
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLong-term ambient air-stable cubic CsPbBr3 perovskite quantum dots using molecular bromine†
Surakcha
Thapa‡
a,
Karishma
Bhardwaj‡
a,
Siddhant
Basel‡
a,
Sajan
Pradhan
a,
Charlotte J.
Eling
b,
Ali M.
Adawi
 c,
Jean-Sebastien G.
Bouillard
c,
Graeme J.
Stasiuk
b,
Peter
Reiss
d,
Anand
Pariyar
a and
Sudarsan
Tamang
*a
c,
Jean-Sebastien G.
Bouillard
c,
Graeme J.
Stasiuk
b,
Peter
Reiss
d,
Anand
Pariyar
a and
Sudarsan
Tamang
*a
aDepartment of Chemistry, School of Physical Sciences, Sikkim University, India 737102. E-mail: stamang@cus.ac.in
bDepartment of Biomedical Sciences, University of Hull, Hull, HU6 7RX, UK
cDepartment of Physics, University of Hull, Hull, HU6 7RX, UK
dUniv. Grenoble Alpes, CEA, CNRS, IRIG/SyMMES/STEP, 38000 Grenoble, France
First published on 12th August 2019
Abstract
We report unprecedented phase stability of cubic CsPbBr3 quantum dots in ambient air obtained by using Br2 as halide precursor. Mechanistic investigation reveals the decisive role of temperature-controlled in situ generated, oleylammonium halide species from molecular halogen and amine for the long term stability and emission tunability of CsPbX3 (X = Br, I) nanocrystals.
High photoluminescence quantum yield (PL QY), narrow emission linewidth, tunable band gap, large diffusion lengths and low exciton binding energies are some of the key attributes of all-inorganic caesium lead halide perovskite nanocrystals (LHP NCs) i.e., CsPbX3, X = I, Br, Cl.1–6 This novel class of NCs has been shown to be highly “defect tolerant”, i.e. defect states are either shallow or localized in the valence or the conduction band.1,2 Unlike conventional semiconductor NCs, the rigorous passivation of their surface via formation of core/shell structures or other methods is not required to achieve high QY. These LHP NCs are promising building blocks for light emitting diode,3,4 solar cell,5,6 laser,7 photocatalysis8 and detector.9 Despite the recent surge of studies on CsPbX3 perovskite NCs, a persisting drawback is their poor phase stability in ambient air. For example, cubic (α) “black” phase CsPbI3 (Eg = 1.73 eV) perovskite NCs undergo rapid phase transformation to non-luminescent orthorhombic (δ) “yellow” phase in ambient condition (Fig. S1†) leading to undesired changes of the band gap, optical and electrical properties.6,10,11 Similarly, cubic (α) CsPbBr3 (Eg = 2.25 eV) is unstable at ambient condition.12 For successful integration of these materials into devices, the issue of long-term phase stability must thus be addressed.6,13 Most of the reported strategies involve the use of additives such as halide salt,12 phosphinic acid,14 ammonium halide,11 2,2′-iminodibenzoic acid15 sulphides and metal ions16 and polymers5 or via special post-synthetic purification step.6,10 Herein, we report the first synthesis of highly stable, cubic α-CsPbBr3 perovskite NCs using Br2 as an independent halide precursor. In a typical synthesis, lead acetate is dissolved in 1-octadecene in the presence of oleyl amine (OAm) and oleic acid (OA). To this solution, Br2 (warning: handle the liquid Br2 in fume hood, Br2 vapors are toxic) and caesium oleate solutions (both dissolved in ODE) are sequentially added. Phase stability and emission colour tunability are achieved by controlling the reaction temperature (75–200 °C) and amount of Br2 (0.6–1.2 mmol) under air-free synthetic condition (cf. ESI; Experimental section†). The “three-precursor” nature16–18 of our synthetic scheme allows for independent tuning of the amount of the individual elements viz., Cs+, Pb2+ and X− ions and in turn, allows for the precise control over the surface chemistry.17 Highly crystalline, monodisperse 7.62 ± 1.0 nm sized cubic α-CsPbBr3 NCs (Fig. 1a) were synthesised under optimized conditions using Cs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb:Br2 ratio of 1:1:6 at 200 °C. High-resolution transmission electron microscopy (HRTEM) and powder X-ray diffraction (PXRD) analyses of the purified sample confirmed the pure cubic perovskite phase. HRTEM reveals (100) lattice fringes with a d-spacing of 0.58 nm (Fig. S2†). PXRD spectra of α-CsPbBr3 NCs sample exposed to ambient air (relative humidity of ∼50–60%) for a period of 60 days showed no alteration (Fig. 1b). The observed stability is much higher than the previously reported (max. 8 days) achieved via passivation of cubic CsPbBr3 NCs with ZnBr2.12 Untreated pristine cubic CsPbBr3 NCs, synthesised via conventional method typically transforms into orthorhombic phase within 1–2 days.12 We reproduced halide salt passivated CsPbBr3 NCs reported by Woo et al. For comparison, we purified and stored the NCs (control) under conditions similar to our cubic CsPbBr3 NCs.
:Pb:Br2 ratio of 1:1:6 at 200 °C. High-resolution transmission electron microscopy (HRTEM) and powder X-ray diffraction (PXRD) analyses of the purified sample confirmed the pure cubic perovskite phase. HRTEM reveals (100) lattice fringes with a d-spacing of 0.58 nm (Fig. S2†). PXRD spectra of α-CsPbBr3 NCs sample exposed to ambient air (relative humidity of ∼50–60%) for a period of 60 days showed no alteration (Fig. 1b). The observed stability is much higher than the previously reported (max. 8 days) achieved via passivation of cubic CsPbBr3 NCs with ZnBr2.12 Untreated pristine cubic CsPbBr3 NCs, synthesised via conventional method typically transforms into orthorhombic phase within 1–2 days.12 We reproduced halide salt passivated CsPbBr3 NCs reported by Woo et al. For comparison, we purified and stored the NCs (control) under conditions similar to our cubic CsPbBr3 NCs.
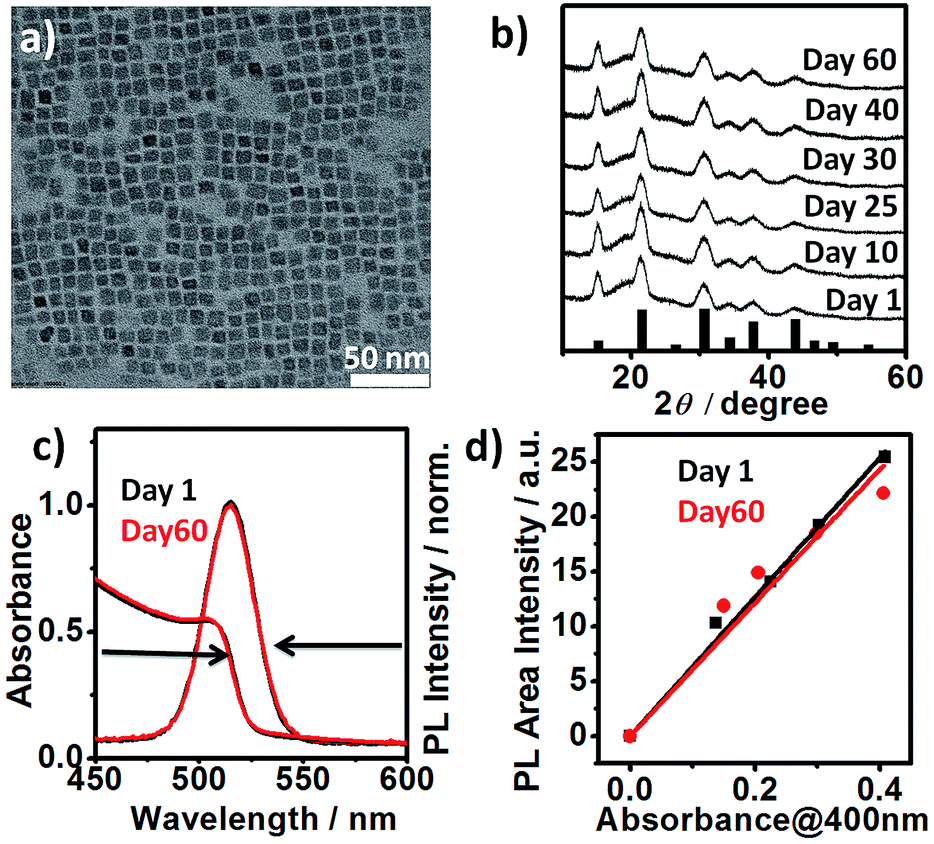 | ||
| Fig. 1 (a) TEM image of cubic CsPbBr3 perovskite NCs (size: 7.62 ± 1.0 nm) (b) XRD patterns showing film stability of CsPbBr3 NCs over a period of 60 days in the air under ambient condition (relative humidity ∼50–60%). The pattern of α-CsPbBr3 (JCPDS 00-054-0752) is indicated as black bars for comparison. (c) UV-vis absorption and normalised PL spectra of as-prepared (day 1) α-CsPbBr3 NCs and the same sample stored in ambient air for 60 days as the colloidal solution (hexane). (d) Integrated photoluminescence vs. absorbance plot29 for day 1 (black) and day 60 samples (red). Absorbance is measured at the excitation wavelength (400 nm). | ||
Consistent with their report, the halide passivation did improve the stability from 2 days to 6 days, thereafter the phase changed to orthorhombic phase (Fig. S3†). In our case, the synthesized α-CsPbBr3 the diffraction peak at 30.5 degrees (2θ) did not split even after 60 days (Fig. S4†) ruling out the presence of the orthorhombic phase. In the case of conventionally prepared CsPbBr3 NCs, the peaks become visibly sharper and split within a week of exposure to ambient air, indicative of the phase transformation into orthorhombic (Pnma) phase.12,19,20 Furthermore, comparison of the integrated photoluminescence vs. absorbance plot for the freshly prepared NCs (day 1) and for the sample stored as the colloidal solution at ambient temperature for 60 days shows the insignificant difference between the two slopes signifying no change in quantum efficiency within the experimental error (Fig. 1c and d). Previous studies have shown that the phase stability of α-CsPbX3 NCs is extremely sensitive to surface chemistry.6,11,17 To understand the cause of the exceptional stability in our case, a series of additional experiments were conducted. FTIR spectra of the purified NCs show the signature of protonated amine groups (–NH3+)21 at 1641 cm−1 in addition to the peak associated to oleate anions (–COO−) at 1576 cm−1 (Fig. 2a).
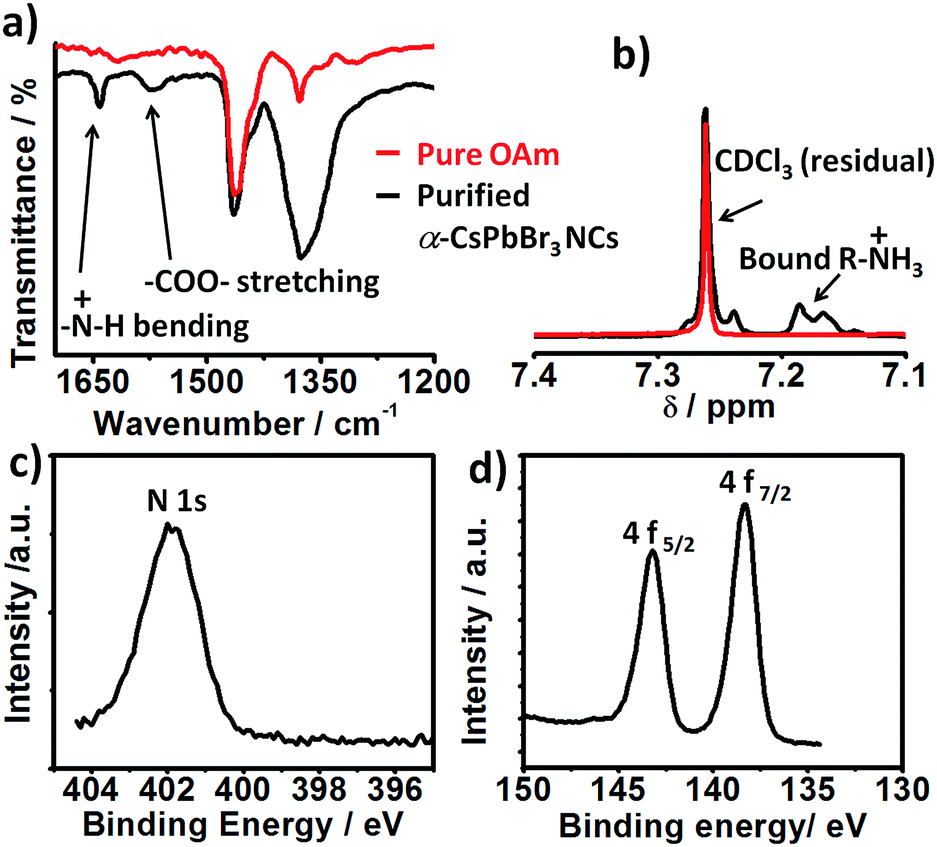 | ||
| Fig. 2 (a) FTIR spectra of the purified CsPbBr3 NCs (black) compared to pure oleyl amine (red). (b) 1H NMR of purified α-CsPbBr3 NCs in CDCl3 (black) (c) XPS N 1s and (d) Pb 4f core level XPS spectra CsPbBr3 NCs. | ||
Furthermore, the 1H NMR shows a multiplet at 7.2 ppm (Fig. 2b) which is uncorrelated with any other proton peak in 1H–1H COrrelated SpectroscopY (COSY) NMR spectra (Fig. S5†). This feature is attributed to diastereotopically coupled protons of ammonium ions bound to the surface of the NCs.11,22 The presence of oleylammonium ions tightly bound to the NC surface is further established with X-ray photoelectron spectroscopy (XPS) analyses. The XPS spectra corrected using the maximum of the C 1s signal at 284.8 eV (Fig. S6†) shows a prominent N 1s peak at 401.8 eV characteristic of bound –NH3+ group (Fig. 2c).21 Furthermore, the Pb 4f core level spectra of ambient stable show two peaks located at 138.4 and 143.2 eV assigned to the Pb 4f7/2 and Pb 4f5/2 levels respectively (Fig. 2d). The observed binding energy value is on the higher side, consistent with observation reported for CsPbBr3 NCs prepared via passivation with ZnBr2 (ref. 12) and tetrafluoroacetate.4 Analysis of the N 1s, Cs 3d, Pb 4f/5d, and Br 3d/3s peaks revealed that the obtained α-CsPbBr3 NCs have bromide ion rich surface (Br/Pb > 3) (Fig. S7†). The estimated N:Cs ratio is 1.56. These results confirmed the presence of ammonium ligand on the lead halide-rich surfaces. The synthetic approach is equally applicable to cubic α-CsPbI3 quantum dots, which are known to be even more sensitive to phase transformation in ambient air (cf. ESI†). In general, under X2-rich condition and with optimized Cs+ ion concentration ([Cs+] ≤ [Pb2+]), the surface of the CsPbX3 NCs can be concomitantly passivated with halide and ammonium ions to achieve long-term stability. Highly crystalline, phase-stable 11.11 nm sized α-CsPbI3 perovskite NCs (Fig. S8†) were obtained using a Cs:Pb:I2 precursor ratio of 1:1:2 at 200 °C. They were stable in ambient air (relative humidity of ∼50–60%, room temperature) for a period of 20 days (Fig. S9†) and for over 4 months in inert condition (Fig. S10†). Similar to the CsPbBr3 NCs surface, the presence of ammonium species (Cs:N ∼ 1.12) and slightly excess halide ions (Cs:Pb:I = 0.8:1:3.4) were confirmed by XPS studies (Fig. S11 and S12†). The presence of surface-bound ammonium ion was further confirmed by FTIR (Fig. S13†) and 1D/2D NMR (Fig. S14 and S15†). The presence of firmly surface-bound oleylammonium ligands is considered important for the in situ stabilisation of the cubic phase of CsPbX3 NCs.11 In fact, the Goldschmidt tolerance factor in cubic CsPbX3 (X = Br, I) perovskite is lower than the ideal value of 1, mainly due to the small size of Cs+ ions compared to the void.23 This effect is accentuated in the case of X = I. Consequently, [PbX6]4− octahedra undergo distortion to decrease the extra space resulting in a transformation from the cubic to the orthorhombic phase. Partial substitution of Cs+ ions on NC surface layers with larger alkyl ammonium ions reduces this distortion, resulting in the observed enhanced stability of the cubic phase. In addition to the presence of ammonium ions, the detection of stoichiometrically-excess halide ions by XPS in our case suggests a possible concomitant role of the halide ions in stabilising the NCs. Kim et al. have demonstrated that halide-amine passivation of fractional dangling electrons satisfies the 8-electron rule on the highly faceted tetrahedral shaped InP NCs surfaces.24 Based on this we propose the co-passivation of α-CsPbX3 NCs with ammonium ions and halide for enhanced stability.
To further gain mechanistic insight into the origin of surface-bound ammonium ions, we examined the fate of OAm in the presence of OA and Br2 at reaction temperature (∼200 °C). In the presence of amine, X2 is reduced to a strong acid HX in situ.25 Therefore, the molecular bromine plays the double role of supplying halide ions and modulating the acid/base (OAm/OA) ligand chemistry via protonation of OAm in the solution. In conventional ligand system, OAm is protonated by OA in solution, resulting in the formation of oleylammonium oleate. As expected Fig. 3a shows the downfield shift of α-CH2 resonance peak of pure OAm from 2.66 to 2.85 ppm (25 °C) on the addition of OA. However, upon heating of the OA/OAm mixture to 200 °C an upfield shift of the α-CH2 resonance peak was observed, which is due to the dissociation of oleylammonium oleate. Its formation from OA and OAm being an exothermic reaction, Le Chatelier's principle predicts indeed that the equilibrium shifts toward the reactants when the temperature is increased.26 The depletion of oleylammonium species at high temperature in typical OA/OAm synthetic methods leads to the instability of the cubic phase. Therefore, CsPbX3 perovskite NCs prepared by conventional methods involving the OA/OAm ligand system is thermally unstable and additional oleylammonium halide ions are required to impede rapid phase change at elevated temperature.11 Typically, synthesis is carried out at 160 °C or lower temperature followed by rapid quenching in an ice bath.11 To the contrary, we found that in the presence of Br2, the OAm/OA equilibria behave differently with a change in temperature. Fig. 3b presents the NMR study of OAm/Br2 mixture at 25 °C and 200 °C. The α-CH2 proton resonance of OAm changes from 2.66 to 2.70 ppm due to protonation of OAm with HBr formed in situ. Importantly, when the OAm/Br2 solution is heated to the reaction temperature (∼200 °C), the α-CH2 (amine) resonance peak shifts further downfield to 2.87 ppm showing the presence of thermally stable ammonium halide ion species. We propose that oleylammonium bromide has been stabilised via self-association or complexation with excess amine present in the mixture.25 The peak broadening at 2.87 ppm also supports that different ammonium halide species (self-associated) are in equilibrium, collectively contributing to the time-averaged NMR spectra. A similar effect of temperature on the NMR chemical shift was reported for pyridine (Py) and HCl mixtures, attributed to the formation of [Py–H–Py]+ and (PyHCl)2 species.27 Furthermore, OAm/I2 mixture exhibited similar characteristics (Fig. S16†), underlining the general nature of the mechanism of phase stabilization in CsPbX3 (X = Br, I) NCs.
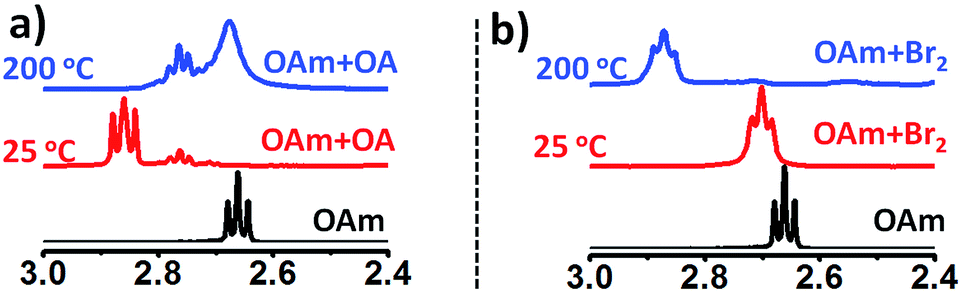 | ||
| Fig. 3 (a) 1H NMR spectra of free OAm (black), OAm/OA mixture at 25 °C (red) and OAm/OA mixture at 200 °C (blue) showing change in α-CH2 (amine) resonance peak. (b) 1H NMR spectra of free OAm (black); OAm/Br2 mixture at 25 °C (red) and at 200 °C (blue). | ||
Both CsPbI3 and CsPbBr3 NCs exhibit emission tunability in a wide spectral range by varying the reaction temperature from 75 °C to 200 °C (Fig. 4). The obtained NCs exhibit narrow PL emission (FWHM: 29–48 nm) and high PL QYs (∼40–79%). Lower emission wavelengths could easily be obtained by simply decreasing the reaction temperature (Fig. 4) or increasing the X2 concentration (Fig. S17†).
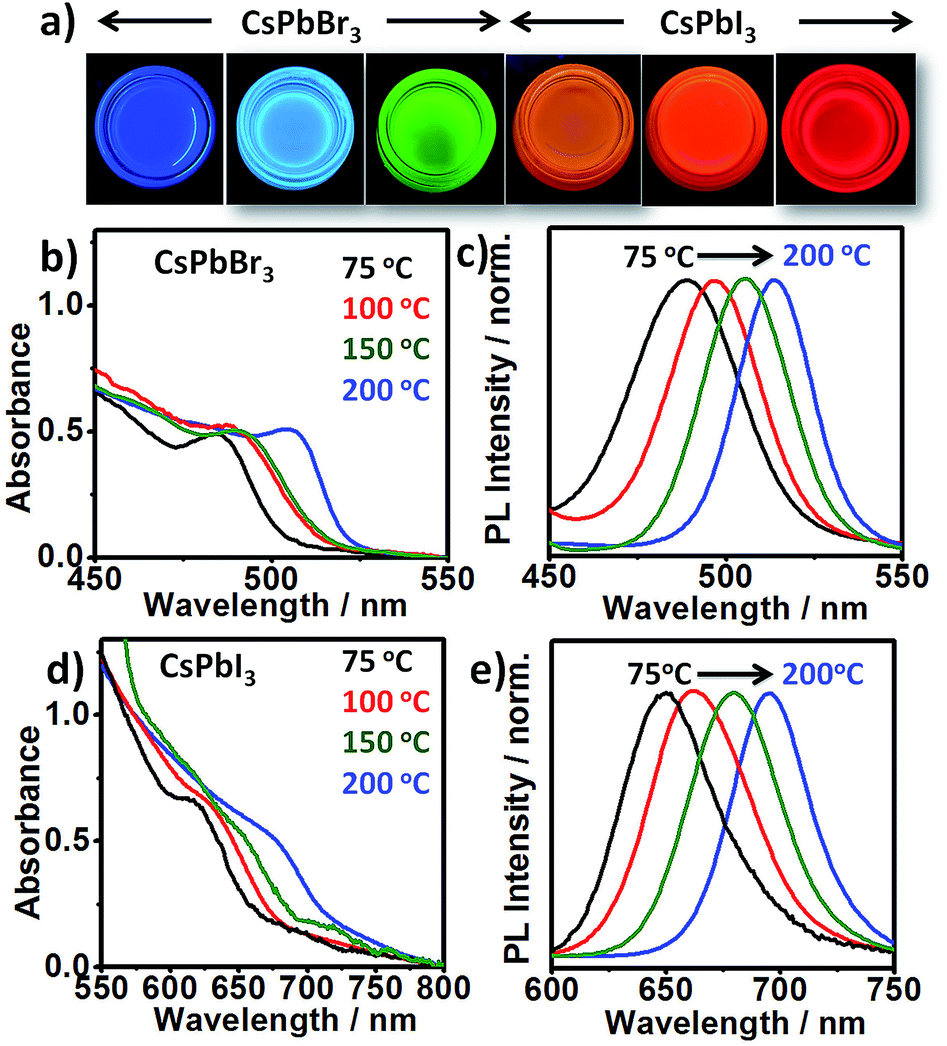 | ||
| Fig. 4 (a) UV illuminated photographs of colloidal solutions of different sized CsPbX3 NCs (X = Br, I). Absorption (b) and emission spectra (c) of CsPbBr3 NCs at different reaction temperatures (75–200 °C). Absorption (d) and emission spectra (e) of CsPbI3 NCs at different reaction temperatures (75–200 °C). | ||
The crystallite sizes calculated from XRD reveals that the change in emission wavelength is due to change in size of the NCs (Fig. S18†). Furthermore, NCs synthesized at higher temperature show lower absorbance (nearly 3-fold) at same photon energy (350 nm), compared to smaller sized NCs synthesized at lower temperature (Fig. S19†). This suggests that the higher temperature produces less nuclei, though of an overall larger final size. On the other hand, at the same temperature increasing the halide concentration leads to increase in supersaturation and hence the increased nucleation rate. By combining the emission ranges of the two materials, the entire visible spectrum from blue to red (478–714 nm) can be accessed. The blue-emitting CsPbBr3 QDs (PL peak 478 nm) obtained at low temperature (∼75 °C) under Br2-rich condition deserve particular attention. Attaining this wavelength is considered difficult due to the small size of CsPbBr3 QDs and their high surface sensitivity.28 It is noteworthy that phase stability of smaller sized NCs synthesized at lower temperature under X2-rich condition were relatively poor compared to ones synthesized at higher temperature (∼200 °C) at same X2 concentration (Table S1†). It has been suggested that due to bulky nature of OAm+ ions, its binding on the surface of the NCs is less feasible at lower temperature.11
In conclusion, we demonstrate a versatile synthetic scheme for achieving long-term ambient stability and emission-tunability of cubic CsPbX3 (X = Br, I) perovskite NCs. The use of molecular X2 allows for the precise control of surface composition, crystal structure and optical properties of the NCs. The key mechanistic insights obtained herein are relevant for the halide perovskite NC synthesis in general.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
ST and KB acknowledge SERB-DST, Government of India for research funding (EEQ/2016/000751 and EMR/2016/002505). SB would like to thank Department of Science and Technology, Government of India (DST/INSPIRE/03/2016/001207) [IF160689] for financial support under DST-INSPIRE Scheme. STh acknowledges Sikkim University for fellowship. AP acknowledges SERB-DST (EEQ/2016/000685) and DST-Inspire (DST/INSPIRE/04/2015/002674), for financial assistance. GJS and ST acknowledge Royal Society (Grant IE160227) for financial support. PR acknowledges funding from the French Research Agency ANR (grants SuperSansPlomb: ANR-15-CE05-0023-01, PERSIL: ANR-16-CE05-0019-02).References
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- D. Aldakov and P. Reiss, J. Phys. Chem. C, 2019, 123, 12527–12541 CAS.
- X. Zhang, H. Lin, H. Huang, C. Reckmeier, Y. Zhang, W. C. H. Choy and A. L. Rogach, Nano Lett., 2016, 16, 1415–1420 CrossRef CAS PubMed.
- H. Wang, X. Zhang, Q. Wu, F. Cao, D. Yang, Y. Shang, Z. Ning, W. Zhang, W. Zheng, Y. Yan, S. V. Kershaw, L. Zhang, A. L. Rogach and X. Yang, Nat. Commun., 2019, 10, 665 CrossRef PubMed.
- B. Li, Y. Zhang, L. Fu, T. Yu, S. Zhou, L. Zhang and L. Yin, Nat. Commun., 2018, 9, 1–8 CrossRef PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS PubMed.
- X. Zhu, Y. Lin, Y. Sun, M. C. Beard and Y. Yan, J. Am. Chem. Soc., 2019, 141, 733–738 CrossRef CAS PubMed.
- X. Chen, H. Hu, Z. Xia, W. Gao, W. Gou, Y. Qu and Y. Ma, J. Mater. Chem. C, 2017, 5, 309–313 RSC.
- A. Dutta and N. Pradhan, ACS Energy Lett., 2019, 4, 709–719 CrossRef CAS.
- A. Dutta, S. K. Dutta, S. Das Adhikari and N. Pradhan, Angew. Chem., Int. Ed., 2018, 57, 9083–9087 CrossRef CAS PubMed.
- J. Y. Woo, Y. Kim, J. Bae, T. G. Kim, J. W. Kim, D. C. Lee and S. Jeong, Chem. Mater., 2017, 29, 7088–7092 CrossRef CAS.
- N. Park, M. Grätzel, T. Miyasaka, K. Zhu and K. Emery, Nat. Energy, 2016, 1, 16152 CrossRef CAS.
- C. Wang, S. R. Chesman and J. J. Jasieniak, Chem. Commun., 2017, 53, 232–235 RSC.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- H. Sun, Z. Li, L. Kong, B. Wang, C. Zhang, Q. Yuan, S. Huang, Y. Liu and L. Li, Chem. Commun., 2018, 54, 9345–9348 RSC.
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed.
- Q. A. Akkerman, L. Mart, L. Goldoni, M. Imran, D. Baranov, H. J. Bolink, F. Palazon and L. Manna, Chem. Mater., 2018, 30, 6915–6921 CrossRef CAS.
- Y. Cai, L. Wang, T. Zhou, P. Zheng, Y. Li and R.-J. Xie, Nanoscale, 2018, 10, 21441–21450 RSC.
- C. Tenailleau, S. Aharon, B.-E. Cohen and L. Etgar, Nanoscale Adv., 2019, 1, 147–153 RSC.
- J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin, L.-J. Xu, H. Yang, A. M. El-Zohry, B. Xin, S. Mitra, M. N. Hedhili, I. S. Roqan, O. F. Mohammed, Y. Han and O. M. Bakr, Chem. Mater., 2019 DOI:10.1021/acs.chemmater.9b00680.
- V. K. Ravi, P. K. Santra, N. Joshi, J. Chugh, S. K. Singh, P. Ghosh and A. Nag, J. Phys. Chem. Lett., 2017, 8, 4988–4994 CrossRef CAS PubMed.
- A. Swarnkar, W. J. Mir and A. Nag, ACS Energy Lett., 2018, 3, 286–289 CrossRef CAS.
- K. Kim, D. Yoo, H. Choi, S. Tamang, J. H. Ko, S. Kim, Y. H. Kim and S. Jeong, Angew. Chem., Int. Ed., 2016, 55, 3714–3718 CrossRef CAS PubMed.
- M. J. Kogan and J. C. Schug, J. Magn. Reson., 1973, 11, 406–415 Search PubMed.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS PubMed.
- C. A. Angell and J. W. Shuppert, J. Chem. Phys., 1977, 67, 3050–3056 CrossRef.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Angew. Chem., Int. Ed., 2018, 57, 1–6 CrossRef.
- S. Tamang, G. Beaune, I. Texier and P. Reiss, ACS Nano, 2011, 5, 9392–9402 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full description of the experimental details and additional experimental data are available. See DOI: 10.1039/c9na00486f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |