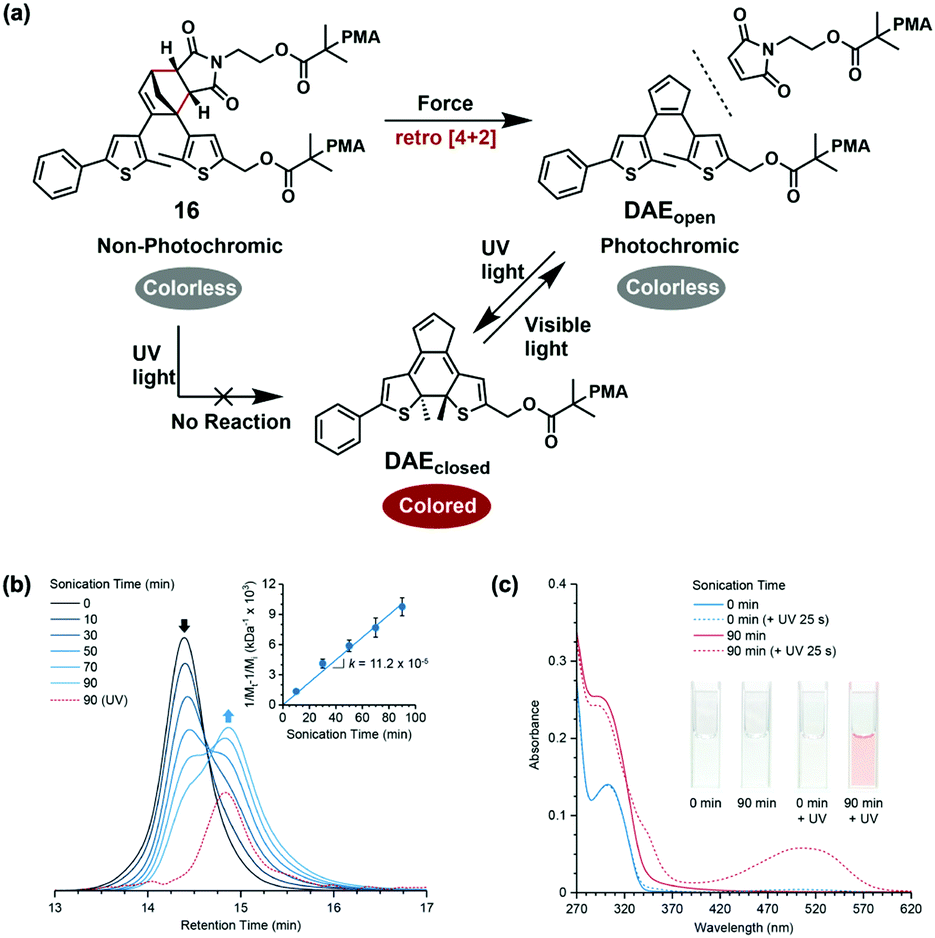 | ||
| Fig. 18 (a) Mechanochemically gated photoswitching using a cyclopentadiene–maleimide mechanophore. (b) GPC traces (RI) and molecular weight data for polymer 16 as a function of ultrasonication time. (c) UV-vis absorption spectra of polymer 16 before and after ultrasonication and UV irradiation demonstrating mechanochemically gated photoswitching. Reproduced with permission.58 Copyright 2018, American Chemical Society. | ||
Very recently, Craig and co-workers continued to include degradable reaction in the scope of mechanochemical gating.59 Acid-responsive ketal was gated by reported cyclobutane to realize more effective degradation on-demand, i.e., mechanically unlocking “locked” degradability if necessary (Fig. 19). They synthesized polymer 17 to confirm its gating features. Mn was slightly decreased from 129 kDa to 120 kDa when adding trifluoroacetic acid (TFA) into the polymer solution, attributed to the cleavage of ketal and indicating that the cyclobutane could protect the polymer from degradation. Then they subjected ultrasound to 17 and both cyclobutane and gDCC were activated. The comparable ring-opening ratio of gDCC per average chain scission between polymer 17 and polybutadiene (PB) containing gDCC based on COGEF simulations reflected mechanochemical inertness of the exposed ketal, which would not influence subsequent degradation. The sonicated solution was further treated with TFA; shorter polymers (similar to acid-treated 17) and some oligomers (∼1 kDa) were recognized both in 1H NMR and GPC measurements. After 240 min of sonication, the ultimate Mn was 2.5 kDa by acid-treatment, which was approximately 1/11 of mechano-treating alone (28 kDa). This gated degradation not only improved resistance to acid triggers in the environment but also was more effective by combining acid- and mechano-treatment. Furthermore, the ketal could be replaced with other degradable moieties to obtain more stimuli-degradable polymers, such as the cyclobutane-lactone (CBL) system reported by Wang et al.60
 | ||
| Fig. 19 Mechanochemically gated degradable reaction using polymer 17. Reproduced with permission.59 Copyright 2020, American Chemical Society. | ||
2.5. Mechanochromism
The visible change in emission colour (spectra) under mechanical load is called “mechanochromism”. In addition to (Cu–NHC)-catalysed fluorescent “click” reaction and “time-stamped” mechanochromism based on proton-releasing from MeO-gDCC mentioned above, two other mechanochromic processes are discussed in this section. It seems to be more intricate for cascade-enabled mechanochromism than one-step mechanochromism, but the cascade process can harvest more marvellous functions (such as electrical conductivity switch) or avoid the laborious design of mechanophores.In 2012, Grzybowski et al. found that macroscopically deformed PDMS could release H2O2 in the presence of water based on mechanoradicals which initiated some chemical reactions, such as nanoparticle synthesis, dye bleaching, or fluorescence activation.61 Inspired by this reaction cascade, in 2014, Goodwin et al. developed a similar process in hydrogels.62 They proposed that the mechanoradicals in polymers reacted with surrounding water molecules to form hydroxyl radicals that activated 3′-(p-aminophenyl) fluorescein (APF) to enhance green emission (Fig. 20a). It was noted that some other active species also appeared such as hydrogen peroxide and hypochlorite anions. All hydrogels were prepared from either 4arm-PEG-SH, 4arm-PEG-acrylate, or both, resulting in PEGs with various crosslinkers (disulfide 18a, thiol–acrylate 18b, acrylate 18c), and 10 μM APF was involved (Fig. 20b). During compression experiments, the green fluorescence intensity increased over strain, and it was more obvious for hydrogels with the smallest content of polymers. Among these hydrogels, activation of APF was the easiest in 18a, followed by 18b, then 18c, fitting well with the order of bond dissociation energy.
 | ||
| Fig. 20 (a) Proposed mechanism of fluorescence activation by mechanical stress. (b) Structures of PEG precursors and synthesized PEGs with different crosslinkers. Reproduced with permission.62 Copyright 2014, American Chemical Society. | ||
Xia and co-workers arduously synthesized insulating polyladderenes 19a through ROMP in 2017.63 It could convert to semiconducting polyacetylene (PA) due to long conjugation length (>100 conjugated CC bonds) revealed by mechanochemical unzipping (Fig. 21a). Mechanochromism detection showed that the pristine colourless polymer solution turned to blue and further to dark blue/purple during sonication (Fig. 21b). Notably, only trans-PA was observed in Resonant Raman spectra of the sonicated solution, potentially attributed to the unique mechanochemical reaction pathway. The unzipping cascade of ladderene was simulated by using force-modified potential energy surface, where two barriers existed corresponding to the structures with 1 or 3 opened rungs without external force. The negligible barriers made the unzipping spontaneous when 3 nN force was exerted, which was consistent with all-or-none unzipping under 3.1 nN in ab initio steered molecular dynamics calculations. In 2019, their group improved the ladderane system by introducing a benzo group, i.e., benzoladderene (Fig. 21a).64 The homopolymers or copolymers of benzoladderenes 19b were easier to synthesize at gram quantities than polyladderenes 19a. Because of the introduction of a benzo group, on one hand, the elimination that could broaden the molecular weight distribution was not needed after ROMP, achieving controlled polymerization. On the other hand, functionalization was also convenient to realize on the benzene ring. The polybenzoladderenes converted to poly(o-phenylene-1,3,5-hexatrienylene) in sonication experiments, along with mechanochromism from colourless to intense yellow (Fig. 21c).
 | ||
| Fig. 21 (a) Mechanochemical formation of conjugated polyacetylene and poly(o-phenylene-1,3,5-hexatrienylene). (b) Photographs of the reaction vessel at different sonication times during sonicating 19a. (c) Photographs of the sonicated 19b solution at different times. (b) Reproduced with permission.63 Copyright 2017, American Association for the Advancement of Science. (c) Reproduced with permission.64 Copyright 2019, American Chemical Society. | ||
3. Summary and outlook
Mechanical forces are secure, attainable and eco-friendly in our daily life. The effect of mechanical energy on chemical reaction remains a challenging problem worth exploring. Polymers are an excellent platform to conduct applied force to the molecular-level mechanophores. Recently, researchers have found that polymer mechanochemistry can be used to generate active species such as radicals, acid, etc. to achieve fantastic functions. Thus, the reaction cascade strategy in polymer mechanochemistry has been attracting tremendous interest in the past several years. This review summarizes the development of polymer mechanochemistry-enabled reaction cascades including mechanocatalysis, cargo-releasing, self-strengthening, gating and mechanochromism. Mechanocatalysis refers to the force-induced latent catalyst activation and the following catalysis reaction. All reported mechanocatalysts are organometallic coordinated complexes and when the coordinate bond is ruptured by force the catalytic performance of the metallic atoms is activated. Cargo-releasing represents small molecules released from the designed mechanophores in the polymers by force. Generally, the generation of cargo undergoes two steps, production of an unstable intermediate under force and then spontaneous decomposition into small molecules. Self-strengthening usually involves the crosslinking of the polymer or formation of new polymer networks triggered by the force-generated active species and then the mechanical property of the polymeric materials is reinforced. Gating means regulating the desired chemical reactions by specific stimuli. Mechanophores embedded in polymers can be used to control other stimuli-responsive reactions, for example, mechanochemical ring-opening, photoswitching or degradable reaction. Cascade-enabled mechanochromism is quite different from one-step mechanochromism and shows some advantages such as harvesting more marvellous functions and avoiding laborious design of mechanophores.Although research on reaction cascades in polymer mechanochemistry has made certain achievements, the related study is still full of challenges but also opportunities. Firstly, the quantity of mechanophores that can realize a reaction cascade is quite limited. Such mechanophores are not only absolutely elaborately designed but also usually laboriously synthesized. Secondly, the force-sensitivity is still quite low. In bulk polymers, the confined environment constricts the mobility of force-generated active species, which greatly limits its practical applications such as self-strengthening, and self-sensing. Improving the free volume and increasing the glass transition temperature of the polymers may be feasible strategies.
Reaction cascade in polymer mechanochemistry is an interesting but challenging research field. It shows new directions for manufacturing novel smart materials and is of great importance for both fundamental research and practical applications in the future.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the Beijing Natural Science Foundation (2182054), the National Natural Science Foundation of China (21704002), the Big Science Project from BUCT (XK180301), and the Fundamental Research Funds for the Central Universities to Z. Y. Ma.References
- M. M. Caruso, D. A. Davis, Q. Shen, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Mechanically-Induced Chemical Changes in Polymeric Materials, Chem. Rev., 2009, 109, 5755–5798 CrossRef CAS PubMed.
- H. Zhang, Y. J. Lin, Y. Z. Xu and W. G. Weng, in Polymer Mechanochemistry, ed. R. Boulatov, 2015, vol. 369, pp. 135–207 Search PubMed.
- J. N. Brantley, K. M. Wiggins and C. W. Bielawski, Polymer mechanochemistry: the design and study of mechanophores, Polym. Int., 2013, 62, 2–12 CrossRef CAS.
- P. A. May and J. S. Moore, Polymer mechanochemistry: techniques to generate molecular force via elongational flows, Chem. Soc. Rev., 2013, 42, 7497–7506 RSC.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Biasing reaction pathways with mechanical force, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- J. M. Lenhardt, M. T. Ong, R. Choe, C. R. Evenhuis, T. J. Martinez and S. L. Craig, Trapping a Diradical Transition State by Mechanochemical Polymer Extension, Science, 2010, 329, 1057–1060 CrossRef CAS PubMed.
- Z. S. Kean, Z. Niu, G. B. Hewage, A. L. Rheingold and S. L. Craig, Stress-Responsive Polymers Containing Cyclobutane Core Mechanophores: Reactivity and Mechanistic Insights, J. Am. Chem. Soc., 2013, 135, 13598–13604 CrossRef CAS PubMed.
- H. M. Klukovich, T. B. Kouznetsova, Z. S. Kean, J. M. Lenhardt and S. L. Craig, A backbone lever-arm effect enhances polymer mechanochemistry, Nat. Chem., 2013, 5, 110–114 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. S. Kean, L. Fan, B. D. Mar, T. J. Martinez and S. L. Craig, A Remote Stereochemical Lever Arm Effect in Polymer Mechanochemistry, J. Am. Chem. Soc., 2014, 136, 15162–15165 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova and S. L. Craig, Reactivity and Mechanism of a Mechanically Activated anti-Woodward-Hoffmann-DePuy Reaction, J. Am. Chem. Soc., 2015, 137, 11554–11557 CrossRef CAS PubMed.
- J. Wang, T. B. Kouznetsova, Z. Niu, M. T. Ong, H. Klukovich, A. L. Rheingold, T. J. Martinez and S. L. Craig, Inducing and quantifying forbidden reactivity with single-molecule polymer mechanochemistry, Nat. Chem., 2015, 7, 323–327 CrossRef CAS PubMed.
- S. Akbulatov, Y. Tian, Z. Huang, T. J. Kucharski, Q. Z. Yang and R. Boulatov, Experimentally realized mechanochemistry distinct from force-accelerated scission of loaded bonds, Science, 2017, 357, 299–303 CrossRef CAS PubMed.
- M. H. Barbee, T. Kouznetsova, S. L. Barrett, G. R. Gossweiler, Y. J. Lin, S. K. Rastogi, W. J. Brittain and S. L. Craig, Substituent Effects and Mechanism in a Mechanochemical Reaction, J. Am. Chem. Soc., 2018, 140, 12746–12750 CrossRef CAS PubMed.
- Y. Lin, Y. Zhang, Z. Wang and S. L. Craig, Dynamic Memory Effects in the Mechanochemistry of Cyclic Polymers, J. Am. Chem. Soc., 2019, 141, 10943–10947 CrossRef CAS PubMed.
- Z. Chen, X. Zhu, J. Yang, J. A. M. Mercer, N. Z. Burns, T. J. Martinez and Y. Xia, The cascade unzipping of ladderane reveals dynamic effects in mechanochemistry, Nat. Chem., 2020, 12, 302–309 CrossRef CAS PubMed.
- D. A. Davis, A. Hamilton, J. Yang, L. D. Cremar, D. Van Gough, S. L. Potisek, M. T. Ong, P. V. Braun, T. J. Martinez, S. R. White, J. S. Moore and N. R. Sottos, Force-induced activation of covalent bonds in mechanoresponsive polymeric materials, Nature, 2009, 459, 68–72 CrossRef CAS PubMed.
- Y. Chen, A. J. H. Spiering, S. Karthikeyan, G. W. M. Peters, E. W. Meijer and R. P. Sijbesma, Mechanically induced chemiluminescence from polymers incorporating a 1,2-dioxetane unit in the main chain, Nat. Chem., 2012, 4, 559–562 CrossRef CAS PubMed.
- G. Hong, H. Zhang, Y. Lin, Y. Chen, Y. Xu, W. Weng and H. Xia, Mechanoresponsive Healable Metallosupramolecular Polymers, Macromolecules, 2013, 46, 8649–8656 CrossRef CAS.
- M. B. Larsen and A. J. Boydston, Successive Mechanochemical Activation and Small Molecule Release in an Elastomeric Material, J. Am. Chem. Soc., 2014, 136, 1276–1279 CrossRef CAS PubMed.
- Z. Wang, Z. Ma, Y. Wang, Z. Xu, Y. Luo, Y. Wei and X. Jia, A Novel Mechanochromic and Photochromic Polymer Film: When Rhodamine Joins Polyurethane, Adv. Mater., 2015, 27, 6469–6474 CrossRef CAS PubMed.
- Z. Li, R. Toivola, F. Ding, J. Yang, P. N. Lai, T. Howie, G. Georgeson, S. H. Jang, X. Li, B. D. Flinn and A. K. Jen, Highly Sensitive Built-In Strain Sensors for Polymer Composites: Fluorescence Turn-On Response through Mechanochemical Activation, Adv. Mater., 2016, 28, 6592–6597 CrossRef CAS PubMed.
- H. Chen, F. Yang, Q. Chen and J. Zheng, A Novel Design of Multi-Mechanoresponsive and Mechanically Strong Hydrogels, Adv. Mater., 2017, 29, 1606900 CrossRef PubMed.
- Y. Sagara, M. Karman, E. Verde-Sesto, K. Matsuo, Y. Kim, N. Tamaoki and C. Weder, Rotaxanes as Mechanochromic Fluorescent Force Transducers in Polymers, J. Am. Chem. Soc., 2018, 140, 1584–1587 CrossRef CAS PubMed.
- J. Park, Y. Lee, M. H. Barbee, S. Cho, S. Cho, R. Shanker, J. Kim, J. Myoung, M. P. Kim, C. Baig, S. L. Craig and H. Ko, A Hierarchical Nanoparticle-in-Micropore Architecture for Enhanced Mechanosensitivity and Stretchability in Mechanochromic Electronic Skins, Adv. Mater., 2019, 31, 1808148 CrossRef PubMed.
- T. Wang, N. Zhang, J. Dai, Z. Li, W. Bai and R. Bai, Novel Reversible Mechanochromic Elastomer with High Sensitivity: Bond Scission and Bending-Induced Multicolor Switching, ACS Appl. Mater. Interfaces, 2017, 9, 11874–11881 CrossRef CAS PubMed.
- C. E. Diesendruck, B. D. Steinberg, N. Sugai, M. N. Silberstein, N. R. Sottos, S. R. White, P. V. Braun and J. S. Moore, Proton-Coupled Mechanochemical Transduction: A Mechanogenerated Add, J. Am. Chem. Soc., 2012, 134, 12446–12449 CrossRef CAS PubMed.
- M. Di Giannantonio, M. A. Ayer, E. Verde-Sesto, M. Lattuada, C. Weder and K. M. Fromm, Triggered Metal Ion Release and Oxidation: Ferrocene as a Mechanophore in Polymers, Angew. Chem., Int. Ed., 2018, 57, 11445–11450 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Polymer Mechanochemistry: From Destructive to Productive, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- C. E. Diesendruck, G. I. Peterson, H. J. Kulik, J. A. Kaitz, B. D. Mar, P. A. May, S. R. White, T. J. Martinez, A. J. Boydston and J. S. Moore, Mechanically triggered heterolytic unzipping of a low-ceiling-temperature polymer, Nat. Chem., 2014, 6, 624–629 CrossRef PubMed.
- S. Akbulatov and R. Boulatov, Experimental Polymer Mechanochemistry and its Interpretational Frameworks, ChemPhysChem, 2017, 18, 1422–1450 CrossRef CAS PubMed.
- L. Anderson and R. Boulatov, in Advances in Physical Organic Chemistry, ed. I. H. Williams and N. H. Williams, 2018, vol. 52, pp. 87–143 Search PubMed.
- N. Willis-Fox, E. Rognin, T. A. Aljohani and R. Daly, Polymer Mechanochemistry: Manufacturing Is Now a Force to Be Reckoned With, Chem, 2018, 4, 2499–2537 CAS.
- E. Izak-Nau, D. Campagna, C. Baumann and R. Goestl, Polymer mechanochemistry-enabled pericyclic reactions, Polym. Chem., 2020, 11, 2274–2299 RSC.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Switchable Polymerization Catalysts, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Activating catalysts with mechanical force, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, Performance of Mechanochemically Activated Catalysts Is Enhanced by Suppression of the Thermal Effects of Ultrasound, ACS Macro Lett., 2012, 1, 1012–1015 CrossRef CAS.
- R. Groote, L. van Haandel and R. P. Sijbesma, The Effect of Molecular Weight and Catalyst Concentration on Catalytic Activity in Mechanochemically Activated Transesterification Using Silver(I)-N-Heterocyclic Carbene Latent Catalysts, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4929–4935 CrossRef CAS.
- R. T. M. Jakobs and R. P. Sijbesma, Mechanical Activation of a Latent Olefin Metathesis Catalyst and Persistence of its Active Species in ROMP, Organometallics, 2012, 31, 2476–2481 CrossRef CAS.
- R. T. M. Jakobs, S. Ma and R. P. Sijbesma, Mechanocatalytic Polymerization and Cross-Linking in a Polymeric Matrix, ACS Macro Lett., 2013, 2, 613–616 CrossRef CAS.
- P. Michael and W. H. Binder, A Mechanochemically Triggered “Click” Catalyst, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS PubMed.
- P. Michael, M. Biewend and W. H. Binder, Mechanochemical Activation of Fluorogenic CuAAC “Click” Reactions for Stress-Sensing Applications, Macromol. Rapid Commun., 2018, 39, 1800376 CrossRef PubMed.
- M. Biewend, S. Neumann, P. Michael and W. H. Binder, Synthesis of polymer-linked copper(I) bis(N-heterocyclic carbene) complexes of linear and chain extended architecture, Polym. Chem., 2019, 10, 1078–1088 RSC.
- M. Biewend, P. Michael and W. H. Binder, Detection of stress in polymers: mechanochemical activation of CuAAC click reactions in poly(urethane) networks, Soft Matter, 2020, 16, 1137–1141 RSC.
- K. Wei, Z. Gao, H. Liu, X. Wu, F. Wang and H. Xu, Mechanical Activation of Platinum-Acetylide Complex for Olefin Hydrosilylation, ACS Macro Lett., 2017, 6, 1146–1150 CrossRef CAS.
- C. Nagamani, H. Liu and J. S. Moore, Mechanogeneration of Acid from Oxime Sulfonates, J. Am. Chem. Soc., 2016, 138, 2540–2543 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, A Latent Mechanoacid for Time-Stamped Mechanochromism and Chemical Signaling in Polymeric Materials, J. Am. Chem. Soc., 2020, 142, 99–103 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, Mechanically Triggered Small Molecule Release from a Masked Furfuryl Carbonate, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- Y. Sha, Y. Zhang, E. Xu, Z. Wang, T. Zhu, S. L. Craig and C. Tang, Quantitative and Mechanistic Mechanochemistry in Ferrocene Dissociation, ACS Macro Lett., 2018, 7, 1174–1179 CrossRef CAS PubMed.
- Y. Sha, Y. Zhang, E. Xu, C. W. McAlister, T. Zhu, S. L. Craig and C. Tang, Generalizing metallocene mechanochemistry to ruthenocene mechanophores, Chem. Sci., 2019, 10, 4959–4965 RSC.
- A. L. Ramirez, Z. S. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Mechanochemical strengthening of a synthetic polymer in response to typically destructive shear forces, Nat. Chem., 2013, 5, 757–761 CrossRef CAS PubMed.
- J. Wang, I. Piskun and S. L. Craig, Mechanochemical Strengthening of a Multi-mechanophore Benzocyclobutene Polymer, ACS Macro Lett., 2015, 4, 834–837 CrossRef CAS.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Mechanochromism and Mechanical-Force-Triggered Cross-Linking from a Single Reactive Moiety Incorporated into Polymer Chains, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- F. Verstraeten, R. Gostl and R. P. Sijbesma, Stress-induced colouration and crosslinking of polymeric materials by mechanochemical formation of triphenylimidazolyl radicals, Chem. Commun., 2016, 52, 8608–8611 RSC.
- M. B. Gordon, S. Wang, G. A. Knappe, N. J. Wagner, T. H. Epps, III and C. J. Kloxin, Force-induced cleavage of a labile bond for enhanced mechanochemical crosslinking, Polym. Chem., 2017, 8, 6485–6489 RSC.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Mechanoresponsive self-growing hydrogels inspired by muscle training, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- J. Kida, K. Imato, R. Goseki, D. Aoki, M. Morimoto and H. Otsuka, The photoregulation of a mechanochemical polymer scission, Nat. Commun., 2018, 9, 3504 CrossRef PubMed.
- J. Wang, T. B. Kouznetsova, R. Boulatov and S. L. Craig, Mechanical gating of a mechanochemical reaction cascade, Nat. Commun., 2016, 7, 13433 CrossRef PubMed.
- X. Hu, M. E. McFadden, R. W. Barber and M. J. Robb, Mechanochemical Regulation of a Photochemical Reaction, J. Am. Chem. Soc., 2018, 140, 14073–14077 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, Mechanically Gated Degradable Polymers, J. Am. Chem. Soc., 2020, 142, 2105–2109 CrossRef CAS PubMed.
- T. G. Hsu, J. Zhou, H. W. Su, B. R. Schrage, C. J. Ziegler and J. Wang, A Polymer with “Locked” Degradability: Superior Backbone Stability and Accessible Degradability Enabled by Mechanophore Installation, J. Am. Chem. Soc., 2020, 142, 2100–2104 CrossRef CAS PubMed.
- H. T. Baytekin, B. Baytekin and B. A. Grzybowski, Mechanoradicals Created in “Polymeric Sponges” Drive Reactions in Aqueous Media, Angew. Chem., Int. Ed., 2012, 51, 3596–3600 CrossRef CAS PubMed.
- K. R. Fitch and A. P. Goodwin, Mechanochemical Reaction Cascade for Sensitive Detection of Covalent Bond Breakage in Hydrogels, Chem. Mater., 2014, 26, 6771–6776 CrossRef CAS.
- Z. Chen, J. A. M. Mercer, X. Zhu, J. A. H. Romaniuk, R. Pfattner, L. Cegelski, T. J. Martinez, N. Z. Burns and Y. Xia, Mechanochemical unzipping of insulating polyladderene to semiconducting polyacetylene, Science, 2017, 357, 475–478 CrossRef CAS PubMed.
- J. Yang, M. Horst, J. A. H. Romaniuk, Z. Jin, L. Cegelski and Y. Xia, Benzoladderene Mechanophores: Synthesis, Polymerization, and Mechanochemical Transformation, J. Am. Chem. Soc., 2019, 141, 6479–6483 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2020 |