DOI:
10.1039/C9NA00431A
(Minireview)
Nanoscale Adv., 2019,
1, 4238-4245
Overall water splitting and hydrogen peroxide synthesis by gold nanoparticle-based plasmonic photocatalysts
Received
9th July 2019
, Accepted 16th September 2019
First published on 16th September 2019
Abstract
Plasmonic photocatalysts driven by the localized surface plasmon resonance excitation of gold nanoparticles (Au NPs) can be efficient solar-to-chemical converters due to their wide spectral response. This review article highlights recent studies on plasmonic water splitting and H2O2 synthesis from water and oxygen (O2) with a particular emphasis placed on the electrocatalysis of Au NPs. The Introduction (Section 1) points to the importance of the establishment of solar hydrogen and oxygen cycles involving hydrogen (H2) and hydrogen peroxide (H2O2) as the key compound, respectively, for realizing a “sustainable society”. Section 2 deals with the basic action mechanisms of Au NP-based plasmonic photocatalysts. Section 3 treats the electrocatalytic activity of Au NPs for the half-reactions involved in the reactions. Section 4 describes recent advances in the plasmonic overall water splitting (4.1) and H2O2 synthesis (4.2). Finally, a summary is presented with the possible development direction in Section 5.
1. Introduction
Hydrogen (H2) and hydrogen peroxide (H2O2) are basic feedstocks in chemical industries, and their use will expand more and more in future due to their environmental benignity. Presently, most H2 is produced by steam reforming of methane at 700–800 °C using a Ni-based catalyst, while H2O2 is industrially produced by the anthraquinone auto-oxidation involving multiple steps.1 Both processes consume large amounts of energy and are undesirable from both environmental and economic aspects. Unambiguously, overall water splitting is the ideal process for H2 production (reaction (1)), while the synthesis of H2O2 from water and oxygen (O2) as well as the direct synthesis from H2 and O2 (ref. 2) is very beneficial (reaction (2)).| | | H2O → H2 + 1/O2 ΔrG0 = +237.13 kJ mol−1 | (1) |
where ΔrG0 is the standard Gibbs energy of reaction.| | | H2O + 1/O2 → H2O2 ΔrG0 = +115.8 kJ mol−1 | (2) |
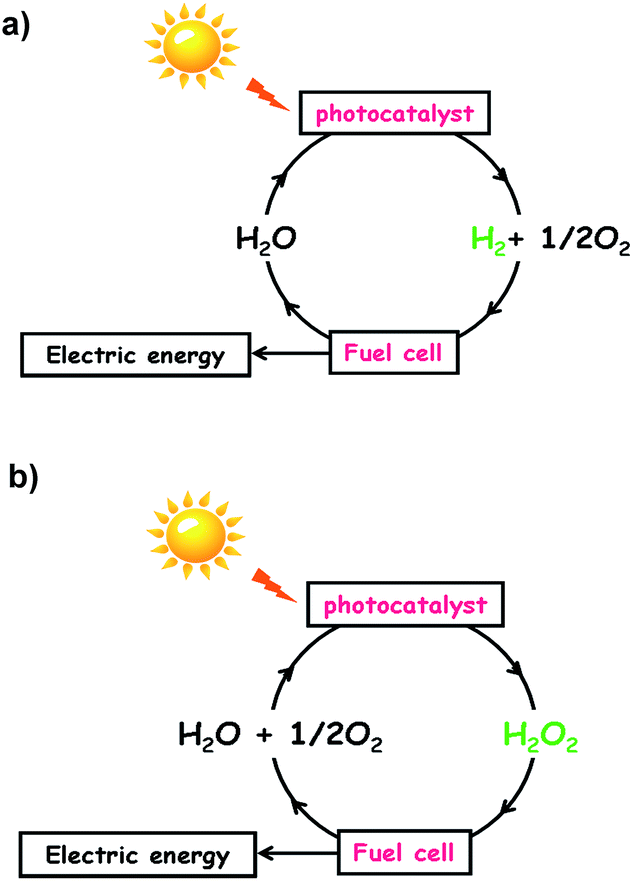
External energy must be supplied to drive the energetically up-hill reactions. Utilization of thermal energy as the driving force is impossible for both the reactions because only 2% of water is decomposed even at 2000 °C, and H2O2 is vulnerable to heat. In principle, reactions (1) and (2) can be driven by photons with energy larger than 1.23 eV and 0.54 eV, respectively, under mild conditions. These considerations motivate one to develop photocatalysts for reactions (1) and (2) by effectively utilizing the visible-to-near infrared light abundantly present in sunlight. If such production of H2 and H2O2 is achieved, input of H2 into H2–O2 fuel cells and input of H2O2 into one-compartment fuel cells3–6 and photofuel cells7 would generate electric energy to complete the solar hydrogen cycle (Scheme 1a) and solar oxygen cycle (Scheme 1b), respectively. So far, many semiconductor photocatalysts for water splitting have been intensively studied; however, a band gap larger than ∼2.5 eV is usually necessary8 because of the large overpotentials for water splitting, particularly the water oxidation reaction (WOR).9 The same is true for the synthesis of H2O2 from water and O2 by semiconductor photocatalysts including graphitic carbon nitride (g-C3N4)10–13 and bismuth vanadate.14 On the other hand, the research on Au NP-based plasmonic photocatalysts with strong and broad absorption in the visible-to-near infrared region due to the localized surface plasmon resonance (LSPR) is rapidly progressing.15–22 The great advantage of plasmonic photocatalysts over semiconductor ones is the availability of “hot carriers” generated by the absorption of such low-energy-photons for chemical reactions,15,17,18,21 which is a prerequisite for achieving high solar-to-chemical conversion efficiency.
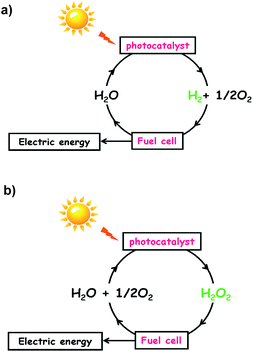 |
| | Scheme 1 (a) Solar hydrogen cycle and (b) solar oxygen cycle. | |
This article reviews recent advances in the plasmonic overall water splitting and H2O2 synthesis from water and O2. This field is still in its infancy, but I believe that this article would contribute to the acceleration and future development in the very important and fascinating topics.
2. Action mechanisms of plasmonic photocatalysts
Plasmonic photocatalysts can be categorized into the hot-electron transfer (HET)-type23,24 and the local electric field-enhanced (LEFE)-type25–28 or the plasmon-resonant energy-transfer-type.29 Here the basic action mechanisms of the HET- and LEFE-type plasmonic photocatalysts are explained for the typical heteronanostructures of Au NP-loaded semiconductors (Au/semiconductor) and Au NP-incorporated semiconductors (Au@semiconductor) including Au NP (core)–semiconductor (shell) nanohybrids, respectively.
2.1 Hot-electron transfer mechanism
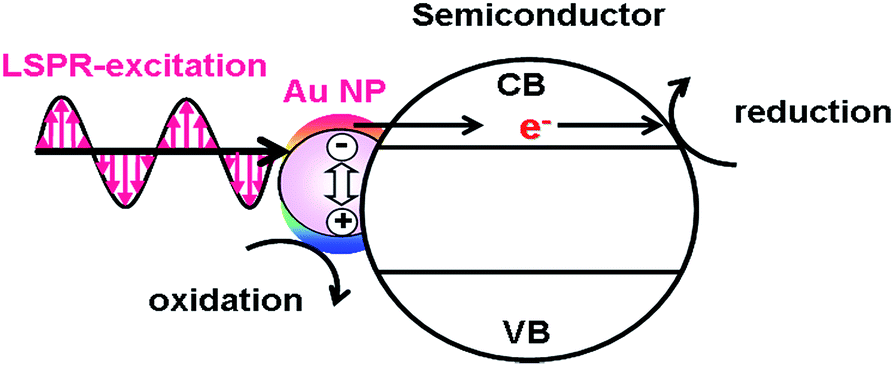
The excitation of the LSPR of Au NP generates excited carriers referred to as “hot carriers” having sufficient energy to drive chemical reactions through the non-radiative decay process or the Landau damping with an efficiency of ϕHCG.21 In the HET-type (Fig. 1), the hot electrons are transferred to the conduction band (CB) of a semiconductor.23,24 The CB-electrons cause a reduction reaction on the semiconductor surface, while an oxidation reaction is induced by the holes left in Au NPs. In this case, solar-to-chemical conversion efficiency (ϕSTC) can be provided by multiplication of the efficiencies of light harvesting by the plasmonic metal (LHEPM), ϕHCG, hot-electron injection from the plasmonic metal to semiconductor (ϕINJ(Au→SEM)), charge separation (ϕCS), and redox reaction efficiency (ϕredox) (eqn (3)).| | | ϕSTC(HET) = LHEPM × ϕHCG × ϕINJ(Au→SEM) × ϕCS × ϕredox | (3) |
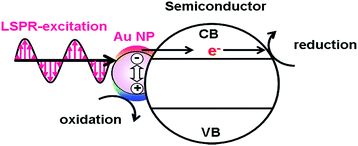 |
| | Fig. 1 Hot electron transfer mechanism. | |
2.2 Local electromagnetic field-enhanced mechanism
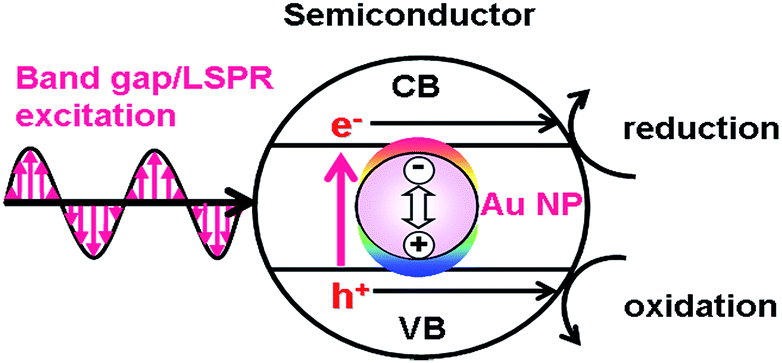
On the other hand, in the LEFE-type (Fig. 2), redox reactions are caused by the band gap excitation of a semiconductor. In this case, the active optical antenna effect of Au NPs enhances the LHE of the semiconductor.30 The local electric field enhancement of Au NPs increases the rate of electron–hole generation in the semiconductor, further promoting the charge separation.26,31 Thus, the ϕSTC is expressed by multiplication of the efficiencies of light harvesting by Au@semiconductor (LHEAu@SEM), carrier generation (ϕCG), ϕCS, and ϕredox (eqn (4)).| | | ϕSTC(LEFE) = LHEAu@SEM × ϕCG × ϕCS × ϕredox | (4) |
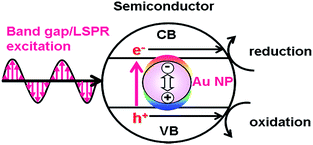 |
| | Fig. 2 Local electromagnetic field-enhanced mechanism. | |
Ag NPs possess a LEFE factor much greater than that of Au NPs, and for example, Ag NP-incorporated AgX (X = Cl, Br, and I) can be a promising plasmonic photocatalyst.32 Excellent review papers on Ag-based plasmonic photocatalysts have recently been published;33,34 however, the photocatalytic reaction is almost limited to the decomposition of organic dyes. It should also be stressed that most features of plasmonic photocatalysts or the wide spectral response can be obtained only for the HET-type. Also, in both types, the reduction in the overpotentials for the redox reactions is pivotal to enhance the photocatalytic activity.
2.3 Photothermal and light-scattering effects
As the secondary effects, photothermal conversion and light scattering can enhance the activity of plasmonic photocatalysts. Generally, radiative damping is the dominant decay mechanism for large Au NPs larger than 50 nm for dipolar plasmon resonances, while nonradiative damping is the dominant decay mechanism for small Au NPs.35
Strong light absorption due to the LSPR of Au NPs followed by nonradiative energy dissipation causes a rise in temperature of the surroundings.36 Particularly, under high-power irradiation, this photothermal effect may boost ϕSTC for the HET-type and LEFE-type plasmonic photocatalysts through the increases in ϕredox shown in eqn (3) and (4). The LSPR extinction band for plasmonic metal NPs consists of the contributions from light absorption and scattering. When the size of Au particles exceeds 50 nm, light scattering becomes dominant.37 In the LEFE-type plasmonic photocatalysts, light scattering effectively increases to enhance the rate of photogenerated charge carriers in the semiconductor.38 Thus, the light-scattering effect can boost the ϕSTC through the increase in LHE shown in eqn (4). However, the light scattering effect would be usually negligible because the particle size of Au NPs used in most studies on plasmonic photocatalysts is significantly smaller than 50 nm.
3. Electrocatalytic activity of gold nanoparticles
The oxidation half-reaction common in overall water splitting and H2O2 synthesis from water and O2 is the WOR (reaction in the reverse direction of reaction (5)), where E0 is the standard electrode potential with respect to the standard hydrogen electrode (SHE).| | | O2 + 4H+ + 4e− → 2H2O E0 = +1.23 V | (5) |
On the other hand, the reduction half-reactions in the former and latter are the hydrogen evolution reaction (HER, reaction (6)) and the two-electron oxygen reduction reaction (ORR) (reaction (7)), respectively. The key aspect in H2O2 synthesis is to restrict the four-electron ORR (reaction (5)), which is thermodynamically more favored than the two-electron ORR. The selectivity for electrochemical H2O2 formation can be quantitatively determined using the rotating ring disk technique.39
| | | 2H+ + 2e− → H2E0 = 0 V | (6) |
| | | O2 + 2H+ + 2e− → H2O2E0 = +0.695 V | (7) |
The electrocatalytic activities of Au NPs for these reactions are described below.
3.1 Electrocatalytic activity for the water oxidation reaction
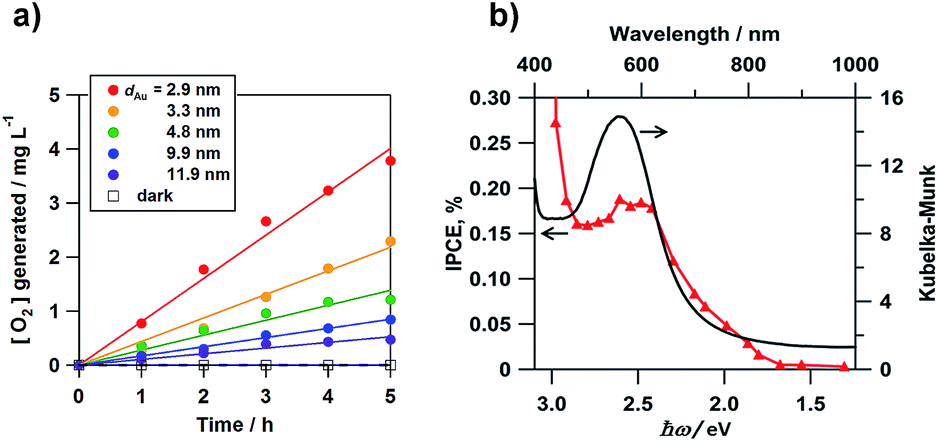
Misawa and co-workers first showed that the WOR occurs on an Au nanorod-loaded TiO2 electrode under illumination of visible and even near infrared light.40 We studied the electrocatalytic activity of Au/TiO2 films coated on a fluorine-doped tin oxide photoanode (Au/TiO2/FTO) for the WOR in a three-electrode photoelectrochemical (PEC) cell with the structure of photoanode|0.1 mol dm−3 NaClO4 aqueous solution|Ag/AgCl (reference electrode)|glassy carbon (cathode) under illumination of simulated sunlight (AM 1.5 one sun, λ > 430 nm).41Fig. 3a shows the time courses for O2 generation in aqueous solution. TiO2/FTO is inactive for the WOR under these conditions. In the Au/TiO2/FTO system, O2 concentration increases in proportion to irradiation time. Also, the rate of O2 generation increases with a decrease in Au particle size (dAu). García and co-workers also reported the same trend in an Au/TiO2 particulate system.42Fig. 3b shows the action spectrum of the incident photon-to-current efficiency (IPCE) for the Au/TiO2/FTO photoanode cell. Surprisingly, the IPCE rises at excitation energy (ħω) ≈ 1.5 eV. As the wavelength of incident light shortens, the IPCE increases with a peak near the LSPR peak. A similar action spectrum was reported for the WOR in a PEC cell employing Au/SrTiO3 as the photoanode.43 Also, the electrocatalytic activity of Au/TiO2 for the WOR can be dramatically enhanced by the formation of a Pb shell on the Au NPs.44
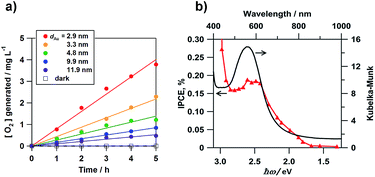 |
| | Fig. 3 (a) Time courses for the rate of O2 generation. (b) IPCE action spectrum for the photoelectrochemical WOR of the Au/TiO2/FTO photoelectrodes with the absorption spectrum for comparison. Figure (a) is taken from ref. 41. | |
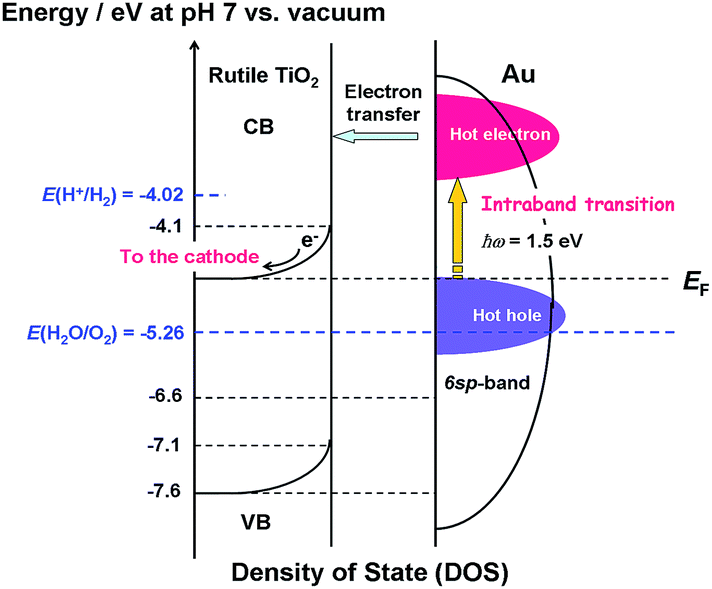
Scheme 2 shows the energy diagram of the Au/TiO2 plasmonic anode with the intraband transition via the surface plasmon decay.45 The excitation by photons with ħω = 1.5 eV promotes the electrons with energy below the Fermi energy (EF) to high energy levels within the 6sp-band. The hot electrons are injected into the CB of TiO2, while the hot holes oxidize water. In this manner, the rise in the IPCE at ħω ≥ 1.5 eV can be explained by the intraband-transition mechanism, and in this scheme, the WOR by the hot holes would determine the rate of the reaction.
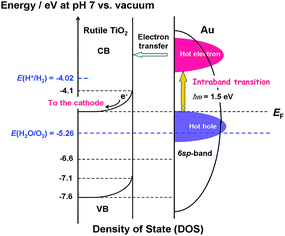 |
| | Scheme 2 Energy diagram of the Au/TiO2 plasmonic electrode with the density of states of TiO2 and energy distribution of the hot carriers in Au NPs with the intraband transition via the surface plasmon decay. | |
3.2 Electrocatalytic activity for the hydrogen evolution reaction
The electrocatalytic activity of bulk-state metal electrodes for the HER can be correlated to the M–H bond energy.46 A clear volcano-shaped curve in the plots of electrocatalytic activity for the HER vs. Gibbs energy of metal–H bond formation indicates that the group of metals with moderate M–H bond energy have high levels of activity for the HER. Among metal electrodes, Au possesses a high electrocatalytic activity for the HER following some rare metals (Pt, Re, Rh, and Ir). Then, to evaluate the electrocatalytic activity of Au NPs, the Au/TiO2-photocatalyzed H2 generation from ethanol aqueous solution was carried out under UV-light irradiation in the absence of O2. UV-light irradiation of Au/TiO2 excites the electrons in the valence band (VB) of TiO2 to the CB (eqn (8)). The CB-electrons in TiO2 are effectively transferred to Au NPs with a large work function to be accumulated in them (eqn (9)).47 The VB-holes in TiO2 easily oxidize ethanol to yield acetaldehyde and protons (eqn (10)). On the other hand, the electrons accumulated in Au NPs reduce water to H2 (eqn (11)). Accordingly, the photocatalytic activity can be a good indicator of the electrocatalytic activity of Au NPs for the HER.| | | Au/TiO2 + hν (λex < 400 nm) → Au/TiO2(e−⋯h+) | (8) |
| | | Au/TiO2(e−⋯h+) → Au(e−)/TiO2(h+) | (9) |
| | | Au(2e−)/TiO2(2h+) + C2H5OH → Au(2e−)/TiO2 + CH3CHO + 2H+ | (10) |
| | | Au(2e−)/TiO2 + 2H+ → Au/TiO2 + H2 | (11) |
Idriss and co-workers reported Au/TiO2-photocatalyzed H2 generation from an aqueous solution of ethanol.48 The authors showed that UV-light irradiation of Au/anatase TiO2 and Au/rutile TiO2 yields H2, while each unmodified TiO2 is inactive. Interestingly, the activity of Au/anatase TiO2 is significantly higher than that of Au/rutile TiO2 at the same Au loading, further exceeding the activity of Pt/TiO2.
