
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrophobic ion pairing: encapsulating small molecules, peptides, and proteins into nanocarriers
Kurt D.
Ristroph
and
Robert K.
Prud'homme
 *
*
Department of Chemical and Biological Engineering, Princeton University, Princeton, New Jersey 08544, USA. E-mail: prudhomm@princeton.edu
First published on 1st October 2019
Abstract
Hydrophobic ion pairing has emerged as a method to modulate the solubility of charged hydrophilic molecules ranging in class from small molecules to large enzymes. Charged hydrophilic molecules are ionically paired with oppositely-charged molecules that include hydrophobic moieties; the resulting uncharged complex is water-insoluble and will precipitate in aqueous media. Here we review one of the most prominent applications of hydrophobic ion pairing: efficient encapsulation of charged hydrophilic molecules into nano-scale delivery vehicles – nanoparticles or nanocarriers. Hydrophobic complexes are formed and then encapsulated using techniques developed for poorly-water-soluble therapeutics. With this approach, researchers have reported encapsulation efficiencies up to 100% and drug loadings up to 30%. This review covers the fundamentals of hydrophobic ion pairing, including nomenclature, drug eligibility for the technique, commonly-used counterions, and drug release of encapsulated ion paired complexes. We then focus on nanoformulation techniques used in concert with hydrophobic ion pairing and note strengths and weaknesses specific to each. The penultimate section bridges hydrophobic ion pairing with the related fields of polyelectrolyte coacervation and polyelectrolyte-surfactant complexation. We then discuss the state of the art and anticipated future challenges. The review ends with comprehensive tables of reported hydrophobic ion pairing and encapsulation from the literature.
1. Introduction, overview, and terminology
In the twenty years since Meyer and Manning's classic 1998 review,1 hydrophobic ion pairing (HIP) has gained prominence as a useful strategy for making charged hydrophilic molecules into hydrophobic complexes. The technique has a number of applications and has been used, among others, to dissolve molecules in supercritical CO2,2 dissolve enzymes in organic solvents without losing activity,3 improve intestinal adsorption4–6 or skin permeation,7,8 or otherwise enhance bioavailability.9This review will focus on one of the most prevalent uses of hydrophobic ion pairing: the complexation and encapsulation of charged hydrophilic small molecule, peptide, or protein therapeutics into drug delivery vehicles. The first section summarizes the general rules for hydrophobic ion pairing. We discuss drug eligibility and class-specific considerations, review commonly-used counterions, and outline key parameters such as counterion pKa. The third section focuses on formulation techniques that have been used to encapsulate hydrophobic complexes into nanoparticles, microparticles, and emulsions for drug delivery. The fourth section discusses how ion paired drug payloads are released from their delivery vehicles. The fifth section bridges the HIP technique with polyelectrolyte–polyelectrolyte complexation (‘coacervation’) and polyelectrolyte–surfactant complexation, related fields that have remained largely unconnected from the hydrophobic ion pairing literature. We do not review another related field, nucleotide complexation with cationic lipids to form lipoplexes or solid lipid nanoparticles, but provide references to a number of excellent reviews. At the end of the article we present tables to organize the reported results of hydrophobic ion pairing used for encapsulation. The tables are sorted by both therapeutic and counterion for easy reference and rapid comparison (Fig. 1).
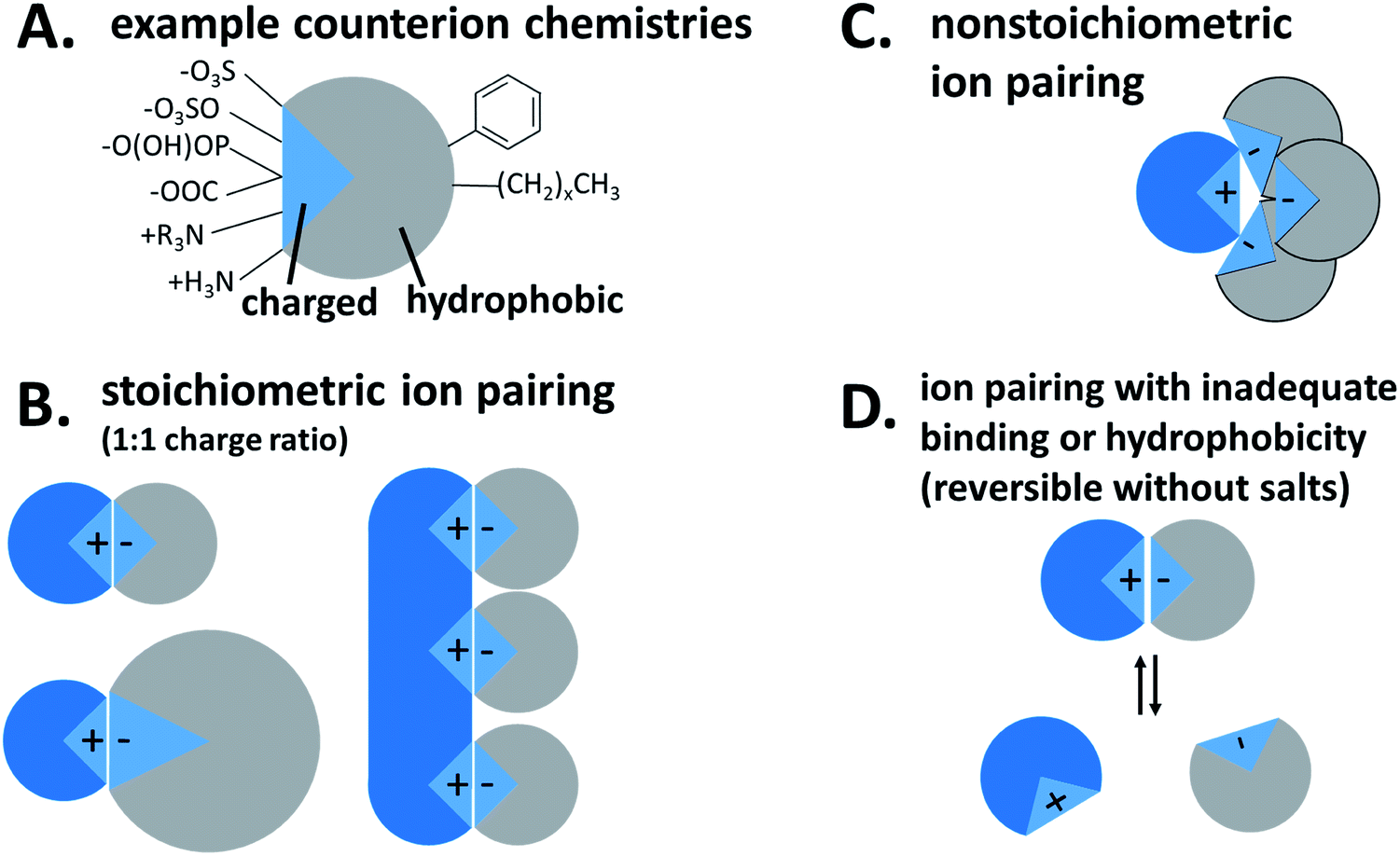 | ||
| Fig. 1 Hydrophobic ion pairing schematic. (A) Possible charged groups (left) and hydrophobic moieties (right) for a counterion. (B) Stoichiometric ion pairing between a cationic API (blue) and anionic counterion. (C) Non-stoichiometric ion pairing. (D) Reversible ion pairing due to inadequate binding or hydrophobicity. | ||
Hydrophobic ion pairing is the process of forming ionic interactions10 between a charged hydrophilic molecule with an oppositely-charged counterion.1 The counterion contains at least one hydrophobic domain such as an alkyl tail or aromatic ring. The complexation increases hydrophobicity by two main mechanisms: first, the molecule's natural charge is masked, mitigating solubility in polar solvents such as water. Second, the hydrophobic groups on the counterion, typically nonpolar aliphatic tails or aromatic groups, help to coat the original molecule's surface area with hydrophobic domains that exclude water.
For our purposes, the charged hydrophilic is a drug or dye and may be referred to as an ‘active pharmaceutical ingredient (API)’ or ‘therapeutic.’ The counterion is referred to in the literature as a ‘hydrophobic counterion,’ ‘ion pair(ing agent) (IP),’ or ‘salt former.’ Due to their amphiphilic chemical nature, many hydrophobic counterions used are surfactants, so the term ‘surfactant’ may be used as well. The act of forming an ionic association between the two species is termed either ‘hydrophobic ion pairing’ or ‘ionic complexation,’ and the resulting paired species is a ‘hydrophobic complex’ or ‘HIP complex.’ We will discuss later why we do not use the term ‘salt.’
Another important piece of terminology is the stoichiometry between the two species. In the HIP literature, there is no standard convention for reporting the ratio of hydrophilic therapeutic to counterion. Molar ratio (reported either as a ratio of x:y or as a fraction), mass ratio, charge ratio, and N/P ratio – i.e. ratio of positive to negative charges, usually reported as a fraction, from the lipoplex literature – have all been used. Consider a 1300 Da peptide with five cationic groups that is paired with five molecules of a monovalent counterion of molecular weight 280 Da (Fig. 2A). Reporting ratios as drug:counterion, this complex has a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 or 0.2, a mass ratio of 0.93, a charge ratio of 1:1, and an N/P ratio of 1. Charge and molar ratios are the most intuitive of these, and the x:y ratio nomenclature is more intuitive than fractions.
:5 or 0.2, a mass ratio of 0.93, a charge ratio of 1:1, and an N/P ratio of 1. Charge and molar ratios are the most intuitive of these, and the x:y ratio nomenclature is more intuitive than fractions.
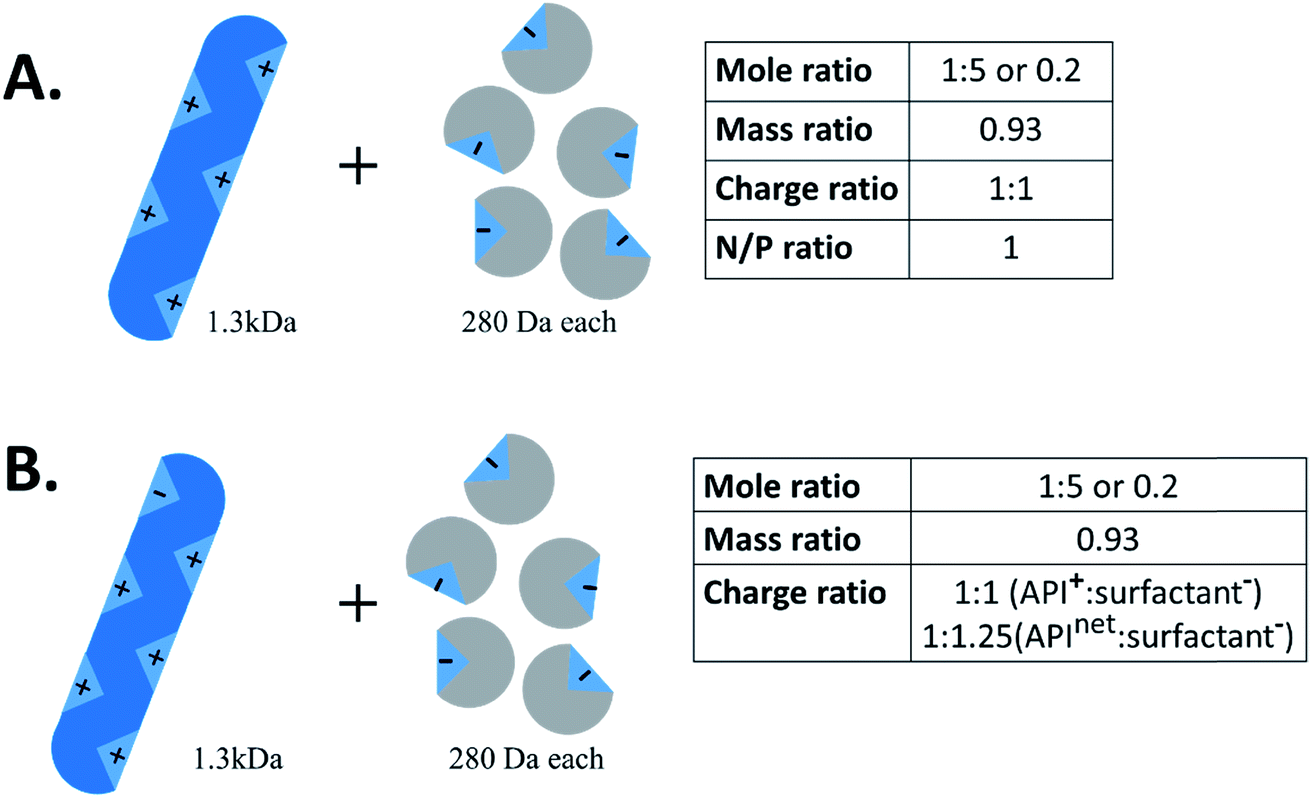 | ||
| Fig. 2 Example schematic of hydrophobic ion pairing between a 1.3 kDa peptide and 280 Da anionic surfactants. When reporting charge ratio, it is helpful to specify if the value given is based on the API's net charge (typical for proteins) or total number of one kind of charge. | ||
We recommend that future researchers in the field use charge ratios and report the ratio as ‘drug:counterion’ rather than as a fraction. Charge ratio is a useful and intuitive parameter in HIP, and should be reported whenever possible. Both molecules' degrees of ionisation may vary with pH; when possible, the charge ratio should be reported at the pH of the complexation.11 When describing the charge ratio of a system where one molecule is zwitterionic, researchers should note whether their reported charge ratio is based on the molecule's net charge or charge of only one type. We recommend the latter, but this is not always possible for large proteins, where only net charge can readily be determined. Consider the example above; if the peptide had one anionic group in addition to five cationic groups, the charge ratio of peptide cations (5) to counterion anions (5) is still 1:1 (Fig. 2B). The peptide:counterion charge ratio calculated from the peptide's net charge of (5−1=)4, though, is 4:5 or 1:1.25, suggesting an excess of counterions when none actually exists. Reporting the molar ratio along with the charge ratio should clarify this point, provided an accurate counting of what charged groups exist on each species is included. In this review, we have converted reported stoichiometries into charge ratios to facilitate comparisons.
2. Hydrophobic ion pairing
Hydrophobic ion pairing is an attractive technique for encapsulating water-soluble therapeutics using formulation strategies optimized for water-insoluble drugs. These strategies are desirable because new strategies to encapsulate hydrophilic molecules in nano-scale delivery vehicles remain challenging.12 Low drug loadings, poor encapsulation efficiencies, and a lack of scalability continue to prevent many liposome and nanoparticle formulations of biologic therapeutics from reaching the market.12 The potential benefits of encapsulation – targeting, protection from enzymatic degradation, improved circulation time, enhanced bioavailability, controlled release, reduced toxicity, and overall improved drug performance – are strong driving motivations to develop scalable, highly-loaded formulations with high encapsulation efficiencies.13,14 This is particularly attractive for biologic (peptide and protein) therapeutics, whose circulation time unprotected in the blood may be as low as minutes.12Nanoparticle formulation strategies for hydrophobic drugs have been developed to address the growing number of new, strongly hydrophobic therapeutics.15,16 These techniques – oil-in-water emulsions, nanoprecipitation, solid lipid nanoparticles, etc. – are designed to take advantage of a drug's hydrophobicity/lipophilicity. They do not translate easily to the encapsulation of hydrophilic therapeutics.17 HIP solves this problem by temporarily modifying the therapeutics to increase their hydrophobicity and allow encapsulation. When the modification is undone, the original hydrophilic therapeutic is regenerated.1 We will discuss ways of controlling dissociation to tune release elsewhere in the review. In many reported cases, the de-complexed released therapeutic remains fully active; this has been shown even for large proteins with tertiary structure-dependent activity.18
Modifying a drug's solubility profile to make it more hydrophobic for encapsulation is also the goal of some prodrug strategies;19 both techniques temporarily add hydrophobic groups to a hydrophilic molecule.20 Unlike prodrug approaches, HIP does not modify any covalent bonds on the original API. This is important from a regulatory standpoint: prodrugs require full FDA approval but the requirements for hydrophobic ion pairs may not be as stringent, depending on the other changes made to the formulation.21
2.1 Thermodynamics
lnxi, where μi is the chemical potential (μi0 is the chemical potential of the pure species) and xi is the molar fraction of the solute. It can be seen that entropy always favours dissolution, i.e. increasing the degrees of freedom in the system is favoured.
There is some subtlety with water as the aqueous solvent, since the hydrogen bonding interactions between water molecules adds an entropy contribution to the water solvent itself.22 That entropic contribution determines observations such as the Hofmeister series, where the specific salt cations and anions influence solubility.23 For this review, we will ignore this effect, since the concept of counter ion binding and precipitation does not require a detailed understanding of water structure.
Water is a unique solvent and is the strongest of the hydrogen bonding fluids. The polarity of the water molecule gives water a high dielectric constant: ε = 80. This is in contrast to the dielectric constant of a hydrophobic oil phase (e.g. dodecane), which will have ε = 2. The dielectric constant determines the strength of electrostatic interactions between elementary charges. The interaction energy between a positive and negative charge in solution is 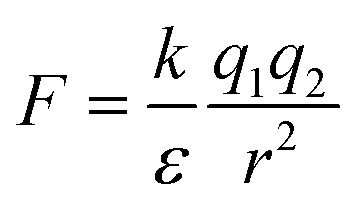 . As the dielectric constant increases, therefore, the force holding ions together decreases. Hydrophobic ion pairs stay insoluble in part because they usually include large nonpolar groups that exclude water from fully solvating the ionic–ionic interaction sites. The hydrophobic ion paired precipitate or core of a NC has a low dielectric constant, which magnifies the strength or the electrostatic attractions. This same concept arises in the protein literature, where the interactions between anionic and cationic peptides in the hydrophobic core of a globular protein enhance its stability. However, the same residues on the surface of a protein would enhance its water solubility. It often remains unclear if any water remains associated with the pair in a nanoparticle core; the best data addressing this question comes from studies of ionomers.24–27
. As the dielectric constant increases, therefore, the force holding ions together decreases. Hydrophobic ion pairs stay insoluble in part because they usually include large nonpolar groups that exclude water from fully solvating the ionic–ionic interaction sites. The hydrophobic ion paired precipitate or core of a NC has a low dielectric constant, which magnifies the strength or the electrostatic attractions. This same concept arises in the protein literature, where the interactions between anionic and cationic peptides in the hydrophobic core of a globular protein enhance its stability. However, the same residues on the surface of a protein would enhance its water solubility. It often remains unclear if any water remains associated with the pair in a nanoparticle core; the best data addressing this question comes from studies of ionomers.24–27
2.2 Eligibility for hydrophobic ion pairing and commonly-used counterions
We typically do not use or recommend using the term ‘salt’ to describe the complexes formed by hydrophobic ion pairing, because ‘salts’ are commonly understood to refer to crystalline assemblies of stoichiometric amounts of oppositely-charged ions. HIP complexes may be less crystalline than the original drug used,28–30 and non-stoichiometric charge ratios are common.
We pause here to briefly address the field of nucleic acid encapsulation and delivery. Nucleic acids – plasmid DNA, linear DNA, siRNA, mRNA, etc. – have been packaged into solid lipid nanoparticles (SLNs) or lipoplexes through ionic complexation between cationic lipids and the nucleic acid's anionic phosphate backbone. This strategy shares a number of similarities with hydrophobic ion pairing, with a few notable exceptions. The most significant is that the regular charge along the phosphate backbone gives nucleic acids a strong, uniform charge density along the molecule. This is different from the small molecule, peptide and protein therapeutics discussed here, which often have less ordered regions of hydrophobicity and hydrophilicity/charge. For the reader familiar with HIP but not SLNs/lipoplexes, we recommend a number of reviews.31–37
| Name | Structure | MW, Da | pKa | logP |
Used to pair with |
|---|---|---|---|---|---|
| 1-Hydroxy-2-naphthoic acid (xinafoic acid) |
|
188.2 | 3.02 | 2.6 | AZD2811 (ref. 38 and 89) |
| 2-Naphthalene sulfonic acid (NSA) |
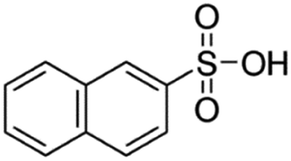
|
208.2 | −1.8 | 2.14 | Atazanavir28 |
| Brilliant blue FCF |
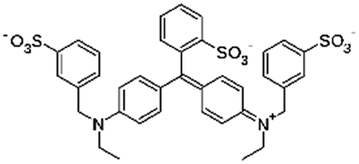
|
792.8 | 5.83 and 6.58 | −1.45 | Atenolol155 |
| Carboxy methyl polyethylene glycol (CM-PEG) |
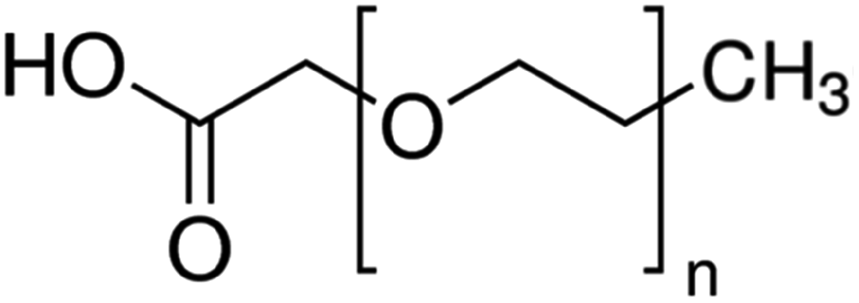
|
PEG length not given | Bovine serum albumin56 | ||
| Lysozyme56 | |||||
| r-met-HuGdNF56 | |||||
| Cholesteryl hemisuccinate |
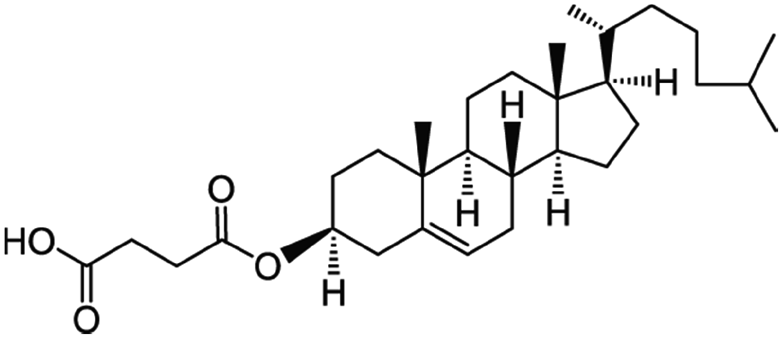
|
486.7 | 5.8 | 8.5 | Colistin156 |
| Doxorubicin112 | |||||
| Cholic acid (sodium cholate) |
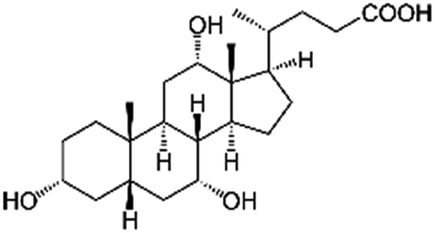
|
408.6 | 4.98 | 2.02 | AZD2811 (ref. 38 and 89) |
| Bovine serum albumin56 | |||||
| Lysozyme56 | |||||
| r-met-HuGdNF56 | |||||
| Insulin157 | |||||
| Decanoic acid (sodium decanoate/sodium caprate) |

|
194.3 | 4.9 | 4.09 | Octreotide9,96 |
| Dimyristoyl phosphatidyl glycerol (DMPG) |
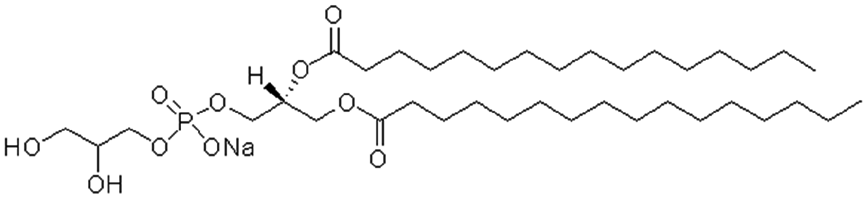
|
666.9 | 1.89 | 9.2 | Insulin42 |
| Salmon calcitonin85 | |||||
| Dioleoyl phosphatidic acid (DOPA) |
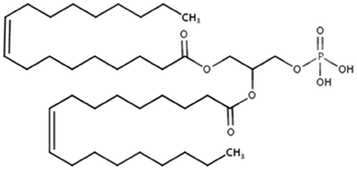
|
701 | 1.3 | 13.2 | Doxorubicin71 |
| Gefitinib30 | |||||
| Docosahexaenoic acid |
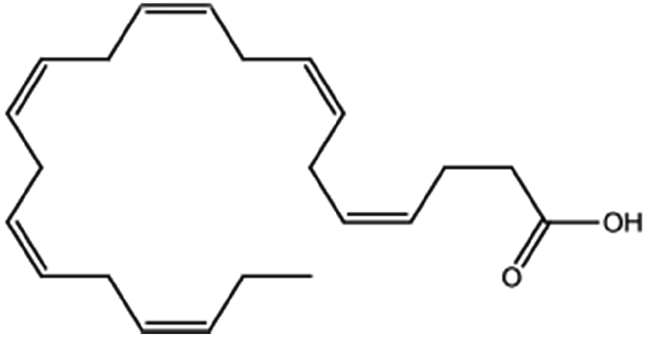
|
328.5 | 4.89 | 6.75 | Doxorubicin70 |
| Hexadecylphosphate |
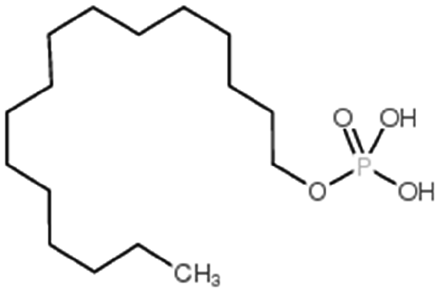
|
320.4 | 6.38 | Doxorubicin158 | |
| Thymopentin159 | |||||
| Tobramycin160 | |||||
| Linoleic acid |
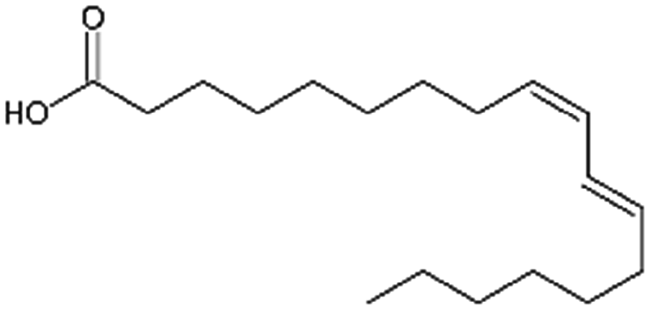
|
280.5 | 4.77 | 6.8 | Vancomycin64 |
| N,N-Dipalmitoyl-L-lysine |
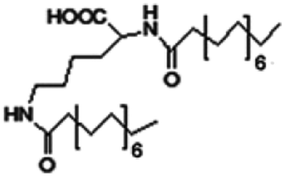
|
Colistin156 | |||
| Oleic acid (sodium oleate also used) |
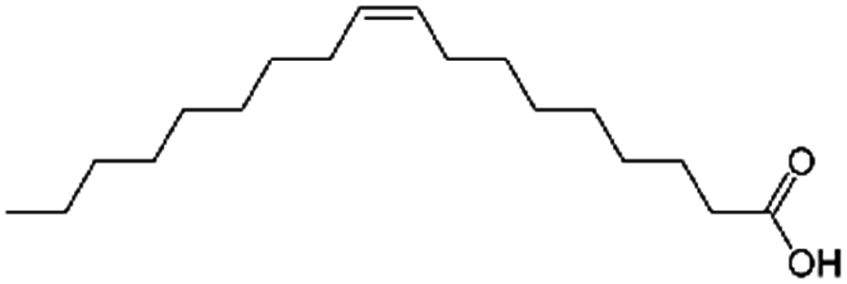
|
282.5 | 5 | 6.78 | AZD2811 (ref. 38 and 89) |
| Berberine161 | |||||
| Desmopressin77,86 | |||||
| Dorzolamide81 | |||||
| Doxorubicin106,121 | |||||
| Insulin86,102,162 | |||||
| Leuprolide86,94,101 | |||||
| Lumefantrine48 | |||||
| Lycobetaine163 | |||||
| Lysozyme57,58 | |||||
| Octreotide96 | |||||
| OZ439 (ref. 43 and 154) | |||||
| Polymyxin B78 | |||||
| Salmon calcitonin85 | |||||
| Vincristine164 | |||||
| Pamoic acid (disodium pamoate also used) |
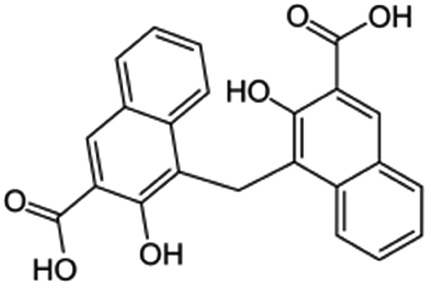
|
388.4 | 2.68 | 6.17 | AZD2811 (ref. 38) |
| Bovine serum albumin165 | |||||
| Cinnarizine47 | |||||
| Clozapine47 | |||||
| Donepezil166 | |||||
| Insulin165 | |||||
| Leuprolide165 | |||||
| Polymyxin B78 | |||||
| Sodium acetate |
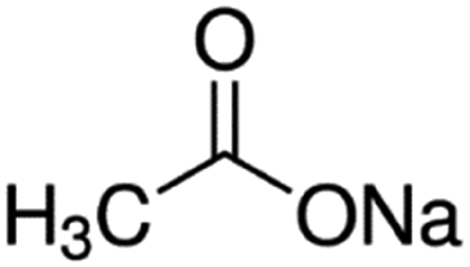
|
82 | 4.7 | −0.2 | Doxorubicin61 |
| Propanolol61 | |||||
| Quinidine sulfate61 | |||||
| Verapamil61 | |||||
| Sodium cholesteryl sulfate |
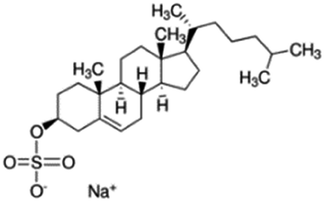
|
466.3 | 3.13 | 4.2 | Colistin156 |
| Sodium decanesulfonate (SDES) |
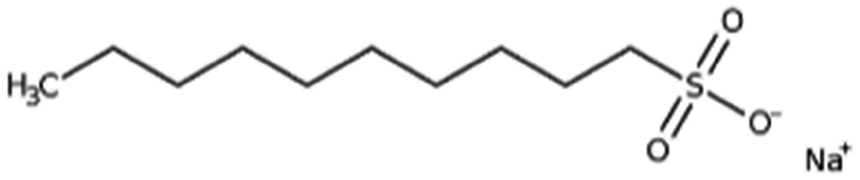
|
244.3 | 3.75 | Doxorubicin116 | |
| Sodium deoxycholate |
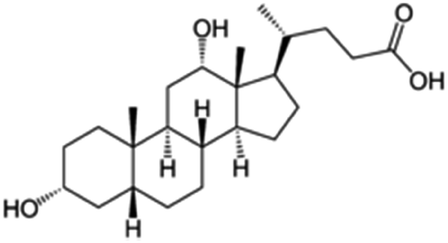
|
392.6 | 4.65 | 3.8 | AZD2811 (ref. 38) |
| Bovine serum albumin165 | |||||
| Ciprofloxacin88 | |||||
| Insulin80,93,165 | |||||
| Lanreotide98 | |||||
| Leuprolide165 | |||||
| Mitoxantrone diHCl79 | |||||
| Octreotide9,96 | |||||
| Papain97 | |||||
| Salmon calcitonin85 | |||||
| Sodium docusate (AOT, sodium dioctyl sulfosuccinic acid, sodium bis-2- ethylhexyl-sulfosuccinate) |
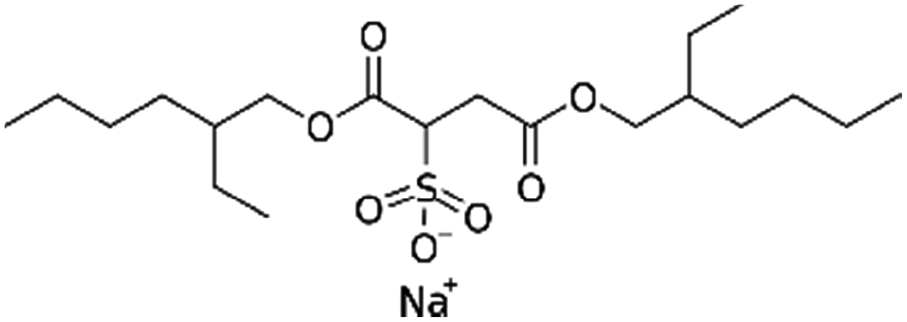
|
444.6 | −0.75 | 5.2 | α-Chymotrypsin167 |
| Atazanavir28 | |||||
| AZD2811 (ref. 38 and 89) | |||||
| Bevacizumab168 | |||||
| Bovine serum albumin56,169 | |||||
| Cisplatin83 | |||||
| Concanavalin A167 | |||||
| Desmopressin45,77,86,170 | |||||
| Doxorubicin116 | |||||
| Gentamycin82,117–119,171 | |||||
| Irinotecan29 | |||||
| Lanreotide98 | |||||
| Leuprolide45,84,86,110,172 | |||||
| Lysozyme56 | |||||
| Minocycline173 | |||||
| Mtb8.4 (ref. 174) | |||||
| Naloxone117 | |||||
| Naltrexone117 | |||||
| Octreotide9 | |||||
| r-met-HuGdNF56 | |||||
| Tobramycin175 | |||||
| Trypsin167 | |||||
| Vancomycin176 | |||||
| Sodium dodecyl benzenesulfonate (SDBS) |
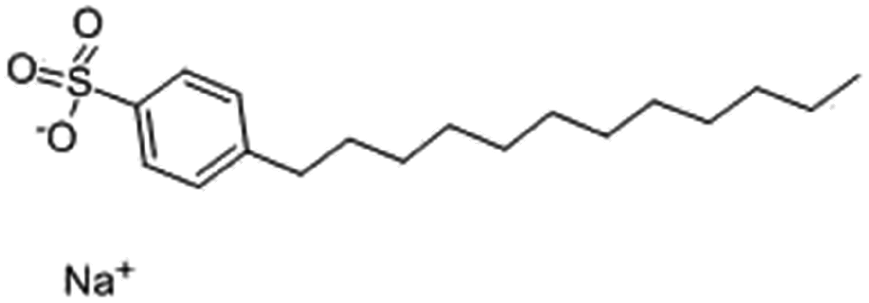
|
348.5 | −1.7 | 3.73 | Polymyxin B78 |
| Sodium dodecyl sulfate (sodium lauryl sulfate) |
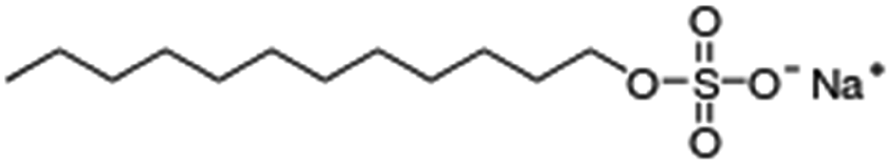
|
288.4 | −1.5 | 1.6 | Bovine serum albumin56 |
| Desmopressin77,86 | |||||
| Dorzolamide81 | |||||
| IGG-Fab fragment90 | |||||
| Insulin49,58,84,86,111,177,178 | |||||
| Irinotecan29 | |||||
| Leuprolide11,84,86 | |||||
| Lysozyme18,56,57 | |||||
| Melittin109 | |||||
| Octreotide92,96 | |||||
| Polymyxin B78 | |||||
| r-met-HuGdNF56 | |||||
| Sodium laurate (sodium dodecanoate) |
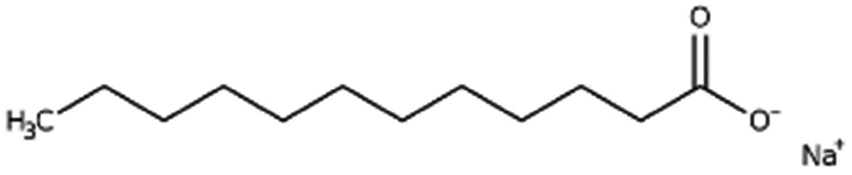
|
222.3 | 4.95 | 5.3 | Bovine serum albumin165 |
| Insulin165 | |||||
| Leuprolide165 | |||||
| Sodium n-octadecyl sulfate (sodium stearyl sulfate) |
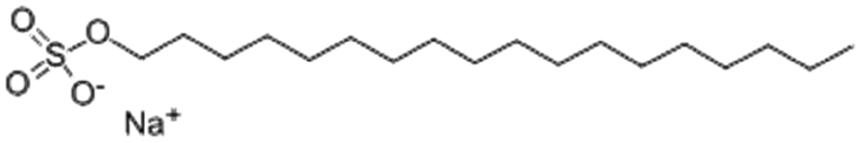
|
372.5 | 6.8 | Desmopressin77 | |
| Lanreotide98 | |||||
| Sodium stearate (stearic acid also used) |
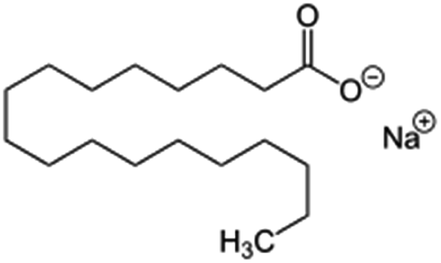
|
306.5 | 4.7 | 8.23 | Desmopressin77 |
| Doxorubicin61 | |||||
| Propanolol61 | |||||
| Quinidine sulfate61 | |||||
| Verapamil61 | |||||
| Sodium stearoyl glutamate (SSG) |
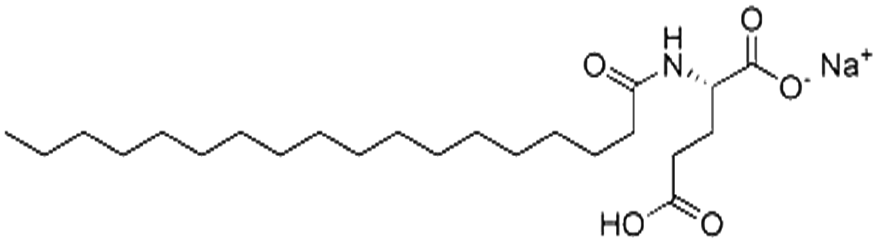
|
435.6 | 6.3 | Bovine serum albumin165 | |
| Insulin165 | |||||
| Leuprolide165 | |||||
| Sodium taurodeoxycholate (STDC) |
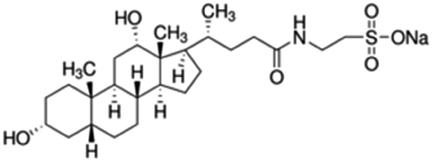
|
499.7 | −0.94 | 4.5 | Doxorubicin72,95,116 |
| Idarubicin95 | |||||
| Sodium tetradecyl sulfate |
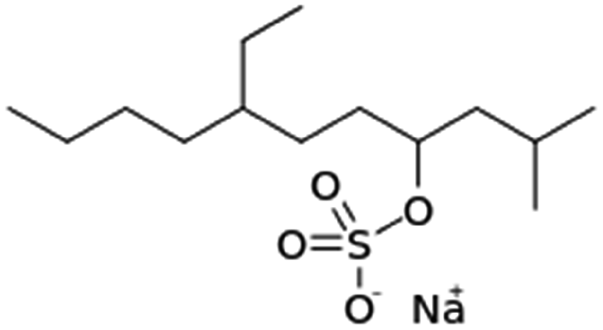
|
316.4 | −1.1 | 5.04 | Doxorubicin95 |
| Idarubicin95 | |||||
| Sodium tripolyphosphate |
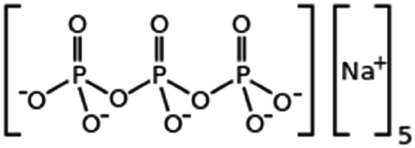
|
367.9 | 0.89 | −1.9 | Irinotecan29 |
| Taurocholic acid (sodium taurocholate also used) |
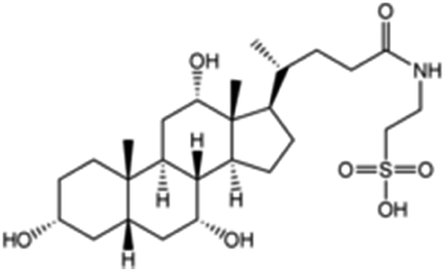
|
515.7 | 1.4 | 0.79 | Bovine serum albumin56 |
| Lanreotide98 | |||||
| Lysozyme56 | |||||
| r-met-HuGdNF56 | |||||
| IGG-Fab fragment90 | |||||
| Vitamin E (α-tocopherol) succinate |
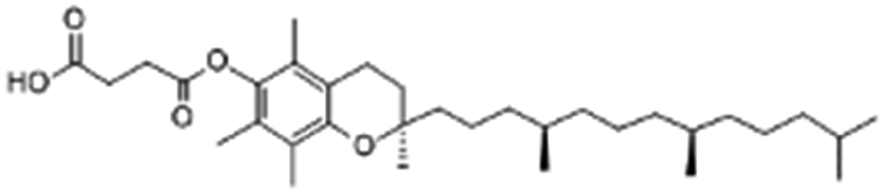
|
530.8 | 4 | 10.2 | Doxorubicin105 |
The most common cations in the HIP literature are quaternary amines and alkylamines (see Table 2). Quaternary amines are permanently charged, so complexation is possible over a wider range of pH values than primary, secondary, or tertiary amines. The permanent charge is usually cytotoxic, and using quatamines adds toxicity to otherwise nontoxic formulations.39 A wide variety of quaternary amines is commercially available, with varying lengths and numbers of alkyl tails that lead to an easily tuneable range of hydrophobicities.40 Researchers have recently reported efforts to synthesize arginine-based cationic surfactants for HIP, which should be both biodegradable and non-cytotoxic.41
| Name | Structure | Mol. wt. | pKa | logP |
Paired with |
|---|---|---|---|---|---|
| Arginine-hexadecanoyl ester (AHE) |
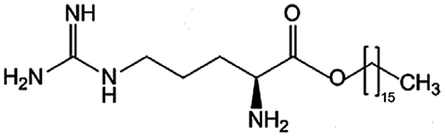
|
398.6 | 0.19 | Daptomycin41 | |
| Heparin41 | |||||
| Arginine-nonyl ester (ANE) |
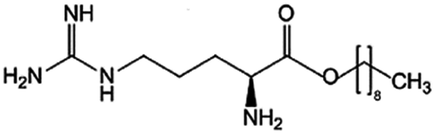
|
300.5 | −0.06 | Daptomycin41 | |
| Heparin41 | |||||
| Benethamine(N-benzyl-2-phenylethanamine) |
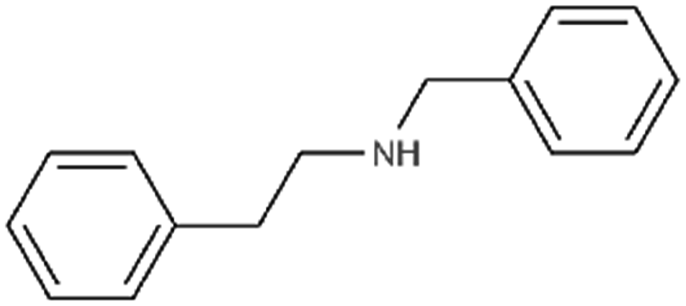
|
211.3 | 3.6 | Retinoic acid65,108,115 | |
| Chitosan |
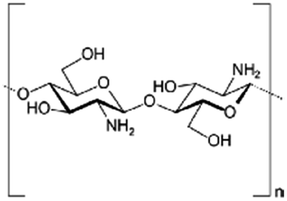
|
Varies | Insulin53 | ||
| Dodecylamine (laurylamine) |

|
185.3 | 10.6 | 5.2 | Retinoic acid46,107,114 |
| Hexadecyl trimethylammonium(cetrimonium) bromide (CTAB) |

|
364.5 | — | 2.69 | Ovalbumin39 |
| Pemetrexed103 | |||||
| Poly(I:C)39 | |||||
| Maprotiline |
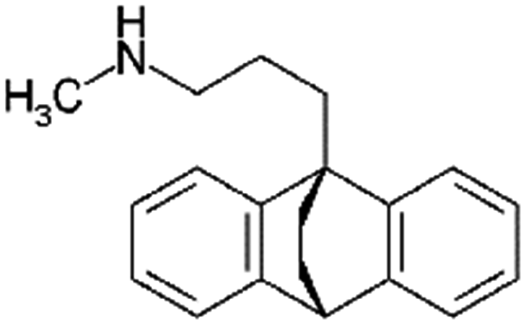
|
277.4 | 10.5 | 5.1 | Retinoic acid115 |
| N α-Deoxycholyl-L-lysyl-methylester |
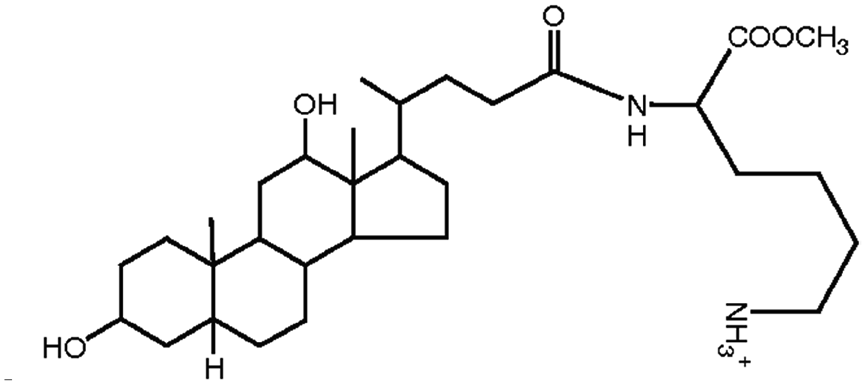
|
534.8 | 3.8 | Pemetrexed4 | |
| N,N′-Dibenzyl ethylenediamine(benzathine) |
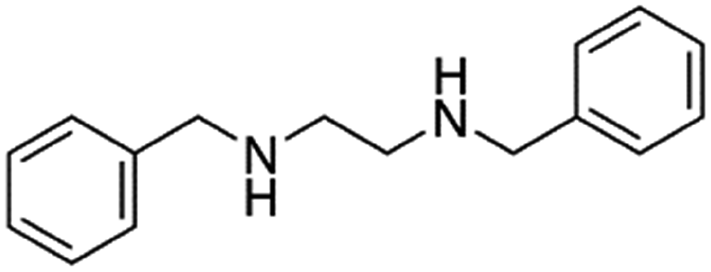
|
240.3 | 2.86 | α-Lipoic acid47 | |
| N,N-Dimethyl dodecylamine (DDA) |
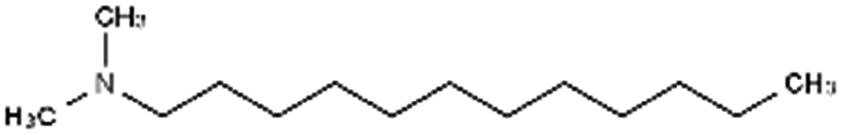
|
213.4 | 9.97 | 5.91 | Am80 (ref. 40) |
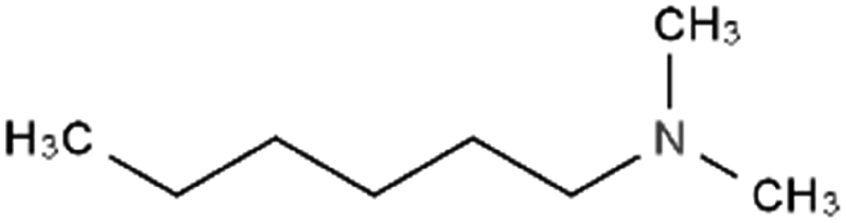
| N,N-Dimethyl hexylamine |
|
129.2 | 10.4 | 2.72 | Am80 (ref. 40) |
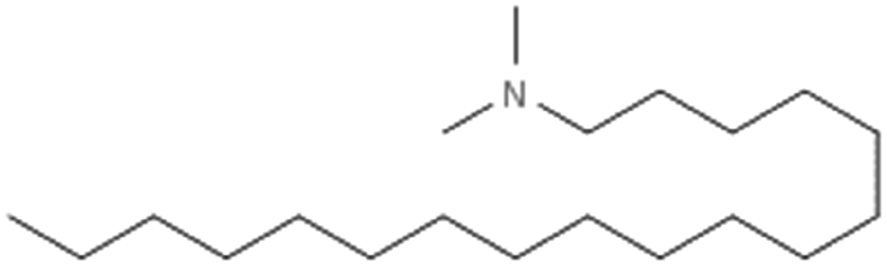
| N,N-Dimethyl octadecylamine(dimethyl stearamine) |
|
297.6 | 8.8 | Am80 (ref. 40) | |
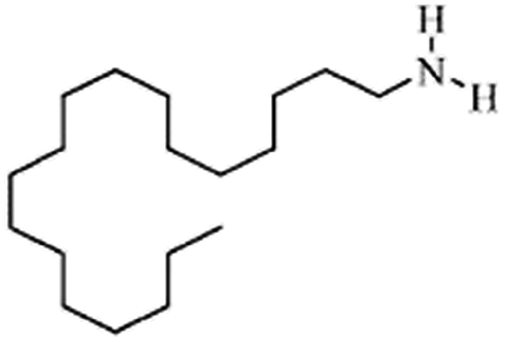
| Stearylamine(octadecylamine) |
|
269.5 | 10.7 | 7.7 | Retinoic acid46,65,107,114 |
| Tetrabutyl ammonium bromide (TBAB) |
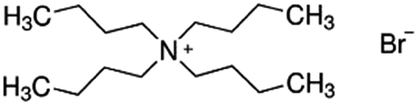
|
322.4 | — | 2.1 | Bromothymol blue66 |
| Rose bengal66 | |||||
| Tetraheptyl ammonium bromide (THA) |
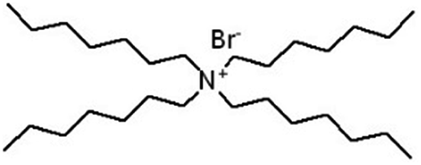
|
490.7 | — | 8.16 | Isoniazid methanesulfonate179 |
| Tetrahexyl ammonium bromide |
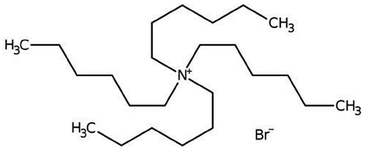
|
434.6 | — | 6.16 | Bromothymol blue66 |
| Rose bengal66 | |||||
| Tetraoctyl ammonium bromide (TOAB) |
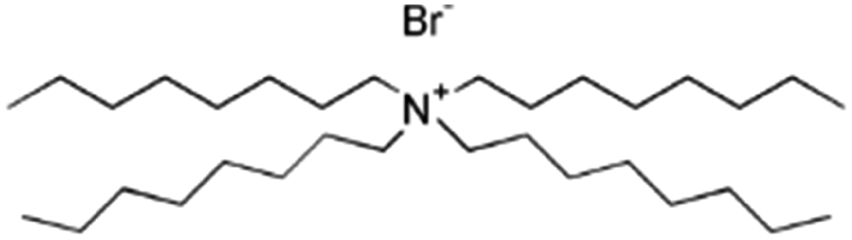
|
546.7 | — | 9.16 | Bromothymol blue66 |
| Rose bengal66 | |||||
| Tetrapentyl ammonium bromide (TPA) |
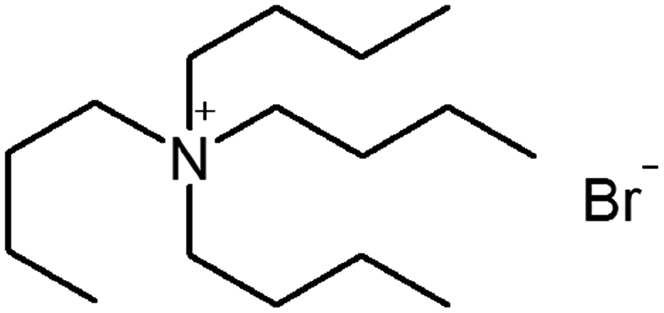
|
378.5 | — | 4.14 | Isoniazid methanesulfonate179 |
| Triethylamine (TEA) |
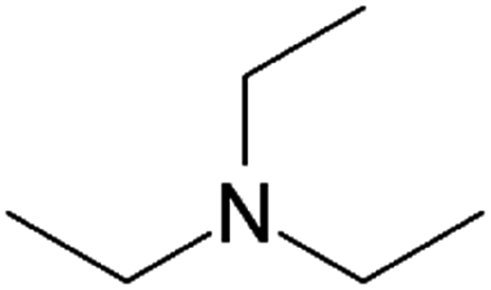
|
101.2 | 10.8 | 1.65 | Retinoic acid46,65,107,114 |
Small molecules. Many small molecule drugs have only one ionic group. Depending on the pKa of the ionic group and the drug's solubility, HIP is relatively straightforward and can be carried out in water. In a typical ‘pre-forming’ scenario for hydrophilic small molecules, the drug and counterion are each dissolved in water and mixed to form a precipitate.11,42 It is worth noting that small molecules with ionizable groups may be manufactured either as a salt or in the free acid/base form. The free acid/base is usually less soluble in water than the salt, but might not be hydrophobic enough for a desired encapsulation strategy.43 Since species must be charged in order to ion pair, salt forms of the drug and hydrophobic counterion may be preferred. When the drug is manufactured in the free acid/base form, conversion to a readily-dissociating salt form (e.g. mesylate, ammonium, or sodium) before HIP may assist complexation. A drawback of this approach is that it increases the solution's overall ionic strength, which can drive decomplexation and drug release from a delivery vehicle by ion exchange.38,44,45 Researchers should examine the effect of ionic strength on their specific systems to determine if one charge equivalent of soluble counterions such as sodium or ammonium will noticeably affect release.
Some ionic small molecule drugs such as lumefantrine (for structure, see Table 3) are already hydrophobic, so it is not possible to form an aqueous solution as the starting point for HIP. Hydrophobic ion pairing an already-hydrophobic drug can be useful – for example, to decrease drug crystallinity30,46,47 – but the complex formation is more challenging. Lumefantrine's tertiary amine has a pKa of 8.7, but the drug's logP of 9.2 severely limits its ability to dissolve, and the amine to become charged, in water.48 Dissolving lumefantrine free base in a nonpolar solvent such as tetrahydrofuran guarantees dissolution, but the extent of the amine's charge is more difficult to control and measure in a non-aqueous environment. As mentioned above, conversion to a salt form before complexation may be useful (Table 4).
| Name | Structure/etc. | Paired with | Formulation technique |
|---|---|---|---|
| α-Chymotrypsin | 25 kDa protein, 241 residues, pI: 8.75 | Sodium docusate167 | Solvent evaporation with polymethyl methacrylate, polystyrene, or poly(vinyl acetate)167 |
| α-Lipoic acid |
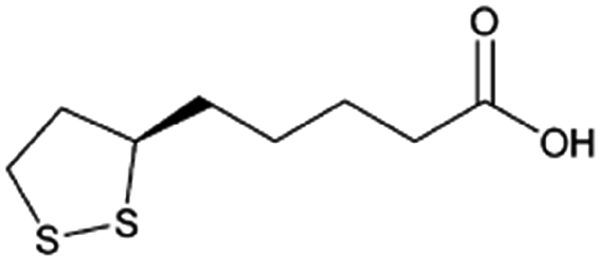
|
N,N′-Dibenzylethylene diamine (DBDA),47 note: included pamoic acid to frustrate αLA:DBDA recrystallization and improve encapsulation |
PLA-b-PEG NPs by Flash NanoPrecipitation, in situ HIP47 |
| Am80 |
|
N,N-Dimethyldodecyl amine (DDA)40 | Block copolymer micelles by evaporation-sonication40 |
| N,N-Dimethylhexyl amine40 | |||
| N,N-Dimethyloctadecyl amine40 | |||
| Atazanavir |
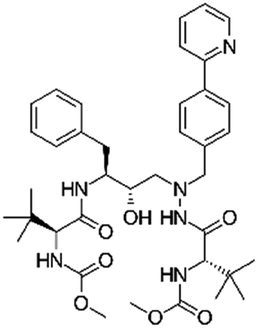
|
2-Naphthalene sulfonic acid28 | SEDDS28 |
| Sodium docusate (AOT)28 | |||
| Atenolol |
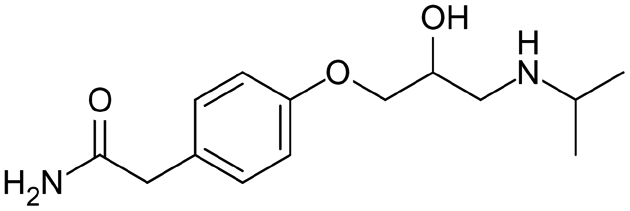
|
Brilliant blue FCF155 | PLGA NPs by nanoprecipitation155 |
| AZD2811 |
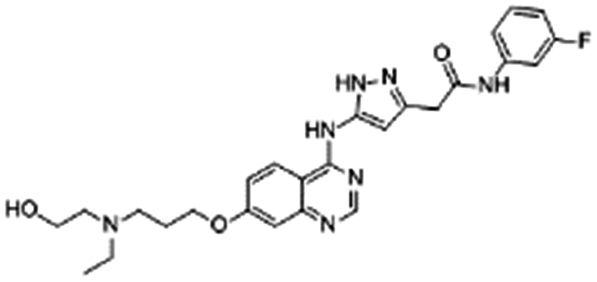
|
Oleic acid38,89 | Oil in water (o/w) nanoemulsification solvent extraction to form PLA-PEG NPs using in situ HIP38,89 |
| 1-Hydroxy-2-naphthoic acid38,89 | |||
| Cholic acid38,89 | |||
| Sodium deoxycholate38 | |||
| Docusate sodium38,89 | |||
| Pamoic acid38 | |||
| Berberine |
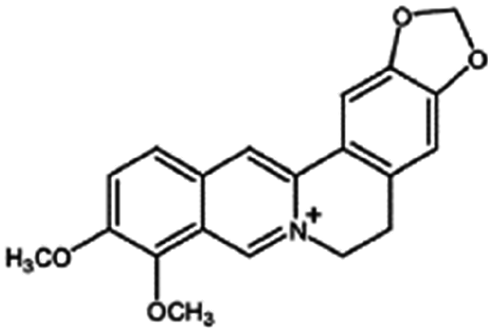
|
Oleic acid161 | Liquid crystalline nanoparticulates by a hydrotrope method161 |
| Bevacizumab | 149 kDa antibody | Docusate sodium168 | Lipid coacervation168 |
| Bovine serum albumin (BSA) | 66.5 kDa protein, 583 residues, pI: 4.7 | Cholic acid56 | Double emulsion56 |
| CM-PEG56 | Single emulsion56 | ||
| Sodium dodecyl sulfate56 | |||
| Taurocholic acid56 | |||
| Sodium docusate56 | Double emulsion56 | ||
| Single emulsion56 | |||
| SEDDS169 | |||
| Dextran sulfate91 | Solid in oil in water (S/O/W) to form PLGA NPs91 | ||
| Sodium deoxycholate165 | SEDDS165 | ||
| Sodium laurate165 | |||
| Sodium stearoyl glutamate165 | |||
| Pamoic acid disodium165 | |||
| Bromothymol blue |

|
Tetrabutylammonium bromide66 | Encapsulated into polystyrene microparticles using compressed carbon dioxide66 |
| Tetrahexylammonium bromide66 | |||
| Tetraoctylammonium bromide66 | |||
| Chlorhexidine |
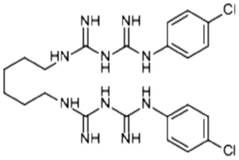
|
Losartan152 | Nanoprecipitation152 |
| Cinnarizine |
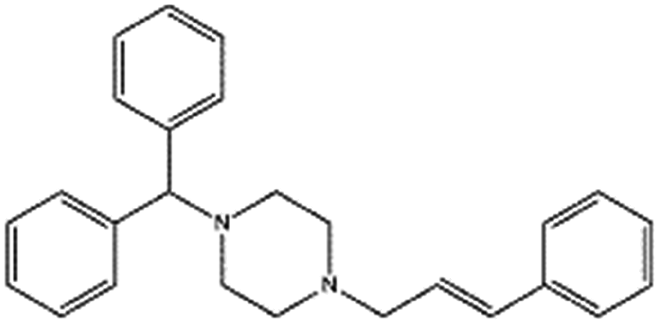
|
Pamoic acid,47 note: Also unsuccessfully tried camphor-10 sulfonic acid (micellized), cinnamic acid, palmitic acid, and oleic acid | PLA-b-PEG NPs by Flash NanoPrecipitation, in situ HIP47 |
| Ciprofloxacin |
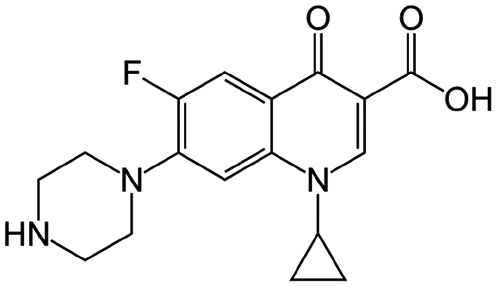
|
Sodium deoxycholate88 | Oil-in-water (o/w) submicron emulsion88 |
| Cisplatin |
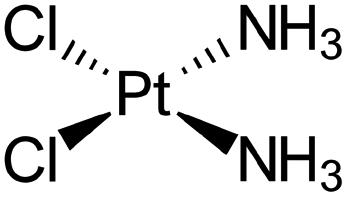
|
Sodium docusate83 | Stearic acid coacervation83 |
| Clozapine |
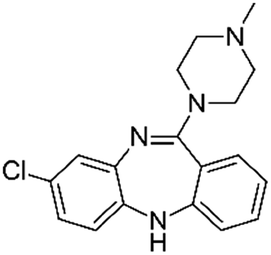
|
Pamoic acid47 | PLA-b-PEG NPs by Flash NanoPrecipitation, in situ HIP47 |
| Colistin |
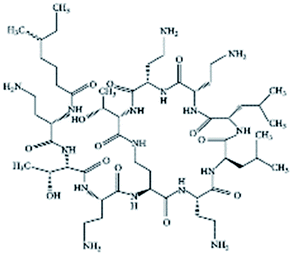
|
Cholesteryl hemisuccinate156 | PLA NPs by emulsion evaporation156 |
| N,N-Dipalmitoyl-L-lysine156 | |||
| Sodium cholesteryl sulfate156 | |||
| Concanavalin A | 104–112 kDa protein (tetramer), pI: 4.5–5.5 | Sodium docusate167 | Solvent evaporation with polymethyl methacrylate, polystyrene, or poly(vinyl acetate)167 |
| Dalargin |
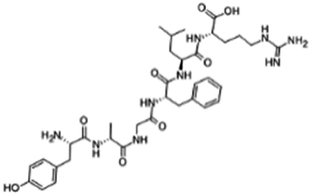
|
Dextran sulfate104 | PLGA-PEG NPs by S/O/W emulsion104 |
| Daptomycin |
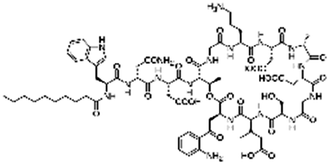
|
Arginine-hexadecanoyl ester41 | N/A; proof-of-concept HIP using novel cationic surfactants demonstrates precipitation and increased logP41 |
| Arginine-nonyl ester41 | |||
| Desmopressin |
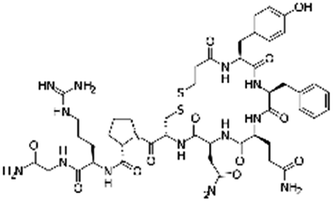
|
Oleic acid77 | SEDDS45,77,86,170 |
| Sodium docusate45,77,86,170 | |||
| Sodium dodecyl sulfate77,86 | |||
| Sodium stearate77 (note: less effective than SDS, AOT, and oleate) | |||
| Sodium stearyl sulfate77 (note: less effective than SDS, AOT, and oleate) | |||
| Dexamethasone valine valine prodrug |
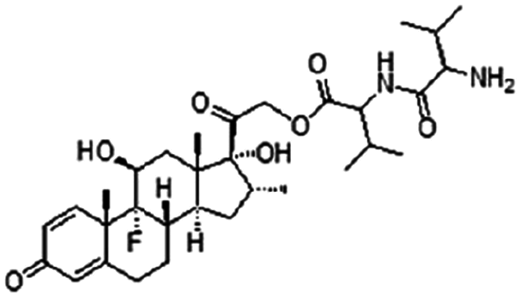
|
Dextran sulfate120 | PLGA NPs by S/O/W emulsion120 |
| Donepezil |
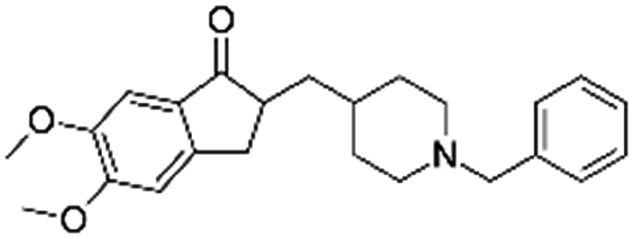
|
Pamoic acid166 | High pressure homogenization with D-α-tocopherol polyethylene glycol 1000 succinate166 |
| Dorzolamide |
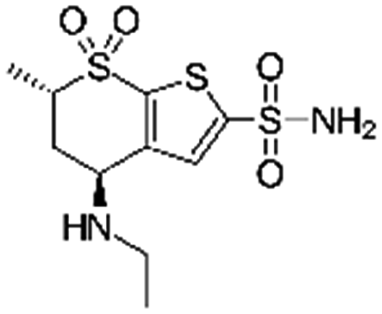
|
Oleic acid81 | PLGA NPs or PEG3-PSA microparticles by S/O/W emulsion81 |
| Sodium dodecyl sulfate81 | |||
| Doxorubicin |
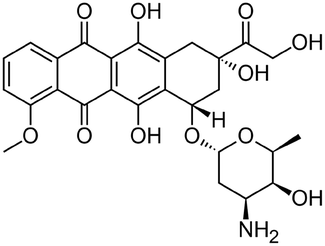
|
Alginic acid61 | Microemulsion by stearic acid coacervation61 |
| Cholesteryl hemisuccinate112 | Thin film dispersion112 | ||
| Dextran sulfate61,95 | Microemulsion by stearic acid coacervation61 | ||
| Warm wax microemulsion solvent evaporation95 | |||
| Dioleoyl phosphatidic acid (DOPA)71 | PLA-b-PEG NPs by nanoprecipitation71 | ||
| Docosahexaenoic acid70 | SLNs by hot melt ultrasound emulsification70 | ||
| Hexadecylphosphate158 | SLNs by warm oil-in-water microemulsion with stearic acid and taurocholate sodium158 | ||
| Hyaluronic acid153 | Thin film dispersion by lipid film hydration with suspended HIP complex and homogenization153 | ||
| Oleic acid106,121 | 70 °C high-pressure homogenization121 | ||
| High-pressure film homogenization106 | |||
| Sodium acetate61 | Microemulsion by stearic acid coacervation61 | ||
| Sodium alginate61 | Microemulsion by stearic acid coacervation61 | ||
| Sodium decanesulfonate116 | Microemulsion by stearic acid coacervation116 | ||
| Sodium docusate116 | Microemulsion by stearic acid coacervation116 | ||
| Sodium stearate61 | Microemulsion by stearic acid coacervation61 | ||
| Sodium taurodeoxycholate72,95,116 | Warm wax microemulsion solvent evaporation95 | ||
| Microemulsion by stearic acid coacervation116 | |||
| Microemulsion by shear and ultrasonic homogenization after drying from molten stearyl alcohol72 | |||
| Sodium tetradecyl sulfate95 | Warm wax microemulsion solvent evaporation95 | ||
| Vitamin E succinate105 | SLNs by hot melt ultrasound emulsification105 | ||
| Gefitinib |
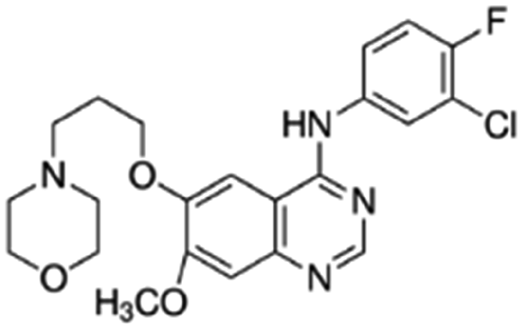
|
Dioleoyl phosphatidic acid (DOPA)30 | Nanoprecipitation with doxorubicin-conjugated PLA-b-PEG NPs30 |
| Gentamicin |
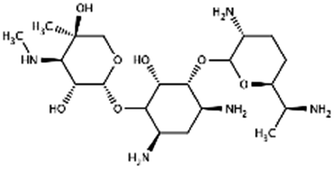
|
Sodium docusate82,117–119,171 | PLA microparticles by precipitation with compressed antisolvent117,171 |
| Microparticles by PCA using stabilizer poly(methyl vinyl ether-co-maleic anhydride)118 | |||
| PCA with no stabilizer82,119 | |||
| PLGA NPs by emulsion solvent evaporation82,119 | |||
| Heparin |
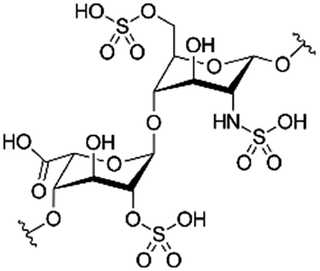
|
Arginine-hexadecanoyl ester41 | N/A; proof-of-concept HIP using novel cationic surfactants demonstrates precipitation and increased logP41 |
| Arginine-nonyl ester41 | |||
| Idarubicin |
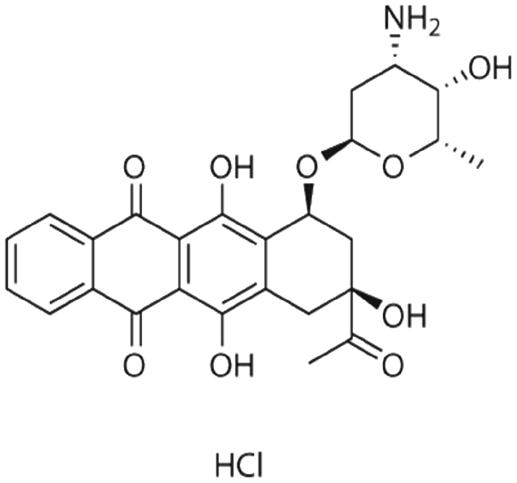
|
Dextran sulfate95 | Warm wax microemulsion solvent evaporation95 |
| Sodium taurodeoxycholate95 | |||
| Sodium tetradecyl sulfate95 | |||
| IGG-Fab fragment | 48 kDa protein | Sodium dodecyl sulfate90 | Modified nanoprecipitation90 |
| Taurocholic acid90 | S/O/W PLGA NPs90 | ||
| Dextran sulfate90 | |||
| Insulin | 5.8 kDa peptide, 51 residues (6 cationic and 6 anionic), pI: 5.3 | Cholic acid157 | Reverse micelle-double emulsion using palmitic and stearic acid157 |
| Chitosan53 | Homogenization and stabilization with SDS53 | ||
| Dimyristoyl phosphatidyl glycerol42 | SNEDDS42 | ||
| Oleic acid58,102,162 | S/O/W emulsion58 | ||
| PLGA NPs by emulsion solvent diffusion102,162 | |||
| Pamoic acid disodium165 | SEDDS165 | ||
| Sodium laurate165 | |||
| Sodium stearoyl glutamate165 | |||
| Sodium deoxycholate80,93,165 | PLGA NPs by emulsion solvent diffusion93 | ||
| S/O/W emulsion80 | |||
| SEDDS165 | |||
| Sodium docusate84,86 | SEDDS86 | ||
| Stearic acid coacervation84 | |||
| Sodium dodecyl sulfate49,84,111,177,178 | Stearic acid coacervation84 | ||
| PLGA NPs by emulsion solvent diffusion111,177,178 | |||
| Electrospray with stearic or pamoic acid49 | |||
| Irinotecan |
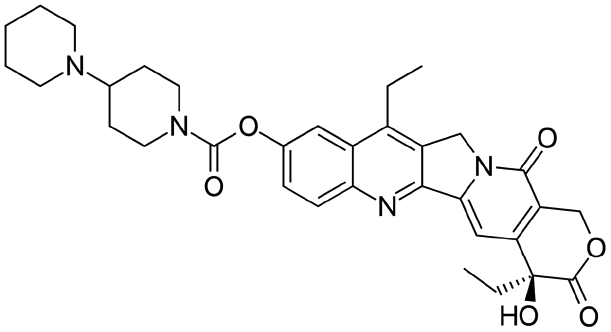
|
Sodium docusate29 | PEG-b-PLGA NPs via water/oil/water double emulsion; in situ HIP29 |
| Sodium dodecyl sulfate29 | |||
| Sodium tripolyphosphate29 | |||
| Isoniazid methanesulfonate |
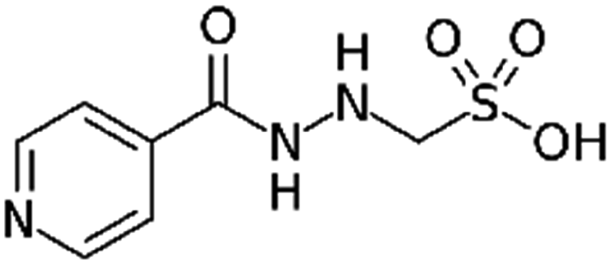
|
Tetraheptylammonium bromide179 | Precipitation with compressed antisolvent (PCA)179 |
| Tetrapentylammonium bromide179 | |||
| Lanreotide |
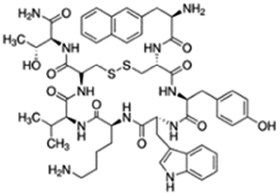
|
Sodium deoxycholate98 | SNEDS98 |
| Sodium docusate98 | |||
| Sodium stearyl sulfate98 | |||
| Taurocholic acid98 | |||
| Leuprolide |
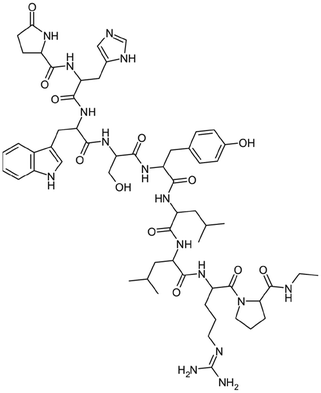
|
Oleic acid94,101 | PLGA microspheres by O/W emulsion101 |
| SMEDDS94 | |||
| Sodium deoxycholate165 | SEDDS165 | ||
| Sodium laurate165 | |||
| Sodium stearoyl glutamate165 | |||
| Pamoic acid disodium165 | |||
| Sodium docusate45,84,86,172 | SEDDS45,86 | ||
| Stearic acid coacervation84 | |||
| Oligosaccharide ester microparticles by spray drying172 | |||
| Solid lipid nanoparticles and nanostructured lipid carriers by high pressure homogenization110 | |||
| Sodium dodecyl sulfate11,84 | Stearic acid coacervation84 | ||
| Hydrogen bonding complexation between polyacrylic acid and Pluronic F68 (ref. 11) | |||
| Sodium stearate87 | Solid lipid NPs by: solvent diffusion87 | ||
| Oil-in-oil (O/O) emulsion-evaporation87 | |||
| Loperamide |
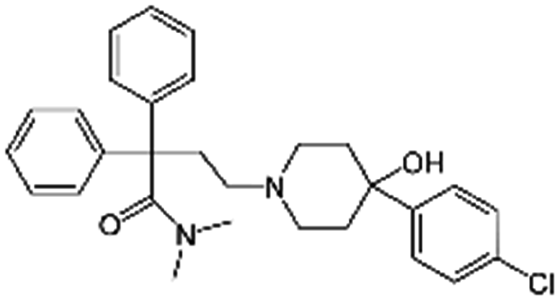
|
Dextran sulfate104 | PLGA-PEG NPs by S/O/W emulsion104 |
| Losartan |
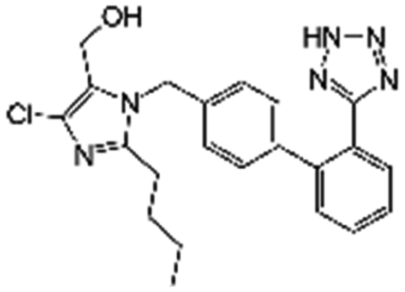
|
Chlorhexidine152 | Nanoprecipitation152 |
| Lumefantrine |
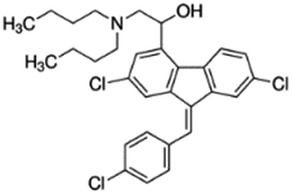
|
Oleic acid48 | SEDDS48 |
| Lycobetaine |
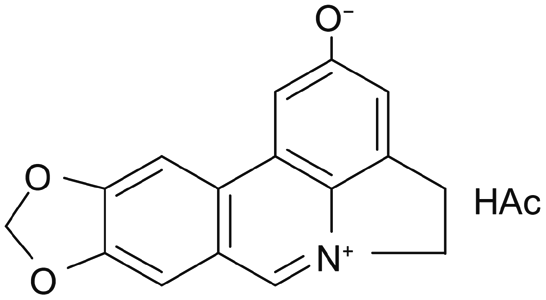
|
Oleic acid163 | Emulsion by lipid film hydration high-pressure homogenization163 |
| Lysozyme | 14.4 kDa protein, 129 residues, pI: 11.35 | Cholic acid56 | Double emulsion56 |
| Single emulsion56 | |||
| CM-PEG56 | Double emulsion56 | ||
| Single emulsion56 | |||
| Dextran sulfate59 | Emulsion solvent diffusion59 | ||
| Oleic acid57,58 | PLGA NPs by emulsion diffusion57 | ||
| S/O/W emulsion58 | |||
| Sodium docusate56 | Double emulsion56 | ||
| Single emulsion56 | |||
| Sodium dodecyl sulfate18,57 | PLGA NPs by emulsion diffusion57 | ||
| S/O/W emulsion: Polymer/lipid NPs18 | |||
| Taurocholic acid56 | Double emulsion56 | ||
| Single emulsion56 | |||
| Melittin | 2.8 kDa peptide, 26 residues, pI: 12.01 | Sodium dodecyl sulfate109 | PLGA nanoparticles by emulsion solvent diffusion109 |
| Minocycline |
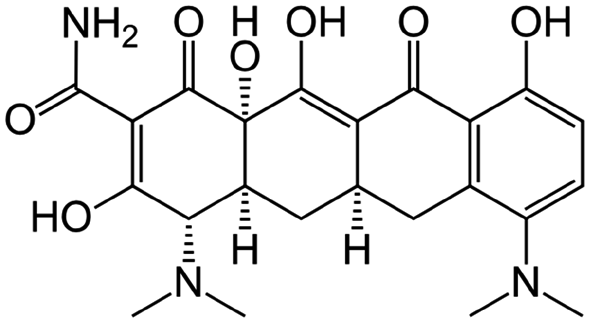
|
Sodium docusate173 | PLGA NPs by emulsion-solvent-diffusion173 |
| Mitoxantrone dihydrochloride |

|
Sodium deoxycholate79 | Nanoprecipitation79 |
| Mtb8.4 | Protein, TB antigen, pI: 6.3 | Sodium docusate174 | PLG microspheres by emulsification174 |
| Naloxone |
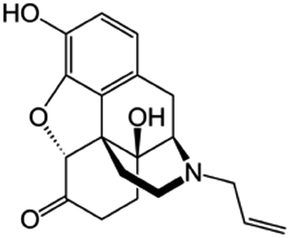
|
Sodium docusate117 | PLA microparticles by precipitation with compressed antisolvent117 |
| Naltrexone |
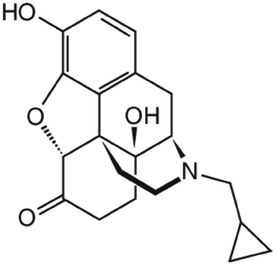
|
Sodium docusate117 | PLA microparticles by precipitation with compressed antisolvent117 |
| Octreotide |
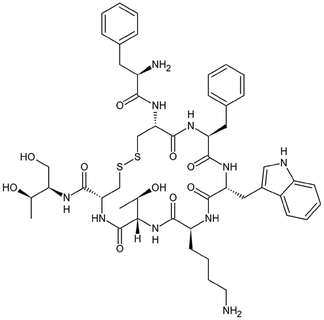
|
Dextran sulfate92 | S/O/W emulsion92 |
| Oleic acid96 | SNEDDS96 | ||
| Sodium decanoate9,96 | SNEDDS96 | ||
| SEDDS9 | |||
| Sodium deoxycholate9,96 | SNEDDS96 | ||
| SEDDS9 | |||
| Sodium docusate9 | SEDDS9 | ||
| Sodium dodecyl sulfate92,96 | S/O/W emulsion92 | ||
| SNEDDS96 | |||
| Ovalbumin (OVA) | 43 kDa protein, 385 residues, pI: 5.19 | Cetrimonium bromide (CTAB)39 | pH-sensitive polyketal microparticles by single emulsion39 |
| OZ439 mesylate (artefenomel) |
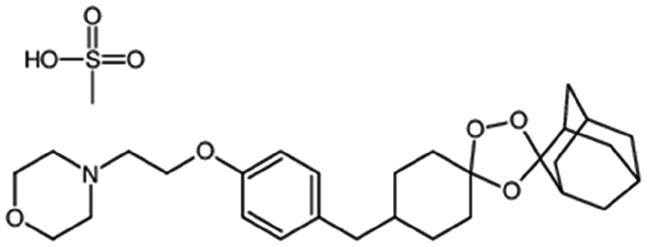
|
Sodium oleate43,154 | HPMCAS NPs by Flash NanoPrecipitation; in situ HIP43,154 |
| Papain | 23.4 kDa protein, 212 residues, pI: 8.8–9.6 | Sodium deoxycholate97 | SEDDS97 |
| Pemetrexed |
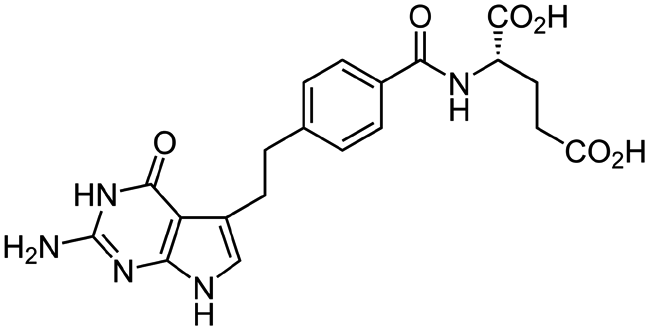
|
Cetrimonium bromide (CTAB)103 | Lyotropic liquid crystalline nanoparticles by homogenization (in situ HIP)103 |
| N α-Deoxycholyl-L-lysyl-methylester4 | W/O/W emulsion4 | ||
| Polymyxin B |
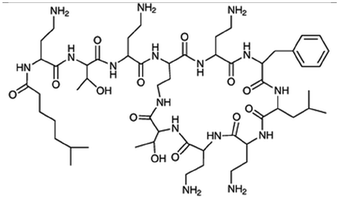
|
Oleic acid sodium salt78 | PCL-b-PEG NPs by Flash NanoPrecipitation (FNP), in situ HIP,78 note: sodium decanoate, myristate, deoxycholate, 2-naphthalenesulfonate, 1-heptanesulfonate, 1-octane-sulfonate, and 1-decanesulfonate formed a precipitate when mixed with polymyxin B at 1:1 charge ratio but did not form NPs by FNP. Sodium hexanoate, benzenesulfonic acid, camphorsulfonic acid, and 1,2-ethanesulfonate did not form a precipitate when mixed with polymyxin B at 1:1 charge ratio. |
| Pamoic acid sodium salt78 | |||
| Sodium dodecyl sulfate78 | |||
| Sodium dodecyl benzenesulfonate78 | |||
| Poly(inosinic acid)-poly (cytidylic acid) (poly(I:C)) |
Double-stranded RNA analog, TLR3 agonist | Cetrimonium bromide (CTAB)39 | pH-sensitive polyketal microparticles by single emulsion39 |
| Propranolol |
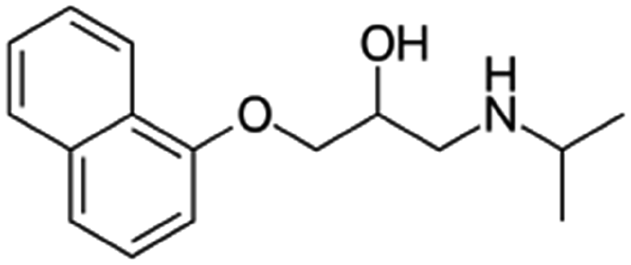
|
Alginic acid61 | Microemulsion by stearic acid coacervation61 |
| Dextran sulfate61 | |||
| Sodium acetate61 | |||
| Sodium stearate61 | |||
| Quinidine sulfate |
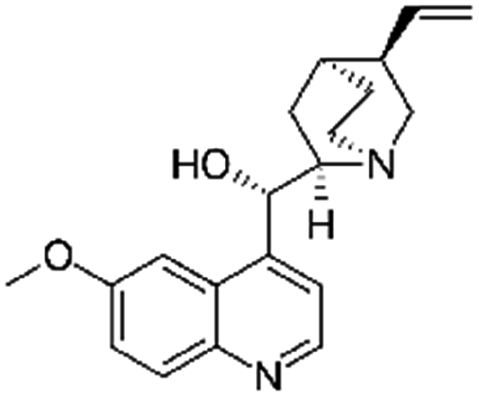
|
Alginic acid61 | Microemulsion by stearic acid coacervation61 |
| Dextran sulfate61 | |||
| Sodium acetate61 | |||
| Sodium stearate61 | |||
| r-met-HuGdNF | Recombinant methionyl human Glial-cell line derived neurotrophic factor | Cholic acid56 | Double emulsion56, single emulsion56 |
| CM-PEG56 | |||
| Sodium docusate56 | |||
| Sodium dodecyl sulfate56 | |||
| Taurocholic acid56 | |||
| Retinoic acid |
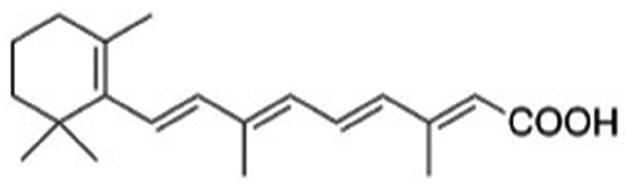
|
Benethamine65,108,115 | Hot melt homogenization using ultrasound emulsification41–43,65,108,115 |
| Laurylamine46,107,114 | |||
| Maprotiline (both HCl and free base)115 | |||
| Stearylamine46,65,107,114 | |||
| Triethylamine46,65,107,114 | |||
| Rose bengal |
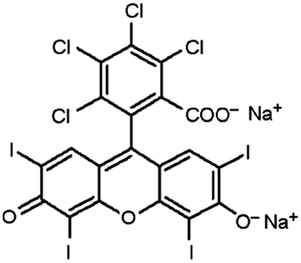
|
Tetrabutylammonium bromide66 | Encapsulated into polystyrene microparticles using compressed carbon dioxide to plasticize polystyrene MPs and allow diffusion in66 |
| Tetrahexylammonium bromide66 | |||
| Tetraoctylammonium bromide66 | |||
| Salmon calcitonin | 3.4 kDa peptide, 32 residues, pI: 8.86 | Dimyristoyl phosphatidyl glycerol (DMPG)85 | PLGA NPs by solvent diffusion85 |
| Oleic acid85 | |||
| Sodium deoxycholate85 | |||
| Thymopentin |
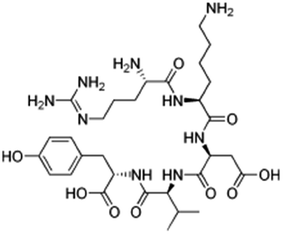
|
Hexadecylphosphate159 | Warm oil in water microemulsion159 |
| Tobramycin |
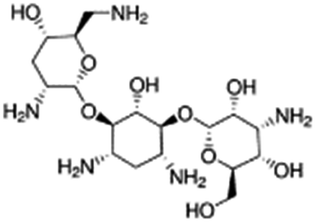
|
Hexadecylphosphate160 | Warm oil in water microemulsion160 |
| Sodium docusate175 | PLGA NPs by O/W emulsion175 | ||
| Trypsin | 23 kDa protein, 220 residues, pI: 10.1–10.5 | Sodium docusate167 | Solvent evaporation with polymethyl methacrylate, polystyrene, or poly(vinyl acetate)167 |
| Vancomycin |
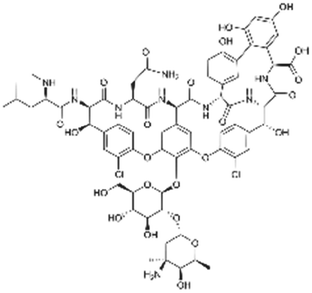
|
Linoleic acid64 | Hot homogenization and ultrasonication64 |
| Sodium docusate176 | SEDDS176 | ||
| Verapamil |
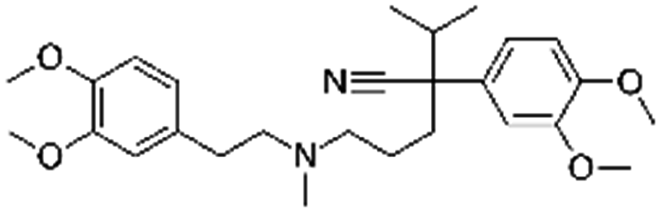
|
Alginic acid61 | Microemulsion by stearic acid coacervation61 |
| Dextran sulfate61 | |||
| Sodium acetate61 | |||
| Sodium stearate61 | |||
| Vincristine |
|
Oleic acid164 | High pressure homogenization164 |
| Name | Structure | MW, Da | pKa | logP |
Used to pair with: |
|---|---|---|---|---|---|
| Anions | |||||
| Alginic acid (sodium alginate also used) |
|
Varies | 1.5–3.5 | −1.5 | Doxorubicin61 |
| Propanolol61 | |||||
| Quinidine sulfate61 | |||||
| Verapamil61 | |||||
| Dextran sulfate |
|
Varies | <2 | Bovine serum albumin91 | |
| Dalargin104 | |||||
| Dexamethasone valine valine prodrug120 | |||||
| Doxorubicin61,95 | |||||
| Idarubicin95 | |||||
| IGG-Fab fragment90 | |||||
| Loperamide104 | |||||
| Lysozyme59 | |||||
| Octreotide92 | |||||
| Propanolol61 | |||||
| Quinidine sulfate61 | |||||
| Verapamil61 | |||||
| Hyaluronic acid |
|
Varies | 2.9 | −8.2 | Doxorubicin153 |
|
|||||
| Cations | |||||
| Chitosan |
|
Varies | Insulin53 | ||
Peptides. Many antibiotic peptides such as nisin and colistin (for structure, see Table 3) are cationic and strongly water soluble, with log
P values less than 0. Basic amino acid residues in the peptide (lysine, histidine, arginine) are positively charged at physiological or acidic pH and are sites for ion pairing. Some cationic peptide drugs are manufactured as sulfate salts that dissociate readily in water and do not have the same solubility and ionization challenges as hydrophobic small molecules. For peptides with only cationic charges such as polymyxin B, aqueous complexation with anionic surfactants is straightforward. Zwitterionic peptides are more challenging, however. If a peptide contains both cationic and anionic groups, it is possible that complexing only the cationic sites and leaving anionic sites charged and exposed (or vice versa) will impart sufficient hydrophobicity for the desired application. This is especially true when one kind of charged site significantly outnumbers the other, as in the case of a COOH-terminated peptide with five cationic sites. Complexing five out of the six charged sites with hydrophobic counterions may reduce water solubility enough to enable encapsulation.
When there are approximately the same number of cationic and anionic sites on a zwitterionic peptide, though, complexing only one charge may not be sufficient. It is preferable to use only one counterion species to complex a molecule, rather than adding both anionic and cationic hydrophobic counterions (which will invariably pair with each other and precipitate, complicating stoichiometry and adding difficult-to-separate insoluble salts to the system) in an attempt to complex every charged site. In this case, shifting the pH to turn off one type of charge is a valid approach. Consider insulin, a 5.8 kDa peptide with 51 residues, 6 of which are cationic and 6 anionic. Insulin has no net charge at its isoelectric point at pH 5.3. Researchers have reported shifting the solution pH either up or down from 5.3 to deprotonate insulin's basic residues or protonate its acidic residues, respectively.42,49–53 With only one type of charge, the peptide can then be hydrophobically ion paired (Fig. 3).
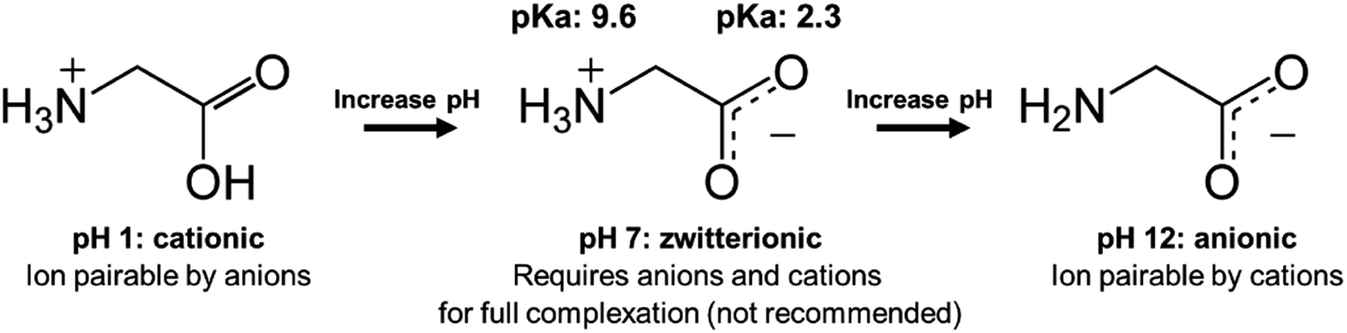 | ||
| Fig. 3 Schematic illustrating the pH-shifting strategy using glycine as a model API. At low pH, carboxylic acid groups are protonated and uncharged. At high pH, animes are deprotonated and uncharged. For some zwitterionic APIs, researchers have reported shifting pH to one extreme to turn off one type of charge prior to ion pairing. | ||
Researchers should consider several factors when using a pH shifting strategy. First, peptides are subject to degradation under basic conditions, so shifting the pH to strongly acidic is likely preferable.54 Second, the complexing counterions are subject to protonation or deprotonation under extreme pH conditions as well. Insulin has only cationic charges at pH 1.5, but an anionic fatty acid counterion such as oleic acid (pKa ∼ 5) will be protonated under those conditions too. A much more acidic counterion such as sodium dodecyl sulfate (pKa −1.5) or sodium docusate (pKa −0.75) must be used. These sulfate surfactants are less biocompatible than fatty acids, in part because of this difference in pKa. The same considerations apply when shifting the pH to basic. Quaternary amines may be the only groups to reliably retain their cationic charge at a high pH, but using these cytotoxic surfactants to complex an anionic peptide presents its own challenges.
Proteins. Protein therapeutics are commonly zwitterionic, and all the considerations of net charge, ratio of basic to acidic residues, pI, and pH shifting that apply to zwitterionic peptides also apply to proteins. An additional complication when complexing proteins is their sensitivity to denaturation. Some surfactants such as sodium dodecyl sulfate disrupt tertiary structure and cause proteins to denature.55 Using ‘gentler’ surfactants such as fatty acids may cause less degradation, but might also prevent the pH shifting approaches discussed above.
A popular model protein for hydrophobic ion pairing and encapsulation is lysozyme, which is cationic at physiological pH.18,56–59 Lysozyme's enzymatic activity can be easily measured via a cell lysis assay; therefore, testing whether or not the protein was denatured during complexation, encapsulation, and release is straightforward. Devrim et al. found that even when using sodium dodecyl sulfate as an ion pairing agent, released lysozyme retained over 80% of its enzymatic activity.18 Yoo et al. reported that the enzyme was more stable in DMSO when ion paired using SDS or oleate, and postulated that HIP complexation could help stabilize a protein's tertiary structure.57 Notably, lysozyme tends to refold into its native active form, so not all techniques that claim to ‘retain’ the protein's activity will do so for all enzymes.
2.3 Key parameters for hydrophobic ion pairing
The following section is intended to guide the reader in choosing an effective hydrophobic counterion for a given encapsulation and/or delivery system. It is important to note that the goals for a given delivery system – e.g. drug chemistry, drug loading, encapsulation technique, biological target, release profile, etc. – are the most important factors when choosing a suitable counterion. This section will overview how parameters such as drug:counterion charge ratio and counterion chemistry affect those goals.
P, the logarithm of the octanol–water partition coefficient, is a typical measure of hydrophobicity that is convenient for HIP. For a given charged head group, the longer or more saturated an alkyl tail, or the more alkyl tails, the higher the logP. Stearic acid (lipid number 18:0), for example, has a higher logP than both capric acid (lipid number 10:0) and oleic acid (lipid number 18:1). Quaternary amines also follow this trend, though their alkyl tails are fully saturated. Dimethyl dihexadecyl ammonium bromide (two methyl tails and two C16 tails) is more hydrophobic than CTAB (three methyl tails and one C16 tail), and tetraheptyl ammonium bromide (four C7 tails) is more hydrophobic than tetrabutyl ammonium bromide (four C4 tails). Note that logP values for a free acid/base or an ionized surfactant may be different when reported from measurements or calculations. In general, the higher the logP of the counterion used, the higher the logP of the resulting complex.40,60
The most hydrophobic counterion is not always the best to use. Increasing alkyl tail length or number of tails increases molecular weight, meaning the final complex will have a lower mass fraction of drug. This drives down drug loading in a delivery vehicle, all else (charge ratio, encapsulation efficiency, etc.) being equal. Availability and cost are another factor, since not all fatty acids or quaternary amines are commercially available at high purity and low cost. Solubility limitations are discussed in the following paragraph. Finally, comparing logP values among fatty acids is straightforward, but it is difficult a priori to compare the effect of a fatty acid vs. a bile acid or other carboxylic acid surfactant (e.g. oleic acid vs. pamoic acid, which is divalent) on complexation.
Extremely hydrophobic counterions, particularly those with protonated (free acid) carboxylic head groups, are difficult to dissolve in water for ion pairing. For the pairing to be effective, care should be taken to ensure that both species are dissolved and ionized prior to complexation. We recommend using a counterion's most water-soluble salt form, usually a sodium salt for anions and a bromide salt for cations.61 For example, oleic acid is sparingly soluble in water, but sodium oleate is water-soluble up to 10 wt%.62,63
When choosing among different counterions with various logP values, it is important to keep in mind why HIP is needed. This will vary by the encapsulation technique used. For example, when using nanoprecipitation, the primary goal of complexation is to decrease water solubility. When using an emulsion or SLN approach, however, the main goal is to increase lipophilicity. These distinctions will be discussed in further detail in Section 3, which focuses on encapsulation strategies, but we will give a brief example here. Consider vancomycin, a 1450 Da peptide with a single ionisable primary amine. We have found that vancomycin cannot be made to precipitate in Flash NanoPrecipitation, even using HIP, due to its low charge density. Kalhapure et al., however, improved vancomycin encapsulation efficiency from 16.8% to and 70.7% by preforming a vancomycin:linoleic acid complex prior to formulation by hot homogenization and ultrasonication using the solid lipid Compritol 888 ATO and additional surfactants.64 It is likely that vancomycin's increased lipophilicity, rather than improved hydrophobicity, led to this result. Adding oleate's 18-carbon tail to vancomycin likely improved the API's ability to interact with and remain associated with Compritol 888's alkyl tail.
A counterion's logP value is therefore not the only factor to consider when considering hydrophobicity.56 It is important to remember that in addition to excluding water, hydrophobic domains on a counterion can interact hydrophobically and sterically with (1) one another, (2) hydrophobic domains on the complexed drug, and (3) the delivery vehicle's polymers, lipids, or surfactants.65,66 Hydrophobic interactions may make a counterion with aromatic groups more suitable for use than one with an aliphatic tail, for example, or give rise to favourable cooperativity between a drug and counterion with an unsaturated aliphatic tail, even though one with a saturated tail may have a higher logP. These interactions remain an active area of research.
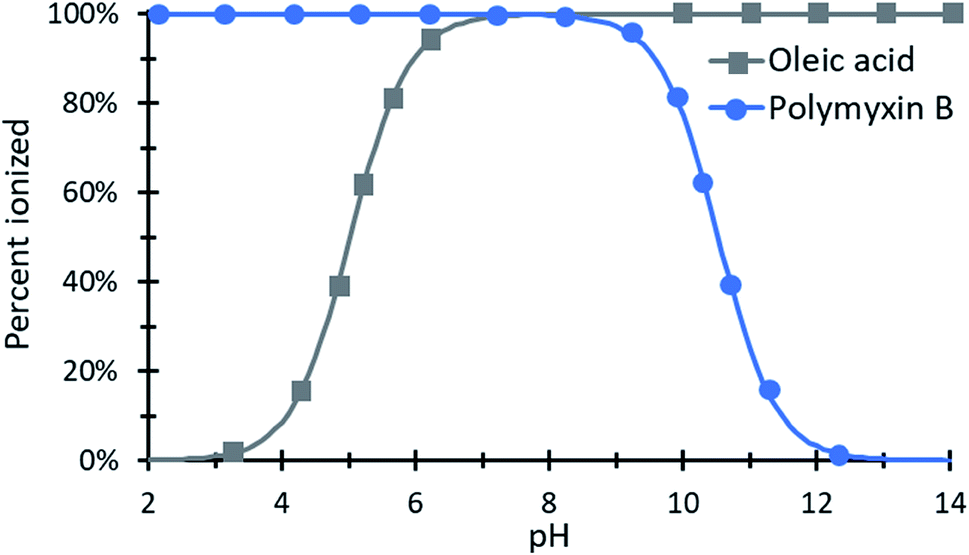 | ||
| Fig. 4 An example species diagram showing the percent ionization of polymyxin B and oleic acid as a function of pH. From approximately pH 6.5 to 9, both species are nearly 100% ionized and could be paired with clear expectations about the resulting complex's charge ratio. | ||
For peptide and protein drugs with many ionizable groups, the isoelectric point pI is a straightforward parameter to use, rather than trying to account for the pKa and ionizable state of each charged residue. As in charged polymers, the curve of charge versus pH for proteins is typically broader about the pI than an individual monomer would be. Curves denoting net charge versus pH are available for many proteins in the literature.67–69
When either the drug or counterion used has a carboxylic acid or non-quaternary amine head group, the resulting complex may demonstrate pH-sensitive dissociation, which can be used to tune drug release. pH-dependent release is useful in drug delivery, for example, for targeting to endosomes or tumors. Cationic peptides are popular in the HIP literature; these are positively charged at physiological and acidic pHs, so pH-dependent release could be accomplished by pairing them with fatty acids, rather than sulfates or phosphates. At a pH below the acid's pKa, carboxylic acid become protonated, forming the uncharged free acid and decomplexing from their cationic counterparts. Hydrophobic and steric interactions from the former ion pair remain effective, but faster drug release can be expected.30,70–72 This will be discussed in more detail in the section on drug release.
Pinkerton et al. note that the pKa values of the two charged species should be different by at least two pH units for an ion pair to reliably form. Importantly, the authors pointed out that solvent quality affects pKa values. Therefore, when complexing in a mixed solvent of water and organics increasing the volume fraction of water may be useful to ensure complexation between an anion and cation with pKa values close to neutral.47,73,74 Other researchers have noted that physical confinement, e.g. in a delivery vehicle, may affect pKa values as well; this phenomenon has the potential to affect HIP, and further study is needed75,76.
An interesting study that to our knowledge has not been carried out in the literature would examine pH-sensitive ion paired drug release behavior as a function of counterion head group. For example, Zupancic et al. paired cationic desmopressin with both sodium n-octadecyl sulfate and sodium stearate.77 The two have aliphatic tails of similar lengths, but the former has a sulfate head group and the latter has a carboxylic acid. If paired with a cationic API and encapsulated (ceteris paribus, and in a system with no other ionic or pH-sensitive components), we would expect the n-octadecyl sulfate system's release profile not to vary between pH values of e.g. 6.5, 4.5, and 2.5. The system containing stearate should release differently at the three pH values, since stearate's pKa is 4.7. Zupancic et al. found that sodium docusate and sodium oleate complexed with and precipitated desmopressin more effectively than either stearate or n-octadecyl sulfate, so the latter two counterions were not examined further. Both counterions must effectively complex with and precipitate the drug of interest. It is possible that desmopressin (1.1 kDa, 1 cationic charge) has too low of a charge density for the experiment proposed above.
We have discussed counterion pKa and logP values independently in the previous two sections. Researchers have noted that for a given counterion, it is prudent to also consider pKa and logP together.56,65,78 Carneiro et al. noted that triethylamine was a worse hydrophobic counterion for pairing with all-trans retinoic acid than both benethamine and stearylamine. Although triethylamine is a stronger base than the other two counterions, and should therefore be able to interact more easily with retinoic acid, it is so much less hydrophobic that the resulting complex does not have the desired lipophilicity.65 Likewise, Lu et al. screened fifteen counterions as candidates to form hydrophobic complexes with the pentacationic peptide polymyxin b.78 We found that at constant counterion pKa, the threshold logP required to form an ion pair hydrophobic enough for their encapsulation method (nanoprecipitation) varied. For aliphatic fatty acid sodium salts such as sodium hexanoate, sodium decanoate, and sodium oleate, precipitates formed at counterion logP values above 4. Only sodium oleate, logP ∼ 6.8, formed complexes that precipitated as required for encapsulation (green box, Fig. 5). Sulfate surfactants formed sufficiently hydrophobic complexes at and above logP values of ∼2 (yellow box, Fig. 5), suggesting that the sulfate surfactants interact more strongly with polymyxin b's cationic charges and form an ion pair more readily than the carboxylic acids. At a counterion logP of 5, dodecylbenzene sulfate formed a sufficiently hydrophobic complex, but fatty acids decanoate and myristate did not (red box, Fig. 5).
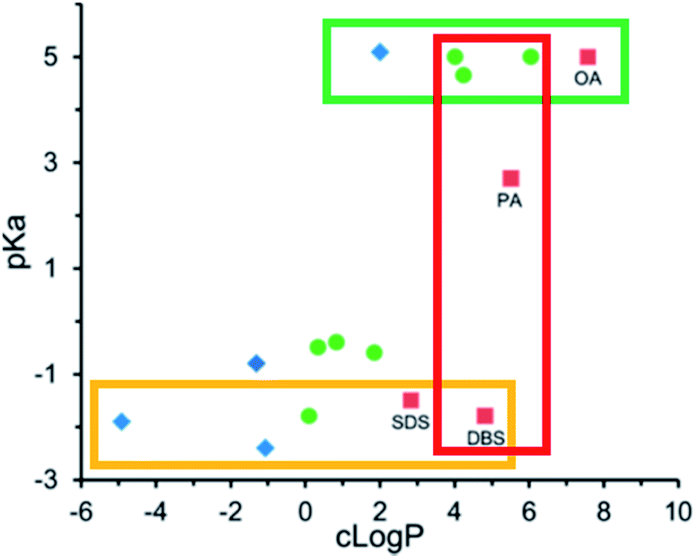 | ||
| Fig. 5 pKa and logP values for various anionic counterions. Complexes were pre-formed in MQ water at a 1:1 charge ratio with polymyxin b. Blue diamonds indicate no precipitate was observed; green circles indicate a precipitate was observed, but was insufficiently hydrophobic for nanoprecipitation; and red boxes indicate a sufficiently hydrophobic precipitate was formed. Adapted with permission from H. Lu, P. Rummaneethorn, K. Ristroph, and R. K. Prud'homme, Hydrophobic Ion Pairing of Peptide Antibiotics for Processing into Controlled Release Nanocarrier Formulations, Mol. Pharmaceutics, 2018, 15(1), 216–225. Copyright (2017) American Chemical Society.78 | ||
We recommend that researchers complex their drug of interest using a suite of counterions at first, noting the pKa and logP values of the counterions used. The resulting complex's aqueous solubility and/or lipophilicity can be measured, and counterion chemistry or charge ratio can be varied to tune these values as desired.
P empirically and fully characterize it using techniques such as differential scanning calorimetry (DSC), X-ray diffraction (XRD), NMR, FTIR, etc.79 Isolated complexes are often loaded into an oil phase or organic solvent (e.g. DCM80,81 or acetone82) and treated as a lipophilic molecule. Since the oils and organics used are aprotic and often nonpolar, dissociation is unlikely.
Pre-formed complexes have a known stoichiometry and are already paired together, meaning electrostatic interactions between the drug and other delivery vehicle components are less likely to occur during encapsulation. This is particularly advantageous when encapsulation relies on charged species, such as lipid pH coacervation to encapsulate a complex. In lipid coacervation, ionized lipids are precipitated by dropping the solution pH below their pKa values.83,84 An undissolved hydrophobic pre-formed complex is less likely to ion pair with the lipids used than a dissolved, charged drug would be.
Many researchers have noted that when pre-forming at drug:surfactant charge ratios above 1:1, excess surfactants form micelles.50,85–88 A solution that is cloudy and has visible precipitates at a 1:1 charge ratio may become clear when more surfactant is added, indicating the presence of micelles that solubilize the hydrophobic complex. When the logP of these micelle-loaded complexes is measured, it is unsurprisingly lower than the complex alone.50,61 For this reason, many studies using the pre-forming approach have stayed at or near a 1:1 drug:surfactant charge ratio to avoid micelles. Using higher charge ratios (e.g. 1:2 drug:surfactant, 1:4, etc.) should not be fully ruled out, though. Drying the pre-formed complex by lyophilization should disrupt micelles and yield a complex with a stoichiometry closer to the desired charge ratio, which may be required to tune release. This will be discussed further in the following section.
In situ ion pair formation is less common but avoids the micellization problem. Ashton et al. and Song et al. successfully paired AZD2811 with anionic surfactants during their nanoemulsion's formation, and Mussi et al. added docosahexaenoic acid to the oily phase of their SLN emulsion to pair with doxorubicin in situ.38,70,89 Pinkerton et al. and Lu et al. complexed small molecule and peptide APIs with counterions during rapid mixing in nanoprecipitation.43,47,78 A comparison between pre-formed and in situ ion pairs at a 1:1 charge ratio found no appreciable difference in the size of NPs formed by nanoprecipitation.78
:counterion molar ratios is a straightforward series of experiments to perform. By doing so, researchers have measured how a number of important parameters vary with charge ratio: complexation efficiency,18,51,57,86–88,90–98 complex logP50,98–100 and zeta potential,93,101,102 drug encapsulation efficiency,30 and even droplet size in a SEDDS (self-emulsifying drug delivery system).94 Complexation efficiency in water is typically measured by centrifuging precipitated complexes and measuring the amount of free drug in the supernatant. When measured this way, efficiency is often reported as going through a maximum near a 1:1 drug:counterion charge ratio because of the solubilization of drug into micelles at higher ratios (more counterion) and insufficient complexation at lower ratios (less counterion). We note again that at the higher charge ratios, complex formation is not less efficient than at the 1:1, but that solubilization into micelles at equilibrium results in less complex settling during centrifugation. The final solution contains solubilized drug in thermodynamically stable micelles, and a second phase which is the drug:counterion complex with a different stoichiometry.
logP measurements as a function of charge ratio are sometimes reported to go through a maximum around 1:1 as well. This is seen particularly if the experiment conducted involved forming an ion pair in water and then adding octanol.50 At charge ratios with higher counterion concentrations, micelles will have already formed in water by the time octanol is added and will be very unlikely to partition into the octanol phase. A better experimental design is to dissolve counterions first in octanol-saturated water and then add the drug of interest,99 or to dry the pre-formed complex before adding it into an octanol–water system.86,98 In this case, logP vs. charge ratio shows asymptotic behavior at higher counterion charge ratios.
Drug release as a function of drug:counterion charge ratio has also been reported.71,78,103 As may be expected, at charge ratios with more equivalents of counterion, the release rate of drug from delivery vehicle slows. This is likely because the complex is more hydrophobic, or slower to dissociate, or both. We will discuss this phenomenon in more detail in the following section.
3. Encapsulation techniques
A number of common encapsulation techniques have been applied to molecules during or after hydrophobic ion pairing. This section discusses specific considerations that should be made when using these techniques to encapsulate an ion paired molecule. A general overview of some processing parameters and important outcomes such as encapsulation efficiency and drug loading is provided.Encapsulation efficiency (EE): the encapsulation efficiency for a given drug formulation is calculated by measuring the amount of free drug (i.e. not encapsulated in or associated with delivery vehicles) after complexation and encapsulation. Separating free drug from delivery vehicles may be done via ultrafiltration or centrifugation, when the free drug and delivery vehicles have very different sizes, or by a technique such as size exclusion chromatography, for separating proteins from nanoparticles where filtration is ineffective. Once the amount of free API is measured, encapsulation efficiency is reported according to the following equation:
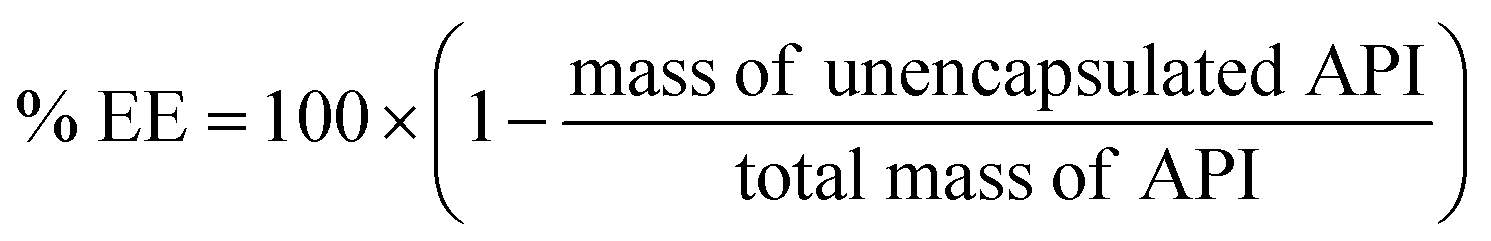
For a formulation technique to be implemented at the industrial scale, high encapsulation efficiency – i.e. less material lost during processing – is desirable. High drug loading and few unit operations are also preferred.
Many papers report that HIP enabled researchers to encapsulate molecules that they previously could not, or that the technique improved their system's encapsulation efficiency, sometimes by more than 50%.18,49,84,87,88,90,101,104–110 Encapsulation efficiencies higher than 90%49,60,84,102,103,105,106,111 and as high as 100%57,78,82,109,112 have been reported.
Drug loading: the mass fraction of API in a nanodelivery vehicle is the drug loading. The rest of the vehicle mass consists of excipients such as lipids, stabilizing polymers, oils, etc. Using HIP, drug loadings ranging from 3–7%,85,106,112 10–20%,57,81,101 and up to 30% (ref. 47, 78 and 113) have been reported. Some formulation strategies have inherent limits on drug loading; for example, S/O/W emulsions form percolation networks when the oil phase containing a hydrophobic drug reaches too high a volume fraction within a single droplet.12 Therefore, in these systems there is an inverse relationship between drug loading and vehicle stability, which is undesirable at scale and for clinical application.
3.1 Emulsions
Single (e.g. oil-in-water, O/W)38,39,56 and double (e.g. solid-in-oil-in-water or water-in-oil-in-water, S/O/W or W/O/W),18,90,91etc. – see Table 3 emulsions have been used to encapsulate ion paired complexes into droplets that may then be dried or otherwise further processed. Both require surfactants to stabilize, and typically a non-ionic species such as PVA is used. Using an ionic surfactant as an emulsion stabilizer may interfere with the hydrophobic complex's formation or stability.Researchers commonly pre-form hydrophobic complexes prior to introducing them into an emulsified system. This has an advantage over in situ formation in that the complex may be added to the oil phase before emulsification, which promotes better encapsulation. If the hydrophilic component were introduced in the aqueous phase and the hydrophobic counterion were introduced via the oil phase, pairing would likely occur at oil-water interfaces if at all (and the degree of counterion ionization in the oil phase would be difficult to determine and control). If both components were added unpaired in a mixed oil phase, pairing would again be limited by ionization. Finally, if API and counterion were introduced in water and allowed to pair, the resulting hydrophobic complex would need to partition into the oil droplets, which would take longer and be less efficient than loading the pre-formed complex into oil, where it will prefer to remain.
A HIP complex's final geometry and possible amphiphilicity are another parameter that should be considered. A hydrophilic API's water-soluble charged group may be complexed by HIP, but other polar regions on the molecule may result in an amphiphilic complex that may tend to accumulate on the emulsion droplets' oil-water interface.97 Using excess counterion, or a counterion with larger hydrophobic regions, may partially mitigate this effect. Proteins may be stabilized from denaturation at oil–water interfaces may be stabilized via ion pairing if they are complexed in such a way that their tertiary structure is largely preserved and their hydrophilic regions, which would lead to interfacial aggregation if exposed, are hidden.12,100
3.2 Lipid nanoparticles
Hydrophobic complexes have been incorporated into solid lipid nanoparticles either by emulsification from a hot melt46,65,70,105,107,108,114,115 or by stearic acid coacervation.83,84,116 In the former, non-ionic lipids and surfactants such as glyceryl behenate were heated and added to an oil phase along with a pre-formed complex. The hot oily phase was added to water and sonicated. With no other charged species present, it is unlikely that the ion pair was disrupted prior to encapsulation. Carneiro et al. note that without complexation, the API of interest, all-trans retinoic acid, resides primarily at the lipid–water interface, and that hydrophobic complexation helps incorporate it more fully in the lipid matrix.65Lipid nanoparticle formation by stearic acid coacervation involves lowering the pH of a solution of water and ethanol containing stearic acid to protonate and precipitate it as a free acid.83,84,116 In these systems, a pre-formed hydrophobic complex was added along with ethanol into the hot aqueous solution of stearic acid. Since ion pair formation and stability vary with ionic strength and pH, it remains unknown if the ion pair remained together during this formulation strategy, or if dissociation (and possibly re-pairing between the drug and stearic acid, before the pH dropped too low) occurred. Therefore the final stoichiometry and identity of the ion pair are difficult to know, even using a pre-formed system in the presence of additional potential ion pairing partners.
3.3 Precipitation
Controlled nanoprecipitation techniques such as Flash NanoPrecipitation take advantage of diffusion-limited aggregation between precipitating molecules in an aqueous or mixed solvent system. Hydrophobic complexes are well-suited for this approach, since their water solubility is very poor and they precipitate quickly. Rapid precipitation followed by stabilization, e.g. surface deposition of the hydrophobic block of a block copolymer, yields kinetically trapped core–shell nanoparticles.47 Rapid, good mixing will result in homogeneous nucleation and growth, which is desirable in nanoprecipitation. Heterogeneous nucleation or poor mixing may allow sufficient time for the formation of a thermodynamically favoured micelle phase from excess hydrophobic counterion. This is undesirable because the hydrophobic polymers or polymer blocks used in nanoprecipitation may not deposit onto a micelle's charged surface as they would onto a hydrophobic surface, and the same kinetically-trapped particle may not be formed.Researchers have demonstrated both in situ and pre-formed ion pairing approaches with nanoprecipitation. Water is typically used as an antisolvent to induce precipitation, so using salt forms of the API is a straightforward method of ensuring an initially ionized state of the API. This means that in situ complex formation, followed immediately by precipitation, is easy to accomplish. Unlike the water–oil systems such as those used for emulsions, nanoprecipitation systems use water-miscible organic solvents, meaning interfacial partitioning is not a factor. The main limit to complexation is therefore diffusion, falling in line with nanoprecipitation's typical diffusion-limited aggregation kinetics. Lu et al. reported that even in a Flash NanoPrecipitation system, where rapid mixing on the order of 2 ms is followed by nucleation, growth, and stabilization by block copolymer adsorption all within about 20 ms, complexes formed in situ were efficiently encapsulated.43,78 This suggests the time scale of complexation and precipitation is less than 20 ms. This is comparable to the precipitation time of a strongly hydrophobic (logP > 5) molecule – or pre-formed hydrophobic complex – in the same system.
Precipitation with a compressed antisolvent (PCA) has also been used to encapsulate HIP complexes into nanoparticles or microparticles.82,117–119 Because the mixing in PCA is between a solvent containing the drug of interest and a chamber of pressurized gas, pre-formation of the hydrophobic complex is required.
3.4 Others
Other formulation strategies can be viewed through a similar lens to the one we have used above. Techniques that treat a pre-formed complex such as a typical hydrophobic molecule are valid provided they have not neglected the complex's sensitivity to salts and pH. For example, self (micro/nano)-emulsifying drug delivery systems (S[M/N]EDDS) may use either ionic or non-ionic surfactants. We described when discussing lipid nanoparticles that ionic surfactants could disrupt ion pairing or exchange with a complex's counterions – these considerations are important to keep in mind when modifying a system usually used to encapsulate a non-ionic hydrophobic molecule to one capable of encapsulating a HIP complex.Systems that require pH modulation are not wholly ineligible for use with HIP, but care should be taken to ensure that the complex is not disrupted if possible. Consider Iqbal et al., who used a unique method of interpolymer complexation between polyethylene glycol and poly(acrylic acid) to form nanoparticles.11 This required adjusting the solution pH to 3 to protonate poly(acrylic acid); the cationic drug of interest was pre-formed with docusate and introduced along with Pluronic F68 in ethanol into an acidic aqueous PAA solution. Docusate has a sulfonate head group that should remain charged at pH 3, and PAA was already protonated and uncharged before the complex was added. Taken together, these suggest the pre-formed leuprolide:docusate complex was likely to survive intact in this formulation technique than in (1) one where it encountered another ionized species (as in the case of stearic acid coacervation discussed above) or (2) a system using a hydrophobic counterion (e.g. a fatty acid) that would be deprotonated at the final pH.
4. Ion paired drug release from a delivery vehicle
Drug release from a delivery vehicle containing a hydrophobic complex varies with the type of vehicle (core–shell nanoparticle, SLN, double emulsion, SEDDS, etc.), but useful similarities exist. The ion paired drug will behave like a hydrophobic molecule as long as it remains complexed. Once complexation is reversed, the original hydrophilic molecule and hydrophobic counterion are regenerated and will usually partition out of the delivery vehicle. De-complexation is driven by one of two main mechanisms: counterion competition by salts or pH-driven charge negation. The former occurs when salts in the surrounding medium are able to access the complex and outcompete the hydrophobic ion pair, leading to dissociation. The high ionic strength in the surrounding medium screens the charges between the two regenerated species, so re-complexation is unlikely. The former follows a similar mechanism, protonating or deprotonating one of the charged species and leading to de-complexation.Both mechanisms depend on water accessing the hydrophobic complex. For this reason, the vehicle's type and geometry are both important. Core–shell nanoparticles have a less water-accessible core than PLGA-stabilized double emulsions, for example, and it may be expected that they release drugs more slowly in similar salt/pH conditions. Because water can access ion pairs at the water–vehicle interface much more readily than complexes deep in the vehicle core, release is expected to occur from the outside in. After ion pairs at the surface have been de-complexed and partition into the bulk phase, water will be able to access the complexes deeper in the vehicle. During this ‘erosion,’ the vehicle itself may lose structural integrity or collapse.61 For vehicles with low drug loading, the hydrophobic complex's location in the vehicle is another factor to consider. A complex with amphiphilic character that resides primarily on the vehicle surface is easily water-accessible, and rapid burst release may be observed.65,97,111
The fact that de-complexation is a precursor to this type of release explains why slower drug release is seen at higher charge ratios.71,78,103 A monovalent drug complexed with a single monovalent counterion should fully dissociate much more quickly than a drug complexed with four, and will not release from the vehicle until it is fully dissociated. In the 1:4 charge ratio case, only one of the counterions can truly form an ion pair with the drug (see the preceding section on drug:counterion charge ratio). The remaining three counterions can remain associated with the complex, though, adsorbing onto the first counterion via tail–tail hydrophobic interactions. The resulting large hydrophobic surface area serves as a mass transfer barrier that slows water diffusion to the site of ion pairing. For this reason, it is more difficult for water to access and dissociate the 1:4 complex than the 1:1 complex, so drug release is slower. The probability of a drug re-complexing with a hydrophobic counterion after salt-driven decomplexation also increases with the number of hydrophobic counterions near a drug molecule.
After dissociation, the therapeutic and counterion are regenerated. Without its hydrophobic counterion, the therapeutic is likely too hydrophilic to remain associated with the vehicle (even if the solution pH has turned off the drug's charge) and will partition into the bulk. Depending on its chemistry and the bulk pH, the counterion may either diffuse into the bulk or remain with the vehicle. For example, we found that at a 1:4 polymyxin B:oleate charge ratio, drug release plateaued around 35%. This suggests that after soluble polymyxin b was released, the poorly-water-soluble oleate fatty acids remain with the nanoparticle and may form an oleate/oleic acid liquid crystal phase in or around the NP core. This type of plateauing release profile was not observed for polymyxin paired at a 1:4 polymyxin:SDS charge ratio, because SDS is more water-soluble and prefers to partition into the bulk.78
Complexation formation, dissociation, and release have all been found to be a function of bulk ionic strength.18,38,44,45,72,120 As expected, at higher ionic strength, it is more difficult to form complexes due to charge screening, and, if formed, complexes dissociate and drugs are released faster at higher salt concentrations. PBS, sodium chloride, and serum are common release media. Researchers have found both complexation18,57,120,121 and release30,38,48,70–72,105,112,121 to be pH-dependent. These assays are usually run at pH values at and below 7.3. In most cases, faster release at lower pH has been observed as expected.
This understanding of the mechanism behind salt- and pH-dependent release is useful. Many nano-scale delivery vehicles are stable in water and can be stored in deionized water without beginning to release their payload. For parenteral or oral formulations, the body's natural ionic strength will trigger release. pH-dependent release could be useful for targeting to endosomes, tumors, or different regions in the intestinal tract. On the other hand, pH sensitivity may rule out some long-term depot delivery strategies; for example, it has been well-documented that PLGA microparticles may acidify as the polymer is degraded over time.12,122 An acid microenvironment could trigger ion pair dissociation and lead to faster release.
The previous several paragraphs have discussed drug release following ion pair dissociation. In general, unless a hydrophobic complex is exposed to salts or pH changes, it should behave like a hydrophobic molecule – depending on the vehicle and the chemistry of the species that make up the pair, the complex could still be released intact from the vehicle. Hydrophobic molecules in nano-scale delivery vehicles can diffuse and, depending on their solubility in the release medium, may still partition into the bulk. The complex's size and hydrophobic interactions with the delivery vehicle are barriers to diffusion.65,66,116 When buffers containing only salts are used as simple release media, this type of release is unlikely. In more complex, more realistic release media – i.e. those containing some kind of hydrophobic sink such as albumins or bile salt micelles – this type of direct release, as well as salt- or pH-driven release, may occur simultaneously. The driving forces for diffusion of all species, including paired and unpaired drugs and paired, unpaired, and uncharged counterions, should be considered.
It is straightforward to see that both major exit routes from a particle – either following dissociation or as an intact complex – depend on the complex itself and the counterion used. Alkyl tail length or hydrophobic group size, for example, affect both. In the case of post-dissociation release, larger hydrophobic surface areas decrease water permeability and slow decomplexation, as discussed in the paragraph about release as a function of charge ratio. And in the case of release as an intact complex, longer alkyl tails both decrease the complex's solubility in the bulk and increase steric and hydrophobic interactions that tend to keep the complex in its vehicle. As counterion hydrophobicity increases, therefore, release tends to decrease (Fig. 6).29,38,65,78,88
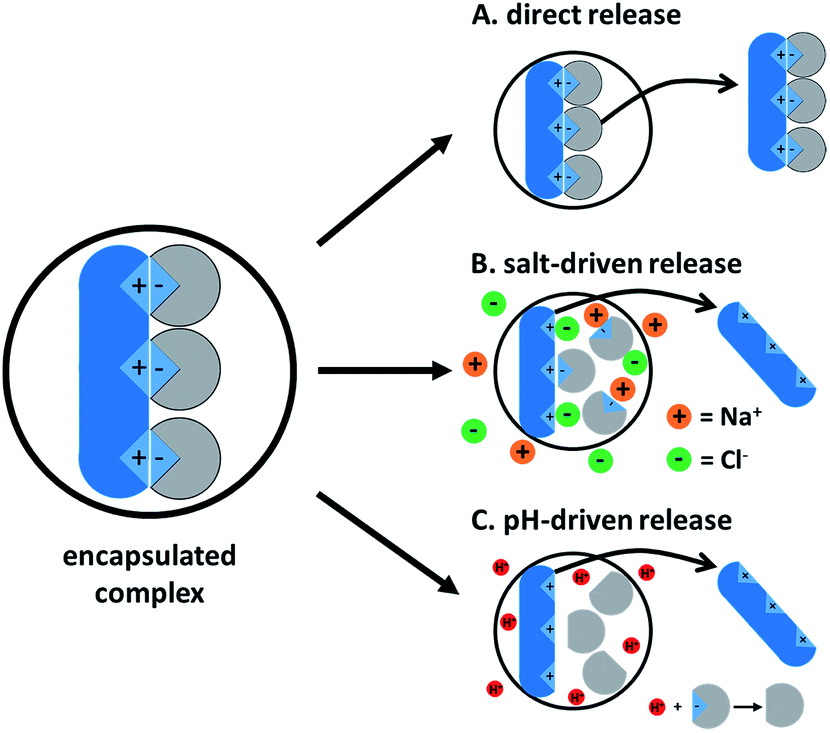 | ||
| Fig. 6 A summary of how complexed drugs may be released from a delivery vehicle. (A) The intact complex may release directly from the particle into the bulk. This type of release is a function of the complex's solubility in the bulk, so hydrophobic sinks such as bile salt micelles or albumins in the bulk phase will provide more of a driving force than simply a buffer. (B) Salts lead to counterion competition and decomplexation, which is followed by release as the water-soluble drug is released into the bulk. (C) When counterions such as fatty acids are used, lowering the pH below their pKa will lead to protonation. The protonated counterions will no longer complex the drug, leading to release as in (B). | ||
5. Bridging polyelectrolyte coacervation, polymer–surfactant complexation, and hydrophobic ion pairing
The HIP literature has generally not overlapped with the literature from the fields of polyelectrolyte–polyelectrolyte coacervation or polyelectrolyte-surfactant complexation. The fields share a number of similarities that we will highlight here. Polyelectrolyte complex coacervation used here refers to the phase separation induced when oppositely-charged polyelectrolytes or ionomers ion pair with one another, and is not to be confused with the acid-induced lipid precipitation technique mentioned earlier (which is also called ‘coacervation’). Polyelectrolyte-surfactant complexation has been studied extensively and is useful in a number of industrial applications, including personal care products and detergents.1235.1 Polyelectrolyte coacervation
Ion pairing between polyelectrolyte species results in the formation of a highly electrostatically crosslinked complex that phase separates from its surrounding media; the new phase may be either a solid (‘precipitate’) or liquid (‘coacervate’), but the term ‘complex coacervation’ is applied in both cases.124–126 Stoichiometric ratios (i.e. 1:1 cation:anion) are common in this literature, since these systems tend to form more distinct coacervate phases from charge neutral conditions – at uneven ratios, electrostatically-stabilized colloidal particles may form.125 Like hydrophobic ion pairs, coacervate phases are sensitive to salt and may be dissociated at sufficient ionic strength or as a function of pH, depending on the chemistry.
The entropic and enthalpic effects of coacervate formation and phase separation have been studied and reported elsewhere.125,127–132 Interestingly, polymers in a complex coacervate may remain mobile, and rearrangement is possible. Many studies focus on the coacervate's phase behaviour133 or rheological134 or thermal135 properties. The final phase's logP (and hydrophobicity in general) is not a major concern in complex coacervation, since the mechanism of phase separation is different and the ultimate goal of coacervation is not necessarily to increase hydrophobicity or modify solubility as in HIP.
Because complex coacervates form distinct phases, the technique has been studied as a possible method of encapsulating polyvalent drugs such as peptides and proteins.136–139 For insufficiently charged proteins, ‘supercharging’ – adding more charged amino acids – increases charge density and can lead to more reliable coacervation.140 Some similarities and differences between encapsulation using coacervation and HIP may be seen. Encapsulation via coacervation is typically carried out near or at a stoichiometric charge ratio.136,138 This differs from the HIP literature, where a screen of different drug:counterion charge ratios is commonly performed and the resulting complex's logP is characterized. Using a complex coacervate as a delivery vehicle introduces problems similar to W/O/W double emulsions; namely, a tradeoff between drug loading and encapsulation efficiency.141 Another notable difference between HIP and PE coacervation other than the molecular weights of the species used is the lack of ionic crosslinking in much of the HIP literature. Most HIP studies use monovalent small molecule counterions (exceptions are discussed in the following paragraph), but PE coacervation depends on polyvalency to form its characteristic crosslinked polymer networks.
Some papers using small-molecule surfactants for HIP have also used polyvalent counterions to complex drugs, and most unfortunately fail to make the distinction between the two techniques in their descriptions of what was done. The most common polyvalent counterion in these cases is dextran sulfate, a polyanionic sugar that does not contain distinct hydrophobic domains.59,91,92,104,120 When it is used to complex multivalent APIs such as proteins, dextran sulfate forms a complex coacervate rather than a true hydrophobic ion pair. The coacervate may still be encapsulated using methods that also encapsulate hydrophobic ion pairs, but drug release from ion pairs compared to coacervates may differ noticeably. Consider Song et al., who found that drug release of an ion paired multivalent small molecule AZD2811 varied significantly whether using pamoic acid (divalent) or xinafoic acid (monovalent, effectively half of pamoic acid) at the same charge ratio.38 The difference in release profile observed was most likely a function of crosslinking between divalent pamoic acid and the multivalent small molecule, which resulted in slower release. It is easy to see that a system containing dextran sulfate, which has much greater protein crosslinking potential than divalent pamoic acid, could be expected to have very different release kinetics from a system using a small molecule surfactant counterion. Understanding and carefully distinguishing between the two approaches will be beneficial for future studies aiming to ionically complex and encapsulate charged therapeutics.
5.2 Polymer–surfactant complexation
Polymer–surfactant complexation and phase behavior have been studied for decades.142,143 The field has carefully examined precipitate formation as a function of many parameters familiar to the hydrophobic ion pairing literature, including charge density,144 stoichiometry, and formation and dissociation in the presence of salts.145 Studies of self-assembly have developed binding isotherms and a thermodynamic understanding of surfactant monolayer and bilayer formation.123,146,147 Important questions in this field include: what are the phase behavior and rheological properties (surface tension, viscosity, etc.) of polymer–surfactant complexes?144,148,149 Do they form one-phase or two-phase systems, and does this change upon dilution – i.e., what are the critical aggregation concentrations? What is the effect of surfactant alkyl tail length on this behavior?146,150 Are the polymers binding to surfactant monomers, or micelles?145,151The polymers used in these studies often have higher molecular weight and more regular charge spacing than the peptides and proteins complexed with surfactants in the HIP literature. As in the coacervation literature, imparting additional hydrophobicity to a polymer is not necessarily the ultimate goal of a study, and drug encapsulation is rarely discussed. These studies often examine wetting or solubilization behavior, and are useful in the development of new detergents and shampoos.
A familiarity with the field of polyelectrolyte-surfactant complexation will help hydrophobic ion pairing researchers appreciate similar sensitivities – e.g. to salts, temperature, or pH – that their own systems might experience. It will also help them develop an appreciation for surfactant phase behavior, which has not been well-characterized in most studies that use HIP to encapsulate a drug molecule.
6. Perspective
Encapsulating hydrophilic therapeutics via hydrophobic ion pairing is a useful technique and offers a number of attractive possibilities. In this section, we highlight four major ones: (1) co-encapsulating hydrophilic and hydrophobic drugs, (2) forming a HIP complex from two therapeutic species, (3) decreasing API crystallinity, and (4) tuning drug release rates by simply altering the counterion used in HIP.Many encapsulation techniques that have been optimized for hydrophobic therapeutics can easily be adapted to work on ion paired hydrophobic complexes. This introduces a unique and powerful possibility: straightforward co-encapsulation of hydrophilic and hydrophobic therapeutics into a single delivery vehicle at high loadings. This is highly desirable, and is not possible using encapsulation techniques developed specifically for water-soluble drugs, e.g. W/O/W emulsions. Using two different encapsulation techniques to prepare two populations of particles encapsulating two therapeutics can be difficult (and expensive), especially if the particle chemistries, sizes, and fates are intended to match. There can be no guarantee of simultaneous delivery or even co-delivery in the body using a mixed population of particles, particularly at the single-cell level where local concentrations of both species may be important for therapeutic synergy.
Using HIP, researchers have co-encapsulated hydrophilic and hydrophobic therapeutics.30,71,103 Tuning release rates of co-encapsulated drugs from these two classes is an exciting prospect for future study. For example, Zhou et al. found their hydrophobic therapeutic released at a pH-independent rate, while the ion paired complex's release varied with pH.30 Future formulations may take advantage of this difference in behaviour while still benefiting from co-localization. Or, if the mismatch is undesirable, researchers could tune parameters discussed above such as charge ratio, counterion chemistry, etc. to make hydrophilic and hydrophobic release rates match.
Another exciting opportunity of hydrophobic ion pairing is the co-delivery of two water-soluble charged APIs by ion pairing the two of them together. Denadai et al. formed a hydrophobic complex from two therapeutic small molecules, cationic chlorhexidine and anionic losartan, but did not encapsulate the result in a separate delivery vehicle.152 Other papers report choosing their counterion based on therapeutic synergy. Kalhapure et al. chose from among several similarly-lipophilic counterions – palmitoleic, oleic, linoleic, linolenic, and arachidonic acid – by checking the ability of each to inhibit S. aureus growth. Linoleic acid's MIC was superior to the others, so it was used to pair with vancomycin.64 Similarly Oliveira et al. ion paired doxorubicin with α-tocopherol succinate to take advantage of the anticancer properties of each,105 and Li et al. paired doxorubicin with hyaluronic acid since the latter is possibly useful for tumor targeting.153
Several papers have reported that ionic complexation with hydrophobic counterions formed an ion pair with lower crystallinity than its individual components.28–30,43,46,47,154 Improved amorphous character may be useful for quickly releasing drugs, particularly as intact complexes, if desired. It may also be helpful for formulating hydrophobic ionic drugs into stable NPs with faster dissolution kinetics.43 In this case, controls with a non-ionic analogue of the counterion (e.g. methyl esters instead of carboxylic acids; see Oliviera and Mussi for examples of this important control experiment) should be used to determine if reduced crystallinity is simply a co-core confinement effect or truly the result of HIP.70,105
For formulations containing a HIP complex, tuning drug release rates by changing the hydrophobic counterion or API:counterion charge ratio used is a straightforward and powerful tool for formulation scientists and drug delivery researchers.71,78,103 Changing the amount or type of counterion used during a formulation technique while holding all other excipients and processing steps constant is relatively easy and should allow for several formulations with different release rates – and therefore possibly different PK profiles – to be developed and tested in rapid succession.
Hydrophobic ion pairing remains an active area of research, and several fundamental aspects of the process remain unclear: the precise dynamics of API–counterion assembly, aggregation/precipitation, and dissociation/release as a function of drug and surfactant chemistry (e.g. head group/pKa, charge density, charge ratio, pH, ionic strength, hydrophobic moieties, etc.) are active areas of research. The phase behavior of surfactants used as hydrophobic counterions once paired is similarly unknown; this may prove to be an important parameter in modulating release.78 Molecular dynamics simulations may be the best way to examine the details of these phenomena, as they have been used extensively to study PE coacervation and PE-surfactant complexation.130,131
We have reviewed how and why drug release from an ion paired system varies with counterion. It remains unknown if a mixture of counterions in a single formulation could lead to two different release profiles simultaneously; e.g. preparing two different pre-formed complexes of a cationic peptide with two different anionic surfactants, then incorporating both into a single formulation to achieve an initial burst release followed by slower, steady release over time.
This review has summarized the many proof-of-concept studies that demonstrate hydrophobic ion pairing as a useful tool for complexing and encapsulating hydrophilic therapeutics across several classes – small molecules, peptides, protein fragments, and full proteins such as antibodies and enzymes – into nano-scale delivery vehicles. The technique is straightforward, uses accessible and inexpensive surfactants, and yields hydrophobic complexes that may be precipitated, emulsified, or otherwise packaged using existing technology. Fundamental questions remain about counterion phase behavior, the details of hydrophobic and ionic interactions during complexation, and the exact mechanism of drug release across several chemistries and external parameters. Moving forward, the field offers exciting possibilities such as co-encapsulation of hydrophobic and hydrophilic therapeutics, collaborations with the related fields of polyelectrolyte coacervation and polyelectrolyte-surfactant complexation, or the co-encapsulation and co-delivery of two ionic APIs by forming a HIP complex from the two of them.
Abbreviations
| a priori | Beforehand |
| API | Active pharmaceutical ingredient |
| ceteris paribus | With all else being equal |
| CTAB | Cetrimonium bromide |
| DCM | Dichloromethane |
| DSC | Differential scanning calorimetry |
| et al. | And the others |
| FDA | United States Food and Drug Administration |
| FTIR | Fourier-transform infrared spectroscopy |
| HIP | Hydrophobic ion pair/pairing |
| in situ | on site/in place |
| IP | Ion pair or ion pairing agent |
| NMR | Nuclear magnetic resonance |
| N/P ratio | Molar ratio of cationic nitrogen in lipids to anionic phosphorous in nucleic acids, used in the lipoplex literature |
| NCs | Nanocarriers |
| NPs | Nanoparticles |
| PBS | Phosphate-buffered saline |
| PLGA | Poly(lactic-co-glycolic acid) |
| SDS | Sodium dodecyl sulfate |
| SLN | Solid lipid nanoparticle |
| S(M/N)EDDS | Self-(micro/nano)emulsifying drug delivery system |
| XRD | X-ray diffraction |
Funding
We acknowledge financial support from the Bill and Melinda Gates Foundation (BMGF OPP1150755), The Princeton SEAS Helen Shipley Hunt Fund, and the National Science Foundation for the research fellowship for K. D. R. (DGE-1656466) and (CBET 1605816).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Nathalie Pinkerton, Dr Robert Pagels, Mr Chester Markwalter, and Mr Douglas Scott for intellectual discussion and for their help reviewing the document.References
- J. D. Meyer and M. C. Manning, Hydrophobic Ion Pairing: Altering the Solubility Properties of Biomolecules, Pharm. Res., 1998, 15(2), 188–193 CrossRef CAS PubMed.
- T. Wang and X. Wang, Solubilization of an indicator dye for optical fiber chemical sensors in supercritical CO2 by ion pairing, Sens. Actuators, B, 2003, 89(1), 144–149 CrossRef CAS.
- S. Wu, A. Buthe and P. Wang, Organic-soluble enzyme nano-complexes formed by ion-pairing with surfactants, Methods Mol. Biol., 2011, 743, 51–63 CrossRef CAS PubMed.
- R. Pangeni, J. U. Choi, V. K. Panthi, Y. Byun and J. W. Park, Enhanced oral absorption of pemetrexed by ion-pairing complex formation with deoxycholic acid derivative and multiple nanoemulsion formulations: preparation, characterization, and in vivo oral bioavailability and anticancer effect, Int. J. Nanomed., 2018, 13, 3329–3351 CrossRef CAS PubMed.
- S. K. You, H. H. Kwon, J. M. Lee, S. C. Shin and C. W. Cho, Studies on the formation of hydrophobic ion-pairing complex of alendronate, Arch. Pharmacal Res., 2009, 32(7), 1055–1060 CrossRef CAS PubMed.
- T. N. Q. Phan, I. Shahzadi and A. Bernkop-Schnürch, Hydrophobic ion-pairs and lipid-based nanocarrier systems: the perfect match for delivery of BCS class 3 drugs, J. Controlled Release, 2019 Search PubMed.
- S. A. Megwa, S. E. Cross, M. W. Whitehouse, H. A. Benson and M. S. Roberts, Effect of ion pairing with alkylamines on the in-vitro dermal penetration and local tissue disposition of salicylates, J. Pharm. Pharmacol., 2000, 52(8), 929–940 CrossRef CAS.
- A. S. Torky, M. S. Freag, M. M. A. Nasra and O. Y. Abdallah, Novel skin penetrating berberine oleate complex capitalizing on hydrophobic ion pairing approach, Int. J. Pharm., 2018, 549(1–2), 76–86 CrossRef CAS PubMed.
- S. Bonengel, M. Jelkmann, M. Abdulkarim, M. Gumbleton, V. Reinstadler, H. Oberacher, F. Prufert and A. Bernkop-Schnurch, Impact of different hydrophobic ion pairs of octreotide on its oral bioavailability in pigs, J. Controlled Release, 2018, 273, 21–29 CrossRef CAS.
- M. Goswami, S. K. Kumar, A. Bhattacharya and J. F. Douglas, Computer Simulations of Ionomer Self-Assembly and Dynamics, Macromolecules, 2007, 40(12), 4113–4118 CrossRef CAS.
- J. Iqbal, C. Vigl, G. Moser, M. Gasteiger, G. Perera and A. Bernkop-Schnurch, Development and in vivo evaluation of a new oral nanoparticulate dosage form for leuprolide based on polyacrylic acid, Drug Delivery, 2011, 18(6), 432–440 CrossRef CAS.
- R. F. Pagels and R. K. Prud'homme, Polymeric nanoparticles and microparticles for the delivery of peptides, biologics, and soluble therapeutics, J. Controlled Release, 2015, 219, 519–535 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard Jr, Nanoparticle-based targeted drug delivery, Exp. Mol. Pathol., 2009, 86(3), 215–223 CrossRef CAS PubMed.
- C. E. Markwalter, R. F. Pagels, B. K. Wilson, K. D. Ristroph and R. K. Prud'homme, Flash NanoPrecipitation for the Encapsulation of Hydrophobic and Hydrophilic Compounds in Polymeric Nanoparticles, JoVE, 2019,(143), e58757 Search PubMed.
- K. T. Savjani, A. K. Gajjar and J. K. Savjani, Drug solubility: importance and enhancement techniques, ISRN Pharm., 2012, 2012, 195727 Search PubMed.
- S. Kumar, D. Bhargava, A. Thakkar and S. Arora, Drug carrier systems for solubility enhancement of BCS class II drugs: a critical review, Crit. Rev. Ther. Drug Carrier Syst., 2013, 30(3), 217–256 CrossRef CAS PubMed.
- C. Wischke and S. P. Schwendeman, Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles, Int. J. Pharm., 2008, 364(2), 298–327 CrossRef CAS PubMed.
- B. Devrim and A. S. Bozkir, in Design and Evaluation of Hydrophobic Ion-Pairing Complexation of Lysozyme with Sodium Dodecyl Sulfate for Improved Encapsulation of Hydrophilic Peptides/Proteins by Lipid-Polymer Hybrid Nanoparticles, 2015 Search PubMed.
- S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prud'homme and L. D. Mayer, Modulating the Therapeutic Activity of Nanoparticle Delivered Paclitaxel by Manipulating the Hydrophobicity of Prodrug Conjugates, J. Med. Chem., 2008, 51(11), 3288–3296 CrossRef CAS PubMed.
- J. S. Sohn, J. I. Jin, M. Hess and B. W. Jo, Polymer prodrug approaches applied to paclitaxel, Polym. Chem., 2010, 1(6), 778–792 RSC.
- V. J. Stella, Prodrugs: Some thoughts and current issues, J. Pharm. Sci., 2010, 99(12), 4755–4765 CrossRef CAS.
- E. E. Dormidontova, Role of Competitive PEO-Water and Water-Water Hydrogen Bonding in Aqueous Solution PEO Behavior, Macromolecules, 2002, 35(3), 987–1001 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, Specific Ion Effects on the Water Solubility of Macromolecules: PNIPAM and the Hofmeister Series, J. Am. Chem. Soc., 2005, 127(41), 14505–14510 CrossRef CAS PubMed.
- R. J. Goddard, B. P. Grady and S. L. Cooper, The Room Temperature Annealing Peak in Ionomers: Ionic Crystallites or Water Absorption?, Macromolecules, 1994, 27(7), 1710–1719 CrossRef CAS.
- B. A. Brozoski, P. C. Painter and M. M. Coleman, Concerning the origin of broad bands observed in the FT-IR spectra of ionomers. Cluster formation or water adsorption?, Macromolecules, 1984, 17(8), 1591–1594 CrossRef CAS.
- J.-S. Kim and A. Eisenberg, Effect of sample preparation conditions and degree of neutralization on the dynamic mechanical properties of poly(styrene-co-sodium methacrylate) ionomers, J. Polym. Sci., Part B: Polym. Phys., 1995, 33(2), 197–209 CrossRef CAS.
- S. Yang, K. Sun and W. M. Risen Jr., Preparation and thermal characterization of the glass transition temperatures of sulfonated polystyrene-metal ionomers, J. Polym. Sci., Part B: Polym. Phys., 1990, 28(10), 1685–1697 CrossRef CAS.
- M. Morgen, A. Saxena, X. Q. Chen, W. Miller, R. Nkansah, A. Goodwin, J. Cape, R. Haskell, C. Su, O. Gudmundsson, M. Hageman, A. Kumar, G. S. Chowan, A. Rao and V. K. Holenarsipur, Lipophilic salts of poorly soluble compounds to enable high-dose lipidic SEDDS formulations in drug discovery, Eur. J. Pharm. Biopharm., 2017, 117, 212–223 CrossRef CAS PubMed.
- B. K. Poudel, B. Gupta, T. Ramasamy, R. K. Thapa, Y. S. Youn, H. G. Choi, C. S. Yong and J. O. Kim, Development of polymeric irinotecan nanoparticles using a novel lactone preservation strategy, Int. J. Pharm., 2016, 512(1), 75–86 CrossRef CAS PubMed.
- Z. Zhou, M. Jafari, V. Sriram, J. Kim and J. Y. Lee, Delayed Sequential Co-Delivery of Gefitinib and Doxorubicin for Targeted Combination, Chemotherapy, 2017, 14(12), 4551–4559 CAS.
- P. R. Cullis and M. J. Hope, Lipid Nanoparticle Systems for Enabling Gene Therapies, Mol. Ther., 2017, 25(7), 1467–1475 CrossRef CAS.
- L. Xu and T. Anchordoquy, Drug delivery trends in clinical trials and translational medicine: Challenges and opportunities in the delivery of nucleic acid-based therapeutics, J. Pharm. Sci., 2011, 100(1), 38–52 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Nonviral Vectors for Gene Delivery, Chem. Rev., 2009, 109(2), 259–302 CrossRef CAS.
- K. A. Whitehead, R. Langer and D. G. Anderson, Knocking down barriers: advances in siRNA delivery, Nat. Rev. Drug Discovery, 2009, 8(2), 129–138 CrossRef CAS.
- W. Li and F. C. Szoka, Lipid-based Nanoparticles for Nucleic Acid Delivery, Pharm. Res., 2007, 24(3), 438–449 CrossRef CAS PubMed.
- S. A. Wissing, O. Kayser and R. H. Muller, Solid lipid nanoparticles for parenteral drug delivery, Adv. Drug Delivery Rev., 2004, 56(9), 1257–1272 CrossRef CAS.
- W. Mehnert and K. Mader, Solid lipid nanoparticles: production, characterization and applications, Adv. Drug Delivery Rev., 2001, 47(2–3), 165–196 CrossRef CAS.
- Y. H. Song, E. Shin, H. Wang, J. Nolan, S. Low, D. Parsons, S. Zale, S. Ashton, M. Ashford, M. Ali, D. Thrasher, N. Boylan and G. Troiano, A novel in situ hydrophobic ion paring (HIP) formulation strategy for clinical product selection of a nanoparticle drug delivery system, J. Controlled Release, 2016, 229, 106–119 CrossRef CAS.
- M. J. Heffernan, S. P. Kasturi, S. C. Yang, B. Pulendran and N. Murthy, The stimulation of CD8+ T cells by dendritic cells pulsed with polyketal microparticles containing ion-paired protein antigen and poly(inosinic acid)-poly(cytidylic acid), Biomaterials, 2009, 30(5), 910–918 CrossRef CAS PubMed.
- T. Satoh, Y. Higuchi, S. Kawakami, M. Hashida, H. Kagechika, K. Shudo and M. Yokoyama, Encapsulation of the synthetic retinoids Am80 and LE540 into polymeric micelles and the retinoids' release control, J. Controlled Release, 2009, 136(3), 187–195 CrossRef CAS PubMed.
- I. Shahzadi, M. H. Asim, A. Dizdarević, J. D. Wolf, M. Kurpiers, B. Matuszczak and A. Bernkop-Schnürch, Arginine-based cationic surfactants: Biodegradable auxiliary agents for the formation of hydrophobic ion pairs with hydrophilic macromolecular drugs, J. Colloid Interface Sci., 2019, 552, 287–294 CrossRef CAS PubMed.
- T. Karamanidou, K. Karidi, V. Bourganis, K. Kontonikola, O. Kammona and C. Kiparissides, Effective incorporation of insulin in mucus permeating self-nanoemulsifying drug delivery systems, Eur. J. Pharm. Biopharm., 2015, 97(Pt A), 223–229 CrossRef CAS PubMed.
- H. D. Lu, K. D. Ristroph, E. L. K. Dobrijevic, J. Feng, S. A. McManus, Y. Zhang, W. D. Mulhearn, H. Ramachandruni, A. Patel and R. K. Prud'homme, Encapsulation of OZ439 into Nanoparticles for Supersaturated Drug Release in Oral Malaria Therapy, ACS Infect. Dis., 2018 Search PubMed.
- L. Feng, A. De Dille, V. J. Jameson, L. Smith, W. S. Dernell and M. C. Manning, Improved potency of cisplatin by hydrophobic ion pairing, Cancer Chemother. Pharmacol., 2004, 54(5), 441–448 CrossRef CAS PubMed.
- J. Chamieh, A. Domènech Tarrat, C. Doudou, V. Jannin, F. Demarne and H. Cottet, Peptide release from SEDDS containing hydrophobic ion pair therapeutic peptides measured by Taylor dispersion analysis, Int. J. Pharm., 2019, 559, 228–234 CrossRef CAS PubMed.
- G. A. Castro, A. L. Coelho, C. A. Oliveira, G. A. Mahecha, R. L. Orefice and L. A. Ferreira, Formation of ion pairing as an alternative to improve encapsulation and stability and to reduce skin irritation of retinoic acid loaded in solid lipid nanoparticles, Int. J. Pharm., 2009, 381(1), 77–83 CrossRef CAS PubMed.
- N. M. Pinkerton, A. Grandeury, A. Fisch, J. Brozio, B. U. Riebesehl and R. K. Prud'homme, Formation of stable nanocarriers by in situ ion pairing during block-copolymer-directed rapid precipitation, Mol. Pharm., 2013, 10(1), 319–328 CrossRef CAS.
- K. Patel, V. Sarma and P. Vavia, Design and evaluation of lumefantrine – oleic acid self nanoemulsifying ionic complex for enhanced dissolution, Daru, J. Fac. Pharm., Tehran Univ. Med. Sci., 2013, 21(1), 27 CrossRef CAS PubMed.
- R. Bussano, D. Chirio, L. Costa, F. Turci and M. Trotta, Preparation and Characterization of Insulin-Loaded Lipid-Based Microspheres Generated by Electrospray, J. Dispersion Sci. Technol., 2011, 32(10), 1524–1530 CrossRef CAS.
- W. G. Dai and L. C. Dong, Characterization of physiochemical and biological properties of an insulin/lauryl sulfate complex formed by hydrophobic ion pairing, Int. J. Pharm., 2007, 336(1), 58–66 CrossRef CAS.
- S. Sun, F. Cui, Y. Kawashima, N. Liang, L. Zhang, K. Shi and Y. Yu, A novel insulin-sodium oleate complex for oral administration: preparation, characterization and in vivo evaluation, J. Drug Delivery Sci. Technol., 2008, 18(4), 239–243 CrossRef CAS.
- F. Cui, K. Shi, L. Zhang, A. Tao and Y. Kawashima, Biodegradable nanoparticles loaded with insulin-phospholipid complex for oral delivery: preparation, in vitro characterization and in vivo evaluation, J. Controlled Release, 2006, 114(2), 242–250 CrossRef CAS PubMed.
- A. Elsayed, M. Al-Remawi, N. Qinna, A. Farouk, K. A. Al-Sou'od and A. A. Badwan, Chitosan-sodium lauryl sulfate nanoparticles as a carrier system for the in vivo delivery of oral insulin, AAPS PharmSciTech, 2011, 12(3), 958–964 CrossRef CAS.
- K. Talley and E. Alexov, On the pH-optimum of activity and stability of proteins, Proteins, 2010, 78(12), 2699–2706 CAS.
- D. Otzen, Protein-surfactant interactions: a tale of many states, Biochim. Biophys. Acta, 2011, 1814(5), 562–591 CrossRef CAS PubMed.
- K. Fu, R. Harrell, K. Zinski, C. Um, A. Jaklenec, J. Frazier, N. Lotan, P. Burke, A. M. Klibanov and R. Langer, A potential approach for decreasing the burst effect of protein from PLGA microspheres, J. Pharm. Sci., 2003, 92(8), 1582–1591 CrossRef CAS PubMed.
- H. S. Yoo, H. K. Choi and T. G. Park, Protein-fatty acid complex for enhanced loading and stability within biodegradable nanoparticles, J. Pharm. Sci., 2001, 90(2), 194–201 CrossRef CAS.
- U. Bilati, E. Allemann and E. Doelker, Nanoprecipitation versus emulsion-based techniques for the encapsulation of proteins into biodegradable nanoparticles and process-related stability issues, AAPS PharmSciTech, 2005, 6(4), E594–E604 CrossRef PubMed.
- R. Gaudana, M. Gokulgandhi, V. Khurana, D. Kwatra and A. K. Mitra, Design and evaluation of a novel nanoparticulate-based formulation encapsulating a HIP complex of lysozyme, Pharm. Dev. Technol., 2013, 18(3), 752–759 CrossRef CAS PubMed.
- H. Zhou, C. Lengsfeld, D. J. Claffey, J. A. Ruth, B. Hybertson, T. W. Randolph, K. Y. Ng and M. C. Manning, Hydrophobic ion pairing of isoniazid using a prodrug approach, J. Pharm. Sci., 2002, 91(6), 1502–1511 CrossRef CAS PubMed.
- H. L. Wong, R. Bendayan, A. M. Rauth and X. Y. Wu, Development of solid lipid nanoparticles containing ionically complexed chemotherapeutic drugs and chemosensitizers, J. Pharm. Sci., 2004, 93(8), 1993–2008 CrossRef CAS PubMed.
- Database., N. C. f. B. I. P. Sodium oleate, CID=23665730. https://pubchem.ncbi.nlm.nih.gov/compound/23665730.
- Database., N. C. f. B. I. P. Oleic acid, CID=445639. https://pubchem.ncbi.nlm.nih.gov/compound/445639.
- R. S. Kalhapure, C. Mocktar, D. R. Sikwal, S. J. Sonawane, M. K. Kathiravan, A. Skelton and T. Govender, Ion pairing with linoleic acid simultaneously enhances encapsulation efficiency and antibacterial activity of vancomycin in solid lipid nanoparticles, Colloids Surf., B, 2014, 117, 303–311 CrossRef CAS PubMed.
- G. Carneiro, E. L. Silva, L. A. Pacheco, E. M. de Souza-Fagundes, N. C. Correa, A. M. de Goes, M. C. de Oliveira and L. A. Ferreira, Formation of ion pairing as an alternative to improve encapsulation and anticancer activity of all-trans retinoic acid loaded in solid lipid nanoparticles, Int. J. Nanomed., 2012, 7, 6011–6020 CAS.
- W. Yin, Z. Dong, X. Chen, N. Finn and M. Z. Yates, Hydrophobic ion pairing to enhance encapsulation of water-soluble additives into CO2-swollen polymer microparticles, J. Supercrit. Fluids, 2007, 41(2), 293–298 CrossRef CAS.
- D. E. Kuehner, J. Engmann, F. Fergg, M. Wernick, H. W. Blanch and J. M. Prausnitz, Lysozyme Net Charge and Ion Binding in Concentrated Aqueous Electrolyte Solutions, J. Phys. Chem. B, 1999, 103(8), 1368–1374 CrossRef CAS.
- V. Spassov and L. Yan, A pH-dependent computational approach to the effect of mutations on protein stability, 2016, vol. 37 Search PubMed.
- G. Yu, J. Liu and J. Zhou, Mesoscopic coarse-grained simulations of hydrophobic charge induction chromatography (HCIC) for protein purification, AIChE J., 2015, 61(6), 2035–2047 CrossRef CAS.
- S. V. Mussi, R. C. Silva, M. C. Oliveira, C. M. Lucci, R. B. Azevedo and L. A. Ferreira, New approach to improve encapsulation and antitumor activity of doxorubicin loaded in solid lipid nanoparticles, Eur. J. Pharm. Sci., 2013, 48(1–2), 282–290 CrossRef CAS.
- Z. Zhou, C. Kennell, M. Jafari, J. Y. Lee, S. J. Ruiz-Torres, S. E. Waltz and J. H. Lee, Sequential delivery of erlotinib and doxorubicin for enhanced triple negative Breast cancer treatment using polymeric nanoparticle, Int. J. Pharm., 2017, 530(1–2), 300–307 CrossRef CAS PubMed.
- A. Siddiqui, V. Gupta, Y. Y. Liu and S. Nazzal, Doxorubicin and MBO-asGCS oligonucleotide loaded lipid nanoparticles overcome multidrug resistance in adriamycin resistant ovarian cancer cells (NCI/ADR-RES), Int. J. Pharm., 2012, 431(1–2), 222–229 CrossRef CAS PubMed.
- J. K. Guillory, Handbook of Pharmaceutical Salts: Properties, Selection, and Use Edited by P. Heinrich Stahl and Camile G. Wermuth. VHCA, Verlag Helvetica Chimica Acta, Zürich, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002. vix + 374 pp. 17.5 × 24.5 cm. ISBN 3-906390-26-8. $130.00, J. Med. Chem., 2003, 46(7), 1277 CrossRef CAS.
- W. Q. Tong and G. Whitesell, In situ salt screening--a useful technique for discovery support and preformulation studies, Pharm. Dev. Technol., 1998, 3(2), 215–223 CrossRef CAS PubMed.
- I. Ghosh and W. M. Nau, The strategic use of supramolecular pKa shifts to enhance the bioavailability of drugs, Adv. Drug Delivery Rev., 2012, 64(9), 764–783 CrossRef CAS PubMed.
- N. i. Saleh, A. L. Koner and W. M. Nau, Activation and Stabilization of Drugs by Supramolecular pKa Shifts: Drug-Delivery Applications Tailored for Cucurbiturils, Angew. Chem., Int. Ed., 2008, 47(29), 5398–5401 CrossRef CAS PubMed.
- O. Zupancic, G. Leonaviciute, H. T. Lam, A. Partenhauser, S. Podricnik and A. Bernkop-Schnurch, Development and in vitro evaluation of an oral SEDDS for desmopressin, Drug Delivery, 2016, 23(6), 2074–2083 CrossRef CAS PubMed.
- H. D. Lu, P. Rummaneethorn, K. D. Ristroph and R. K. Prud’homme, Hydrophobic Ion Pairing of Peptide Antibiotics for Processing into Controlled Release Nanocarrier Formulations, Mol. Pharm., 2018, 15(1), 216–225 CrossRef CAS PubMed.
- S. Zafar, L. M. Negi, A. K. Verma, V. Kumar, A. Tyagi, P. Singh, Z. Iqbal and S. Talegaonkar, Sterically stabilized polymeric nanoparticles with a combinatorial approach for multi drug resistant cancer: in vitro and in vivo investigations, Int. J. Pharm., 2014, 477(1–2), 454–468 CrossRef CAS PubMed.
- R. Rastogi, S. Anand and V. Koul, Evaluation of pharmacological efficacy of 'insulin-surfoplex' encapsulated polymer vesicles, Int. J. Pharm., 2009, 373(1–2), 107–115 CrossRef CAS PubMed.
- J. Fu, F. Sun, W. Liu, Y. Liu, M. Gedam, Q. Hu, C. Fridley, H. A. Quigley, J. Hanes and I. Pitha, Subconjunctival Delivery of Dorzolamide-Loaded Poly(ether-anhydride) Microparticles Produces Sustained Lowering of Intraocular Pressure in Rabbits, Mol. Pharm., 2016, 13(9), 2987–2995 CrossRef CAS PubMed.
- E. Imbuluzqueta, E. Elizondo, C. Gamazo, E. Moreno-Calvo, J. Veciana, N. Ventosa and M. J. Blanco-Prieto, Novel bioactive hydrophobic gentamicin carriers for the treatment of intracellular bacterial infections, Acta Biomater., 2011, 7(4), 1599–1608 CrossRef CAS.
- M. Gallarate, M. Trotta, L. Battaglia and D. Chirio, Cisplatin-loaded SLN produced by coacervation technique, J. Drug Delivery Sci. Technol., 2010, 20(5), 343–347 CrossRef CAS.
- M. Gallarate, L. Battaglia, E. Peira and M. Trotta, Peptide-Loaded Solid Lipid Nanoparticles Prepared through Coacervation Technique, Int. J. Chem. Eng., 2011, 2011, 6 Search PubMed.
- H. Sang Yoo and T. Gwan Park, Biodegradable nanoparticles containing protein-fatty acid complexes for oral delivery of salmon calcitonin, J. Pharm. Sci., 2004, 93(2), 488–495 CrossRef PubMed.
- J. Griesser, G. Hetenyi, M. Moser, F. Demarne, V. Jannin and A. Bernkop-Schnurch, Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems, Int. J. Pharm., 2017, 520(1–2), 267–274 CrossRef CAS PubMed.
- H. Yuan, S. P. Jiang, Y. Z. Du, J. Miao, X. G. Zhang and F. Q. Hu, Strategic approaches for improving entrapment of hydrophilic peptide drugs by lipid nanoparticles, Colloids Surf., B, 2009, 70(2), 248–253 CrossRef CAS PubMed.
- V. Jain, D. Singodia, G. K. Gupta, D. Garg, G. B. S. Keshava, R. Shukla, P. K. Shukla and P. R. Mishra, Ciprofloxacin surf-plexes in sub-micron emulsions: A novel approach to improve payload efficiency and antimicrobial efficacy, Int. J. Pharm., 2011, 409(1), 237–244 CrossRef CAS.
- S. Ashton, Y. H. Song, J. Nolan, E. Cadogan, J. Murray, R. Odedra, J. Foster, P. A. Hall, S. Low, P. Taylor, R. Ellston, U. M. Polanska, J. Wilson, C. Howes, A. Smith, R. J. A. Goodwin, J. G. Swales, N. Strittmatter, Z. Takáts, A. Nilsson, P. Andren, D. Trueman, M. Walker, C. L. Reimer, G. Troiano, D. Parsons, D. De Witt, M. Ashford, J. Hrkach, S. Zale, P. J. Jewsbury and S. T. Barry, Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo, Sci. Transl. Med., 2016, 8(325), 325ra17 CrossRef PubMed.
- A. Patel, R. Gaudana and A. K. Mitra, A novel approach for antibody nanocarriers development through hydrophobic ion-pairing complexation, J. Microencapsulation, 2014, 31(6), 542–550 CrossRef CAS PubMed.
- R. Gaudana, V. Khurana, A. Parenky and A. K. Mitra, Encapsulation of Protein-Polysaccharide HIP Complex in Polymeric Nanoparticles, J. Drug Delivery, 2011, 2011 Search PubMed.
- R. D. Vaishya, A. Mandal, M. Gokulgandhi, S. Patel and A. K. Mitra, Reversible hydrophobic ion-paring complex strategy to minimize acylation of octreotide during long-term delivery from PLGA microparticles, Int. J. Pharm., 2015, 489(1–2), 237–245 CrossRef CAS PubMed.
- S. Sun, N. Liang, Y. Kawashima, D. Xia and F. Cui, Hydrophobic ion pairing of an insulin-sodium deoxycholate complex for oral delivery of insulin, Int. J. Nanomed., 2011, 6, 3049–3056 CAS.
- F. Hintzen, G. Perera, S. Hauptstein, C. Muller, F. Laffleur and A. Bernkop-Schnurch, In vivo evaluation of an oral self-microemulsifying drug delivery system (SMEDDS) for leuprorelin, Int. J. Pharm., 2014, 472(1–2), 20–26 CrossRef CAS PubMed.
- P. Ma, X. Dong, C. L. Swadley, A. Gupte, M. Leggas, H. C. Ledebur and R. J. Mumper, Development of idarubicin and doxorubicin solid lipid nanoparticles to overcome Pgp-mediated multiple drug resistance in leukemia, J. Biomed. Nanotechnol., 2009, 5(2), 151–161 CrossRef CAS PubMed.
- R. Mahjub, F. A. Dorkoosh, M. Rafiee-Tehrani and A. Bernkop Schnurch, Oral self-nanoemulsifying peptide drug delivery systems: impact of lipase on drug release, J. Microencapsulation, 2015, 32(4), 401–407 CrossRef CAS PubMed.
- C. Leichner, C. Menzel, F. Laffleur and A. Bernkop-Schnurch, Development and in vitro characterization of a papain loaded mucolytic self-emulsifying drug delivery system (SEDDS), Int. J. Pharm., 2017, 530(1–2), 346–353 CrossRef CAS.
- M. Ijaz, S. Bonengel, O. Zupancic, M. Yaqoob, M. Hartl, S. Hussain, C. W. Huck and A. Bernkop-Schnurch, Development of oral self nano-emulsifying delivery system(s) of lanreotide with improved stability against presystemic thiol-disulfide exchange reactions, Expert Opin. Drug Delivery, 2016, 13(7), 923–929 CrossRef CAS PubMed.
- A. Adjei, S. Rao, J. Garren, G. Menon and M. Vadnere, Effect of ion-pairing on 1-octanol-water partitioning of peptide drugs. I: the nonapeptide leuprolide acetate, Int. J. Pharm., 1993, 90(2), 141–149 CrossRef CAS.
- M. E. Powers, J. Matsuura, J. Brassell, M. C. Manning and E. Shefter, Enhanced solubility of proteins and peptides in nonpolar solvents through hydrophobic ion pairing, Biopolymers, 1993, 33(6), 927–932 CrossRef CAS.
- S. H. Choi and T. G. Park, Hydrophobic ion pair formation between leuprolide and sodium oleate for sustained release from biodegradable polymeric microspheres, Int. J. Pharm., 2000, 203(1–2), 193–202 CrossRef CAS PubMed.
- S. Sun, N. Liang, H. Piao, H. Yamamoto, Y. Kawashima and F. Cui, Insulin-S.O (sodium oleate) complex-loaded PLGA nanoparticles: formulation, characterization and in vivo evaluation, J. Microencapsulation, 2010, 27(6), 471–478 CrossRef CAS PubMed.
- H. M. Abdelaziz, A. O. Elzoghby, M. W. Helmy, M. W. Samaha, J. Y. Fang and M. S. Freag, Liquid crystalline assembly for potential combinatorial chemo-herbal drug delivery to lung cancer cells, Int. J. Nanomed., 2019, 14, 499–517 CrossRef CAS.
- G. Dalwadi and B. Sunderland, An ion pairing approach to increase the loading of hydrophilic and lipophilic drugs into PEGylated PLGA nanoparticles, Eur. J. Pharm. Biopharm., 2009, 71(2), 231–242 CrossRef CAS PubMed.
- M. S. Oliveira, S. V. Mussi, D. A. Gomes, M. I. Yoshida, F. Frezard, V. M. Carregal and L. A. M. Ferreira, alpha-Tocopherol succinate improves encapsulation and anticancer activity of doxorubicin loaded in solid lipid nanoparticles, Colloids Surf., B, 2016, 140, 246–253 CrossRef CAS PubMed.
- X. Zhang, X. Sun, J. Li, X. Zhang, T. Gong and Z. Zhang, Lipid nanoemulsions loaded with doxorubicin-oleic acid ionic complex: characterization, in vitro and in vivo studies, Pharmazie, 2011, 66(7), 496–505 CAS.
- G. A. Castro, R. L. Orefice, J. M. Vilela, M. S. Andrade and L. A. Ferreira, Development of a new solid lipid nanoparticle formulation containing retinoic acid for topical treatment of acne, J. Microencapsulation, 2007, 24(5), 395–407 CrossRef CAS PubMed.
- E. L. Silva, G. Carneiro, P. A. Caetano, G. Goulart, D. Ferreira Costa, E. M. de Souza-Fagundes, D. A. Gomes and L. A. Ferreira, Nanostructured lipid carriers loaded with tributyrin as an alternative to improve anticancer activity of all-trans retinoic acid, Expert Rev. Anticancer Ther., 2015, 15(2), 247–256 CrossRef CAS PubMed.
- L. Yang, F. Cui, K. Shi, D. Cun and R. Wang, Design of high payload PLGA nanoparticles containing melittin/sodium dodecyl sulfate complex by the hydrophobic ion-pairing technique, Drug Dev. Ind. Pharm., 2009, 35(8), 959–968 CrossRef CAS.
- C. Dumont, S. Bourgeois, H. Fessi, P. Y. Dugas and V. Jannin, In-vitro evaluation of solid lipid nanoparticles: Ability to encapsulate, release and ensure effective protection of peptides in the gastrointestinal tract, Int. J. Pharm., 2019, 565, 409–418 CrossRef CAS PubMed.
- K. Shi, F. Cui, H. Yamamoto and Y. Kawashima, Investigation of drug loading and in vitro release mechanisms of insulin-lauryl sulfate complex loaded PLGA nanoparticles, Pharmazie, 2008, 63(12), 866–871 CAS.
- H. Xu, L. Zhang, L. Li, Y. Liu, Y. Chao, X. Liu, Z. Jin, Y. Chen, X. Tang, H. He, Q. Kan and C. Cai, Membrane-Loaded Doxorubicin Liposomes Based on Ion-Pairing Technology with High Drug Loading and pH-Responsive Property, AAPS PharmSciTech, 2017, 18(6), 2120–2130 CrossRef CAS PubMed.
- H. D. Lu, K. D. Ristroph, E. L. K. Dobrijevic, J. Feng, S. A. McManus, Y. Zhang, W. D. Mulhearn, H. Ramachandruni, A. Patel and R. K. Prud'homme, Encapsulation of OZ439 into Nanoparticles for Supersaturated Drug Release in Oral Malaria Therapy, ACS Infect. Dis., 2018, 4(6), 970–979 CrossRef CAS PubMed.
- G. A. Castro, L. A. M. Ferreira, R. L. Oréfice and V. T. L. Buono, Characterization of a new solid lipid nanoparticle formulation containing retinoic acid for topical treatment of acne, Powder Diffr., 2012, 23(S1), S30–S35 CrossRef.
- E. L. Silva, F. A. Lima, G. Carneiro, P. Ramos Jonas, D. A. Gomes, E. M. de Souza-Fagundes and L. A. Ferreira, Improved In Vitro Antileukemic Activity of All-Trans Retinoic Acid Loaded in Cholesteryl Butyrate Solid Lipid Nanoparticles, J. Nanosci. Nanotechnol., 2016, 16(2), 1291–1300 CrossRef CAS PubMed.
- L. Battaglia, M. Gallarate, E. Peira, D. Chirio, E. Muntoni, E. Biasibetti, M. T. Capucchio, A. Valazza, P. P. Panciani, M. Lanotte, D. Schiffer, L. Annovazzi, V. Caldera, M. Mellai and C. Riganti, Solid lipid nanoparticles for potential doxorubicin delivery in glioblastoma treatment: preliminary in vitro studies, J. Pharm. Sci., 2014, 103(7), 2157–2165 CrossRef CAS PubMed.
- R. Falk, T. W Randolph, J. D Meyer, R. M. Kelly and M. Manning, Controlled release of ionic compounds from poly (L-lactide) microspheres produced by precipitation with a compressed antisolvent, 1997, vol. 44, pp. 77–85 Search PubMed.
- E. Elizondo, S. Sala, E. Imbuluzqueta, D. Gonzalez, M. J. Blanco-Prieto, C. Gamazo, N. Ventosa and J. Veciana, High loading of gentamicin in bioadhesive PVM/MA nanostructured microparticles using compressed carbon-dioxide, Pharm. Res., 2011, 28(2), 309–321 CrossRef CAS PubMed.
- E. Imbuluzqueta, S. Lemaire, C. Gamazo, E. Elizondo, N. Ventosa, J. Veciana, F. Van Bambeke and M. J. Blanco-Prieto, Cellular pharmacokinetics and intracellular activity against Listeria monocytogenes and Staphylococcus aureus of chemically modified and nanoencapsulated gentamicin, J. Antimicrob. Chemother., 2012, 67(9), 2158–2164 CrossRef CAS PubMed.
- R. Gaudana, A. Parenky, R. Vaishya, S. K. Samanta and A. K. Mitra, Development and characterization of nanoparticulate formulation of a water soluble prodrug of dexamethasone by HIP complexation, J. Microencapsulation, 2011, 28(1), 10–20 CrossRef CAS PubMed.
- S. Zhao, L. V. Minh, N. Li, V. M. Garamus, U. A. Handge, J. Liu, R. Zhang, R. Willumeit-Romer and A. Zou, Doxorubicin hydrochloride-oleic acid conjugate loaded nanostructured lipid carriers for tumor specific drug release, Colloids Surf., B, 2016, 145, 95–103 CrossRef CAS PubMed.
- K. Fu, D. W. Pack, A. M. Klibanov and R. Langer, Visual Evidence of Acidic Environment Within Degrading Poly(lactic-co-glycolic acid) (PLGA) Microspheres, Pharm. Res., 2000, 17(1), 100–106 CrossRef CAS PubMed.
- E. D. Goddard, Polymer/surfactant interaction—Its relevance to detergent systems, J. Am. Oil Chem. Soc., 1994, 71(1), 1–16 CrossRef CAS.
- J. T. G. Overbeek and M. J. Voorn, Phase separation in polyelectrolyte solutions. Theory of complex coacervation, J. Cell. Comp. Physiol., 1957, 49(S1), 7–26 CrossRef CAS.
- J. v. d. Gucht, E. Spruijt, M. Lemmers and M. A. Cohen Stuart, Polyelectrolyte complexes: Bulk phases and colloidal systems, J. Colloid Interface Sci., 2011, 361(2), 407–422 CrossRef PubMed.
- N. R. Johnson and Y. Wang, Coacervate delivery systems for proteins and small molecule drugs, Expert Opin. Drug Delivery, 2014, 11(12), 1829–1832 CrossRef CAS PubMed.
- J. Lee, Y. O. Popov and G. H. Fredrickson, Complex coacervation: A field theoretic simulation study of polyelectrolyte complexation, J. Chem. Phys., 2008, 128(22), 224908 CrossRef PubMed.
- K. T. Delaney and G. H. Fredrickson, Theory of polyelectrolyte complexation—complex coacervates are self-coacervates, J. Chem. Phys., 2017, 146(22), 224902 CrossRef PubMed.
- P. Pincus, Colloid stabilization with grafted polyelectrolytes, Macromolecules, 1991, 24(10), 2912–2919 CrossRef CAS.
- Y. O. Popov, J. Lee and G. H. Fredrickson, Field-theoretic simulations of polyelectrolyte complexation, J. Polym. Sci., Part B: Polym. Phys., 2007, 45(24), 3223–3230 CrossRef CAS.
- M. Muthukumar, 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions, Macromolecules, 2017, 50(24), 9528–9560 CrossRef CAS PubMed.
- P. Guenoun, H. T. Davis, M. Tirrell and J. W. Mays, Aqueous Micellar Solutions of Hydrophobically Modified Polyelectrolytes, Macromolecules, 1996, 29(11), 3965–3969 CrossRef CAS.
- Q. Wang and J. B. Schlenoff, The Polyelectrolyte Complex/Coacervate Continuum, Macromolecules, 2014, 47(9), 3108–3116 CrossRef CAS.
- W. Xiong, C. Ren, W. Jin, J. Tian, Y. Wang, B. R. Shah, J. Li and B. Li, Ovalbumin-chitosan complex coacervation: Phase behavior, thermodynamic and rheological properties, Food Hydrocolloids, 2016, 61, 895–902 CrossRef CAS.
- Y. Zhang, F. Li, L. D. Valenzuela, M. Sammalkorpi and J. L. Lutkenhaus, Effect of Water on the Thermal Transition Observed in Poly(allylamine hydrochloride)–Poly(acrylic acid) Complexes, Macromolecules, 2016, 49(19), 7563–7570 CrossRef CAS.
- H. K. Awada, N. R. Johnson and Y. Wang, Dual Delivery of Vascular Endothelial Growth Factor and Hepatocyte Growth Factor Coacervate Displays Strong Angiogenic Effects, Macromol. Biosci., 2014, 14(5), 679–686 CrossRef CAS PubMed.
- H. Bysell, P. Hansson and M. Malmsten, Effect of Charge Density on the Interaction between Cationic Peptides and Oppositely Charged Microgels, J. Phys. Chem. B, 2010, 114(21), 7207–7215 CrossRef CAS PubMed.
- A. Nolles, A. H. Westphal, J. A. de Hoop, R. G. Fokkink, J. M. Kleijn, W. J. H. van Berkel and J. W. Borst, Encapsulation of GFP in Complex Coacervate Core Micelles, Biomacromolecules, 2015, 16(5), 1542–1549 CrossRef CAS PubMed.
- C. B. Packhaeuser and T. Kissel, On the design of in situ forming biodegradable parenteral depot systems based on insulin loaded dialkylaminoalkyl-amine-poly(vinyl alcohol)-g-poly(lactide-co-glycolide) nanoparticles, J. Controlled Release, 2007, 123(2), 131–140 CrossRef CAS PubMed.
- A. Obermeyer, E. C. Mills, X.-H. Dong, R. J. Flores and B. Olsen, Complex coacervation of supercharged proteins with polyelectrolytes, 2016, vol. 12 Search PubMed.
- K. A. Black, D. Priftis, S. L. Perry, J. Yip, W. Y. Byun and M. Tirrell, Protein Encapsulation via Polypeptide Complex Coacervation, ACS Macro Lett., 2014, 3(10), 1088–1091 CrossRef CAS.
- M. N. Jones, The interaction of sodium dodecyl sulfate with polyethylene oxide, J. Colloid Interface Sci., 1967, 23(1), 36–42 CrossRef CAS.
- E. D. Goddard and R. B. Hannan, Cationic polymer/anionic surfactant interactions, J. Colloid Interface Sci., 1976, 55(1), 73–79 CrossRef CAS.
- R. Y. Lochhead and L. R. Huisinga, A brief review of polymer/surfactant interaction, Feb 2004 Search PubMed.
- M. S. Johal and P. A. Chiarelli, Polymer–surfactant complexation in polyelectrolyte multilayer assemblies, Soft Matter, 2007, 3(1), 34–46 RSC.
- E. D. Goddard and R. B. Hannan, Polymer/surfactant interactions, J. Am. Oil Chem. Soc., 1977, 54(12), 561–566 CrossRef CAS.
- M. Goswami, J. M. Borreguero, P. A. Pincus and B. G. Sumpter, Surfactant-Mediated Polyelectrolyte Self-Assembly in a Polyelectrolyte–Surfactant Complex, Macromolecules, 2015, 48(24), 9050–9059 CrossRef CAS.
- Y. Wang, K. Kimura, Q. Huang, P. L. Dubin and W. Jaeger, Effects of Salt on Polyelectrolyte–Micelle Coacervation, Macromolecules, 1999, 32(21), 7128–7134 CrossRef CAS.
- M. Manuszak Guerrini, R. Lochhead and W. Daly, Interactions of aminoalkylcarbamoyl cellulose derivatives and sodium dodecyl sulfate. 2. Foam stabilization, 1999, vol. 147, pp. 67–78 Search PubMed.
- M. Antonietti, J. Conrad and A. Thuenemann, Polyelectrolyte-Surfactant Complexes: A New Type of Solid, Mesomorphous Material, Macromolecules, 1994, 27(21), 6007–6011 CrossRef CAS.
- J. Fundin and W. Brown, Polymer/Surfactant Interactions. Sodium Poly(styrene sulfonate) and CTAB Complex Formation. Light Scattering Measurements in Dilute Aqueous Solution, Macromolecules, 1994, 27(18), 5024–5031 CrossRef CAS.
- Â. M. L. Denadai, A. M. de Oliveira, I. M. P. Daniel, L. A. Carneiro, K. C. Ribeiro, H. d. O. Beraldo, K. J. R. da Costa, V. C. da Cunha, M. E. Cortés and R. D. Sinisterra, Chlorhexidine/losartan ionic pair binding and its nanoprecipitation: physico-chemical characterisation and antimicrobial activity, Supramol. Chem., 2012, 24(3), 204–212 CrossRef.
- W. Li, X. Yi, X. Liu, Z. Zhang, Y. Fu and T. Gong, Hyaluronic acid ion-pairing nanoparticles for targeted tumor therapy, J. Controlled Release, 2016, 225, 170–182 CrossRef CAS PubMed.
- K. D. Ristroph, J. Feng, S. A. McManus, Y. Zhang, K. Gong, H. Ramachandruni, C. E. White and R. K. Prud’homme, Spray drying OZ439 nanoparticles to form stable, water-dispersible powders for oral malaria therapy, J. Transl. Med., 2019, 17(1), 97 CrossRef PubMed.
- I. Lozoya-Agullo, M. Planelles, M. Merino-Sanjuán, M. Bermejo, B. Sarmento, I. González-Álvarez and M. González-Álvarez, Ion-pair approach coupled with nanoparticle formation to increase bioavailability of a low permeability charged drug, Int. J. Pharm., 2019, 557, 36–42 CrossRef CAS PubMed.
- C. Tang, E. Zhang, Y. Li and L. Yang, An innovative method for preparation of hydrophobic ion-pairing colistin entrapped poly(lactic acid) nanoparticles: Loading and release mechanism study, Eur. J. Pharm. Sci., 2017, 102, 63–70 CrossRef CAS PubMed.
- J. Liu, T. Gong, C. Wang, Z. Zhong and Z. Zhang, Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: preparation and characterization, Int. J. Pharm., 2007, 340(1–2), 153–162 CrossRef CAS PubMed.
- G. P. Zara, R. Cavalli, A. Fundaro, A. Bargoni, O. Caputo and M. R. Gasco, Pharmacokinetics of doxorubicin incorporated in solid lipid nanospheres (SLN), Pharmacol. Res., 1999, 40(3), 281–286 CrossRef CAS PubMed.
- S. Morel, E. Ugazio, R. Cavalli and M. R. Gasco, Thymopentin in solid lipid nanoparticles, Int. J. Pharm., 1996, 132(1), 259–261 CrossRef CAS.
- R. Cavalli, M. R. Gasco, P. Chetoni, S. Burgalassi and M. F. Saettone, Solid lipid nanoparticles (SLN) as ocular delivery system for tobramycin, Int. J. Pharm., 2002, 238(1–2), 241–245 CrossRef CAS PubMed.
- M. S. Freag, A. S. Torky, M. M. Nasra, D. A. Abdelmonsif and O. Y. Abdallah, Liquid crystalline nanoreservoir releasing a highly skin-penetrating berberine oleate complex for psoriasis management, Nanomedicine, 2019, 14(8), 931–954 CrossRef CAS PubMed.
- S. Sun, N. Liang, H. Yamamoto, Y. Kawashima, F. Cui and P. Yan, pH-sensitive poly(lactide-co-glycolide) nanoparticle composite microcapsules for oral delivery of insulin, Int. J. Nanomed., 2015, 10, 3489–3498 CrossRef CAS PubMed.
- H. Zhao, H. Lu, T. Gong and Z. Zhang, Nanoemulsion loaded with lycobetaine-oleic acid ionic complex: physicochemical characteristics, in vitro, in vivo evaluation, and antitumor activity, Int. J. Nanomed., 2013, 8, 1959–1973 CrossRef PubMed.
- T. Zhang, Y. Zheng, Q. Peng, X. Cao, T. Gong and Z. Zhang, A novel submicron emulsion system loaded with vincristine-oleic acid ion-pair complex with improved anticancer effect: in vitro and in vivo studies, Int. J. Nanomed., 2013, 8, 1185–1196 CrossRef PubMed.
- I. Nazir, M. H. Asim, A. Dizdarevic and A. Bernkop-Schnurch, Self-emulsifying drug delivery systems: Impact of stability of hydrophobic ion pairs on drug release, Int. J. Pharm., 2019, 561, 197–205 CrossRef CAS PubMed.
- N. Mittapelly, M. Thalla, G. Pandey, V. T. Banala, S. Sharma, A. Arya, S. Mishra, K. Mitra, S. Shukla and P. R. Mishra, Long Acting Ionically Paired Embonate Based Nanocrystals of Donepezil for the Treatment of Alzheimer's Disease: a Proof of Concept Study, Pharm. Res., 2017, 34(11), 2322–2335 CrossRef CAS PubMed.
- S. J. Novick and J. S. Dordick, Protein-containing hydrophobic coatings and films, Biomaterials, 2002, 23(2), 441–448 CrossRef CAS PubMed.
- L. Battaglia, M. Gallarate, E. Peira, D. Chirio, I. Solazzi, S. Marzia Adele Giordano, C. Gigliotti, C. Riganti and C. Dianzani, Bevacizumab loaded solid lipid nanoparticles prepared by the coacervation technique: Preliminary in vitro studies. 2015, vol. 26, p. 255102 Search PubMed.
- N. Lupo, V. N. Tkadlečková, M. Jelkmann, F. Laffleur, G. Hetényi, K. Kubová and A. Bernkop-Schnürch, Self-emulsifying drug delivery systems: In vivo evaluation of their potential for oral vaccination, Acta Biomater., 2019 Search PubMed.
- J. Griesser, G. Hetenyi, H. Kadas, F. Demarne, V. Jannin and A. Bernkop-Schnurch, Self-emulsifying peptide drug delivery systems: How to make them highly mucus permeating, Int. J. Pharm., 2018, 538(1–2), 159–166 CrossRef CAS PubMed.
- R. Falk and T. W. Randolph, Process Variable Implications for Residual Solvent Removal and Polymer Morphology in the Formation of Gentamycin-Loaded Poly (L-lactide) Microparticles. 1998; vol. 15, pp. 1233–7 Search PubMed.
- R. Alcock, J. A. Blair, D. J. O'Mahony, A. Raoof and A. V. Quirk, Modifying the release of leuprolide from spray dried OED microparticles, J. Controlled Release, 2002, 82(2–3), 429–440 CrossRef CAS PubMed.
- A. D. Holmkvist, A. Friberg, U. J. Nilsson and J. Schouenborg, Hydrophobic ion pairing of a minocycline/Ca2+/AOT complex for preparation of drug-loaded PLGA nanoparticles with improved sustained release, Int. J. Pharm., 2016, 499(1), 351–357 CrossRef CAS PubMed.
- J. T. Evans, J. R. Ward, J. Kern and M. E. Johnson, A single vaccination with protein-microspheres elicits a strong CD8 T-cell-mediated immune response against Mycobacterium tuberculosis antigen Mtb8.4, Vaccine, 2004, 22(15–16), 1964–1972 CrossRef CAS PubMed.
- M. Hill, R. N. Cunningham, R. M. Hathout, C. Johnston, J. G. Hardy and M. E. Migaud, Formulation of Antimicrobial Tobramycin Loaded PLGA Nanoparticles via Complexation with AOT, J. Funct. Biomater., 2019, 10(2), 26 CrossRef PubMed.
- N. A. Efiana, A. Dizdarevic, C. W. Huck and A. Bernkop-Schnurch, Improved intestinal mucus permeation of vancomycin via incorporation into nanocarrier containing papain-palmitate, J. Pharm. Sci., 2019 Search PubMed.
- K. Shi, F. Cui, H. Yamamoto and Y. Kawashima, Optimized preparation of insulin-lauryl sulfate complex loaded poly (lactide-co-glycolide) nanoparticles using response surface methodology, Pharmazie, 2008, 63(10), 721–725 CAS.
- K. Shi, F. Cui, H. Yamamoto and Y. Kawashima, Optimized formulation of high-payload PLGA nanoparticles containing insulin-lauryl sulfate complex, Drug Dev. Ind. Pharm., 2009, 35(2), 177–184 CrossRef CAS PubMed.
- H. Zhou, Y. Zhang, D. L. Biggs, M. C. Manning, T. W. Randolph, U. Christians, B. M. Hybertson and K. Y. Ng, Microparticle-based lung delivery of INH decreases INH metabolism and targets alveolar macrophages, J. Controlled Release, 2005, 107(2), 288–299 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |