
Synergy effect between photocatalysis and heterogeneous photo-Fenton catalysis on Ti-doped LaFeO3 perovskite for high efficiency light-assisted water treatment†
Patricia
Garcia-Muñoz
 *a,
Fernando
Fresno
b,
Christophe
Lefevre
c,
Didier
Robert
a and
Nicolas
Keller
*a
*a,
Fernando
Fresno
b,
Christophe
Lefevre
c,
Didier
Robert
a and
Nicolas
Keller
*a
aInstitut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé (ICPEES), CNRS/University of Strasbourg, 25 rue Becquerel, Strasbourg, France. E-mail: garciamunoz@unistra.fr; nkeller@unistra.fr
bPhotoactivated Processes Unit, IMDEA Energy Institute. Avda. Ramón de la Sagra, 3, Parque Tecnológico de Móstoles, 28935 Móstoles, Madrid, Spain
cInstitut de Physique et de Chimie des Matériaux de Strasbourg (IPCMS), CNRS/University de Strasbourg, 23 rue du Loess, Strasbourg, France
First published on 17th January 2020
Abstract
A strategy combining both heterogenized photo-Fenton and photocatalysis advanced oxidation processes for a single dual catalyst is developed simultaneously benefiting from the higher reaction rate of photo-Fenton and the higher mineralization yield of photocatalysis in water treatment, while overcoming the strong drawbacks still limiting their viability as single processes. We prepared via a facile Pechini sol–gel route a highly efficient and stable Ti-substituted LaFeO3 dual catalyst, acting simultaneously as a reusable photo-Fenton solid catalyst and a photocatalyst in water treatment. The partial substitution of La by Ti ions in the crystalline network led to pure heterogeneous surface reactions with an increase in the catalyst robustness by more than two orders of magnitude, indicating the absence of any Fe released and the stability of catalytic performance with test cycles. It further strongly enhanced the reaction rates, due to both increased availability of the photogenerated charge carriers at the surface and the electronic enrichment of surface Fe, and allowed full mineralization of a pollutant to be achieved at circumneutral pH through combined advanced oxidation processes.
Introduction
In addition to increasingly restrictive environmental government regulations, the emergence of biorecalcitrant refractory pollutants in water, the heterogeneity of the targets and the increase in the volume and concentration of polluted water streams have attracted strong interest for the elaboration of new sustainable and cost-effective water treatment technologies.1–4 In this frame, advanced oxidation processes (AOPs), based on the generation of highly active HOx˙ oxidative radicals that react with pollutants and further with reaction intermediates to achieve substrate mineralization, have been developed for removing a large class of refractory substances that cannot be treated through conventional mechanical, biological or physico-chemical water treatments.5,6Among them, (i) the homogeneous Fenton process,7 or heterogeneous catalytic wet peroxide oxidation (CWPO),8 that is based on the catalytic decomposition of H2O2 in the presence of metallic ions, in most of the cases Fe, and (ii) photocatalysis,9 that uses the redox surface properties developed by an adequately-irradiated semiconductor, have aroused particular interest because of their high efficiency as well as relatively low investment/running costs, although they operate via different mechanisms and under different conditions. However, both processes still suffer from strong limitations that reduce their viability.
While heterogeneous semiconductor photocatalysis is an AOP that takes advantage of its ability to achieve total mineralization of a large variety of refractory organic substances and works within a large pH range, it unfortunately suffers from far lower reaction rates than homogeneous Fenton-like AOPs, and accordingly from far longer process durations.5,10,11
By contrast, the viability of the Fenton AOP is strongly reduced by the requirement to operate at acidic pH and by the complexation of Fe with short-chain acids which leads to the generation/precipitation of sludge, with the consequent continuous loss of the active phase and the non-sustainable production of waste.12 Heterogenization of the Fenton AOP by incorporating Fe in a catalytic body partially overcomes these issues, but at the expense of a strong reduction in the reaction rates in comparison to the homogeneous Fenton counterpart, with a drastic decrease of the process ability to achieve complete pollutant mineralization and continuous Fe release in water by leaching.7,12 Furthermore, although performing CWPO under light in the so-called photo-assisted CWPO or heterogenized photo-Fenton process is a promising approach for increasing the catalyst reactivity by further boosting the rate-limiting FeIII → FeII conversion in the FeIII/FeII redox cycle, the above-mentioned drawbacks still limit the viability of the heterogenized catalytic process.5,7,13
Therefore, a strategy consisting in combining both heterogenized photo-Fenton and photocatalysis AOPs with a unique catalyst is considered as a promising approach simultaneously benefiting from a higher mineralization yield and a higher reaction rate, with as key-features that this unique dual catalyst works under similar reaction conditions as a heterogeneous photocatalyst and a Fe-based photo-Fenton catalyst without any loss of the Fe active phase with time-on-run. Meanwhile, physical mixtures of catalysts have been tested,14–17 and the design of a single catalyst combining both AOPs has been scarcely reported, notably using ZnFe2O4 spinel ferrite, natural Fe oxide or Ba-doped BiFeO3 perovskite.18–20
Here, we report on the use of Ti-substituted lanthanum ferrite (LaFeO3) perovskite catalysts in water treatment acting simultaneously as a heterogenized photo-Fenton catalyst and a photocatalyst under similar circumneutral pH reaction conditions for achieving complete mineralization of a substrate under UV-A light with strongly enhanced reaction rates, while improving the catalyst stability by totally inhibiting the Fe release in water. This new dual catalyst is based on a LaFeO3 semiconductor that is one of the most common ABO3 perovskite oxides studied in many research fields such as solid oxide fuel cells, opto-electronics, magnetism, gas sensors or heterogeneous thermal catalysis, because it takes advantage of its 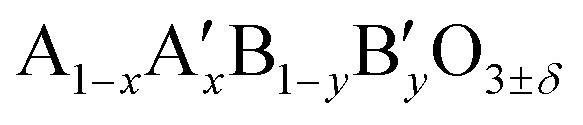 composition versatility that allows notably its textural, structural or electronic properties to be modified, and as a result, its redox and catalytic ability to be tuned through A- and/or B-site cationic partial substitution.21 Its composition is consequently in agreement with the sustainability need currently in force that requires the use of cheap, abundant, and environmentally non-toxic elements in chemical processes.22 While cation-doped LaFeO3 ferrites have been mainly used till now as photo Fenton catalysts, their use also as photocatalysts remained scarce. Here, the removal of 4-chorophenol (4-CP) has been selected as the test reaction in water under UV-A light, as 4-CP is a model molecule used in AOP studies as a representative of industrial waste-water refractory pollutants,23–25 while some mineralization tests were also performed using the myclobutanil C15H17ClN4 triazole as an antifungal substrate in order to broaden the scope of the work.26,27
composition versatility that allows notably its textural, structural or electronic properties to be modified, and as a result, its redox and catalytic ability to be tuned through A- and/or B-site cationic partial substitution.21 Its composition is consequently in agreement with the sustainability need currently in force that requires the use of cheap, abundant, and environmentally non-toxic elements in chemical processes.22 While cation-doped LaFeO3 ferrites have been mainly used till now as photo Fenton catalysts, their use also as photocatalysts remained scarce. Here, the removal of 4-chorophenol (4-CP) has been selected as the test reaction in water under UV-A light, as 4-CP is a model molecule used in AOP studies as a representative of industrial waste-water refractory pollutants,23–25 while some mineralization tests were also performed using the myclobutanil C15H17ClN4 triazole as an antifungal substrate in order to broaden the scope of the work.26,27
Experimental
Synthesis of Ti-substituted LaFeO3
La1−xTixFeO3 catalysts were prepared by modifying the sol–gel Pechini synthesis. La(NO3)3 lanthanum nitrate hexahydrate (108.3 g L−1, 99.9%, Sigma Aldrich) and Fe(NO3)3 iron nitrate nonahydrate (133 g L−1, 99.95%, Sigma Aldrich) were dissolved in stoichiometric amounts in distilled water (30 mL), and the metallic ions underwent complexation by subsequent addition of citric acid (C6H8O7xH2O, 70 g L−1, 99.5%, Sigma Aldrich) in a stoichiometric molar ratio with respect to the metallic ions.A titanium source was introduced under stirring into the aqueous sol and the resulting suspensions were maintained at 80 °C for 48 h (2 °C min−1 heating rate) in order to induce the complete gel formation. The calcination of the dried gel was performed using a heating cycle as reported by Gosavi et al.28 with a heating rate of 5 °C min−1, holding the temperature at 600 °C for 2 h, before subsequently maintaining a temperature plateau at 800 °C for 2 h.
In the synthesis of the P25-LFO catalyst, commercial TiO2 Aeroxide© P25 powder (hydrophilic fumed titanium dioxide, ≥99.50%, Evonik, Germany) that consists of a mixture of anatase and rutile crystalline phases in a 80/20 molar ratio was used as a titanium source.
In the synthesis of the SG-LFO catalyst, dried amorphous titania was used as a titanium source. It was obtained according to a sol–gel synthesis method under basic conditions using titanium tetraisopropoxide (TTIP, 97%, Sigma-Aldrich) as a precursor. In a typical synthesis, 17.8 g of TTIP was first added to a 40 mL ethanolic solution, before a similar volume of water was added dropwise to the solution. After rectification of the pH to 9 by adding ammonia (Carlo Erba, 30% aqueous solution), the solution was left under agitation at room temperature until a dry paste was obtained. The resulting powder was further dried at 110 °C for 12 h to obtain the dried amorphous titania used as a titanium source in the modified sol–gel Pechini synthesis.
Regardless of the titanium source, the titanium added during the Pechini synthesis corresponded to a nominal amount of 10 wt% in terms of TiO2 with respect to the theoretical amount of LaFeO3 perovskite obtained. Non-modified lanthanum ferrite powders have been synthesized as reference materials via the same protocol without any addition of titanium source, with a similar calcination procedure.
The stoichiometric reaction forming the LaFeO3 perovskite is as follows (eqn (1)):29
| La(NO3)3x6H2O + Fe(NO3)3x9H2O + C6H8O7xH2O → LaFeO3 + 6CO2 + 3N2 + nH2O | (1) |
The reference titanium-free material was labelled as LFO, while the Ti-modified materials were labelled as P25-LFO and SG-LFO when using Aeroxide© TiO2 P25 and dried amorphous titania as titanium sources, respectively.
Characterisation techniques
The reference X-ray diffraction (XRD) patterns were recorded at room temperature in a θ/θ mode on a Bruker D8 Advance diffractometer equipped with a monochromatic copper radiation source (Kα = 1.54056 Å) with a scan step of 0.02°. Some patterns have been investigated by Rietveld refinement with the Fullprof software for which the modified Thompson–Cox–Hasting function was chosen to generate the line shape of the diffraction peaks. Instrumental broadening was previously determined by measuring the scattering from corundum (NIST standard SRM 1976b).30,31Scanning electron microscopy (SEM) was performed in secondary electron mode on a JEOL JSM-6700 F FEG microscope.
Transmission electron microscopy (TEM) was performed using a JEOL 2100F with a 0.2 nm point resolution. A copper grid covered by a holey carbon membrane was used for observation. The interplanar spacings were calculated using ImageJ software.
UV-vis diffuse reflectance spectra were acquired using a UV/vis/NIR Perkin Elmer Lambda 1050 spectrometer, recording the reflectance between 250 and 800 nm. Band gaps were estimated from Tauc plots represented by (αhν)2vs. photon energy, where α = −ln(reflectance), in accordance with the direct semiconductor character of LaFeO3.32
The fluorescence spectra of the powdered samples were recorded with a fluorescence spectrometer Perkin Elmer LS55, using an excitation wavelength of 450 nm and a cut-off filter at 515 nm. Fluorescence lifetime measurements by time-correlated single photon counting (TCSPC) were carried out with Mini-τ equipment from Edinburgh Instruments using a laser diode (model EPL375) as an excitation source with a wavelength of 445.2 nm and a pulse width of 90.7 ps, at a repetition rate of 1 MHz, with a band pass filter at 600 ± 25 nm. The decay profiles were fitted to a bi-exponential decay curve, with lifetimes obtained as the average of both components, calculated as <τ> = [∑(Aiτi2)]/∑(Aiτi), where Ai is the amplitude and τi the lifetime of each component.
X-ray photoelectron spectroscopy (XPS) analysis was performed on a ThermoVGMultilabESCA3000 spectrometer (Al Kα anode at hλ = 1486.6 eV). The energy shift due to electrostatic charging was subtracted using the contamination sp2 carbon C 1s band at 284.6 eV. Contributions with a Doniach–Sunjic shape33 and an S-shaped Shirley type background34 were used.
Degradation test protocol
The 4-CP and myclobutanil degradation tests were performed in a batch beaker-type glass reactor at atmospheric pressure, at a constant temperature of 20 °C ± 2 °C (using a cooling bath) with H2O2 as the main oxidant. In a typical reaction procedure, 50 mg of catalyst was dispersed under stirring in a 100 mL aqueous solution of 4-chlorophenol at a concentration of 25 mg L−1, corresponding to an initial organic carbon concentration of 14.8 ppm and to a catalyst load of 0.5 g L−1. For the reactions with myclobutanil, a starting concentration of 20 mg L−1 of the pollutant was selected. Prior to irradiation, the catalyst suspension was stirred for 1 h in the dark to ensure the establishment of the adsorption/desorption equilibrium. After adding H2O2 at the stoichiometric concentration corresponding to the complete mineralization of the initial pollutant (125 mg L−1), or substoichiometric in some cases (31.25 and 62.50 mg L−1), the tests were performed under UV-A (60 W m−2; λ = 365 nm, Philips 24 W/10/4P lamps). At each time interval, 8 mL of solution was sampled and then filtered through a 0.20 μm porosity filter to remove the photocatalyst powder if any, before the concentration of 4-chlorophenol or myclobutanil was determined by UV-visible spectrophotometry by following the disappearance of the main absorption peak at λ = 224 nm.35 Total organic carbon measurements were carried out using a Shimadzu TOC-L analyzer to determine the organic carbon load. The H2O2 concentration was measured by colorimetric titration using the TiOSO4 method36 and the Fe leached during the time-course of the reaction was quantified by the ortho-phenanthroline method.37Tests with myclobutanil as the substrate were performed following a similar test protocol, at a myclobutanil concentration of 20 mg L−1. The myclobutanil concentration was determined by UV-visible spectrophotometry by monitoring the disappearance of the main absorption peak at λ = 220 nm.
Results and discussion
Catalyst characterization
Fig. 1 shows the XRD patterns of the pristine and Ti-modified LaFeO3 materials with their corresponding Rietveld refinement profiles. Tables 1 and S1† show the refined cationic distribution in the LaFeO3 structure and the crystallographic data, respectively. The XRD patterns of the pristine catalyst exhibited the most intense diffraction peaks at 22.6°, 32.2°, 39.7°, 46.1°, 57.4° and 67.3° in 2θ, corresponding to the diffraction of the (100), (110), (111), (200), (210) and (220) planes of the LaFeO3 phase, respectively. α-Fe2O3 in a small amount (11%) with a mean crystallite size of 35 nm was observed as the extra phase. In the Ti-doped samples, independent of the Ti source, all the patterns can be indexed to the Pbnm orthorhombic unit cell of LaFeO3, with no change in terms of α-Fe2O3 additional phase. Neither La2O3 nor TiO2 is observed as the extra phase. Elemental chemical analysis reveals a 3.2 at% Ti content in the Ti-modified LaFeO3 catalyst prepared using the crystalline TiO2 nanopowder as the precursor, which further increased up to 4.0 at% using the dried amorphous precursor, while the Ti/Fe molar ratio remains unchanged (Table 2).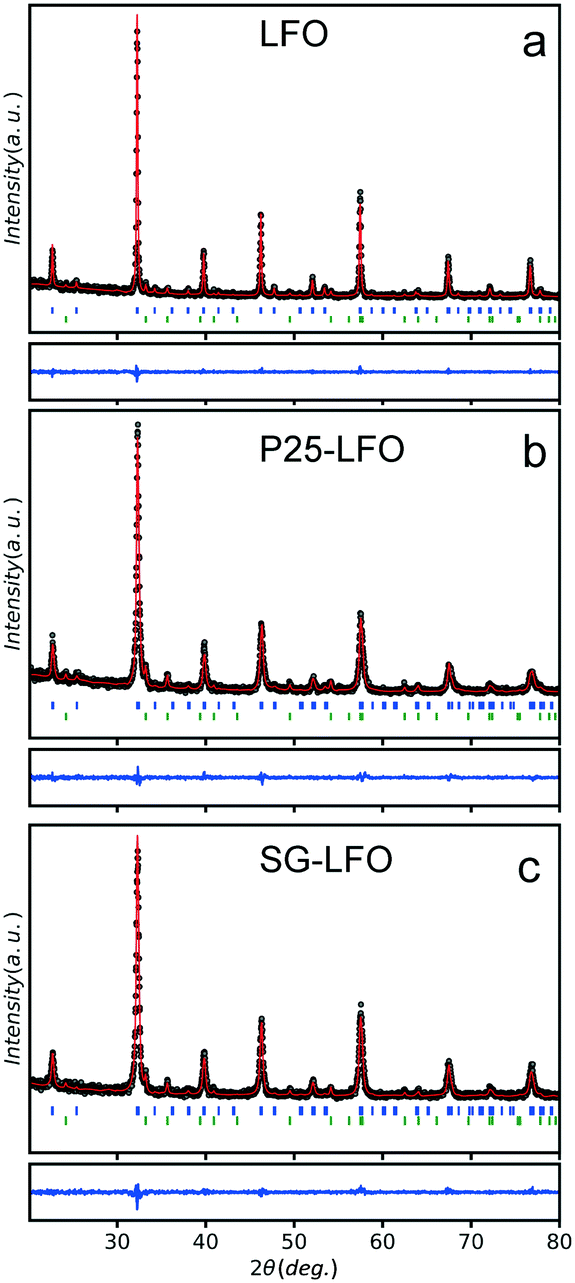 | ||
| Fig. 1 Observed (black), calculated (red full line), and difference (blue full line) X-ray powder diffraction patterns of (a) LFO, (b) P25-LFO and (c) SG-LFO catalysts recorded on a D8 diffractometer (λ = 1.54056 Å). The positions of the Bragg reflections are represented by vertical bars, in blue for the reflections indexed in the Pbnm orthorhombic unit cell of LaFeO3 and in green for those of the α-Fe2O3 extra phase. | ||
| Catalyst | La site distributiona | Fe site distributiona | α-Fe2O3, % | TiO2, % | |
|---|---|---|---|---|---|
| La | Ti | Fe | |||
| a The determination of the cationic distribution within the structures was performed considering that titanium can move towards both cationic sites. b SG-LFO #1, #3 and #5 correspond to the SG-LFO catalyst after tests #1, #3 and #5, respectively. | |||||
| LFO | 1 | 0 | 1 | 11 | 0 |
| P25-LFO | 0.95 | 0.05 | 1 | 15 | 0 |
| SG-LFO | 0.89 | 0.11 | 1 | 18 | 0 |
| SG-LFO #1b | 0.88 | 0.12 | 1 | 12 | 0 |
| SG-LFO #3b | 0.88 | 0.12 | 1 | 11 | 0 |
| SG-LFO #5b | 0.88 | 0.12 | 1 | 12 | 0 |
| Catalyst | TiO2 precursor | Fea (at%) | Tia (at%) | Ti/Fe molar ratio | S BET (m2 g−1) | D (nm) |
|---|---|---|---|---|---|---|
| a The iron and titanium contents in the catalysts were determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) carried out on an Optima 7000 DV spectrometer (Perkin Elmer), after microwave-assisted acidic dissolution in aqua regia at 185 °C under autogenic pressure. b The Brunauer–Emmett–Teller (BET) specific surface area was calculated from the N2 adsorption isotherm recorded on a Micrometrics Tristar 3000 using N2 as the adsorbent at −196 °C with prior outgassing at 250 °C overnight to desorb the impurities or moisture. c The mean crystallite size defined as the average size of the coherent diffracting domains was derived from the whole XRD patterns after refinement and determined using the Scherrer equation with the usual assumption of spherical crystallites taking into account the intrinsic broadening of the peaks due to the instrumentation. | ||||||
| LFO | — | 23.0 | — | — | 8 | 46(1) |
| P25-LFO | Crystalline anatase/rutile | 16.0 | 3.2 | 0.23 | 28 | 21(1) |
| SG-LFO | Amorphous hydroxide | 22.7 | 4.0 | 0.21 | 5 | 20(1) |
The separate refinement of both Ti/Fe and Ti/La site occupations evidences that titanium atoms preferentially occupy exclusively the La A-site and substitute only the La atoms, so that the catalysts can be labelled as La1−xTixFeO3. A partial Ti → La substitution rate of 5 at% was achieved within the perovskite network in the A-site using the crystalline TiO2 nanopowder as the titanium precursor, while it increases up to 11 at% using the dried amorphous precursor (Table 1). The exclusive La A-site occupation is rather unexpected when considering the involved cationic sizes – (rFe+III)CN=6 = 0.64, (rLa+III)CN12 = 1.46 and (rTi+III)CN=6 = 0.76.38 Both A- and B-site substitutions have been reported for the LaFeO3 structure depending on the doping element, although high valence metal ions usually occupy the Fe3+ sites.39,40 However, there is no empirical rule on the location of the substituting element depending on the electronic or steric criterion, and many extrinsic parameters are known to affect the cationic distribution in oxides, e.g. the cooling rate or the synthesis temperature.41,42 Here, we suggested that the partial substitution resulted from temperature-driven solid–solid diffusion that is proposed to occur between the amorphous LaFeO3 network – before it crystallizes – and the TiO2 crystallites, allowing the Ti atoms to distribute in the LaFeO3 network. Consequently, replacing the well-crystallized TiO2 nanoparticles with a 22 nm average size by an amorphous network increased further the substitution rate by favouring the atomic titanium diffusion in the amorphous LaFeO3 perovskite network. In addition, this partial substitution hindered the growth of large crystallites during the temperature-driven crystallization at 800 °C when compared to the undoped counterpart (20–21 nm vs. 46 nm), as previously observed for Li- or Cu-doped LaFeO3 materials.43,44 An increase in the specific surface area from 8 to 28 m2 g−1 was simultaneously observed for the P25-LFO catalyst as a result of the decrease in mean crystallite size, as shown in Table 2. However, the SG-LFO catalyst displayed a low specific surface area of 5 m2 g−1, although it displayed a smaller mean crystallite size of 20 nm. This was attributed to the existence of more grain boundaries within the SG-LFO materials compared to the P25-LFO counterpart.
While the SEM images shown in Fig. S1† evidenced the overall powder morphology of the catalysts, the TEM images shown in Fig. S2† and in Fig. 2 confirmed the structural features of both the pristine LFO and the SG-LFO catalysts, respectively. Whether the catalyst was doped or not with titanium, interplanar spacings of 0.39 nm, 0.28 nm and 0.23 nm consistent with the (110), (100) and (111) planes characteristic of the LaFeO3 structural crystallographic features were observed.
 | ||
| Fig. 2 TEM images of the SG-LFO catalyst. The interplanar distances of 0.39 nm, 0.28 nm and 0.23 nm corresponded to the (110), (100) and (111) planes of the LFO structure, respectively. | ||
The ability of the La1−xTixFeO3 materials to be used as photocatalysts under UV-A light is confirmed by the UV-vis absorbance spectra, that reveal a broad absorption band in the UV-vis range, and a band-gap estimated graphically from the Tauc plots at 2.1 eV whether the catalyst contained substitutional titanium or not (Fig. 3).
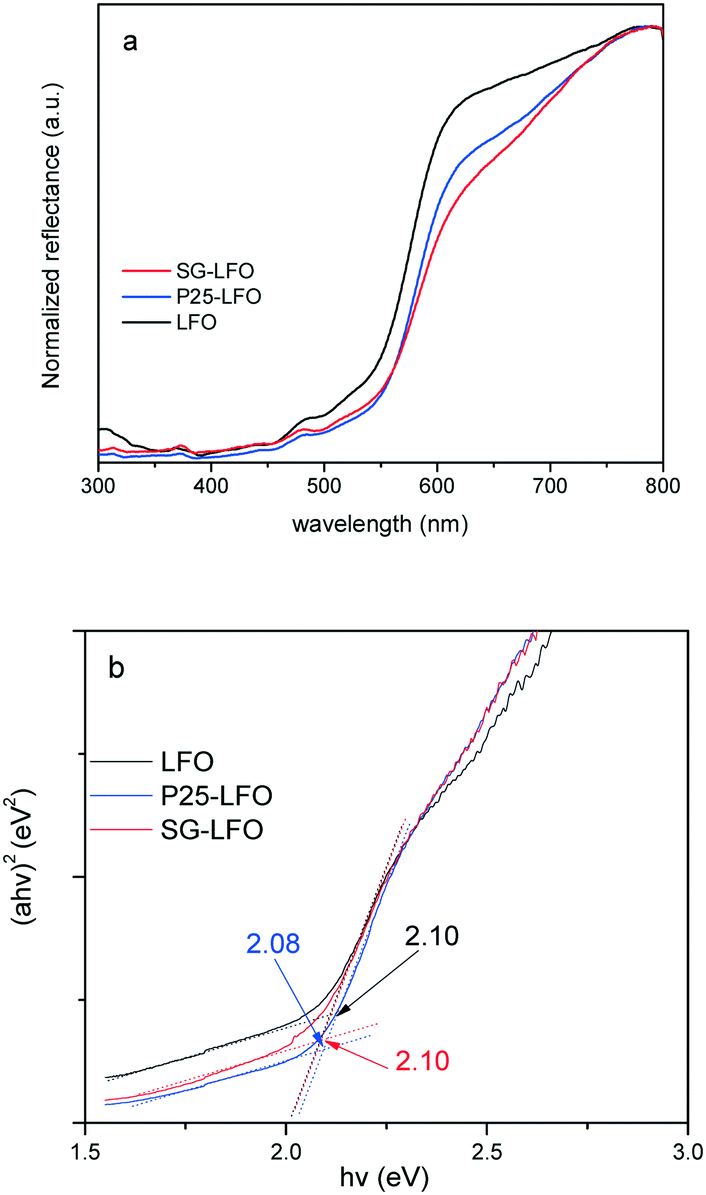 | ||
| Fig. 3 UV-vis diffuse reflectance spectra (a) and Tauc plots (b) of the LFO, P25-LFO and SG-LFO catalysts. The numerical values in (b) indicate the band gap energies estimated from the Tauc plots. | ||
In order to shed light on the effects of Ti → La substitution, further characterization of the doped and undoped samples was performed. The fluorescence spectra shown in the inset of Fig. 4 showed the emission profile of LaFeO3 with a main contribution centered at ca. 485 nm, similar to what has been reported for lanthanum ferrite,45 which can be associated with the direct radiative de-excitation of electrons from high-energy levels within the conduction band to the valence band.45,46 Lower intensity emission at energies lower than the band gap reflects excitonic photoluminescence which is usually associated with sub-band-gap transitions from electronic states related to surface defects or oxygen vacancies.46
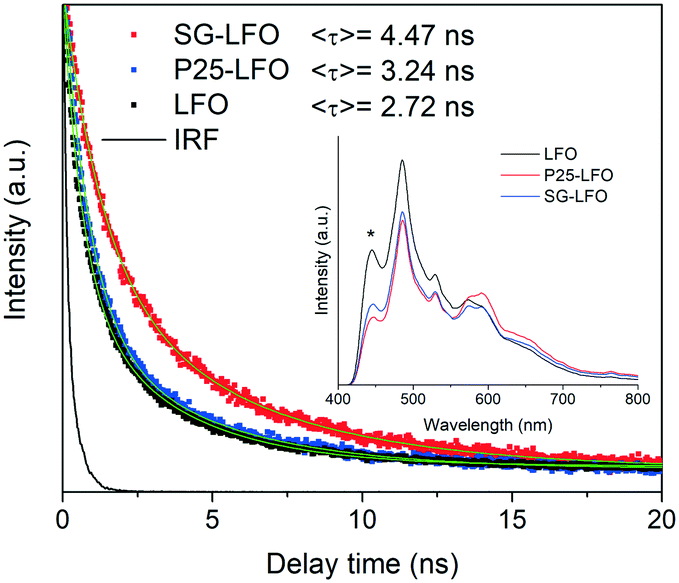 | ||
| Fig. 4 Fluorescence decay curves of the pristine and Ti-doped LaFeO3 samples between 575 and 625 nm. The green lines correspond to the decay curves fitted to each dataset. <τ> represents the average fluorescence lifetimes. The instrument response function (IRF) has been included for comparison. Inset: Fluorescence spectra of the same samples; the signal marked with (*) corresponds to the lamp emission as confirmed by recording the emission spectra with different excitation wavelengths. | ||
Upon titanium doping, the positions of the emission bands remain unchanged, while some changes in the intensity are visible, depending on the involved feature. Thus, the high-energy emission is somewhat quenched in the presence of the dopant, which has been associated to a first approximation with a decreased direct recombination rate due to improved charge separation within the semiconductor structure, and consequently with enhanced photocatalytic activity.47,48 In contrast, the reverse effect of doping is found on the intensity the sub-band-gap features, which can be related to a higher content of surface vacancies/defects in the doped systems.46 The fluorescence lifetime should nevertheless give more accurate information than the steady-state spectra about recombination kinetics.49 The fluorescence decay curves for the different samples between 550 and 600 nm, corresponding to the band gap energy shown in Fig. 4, give fluorescence lifetimes of the same order as previously reported values for LaFeO3 and related structures.47,50 As observed in the curves and the average lifetime values from curve fittings, the P25-LFO sample gives rise to a slightly higher lifetime than the pristine LFO ferrite, while the SG-LFO sample is characterized by a considerably longer fluorescence lifetime, indicative of slower radiative recombination kinetics, which can be ascribed to enhanced charge separation and therefore higher availability of photogenerated electrons and holes for the subsequent surface redox reactions.47,49,51
Fig. 5 shows the influence of the partial substitution of La ions by Ti ions in the LFO structure on the surface state of the SG-LFO catalyst. The XPS analysis revealed the presence of both cations as La3+ and Fe3+ ions at the surface of the pristine LFO and the SG-LFO catalyst. Indeed, the La 3d core level spectra recorded for the pristine LFO material exhibited a La 3d5/2–3/2 orbital doublet contribution at 833.4 eV and 850.1 eV, with a 16.7 eV spin orbit splitting52–54 and a 3.8 eV multiplet splitting,55 that is characteristic of La3+ species. The binding energies of the Fe 2p3/2–1/2 orbital doublet contribution at 710.3 eV and 723.7 eV, with a spin orbit coupling of 13.4 eV54,55 and with the broad and low intensity shake-up satellite peak at about 718 eV characteristic of Fe3+,56,57 confirmed the presence of Fe3+ ions at the surface of the pristine LFO.
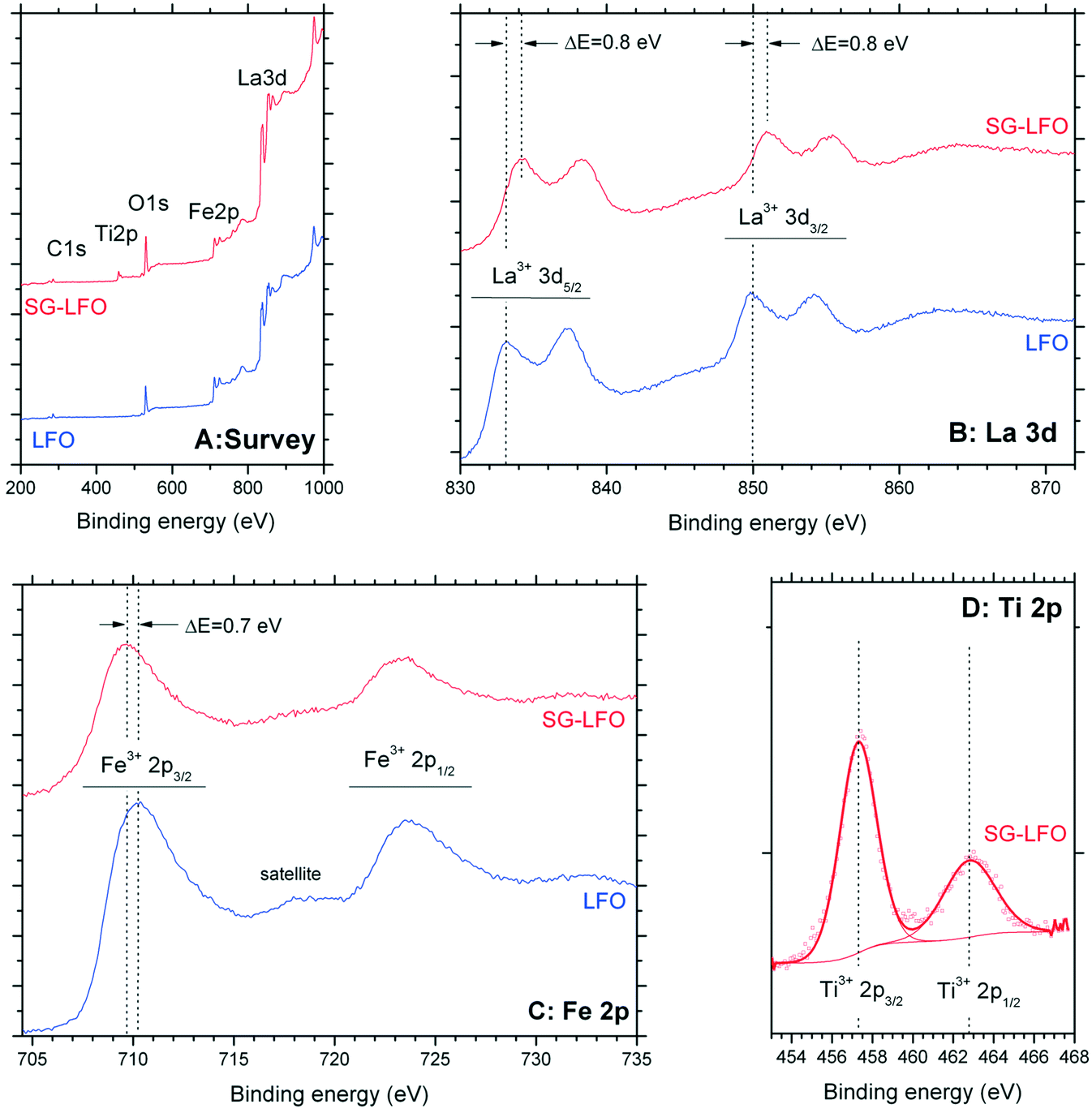 | ||
| Fig. 5 XPS profiles recorded for both the Ti-free LFO and the SG-LFO catalysts: (A) wide scan survey spectrum, (B) La 3d, (C) Fe 2p and (D) Ti 2p orbitals. | ||
After the Ti → La partial substitution in the LFO perovskite, the XPS Ti 2p profile of the SG-LFO catalyst displayed a single contribution, with the doublet related to Ti 2p3/2–1/2 spin–orbit components at 457.3 eV and 463.0 eV. This revealed the stabilization of Ti cations at the surface as lower oxidation state Ti3+ in the substitution of La3+ in the perovskite structure,55 or as Ti4+–O–M with a strong lower binding energy than Ti4+ in an usual TiO2 network environment (Fig. 5).58 Further, this led also to a slight downward (−0.7 eV) shift for the binding energies of the Fe 2p orbital doublet contribution at 709.7 eV and 723.2 eV, and simultaneously in a slight upward (+0.8 eV) shift for the La 3d orbital doublet contribution at 834.2 eV and 850.9 eV, both cations remain present at the surface in the formal +3 oxidation state.
According to the electrostatic model, in the Ti–O–La configuration, the upward shift observed for La3+ compared to the La–O–La reference configuration results from the higher electronegativity of Ti vs. La, that causes a O → Ti shift of the electronic density, and consequently a La → O shift of the La–O bond electronic density, that corresponds to a higher binding energy of La core electrons. So, in contrast, the downward shift of the Fe3+ binding energy was assigned to the electronic enrichment of Fe cations, resulting in the lower binding energy of Fe core electrons.
In addition, by combining the UV-vis diffuse reflectance and UPS data, it can be assumed that the partial cationic substitution did not significantly alter neither the potential of the conduction band edge, nor the valence band maximum, determined by UPS (He I, hλ = 21.2 eV) at ca. 2.2 ± 0.1 eV vs. Fermi level whether the surface is modified or not by titanium (Fig. S3†).
Catalytic activity
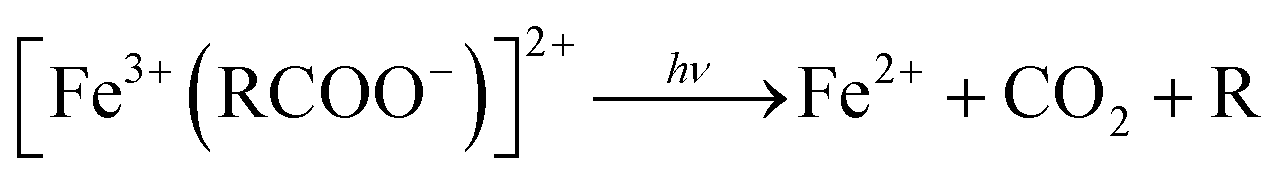 | (2) |
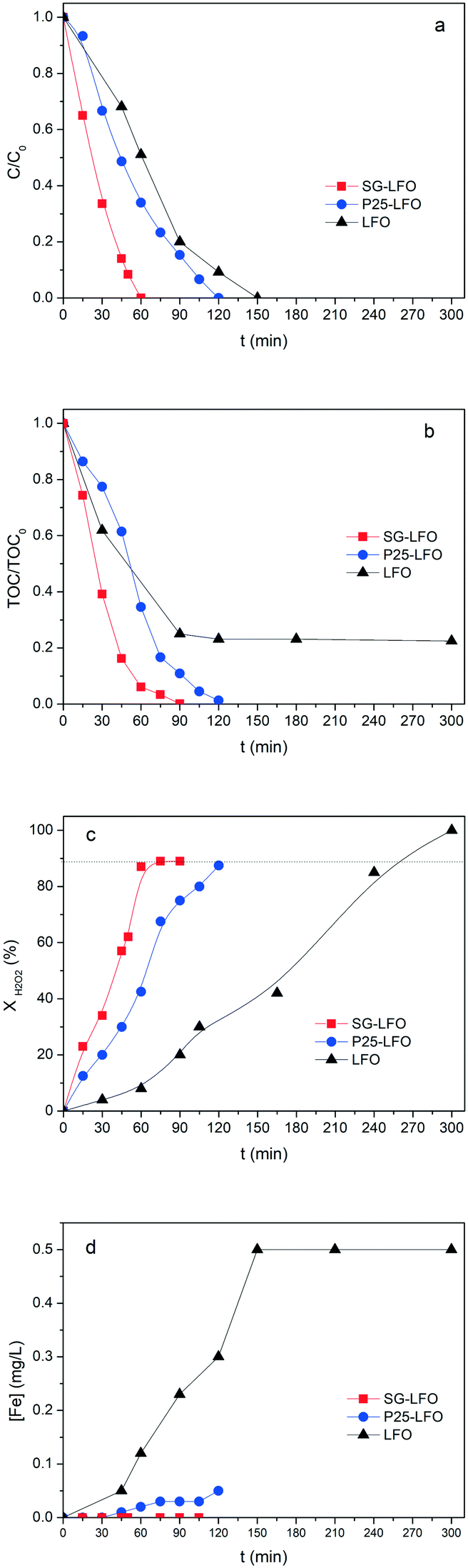 | ||
| Fig. 6 Evolution with time under irradiation of a) the relative 4-CP concentration, b) the relative TOC concentration, c) the H2O2 decomposition yield and d) the concentration of leached Fe in the solution, during the combined photo-Fenton and photocatalytic degradation of 4-CP over the pristine and the Ti-substituted LaFeO3 catalysts. Conditions: [4-CP]0 = 25 mg L−1; [H2O2]0 = 125 mg L−1; T = 25 °C; [cat] = 0.5 g L−1; UV-A irradiance of 60 W m−2. | ||
In the case of the La1−xTixFeO3 catalysts, the incorporation of titanium atoms into the perovskite network with the Ti → La partial substitution increases considerably the robustness of the catalyst by more than two orders of magnitude. This was evidenced by a strong decrease in the iron release to 0.05 mg L−1 for a 5% substitution rate (P25-LFO), and by a further reduction down to the non-detectable level (below 0.001 mg L−1, i.e. lower than 0.0009% relative to the catalyst Fe content) for the SG-LFO catalyst with a 11% substitution rate. This increase in stability is accompanied by strong enhancement of the catalytic activity, more pronounced as the substitution rate increases. Thus the duration necessary for achieving the full removal of 4-CP is reduced to 120 min and 60 min for the P25-LFO and SG-LFO catalysts, respectively, while both catalysts achieve complete mineralization within 125 min and 90 min of reaction, respectively. This very good behaviour is reflected in a strong increase in the apparent kinetic constants for 4-CP degradation, TOC conversion and H2O2 decomposition (Table 3). The absence of any detectable released iron demonstrates that the strong increase in the catalytic activity can be ascribed to pure heterogeneous surface reactions, without any homogeneous contribution.
| Catalyst | k (H2O2),a min−1 | k (4-CP),a min−1 | r 0 (TOC),b mg L−1 min−1 | [Fe] leached, mg L−1 | X (H2O2), % | X (TOC), % | ξ (H2O2)c |
|---|---|---|---|---|---|---|---|
| a The H2O2 and 4-CP concentration evolution curves were fitted by a pseudo first-order kinetic model as usually applied. b The TOC concentration evolution curve was fitted with a zero-order kinetic model as usually applied. c A theoretical ξ = 1 efficiency of H2O2 used in Fenton-like processes reflects the fact that H2O2 and the formed radicals do not suffer from any detrimental scavenger reactions. d The detection limit of Fe leached in the solution was measured at 0.001 ppm. | |||||||
| LFO | 0.010 | 0.020 | 0.12 | 0.5 | 100 | 75 | 0.75 |
| P25-LFO | 0.017 | 0.017 | 0.18 | 0.05 | 90 | 100 | 1.11 |
| SG-LFO | 0.043 | 0.033 | 0.28 | <0.001d | 89 | 100 | 1.12 |
| 1–5# reuse of SG-LFO | — | 33.3 ± 0.3 10−3 | 0.28 ± 0.01 | <0.001d | 100 | ||
Further, both La1−xTixFeO3 catalysts exhibit a H2O2 efficiency ξ greater than 1, which reflects the fact that they allow full TOC conversion to be achieved at incomplete H2O2 conversion. This evidences that the La1−xTixFeO3 catalysts can act as a dual catalyst, by working simultaneously as a heterogenized photo-Fenton catalyst and a photocatalyst. This dual behaviour was confirmed by the evolution of the TOC conversion with the H2O2 conversion shown in Fig. 7a. While the curve obtained for the titanium-free LaFeO3 catalyst is distorted by the strong homogeneous Fenton contribution, the clear distribution of the whole data set for both catalysts above the unity diagonal confirms that the heterogeneous photo-Fenton process cannot be considered as the only AOP operating for converting the TOC. Additional experiments performed using non-stoichiometric H2O2 doses underlined further that the TOC conversion is obtained simultaneously through both photo-Fenton and photocatalysis AOPs (Fig. 7b). Indeed, complete TOC conversion was achieved following zero-order kinetics with a similar apparent kinetic rate constant independent of the H2O2 dose at 25% or 50%, while the decomposition of H2O2 was already completed. This confirms that pure photocatalysis took place allowing full TOC conversion to be obtained.
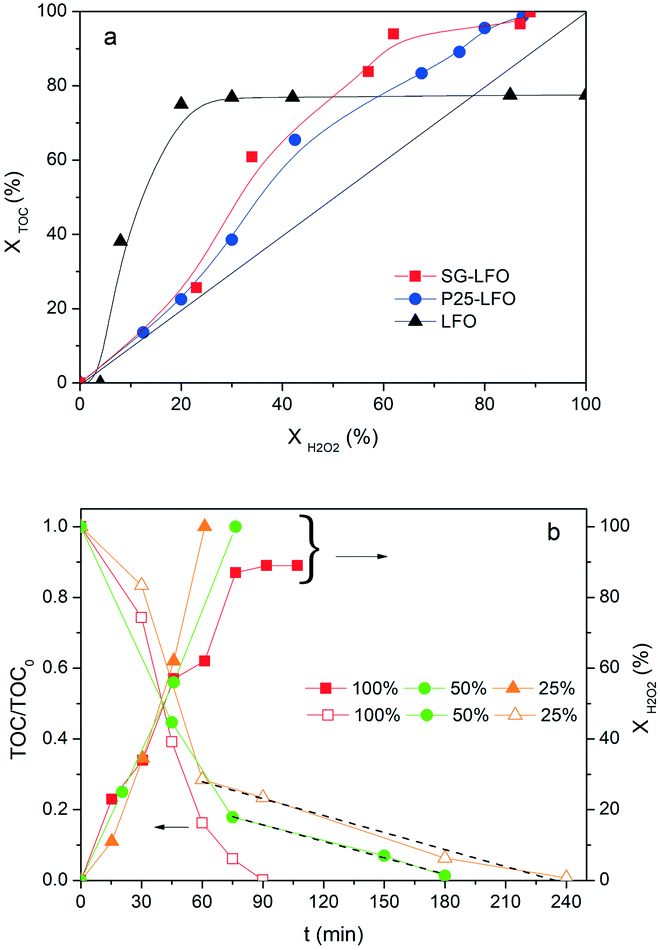 | ||
| Fig. 7 a) TOC conversion vs. H2O2 conversion for the combined photo-Fenton and photocatalytic degradation of 4-CP over the pristine and the Ti-substituted LaFeO3 catalysts. Conditions: [4-CP]0 = 25 mg L−1; [H2O2]0 = 125 mg L−1; T = 25 °C; [cat] = 0.5 g L−1; UV-A irradiance of 60 W m−2. b) Influence of the H2O2 dose on the relative TOC concentration time-evolution and the H2O2 conversion obtained for the SG-LFTO catalyst. Conditions: [4-CP]0 = 25 mg L−1; [H2O2]0 = 31.25–125.00 mg L−1, corresponding to the 25–100% range; T = 25 °C; [cat] = 0.5 g L−1; UV-A irradiance of 60 W m−2. Taking into account that the experiments were carried out using the stoichiometric amount of H2O2 to mineralize TOC, the linear curve with a unity slope reflects the implementation of an heterogenized photo-Fenton process with no occurrence of scavenging reactions. | ||
Although the incorporation of Ti atoms into the LFO network strongly increases the activity in pure photocatalysis, more pronounced as the Ti → La partial substitution increases, the activity remains far lower than that obtained using H2O2 (Fig. 8 and Table 4). This corroborates the fact that, in the combined AOPs, the dual La1−xTixFeO3 catalyst acts predominantly as a heterogeneous photo-Fenton catalyst at the beginning of the process, so that high reaction rates can be obtained, while it subsequently operates as a photocatalyst for mineralizing the short chain acids, and consequently allows full TOC conversion to be achieved by hindering the detrimental formation of stable Fe complexes.
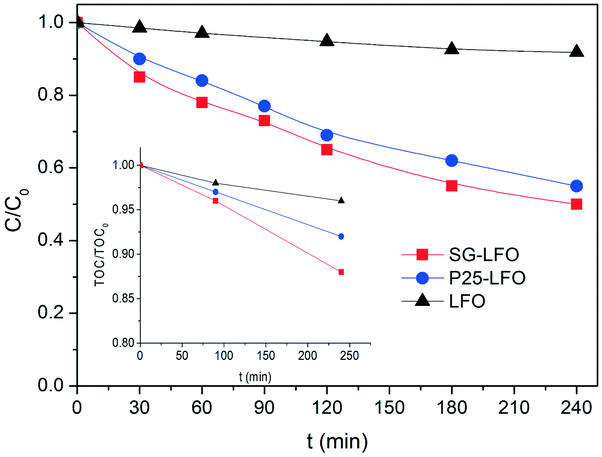 | ||
| Fig. 8 Time-evolution of both the C/C0 and TOC/TOC0 relative concentrations during the pure photocatalytic degradation of 4-CP over the pristine and the Ti-substituted LaFeO3 catalysts. Conditions: [4-CP]0 = 25 mg L−1; T = 25 °C; [cat] = 0.5 g L−1; UV-A irradiance of 60 Wm−2. | ||
| Catalyst | k (4-CP),a min−1 | r 0 (TOC),b mg L−1 min−1 |
|---|---|---|
| a The 4-CP concentration evolution curve was fitted by a pseudo first-order kinetic model as usually applied. b The TOC concentration evolution curve was fitted with a zero-order kinetic model as usually applied. | ||
| LFO | 0.4 10−3 | 0.2 10−3 |
| P25-LFO | 1.8 10−3 | 0.3 10−3 |
| SG-LFO | 2.8 10−3 | 0.5 10−3 |
It is worth noting that, although the P25-LFO catalyst might take advantage the enhanced surface area of 28 m2 g−1 for exposing more surface iron active in the decomposition of H2O2, the specific surface area of the catalysts cannot be the key factor driving the catalytic activity, since the most active SG-LFO catalyst displays a low surface area of 5 m2 g−1. We propose therefore that a combination of surface and electronic modifications modulates the activity of the La1−xTixFeO3 catalysts and leads the SG-LFO catalyst to exhibit the best catalytic results. On the one hand, electron-rich surface iron enables faster H2O2 decomposition by the Fe surface sites, and therefore the production of higher concentrations of active radicals during the FeIII/FeII redox cycle (eqn (3)–(5)). We may assume that the photoassisted Fe3+ to Fe2+ reduction in eqn. (4) might be accompanied by Ti3+ to Ti4+ oxidation as a plausible way of compensating the charge. The Ti4+ species might further be reduced back to Ti3+ by a photogenerated electron.
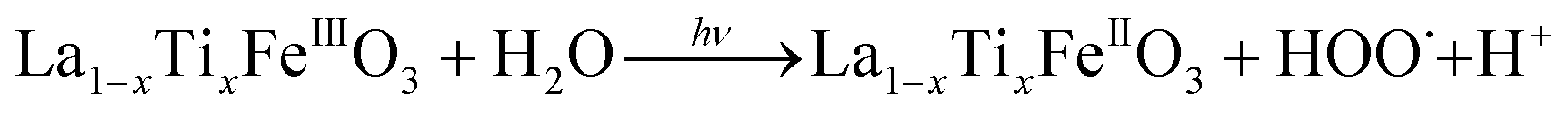 | (3) |
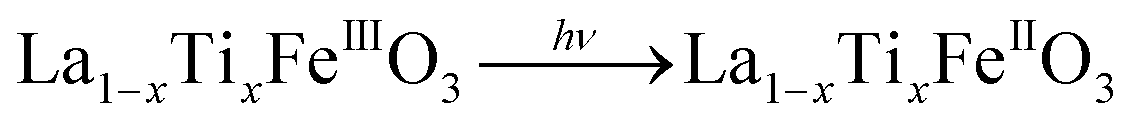 | (4) |
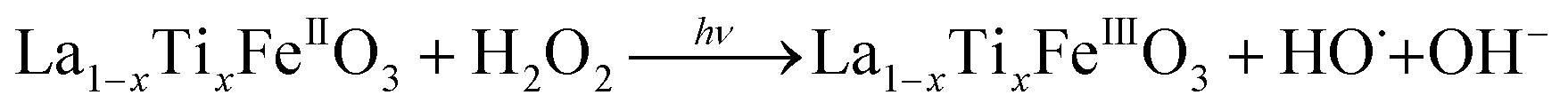 | (5) |
Further, the degradation of the short-chain acids and consequently the mineralization of the carboxylic acids might be favored by the electronic enrichment of the surface Fe according to eqn (6), with the simultaneous FeIII → FeII surface reduction, known as the rate limiting step in the Fe redox cycle.
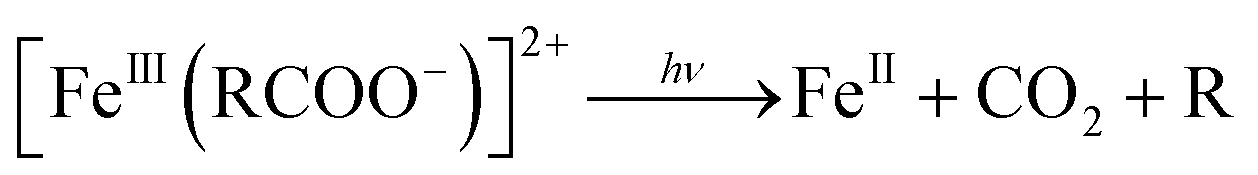 | (6) |
On the other hand, the enhanced availability of the photogenerated charge carriers also enables more efficient photocatalytic decomposition of H2O2, with the production of HOx˙ radicals (eqn (7)–(9)), as well as more efficient production of OH˙ radicals from adsorbed water (eqn (10)).
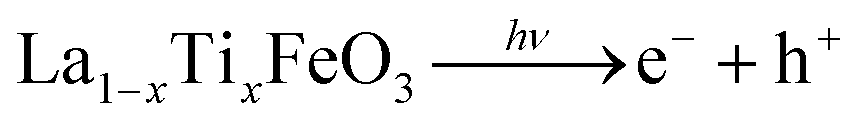 | (7) |
| e− + H2O2 → HO˙ + OH− | (8) |
| h+ + H2O2 → HOO˙ + H+ | (9) |
| h+ + H2O → HO˙ + H+ | (10) |
The stabilization of Fe3+ cations within the orthorhombic La1−xTixFeO3 nanocrystals – both in the bulk and at the surface, as revealed by XRD and XPS – explains the strong increase in the robustness of the substituted catalysts that leads essentially to the suppression of iron leaching. We cannot rule out that the α-Fe2O3 minority phase is subjected to negligible Fe leaching,62,63 nevertheless remaining below the detection threshold, so that its role in the catalytic performance can be discarded.
Finally, degradation of myclobutanil was studied in order to broaden the scope of the study and confirm further the interest of this new dual SG-LFO catalyst. Similarly, Table 5 clearly shows the superiority of the SG-LFO catalyst in terms of both myclobutanil and TOC removal rates, as well as catalyst robustness.
| Catalyst | k (myclobutanil),a min−1 | r 0 (TOC),a mg L−1 min−1 | [Fe] leached, mg L−1 |
|---|---|---|---|
| a The myclobutanil and the TOC concentration evolution curves were fitted by a pseudo first-order model and a zero-order kinetic model as usually applied. b The detection limit of Fe leached in the solution was measured at 0.001 ppm. | |||
| LFO | 0.18 | 2.3 10−2 | 2.4 |
| P25-LFO | 0.25 | 2.4 10−2 | 0.10 |
| SG-LFO | 0.38 | 5.8 10−2 | <0.001b |
Stability tests of the SG-LFO catalyst
Considering the robustness of the SG-LFO catalyst in the first test cycle, its stability was further studied in terms of activity, iron release and structural features during recycling tests. Actually, the refinement of the XRD patterns of the used catalysts reveals the structural stability of the La1−xTixFeO3 catalysts during recycling tests (Table 1 and Fig. S4 and Table S2†). Thus, no extra-phase formation occurred under UV-A during the reaction, and no change in terms of the Ti → La substitution rate within the LaFeO3 network and the mean crystallite size has been observed. Regarding the activity, the results of the recycling tests shown in Fig. 9 and Table 3 demonstrate the very good stability and consequently the reusability of the SG-LFO catalyst. Indeed, the apparent kinetic constants for 4-CP and TOC removal remained essentially unchanged after five consecutive runs, and the Fe release in the solution was below the detection threshold (i.e. lower than 0.0009% relative to the catalyst Fe content).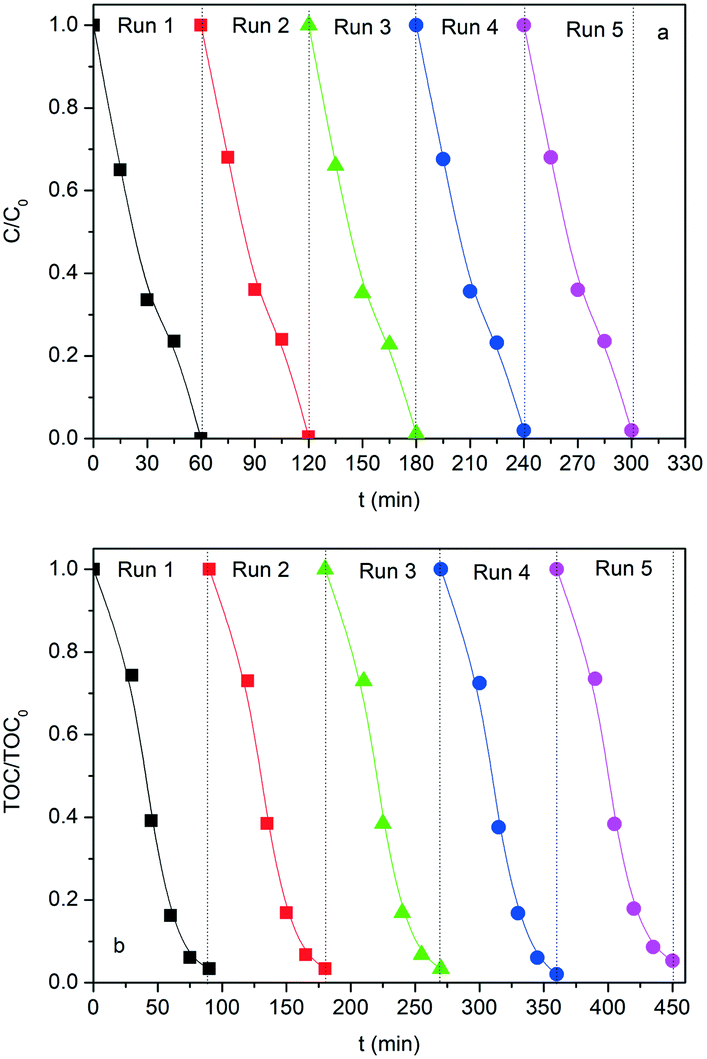 | ||
| Fig. 9 Catalyst recycling efficiency expressed in terms of time-evolution of both (a) the C/C0 and (b) the TOC/TOC0 relative concentrations for 5 consecutive runs in the combined photo-Fenton and photocatalytic degradation of 4-CP with the reused SG-LFO catalyst. Conditions: [4-CP]0 = 25 mg L−1; [H2O2]0 = 125 mg L−1; T = 25 °C; [cat] = 0.5 g L−1; UV-A irradiance of 60 W m−2. The catalyst recovery was achieved by powder filtration. | ||
Conclusions
To sum up, we have highlighted the interest of using a new and highly robust Ti-substituted LaFeO3 dual catalyst, active in water treatment as a reusable heterogenized photo-Fenton catalyst and a photocatalyst. The La1−xTixFeO3 dual catalyst with a 11% substitution degree was prepared by modifying a facile Pechini sol–gel route using a dried amorphous titania as a titanium source. The strategy combining both AOPs with a single dual catalyst allowed the La1−xTixFeO3 catalyst to simultaneously take advantage the higher reaction rate of photo-assisted CWPO and the higher mineralization yield of photocatalysis. The partial Ti → La cation substitution in the LaFeO3 perovskite network led to pure heterogeneous surface reactions with an increase in the catalyst robustness by more than two orders of magnitude, evidenced by the complete inhibition of the Fe release in water and the stability of the catalytic performance over five test cycles. It further strongly increased the reaction rates, due to both the enhanced availability of the photogenerated charge carriers at the surface and the electronic enrichment of the surface iron, and allowed full mineralization of a pollutant to be achieved at circumneutral pH through combined AOPs.Forthcoming studies will be concerned with the use of solar light for taking advantage of the narrow band gap of the dual La1−xTixFeO3 catalysts, as well as with downsizing the La1−xTixFeO3 crystallites for increasing further the availability of Fe ions at the catalyst surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors want to thank the European Fund for Regional Development (EFRE/FEDER) for funding the project PHOTOPUR which is performed within the framework of the Interreg V program and Offensive Sciences call. F. Fresno thanks financial support from project SOLPAC: ENE2017-89170-R from the Ministry of Science, Innovation and Universities. T. Dintzer (ICPEES) and D. Ihiawakrim (IPCMS) are thanked for performing the SEM and TEM characterization, respectively. V. Papaefthimiou and S. Zafeiratos (ICPEES) are thanked for performing the XPS and UPS measurements.Notes and references
- Y. Chen, G. Zhang, H. Liu and J. Qu, Confining Free Radicals in Close Vicinity to Contaminants Enables Ultrafast Fenton-like Processes in the Interspacing of MoS2 Membranes, Angew. Chem., Int. Ed., 2019, 58, 8134–8138 CrossRef CAS PubMed.
- A. M. Braun, Photochemical Purification of Water and Air, Angew. Chem., Int. Ed., 2003, 42, 5117–5117 CrossRef CAS.
- L. Ma, Y. Liu, Y. Liu, S. Jiang, P. Li, Y. Hao, P. Shao, A. Yin, X. Feng and B. Wang, Ferrocene-Linkage-Facilitated Charge Separation in Conjugated Microporous Polymers, Angew. Chem., Int. Ed., 2019, 58, 4221–4226 CrossRef CAS PubMed.
- S. Navalon, R. Martin, M. Alvaro and H. Garcia, Gold on Diamond Nanoparticles as a Highly Efficient Fenton Catalyst, Angew. Chem., Int. Ed., 2010, 49, 8403–8407 CrossRef CAS PubMed.
- M. Sun, C. Chu, F. Geng, X. Lu, J. Qu, J. Crittenden, M. Elimelech and J. Kim, Reinventing Fenton Chemistry: Iron Oxychloride Nanosheet for pH-Insensitive H2O2 Activation, Environ. Sci. Technol. Lett., 2018, 5, 186–191 CrossRef CAS.
- P. Bautista, A. F. Mohedano, J. A. Casas, J. A. Zazo and J. J. Rodriguez, An overview of the application of Fenton oxidation to industrial wastewaters treatment, J. Chem. Technol. Biotechnol., 2008, 83, 1323–1338 CrossRef CAS.
- R. Chen and J. J. Pignatello, Role of Quinone Intermediates as Electron Shuttles in Fenton and Photoassisted Fenton Oxidations of Aromatic Compounds, Environ. Sci. Technol., 1997, 31, 2399–2406 CrossRef CAS.
- J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz and J. J. Rodríguez, Chemical pathway and kinetics of phenol oxidation by Fenton's reagent, Environ. Sci. Technol., 2005, 39, 9295–9302 CrossRef CAS PubMed.
- S. Malato, P. Fernández-Ibáñez, M. I. Maldonado, J. Blanco and W. Gernjak, Decontamination and disinfection of water by solar photocatalysis: Recent overview and trends, Catal. Today, 2009, 147, 1–59 CrossRef CAS.
- A. Verma, D. Dixit, A. Toor and J. Srivastava, Heterogeneous photocatalytic degradation of 2-chloro-4-nitrophenol using slurry and fixed bed reactor, Environ. Prog. Sustainable Energy, 2015, 34, 380–386 CrossRef CAS.
- A. Goi and M. Trapido, Hydrogen peroxide photolysis, Fenton reagent and photo-Fenton for the degradation of nitrophenols: a comparative study, Chemosphere, 2002, 46, 913 CrossRef CAS PubMed.
- J. A. Zazo, J. A. Casas, A. F. Mohedano and J. J. Rodríguez, Catalytic wet peroxide oxidation of phenol with a Fe/active carbon catalyst, Appl. Catal., B, 2006, 65, 261–268 CrossRef CAS.
- M. Hartmann, S. Kullmann and H. Keller, Wastewater treatment with heterogeneous Fenton-type catalysts based on porous materials, J. Mater. Chem., 2010, 20, 9002–9017 RSC.
- M. E. Zarazua-Morin, L. M. Torres-Martinez, E. Moctezuma, I. Juarez-Ramirez and B. B. Zermeno, Synthesis, characterization, and catalytic activity of FeTiO3/TiO2 for photodegradation of organic pollutants with visible light, Res. Chem. Intermed., 2016, 42, 1029–1043 CrossRef CAS.
- A. Sivakumar, A. Selvaraj, A. Ramasamy and V. Balasubramanian, Enhanced Photocatalytic Degradation of Reactive Dyes over FeTiO3/TiO2 Heterojunction in the Presence of H2O2, Water, Air, Soil Pollut., 2013, 224, 5 CrossRef.
- R. Dhinesh Kumar, R. Thangappan and R. Jayavel, Synthesis and characterization of LaFeO3/TiO2 nanocomposites for visible light photocatalytic activity, J. Phys. Chem. Solids, 2017, 101, 25–33 CrossRef CAS.
- S. Yu, Y. Wang, F. Sun, R. Wang and Y. Zhou, Novel mpg-C3N4/TiO2 nanocomposite photocatalytic membrane reactor for sulfamethoxazole photodegradation, Chem. Eng. J., 2018, 337, 183–192 CrossRef CAS.
- C. Cai, Z. Zhang, J. Liu, N. Shan, H. Zhang and D. D. Dionysiou, Visible light-assisted heterogeneous Fenton with ZnFe2O4 for the degradation of Orange II in water, Appl. Catal., B, 2016, 182, 456–468 CrossRef CAS.
- H. Mechakra, T. Sehili, M. A. Kribeche, A. A. Ayachi, S. Rossignol and C. George, Use of natural iron oxide as heterogeneous catalyst in photo-Fenton-like oxidation of chlorophenylurea herbicide in aqueous solution: Reaction monitoring and degradation pathways, J. Photochem. Photobiol., A, 2016, 317, 140–150 CrossRef CAS.
- T. Soltani and B. Lee, Improving heterogeneous photo-Fenton catalytic degradation of toluene under visible light irradiation through Ba-doping in BiFeO3 nanoparticles, J. Mol. Catal. A: Chem., 2016, 425, 199–207 CrossRef CAS.
- W. Wang, M. Xu, X. Xu, W. Zhou and Z. Shao, Perovskite Oxide-Based Electrodes for High-Performance Photoelectrochemical Water Splitting: A Review, Angew. Chem., Int. Ed., 2020, 59(1), 136–152 CrossRef CAS PubMed.
- M. Monai, M. Melchionna and P. Fornasiero, Chapter One - From metal to metal-free catalysts: Routes to sustainable chemistry, Academic Press, 2018 Search PubMed.
- E. J. Rosenfeldt and K. G. Linden, Degradation of Endocrine Disrupting Chemicals Bisphenol A, Ethinyl Estradiol, and Estradiol during UV Photolysis and Advanced Oxidation Processes, Environ. Sci. Technol., 2004, 38, 5476–5483 CrossRef CAS PubMed.
- J. Matos, J. Laine and J.-M. Herrmann, Effect of the Type of Activated Carbons on the Photocatalytic Degradation of Aqueous Organic Pollutants by UV-Irradiated Titania, J. Catal., 2001, 200(1), 10–20 CrossRef CAS.
- E. Tütem, R. Apak and Ç F. Ünal, Adsorptive removal of chlorophenols from water by bituminous shale, Water Res., 1998, 32, 2315–2324 CrossRef.
- D. A. Haith, Ecological risk assessment of pesticide runoff from grass surfaces, Environ. Sci. Technol., 2010, 44, 6496–6502 CrossRef CAS PubMed.
- P. Garcia-Muñoz, W. Dachtler, B. Altmayer, R. Schulz, D. Robert, F. Seitz, R. Rosenfeldt and N. Keller, Reaction pathways, kinetics and toxicity assessment during the photocatalytic degradation of glyphosate and myclobutanil pesticides: Influence of the aqueous matrix, Chem. Eng. J., 2020, 384, 1233152020 CrossRef.
- P. V. Gosavi and R. B. Biniwale, Pure phase LaFeO3 perovskite with improved surface area synthesized using different routes and its characterization, Mater. Chem. Phys., 2010, 119, 324–329 CrossRef CAS.
- X. Qi, J. Zhou, Z. Yue, Z. Gui and L. Li, A simple way to prepare nanosized LaFeO3 powders at room temperature, Ceram. Int., 2003, 29, 347–349 CrossRef CAS.
- L. B. McCusker, R. B. Von Dreele, D. E. Cox, D. Louër and P. Scardi, Rietveld refinement guidelines, J. Appl. Crystallogr., 1999, 32, 36–50 CrossRef CAS.
- J. Rodríguez-Carvajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192(1), 55–69 CrossRef.
- J. Luo, R. Li, Y. Chen, X. Zhou, X. Ning, L. Zhan, L. Ma, X. Xu, L. Xu and L. Zhang, Rational design of Z-scheme LaFeO3/SnS2 hybrid with boosted visible light photocatalytic activity towards tetracycline degradation, Sep. Purif. Technol., 2019, 210, 417–430 CrossRef CAS.
- M. S. S. Doniach, Many-electron singularity in X-ray photoemission and X-ray line spectra from metals, J. Phys. C: Solid State Phys., 1970, 3, 285 CrossRef.
- D. A. Shirley, High-Resolution X-Ray Photoemission Spectrum of the Valence Bands of Gold, Phys. Rev. B: Solid State, 1972, 5, 4709–4714 CrossRef.
- M. Pera-Titus, V. García-Molina, M. A. Baños, J. Giménez and S. Esplugas, Degradation of chlorophenols by means of advanced oxidation processes: a general review, Appl. Catal., B, 2004, 47, 219–256 CrossRef CAS.
- G. Eisenberg, Colorimetric Determination of Hydrogen Peroxide, Ind. Eng. Chem., Anal. Ed., 1943, 15(5), 327–328 CrossRef CAS.
- E. B. Sandell, Colorimetric Determination of Traces of Metals, J. Phys. Chem., 1945, 49(3), 263–264 CrossRef.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- T. S. Jamil, H. A. Abbas, R. A. Nasr and R. Vannier, Visible light activity of BaFe1-xCuxO3-δ as photocatalyst for atrazine degradation, Ecotoxicol. Environ. Saf., 2018, 163, 620–628 CrossRef CAS PubMed.
- S. K. Rashmi, H. S. Bhojya Naik, H. Jayadevappa, R. Viswanath, S. B. Patil and M. Madhukara Naik, Solar light responsive Sm-Zn ferrite nanoparticle as efficient photocatalyst, Mater. Sci. Eng., B, 2017, 225, 86–97 CrossRef CAS.
- M. R. De Guire, R. C. O'Handley and G. Kalonji, The cooling rate dependence of cation distributions in CoFe2O4, J. Appl. Phys., 1989, 65, 3167–3172 CrossRef CAS.
- Z. J. Zhang, Z. L. Wang, B. C. Chakoumakos and J. S. Yin, Temperature Dependence of Cation Distribution and Oxidation State in Magnetic Mnâ'Fe Ferrite Nanocrystals, J. Am. Chem. Soc., 1998, 120, 1800–1804 CrossRef CAS.
- T. T. N. Phan, A. N. Nikoloski, P. A. Bahri and D. Li, Heterogeneous photo-Fenton degradation of organics using highly efficient Cu-doped LaFeO3 under visible light, J. Ind. Eng. Chem., 2018, 61, 53–64 CrossRef CAS.
- L. Hou, G. Sun, K. Liu, Y. Li and F. Gao, Preparation, characterization and investigation of catalytic activity of Li-doped LaFeO3 nanoparticles, J. Sol-Gel Sci. Technol., 2006, 40, 9–14 CrossRef CAS.
- Y. Ye, H. Yang and R. Li, Enhanced photocatalytic performance and mechanism of Ag-decorated LaFeO3 nanoparticles, J. Sol-Gel Sci. Technol., 2017, 82, 509–518 CrossRef CAS.
- J. Liqiang, Q. Yichun, W. Baiqi, L. Shudan, J. Baojiang, Y. Libin, F. Wei, F. Honggang and S. Jiazhong, Review of photoluminescence performance of nano-sized semiconductor materials and its relationships with photocatalytic activity, Sol. Energy Mater. Sol. Cells, 2006, 90(12), 1773–1787 CrossRef.
- S. Bharathkumar, M. Sakar and S. Balakumar, Experimental Evidence for the Carrier Transportation Enhanced Visible Light Driven Photocatalytic Process in Bismuth Ferrite (BiFeO3) One-Dimensional Fiber Nanostructures, J. Phys. Chem. C, 2016, 120, 18811–18821 CrossRef CAS.
- L. Collado, A. Reynal, F. Fresno, M. Barawi, C. Escudero, V. Perez-Dieste, J. M. Coronado, D. P. Serrano, J. R. Durrant and L. P. de, Unravelling the effect of charge dynamics at the plasmonic metal/semiconductor interface for CO2 photoreduction, Nat. Commun., 2018, 9, 4986 CrossRef PubMed.
- A. Charanpahari, S. S. Umare and R. Sasikala, Effect of Ce, N and S multi-doping on the photocatalytic activity of TiO2, Appl. Surf. Sci., 2013, 282, 408–414 CrossRef CAS.
- S. Y. Smolin, A. K. Choquette, J. Wang, S. J. May and J. B. Baxter, Distinguishing Thermal and Electronic Effects in Ultrafast Optical Spectroscopy Using Oxide Heterostructures, J. Phys. Chem. C, 2018, 122, 115–123 CrossRef CAS.
- R. Sasikala, A. R. Shirole, V. Sudarsan, K. G. Girija, R. Rao, C. Sudakar and S. R. Bharadwaj, Improved photocatalytic activity of indium doped cadmium sulfide dispersed on zirconia, J. Mater. Chem., 2011, 21, 16566–16573 RSC.
- S. Phokha, S. Pinitsoontorn, S. Rujirawat and S. Maensiri, Polymerized complex synthesis and effect of Ti dopant on magnetic properties of LaFeO3 nanoparticles, J. Nanosci. Nanotechnol., 2015, 15, 9171–9177 CrossRef CAS.
- S. Thirumalairajan, K. Girija, V. Ganesh, D. Mangalaraj, C. Viswanathan and N. Ponpandian, Novel Synthesis of LaFeO3 Nanostructure Dendrites: A Systematic Investigation of Growth Mechanism, Properties, and Biosensing for Highly Selective Determination of Neurotransmitter Compounds, Cryst. Growth Des., 2013, 13, 291–302 CrossRef CAS.
- M. Humayun, N. Sun, F. Raziq, X. Zhang, R. Yan, Z. Li, Y. Qu and L. Jing, Synthesis of ZnO/Bi-doped porous LaFeO3 nanocomposites as highly efficient nano-photocatalysts dependent on the enhanced utilization of visible-light-excited electrons, Appl. Catal., B, 2018, 231, 23–33 CrossRef CAS.
- S. Petrović, A. Terlecki-Baričević, L. Karanović, P. Kirilov-Stefanov, M. Zdujić, V. Dondur, D. Paneva, I. Mitov and V. Rakić, LaMO3 (M=Mg, Ti, Fe) perovskite type oxides: Preparation, characterization and catalytic properties in methane deep oxidation, Appl. Catal., B, 2008, 79(2), 186–198 CrossRef.
- T. Yamashita and P. Hayes, Analysis of XPS spectra of Fe2+ and Fe3+ ions in oxide materials, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- D. D. Hawn and B. M. DeKoven, Deconvolution as a correction for photoelectron inelastic energy losses in the core level XPS spectra of iron oxides, Surf. Interface Anal., 1987, 10, 63–74 CrossRef CAS.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Effects of F- Doping on the Photocatalytic Activity and Microstructures of Nanocrystalline TiO2 Powders, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- D. L. Sedlak and A. W. Andren, Oxidation of Chiorobenzene with Fenton's Reagent, Environ. Sci. Technol., 1991, 25, 777–782 CrossRef CAS.
- M. E. Balmer and B. Sulzberger, Atrazine degradation in irradiated iron/oxalate systems: Effects of pH and oxalate, Environ. Sci. Technol., 1999, 33, 2418–2424 CrossRef CAS.
- G. Pliego, J. A. Zazo, P. Garcia-Muñoz, M. Munoz, J. A. Casas and J. J. Rodriguez, Trends in the Intensification of the Fenton Process for Wastewater Treatment: An Overview, Crit. Rev. Environ. Sci. Technol., 2015, 45, 2611–2692 CrossRef CAS.
- R. Prucek, M. Hermanek and R. Zbořil, An effect of iron(III) oxides crystallinity on their catalytic efficiency and applicability in phenol degradation—A competition between homogeneous and heterogeneous catalysis, Appl. Catal., A, 2009, 366(2), 325–332 CrossRef CAS.
- G. Carraro, C. Maccato, A. Gasparotto, T. Montini, S. Turner, O. I. Lebedev, V. Gombac, G. Adami, G. Van Tendeloo, D. Barreca and P. Fornasiero, Enhanced Hydrogen Production by Photoreforming of Renewable Oxygenates Through Nanostructured Fe2O3 Polymorphs, Adv. Funct. Mater., 2014, 24, 372–378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM characterization; UPS spectra; crystallographic data of the refined samples (fresh and used); XRD patterns of used samples. See DOI: 10.1039/c9cy02269d |
| This journal is © The Royal Society of Chemistry 2020 |