3.3 Electrocatalytic activity for the oxygen reduction reaction
In bulk-state electrodes, the two-electron ORR preferentially proceeds on Au, while the four-electron ORR occurs on Pt.49 The photocatalytic activity of metal NP-loaded TiO2 (M/TiO2, M = Au and Pt) for the ORR was carried out in aerated ethanol aqueous solution under UV-light irradiation (M/TiO2, M = Au and Pt).50 In this case, the electrons in metal NPs can be used for the two-electron and/or four-electron ORR (reactions (12) and (13)), and the photocatalytic activity can be a good indicator of the electrocatalytic activity of metal NPs for the ORR.| | | M(2e−)/TiO2 + O2 + 2H+ → M/TiO2 + H2O2 | (12) |
| | | M(4e−)/TiO2 + O2 + 4H+ → M/TiO2 + 2H2O | (13) |
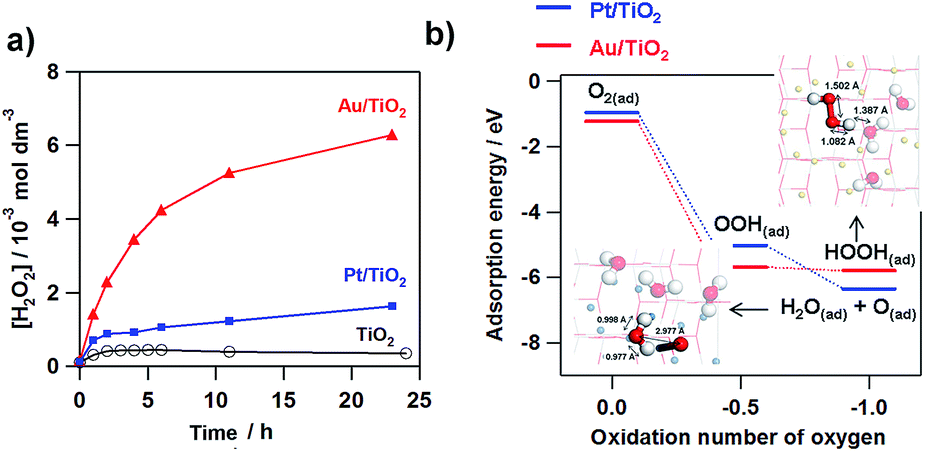
Fig. 4a shows the comparison of the photocatalytic activity of TiO2, Au/TiO2, and Pt/TiO2 for the two-electron ORR. In the TiO2 system, only micromolar H2O2 is generated as previously reported.51–53 Loading Pt NPs on TiO2 increases the saturated H2O2 concentration to ∼1 mmol dm−3. Strikingly, Au/TiO2 exhibits much higher activity than Pt/TiO2, and the H2O2 concentration reaches ∼6 mmol dm−3 at irradiation time = 24 h. To gain information about the origin of the great difference of the activity, density functional theory (DFT) simulations were performed for model systems (Au28/(TiO2)32 and Pt28/(TiO2)32).54Fig. 4b shows the comparison of the adsorption energy of the sequentially reduced O2 species and the optimized structures. The one-electron ORR to OOH(ad) is an energetically large downhill process, and the structure is the same in each system. In the two-electron ORR, the OOH(ad) can be reduced to HOOH in the Au28/(TiO2)32 system, whereas H2O(ad) and O(ad) are produced with the O–O bond cleavage in the Pt28/(TiO2)32 system. In the electrode systems, the selectivity for electrochemical H2O2 formation was reported to be 6.3% for Au/TiO2 and <0.1% for Pt/TiO2.10 These experimental and theoretical calculation results indicate that the Au NP catalyst is a good electrocatalyst for the two-electron ORR, while Pt NPs accelerate the four-electron ORR.
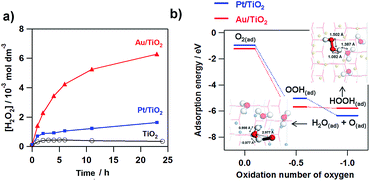 |
| | Fig. 4 Time courses for the M(Au and Pt)/TiO2-photocatalyzed ORR to H2O2 under UV-light irradiation (λ > 300 nm, light intensity I290–390 nm = 3 mW cm−2) and unmodified TiO2 system for comparison. (b) Adsorption energy change with the M28/TiO2-photocatalyzed two-electron ORR (M = Au and Pt. Figure (b) is taken from ref. 54. | |
4. Photocatalytic reactions
Usually, the photocatalytic activity can be evaluated using the external quantum yield (ϕex) defined by eqn (1).| | | ϕex = {n × (number of product molecules)/number of incident photons} | (14) |
where n = 2 for the HER (product = H2) and the 2-electron ORR (product = H2O2), and n = 4 for the WOR (O2).
4.1 Overall water splitting
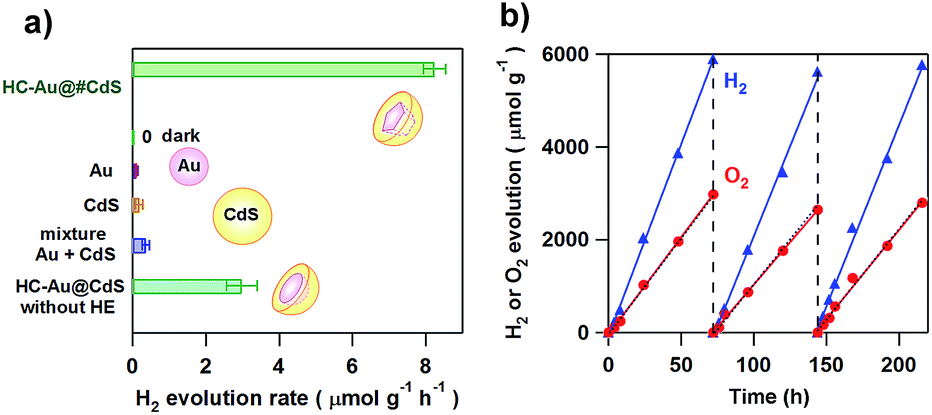
Moskovits and co-workers were the first to show plasmonic overall water splitting using TiO2-capped Au nanorods with Pt as the hydrogen evolution catalyst on TiO2 and a Co-based oxygen evolution catalyst on Au (ϕex = ∼0.1%).55 The Au/TiO2–NiOx plasmonic photocatalyst has also been reported to be capable of splitting water with ϕex = 0.013% at ħω = 2.1 eV.56 As shown in Scheme 2, the low CB minimum of TiO2 (ECBM = −4.1 eV for rutile TiO2 and −3.9 eV for anatase TiO2 at pH 7 vs. vacuum level) which is insufficient for the HER (ECBM = −4.02 eV at pH 7) is mainly responsible for the limited efficiencies. On the other hand, CdS possesses a much higher CB minimum (ECBM = −3.28 eV).57 Unique asymmetrical nanohybrids referred to as half-cut Au (core) and CdS (shell) nanoeggs without and with a heteroepitaxial junction (HC-Au@CdS and HC-Au@#CdS) were synthesized using a modified photodeposition technique.58 Water splitting experiments were carried out under irradiation of red-light.59Fig. 5a shows the comparison of the photocatalytic activity of the nanohybrids and the single components. In this case, the Au particle size was fixed at ∼5.5 nm. Au and CdS are almost inactive, but the physical mixture shows a low activity. Interestingly, non-heteroepitaxial junction HC-Au@CdS has much higher activity, which further increases more than twice in the heteroepitaxial junction sample. As shown in Fig. 5b, the photocatalytic activity increases by about one-order of magnitude with an increase in Au particle size from 5.5 nm to 12.1 nm, and ϕex = 0.24% at ħω = 1.9 eV has been achieved. Further, even after three repetitions of the 3 day-reaction, no decay in the activity is observed with the continuous stoichiometric generation of H2 and O2.
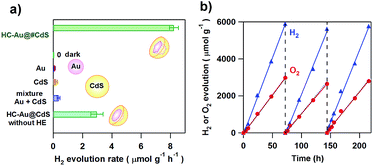 |
| | Fig. 5 (a) The comparison of the H2 evolution rate for HC-Au(dAu = 5.5 nm)@#CdS(lCdS = 1.9 nm), Au colloid, CdS, and a mixture of Au and CdS under red-light illumination (λex = 640 nm) or dark conditions. (b) Repeated water splitting by HC-Au(dAu = 12.1 nm)@#CdS(lCdS = 2.1 nm) under red-light illumination. Figures are taken from ref. 59. | |
Scheme 3 illustrates the basic reaction scheme of water splitting by the HC-Au@#CdS plasmonic photocatalyst. HC-Au@#CdS efficiently absorbs sunlight. The hot-electrons generated by the LSPR excitation can be effectively injected into the CB of CdS60–62 through the large area and high-quality junction.59 The high-energy electrons in the CB of CdS enable a smooth HER, while the hot holes left in Au NPs oxidize water with the aid of the electrocatalytic activity for the WOR.63 Importantly, selective excitation of the Au NP-LSPR suppresses the photodissolution of CdS,64 so far hampering its use as a water splitting photocatalyst.
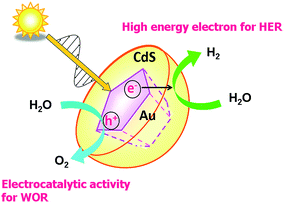 |
| | Scheme 3 Schematic representation of water splitting by the HC-Au@#CdS/TiO2 plasmonic photocatalyst. | |
Tan and co-workers have recently prepared a Pt NP-loaded TiO2 hierarchical nano-architecture (Pt/TiO2-HA), showing that it has a high level of visible-light activity for overall water splitting (ϕex = 0.23% at ħω = 2.25 eV).65 The authors postulated the HET mechanism described in Section 2.1 for the Pt/TiO2-HA-photocatalyzed water splitting.
4.2 Hydrogen peroxide synthesis
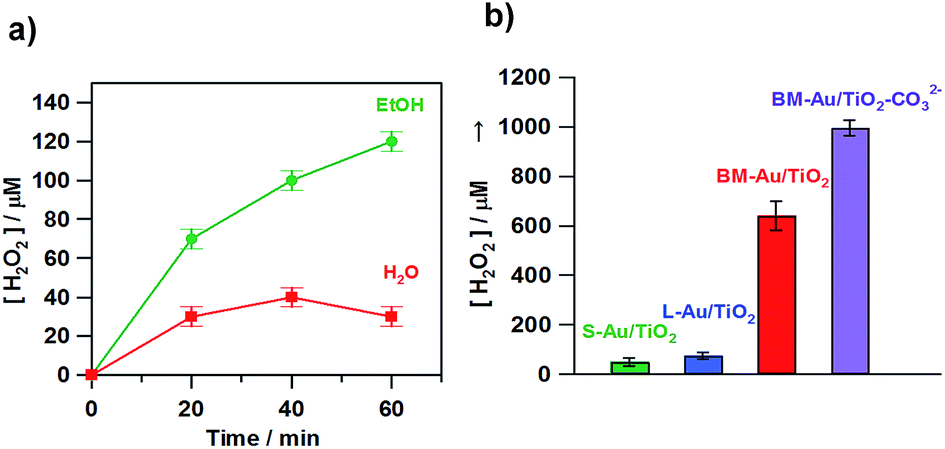
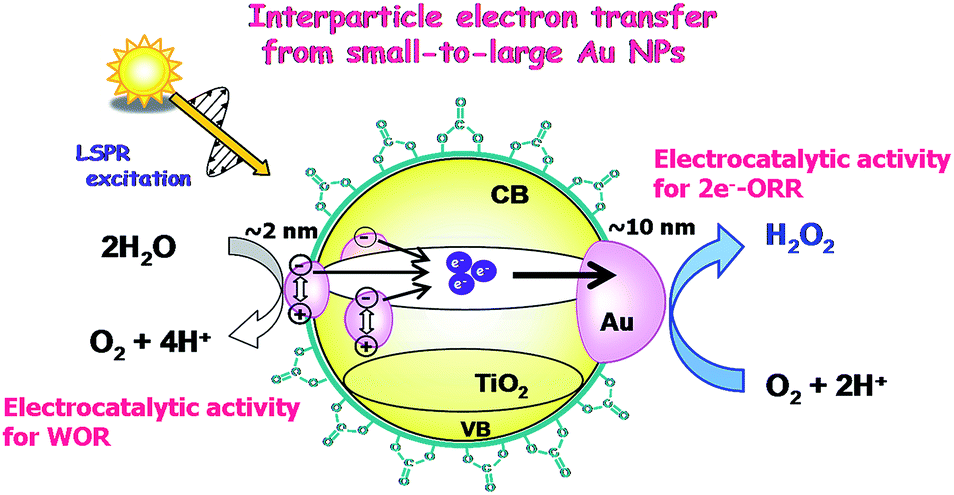
Au/TiO2 plasmonic photocatalysts have been applied to various important oxidations.66 This is probably because the electrocatalytic activity of Au NPs for the reduction reaction cannot be utilized in the usual HET-type Au/TiO2 plasmonic photocatalyst, where Au and TiO2 act as oxidation and reduction sites, respectively (Fig. 1). We have shown that visible-light irradiation of small- (dAu ≈ 2 nm) and large- (dAu ≈ 10 nm) Au NP-loaded TiO2 referred to as bimodal (BM)-Au/TiO2 induces the interfacial electron transfer from small Au NPs to large Au NPs through the CB of TiO2.67 This phenomenon was rationalized in terms of the entropic driving force for the interfacial electron transfer.33Fig. 6a shows the time courses for BM-Au/TiO2-photocatalyzed H2O2 generation from aerated water in the absence and presence of ethanol under visible-light irradiation.68 Even without ethanol, ∼40 μmol dm−3 H2O2 is generated at irradiation time = 40 min. Fig. 6b shows the comparison of the photocatalytic activity of various catalysts for H2O2 generation from aerated aqueous ethanol solution. BM-Au/TiO2 exhibits much higher activity than small-Au/TiO2 and large-Au/TiO2.
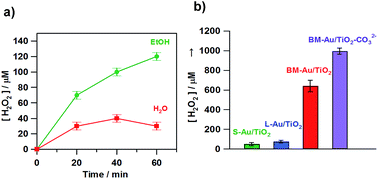 |
| | Fig. 6 (a) Time courses for the BM-Au/TiO2-photocatalyzed H2O2 generation in the absence (red) and presence of 4% C2H5OH (green) under irradiation of visible-light (λex > 430 nm, light intensity integrated from 420 to 485 nm = 4.0 mW cm−2) at 298 K. (b) Photocatalytic activity of S-Au/TiO2, L-Au/TiO2, BM-Au/TiO2, and BM-Au/TiO2–CO32− for the reduction of O2 to H2O2 in an aqueous solution containing 4% HCOOH (pH 1.7) in the dark and under irradiation of visible-light (λex > 430 nm, light intensity integrated from 420 to 485 nm = 4.0 mW cm−2) at 298 K. Figures are taken from ref. 68. | |
It is known that H2O2 strongly adsorbs on TiO2 to form a surface complex (eqn (15)). The surface complex undergoes reductive decomposition by the CB-electrons in TiO2 (eqn (16)).69 Then, it is also important to suppress this degradation pathway to increase the yield of H2O2. An effective way is the surface-fluorination of TiO2 (ref. 70 and 71) enabling the production of H2O2 at a millimolar level under UV-light irradiation.72
| | | Tis–OH + H2O2 → Tis–OOH + H2O | (15) |
where the subscript s denotes the surface atom.
| | | Tis–OOH + 2H+ + 2e−CB → Tis–OH + H2O | (16) |
Then, the effect of the surface modification of BM-Au/TiO2 with carbonate ions (BM-Au/TiO2–CO32−) on the photocatalytic activity was examined. As shown in Fig. 6b, the surface modification drastically increases the photocatalytic activity, and the ϕex reached 5.4% at ħω = 2.3 eV. In the H2O2 synthesis from water and O2 using semiconductor photocatalysts, a ϕex value of 0.24% at ħω = 2.95 eV was reported for Au/BiVO4.14 Importantly, Shiraishi and co-workers have found that g-C3N4 possesses an extremely high selectivity of ∼90% for electrochemical H2O2 formation,10 and the efficiency of photocatalytic H2O2 synthesis was greatly improved by using it as the photocatalyst (ϕex = 2.6% at ħω = 2.95 eV).73 In a holey defective g-C3N4 photocatalytic system, an extremely high ϕex value of ∼16% was achieved in the presence of 2-propanol as an electron donor at ħω = 3.26 eV.13
The high photocatalytic activity of BM-Au/TiO2 for H2O2 synthesis from water and O2 can be rationalized as follows (Scheme 4). Visible-light irradiation of BM-Au/TiO2 gives rise to the net electron transport from small Au NPs to large Au NPs, accumulating electrons and holes in large and small Au NPs, respectively. As a result, water is oxidized on small Au NPs (Fig. 3a), while the two-electron ORR occurs on large Au NPs. Eventually, the high photocatalytic activity of BM-Au/TiO2 for H2O2 synthesis can stem from the effective charge separation through the interfacial electron transfer from small Au NPs to large Au NPs, the former’s excellent electrocatalytic activity for the WOR, and the low catalytic activity of the small and large Au NPs for H2O2 decomposition.50 Further, the surface modification with CO32− ions of BM-Au/TiO2 is effective in suppressing the reductive decomposition of H2O2 to increase its yield.
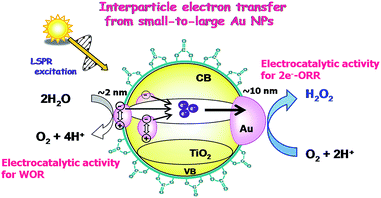 |
| | Scheme 4 Schematic representation of H2O2 synthesis from water and O2 by the BM-Au/TiO2 plasmonic photocatalyst. | |
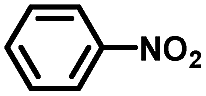
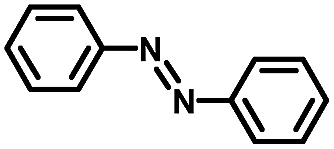
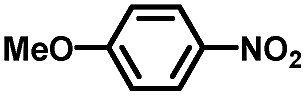
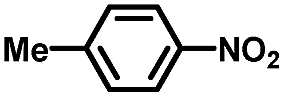
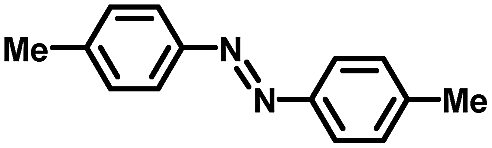
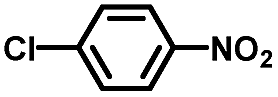
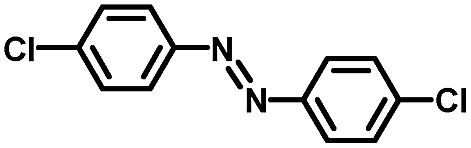
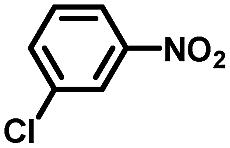
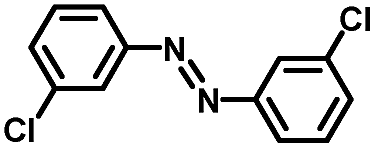
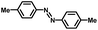
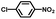
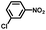
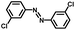
From a viewpoint of organic synthesis, BM-Au/TiO2 has also paved a way for the application of plasmonic photocatalysts in reductive chemical transformations. As an example, Table 1 summarizes the BM-Au/TiO2-photocatalyzed reduction of nitrobenzene and its derivatives.67 BM-Au/TiO2 exhibits a high level of visible-light activity for the one-step synthesis of azobenzenes from nitrobenzenes at 25 °C with a high yield of >95% and selectivity > 99%, whereas unimodal Au/TiO2 is photocatalytically inactive.
Table 1 BM-Au/TiO2 photocatalyzed reduction of nitrobenzenesa
| Substrate |
Product |
Time |
Conversion |
Selectivity |
|
Reaction conditions: BM-Au/TiO2 10 mg, nitrobenzene (10 mM) solution (10 mL, 2-propanol) with KOH (10 mM), visible-light (λex > 430 nm, 10 mW cm−2) irradiation at 25 °C under anaerobic conditions. The table is taken from ref. 67.
|
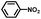
|
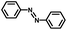
|
12 h |
95% |
>99% |

|
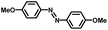
|
24 h |
>99% |
>99% |
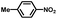
|
|
20 h |
98% |
>99% |
|
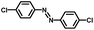
|
24 h |
>99% |
>99% |
|
|
12 h |
>99% |
>99% |
5. Summary and perspectives
H2 and H2O2 are not only basic chemical feedstocks but also the key compounds for the energy cycles. A big challenge in chemistry is the development of highly active photocatalysts for the overall water splitting and H2O2 synthesis from water and O2. The HET-type Au NP-based plasmonic photocatalysts can generate “hot carriers” with potential to drive the energetically uphill reactions through the excitation by low-energy photons which the usual semiconductor photocatalysts cannot utilize. The overall water splitting and H2O2 synthesis comprise the half-reactions of the WOR, HER, and two-electron ORR. Au NPs possess high levels of electrocatalytic activities for these reactions strongly depending on the size, shape, and composition.74 Consequently, the Au NP-based HET-type plasmonic photocatalyst can be a very promising solar-to-chemical converter.
However, there is plenty of room for the enhancement of the photocatalytic activity by the judicious selection of semiconductors and the optimization of Au NPs corresponding to the targeted reaction. First, the information about the influence of the band structure of semiconductors on the activity is a prerequisite for the rational design of plasmonic photocatalysts. Second, the bimetallization of Au NPs has great potential for improving the photocatalytic activity, while the effects of Au NP-size and shape on the activity have been reviewed.22 Alloying of Au NPs with Pd75 and Pt,76 and Pd/Pt-shell formation on the surface of Au NPs77 have been reported to boost the activity of the Au/TiO2 plasmonic photocatalyst for the HER from water containing methanol as a sacrificial electron donor. These effects would partly result from the excellent electrocatalytic activity of Pt and Pd for the HER. Third, as a common subject in heteronanostructured photocatalysts, the importance of devising a general mechanism for enhancing the charge separation between the components should also be stressed. An effective approach to achieve this is the formation of an atomically commensurate heteroepitaxial junction.59,78,79 In addition, the construction of multi-component plasmonic photocatalysts is also very promising. Recently, three-component plasmonic photocatalysts such as Au and Pt NP-codeposited TiO2 (ref. 80) and Au/TiO2–graphitic carbon nitride (g-C3N4)81 have been reported to show much higher photocatalytic activity than Au/TiO2 for the HER from methanol aqueous solution. The effective charge separation through the interfacial electron transfer from Au NPs to Pt NPs in the former system, and from g-C3N4 to TiO2 in the latter system would be responsible for the high photocatalytic activities. Meanwhile, the research on the catalysis of supported Au NPs for various reactions is intensively and constantly continued.82 I anticipate that the combination of the research on the catalysis of Au NPs and Au NP-based plasmonic photocatalysts can effect further development of various “green” processes for solar-to-chemical transformations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author expresses sincere gratitude to Emeritus Professor Dr Hisayoshi Kobayashi (Kyoto Institute of Technology), Dr Musashi Fujishima, Dr Shin-ichi Naya, Ms Miwako Teranishi, T. Onishi, S. Kawano, and Y. Motokawa (Kindai University). This work was supported by the Ministry of Education, Science and Culture, Grant-in-Aid for Scientific Research (C) no. 15K05654, and MEXT-Supported Program for the Strategic Research Foundation at Private Universities.
Notes and references
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962 CrossRef CAS PubMed.
- J. K. Edwards, B. Solsona, E. Ntainjua, N. A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037 CrossRef CAS PubMed.
- S.-i. Yamazaki, Z. Siroma, H. Senoh, T. Iori, N. Fujiwara and K. Yasuda, J. Power Sources, 2008, 178, 20 CrossRef CAS.
- Y. Yamada, S. Yoshida, T. Honda and S. Fukuzumi, Energy Environ. Sci., 2011, 4, 2822 RSC.
- A. M. Shaegh, N.-T. Nguyen, S. M. M. Ehteshami and S. H. Chan, Energy Environ. Sci., 2012, 5, 8225 RSC.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- T. Onishi, M. Fujishima and H. Tada, ACS Omega, 2018, 3, 12099 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253 RSC.
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterlohf and S. Ardo, Energy Environ. Sci., 2015, 8, 2825 RSC.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774 CrossRef CAS.
- L. Yang, G. Dong, D. L. Jacobs, Y. Wang, L. Zang and C. Wang, J. Catal., 2017, 352, 274 CrossRef CAS.
- Y. Peng, L. Wang, Y. Liu, H. Chen, J. Lei and J. Zhang, Eur. J. Inorg. Chem., 2017, 4976 Search PubMed.
- L. Shi, L. Yang, W. Zhou, Y. Liu, L. Yin, X. Hai, H. Song and J. Ye, Small, 2018, 14, 1703142 CrossRef PubMed.
- H. Hirakawa, S. Shiota, Y. Shiraishi, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2016, 6, 4976 CrossRef CAS.
- K. Ueno and H. Misawa, J. Photochem. Photobiol., C, 2013, 15, 31 CrossRef CAS.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC; C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188 RSC.
- H. Cheng, K. Fuku, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2015, 3, 5244 RSC.
- X. Li, J. Zhu and B. Wei, Chem. Soc. Rev., 2016, 45, 3145 RSC.
- P. Lianos, Appl. Catal., B, 2017, 210, 235 CrossRef CAS.
- S. Sahai, A. Ikram, S. Rai, R. Shrivastav, S. Dass and V. R. Satsangi, Renewable Sustainable Energy Rev., 2017, 68, 19 CrossRef CAS.
- Y. Zhang, S. He, W. Guo, Y. Hu, J. Huang, J. R. Mulcahy and W. D. Wei, Chem. Rev., 2018, 118, 2927 CrossRef CAS PubMed.
- H. Tada, Dalton Trans., 2019, 48, 6308 RSC.
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632 CrossRef CAS PubMed.
- A. Furube, L. Du, K. Hara, R. Katoh and M. Tachiya, J. Am. Chem. Soc., 2007, 129, 14852 CrossRef CAS PubMed.
- G. Zhao, H. Kozuka and T. Yoko, Thin Solid Films, 1996, 277, 147 CrossRef CAS.
- K. Awazu, M. Fujimaki, C. Rockstuhl, J. Tominaga, H. Murakami, Y. Ohki, N. Yoshida and T. Watanabe, J. Am. Chem. Soc., 2008, 130, 1676 CrossRef CAS PubMed.
- M. Fujishima, T. Ikeda, R. Akashi and H. Tada, ACS Omega, 2018, 3, 6104 CrossRef CAS PubMed.
- R. Asapu, N. Claes, R.-G. Ciocarlan, M. Minjauw, C. Detavernier, P. Cool, S. Bals and S. W. Verbruggen, ACS Appl. Nano Mater., 2019, 2, 4067 CrossRef CAS.
- K. Ueno, T. Oshikiri and H. Misawa, ChemPhysChem, 2016, 17, 199 CrossRef CAS PubMed; N. Wu, Nanoscale, 2018, 10, 2679 RSC.
- M. W. Knight, H. Sobhani, P. Norrdlander and N. J. Halas, Science, 2011, 332, 702 CrossRef CAS PubMed.
- Y. Hayashido, S. Naya and H. Tada, J. Phys. Chem. C, 2016, 120, 19663 CrossRef CAS.
- Y. Hayashido, S. Naya and H. Tada, J. Phys. Chem. C, 2016, 120, 19663 CrossRef CAS.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813 RSC.
- K. H. Leong, A. A. Aziz, L. C. Sim, P. Saravanan, M. Jang and D. Bahnemann, Beilstein J. Nanotechnol., 2018, 9, 628 CrossRef CAS PubMed.
- N. J. Halas and S. Lal, Chem. Rev., 2011, 111, 3913 CrossRef CAS PubMed.
- N. S. Abadeer and C. J. Murphy, J. Phys. Chem. C, 2016, 120, 4691 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578 CrossRef CAS PubMed.
- C. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188 RSC.
- Y. Shiraishi, Y. Kofuji, H. Sakamoto, S. Tanaka, S. Ichikawa and T. Hirai, ACS Catal., 2015, 5, 3058 CrossRef CAS.
- Y. Nishijima, K. Ueno, Y. Yokota, K. Murakoshi and H. Misawa, J. Phys. Chem. Lett., 2010, 1, 2031 CrossRef CAS.
- M. Teranishi, M. Wada, S. Naya and H. Tada, ChemPhysChem, 2016, 17, 2813 CrossRef CAS PubMed.
- C. G. Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595 CrossRef PubMed.
- L. Liu, P. Li, B. Adisak, S. Ouyang, N. Umezawa, J. Ye, R. Kodiyath, T. Tanabe, G. V. Ramesh, S. Ueda and H. Abe, J. Mater. Chem. A, 2014, 2, 9875 RSC.
- R. Negishi, S. Naya, H. Kobayashi and H. Tada, Angew. Chem., Int. Ed., 2017, 56, 10347 CrossRef CAS PubMed.
- R. Sundararaman, P. Narang, A. S. Jermyn, W. A. Goddard III and H. A. Atwater, Nat. Commun., 2014, 5, 5788 CrossRef CAS PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571 CrossRef CAS.
- H. Tada, T. Kiyonaga and S. Naya, Chem. Soc. Rev., 2009, 38, 1849 RSC.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, Nat. Chem., 2011, 3, 489 CrossRef CAS PubMed.
- C. M. Sánchez-Sánchez and A. J. Bard, Anal. Chem., 2009, 81, 8094 CrossRef PubMed.
- M. Teranishi, S. Naya and H. Tada, J. Am. Chem. Soc., 2010, 132, 7850 CrossRef CAS.
- R. Cai, Y. Kubota and A. Fujishima, J. Catal., 2003, 219, 214 CrossRef CAS.
- H. Goto, Y. Hanada, T. Ohno and M. Matsumura, J. Catal., 2004, 225, 223 CrossRef CAS.
- T. Hirakawa and Y. Nosaka, J. Phys. Chem. C, 2008, 112, 15818 CrossRef CAS.
- H. Kobayashi, M. Teranishi, R. Negishi, S. Naya and H. Tada, J. Phys. Chem. Lett., 2016, 7, 5002 CrossRef CAS PubMed.
- S. Mubeen, J. Lee, N. Singh, S. Krämer, G. D. Stucky and M. Moskovits, Nat. Nanotechnol., 2015, 8, 247 CrossRef PubMed.
- A. Tanaka, K. Teramura, S. Hosokawa, H. Kominami and T. Tanaka, Chem. Sci., 2017, 8, 2574 RSC.
- N. Buhker, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261 CrossRef.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782 CrossRef CAS.
- S. Naya, T. Kume, R. Akashi, M. Fujishima and H. Tada, J. Am. Chem. Soc., 2018, 140, 1251 CrossRef CAS.
- K. Wu, W. E. Rodríguez-Córdoba, Y. Yang and T. Lian, Nano Lett., 2013, 13, 5255 CrossRef CAS PubMed.
- J. W. Ha, T. P. A. Ruberu, R. Han, B. Dong, J. Vela and N. Fang, J. Am. Chem. Soc., 2014, 136, 1398 CrossRef CAS PubMed.
- K. Takayama, K. Fujiwara, T. Kume, S. Naya and H. Tada, J. Phys. Chem. Lett., 2017, 8, 86 CrossRef CAS PubMed.
- P. Reineck, G. P. Lee, M. Karg, P. Mulvaney and U. Bach, Adv. Mater., 2012, 24, 4750 CrossRef CAS PubMed.
- D. Meissner, R. Memming, B. Kastening and D. Bahnemann, Chem. Phys. Lett., 1986, 127, 419 CrossRef CAS.
- L. Qin, G. Wang and Y. Tan, Sci. Rep., 2018, 8, 16198 CrossRef PubMed.
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC; W. Wang and D. Astruc, Chem. Soc. Rev., 2014, 43, 7188 RSC.
- S. Naya, T. Niwa, T. Kume and H. Tada, Angew. Chem., Int. Ed., 2014, 53, 7305 CrossRef CAS PubMed.
- M. Teranishi, R. Hoshino, S. Naya and H. Tada, Angew. Chem., Int. Ed., 2016, 55, 12773 CrossRef CAS.
- X. Li, C. Chen and J. Zhao, Langmuir, 2001, 17, 4118 CrossRef CAS.
- C. Minero, G. Mariella, V. Maurino and E. Pelizzetti, Langmuir, 2000, 16, 2632 CrossRef CAS.
- H. Park and W. Choi, J. Phys. Chem. B, 2004, 108, 4086 CrossRef CAS.
- V. Maurino, C. Minero, G. Mariella and E. Pelizzetti, Chem. Commun., 2005, 2627 RSC.
- Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, Angew. Chem., Int. Ed., 2014, 53, 13454 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137 CrossRef CAS.
- R. S. Moakhar, M. Jalali, A. Kushwaha, G. K. L. Goh, N. Riahi-Noori, A. Dolati and M. Ghorbani, J. Appl. Electrochem., 2018, 48, 995 CrossRef.
- W. Shi, C. Ma, H. Wang, D. Duan, Z. Sun and S. Yang, Int. J. Hydrogen Energy, 2018, 43, 18850 CrossRef CAS.
- A. Malankowska, M. P. Kobylański, A. Mikolajczyk, O. Cavdar, G. Nowaczyk, M. Jarek, W. Lisowski, M. Michalska, E. Kowalska, B. Ohtani and A. Zaleska-Medynska, ACS Sustainable Chem. Eng., 2018, 6, 16665 CrossRef CAS.
- K. Kitazono, R. Akashi, K. Fujiwara, A. Akita, S. Naya, M. Fujishima and H. Tada, ChemPhysChem, 2017, 18, 2840 CrossRef CAS PubMed.
- K. Awa, R. Akashi, A. Akita, S. Naya, H. Kobayashi and H. Tada, ChemPhysChem, 2019, 20, 2155 CrossRef CAS PubMed.
- L. Wen, R. Xu, C. Cui, W. Tang, Y. Mi, X. Lu, Z. Zeng, S. L. Suib, P.-X. Gao and Y. Lei, Nano Lett., 2018, 18, 4914 CrossRef CAS PubMed.
- C. Marchal, T. Cottineau, M. G. Méndez-Medrano, C. Colbeau-Justin, V. Caps and V. Keller, Adv. Energy Mater., 2018, 1702142 CrossRef.
- D. A. Panayotov and J. R. Morris, Surf. Sci. Rep., 2016, 71, 77 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence