
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAll ceramic cathode composite design and manufacturing towards low interfacial resistance for garnet-based solid-state lithium batteries†
Kun Joong
Kim
 a and
Jennifer L. M.
Rupp
*ab
a and
Jennifer L. M.
Rupp
*ab
aElectrochemical Materials Laboratory, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. E-mail: jrupp@mit.edu
bElectrochemical Materials Laboratory, Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 5th October 2020
Abstract
The critical factors that determine the performance and lifetime of solid-state batteries (SSBs) are driven by the electrode–electrolyte interfaces. The main challenge in fabricating all-oxide cathode composites for garnet-based SSBs has been lowering the thermal processing window in which both good contact and low interfacial resistance can be achieved. Here, we report an alternative ceramic processing strategy that enables the fabrication of all-oxide composite cathodes at an unusually low processing temperature without the use of extra sintering additives or a fluid electrolyte (polymer-gel or liquid electrolyte). We present specific examples of the most common LiFePO4 and LiCoO2 cathodes with a Li-garnet (Li7La3Zr2O12, LLZO) solid-electrolyte. We demonstrate an infiltration step to directly synthesize the LiCoO2 cathode from metal salts in a porous LLZO scaffold, resulting in the formation of a composite cathode such as LiCoO2–LLZO on top of a dense LLZO solid electrolyte at a low processing temperature of 700 °C. A promising discharge capacity of 118 mA h g−1 (3–4.05 V) with a low interfacial resistance of 62 Ohm cm2 is realized for LiCoO2 with a lithium anode, whereas critical phase instabilities for LiFePO4 are uncovered. Our findings encourage a move away from synthesis techniques that employ particle mixing and sintering to fabricate composites. We provide a blueprint for circumventing adverse interphase reactions according to chemistry and ceramic thermal processing budgets in the preparation of these ceramic interfaces as well as for increasing the number of reaction sites for high-performing composite cathodes for Li-garnet SSBs. In addition, the ceramic methods presented are scalable and mass manufacturable for the large-scale production of such composite cathodes for future industry.
Broader contextSolid-state batteries based on inorganic solid electrolytes offer safer alternatives to classical lithium-ion batteries due to their non-flammable nature and give prospects of both high energy and power densities. Among them, sulfide- and oxide-based solid-state battery architectures have attracted continuing attention due to their high ionic conductivity and stability with Li metal. Despite the promise of superior chemical and electrochemical stability of oxide-type batteries during cell operation, fabrication difficulties in achieving mechanically rigid and chemically pure interfaces between the cathode and oxide electrolyte have been serious challenges. Oxide batteries typically reveal high interfacial impedance and readily fail to reach the theoretical capacity of the active cathode material. Here, we introduce a low-temperature fabrication route for an oxide-type cathode composite that can enhance the specific capacity and interfacial resistance by avoiding detrimental interfacial reactions and maximizing active reaction sites. We hope to provide the reader with an overview of existing cathode preparation options and new insights that constitute a step forward in unravelling the complex interplay between ceramic processing routes and performance during oxide battery fabrication. |
Introduction
Solid-state batteries (SSBs) based on solid electrolytes and Li metal anodes are considered safer and higher energy alternatives to conventional batteries operating with combustible liquid electrolytes.1 After sustained research efforts, solid electrolytes now possess conductivities that are competitive with those of liquid electrolytes.2 For example, oxide-type Li garnets such as Li7La3Zr2O12 (LLZO) have exhibited conductivities greater than 1 mS cm−1 and can be processed as ceramics in the form of pellets, tapes, and films over a wide temperature range with a wide variety of thicknesses.3,4 Among SSB electrolyte materials, LLZO5,6 offers a relatively wide electrochemical stability window (along with LIPON7–9) as well as compatibility with the high-capacity lithium anode.10 Promising critical current densities as high as 10 mA cm−2 have been demonstrated in lithium symmetrical cells based on tri-layer LLZO electrolytes,11 resulting in their ranking among promising future SSB architectures. From a processing perspective, there have been numerous attempts to achieve good contact between the lithium anode and LLZO electrolyte. Thanks to these efforts, novel strategies to achieve a LLZO–Li interfacial resistance as low as several Ω cm2 are available.3,6,12–16 However, interfacial design and fabrication strategies that result in low interfacial resistance across the LLZO–cathode interface and within the cathode composite require further attention.17,18 For instance, the interfacial resistance for Li-garnet SSB cells has been reported to be readily over thousands of Ω cm2. The state-of-the-art cell consisting of LiCoO2, Li2.3C0.7B0.3O3, and LLZO as the cathode composite, LLZO as the electrolyte, and a Li metal anode exhibits a total interfacial resistance of approximately 270 Ω cm2 at 100 °C, which is predominantly attributed to the resistance of the LLZO–cathode interface.19 The origin of the high interfacial resistance can be generally explained by either the formation of insulating phases at the interfaces at the cell fabrication temperature (700 °C) or weak mechanical bonding between the cathode and LLZO electrolyte if the co-sintering temperature was too low. Nonetheless, recent holistic analysis on the performance of state-of-the-art SSBs has suggested that a high-specific-energy cell requires less than approximately 40 Ω cm2 internal resistance to allow targeted cycling at 1C with more than 90% energy efficiency.20,21 To commercialize these generally established active materials of Li cobaltite, Li phosphate, and Li titanate for future Li-garnet SSB products, appropriate ceramic synthesis routes to prepare suitable LLZO–cathode composites need to be investigated.Recent progress on such cathode composite processing in Li-garnet SSBs has led to the identification of three major strategies for their assembly that define the thermal processing window, tested active materials and loading (wt%) of the active cathode storage material: (i) hybrid cathodes, (ii) all-oxide cathodes with additives, and (iii) all-oxide cathodes without additives, as illustrated in Fig. 1a.
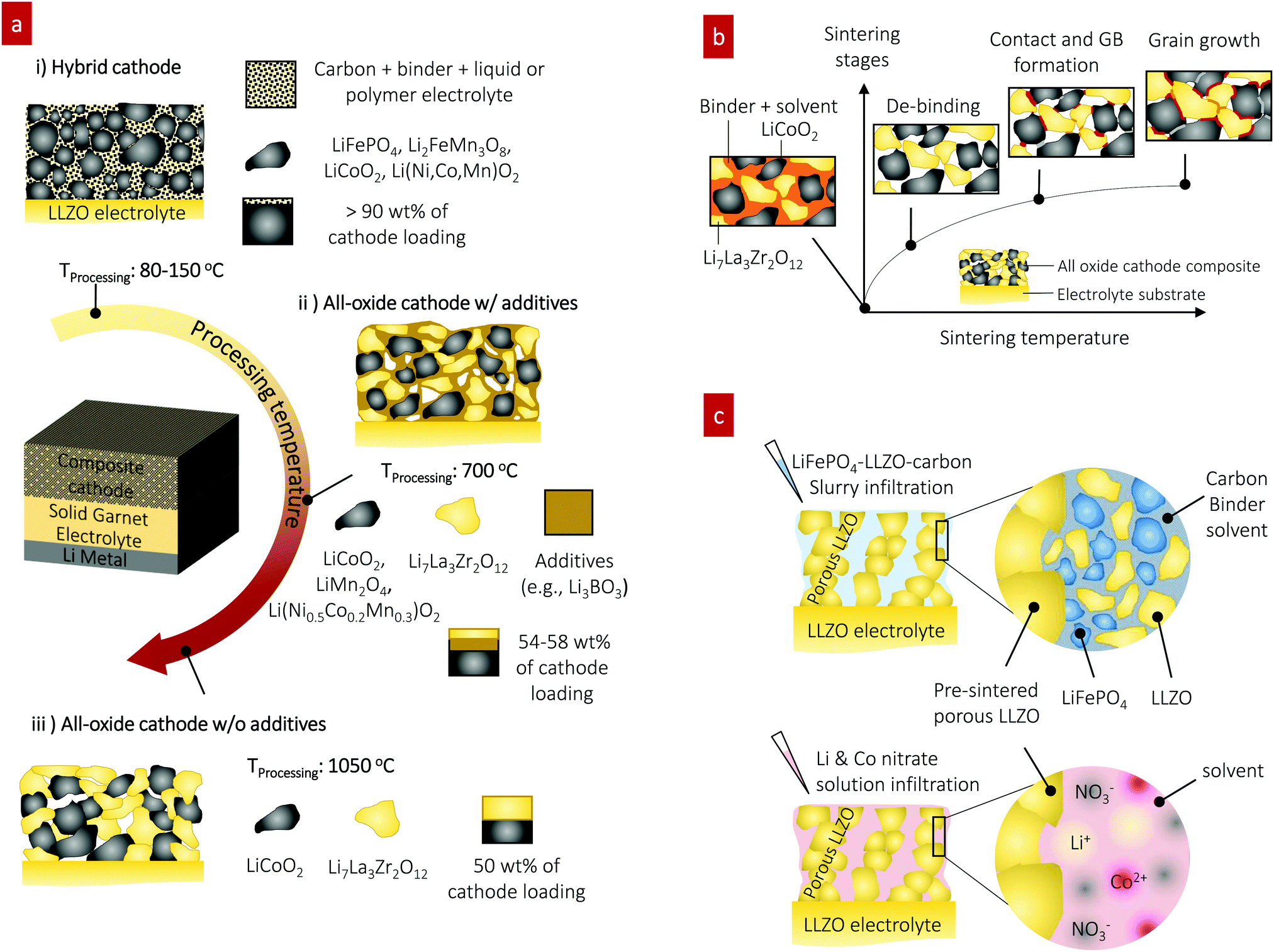 | ||
| Fig. 1 Preparation of cathode composites for Li-garnet SSBs. (a) Overview of the cathode composite processing, temperature and components for each choice of design. (i) Hybrid cathodes using a conventional catholyte (liquid or polymer electrolyte) and tested with active materials of LiCoO2,22 LiFePO4,23–25 Li(Ni,Co,Mn)O2,26,27 Li2FeMn3O8,12 and sulfur11,28 and prepared at 80–150 °C. The cathode loading can easily increase up to 90 wt% due to the ease of processing by infiltrating a fluid-type organic electrolyte (i), whereas all-oxide cathode composites require higher processing temperature to form strong interfacial bonding without the use of organic electrolytes (ii) and (iii). Cathode composites including LiCoO2–LLZO19,32–35 and Li(Ni0.5Co0.2Mn0.3)O2–LLZO36 are fabricated with additives at 700 °C (ii) and LiCoO2–LLZO37 is fabricated without additives at 1050 °C (iii). The use of a sintering additive allows reduction of the processing temperature, whereas the active cathode loading is reduced (54–58 wt%). Sintering over 1000 °C produces possible side reactions, resulting in high interfacial resistance. Currently, 50 wt% cathode loading is reported. (b) Evolution of the microstructure and interface formation during conventional solid-state sintering as exemplified by the LiCoO2–LLZO composite cathode. (c) Preparation of the cathode composite used in this study. | ||
Hybrid cathodes composed of liquid- or polymer-based catholytes (the part of the electrolyte on the cathode side of an electrochemical cell) mixed with active materials and carbon (Fig. 1a) are in principle a copy-cat cathode design of the cathodes used in traditional lithium-ion batteries, with the porous polymer separator simply replaced by a LLZO electrolyte. Cells with hybrid cathodes based on LiCoO2,22 LiFePO4,23–25 Li(Ni,Co,Mn)O2,26,27 Li2FeMn3O8,12 and sulfur11,28 have been reported, and various forms of catholytes consisting of Li salts in an organic solvent or polymer matrix, namely liquid electrolytes,12 polymer-gels,23,24 ionic liquids,22,26 and plastic crystals,27 have been tested. The liquid catholyte penetrates the as-cast porous cathode sheet, Fig. 1a(i), allowing sufficiently large contact area on its surface and forming a percolating network for sufficient ion transport. Typically, the hybrid cathode processing includes casting of a slurry prepared by mixing the active materials (crystallized oxide particles), a binder, and carbon as the conducting agent in a solvent and drying at approximately 80–150 °C, followed by infiltration of the catholyte. Such easy processing at low temperature (no sintering required) makes the hybrid cathode design attractive with a high cathode loading of >90 wt%. The reduced amount of flammable or less thermodynamically stable catholyte in hybrid-cathode Li-garnet SSBs compared with that in a typical liquid battery system may be advantageous for safety.2,3 For these reasons, such a hybrid SSB design is gaining momentum; however, the underlying interfacial reaction requires further investigation29,30 and practical demonstrations targeting cycle numbers >500 and C-rates >1C are needed.3
In contrast, an all-oxide cathode uses purely inorganic ceramic materials for the electrolyte and cathode constituents forming a composite cathode, as shown in Fig. 1a(ii) and (iii). The main merit of all-oxide cathodes over hybrid cathodes is overall battery safety against thermal runaway by completely avoiding flammable liquid catholytes or organic electrolytes.31 However, it is difficult to realize strong bonding within the composite network with low interfacial resistance. Furthermore, high active material loading is difficult to achieve as shown by recent reports with only 50–58 wt%,19,32–37 which makes the realization of high energy density difficult.
With or without additives, all-oxide cathode composite fabrication involves the preparation of a slurry solution (composed of the solid electrolyte and electrode particles, binder, and solvent), followed by screen printing of the composite slurry onto the LLZO electrolyte and sintering (Fig. 1b). With increasing temperature, the organic binder is first burned out, and three different types of solid–solid contacts are made (i.e., LiCoO2–LiCoO2, LLZO–LiCoO2, and LLZO–LLZO), followed by grain growth and neck growth among the constituent phases. For instance, a cathode composite with Ta-doped LLZO and LiCoO2 can be made with mechanically strong bonding via solid-state diffusion at 1050 °C,37 thereby meeting one of the requirements for low interfacial resistance. However, possible side reactions can occur during such high-temperature processes and may increase the interfacial resistance (1138 Ω cm2), reducing the specific capacity below the theoretical one.37 Furthermore, this approach would be exclusive to these phases and would not apply for many other oxide cathodes because of their higher reactivity with LLZO.38 For instance, a pellet-based compatibility study39 shows that Li-insulating interphases begin to form at above 600 °C in LLZO cathode composites with high-voltage active materials of Li2NiMn3O8, Li2FeMn3O8, and LiCoMnO4. In addition, the chemical reactivity and decomposition of composite cathode pellets were confirmed for LLZO–LiCoO2 and LLZO–LiNi0.33Co0.33Mn0.33O2 at 700 °C and even at 500 °C for LLZO–LiMn2O4 and LLZO–LiFePO4.40,41
Sintering additives can be used to facilitate the particle interconnection and densification of the cathode and electrolyte interface at reduced processing temperature, as shown in Fig. 1a(ii). For instance, the introduction of lithium borate (Li–B–O system34,42) in LiCoO2-19,32–35 and Li(Ni0.5Co0.2Mn0.3)O236-based cathode composites significantly reduces the cathode composite synthesis temperature to approximately 700 °C from 1050 °C, Fig. 1a(ii). The oxide sintering additive promotes liquid-phase sintering, accelerating the kinetics of densification compared with that of pure solid-state sintering.43,44 The sintering additive Li3BO3 (LBO) melts at approximately 800 °C,45,46 whereby its liquid flows into the ceramic cathode composite matrix due to the capillary pressure difference between the fine and coarse channels of the solid cathode and electrolyte particle constituents. The capillary forces give rise to strong attractive forces between neighboring particles and rearrangement. As sintering proceeds, elimination of pores and the growth of grains occur simultaneously in a liquid matrix, leading to faster densification. In the as-sintered cathode composite, the residual LBO phase prevails as an amorphous coating layer at the particle boundary between the electrolyte and cathode phases within the microstructure. A critical point for the SSB performance concerning the use of additives is that despite the advantage of lowering the co-sintering temperature, this residual LBO phase in the microstructure will remain as “inactive” constituent volume of the cathode composite, not contributing to the Li storage capacity. Thus, the associated capacity loss must be considered. For example, 12–35 wt% LBO additive has been used for the cathode composite, implying that valuable capacity volume is occupied by the inactive additive.19,34 More importantly, LBO has a two-orders-of-magnitude-lower ionic conductivity than LLZO, thereby limiting the overall charge transfer and slowing down the battery performance.42 These results highlight needs concerning new ceramic processing strategies for cathode composites targeting low temperature for the assembly of the constituents while keeping the strong bonding without introducing inactive additives for high active storage capacity. In parallel, the development of protective coating materials to avoid the interfacial reaction and Li-insulating phase formation needs to be explored.38,47
Instead of co-sintering of cathode constituents, the pre-fabrication of a 3D-connected, Li-conductive structure onto a LLZO electrolyte has been suggested; this process is achieved by screen-printing or tape-casting followed by sintering into a ‘porous LLZO’ framework as an ionic scaffold.11,27,48,49 Therein, the porous scaffold is prepared by casting LLZO slurries on top of a dense LLZO pellet or co-sintering LLZO tapes,50 where the LLZO slurries and tapes use pore-forming agents that are subsequently burned off, such as cornstarch,49 PMMA spheres,11 or ice crystals.27 Sintering leaves well-connected LLZO porous structures with pore volume, which can be filled with the active cathode material via infiltration at rather low synthesis temperature, allowing one to potentially integrate a wider range of cathodes phase-stability wise. Pioneering studies based on such porous LLZO scaffolds, however, have only mainly targeted hybrid cathode composites such as sulfur–carbon–ionic liquid11 and NMC–carbon–plastic crystal.27 An all-oxide cathode composite has been attempted but requires more attention to optimize the electrochemical performance.51
In conclusion, in recent reports on ceramic manufacturing strategies for all-oxide Li-garnet SSBs, none of the approaches resulted in the realization of the theoretical discharge capacities of the tested cathode composites (LiCoO2–LLZO, Li(Ni,Co,Mn)O2–LLZO, and LiMn2O4–LLZO) or the target interfacial resistance of 40 Ω cm2 to cycle SSBs;20 thus, further attention is required for the realization of all-oxide Li-garnet SSBs. The limiting factors, including the poor contact and limited contact area between LLZO and the cathode materials resulting from the ceramic microstructure design, interfacial instabilities during co-sintering, and inactive constituent phases (i.e., LBO) in the cathode composite, increase the interfacial resistance and impede stable battery performance. Although there have been promising demonstrations of the use of porous LLZO scaffolds as a host for various active materials, a specific example of all-oxide Li-garnet SSB technology exhibiting close to the theoretical capacity with the targeted interfacial resistance has yet to be reported. A blueprint is needed for controlling the interfacial stability and microstructure by selecting an appropriate ceramic synthesis route and thermal processing windows, ideally allowing for mass manufacturing in industry as well.
In the present study, we present a ceramic processing route for an all-oxide cathode composite with the lowest reported interfacial resistance for LLZO-based SSBs with a LiCoO2 cathode and lithium anode. Unlike earlier attempts predominantly relying on classic high-temperature sintering routes to prepare a composite via mixing of cathode/electrolyte particles, we turn to a careful discussion on how to circumvent the interfacial phase stability problems through a phase evolution study for the LiFePO4 cathode and the adoption of a new ceramic processing protocol whereby a LiCoO2 cathode precursor solution is directly infiltrated into a porous LLZO scaffold prior to sintering (Fig. 1c). The key to the success of this approach is to keep the processing temperatures lower than those for the conventional solid-state sintering of crystalline particle constituents to balance the interfacial phase stability and bonding. The results have implications for our understanding of phase evolution and the ability of LiFePO4 and LiCoO2 to form stable composite cathodes with Li garnet as tandem materials and provide guidelines for avoiding interface reactions for future composite cathodes in Li-garnet SSBs while introducing a novel ceramic processing technique, borrowed from other fields such as solid-oxide fuel cells, now to all-oxide SSB manufacturing.
Results
A promising ceramic manufacturing route for composite cathodes for SSBs is to fill a porous solid-electrolyte scaffold (LLZO in this work) with a cathode slurry or precursor solution of active materials. We selected LiFePO4 and LiCoO2 as the cathode materials and investigated the optimal cathode composite ceramic arrangements to achieve improved phase stability, interface stability, microstructure, and electrochemical performance for SSBs.LLZO scaffold design for the cathode composite on a dense electrolyte
To study the effect of infiltration into the porous structure for all-oxide cathode formation, we fabricated a dense LLZO electrolyte pellet and a porous LLZO scaffold on the LLZO pellet. The LLZO pellet was prepared from our own calcined powder with a 7.2 mol% Li excess via solid-state synthesis. The LLZO powder was pressed into 10 mm-diameter pellets, sintered to 95% of the theoretical density, and polished down to 300 μm thickness (Fig. 2a). Scanning electron microscopy (SEM) was used to probe the dense pellet and porous LLZO cathode scaffold at all stages. The average grain size of the initial pellet was 3 ± 0.23 μm with fairly straight grain boundaries, reflecting the rather late stage of sintering (Fig. 2b). The X-ray diffraction (XRD) pattern of the as-sintered LLZO pellet exhibited strong (321), (420), and (422) peaks, indicative of a high-purity cubic garnet phase (Fig. 2c). We sputtered both sides of the pellet with a gold electrode and then performed electrochemical impedance spectroscopy (EIS) analysis to measure the total resistance of the pellet and evaluate the ionic conductivity (Fig. 2d). The EIS patterns revealed vertical capacitive behavior at low frequencies (indicative of ion blocking) and one semicircle at high frequencies corresponding to the sum of LLZO bulk and grain boundary contributions.52,53 Consequently, a total Li-ion conductivity of 0.53 mS cm−1 at 25 °C and 6.4 mS cm−1 at 100 °C with an activation energy of 0.35 eV was calculated (Fig. 2e), which is in good agreement with literature values for cubic Li-garnet electrolytes.52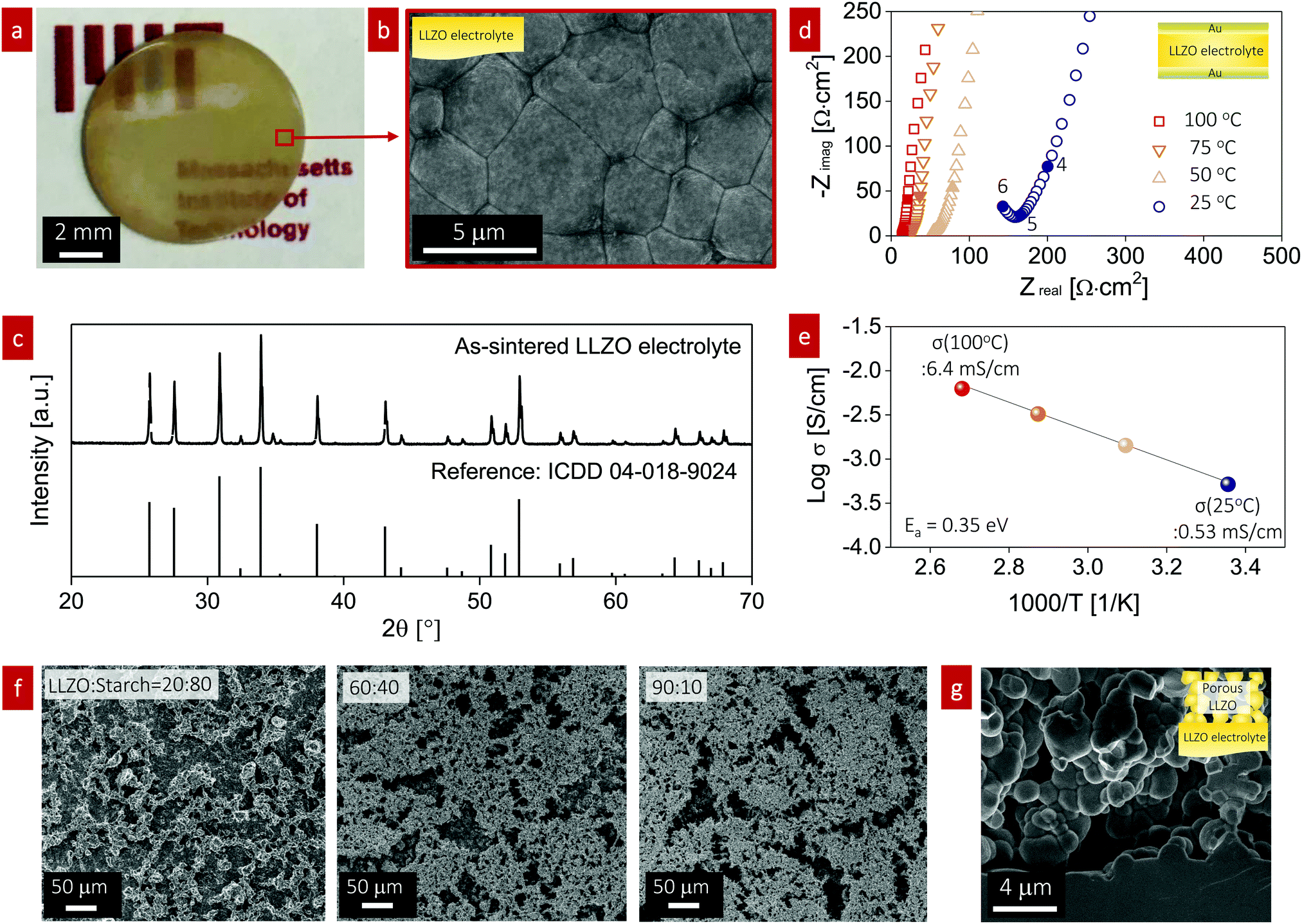 | ||
Fig. 2 Composite cathode scaffold ceramic design. (a) Optical image of the Li6.5La3Zr1.5Ta0.5O12 (LLZO) electrolyte with a thickness of 300 μm. (b) SEM image of the top surface of the LLZO pellet after thermal etching. (c) XRD pattern of the as-sintered LLZO electrolyte pellet. (d) EIS spectra of the LLZO electrolyte pellet in the temperature range of 25–100 °C in dry air. The labels on the curve are log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (frequency) [Hz]. (e) Temperature dependence of the total ionic conductivity of the LLZO electrolyte pellet as a function of the temperature in an Arrhenius-type representation. The activation energy values are determined from the slope of the logσT vs. 1/T plot. (f) Top-view SEM images of the porous LLZO scaffold on the dense LLZO pellet. From left to right, the porous structure is created by varying the ratio of LLZO:corn starch from 20:80 to 90:10 in wt%. (g) Cross-sectional SEM image of porous LLZO for a ratio of LLZO:corn starch of 90:10. (frequency) [Hz]. (e) Temperature dependence of the total ionic conductivity of the LLZO electrolyte pellet as a function of the temperature in an Arrhenius-type representation. The activation energy values are determined from the slope of the logσT vs. 1/T plot. (f) Top-view SEM images of the porous LLZO scaffold on the dense LLZO pellet. From left to right, the porous structure is created by varying the ratio of LLZO:corn starch from 20:80 to 90:10 in wt%. (g) Cross-sectional SEM image of porous LLZO for a ratio of LLZO:corn starch of 90:10. | ||
In the second set of experiments, the porous LLZO scaffold was synthesized as a film on one side of the sintered LLZO pellet surface by slurry coating followed by sintering (Fig. 2f and g). To demonstrate the tunable pore volume for cathode loading, we systematically manipulated the porosity of the scaffold by varying the ratio of LLZO:corn starch from 20:80 to 90:10, as shown in Fig. 2f. The slurry solution consisted of calcined LLZO powder (<1 μm, Fig. S1, ESI†), corn starch (20–30 μm, 26 vol%) as the pore-forming agent, ethyl cellulose binder, and solvent. With increasing temperature below 500 °C, all the organic compounds including the pore formers were burned out in an oxidizing atmosphere; then, sintering of the porous LLZO scaffold was performed at 1100 °C for 2 h at a heating rate of 10 °C min−1, leading to strong interparticle neck formation, a phase transition to the desired fast-conducting cubic phase, and anchoring of the scaffold structure to the dense LLZO pellet. The top surface of the as-sintered porous LLZO scaffold is shown in the SEM images in Fig. 2f. With increasing solid loading of LLZO, the 2D porosity decreased as follows: 72%, 62%, and 38% porosity for LLZO:starch ratios of 20:80 wt%, 60:40 wt%, and 90:10 wt%, respectively. In general, a bimodal pore size distribution with large pores of 10–30 μm diameter and small pores of 1–5 μm diameter was observed for the LLZO scaffolds. The former were created mainly by the starch, whereas the latter arose from incomplete densification due to restricted sintering (pore former) and constrained sintering conditions. We represent the sintered porous LLZO scaffold by a network of 3D-connected LLZO particles with wide neck growth and approximately 38% porosity for a weight ratio of LLZO to initial starch of 90:10 during processing (Fig. 2g), ready for filling with cathode materials in the next steps. We selected these scaffolds because of their robust microstructure for infiltration of the active cathode phases, defined either as LiFePO4 or LiCoO2, in the next synthesis steps for the fabrication of all-oxide cathode composites for SSBs.
LiFePO4–LLZO composite cathodes for SSBs
LiFePO4 is a representative cathode material known for its long-term cycle life and high power capability in conventional lithium-ion batteries54 that has never been investigated as a composite cathode for all-oxide SSBs with Li garnets.In an initial attempt, prior to the fabrication of a full cell with a LiFePO4-based composite cathode (LiFePO4–LLZO–carbon), we first explored the phase compatibility between LiFePO4 and LLZO using composite pellets (50:50 wt%) fired over a wide temperature range from 250 °C to 1000 °C. XRD patterns were obtained and the pellet color was monitored using light microscopy for each firing temperature to analyze the phase constituents (Fig. 3a). With increasing firing temperature, we observed changes in the apparent XRD phases and sample color of the LiFePO4–LLZO composites. For the composite pellet with no heat treatment, two separated XRD signatures consistent with the LLZO and LiFePO4 phases were observed.41 At 250 °C, little changed in the X-ray patterns and the pellet color. After firing to 300 °C, we detected minor XRD peaks at 23.2° and 36.1°, corresponding to small quantities of the Li3Fe2(PO4)3 phase. We observed more pronounced peaks indicative of the Li3Fe2(PO4)3 and Fe2O3 phases and a color change at 350–400 °C. Firing between 600 °C and 1000 °C prompted the decomposition of Li3Fe2(PO4)3 and LLZO, leaving detectable quantities of LaPO4, Li3PO4, La2Zr2O7, and Fe2O3 in the XRD pattern. According to this phase compatibility analysis, we concluded that the desired LiFePO4 phase only allowed for a very narrow processing window of approximately up to 300 °C for co-sintering with LLZO without sacrificing interfacial reactions. The reaction temperature is an even lower than that reported in recent compatibility study, as they performed sintering of a LiFePO4–LLZO pellet in a reducing atmosphere.41
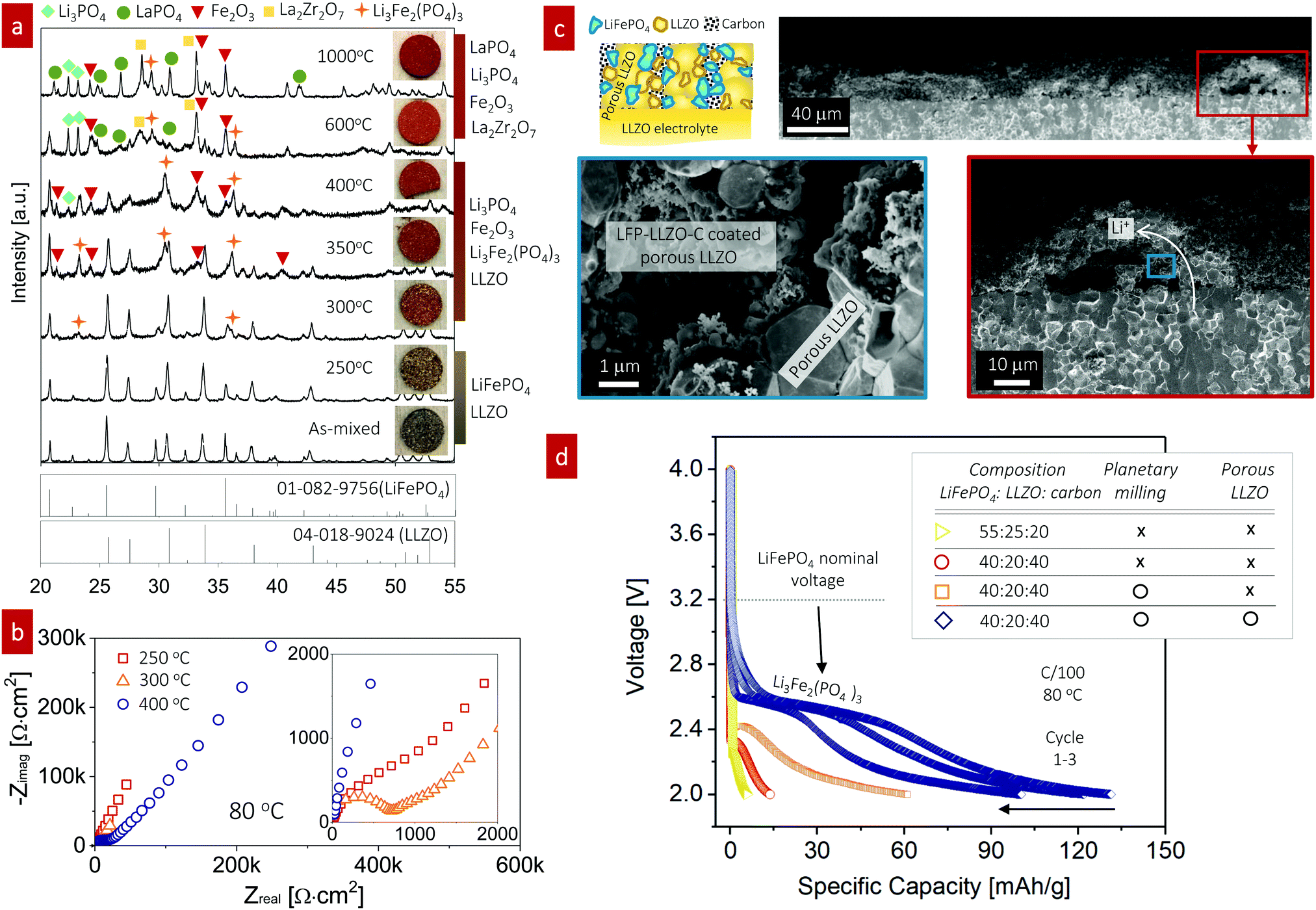 | ||
| Fig. 3 Characterization and fabrication of LiFePO4-based cathode composite cathodes for all-oxide Li-garnet SSBs. (a) Chemical compatibility study of the LiFePO4 and LLZO interface with the cathode firing temperature ranging from room temperature to 1000 °C. The inset presents images of the as-fired composite model pellet. (b) Impedance spectra of the full cell (LiFePO4–LLZO–carbon|LLZO|Li) with the cathode firing temperature ranging from 250 °C to 350 °C. The cathode fired at 300 °C exhibited the lowest total cell resistance among the tested cells. (c) Schematic illustration and cross-sectional SEM images of the interface-engineered cathode composite. A schematic image and associated cross-sectional SEM images captured at low, moderate (red box), and high magnification (blue box) are shown, confirming the good contact between the cathode compounds and porous LLZO scaffold layer (blue box). (d) Discharge capacities of full cells with the cathode composite as a function of composition, planetary milling, and interface engineering. | ||
We assembled composite cathodes based on LiFePO4–LLZO–carbon (with 55:25:20 wt%) in the LLZO scaffold-pellet assembly and integrated a Li metal anode to investigate the electrochemistry and interfacial resistance by varying the cathode firing temperature from 250 °C to 400 °C (Fig. 3b). The cathode composite thickness was approximately 42 μm (Fig. S2, ESI†). The cathode composites fired at 300 °C exhibited the lowest interfacial impedance, indicating a minimum insulating product at the LiFePO4–LLZO interface (Fig. 3b, inset). We then applied the same condition for LiFePO4–LLZO–carbon with a LLZO scaffold-pellet, as shown in Fig. 3c. A schematic image and associated cross-sectional SEM images captured at low, moderate (red box), and high magnification (blue box) are shown, confirming the good contact between the cathode compounds and porous LLZO scaffold layer (blue box). The existence of a LiFePO4–LLZO–carbon composite was further confirmed by energy-dispersive X-ray spectroscopy (EDS), with the elemental mapping for carbon, La, and Fe shown in Fig. S3 (ESI†). However, voids greater than 10 μm in size were often observed, possibly because of the limited access for infiltrating the cathode solution into the all-accessible pore volume. Engineering of the porous structure is required to further optimize the uniformity and degree of ordering toward efficient utilization of the pore volume. The optimum in this processing temperature for the composite cathode was determined by studying the microstructures and phase evolution. The composite cathode annealed at 250 °C presumably lacked the thermal energy for sufficient particle contact and densification, whereas firing above 400 °C resulted in phase decomposition and interfacial reactions.
Next, we characterized full SSBs (LiFePO4–LLZO–carbon|LLZO|Li), where the composite cathode was fired at 300 °C. Under a fixed firing temperature, we varied the cathode composition ratio and reduced the particle size to increase the reaction area to achieve improved electrochemical performance (Fig. 3d). The batteries were tested from 2 to 4 V to probe the redox reaction for LiFePO4 and potential Li3Fe2(PO4)3 phases and the performance of the composite cathode.
For the first co-assembly without further optimization strategies toward increased reaction sites, including particle size reduction and interface modification, the discharge capacity of the LiFePO4–LLZO–carbon cathode composite (55:25:20 wt%) was extremely poor (5.64 mA h g−1) with very limited electrochemical redox reaction at approximately 2.25 V, which is far from either the nominal voltage or theoretical capacity of LiFePO4,55 approximately 170 mA h g−1. Thus, in the following assemblies of the cathode composites, we varied the ratios between LiFePO4, LLZO, and carbon. Increasing the carbon content with decreasing the LLZO content of the composite to LiFePO4:LLZO:carbon = 40:20:40 wt% led to a slightly increased discharge capacity of 13.6 mA h g−1 and elevation of the slight voltage plateau to 2.32 V. In this case, the poor electronic conductivity of LiFePO4, typically approximately 10−9 S cm−1, was positively compensated by the higher carbon content,56,57 whereas the role of the LLZO content inside the cathode composite remains unclear. In the current design, the ion transport inside the cathode composite is most likely dominated by the porous LLZO network because of the high sintering temperature of ∼1100 °C compared with the LLZO particles inside the cathode composite. The LLZO particles inside cathode are fired at a low temperature of only ∼300 °C; thus, strong neck formation and grain growth of LLZO was not expected, whereas LiFePO4/LLZO exhibited good bonding. To increase the reaction length for charge-transfer reactions to occur, we further reduced the particle size using planetary ball milling, which resulted in a further increase of the discharge capacity to 60.8 mA h g−1 at a redox-potential of 2.41 V. SEM imaging further confirmed that fewer agglomerates were present after milling (Fig. S4, ESI†). Among the SSBs tested here, the highest discharge capacity (131.1 mA h g−1 at 2.6 V) was achieved for the LiFePO4–LLZO–carbon cathode composite with an LLZO scaffold structure together with the planetary-ball-milled cathode composite. The discharge curves clearly point to the Li insertion/extraction reaction of monoclinic Li3Fe2(PO4)3, which is consistent with ref. 58, measured for conventional liquid-type batteries. It is speculated that the interfacial decomposition reaction initially progressed from the particle surface into the bulk; thus, the LiFePO4 phase may not be detectable inside the grains, as the concentration is too low to be detectable by XRD or electrochemical cycling at this stage (Fig. 3a).
Based on the phase stability and ceramic processing, we conclude that LiFePO4–LLZO–carbon composite cathodes prepared using a LLZO scaffold and cathode slurry casting possess a cathode loading of 40 wt% and deliver a competitive discharge capacity near the theoretical value of 128 mA h g−1 for the Li3Fe2(PO4)3 phase59 when processed at temperatures as low as 300 °C. Therefore, the thermal processing window, when manufacturing and working with LiFePO4 in combination with LLZO, is very narrow to achieve the expected cathode performance. Our findings contribute to ceramic processing science and understanding, regardless of whether LiFePO4 is ready to integrate as a cathode for all-oxide Li-garnet SSBs, which appears limited as options to stabilize the LiFePO4 phase with LLZO are marginal, as indicated by the summary in Fig. 6. Nevertheless, to the best of our knowledge, these synthesis protocols and data are among the first attempts to make all-oxide SSBs based on an inorganic solid electrolyte (i.e., Li garnet), LiFePO4 as a cathode, and a Li metal anode without using a liquid or polymer-based electrolyte.
LiCoO2–LLZO composite cathodes for SSBs
LiCoO2 cathodes have attracted the most attention for all-oxide SSBs as they possess the highest electrochemical and chemical stability against a Li-garnet electrolyte according to theory39,60 and experiments.32,37,47 However, how to optimize the ceramic processing route of the tandem material while maintaining the interfacial stability remains unclear for composite cathodes. The limited SSB performance is a consequence of managing the interfacial stability of the tandem material as a function of the ceramic processing conditions, as demonstrated by various reports showing the dependence of the interfacial reaction products on the thermal range (300–900 °C) such as for LiCoO2 thin films prepared on LLZO pellets by RF sputtering61 and in LLZO–LiCoO2 powder mixture studies.62 There have also been conflicting reports, claiming relatively high thermal processing windows up to 1050 °C without any sign of interfacial reaction products of LiCoO2–LLZO cathode composites prepared using conventional screen printing and firing starting from crystalline powders.37It is important to emphasize that all of these prior examples involved either vacuum-based thin-film or ceramic processing of crystalline cathode particles.37,61,62 In contrast, in our approach, we use cathode precursor solution infiltration into a porous LLZO scaffold followed by crystallization to the oxide cathode directly from the metal salt constituents. Using such altered pathways in ceramic processing and shaping the cathode composites has the significant advantage of strongly reducing the temperature at which good bonding is achieved without adverse chemical reactions. Importantly, the cathode is directly crystallized on the large surface area of porous LLZO; thus, the active reaction area can be controlled dependent on the microstructure (porosity and thickness) of the porous LLZO. As demonstrated in the previous section with our proposed infiltration using the LLZO scaffold method for the composite cathode on LiFePO4–LLZO, we now turn to the LiCoO2–LLZO system as a tandem material for further exploration and adaption of the technique.
In line with this ceramic process design for the composite, we selected a firing temperature of 700 °C (Fig. 4a) as it is the minimum temperature that results in crystallization of LiCoO2 particles from the Li–Co–O precursor solution without any sign of an amorphous phase (Fig. S5, ESI†). Cross-sectional SEM analysis revealed a well-connected network composed of LLZO grains with a diameter of 2.3 ± 0.1 μm (Fig. 4b) and LiCoO2 grains with a diameter of 0.29 ± 0.020 μm with fairly good coverage on the surface of porous LLZO (Fig. 4c). EDS mapping of the interface at LiCoO2–LLZO revealed separated Co- and La-rich regions across the microstructure of the composite cathode (Fig. 4d and Fig. S6, ESI†). To explore the local phase stability after processing of the cathode composites, we performed Raman spectroscopy analysis across the interface region of the dense LLZO pellet and toward the cathode composite (Fig. 4e). The Raman spectra for LLZO contained characteristic peaks at 120, 210, 250, 360, 420, 640, and 730 cm−1, which are commonly attributed to Eg for La, T2g and A1g for O, T2g and Eg for Li, A1g for Zr, and A1g for Ta vibrational modes, respectively, indicative of the cubic LLZO phase.63,64 We confirmed the presence of the rock-salt LiCoO2 phase,65 with its two main characteristic peaks detected at 485 and 594 cm−1, indicative of Eg (O–Co–O bending) and A1g (O–Co–O stretching) vibrations, respectively. The chemistry, phase, and structure analyses confirm that this ceramic processing route involving the direct transfer of metal salts to a metal oxide for the LiCoO2 cathode constituent within the pre-made LLZO scaffold resulted in successful phase stabilization at the unusually low temperature of 700 °C with no evidence of secondary phase formation, particularly in the interface region (see the LiCoO2/LLZO Raman spectra in Fig. 4e). Thus, this processing route with a low-thermal budget is an attractive option that extends the opportunity to prepare composite cathodes for future all-oxide SSBs.
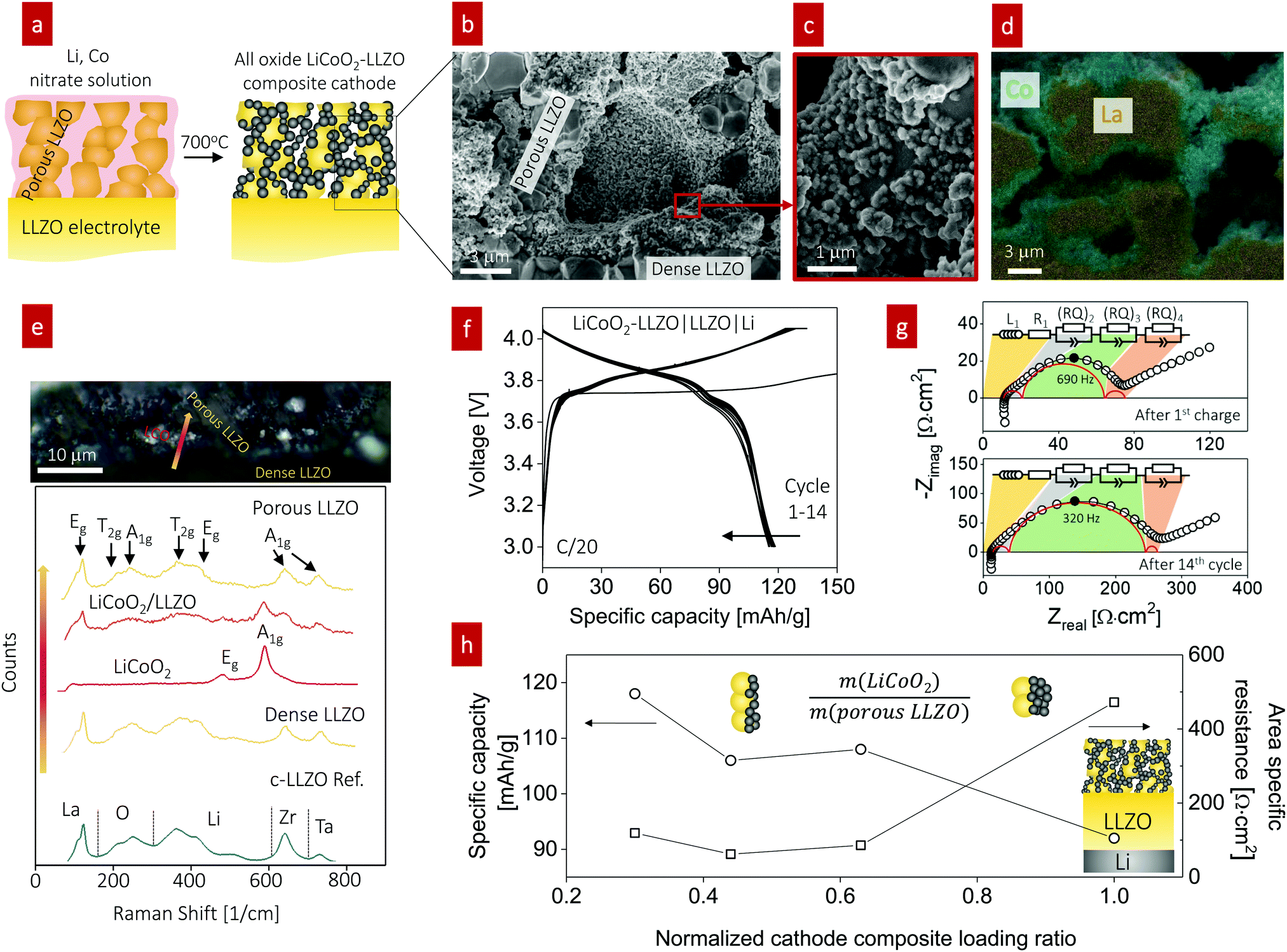 | ||
Fig. 4 Characterization and fabrication of LiCoO2-based composite cathodes for all-oxide Li-garnet SSBs. (a) Schematic illustration of the composite cathode prepared by direct infiltration of Li and Co metal salts into a pre-made porous LLZO scaffold and subsequent firing at 700 °C, resulting in the formation of the composite on top of a dense LLZO solid electrolyte. (b–d) SEM images and EDS mapping of the LiCoO2–LLZO cathode composite. (e) Optical image and corresponding Raman spectra with different points at the interface through the electrolyte pellet, LiCoO2, and porous LLZO after firing at 700 °C. (f) Cycling performance of the full cell (LiCoO2–LLZO|LLZO|Li) at 0.05C and 80 °C. (g) Impedance spectra of the full cell at open-circuit potential after charging. (h) Change in specific capacity (mA h g−1) and area specific resistance (ASR, Ω cm2) with regard to the normalized composite loading ratio 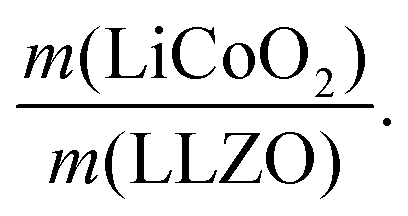 The inset presents a schematic illustration of the cathode microstructure. With decreased loading ratio, improved cathode utilization and ASR are achieved due to the increased surface area of LLZO and shorter Li-ion diffusion distance. The inset presents a schematic illustration of the cathode microstructure. With decreased loading ratio, improved cathode utilization and ASR are achieved due to the increased surface area of LLZO and shorter Li-ion diffusion distance. | ||
For battery testing, we measured the charge and discharge characteristics of full cells of LiCoO2–LLZO|LLZO|Li metal at 0.05C, as shown in Fig. 4f. The discharge capacity was 118 mA h g−1 between 3.0 and 4.05 V with good initial cyclability and a slight decay after 14 cycles (115 mA h g−1). The voltage profiles barely changed apart from the initial charging, indicating the reversibility of the reaction without any major structural changes. The abnormal initial charging behavior (the long plateau at 3.75 V) can be attributed to the electrochemical oxidation (decomposition) of the organolithium compounds that were generated during cathode synthesis and added to the first charging capacity.19 In addition, the longer decomposition plateau indicates a higher first charging capacity; however, it was irreversible, and the compound was not detectable using XRD or Raman spectroscopy. The impedance spectra of the full cell before and after the 1st charging are compared for further discussion (Fig. S7, ESI†). To identify the contributions from Ohmic loss (electrolyte pellet) and polarization loss (electrolyte–electrode interface) with cycling, EIS analysis was performed at open-circuit voltage after charging (Fig. 4g). The typical battery impedance spectra confirm an Ohmic loss of around 10 Ω cm2, corresponding to a typical Li conductivity of 7 mS cm−1. The impedance spectra reveal Warburg-type capacitive behavior at low frequency, interpreted as solid-state Li diffusion contributions inside LiCoO2 particles.66 We note that at moderate frequency, other electrochemical processes (indicated by semicircles) are active. These processes can be seen as a summation of several physical processes for electrochemical reactions. Despite different individual contributions typically being overlapped in the Nyquist diagram, the number of processes can be estimated by fitting appropriate equivalent circuits. This process allows us to deduce the change in the performance (i.e., capacity) by monitoring the change in the physical process (series R or parallel RQ circuit) involved in the impedance spectra, where R is the resistance and Q is the constant phase element. Of several equivalent circuits of two to four RQ elements, that with three RQ elements showed the best fitting result (Fig. 4g and Table S1, ESI†) for this study, whereas two21,67–69 or three70,71RQ elements have been employed for the fitting of other battery systems. Our spectra contain two small semicircles at high ((RQ)2) and low frequency ((RQ)4) and one larger semicircle at mid frequency ((RQ)3), which all increased; however, the main degradation originated from the process at mid frequency, (RQ)3. Bode plots of the imaginary part of the impedance vs. frequency were plotted to obtain further insight (Fig. S8, ESI†). The Bode plots also reveal one clear peak and the peak at ∼690 Hz at the 1st cycle shifted to lower frequency (∼320 Hz) and increased continuously in magnitude up to the 14th cycle; this trend corresponds to a notable increase of (RQ)3 (Fig. 4g), which is the rate-limiting step of the overall battery performance.
Two RQ elements have been suggested for sulfide-based Li metal SSBs: the anode/electrolyte interfacial impedance at low frequency and the cathode/electrolyte interfacial impedance at high frequency.20,68 An additional semicircle can be evolved at high frequency once serious decomposition of the solid electrolyte occurs.67 Unfortunately, no specific impedance models have been proposed for the Li-garnet-based full-cell system consisting of a LCO–LLZO cathode composite, LLZO electrolyte, and Li-metal anode, requiring further systematic investigation to rule out these physical processes, for example, by studying symmetrical cells with distribution of relaxation time (DRT) analysis71 or three-electrode cell tests.72 We checked the Nyquist plots for a symmetrical Li/LLZO cell to determine the potential contribution toward the (RQ)3 process. The Li/LLZO interfaces exhibited low impedance (as low as 2 Ω cm2), and the characteristic frequency (∼26 Hz) of the symmetric Li/LLZO/Li cell (Fig. S9, ESI†) was much smaller than that from the full cell (62 Ω cm2, 320–690 Hz) with stable spectra for 100 cycles under a current density of 0.1 mA mA cm−2. Therefore, (RQ)3 most likely originates from the cathode rather than the anode interfaces.
In general, the irreversible capacity (reduced discharge capacity compared to the charge capacity) in solid-state batteries originates from the complex interplay between the cell components during battery operation. According to the literature on both sulfide- and oxide-based SSBs, it is often explained to result from the increased interfacial impedance due to reactions73 or cracking37,68 at the electrolyte/electrode interface that slow down charge transport. In our study, the interfacial impedance at the LCO or Li/LLZO interface is driven by (i) the fabrication conditions (i.e. contact, heating) and/or (ii) the electrochemical potential (cell voltage). Interphases were observed in several experiments and theoretical calculations:47 La2Zr2O7, La2O3, and Li2CoO3 when LLZO contacted half-lithiated LCO (i); tetragonal LLZO when LLZO contacted Li metal (ii); and La2O3, La2Zr2O7, and Li2CoO4 at 3 V60 and La2Zr2O7 and LaCoO3 above 4 V (iii).74 These interphases are typically insulating; thus, charge transport at the interface becomes sluggish. We tried to identify such features (interphases and interfacial gap formation) for the as-cycled LCO/LLZO and Li/LLZO interface in Raman spectra and cross-sectional SEM images (Fig. S10–S12, ESI†). Neither detectable crystalline interphases nor an obvious interfacial gap were observed. To provide direct evidence of the growing impedance at the LCO/LLZO interfaces, it is important to employ high-resolution or in situ characterization techniques to elucidate the changes in the microstructure and chemistry at the interfaces75,76 together with in situ impedance studies. In addition to understanding the degradation mechanism, further minimization of the interfacial reaction down to the target value of ∼40 Ω cm2 is needed for enhanced performance.20 The low self-discharge rate during the rest (Fig. S13, ESI†) suggests limited electronic leakage (1.14%) in the full cell, potentially indicating that cobalt contamination in the LLZO electrolyte during cell fabrication can be excluded. However, a slight increase of the self-discharge up to 1.24% with cycling was observed.
In the next step, we highlight one of the measures for understanding the relationship between cathode microstructure and performance (Fig. 4h). Assuming that the LiCoO2 cathode covers the entire surface area of the porous LLZO, the number of active reaction sites can be controlled either by adjusting the solid loading (or porosity) of the porous LLZO or the amount of LiCoO2 loading, which directly affects the cathode performance and interfacial resistance. To test this scenario, we varied the solid loading of the porous LLZO (6.41–18.6 mg cm−2) under a rather fixed amount of LiCoO2 (0.73–1.4 mg cm−2) and evaluated the effect on the performance. Microscopically, this translates to changes in the arrangement and coverage of LiCoO2 particles on the porous LLZO backbone. Notably, we observed that both the specific capacity and total area specific resistance (ASR) were largely affected by the composite loading ratio, defined as 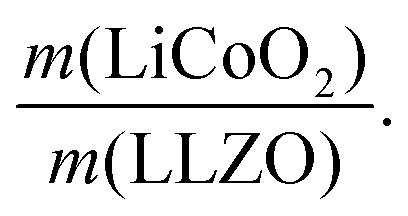 For simplification, the change was normalized from 0 to 1. When this normalized composite ratio decreased from 1 to 0.3, the specific capacities increased from 91 to 118 mA h g−1 with decreasing total ASR from 472 to 114 Ω cm2. A decreased loading ratio implies that there is more empty area for LiCoO2 to directly contact with the porous LLZO scaffold instead of a LiCoO2–LiCoO2 contact as schematically illustrated as an inset. An increase in the LLZO loading also leads to lower current densities in the cathode composite (at the porous LLZO and at the LCO/LLZO interfaces) owing to the increased ionic surface area or LCO/LLZO interfacial length. The number of reaction sites at electrode/electrolyte interfaces is of vital importance to achieve reduced cell polarization and improved capacity. In this work, the cathode microstructure with an optimized solid loading of
For simplification, the change was normalized from 0 to 1. When this normalized composite ratio decreased from 1 to 0.3, the specific capacities increased from 91 to 118 mA h g−1 with decreasing total ASR from 472 to 114 Ω cm2. A decreased loading ratio implies that there is more empty area for LiCoO2 to directly contact with the porous LLZO scaffold instead of a LiCoO2–LiCoO2 contact as schematically illustrated as an inset. An increase in the LLZO loading also leads to lower current densities in the cathode composite (at the porous LLZO and at the LCO/LLZO interfaces) owing to the increased ionic surface area or LCO/LLZO interfacial length. The number of reaction sites at electrode/electrolyte interfaces is of vital importance to achieve reduced cell polarization and improved capacity. In this work, the cathode microstructure with an optimized solid loading of 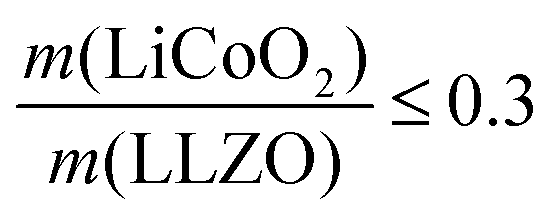 is expected to show a shorter Li-ion diffusion distance with lowered current densities in LLZO during the electrochemical reaction. Therefore, improved cathode utilization and interfacial resistance are achieved. It is, however, important to note that optimization can also be achieved by manipulating another descriptor such as the porosity under the consideration of effective reaction sites. We successfully demonstrate that by using the proposed ceramic synthesis route it is indeed possible to fabricate LiCoO2–LLZO composites that deliver a discharge capacity of 118 mA h g−1, which is near the theoretical capacity of LiCoO2 (115 mA h g−1, 3–4.05 V), with an exceptionally low interfacial resistance of 62 Ω cm2.
is expected to show a shorter Li-ion diffusion distance with lowered current densities in LLZO during the electrochemical reaction. Therefore, improved cathode utilization and interfacial resistance are achieved. It is, however, important to note that optimization can also be achieved by manipulating another descriptor such as the porosity under the consideration of effective reaction sites. We successfully demonstrate that by using the proposed ceramic synthesis route it is indeed possible to fabricate LiCoO2–LLZO composites that deliver a discharge capacity of 118 mA h g−1, which is near the theoretical capacity of LiCoO2 (115 mA h g−1, 3–4.05 V), with an exceptionally low interfacial resistance of 62 Ω cm2.
Perspectives on cathode composite design for garnet-based SSBs
For the successful design of cathode composites in SSBs, a delicate balance must be maintained between the phase stability of all the cathode constituents and an optimal microstructure (here, LLZO vs. LiFePO4 or LiCoO2 phases) to ensure sufficient electronic and ionic transport, a sufficiently large active reaction area, and a high cathode loading. These properties are largely determined by the specific ceramic processing route selected as well as the thermal processing range. We demonstrate here that when taking all of these factors into account, LiCoO2 provides more opportunities for the successful preparation of cathode composites with LLZO than LiFePO4 owing to the improved interfacial stability of the tandem material if precautions are taken to use a ceramic processing route from the direct transfer of a metal salt to an oxide remaining in the low temperature processing regime. The LiCoO2–LLZO composite cathodes in the current work, prepared by precursor infiltration into a porous LLZO scaffold using direct metal salt-to-oxide cathode crystallization, clearly offer an improved capacity, degradation rate, and interfacial resistance compared with those of ceramic composite cathodes prepared via classic solid-state mixing from crystalline powders with alteration of the sintering protocol using a Li–B–O sintering agent (Fig. 1a and 5 and Table 1).19,32,33,36,37,51 To the best of our knowledge, the interfacial resistance of the present LiCoO2–LLZO composite cathodes is the lowest among reported all-oxide Li-garnet SSBs.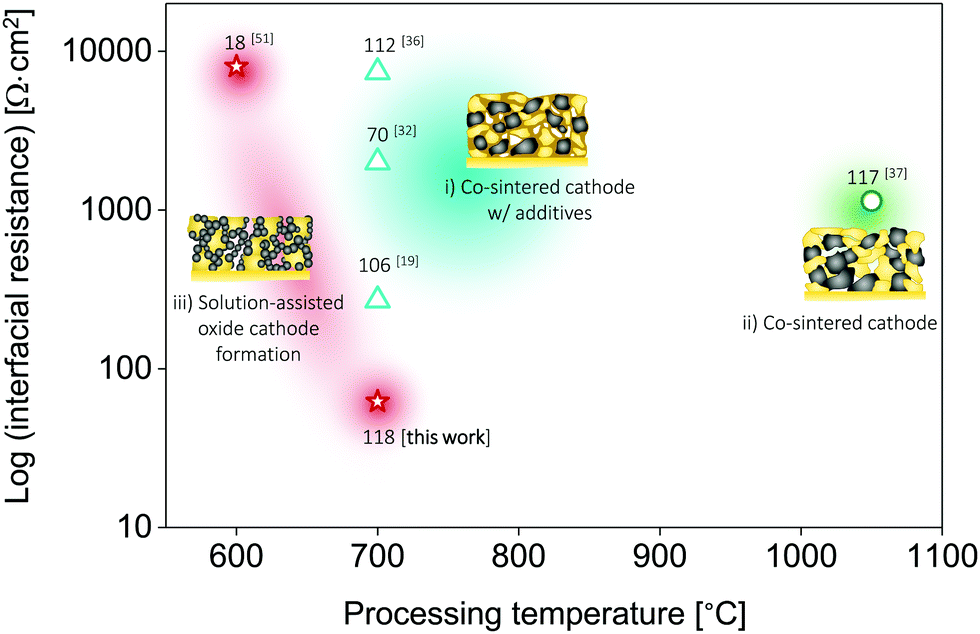 | ||
| Fig. 5 Comparison of the total interfacial resistance vs. processing temperature of all-oxide Li-garnet SSBs based on layered cathode composites. Three different types of all-oxide cathodes are categorized with respect to the processing strategies: (i) all-oxide cathode w/additives,19,32,36 (ii) all-oxide cathode without porous LLZO,37 and (iii) all-oxide cathode with porous LLZO.51 The numbers indicate the specific capacity (mA h g−1). The conditions for processing and testing are based on Table 1. This study presents the lowest interfacial resistance with the desired specific capacity among reported all-oxide Li-garnet SSBs. | ||
| Chemistry and processing | Electrochemical performance | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Description | Cathode composition | Processing method | Temp. [°C] | Volt. range [V] | C-Rate or current density | Initial interfacial resistance [Ohm cm2] | First discharge capacity [mA h g−1] | Capacity loss [mA h g−1/cycles] | Temp. [°C] | Active material loading [mg cm−2] | Ref. |
| All-oxide cathode with additives | LiCoO2 + LLZO + Li2.3C0.7B0.3O3 | Solid-state sintering | 700 | 3–4.05 | 0.05C | 270 | 106 | 39/40 | 100 | 1 | 19 |

|
LiCoO2 + Li3BO3 + In2SnO5 (ITO) | Solid-state sintering | 700 | 2.8–4.3 | 0.007C | >2000 | 69.6 | n.a. | 80 | 4 | 32 |
| LiCoO2 + LLZO + Li3BO3 | Sol–gel mixed w/LBO | 700 | 2.5–4.2 | 14 mA cm−2 | n.a. | 0.6–7 | 2/5 | 60 | n.a. | 33 | |
| NMC + ITO + Li3BO3 w Li–Ti–O coating | Solid-state sintering | 700 | 3–4.6 | 5 mA cm−2 | 7400 | 112 | 65.3/5 | 80 | 1 | 36 | |
| All-oxide cathode | |||||||||||
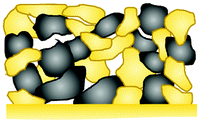
|
LiCoO2 + LLZO | Solid-state sintering | 1050 | 2.4–3.6 | 50 mA cm−2 | 1138 | 117 | 81/100 | 50 | 12.6 | 37 |
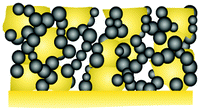
|
LiCoO2 + porous LLZO scaffold | Solution infiltration | 600 | 3–4.2 | 0.016C | >8000 | 18 | 6/10 | 80 | 2.9 | 51 |
| LiCoO2 + porous LLZO scaffold | Solution infiltration | 700 | 3–4.05 | 0.05C | 62 | 118 | 3/14 | 80 | 0.73 | This study | |
From a ceramic manufacturing viewpoint, it is significant to stress that our cathode composite design was achieved without the need for any sintering additives. This translates into more “active” volume being available for filling cathode materials, thereby opening further engineering opportunities for optimization by material processing design. Ideally, an increase of the cathode loading and decrease of porous LLZO are required toward practical application (e.g. areal cathode loading ∼3 mA h cm−2 for a ∼120 μm-thick cathode composite77,78). The areal cathode loading in the tested LCO–LLZO cathode composite (Fig. 4f) was 0.73 mg cm−2 (0.084 mA h cm−2) (Table 1), which is below the level required for practical application. In recent studies in which a similar concept of an infiltrated all-oxide cathode in all-solid-state sodium batteries (ASSNBs) was implemented, a high cathode loading up to 6.2 mg cm−2 (0.6 mA h cm−2) was achieved.79,80 However, this approach required several repeated steps of infiltration and heat treatment and an ultra-thick ∼1 mm-thick porous ionic scaffold, thus still limiting practical application. The fabrication of a highly porous ionic scaffold that can afford more than 90 wt% or 80 vol% of active materials using freeze-tape-casting or inverse-opal-type template methods appears to be a promising option.27,81,82 In all of the methods, the use of a dispersant would increase the uniformity and ordering of the pore distribution further and may be considered for future improvement. A more practical strategy toward a cathode with high LiCoO2 loading would be to reconstruct the current cathode composite preparation by simply reversing the preparation order between LiCoO2 and LLZO (Fig. S14, ESI†), with LiCoO2 as the sintered porous scaffold and direct synthesis of the LLZO network onto the porous surface of LiCoO2. Regarding the rate performance, operating at room temperature under higher current (>0.05C) is desired. We propose that the rate-limiting step in a full cell (LiCoO2–LLZO|LLZO|Li) is either charge transport or electronic conduction at the LCO/LLZO interface, which is from the polarization process, (RQ)4, at mid frequencies (∼320 Hz) (Fig. 4g). Nonetheless, purely by adapting the ceramic synthesis approach to move radically away from previous attempts using crystalline powder constituents of the cathode via casting or other routes but with the direct formation of the active cathode from metal salt to oxide via a precursor solution and infiltration within the porous LLZO scaffold, good bonding and low interfacial resistance between the active materials and LLZO electrolyte were achieved at the relatively low processing temperature of 700 °C.
In summary, using crystalline powders of LLZO and LiCoO2, good mechanical bonding is hardly achieved at 700 °C,35 and high interfacial resistance is inevitable if the temperature is higher than that required for good solid–solid mechanical bonding (i.e., >1000 °C) because of possible side reactions.37 With additives, there has been a promising report;19 however, further engineering to achieve reduced additive loading or alternative processing such as additive coatings on cathode particles is required. Regardless of the cathode preparation methods, ultra-fast sintering strategies83,84 can further help to achieve chemically sharp but metallically strong-enough interfacial bonding for enhanced performance especially for all-oxide cathode composites for SSBs. With society's need for low-cost and mass-manufacturable processing for cathode composites, we conclude that the presented procedure is inexpensive, rapid, and potentially adaptable for large-scale implementation, as it is widely used in the fields of supported catalysts and solid-oxide fuel cells.85
Conclusion
The performance of all-oxide SSBs is often limited by the poor cathode–electrolyte contact and high interfacial impedance arising during fabrication. There are limited reports available on the realization of Li-garnet-based SSBs without the use of a high-temperature process (>1000 °C), sintering additives, or a fluid electrolyte (polymer-gel, liquid electrolyte), which leads to concerns about the interfacial stability, cathode loading, and safety. In this work, we proposed an alternative ceramic processing strategy to assemble an oxide-based cathode composite based on the most common cathodes of LiFePO4 and LiCoO2 (Fig. 6). Thermal heating of the LiFePO4–Li7La3Zr2O12 (LLZO) interface over 300 °C leads to interfacial decomposition and phase changes due to the poor thermal stability. As a result, the Li-garnet SSB starting with the LiFePO4 cathode exhibited a reduction of the voltage plateau from 3.2 V (LiFePO4) to 2.5 V (Li3Fe2(PO4)3). With such a limited thermal processing window, we propose that the use of a hybrid cathode is currently a feasible option but recommend all-oxide processing with a suitable cathode coating47 and cathode firing in a low oxygen partial pressure (e.g., H2/Ar gas).41 Taking key processing factors such as interfacial stability vs. temperature into account, we see that LiCoO2 provides more opportunities for successful preparation and a wider degree of freedom in the processing window of cathode composites with LLZO than LiFePO4. In particular, the presented LiCoO2–LLZO composite cathodes prepared through direct synthesis from metal salts to the oxide cathode in a porous LLZO scaffold clearly demonstrate processing capability that allows good mechanical contact without adverse interfacial reactions at the unusually low processing temperature of 700 °C. Importantly, the additive-free (resp. sinter-agent free) cathode composite results in the lowest interfacial resistance among reported all-oxide Li-garnet SSBs. We can widely adapt this methodology to the exploration of other cathode materials or electrolyte preparation for cathode composites. For example, altered strategies toward higher cathode loading may be the direct synthesis of a Li-ion conductor (e.g., LLZO) in a sintered porous LiCoO2 scaffold (Fig. S7, ESI†). In addition, we foresee that the processing temperature of the method we present here can potentially be reduced further by controlling the crystallization temperature in the metal salts to oxide transfer of the cathode phase through careful selection of metal salts and their melting points, and employment of organics with altered chain length (Fig. 6). With the help of crystallization kinetics information, gained by constructing time–temperature–transformation (TTT)86 diagrams using differential scanning calorimetry, we believe that novel cathode processing guidelines can be further developed.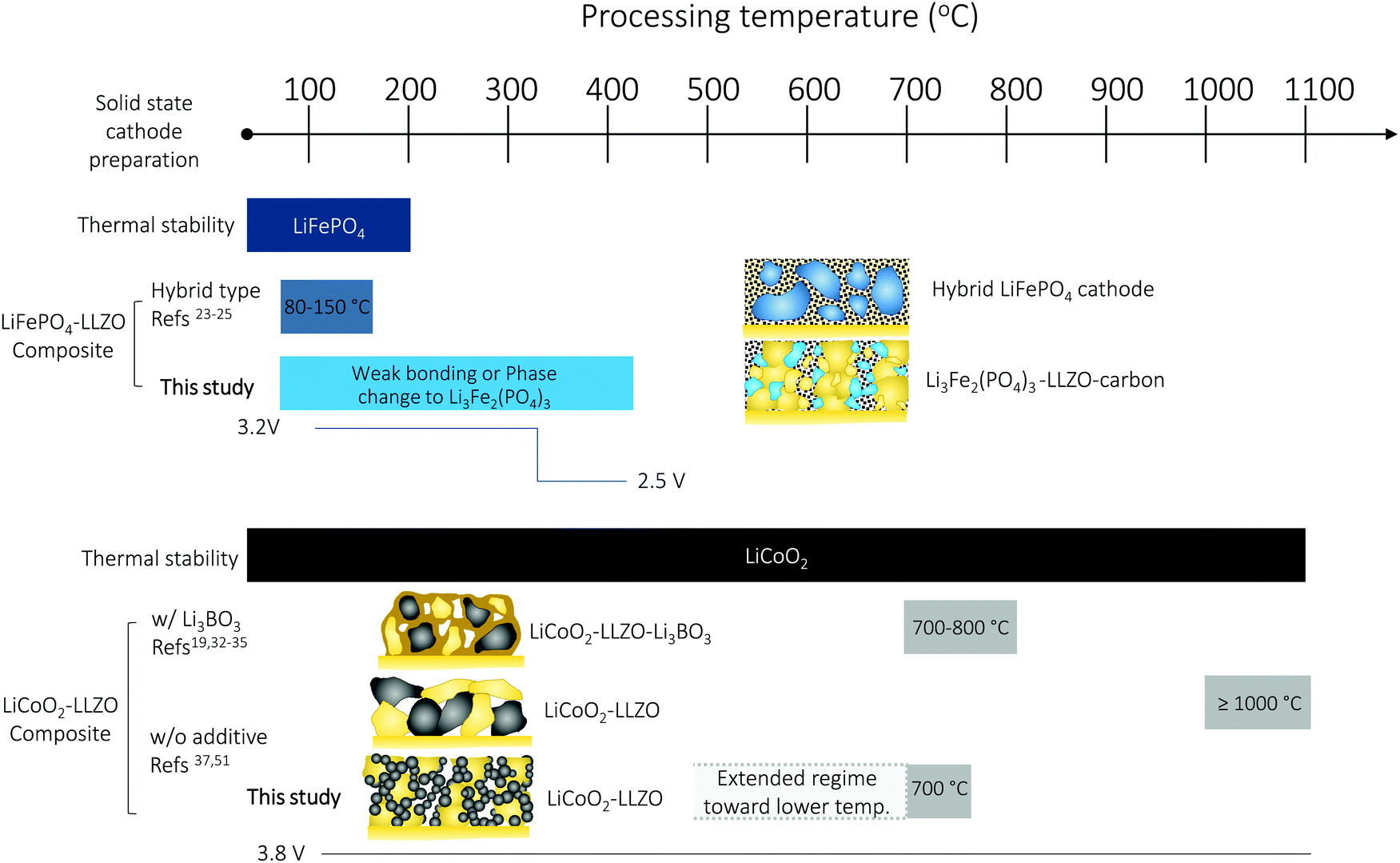 | ||
| Fig. 6 Overview of the cathode composite design and ceramic processing choices of all-oxide Li-garnet SSBs based on LiFePO4- and LiCoO2-based cathode composites: processing temperature vs. chemical and mechanical stability. | ||
Experimental details
Li-garnet electrolyte and porous Li-garnet scaffold
:50. Corn starch (Sigma-Aldrich) was added as a pore former. The prepared LLZO paste was printed on one side of the sintered LLZO pellet and fired at 1100 °C for 2 h under an oxygen flow. The active area of the porous LLZO layer was ∼0.56 cm2 after firing. The loading of porous LLZO achieved was varied between 3.5 and 10 mg depending on the number of screen-printings, resulting in different thicknesses.
All-solid-state battery
:50 to form a powder mixture pellet. Here, cubic-phase LLZO powder, obtained by annealing the second-calcined LLZO at 1100 °C for 1 h, was used. The powder mixture was pressed into 12 mm-diameter pellets. Firing of the composite pellets was performed on MgO plates at 250 °C, 300 °C, 350 °C, 400 °C, 600 °C, and 1000 °C for 1 h each at a heating rate of 10 °C min−1. For full-cell fabrication, three different composite cathode powder mixtures were prepared from LiFePO4 powder, cubic LLZO powder, and carbon-black with different weight ratios and milling processes: (i) 55:25:20 wt% without planetary milling (PM), (ii) 40:20:40 wt% without PM, and (iii) 40:20:40 wt% with PM for 2 h at 500 rpm. The cathode slurry paste was prepared with the powder mixture and binder solution and then infiltrated into the sintered porous LLZO layer. The prepared composite cathode on the LLZO electrolyte pellet was fired at 200 °C, 250 °C, and 400 °C for 1 h each at a heating rate of 10 °C min−1 in an oxygen atmosphere. After cooling, the cathode loading was estimated from the difference in weight before and after the composite cathode preparation. The typical LiFePO4 loading achieved was approximately 0.3–0.6 mg.
After the cathode preparation, gold paste (VWR International) was painted on top of the cathode and annealed at 300 °C for 2 h in an oxygen atmosphere. Subsequently, the prepared half-cells were introduced into an Ar-filled glove box (O2, H2O < 0.5 ppm) to prepare Li metal as the anode. Li foils (9 mm diameter) were punched and physically attached onto the other side of the LLZO electrolyte pellet. Then, the entire cells were heated on a hot plate at 180 °C for 12 h to achieve uniform contact between the LLZO electrolyte pellet and Li metal. The full cells were then assembled in an air-tight stainless steel holder (Pred Materials International).
Structural characterization
Raman spectra were obtained using a confocal WITec alpha300 R Raman microscope (WITec, Germany) with a 532 nm excitation wavelength. All the Raman measurements were performed in ambient air. SEM (Carl Zeiss Merlin HR-SEM, Germany) was employed to characterize the microstructure of the pellet, porous LLZO, and composite cathode after preparation. Image analysis for 2D porosity was performed using ImageJ 1.52a.87 XRD was used to investigate the electrolyte and electrode materials using a Panalytical X’Pert Pro diffractometer equipped with an X-ray tube using Ni-filtered Cu-Kα radiation (λ1 = 1.54056 Å and λ2 = 1.54439 Å). An X’Celerator 1D detector and a fixed divergence slit (1/2°) were used.Battery testing
Electrochemical cycling of the all-solid-state batteries was conducted at 80 °C using a BioLogic VSP-300 potentiostat/galvanostat (Bio-Logic, Knoxville, TN, USA). The current density and specific capacity were calculated based on the weight of LiFePO4 and LiCoO2 in the cathode. EIS measurements were performed over a frequency range of 7 MHz to 0.1 Hz using a 100 mV AC amplitude. EIS was analyzed using an equivalent circuit and fitting software (ZFit, Bio-logic).Author contributions
K. J. K and J. L. M. R. proposed the ceramic processing concepts and designed the experiments. K. J. K performed the experiments. K. J. K and J. L. M. R. analyzed the data and wrote the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This publication is based on work funded by the Skolkovo Institute of Science and Technology (Skoltech) through the MIT Skoltech program under contract number 6937431. Structural characterization of the materials was conducted under the MRSEC Program of the National Science Foundation under award DMR-1419807. J. L. M. R. thanks the Thomas Lord Foundation for financial support. We thank Prof. Michal Struzik and Dr. Juan Carlos Gonzalez-Rosillo for the initial help on solid electrolyte synthesis and current collector preparation.References
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 CrossRef.
- K. Kerman, A. Luntz, V. Viswanathan, Y.-M. Chiang and Z. Chen, J. Electrochem. Soc., 2017, 164, A1731–A1744 CrossRef CAS.
- A. J. Samson, K. Hofstetter, S. Bag and V. Thangadurai, Environ. Sci., 2019, 12, 2957–2975 CAS.
- R. Pfenninger, M. Struzik, I. Garbayo, E. Stilp and J. L. M. Rupp, Nat. Energy, 2019, 4, 475–483 CrossRef CAS.
- C. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- B. J. Neudecker, N. J. Dudney and J. B. Bates, J. Electrochem. Soc., 2000, 147, 517 CrossRef CAS.
- J. B. Bates, N. J. Dudney, B. Neudecker, A. Ueda and C. D. Evans, Solid State Ionics, 2000, 135, 33–45 CrossRef CAS.
- J. Li, C. Ma, M. Chi, C. Liang and N. J. Dudney, Adv. Energy Mater., 2015, 5, 1–6 Search PubMed.
- Y. Zhu, M. Gonzalez-Rosillo, J. C. Balaish, Z. D. Hood, K. J. Kim and J. L. M. Rupp, Nat. Rev. Mater., 2020 Search PubMed , in Press.
- G. T. Hitz, D. W. McOwen, L. Zhang, Z. Ma, Z. Fu, Y. Wen, Y. Gong, J. Dai, T. R. Hamann, L. Hu and E. D. Wachsman, Mater. Today, 2019, 22, 50–57 CrossRef CAS.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS.
- N. J. Taylor, S. Stangeland-Molo, C. G. Haslam, A. Sharafi, T. Thompson, M. Wang, R. Garcia-Mendez and J. Sakamoto, J. Power Sources, 2018, 396, 314–318 CrossRef CAS.
- F. Flatscher, M. Philipp, S. Ganschow, H. M. R. Wilkening and D. Rettenwander, J. Mater. Chem. A, 2020, 8, 15782–15788, 10.1039/c9ta14177d.
- T. Deng, X. Ji, Y. Zhao, L. Cao, S. Li, S. Hwang, C. Luo, P. Wang, H. Jia, X. Fan, X. Lu, D. Su, X. Sun, C. Wang and J. Zhang, Adv. Mater., 2020, 2000030, 2000030 CrossRef.
- J. Duan, L. Huang, T. Wang, Y. Huang, H. Fu, W. Wu, W. Luo and Y. Huang, Adv. Funct. Mater., 2020, 30, 1–7 Search PubMed.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 10, 4257–4300 CrossRef.
- F. Hao, F. Han, Y. Liang, C. Wang and Y. Yao, MRS Bull., 2018, 43, 746–751 CrossRef.
- F. Han, J. Yue, C. Chen, N. Zhao, X. Fan, Z. Ma, T. Gao, F. Wang, X. Guo and C. Wang, Joule, 2018, 2, 497–508 CrossRef CAS.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Nat. Energy, 2020, 5, 1–12 CrossRef.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- Z. Bi, N. Zhao, L. Ma, Z. Fu, F. Xu, C. Wang and X. Guo, Chem. Eng. J., 2020, 387, 124089 CrossRef CAS.
- F. Du, N. Zhao, Y. Li, C. Chen, Z. Liu and X. Guo, J. Power Sources, 2015, 300, 24–28 CrossRef CAS.
- Y. Li, B. Xu, H. Xu, H. Duan, X. Lü, S. Xin, W. Zhou, L. Xue, G. Fu, A. Manthiram and J. B. Goodenough, Angew. Chem., Int. Ed., 2017, 56, 753–756 CrossRef CAS.
- W. Luo, Y. Gong, Y. Zhu, Y. Li, Y. Yao, Y. Zhang, K. K. Fu, G. Pastel, C. F. Lin, Y. Mo, E. D. Wachsman and L. Hu, Adv. Mater., 2017, 29, 1606042, DOI:10.1002/adma.201606042.
- Y. Shao, H. Wang, Z. Gong, D. Wang, B. Zheng, J. Zhu, Y. Lu, Y. S. Hu, X. Guo, H. Li, X. Huang, Y. Yang, C. W. Nan and L. Chen, ACS Energy Lett., 2018, 3, 1212–1218 CrossRef CAS.
- E. Yi, H. Shen, S. Heywood, J. Alvarado, D. Y. Parkinson, G. Chen, S. W. Sofie, M. M. Doeff, S. W. So and M. M. Doe, ACS Appl. Energy Mater., 2020, 3, 170–175 CrossRef CAS.
- Y. Lu, X. Huang, Z. Song, K. Rui, Q. Wang, S. Gu, J. Yang, T. Xiu, M. E. Badding and Z. Wen, Energy Storage Mater., 2018, 15, 282–290, DOI:10.1016/j.ensm.2018.05.018.
- J. Liu, X. Gao, G. O. Hartley, G. J. Rees, C. Gong, F. H. Richter, J. Janek, Y. Xia, A. W. Robertson, L. R. Johnson and P. G. Bruce, Joule, 2020, 4, 101–108 CrossRef CAS.
- M. Naguib, A. Sharafi, E. C. Self, H. M. Meyer, J. Sakamoto and J. Nanda, ACS Appl. Mater. Interfaces, 2019, 11, 42042–42048, DOI:10.1021/acsami.9b11439.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820, DOI:10.1126/sciadv.aas9820.
- T. Liu, Y. Ren, Y. Shen, S.-X. X. Zhao, Y. Lin and C.-W. W. Nan, J. Power Sources, 2016, 324, 349–357 CrossRef CAS.
- M. Shoji, H. Munakata and K. Kanamura, Front. Energy Res., 2016, 4, 1–7 Search PubMed.
- S. Ohta, J. Seki, Y. Yagi, Y. Kihira, T. Tani and T. Asaoka, J. Power Sources, 2014, 265, 40–44 CrossRef CAS.
- K. Park, B. C. Yu, J. W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS.
- L. Ting, Z. Yibo, Z. Xue, W. Lei, Z. Shi-Xi, L. Yuan-Hua, S. Yang, L. Jun, L. Liangliang, N. Ce-Wen, T. Liu, Y. Zhang, X. Zhang, L. Wang, S. X. Zhao, Y. H. Lin, Y. Shen, J. Luo, L. Li and C. W. Nan, J. Mater. Chem. A, 2018, 6, 4649–4657 RSC.
- C. L. Tsai, Q. Ma, C. Dellen, S. Lobe, F. Vondahlen, A. Windmüller, D. Grüner, H. Zheng, S. Uhlenbruck, M. Finsterbusch, F. Tietz, D. Fattakhova-Rohlfing, H. P. Buchkremer and O. Guillon, Sustainable Energy Fuels, 2019, 3, 280–291 RSC.
- Y. Xiao, Y. Wang, S. H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2019, 5, 105–126 CrossRef.
- L. Miara, A. Windmüller, C. L. Tsai, W. D. Richards, Q. Ma, S. Uhlenbruck, O. Guillon and G. Ceder, ACS Appl. Mater. Interfaces, 2016, 8, 26842–26850 CrossRef CAS.
- J. Wakasugi, H. Munakata and K. Kanamura, Electrochemistry, 2017, 85, 77–81 CrossRef CAS.
- Y. Ren, T. Liu, Y. Shen, Y. Lin and C. W. Nan, J. Mater., 2016, 2, 256–264 Search PubMed.
- S. Ohta, S. Komagata, J. Seki, T. Saeki, S. Morishita and T. Asaoka, J. Power Sources, 2013, 238, 53–56 CrossRef CAS.
- N. C. Rosero-Navarro and K. Tadanaga, Sintering Additives for Garnet-Type Electrolytes, Springer, 2000 Search PubMed.
- W. D. Kingery and M. D. Narasimhan, J. Appl. Phys., 1959, 30, 307–310 CrossRef CAS.
- R. A. Jonson and P. J. McGinn, Solid State Ionics, 2018, 323, 49–55 CrossRef CAS.
- R. H. Shin, S. I. Son, Y. S. Han, Y. Do Kim, H. T. Kim, S. S. Ryu and W. Pan, Solid State Ionics, 2017, 301, 10–14 CrossRef CAS.
- Y. Xiao, L. J. Miara, Y. Wang and G. Ceder, Joule, 2019, 3, 1252–1275 CrossRef CAS.
- S. Xu, D. W. McOwen, C. Wang, L. Zhang, W. Luo, C. Chen, Y. Li, Y. Gong, J. Dai, Y. Kuang, C. Yang, T. R. Hamann, E. D. Wachsman and L. Hu, Nano Lett., 2018, 18, 3926–3933 CrossRef CAS.
- J. van den Broek, S. Afyon and J. L. M. Rupp, Adv. Energy Mater., 2016, 6, 1–11 Search PubMed.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- Y. Ren, T. Liu, Y. Shen, Y. Lin and C. W. Nan, Ionics, 2017, 23, 2521–2527 CrossRef CAS.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS.
- D. Rettenwander, G. Redhammer, F. Preishuber-Pflügl, L. Cheng, L. Miara, R. Wagner, A. Welzl, E. Suard, M. M. Doeff, M. Wilkening, J. Fleig and G. Amthauer, Chem. Mater., 2016, 28, 2384–2392 CrossRef CAS.
- N. Nitta, F. Wu, J. T. Lee and G. Yushin, Mater. Today, 2015, 18, 252–264 CrossRef CAS.
- B. Lung-Hao, Hu, F. Y. Wu, C. Te Lin, A. N. Khlobystov and L. J. Li, Nat. Commun., 2013, 4, 1–7 Search PubMed.
- C. Wang and J. Hong, Electrochem. Solid-State Lett., 2007, 10, A65 CrossRef CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS.
- J. Ni and Y. Wang, RSC Adv., 2015, 5, 30537–30541 RSC.
- C. Masquelier, A. K. Padhi, K. S. Nanjundaswamy and B. Goodenough, J. Solid State Chem., 1998, 135, 228–234 CrossRef CAS.
- L. J. Miara, W. D. Richards, Y. E. Wang and G. Ceder, Chem. Mater., 2015, 27, 4040–4047 CrossRef CAS.
- G. Vardar, W. J. Bowman, Q. Lu, J. Wang, R. J. Chater, A. Aguadero, R. Seibert, J. Terry, A. Hunt, I. Waluyo, D. D. Fong, A. Jarry, E. J. Crumlin, S. L. Hellstrom, Y. M. Chiang and B. Yildiz, Chem. Mater., 2018, 30, 6259–6276 CrossRef CAS.
- Y. Ren, T. Liu, Y. Shen, Y. Lin and C. Nan, J. Mater., 2016, 2, 256–264 Search PubMed.
- G. Larraz, A. Orera and M. L. Sanjuán, J. Mater. Chem. A, 2013, 1, 11419–11428 RSC.
- F. Tietz, T. Wegener, M. T. Gerhards, M. Giarola and G. Mariotto, Solid State Ionics, 2013, 230, 77–82 CrossRef CAS.
- T. Gross and C. Hess, J. Power Sources, 2014, 256, 220–225 CrossRef CAS.
- M. D. Levi, J. Electrochem. Soc., 1999, 146, 1279 CrossRef CAS.
- W. Zhang, F. H. Richter, S. P. Culver, T. Leichtweiss, J. G. Lozano, C. Dietrich, P. G. Bruce, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2018, 10, 22226–22236 CrossRef CAS.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS.
- T. Yoshinari, R. Koerver, P. Hofmann, Y. Uchimoto, W. G. Zeier and J. Janek, ACS Appl. Mater. Interfaces, 2019, 11, 23244–23253 CrossRef CAS.
- I. A. J. Gordon, S. Grugeon, H. Takenouti, B. Tribollet, M. Armand, C. Davoisne, A. Débart and S. Laruelle, Electrochim. Acta, 2017, 223, 63–73 CrossRef CAS.
- J. Illig, M. Ender, T. Chrobak, J. P. Schmidt, D. Klotz and E. Ivers-Tiffée, J. Electrochem. Soc., 2012, 159, A952–A960 CrossRef CAS.
- Y. J. Nam, K. H. Park, D. Y. Oh, W. H. An and Y. S. Jung, J. Mater. Chem. A, 2018, 6, 14867–14875 RSC.
- S. P. Culver, R. Koerver, W. G. Zeier and J. Janek, Adv. Energy Mater., 2019, 9, 1–14 Search PubMed.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- J. Lu, T. Wu and K. Amine, Nat. Energy, 2017, 2, 17011 CrossRef CAS.
- D. H. S. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, Nat. Nanotechnol., 2020, 15, 1–11 CrossRef.
- Y. G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D. S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308, DOI:10.1038/s41560-020-0575-z.
- P. Albertus, S. Babinec, S. Litzelman and A. Newman, Nat. Energy, 2018, 3, 16–21 CrossRef CAS.
- C. L. Tsai, T. Lan, C. Dellen, Y. Ling, Q. Ma, D. Fattakhova-Rohlfing, O. Guillon and F. Tietz, J. Power Sources, 2020, 476, 228666 CrossRef CAS.
- T. Lan, C.-L. Tsai, F. Tietz, X.-K. Wei, M. Heggen, R. E. Dunin-Borkowski, R. Wang, Y. Xiao, Q. Ma and O. Guillon, Nano Energy, 2019, 65, 104040 CrossRef CAS.
- J. S. Sakamoto and B. Dunn, J. Mater. Chem., 2002, 12, 2859–2861 RSC.
- M. Hara, H. Nakano, K. Dokko, S. Okuda, A. Kaeriyama and K. Kanamura, J. Power Sources, 2009, 189, 485–489 CrossRef CAS.
- G. Zhong, C. Wang, R. Wang, W. Ping, S. Xu, H. Qiao, M. Cui, X. Wang, Y. Zhou, D. J. Kline, M. R. Zachariah and L. Hu, Energy Storage Mater., 2020, 30, 385–391 CrossRef.
- C. Wang, W. Ping, Q. Bai, H. Cui, R. Hensleigh, R. Wang, A. H. Brozena, Z. Xu, J. Dai, Y. Pei, C. Zheng, G. Pastel, J. Gao, X. Wang, H. Wang, J.-C. Zhao, B. Yang, X. (Rayne) Zheng, J. Luo, Y. Mo, B. Dunn and L. Hu, Science, 2020, 368, 521–526 CrossRef CAS.
- J. T. S. Irvine, D. Neagu, M. C. Verbraeken, C. Chatzichristodoulou, C. Graves and M. B. Mogensen, Nat. Energy, 2016, 1, 1–13 Search PubMed.
- J. L. M. Rupp, B. Scherrer, N. Schäuble and L. J. Gauckler, Adv. Funct. Mater., 2010, 20, 2807–2814 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ee02062a |
| This journal is © The Royal Society of Chemistry 2020 |