
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of Au@TiO2 core–shell nanoparticles with tunable structures for plasmon-enhanced photocatalysis†
Tian-Ming
Chen
a,
Ge-Yang
Xu
a,
He
Ren
a,
Hua
Zhang
a,
Zhong-Qun
Tian
 a and
Jian-Feng
Li
*ab
a and
Jian-Feng
Li
*ab
aMOE Key Laboratory of Spectrochemical Analysis and Instrumentation, State Key Laboratory of Physical Chemistry of Solid Surfaces, iChEM, College of Chemistry and Chemical Engineering, College of Materials, College of Energy, Xiamen University, Xiamen 361005, China. E-mail: Li@xmu.edu.cn
bShenzhen Research Institute of Xiamen University, Shenzhen 518000, China
First published on 10th October 2019
Abstract
Plasmonic metal–semiconductor nanocomposites, especially those with core–shell nanostructures, have received extensive attention as they can efficiently expand light absorption and accelerate electron–hole separation thus improving the photocatalytic efficiency. However, controlled synthesis and structure manipulation of plasmonic metal–semiconductor nanocomposites still remain a significant challenge. Herein, a simple and universal method has been developed for the preparation of plasmonic Au@TiO2 core–shell nanoparticles. Using such a method, uniform TiO2 shells are successfully coated on Au nanoparticles with various morphologies including nanorods, nanocubes, and nanospheres, and the thickness and crystallinity of the TiO2 shell can be simply tuned by adjusting the pH value and thermal treatment, respectively. Furthermore, the influence of the morphology of the Au core and the thickness and crystallinity of the TiO2 shell on the photocatalytic performance of Au@TiO2 towards the photodegradation of methylene blue is systematically explored. It is found that Au@TiO2 NPs with nanorod morphology and crystalline TiO2 shells display the best performance, which is 5 times higher than that of bare Au nanoparticles. This work provides a facile strategy for the fabrication of plasmonic core–shell nanostructures that show excellent performance in plasmon-enhanced photocatalysis.
Introduction
With the depletion of fossil fuels and increasing serious environmental pollution, research on photocatalysis using solar energy has intensified.1–5 TiO2 has emerged as a good photocatalyst candidate owing to its abundance in nature, low cost, non-toxicity, and photochemical stability. However, TiO2 has a wide bandgap of 3.2 eV, thus its photoresponse is limited to the ultraviolet region, significantly hindering its solar energy conversion efficiency.6–9In the past few decades, various strategies have been proposed to improve the photocatalytic activity of TiO2, such as doping with metal/non-metal elements or ions,10–13 organic dye sensitization,14,15 and forming nanocomposites with other semiconductors.16,17 However, most of these proposed solutions only slightly extend the photoresponse of TiO2 to the visible region, and the absorption coefficient still remains small.
Plasmonic nanostructures, such as Au, Ag, and Cu, have extremely large absorption/scattering cross sections in the visible light region due to the surface plasmon resonance (SPR) effect,18–27 and have been applied in many fields, especially in plasmon-enhanced spectroscopy (PES).28–30 The SPR effect enables the injection of hot electrons generated on these noble metals into nearby semiconductors thus expanding the light absorption range of the semiconductor.20,22,31 Besides the hot electron injection mechanism, the SPR effect also benefits photocatalysis by generating strong electromagnetic fields in the near field of metal nanoparticles (NPs) to accelerate electron–hole separation and increasing light scattering for the reuse of unabsorbed light.18,19,27 These advantages make plasmon-enhanced photocatalysis a research hotspot.
However, these mechanisms for boosting photocatalytic activity rely on the synthesis and manipulation of metal–semiconductor composites. Therefore, rational design and construction of metal–semiconductor nanostructures are particularly important. A common nanostructure is constructed by loading noble metal nanoparticles (NPs) onto semiconductor surfaces, such as Au–TiO2,32,33 Au–CdS,34,35 Au–ZnO,36,37 Au–Fe2O3,38etc. In this case, the photocatalytic efficiency can be improved to a certain level. However, it is difficult to fully utilize the SPR properties of the metal NPs due to the very limited interaction between the metal and the semiconductor in such nanocomposites. Furthermore, they would also suffer from metal corrosion under working conditions.
To overcome the above disadvantages, a plasmonic metal@semiconductor core–shell nanostructure is proposed.39–46 For photocatalysis, plasmonic metal@semiconductor core–shell nanostructures have various advantages. First, an enhanced electromagnetic field is generated around the metal nanoparticles, which can improve the excitation efficiency of the semiconductor. Furthermore, the plasmonic cores can generate hot electrons and holes during the dephasing process of the surface plasmons, while the semiconductor shells can promote the separation of the hot electrons and holes. And this structure is advantageous due to the extremely high contact area between the plasmonic metal and the semiconductor. The semiconductor shell also protects the metal core from external interference. The Au@TiO2 core–shell structure has been extensively studied in recent years.46–50 However, Ti precursors such as TiF4 and titanium alkoxides hydrolyze too fast to effectively control the thickness and uniformity of the shell. Moreover, spherical Au NPs are usually used with amorphous TiO2 shells, which may not be ideal for photocatalysis, as anisotropic Au NPs and crystalline TiO2 usually exhibit better plasmonic and photocatalytic properties, respectively. Therefore, the synthesis of Au@TiO2 core–shell nanostructures with various core morphologies, as well as tunable shell thickness and crystallinity, is of great significance.
Herein, we report a simple and universal method to synthesize Au@TiO2 core–shell nanostructures. Au cores with three morphologies including nanocubes, nanorods, and nanospheres are successfully synthesized, and the shell thickness is well controlled with the choice of the Ti precursor and adjustment of the pH value. Additionally, annealing treatment could transform the amorphous TiO2 shell into a crystalline one. Finally, the photocatalytic performance of the Au@TiO2 core–shell nanoparticles towards the degradation of methylene blue is investigated. The results show that the photocatalytic activity is mainly dependent on the SPR effect and that the crystalline shell can greatly improve the catalytic activity. This strategy provides a platform for constructing plasmonic core–shell photocatalysts with improved performance.
Experimental section
Materials
Chloroauric acid tetrahydrate (HAuCl4·4H2O, AR), hexadecyl trimethyl ammonium chloride (CTAC, >99%), hexadecyl trimethyl ammonium bromide (CTAB, >99%), sodium dodecyl sulfate (SDS), NaHCO3 (>99%), NaBH4 (98%), ascorbic acid (AA, AR), AgNO3 (>99%), and HCl (30%) were purchased from Sinopharm Reagents Co. (China). Titanium trichloride (TiCl3, 15–20%) was purchased from Sigma Aldrich. Ultra-pure Milli-Q water (18.2 MΩ cm) was used throughout the study.Synthesis of Au nanospheres (NSs)
The Au nanospheres were synthesized using the seed-mediated method.51 First, 0.25 mL of HAuCl4 solution (0.01 M) was added to 9.75 mL of CTAB solution (0.1 M). After mixing uniformly, 0.6 mL of fresh NaBH4 solution (0.01 M) was quickly added to the mixture. The resultant solution was mixed by rapid inversion for 30 s and then kept at room temperature for 1 h to obtain seed 1.Second, 9.75 mL of CTAB solution (0.1 M), 4 mL of HAuCl4 solution (0.01 M) and 15 mL of ascorbic acid solution (0.1 M) were added to 190 mL of Milli-Q water. 0.12 mL of the seed 1 solution was then added and left to stand at 30 °C for 30 min. This was then centrifuged once and concentrated three times to prepare seed 2.
To synthesize Au nanospheres (NSs), 30 mL of CTAC solution (0.025 M), 0.75 mL of AA solution (0.1 M), 1.5 mL of HAuCl4 solution (0.01 M) and 1.5 mL of seed 2 solution were then mixed uniformly and placed in a 45 °C water bath for 3 hours. This solution was then centrifuged and redispersed in 30 mL of CTAB solution (0.02 M) and 1.5 mL of HAuCl4 solution (0.01 M), and then allowed to react by putting in a 45 °C water bath for 2 hours to obtain the Au NSs.
Synthesis of Au nanorods (NRs)
Typically, 147.5 mL of Milli-Q water was mixed with 75 mL of 0.2 M CTAB solution and 12.5 mL of 10 mM HAuCl4 solution and then placed in a 30 °C water bath for half an hour. Concentrated hydrochloric acid was then added to the solution to adjust the pH value. After 15 min, 2 mL of 0.01 M AgNO3 solution and 14.3 mL of 0.1 M AA solution were added. Finally, 200 μL of seed 1 solution was added and the reaction was allowed to stand for 12 h to obtain Au NRs.Synthesis of Au nanocubes (NCs)
The synthesis of Au nanocubes (NCs) was performed by mixing 6.4 mL of 0.1 M CTAB solution, 3.8 mL of 0.1 M AA solution, and 5 μL of seed 1 solution with 32 mL of Milli-Q water. Then 0.8 mL of 0.01 M HAuCl4 solution was added dropwise in a 35 °C water bath to obtain Au NCs.Synthesis of Au@TiO2 core–shell NPs
Ligand exchange of Au nanostructures was first performed using SDS in the following steps: typically, a solution (10 mL) of an as-prepared metal nanocrystal sample that was stabilized with either CTAB or CTAC was washed once by centrifugation to remove the excess surfactant and then redispersed in water (10 mL). The resultant metal nanocrystal solution was added dropwise under vigorous stirring to an aqueous SDS solution (10 mL, 0.1 M). SDS adsorption was allowed for at least 0.5 h at room temperature. After the excess SDS was removed by centrifugation, the SDS-encapsulated metal nanocrystals were redispersed in water (50 μL). It should be noted that the concentration of SDS remaining in the nanocrystal solution should be as low as possible to avoid the self-growth of TiO2. In a typical synthesis, 50 μL of TiCl3 solution was mixed with 6 mL of Milli-Q water, and 0.1 M NaHCO3 solution was added under stirring. Then, the concentrated Au–SDS solution was quickly mixed with the TiCl3–NaHCO3 solution, stirred for 10 min, and washed twice with ethanol to obtain Au@TiO2 core–shell nanostructures.Synthesis of crystallized Au@TiO2 core–shell NPs
The as-prepared Au@TiO2 core–shell nanostructure sample was washed and transferred into a quartz crucible with absolute alcohol (1 mL). Then the quartz crucible was placed in a muffle furnace and annealed at 300 °C or 500 °C for 2 hours. Finally, the calcined core–shell nanostructure sample was redispersed in water (2 mL) by ultrasonication.Photocatalytic measurements
A 150 W Xe lamp with a 420 nm filter channel was used as the light source. 5 mg of TiO2 or Au@TiO2 photocatalyst were dispersed in 30 mL of MB (5 ppm) aqueous solution. The solution was continuously stirred in the dark for about 1 hour to establish an adsorption–desorption equilibrium between the photocatalyst and the dye. The suspension was then irradiated with the 150 W Xe lamp or with light having a wavelength longer than 420 nm. During the irradiation, the solution was stirred to maintain the suspension. After 2 hours, an aliquot of the suspension was taken and centrifuged. The residual concentration of MB was monitored using a UV-vis spectrophotometer.Characterization
The morphology of the Au@TiO2 NPs was characterized by transmission electron microscopy (TEM, JEOL JEM 1400; HR-TEM, JEOL JEM 2100). EDS mapping analysis was performed on a field emission transmission electron microscope (TEM, Philips TECNAI F30). Absorption spectra were collected on an ultraviolet-visible spectrophotometer (UNICO, UV2012C/PC/PCS). X-ray diffraction (XRD) studies were carried out on an X-ray diffractometer (Rigaku IV) using Cu K radiation (λ = 1.5418 Å). A confocal Raman spectrometer (HORIBA, X-plora) was used to record Raman signals.Results and discussion
Fig. 1 illustrates the formation process of TiO2 shell-coated Au NPs, using Au NR@TiO2 core–shell nanostructures as an example. First, Au NRs are prepared by the seed growth method, and the NRs are stabilized with cetyltrimethylammonium bromide (CTAB) and positively charged. Next, the negative charge modifier, SDS, is added to modify the surface of Au NRs by electrostatic attractions. TiCl3 is chosen as the titanium dioxide precursor because of its relatively low hydrolysis rate. By controlling the pH value, TiCl3 is then hydrolyzed to TiOH2+ and adsorbed by the negatively charged SDS layer. Finally, the dissolved oxygen in the solution oxidized the Ti3+ species to the TiO2 shell at room temperature.49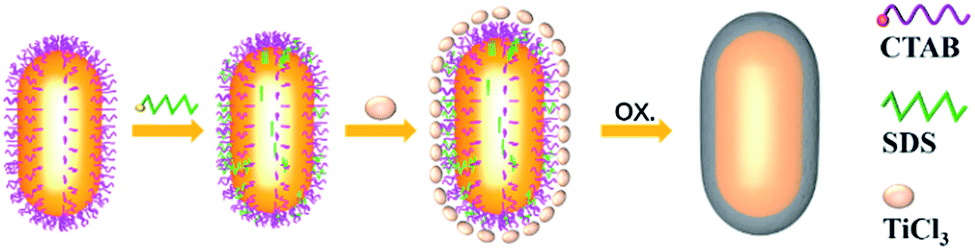 | ||
| Fig. 1 Schematic diagram of the preparation of Au NR@TiO2 nanostructures by coating TiO2 on Au NRs. | ||
The as-prepared core–shell NPs are characterized by TEM (Fig. 2a–l). To demonstrate the universal applicability of this method, we have prepared three different nanostructure Au cores: Au NRs (length: 120 ± 10 nm; diameter: 60 ± 5 nm), Au NCs (diameter: 50 ± 5 nm) and Au NSs (diameter: 65 ± 5 nm). It can be seen from the TEM images that the as-prepared core–shell NPs show uniform morphologies and good dispersion. By controlling the pH below 2.5, the TiO2 shell is formed by close aggregation of many small amorphous TiO2 NPs, and no self-nucleation of TiO2 is found. Different TiO2 shell thicknesses are obtained by controlling the reaction time and pH of the solution. As shown in Fig. 2, the thickness of the TiO2 shells of the three kinds of Au@TiO2 nanostructures can be controlled between 4 and 30 nm. The shell layer only grows along the surface of the core, keeping the shell morphology the same as that of the core.
 | ||
| Fig. 2 (a) TEM image of Au NRs (length: 120 ± 10 nm; diameter: 60 ± 5 nm), (b–d) TEM images of Au NR@TiO2 with different coating thicknesses (shell thickness: 4 ± 1 nm, 15 ± 2 nm, and 30 ± 4 nm). (e) TEM images of Au NCs (diameter: 50 ± 5 nm), (f–h) TEM images of Au NC@TiO2 with different coating thicknesses (shell thickness: 4 ± 1 nm, 10 ± 2 nm, and 20 ± 4 nm). (i) TEM images of Au NSs (diameter: 65 ± 5 nm). (j–l) TEM images of Au NS@TiO2 with different coating thicknesses (shell thickness: 5 ± 1 nm, 15 ± 2 nm, and 30 ± 4 nm). (m) HAADF-STEM image of a single Au NR@TiO2 nanostructure. (n–p) Elemental distribution map of Au, Ti and O. (q) UV-visible absorption spectra of uncoated Au NRs, Au NCs, Au NSs (dashed lines), Au NR@TiO2 (shell thickness: 15 nm), Au NC@TiO2 (shell thickness: 10 nm), and Au NS@TiO2 (shell thickness: 15 nm) core–shell nanostructures (solid-lines). | ||
The structure and composition of Au NR@TiO2 were further investigated by high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping analysis (Fig. 2m–p). These results show that the core is composed of Au, while the shell layer is composed of Ti and O, confirming the formation of the Au@TiO2 core–shell nanostructures. It is well known that the SPR of Au NPs is highly sensitive to the size, shape, structure, and surrounding dielectric environment.27,52 To study how the TiO2 shell affects the SPR of Au NPs, we have examined the UV-vis absorption spectra of bare Au and the corresponding Au@TiO2 nanostructures. Fig. 2q displays the change in the LSPR band of three kinds of Au@TiO2 nanostructures. For the nanorod-core nanostructures, the transverse plasmon peak red-shifts from 514 nm to 527 nm and the longitudinal plasmon peak red-shifts from 672 nm to 721 nm. For the nanocube-core one, the plasmon peak red-shifts from 540 nm to 566 nm. For the nanosphere-core, the plasmon peak red-shifts from 535 nm to 561 nm. And the UV-vis absorption spectra of Au NRs@TiO2 NPs with different TiO2 shell thickness are measured as shown in Fig. S1.† It can be seen that a red shift is observed when the TiO2 shell becomes thicker. The red-shift of the plasmon band in the UV-vis absorption spectra after TiO2 coating is caused by the increase of the refractive index of the surrounding medium.49
We find that the structure and crystallinity of Au@TiO2 nanoparticles are largely affected by the thermal treatment temperature. As shown in Fig. 3a, before thermal treatment, the TiO2 shell shows an amorphous and porous structure. Whereas in Fig. 3b, after annealing at 300 °C, the shell is visibly thinner and formed of small TiO2 grains. This indicates that the shell changes while the size and shape of the Au core remain stable. Increasing the temperature to 500 °C makes the shell denser and smoother than at 300 °C. However, the Au core also deforms (Fig. 3c). HR-TEM imaging clearly shows changes in the TiO2 lattice due to crystallization (Fig. 3d–f). No obvious lattice fringes are found before annealing; however, after 300 °C treatment, lattice fringes appeared, with a lattice spacing of 0.35 nm, corresponding to the (200) plane of anatase (Fig. 3e). After annealing at 500 °C, the lattice fringes are clearer, while the lattice spacing is unchanged at 0.35 nm (Fig. 3f).
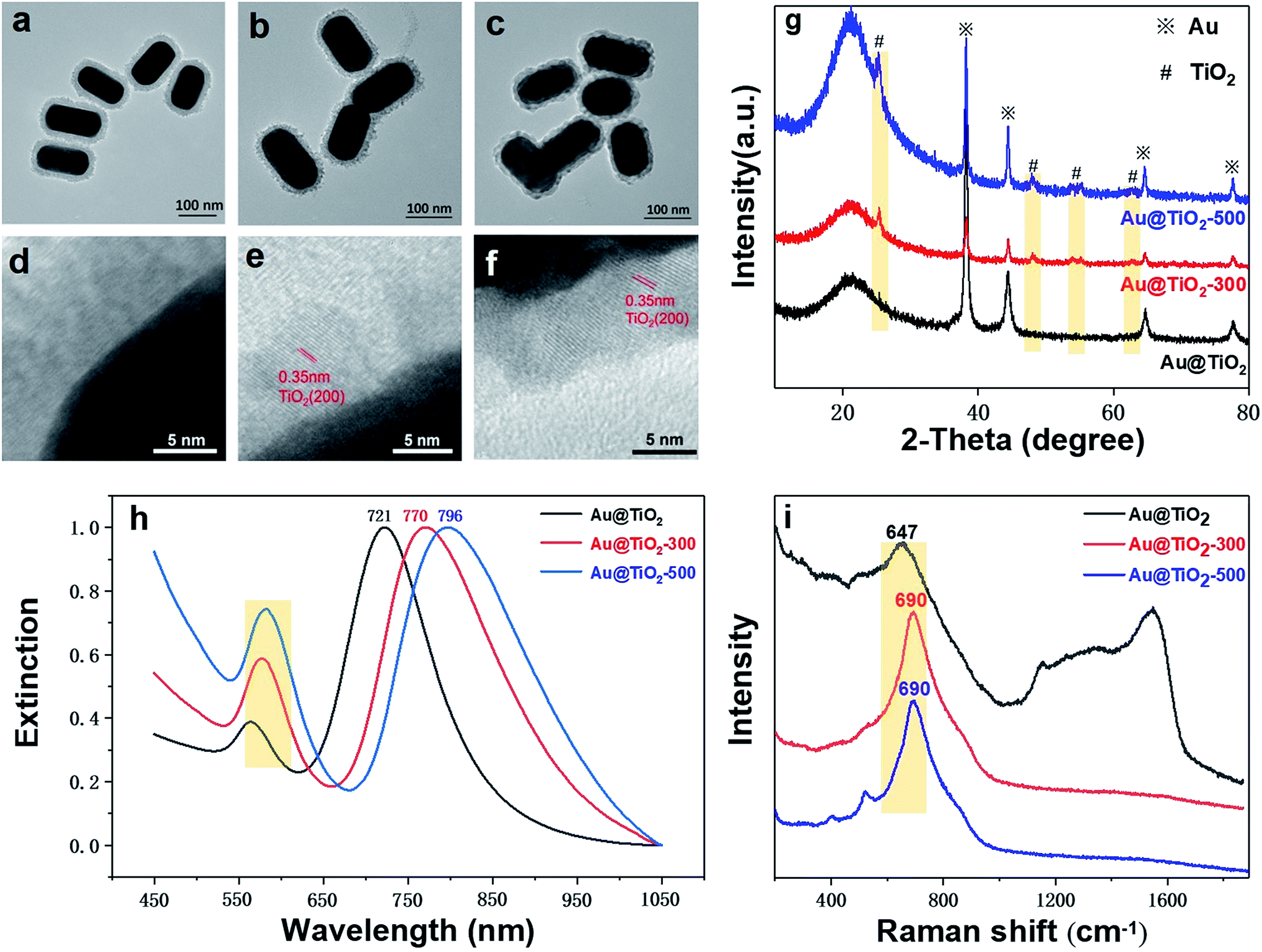 | ||
| Fig. 3 (a and d) TEM images and HR-TEM images before annealing and at (b and e) 300 °C and (c and f) 500 °C. (g) XRD of Au@TiO2 nanostructures before and after annealing. (h) UV-visible spectroscopy of Au@TiO2 nanostructures before and after annealing. (i) Raman spectroscopy of Au@TiO2 nanostructures before and after annealing. | ||
The XRD pattern provides further evidence for the formation of TiO2 shell crystals (Fig. 3g). Before the thermal treatment, only the typical Au (111) and Au (220) diffraction peaks (JCPDS card no. 65-8601) can be seen, indicating that the TiO2 shell is amorphous. However, after annealing at 300 °C, diffraction peaks at 25.3°, 48.1°, 54.0°, 55.1°, and 62.8° appear, which can be assigned to anatase TiO2 (JCPDS card no. 01-0562). After annealing at 500 °C, the characteristic peak of TiO2 shows higher intensities and clearer contours, indicating a more crystalline structure, consistent with TEM observations. These results verify that the structure of Au@TiO2 and the TiO2 shell (amorphous and crystalline) can be tuned through thermal treatment.
Fig. 3h displays the UV-vis spectra of Au NR@TiO2 nanostructures before and after crystallization. After thermal treatment at 300 °C, the longitudinal plasmon peak red-shifts from 721 nm to 770 nm. As the thermal temperature rises to 500 °C, the longitudinal plasmon peak continues to red-shift to 796 nm. Besides, we can clearly find that the Au NR@TiO2 sample after thermal treatment shows a significant increase in absorbance at around 400 nm, which is caused by crystalline TiO2. The possible causes of the red-shift of the plasmon peak are summarized through these experimental phenomena. First, the thermal treatment of the Au@TiO2 nanostructure results in a phase transition of the TiO2 shell from the amorphous phase to the anatase phase. The increase in the crystallinity of TiO2 leads to an increase in the dielectric constant, resulting in a redshift of the SPR absorption band. Second, it has been reported early that when the contact area of Au and TiO2 increases, the peak of the SPR band shifts to a longer wavelength.32 As shown in the TEM image, the heat treatment increases the contact area between Au and TiO2, thus causing a redshift in the SPR band.
Furthermore, Fig. 3i shows the Raman spectra of the sample before and after annealing. It can be found that after annealing, the peak shape at 690 cm−1 for TiO2 becomes sharper and red-shifts, indicating that annealing changes its crystallinity. The Raman signals for carbon deposition between 1200 cm−1 and 1600 cm−1 resulting from the CTAB impurity disappear, indicating that annealing removes impurities of the core–shell structure. These results confirm that the crystallization and porous state of the shell layer in the Au@TiO2 core–shell structure can be adjusted by thermal treatment.
The influence of the Au core morphology and the shell crystallinity and thickness on the photocatalytic catalytic activity of different Au@TiO2 core–shell NPs is evaluated using the photocatalytic degradation of methylene blue, a common environmental pollutant. Fig. 4a and S2† show the photocatalytic performance of Au NR@TiO2 core–shell nanostructures treated at different temperatures. Blank experiments show that self-degradation of methylene blue is minimal. Au NRs or TiO2 before and after annealing treatment also shows very weak catalytic activity under visible light illumination. The Au@TiO2 activity without thermal treatment is slightly better than that of TiO2, indicating that the SPR effect of Au has a positive effect on the photocatalytic activity of TiO2. However, the activity of Au@TiO2 is significantly enhanced after annealing. The degradation efficiencies of Au@TiO2-300 and Au@TiO2-500 in 2 h are 33% and 26%, respectively, which are 5 times and 4 times higher than that of the unannealed sample. The annealing process facilitates the transportation of hot electrons to the crystalline shell. Additionally, it eliminates impurities in the materials so that there are better contact and stronger interactions between the Au core and TiO2 shell. Further increasing the temperature to 500 °C decreases the catalytic activity, probably due to the loss of active sites and the destruction of the porous shell at high temperature. The Au core also shows some deformation at 500 °C, which can weaken its SPR performance.
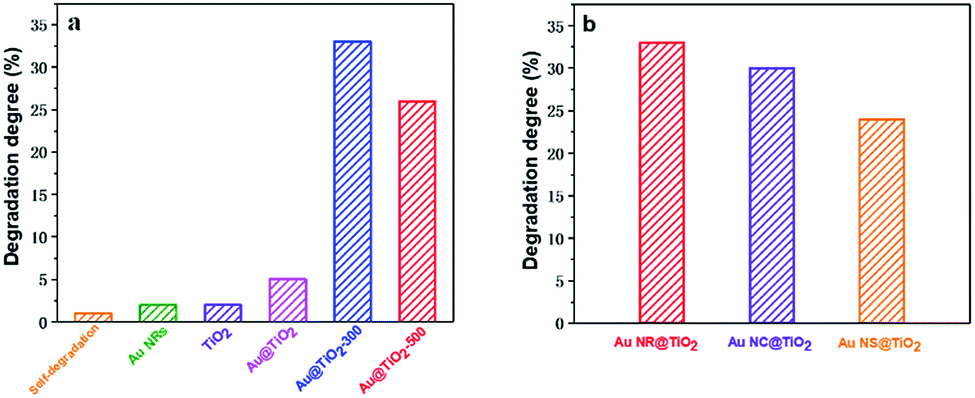 | ||
| Fig. 4 (a) Photocatalytic degradation of methylene blue by Au NRs, TiO2 and annealed Au@TiO2 core–shell nanostructures under visible light. (b) Photocatalytic degradation of methylene blue by Au NR@TiO2, Au NC@TiO2 and Au NS@TiO2 core–shell nanostructures after annealing under visible light. | ||
The effects of Au-core morphology on the photocatalytic activities are also investigated. In this case, all the samples are calcined at 300 °C, and the quantity of the catalyst and thickness of the TiO2 shell are kept the same. As shown in Fig. 4b, nanorods have superior activity over the other two types of NPs. This is likely due to nanorods having two plasmonic modes, i.e. transverse and longitudinal modes, which both contribute to light capturing.5
To better understand how the SPR effect affects the photocatalytic activity, first, each core–shell nanostructure is annealed at 300 °C, and the photocatalytic degradation of methylene blue using Au NR@TiO2 NPs with the same amount of Au cores and different shell thicknesses under full and visible light illumination is studied (Fig. 5a). Under visible light illumination, the 5 nm shell shows the highest activity, achieving a photodegradation efficiency of 46%. As the shell thickness increases, the catalytic activity decreases rapidly corresponding to the decreased SPR intensity and a reduction of hot electron injection rates, with the photodegradation efficiency of the 20 nm shell NPs being only 12%. Under full light illumination, the photodegradation efficiency also decreases as the shell thickness increases. However, the rate of decrease is more moderate than that with visible light. The photodegradation efficiency of samples with 5 nm and 20 nm shells is 65% and 47%, respectively. Furthermore, bare Au NRs show much lower activity compared to the Au NR@TiO2 NPs. These results illustrate that the SPR effect is the dominant factor causing the higher photocatalytic activity.
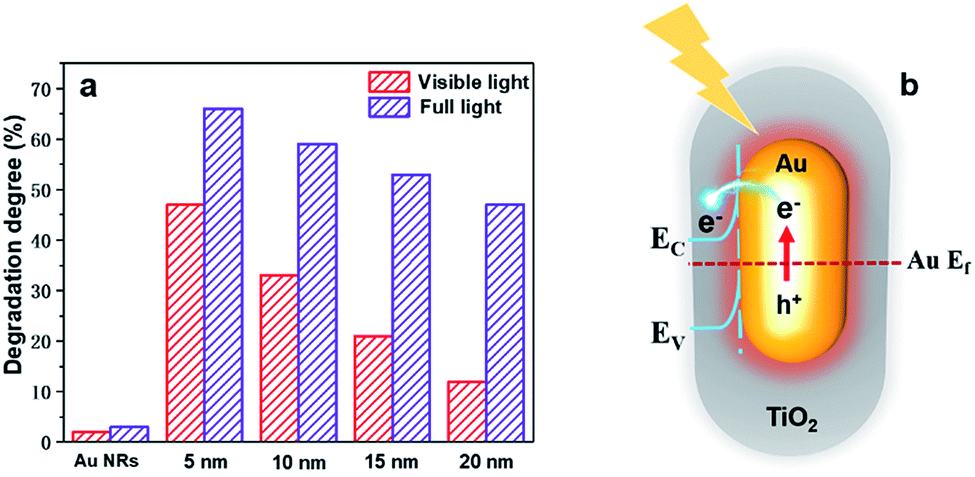 | ||
| Fig. 5 (a) Photocatalytic degradation of methylene blue by Au NR@TiO2 core–shell nanostructures with different shell thicknesses under full light and visible light illumination. (b) Mechanistic diagram of Au NR@TiO2 core–shell nanostructure enhanced photocatalysis under illumination. | ||
Based on the analysis of experimental results, the mechanism of Au@TiO2 core–shell nanostructure enhanced photocatalysis is summarized in Fig. 5b. Au NRs standards alone also show very weak catalytic activity under full light or visible light. Under visible light, although TiO2 is inactive, Au NPs can generate hot carriers which can induce photocatalysis. Obviously, the thicker the shell, the harder it is for hot carriers to reach the TiO2 surface and the lower the catalytic activity.21 Under full light illumination, photoelectrons can be generated in TiO2 and participate in the photocatalytic reaction. In this case, nanoparticles have better catalytic activities when they have thicker shells, i.e., a lager amount of TiO2. At the same time, hot carriers and an electromagnetic field are also generated by the Au nanoparticles under such a condition, which would benefit the reaction. However, such an influence would decrease with the increase of shell thickness. As a result, a balance is achieved when the photocatalytic performance under full light illumination shows a moderate decrease as the shell thickness increases. According to previous studies, the electron transfer from Au to TiO2 plays a primary role in enhancing the photocatalytic activity under full light.22 Therefore, the change of photocatalytic activity is consistent with the trend of the decreased SPR intensity under full light and visible light illumination, indicating that the SPR effect is the main factor to improve the photocatalytic performance.
Conclusions
In summary, a simple, repeatable, and universal method for coating TiO2 onto different Au core nanostructures has been developed. After shell coating, the morphology of the Au core is preserved and the structure of the photocatalysts is well controlled with good dispersibility. The effects of the crystallinity and thickness of the TiO2 shell and the Au core morphology on the photocatalytic activity of the catalysts are also studied in detail. Owing to better transportation of hot electrons, annealing the amorphous shell to form a crystalline TiO2 shell considerably improved the photocatalytic activity of the NPs. Among the different morphologies screened, Au NR core photocatalysts showed the highest activity due to better light absorption. Finally, by comparing photocatalysts with different shell thicknesses, we propose that hot electron transfer from the Au core is the main factor dictating the photocatalytic activity, regardless of whether under full light or visible light illumination. We believe that the plasmonic core-semiconductor shell nanocomposite can play an important role in photocatalysis by manipulating the structure and composition in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21775217, 21703181, 21427813, and 21521004), the Fundamental Research Funds for the Central Universities (20720190044), the Natural Science Foundation of Guangdong Province (2016A030308012), the Basic Research Projects of Shenzhen Research & Development Fund (JCYJ20170306140934218), and the Open Fund of the State Key Laboratory of Luminescent Materials and Devices (South China University of Technology).Notes and references
- C. C. Chen, W. H. Ma and J. C. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- X. X. Chang, T. Wang and J. L. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 RSC.
- Y. Ma, X. L. Wang, Y. S. Jia, X. B. Chen, H. X. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133–5146 RSC.
- W. B. Hou and S. B. Cronin, Adv. Funct. Mater., 2013, 23, 1612–1619 CrossRef CAS.
- D. J. Yang, H. W. Liu, Z. F. Zheng, Y. Yuan, J. C. Zhao, E. R. Waclawik, X. B. Ke and H. Y. Zhu, J. Am. Chem. Soc., 2009, 131, 17885–17893 CrossRef CAS PubMed.
- X. B. Chen, L. Liu and F. Q. Huang, Chem. Soc. Rev., 2015, 44, 1861–1885 RSC.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS.
- W. E. Kaden, T. P. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- A. P. Bhirud, S. D. Sathaye, R. P. Waichal, J. D. Ambekar, C. J. Park and B. B. Kale, Nanoscale, 2015, 7, 5023–5034 RSC.
- G. S. Zhang, Y. C. Zhang, M. Nadagouda, C. Han, K. O'Shea, S. M. El-Sheikh, A. A. Ismail and D. D. Dionysiou, Appl. Catal., B, 2014, 144, 614–621 CrossRef CAS.
- A. Yella, H. W. Lee, H. N. Tsao, C. Y. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Gratzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- G. S. Li, B. Jiang, S. N. Xiao, Z. C. Lian, D. Q. Zhang, J. C. Yu and H. X. Li, Environ. Sci.: Processes Impacts, 2014, 16, 1975–1980 RSC.
- C. Eley, T. Li, F. L. Liao, S. M. Fairclough, J. M. Smith, G. Smith and S. C. E. Tsang, Angew. Chem., Int. Ed., 2014, 53, 7838–7842 CrossRef CAS PubMed.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- V. Giannini, A. I. Fernandez-Dominguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912 CrossRef CAS PubMed.
- S. Linic, P. Christopher and D. B. Ingram, Nat. Mater., 2011, 10, 911–921 CrossRef CAS PubMed.
- C. Boerigter, R. Campana, M. Morabito and S. Linic, Nat. Commun., 2016, 7, 10545 CrossRef CAS PubMed.
- M. L. Brongersma, N. J. Halas and P. Nordlander, Nat. Nanotechnol., 2015, 10, 25–34 CrossRef CAS PubMed.
- K. Wu, J. Chen, J. R. McBride and T. Lian, Science, 2015, 349, 632–635 CrossRef CAS PubMed.
- A. Sanchez-Iglesias, A. Chuvilin and M. Grzelczak, Chem. Commun., 2015, 51, 5330–5333 RSC.
- N. Zhou, V. Lopez-Puente, Q. Wang, L. Polavarapu, I. Pastoriza-Santos and Q. H. Xu, RSC Adv., 2015, 5, 29076–29097 RSC.
- J.-F. Li, Y.-J. Zhang, S.-Y. Ding, R. Panneerselvam and Z.-Q. Tian, Chem. Rev., 2017, 117, 5002–5069 CrossRef CAS PubMed.
- C. G. Silva, R. Juarez, T. Marino, R. Molinari and H. Garcia, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Kramer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2013, 8, 247–251 CrossRef CAS PubMed.
- J. F. Li, Y. F. Huang, Z. L. Yang, D. Y. Wu, B. Ren, Z. L. Wang and Z. Q. Tian, Nature, 2010, 464, 392–395 CrossRef CAS PubMed.
- C.-Y. Li, J.-B. Le, Z.-L. Yang, J.-F. Li, J. Cheng and Z.-Q. Tian, Nat. Mater., 2019, 18, 697–701 CrossRef CAS PubMed.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- M. J. Kale, T. Avanesian and P. Christopher, ACS Catal., 2014, 4, 116–128 CrossRef CAS.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489–492 CrossRef CAS PubMed.
- A. Luken, M. Muhler and J. Strunk, Phys. Chem. Chem. Phys., 2015, 17, 10391–10397 RSC.
- J. Xu, W. M. Yang, S. J. Huang, H. Yin, H. Zhang, P. Radjenovic, Z. L. Yang, Z. Q. Tian and J. F. Li, Nano Energy, 2018, 49, 363–371 CrossRef CAS.
- K. F. Wu, W. E. Rodriguez-Cordoba, Y. Yang and T. Q. Lian, Nano Lett., 2013, 13, 5255–5263 CrossRef CAS PubMed.
- J. Strunk, K. Kaehler, X. Y. Xia, M. Comotti, F. Schuth, T. Reinecke and M. Muhler, Appl. Catal., A, 2009, 359, 121–128 CrossRef CAS.
- P. Li, Z. Wei, T. Wu, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2011, 133, 5660–5663 CrossRef CAS PubMed.
- H. Yu, M. Chen, P. M. Rice, S. X. Wang, R. L. White and S. H. Sun, Nano Lett., 2005, 5, 379–382 CrossRef CAS PubMed.
- B. X. Li, T. Gu, T. Ming, J. X. Wang, P. Wang, J. F. Wang and J. C. Yu, ACS Nano, 2014, 8, 8152–8162 CrossRef CAS PubMed.
- K. S. Mayya, D. I. Gittins and F. Caruso, Chem. Mater., 2001, 13, 3833–3836 CrossRef CAS.
- D. Y. Liu, S. Y. Ding, H. X. Lin, B. J. Liu, Z. Z. Ye, F. R. Fan, B. Ren and Z. Q. Tian, J. Phys. Chem. C, 2012, 116, 4477–4483 CrossRef CAS.
- T. Hirakawa and P. V. Kamat, J. Am. Chem. Soc., 2005, 127, 3928–3934 CrossRef CAS PubMed.
- F. Bao, J. L. Yao and R. A. Gu, Langmuir, 2009, 25, 10782–10787 CrossRef CAS PubMed.
- X. N. Fu, J. Liu, H. Yang, J. C. Sun, X. Li, X. K. Zhang and Y. X. Jia, Mater. Chem. Phys., 2011, 130, 334–339 CrossRef CAS.
- I. Lee, J. B. Joo, Y. D. Yin and F. Zaera, Angew. Chem., Int. Ed., 2011, 50, 10208–10211 CrossRef CAS PubMed.
- N. Zhou, L. Polavarapu, N. Y. Gao, Y. L. Pan, P. Y. Yuan, Q. Wang and Q. H. Xu, Nanoscale, 2013, 5, 4236–4241 RSC.
- W. L. Liu, F. C. Lin, Y. C. Yang, C. H. Huang, S. Gwo, M. H. Huang and J. S. Huang, Nanoscale, 2013, 5, 7953–7962 RSC.
- X. Li, X. N. Fu and H. Yang, Phys. Chem. Chem. Phys., 2011, 13, 2809–2814 RSC.
- C. H. Fang, H. L. Jia, S. Chang, Q. F. Ruan, P. Wang, T. Chen and J. F. Wang, Energy Environ. Sci., 2014, 7, 3431–3438 RSC.
- B. Liu, Y. Jiang, Y. Wang, S. Shang, Y. Ni, N. Zhang, M. Cao and C. Hu, Catal. Sci. Technol., 2018, 8, 1094–1103 RSC.
- Q. F. Ruan, L. Shao, Y. W. Shu, J. F. Wang and H. K. Wu, Adv. Opt. Mater., 2014, 2, 65–73 CrossRef.
- Z. F. Bian, T. Tachikawa, P. Zhang, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2014, 136, 458–465 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c9na00548j |
| This journal is © The Royal Society of Chemistry 2019 |