
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsocyanic acid (HNCO) and its fate in the atmosphere: a review
Michael David
Leslie
 a,
Melanie
Ridoli
b,
Jennifer Grace
Murphy
a and
Nadine
Borduas-Dedekind
*b
a,
Melanie
Ridoli
b,
Jennifer Grace
Murphy
a and
Nadine
Borduas-Dedekind
*b
aDepartment of Chemistry, University of Toronto, 80 St George Street, Toronto, Ontario M5S 3H6, Canada
bDepartment of Environmental Science Systems, ETH Zürich, Universitätstrasse 16, Zürich, 8092, Switzerland. E-mail: nadine.borduas@usys.ethz.ch; Web: http://www.twitter.com/nadineborduas
First published on 26th March 2019
Abstract
Isocyanic acid (HNCO) has recently been identified in ambient air at potentially concerning concentrations for human health. Since its first atmospheric detection, significant progress has been made in understanding its sources and sinks. The chemistry of HNCO is governed by its partitioning between the gas and liquid phases, its weak acidity, its high solubility at pH above 5, and its electrophilic chemical behaviour. The online measurement of HNCO in ambient air is possible due to recent advances in mass spectrometry techniques, including chemical ionization mass spectrometry for the detection of weak acids. To date, HNCO has been measured in North America, Europe and South Asia as well as outdoors and indoors, with mixing ratios up to 10s of ppbv. The sources of HNCO include: (1) fossil fuel combustion such as coal, gasoline and diesel, (2) biomass burning such as wildfires and crop residue burning, (3) secondary photochemical production from amines and amides, (4) cigarette smoke, and (5) combustion of materials in the built environment. Then, three losses processes can occur: (1) gas phase photochemistry, (2) heterogenous uptake and hydrolysis, and (3) dry deposition. HNCO lifetimes with respect to photolysis and OH radical oxidation are on the order of months to decades. Consequently, the removal of HNCO from the atmosphere is thought to occur predominantly by dry deposition and by heterogeneous uptake followed by hydrolysis to NH3 and CO2. A back of the envelope calculation reveals that HNCO is an insignificant global source of NH3, contributing only around 1%, but could be important for local environments. Furthermore, HNCO can react due to its electrophilic behaviour with various nucleophilic functionalities, including those present in the human body through a reaction called protein carbamoylation. This protein modification can lead to toxicity, and thus exposure to high concentrations of HNCO can lead to cardiovascular and respiratory diseases, as well as cataracts. In this critical review, we outline our current understanding of the atmospheric fate of HNCO and its potential impacts on outdoor and indoor air quality. We also call attention to the need for toxicology studies linking HNCO exposure to health effects.
 Michael Leslie | Michael Leslie is in the second year of his Master's degree in environmental chemistry at the University of Toronto. He received his honours B.Sc. from the University of Toronto in 2017. Currently, he is working on a project studying the removal processes and specifically the aqueous reactivity of isocyanic acid (HNCO) in the atmosphere. |
 Melanie Ridoli | Melanie Ridoli obtained her Master's degree in environmental science from ETH Zürich in 2018, majoring in biogeochemistry and pollutant dynamics. Her Master's research focused on methane entrapped in calcareous glacier forefields. She is interested in natural hazard management and is currently working as a trainee in an insurance company in Switzerland. |
 Jennifer Murphy | Jennifer Murphy is a professor in the Department of Chemistry at the University of Toronto, where she held a Canada Research Chair in Atmospheric and Environmental Chemistry from 2007–2016. She completed her BSc in Chemistry at McGill University and her PhD in Chemistry at the University of California, Berkeley. Her research program focuses mainly on understanding the sources and sinks of reactive nitrogen in the atmosphere and on biosphere–atmosphere exchange. She serves as a member of the Scientific Steering Committee of the International Global Atmospheric Chemistry project. |
 Nadine Borduas-Dedekind | Nadine Borduas-Dedekind is an SNF Ambizione Fellow in the Department of Environmental Systems Science at ETH Zürich where she leads a group in organic aerosol chemistry. She completed her PhD as a Vanier Scholar at the University of Toronto in 2015, working on the oxidative fate of organic nitrogen in the atmosphere. She subsequently completed an NSERC postdoctoral fellowship in organic aerosol photochemistry at ETH Zürich. Her research interests include bringing an organic chemistry perspective to atmospheric processes. |
Environmental significanceNew advances in instrumentation has recently allowed for the detection of isocyanic acid (HNCO) in ambient air at concentrations potentially able to cause adverse health effects. We review the current knowledge of the atmospheric fate of HNCO by discussing and synthesizing its sources, sinks and chemical transformations in the atmosphere. Recent research has identified coal combustion, biomass burning, photochemical transformation of amines and amides, cigarette smoke and building material combustion as sources of HNCO to the atmosphere. HNCO is a weak acid and can partition to the aqueous particle phase and undergo hydrolysis. Based on its thermodynamic data, its atmospheric lifetime can range from hours to months and may contribute to a small local effect on the N budget. |
1. Introduction
Isocyanic acid (HNCO) was first identified and characterized in the 19th century when Liebig and Wöhler investigated its stability, structure and chemical production pathways in solution.1 They speculated on two tautomers, NCOH and HNCO, and proposed the latter formula of HNCO based on its reactivity with ammonia (NH3) to form urea.HNCO is an organic acid with a pKa of 3.7 (at 298 K)2–4 and a nearly-linear structure through the π-system of the N, C and O atoms, experimentally confirmed by microwave and infrared absorption spectroscopy and molecular modelling (Fig. 1).5–8 Until recently, there was limited knowledge regarding the behaviour of HNCO in the atmosphere including its sources, sinks, ambient concentrations, and exposure and risks related to human health.
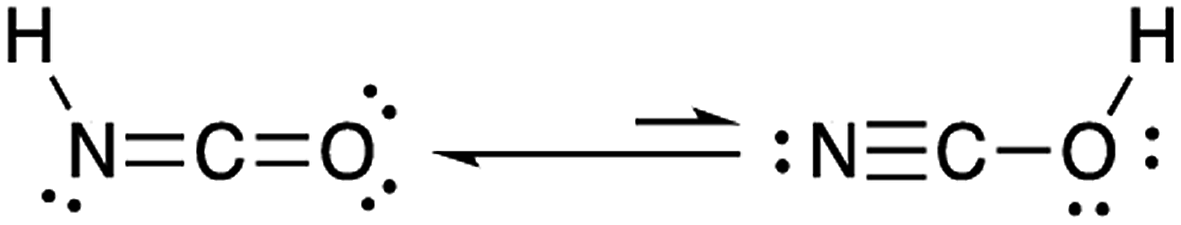 | ||
| Fig. 1 Lewis and resonance structures of HNCO. | ||
Biochemical studies identify NCO− as a toxic biomolecule due to post-translational non-enzymatic protein modifications called carbamoylation.9–15 Both HNCO and its conjugate base, NCO−, have been observed to be intermediates of carbamoylation reactions, the reaction of a nucleophile, such as an amine on a lysine residue, with the electrophilic C atom of HNCO.9,16,17 Note that the carbamylation term is specific for a reaction between an amine and CO2 yielding carbamates whereas the carbamoylation term involves the reaction between an amine and other electrophilic C substrates such as HNCO.15,18 For instance, a nucleophilic amine could be at the terminal end of a protein or on the side chain of a lysine residue. Carbamoylation can result in the development of cataracts, cardiovascular disease, and rheumatoid arthritis and can thus be potentially dangerous to humans.9–13 However, no study directly links inhalation exposure of HNCO to adverse health effects. Nonetheless, using Henry's law constant of HNCO, an estimated exposure to a mixing ratio of 1 ppbv (parts per billion by volume) through inhalation could lead to blood concentrations of 100 μM,19 equivalent to threshold concentrations for protein carbamoylation in vitro.14 Despite the potential for toxicity, no ambient air quality standards or exposure limits for HNCO currently exist.
The recent development of both chemical ionization mass spectrometry (CIMS) using reagent ions (e.g. acetate or iodide) capable of detecting acids and proton transfer reaction-mass spectrometry has allowed for the measurement and quantification of HNCO in the atmosphere.20–25 With a growing dataset of HNCO measurements, atmospheric chemists are able to investigate the sources and sinks of HNCO. This weak acid is a tracer of the oxidation of organo-nitrogen compounds as well as a potential source of NH3 to the atmosphere. An improved understanding of HNCO in the atmosphere will better inform local and regional estimates of potential risks of exposure.
HNCO is emitted into the atmosphere from primary and secondary processes. Many forms of combustion including biomass burning,19,20,22,26–29 fossil fuel combustion,19,30–37 and cigarette smoke;19,38,39 are all known to produce HNCO. Secondary sources such as the oxidation of atmospheric amines and amides have also been identified as sources of HNCO.40–45
There is little direct evidence for the loss processes of HNCO in the atmosphere. The atmospheric lifetime of HNCO by photolysis and by oxidation by OH radicals is on the order of months to decades.46,47 We thus expect that the main removal processes in the atmosphere are dry deposition and heterogeneous uptake to water followed by either hydrolysis or wet deposition.2
This review synthesizes the existing data on the atmospheric fate of HNCO. Ambient measurement data are summarized and the individual processes contributing to the sources and sinks are discussed. In addition, modelling simulations looking at potential atmospheric concentrations are described. Finally, the toxicology and risks of exposure to HNCO are discussed with a call for further investigations.
2. Ambient measurements
Prior to 2010, the detection and measurement of atmospheric HNCO relied on methods such as sample derivatization,48 selective hydrolysis followed by the detection of NH3 (ref. 49) or FTIR.27 While these methods are able to detect gas phase HNCO, they are neither fast nor sensitive enough to measure real-time concentrations. The recent development of the chemical ionization mass spectrometer (CIMS) enabled the selective detection of organic acids by using acetate as the reagent ion. This soft ionization through a proton transfer reaction led to the detection and quantification as NCO−.20 This method is sufficiently fast, selective, and sensitive (detection limits reaching 0.005 ppbv), while not requiring labour-intensive collection or preparation steps. An iodide reagent ion CIMS technique has also been developed for HNCO.50 It allows for greater signal-to-noise ratios by providing a lower and more stable background signal, yet at the expense of lower sensitivities.50 In parallel to the development of CIMS techniques for organic acids, a technique employing a proton-transfer reaction mass spectrometer (PTR-MS) was also developed for detecting high concentrations of HNCO within a health and safety context for workplace exposure, particularly in the metal work and welding industries. This instrument also uses soft ionisation through proton transfer but from hydronium ions (H3O+) and can detect HNCO mixing ratios from low ppbv up to 1000s of ppbv.23–25,51–53Online CIMS techniques with acetate and iodide reagent ions and PTR-MS instruments have facilitated atmospheric measurements of HNCO providing observations from field campaigns from 10 locations across three continents (Pasadena, California;19 Erie, Colorado;26,54 La Jolla, California;55 Fort Collins, Colorado;26 Toronto, Ontario;30,56 Calgary, Alberta;50 the marine boundary layer in the Canadian Arctic Archipelago;57 Mohali, India;51 Kathmandu Valley, Nepal;52 and Manchester, UK58) (Table 1). The number of ambient measurements continues to grow, yet long term datasets are still missing (Fig. 2).
| Location | Reagent ion/MS | Ave. Mixing Ratio (ppbv) | Reference |
|---|---|---|---|
| Mohali, India | H3O+/quad | 0.940 (mean) | Chandra et al. 201651 |
| Kathmandu Valley, Nepal | H3O+/ToF | 0.900 (mean) | Sarkar et al. 201652 |
| Toronto, Ontario | Acetate/ToF | Fall: 0.100 (mean), 0.080 (median) | Hems et al. 2019104 |
| Acetate/quad | Summer: 0.085 (mean) | Wentzell et al. 201330 | |
| Iodide/ToF | Summer: 0.045 (mean), 0.039 (median) | Wren et al. 201856 | |
| Winter: 0.025 (mean), 0.015 (median) | |||
| La Jolla, California | Acetate/quad | 0.081 (mean) | Zhao et al. 201455 |
| Erie, Colorado | Acetate/quad | Winter: 0.072 (mean), 0.065 (median) | Roberts et al. 201426 |
| Acetate/ToF | Summer: 0.030 (mean) | Mattila et al. 201854 | |
| Fort Collins, Colorado | Acetate/quad | 0.055 (mean), 0.050 (median) | Roberts et al. 201426 |
| Calgary, Alberta | Acetate & iodide/quad | 0.036 (mean), 0.034 (median) | Woodward-Massey et al. 201450 |
| Winter: 0.028 (mean), 0.033 (median) | |||
| Pasadena, California | Acetate/quad | Summer: 0.025 (mean), 0.022 (median) | Roberts et al. 2011, 201419,26 |
| Canadian Arctic Archipelago | Acetate/ToF | 0.020 (mean), 0.016 (median) | Mungall et al. 201757 |
| Manchester, UK | Iodide/ToF | 0.012 (mean) | Priestley et al. 201858 |
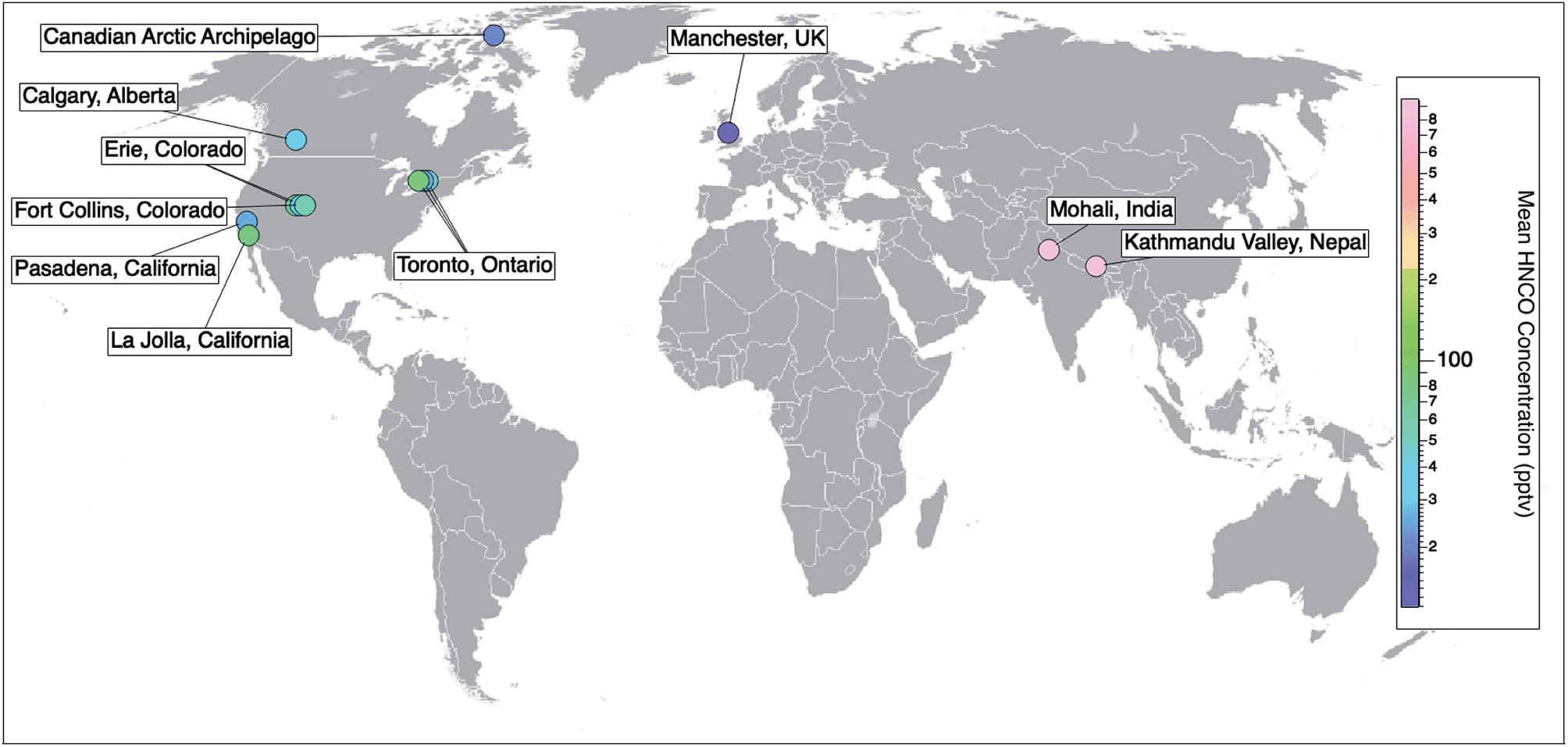 | ||
| Fig. 2 Spatial distribution of ambient measurements of HNCO from currently published data. Field campaigns reporting HNCO concentrations include 10 locations across three continents (Pasadena, California;15 Erie, Colorado;19,47 La Jolla, California;48 Fort Collins, Colorado;19 Toronto, Ontario;23,49 Calgary, Alberta;43 the marine boundary layer in the Canadian Arctic Archipelago;50 Mohali, India;51 Kathmandu Valley, Nepal;52 and Manchester, UK53). Markers are coloured with respect to their measured mean HNCO concentrations (Table 1). To add to this compilation, there is also the opportunity to look back at existing CIMS datasets and quantify HNCO to gather a more extensive network of data. | ||
Furthermore, HNCO ambient measurements by mass spectrometry present challenges particularly in its calibration. Indeed, HNCO is not commercially available as it polymerizes at high concentrations. It therefore is synthesized in house from thermolyzing solid cyanuric acid at 250 °C.19 Nonetheless, it is well behaved inside Teflon tubing and does not sorb to surfaces compared to amines.43 HNCO also does not require a heated inlet for detection. Both iodide CIMS and PTR-MS have an HNCO signal that is humidity dependent, which can complicate the quantification of the signal particularly while working outdoors.50,59 In contrast, acetate CIMS has a negligible humidity dependence and is thus a preferred measurement technique for applications to ambient air.20
HNCO exhibits different diurnal profiles at different measurement locations, and Roberts et al. 2014 specifically compare diurnal profiles from three data sets.26 In Pasadena, the maximum mixing ratio is observed in the early afternoon (1–2 pm), with a steady decrease into a minimum in the early morning (4–6 am).19,26 At this site, a midday peak is common for many pollution tracers because of the time required to transport air masses influenced by morning rush hour emissions in downtown Los Angeles.60 In the Fort Collins and Manchester data sets, there is an early morning minimum with increasing concentrations throughout the day to a late afternoon maximum.26,58 One hypothesis for the late afternoon peak observed in some datasets is the production of HNCO through secondary photochemical sources.26 Support for secondary production comes from two lines of evidence. First, the diurnal profiles of HNCO and common photochemically produced species, such as HNO3, O3, and oxygenated VOCs,19,55 correlate well with each other. Second, laboratory experiments have observed HNCO production during the oxidation of amines and amides in the gas phase.40–45 Measurements made in La Jolla, Mohali, and Bode all have similar diurnal profiles with early morning minimum (4–6 am) and late morning (10–12 am) maximum mixing ratios.51,52,55 The summertime data set from the Boulder Atmospheric Observatory tower in Erie, CO54 shows a diurnal cycle similar to that of Pasadena, with an early morning minimum and early afternoon maximum.54 The Toronto data similarly has minimum mixing ratios in the early morning but shows two distinct maxima throughout the day; one in mid-morning (8 am, local max) and one in the early evening (7 pm, absolute max).30 These measurements were made along the roadside and suggest vehicle emission as a primary source of HNCO. These measurements are in contrast to the measurements in the winter in Calgary, in the winter at the Boulder Atmospheric Observatory tower in Erie, CO26 and in the Canadian Arctic, where no clear diurnal trend has been observed.26,50,57
A common theme among data collected at ground sites is that observations made during the summer and fall months all had diurnally varying concentration profiles,26,30,51,55 while observations during the winter months did not.26 Reduced photochemistry in the winter months could explain the lack of diurnal variation observed during measurements. Further discussion of secondary HNCO production is in Section 3.4.
In comparison to outdoor air, less is known about HNCO in indoor air. However, larger isocyanates such as toluene diisocyanate and 4,4′-methylene diphenyl diisocyanate have known serious health effects such as asthma, methemoglobinemia and respiratory cancers and are thus regulated for occupational and health in the workplace.61–63 HNCO is the smallest molecule within the class of isocyanates and it is unclear if it triggers similar health effects as its phenyl counterparts. Nonetheless, HNCO has been quantified in the workplace particularly in the welding, paint, and transport industry, where mixing ratios up to 4.4 ppbv have been measured (Table 2).64 Mixing ratios as high as 940 ppbv have been measured indoors from heating polyurethane paint used in marine applications.25
| Indoor activity | Reagent ion/MS | Ave. Mix. Rat. (ppbv) | Reference |
|---|---|---|---|
| Thermal decomposition of polymer paint | H3O+/quad | <940 | Jankowski et al. 201625 |
| Plasma welding of HDI-painted sheets of steel | H3O+/quad | 3.440 (mean) | Jankowski et al. 201424 |
| Foundry | H3O+/quad | 0.140–4.400 (range) | Jankowski et al. 201764 |
| Residential home, Toronto, Ontario | Acetate/ToF | 0.150 (mean), 0.150 (median) | Hems et al. 2019104 |
Furthermore, growing interest in indoor air chemistry has led to the recent detection of HNCO in residential indoor air suggesting that HNCO exposure may not only be an occupational work place hazard (Table 2). Indeed, concentrations of HNCO in a residential kitchen in Toronto, Ontario were up to four times higher than outdoor concentrations.104 It is thus clear that we as humans are exposed to HNCO outdoors and indoors.
3. Sources
Emissions of HNCO into the atmosphere have been quantified from a variety of anthropogenic and natural sources through laboratory experiments and field measurements. The primary emission sources include biomass burning,19,20,22,26–29 fossil fuel combustion (coal, gasoline, and diesel),19,30–37 and cigarette smoke.19,38,39 The secondary production of HNCO, through the oxidation of amines and amides is also a potential source.40–45,65 We first summarize the studies of HNCO direct emissions in Sections 3.1 and 3.2, and then describe HNCO secondary production in Section 3.3. The contributions of cigarette smoke to HNCO are described in Section 3.4. In Section 3.5, we describe the sources of HNCO from the combustion of materials in the built environment.3.1. Fossil fuel combustion
HNCO has been detected in exhaust gases from three major fossil fuels: coal,19,31,32 gasoline,33,37 and diesel30,34,36,37 (Table 3).| Fuel | Engine tested | Type of study | Engine conditions | Rate [mg HNCO per kg fuel] | Reference |
|---|---|---|---|---|---|
| Laboratory measurements | |||||
| Diesel | Direct injection turbo diesel IVECO, type F1C | Engine dynamometer | ISO 8178/4 cycle – no SCR | 28 ± 1 mg HNCO per h | Heeb et al. 2011, 201236,76 |
| Diesel | ISO 8178/4 cycle – SCR only | 282 ± 7 mg HNCO per h | |||
| Diesel | ISO 8178/4 cycle – DPF + SCR | 245 ± 6 mg HNCO per h | |||
| Diesel | Direct injection turbo diesel engine from a Volkswagen Jetta 2001 | Engine dynamometer | Aggressive driving | 3.96 ± 1.1 | Wentzell et al. 201330 |
| Diesel | Highway economy driving | 3.49 ± 1.1 | |||
| Diesel | City driving | 0.21 ± 0.14 | |||
| Diesel | Idle state | 0.69 ± 0.06 | |||
| Diesel | Heavy-duty diesel engine John Deere 4045H | Engine dynamometer | Idle state | 54 ± 3 | Link et al. 201634 |
| Biodiesel | Idle state | 54 ± 1 | |||
| Diesel | 50% load | 17 ± 1 | |||
| Biodiesel | 50% load | 17 ± 2 | |||
| Diesel/biodiesel | Averaged | 35 ± 2 | |||
| Diesel | Heavy-duty diesel engine John Deere 4045H | Engine dynamometer | 1500 rpm at 45 kW | 38.80 | Jathar et al. 201735 |
| Diesel | 2400 rpm at 11 kW | 56.20 | |||
| Diesel | 2400 rpm at 57 kW | 30.90 | |||
| Diesel | Euro 5 light duty diesel vehicle | Chassis dynamometer | WLTC | 37 | Suarez-Bertoa et al. 201637 |
| Gasoline/hybrid | Plug in hybrid electric vehicle with gasoline engine | WLTC | 2.4 mg HNCO per km | ||
| Gasoline | Average of a Euro 5 and Euro 6 LDGV | WLTC | 93 | ||
| Gasoline | 8 different light duty gasoline-powered vehicles | Chassis dynamometer | Engine start | 0.46 ± 0.13 | Brady et al. 201433 |
| Gasoline | Hard acceleration after warming up | 1.70 ± 1.77 | |||
| Gasoline | Averaged | 0.91 ± 0.58 | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Mobile measurements | |||||
| Diesel | Off-road diesel engines | Aircraft | Oil sands fleet | 9.2 | Liggio et al. 201772 |
| Diesel/gasoline | On-road vehicles | Mobile | Winter fleet, plume based | 2.3 (mean), 3.3 (median) | Wren et al. 201856 |
| Winter fleet, time based | 2.6 (mean), 4.0 (median) | ||||
| Summer fleet, time based | 3.1 (mean), 5.4 (median) | ||||
HNCO emissions are dependent on both the quantity and the functionalities of nitrogen-containing species within the coal. Nitrogen-containing functional groups within coal often include pyrrolic and pyridinic compounds, amines, aminium salts, and pyridones. During pyrolysis, pyrrolic and pyridinic compounds produce hydrogen cyanide (HCN); amines and aminium salts produce NH3; and pyridone produces HNCO and HCN.31,67,68 These functional groups can be quantified using a parameter called the rank of coal, which represents the O/N ratio in coal.69 Lower ranking coals (low O/N ratios) contain higher concentrations of amine, aminium salts, and pyridones and give rise to higher yields of HNCO and NH3 upon combustion.68,69
The formation of HNCO from pyrolysis of coal is complex due to the large number of precursor constituents in coal. Coal can be simplified into a material containing aromatic clusters held together through intermolecular forces.68 Upon heating, these weaker forces break and produce tars with less diverse chemical functionalities. While the mechanism is not fully understood, the cracking of cyclic amides (pyridones) seems to be a likely pathway for HNCO production from coal burning.68
In addition to the elemental nitrogen fraction in coal, the temperature of pyrolysis strongly influences the release of nitrogen species. When coal is pyrolyzed over a range of temperatures, the HNCO/HCN emission ratios decrease with temperature up to 1100 °C.68 In other words, high temperatures lead to the preferential formation of HCN whereas lower temperatures lead to higher concentrations of HNCO.68
The mechanism for HNCO formation is thought to occur through a series of heterogenous reactions involving NO, CO, H2, and NH3 (R1–R6).71 The asterisks (*) in the following reactions indicate that the species is surface-bound.
NO and CO are thought to interact with the surface of the catalyst creating surface-bound N and CO; which combine to form surface-bound NCO (R1–R3).
| NO(g) ⇌ *NO → *N + *O | (R1) |
| CO(g) ⇌ *CO | (R2) |
| *N + *CO ⇌ *NCO | (R3) |
In another set of heterogeneous reactions, surface-bound hydrogen atoms are formed through the dissociation of either hydrogen or NH3 (R4 and R5a–d).
| H2(g) ⇌ 2*H | (R4) |
| NH3(g) ⇌ *NH3 | (R5a) |
| NH3(g) ⇌ *NH2 + *H | (R5b) |
| NH3(g) ⇌ *NH + 2*H | (R5c) |
| NH3(g) ⇌ *N + 3*H | (R5d) |
Lastly, the surface-bound hydrogen can combine with the surface-bound NCO, forming HNCO (R6) that can subsequently desorb to the gas phase. We further acknowledge that a water molecule or a volatile organic compound could diffuse to the surface and react with the surface-bound NCO radical producing HNCO.
| *H + *NCO → HNCO(g) | (R6) |
From the catalytic converter, the gas phase HNCO is then propelled out of the tailpipe and into the atmosphere.
To better quantify the contributing role of traffic emission to the HNCO atmospheric burden, emission factors for HNCO from light-duty gasoline-powered vehicles have been developed. HNCO emission factors are known for different phases of a driving cycle (Table 3).33,37 Driving cycles are simulated by engine programs aimed at reproducing on-road vehicle conditions in a laboratory setting. HNCO was emitted from all engines tested and the emission factors varied depending on the stage of the driving cycle from engine start-up, warm-up, to hard acceleration. The lowest emission ratios were observed during start-up (0.46 ± 0.13 mg HNCO per kg fuel) and HNCO concentrations steadily increased until the catalytic converter reached operating temperature. Furthermore, the maximum emission ratios observed were during hard acceleration (1.70 ± 1.77 mg HNCO per kg fuel). An average HNCO emission ratio for light-duty gasoline-powered vehicles is proposed to be 0.91 ± 0.58 mg HNCO per kg fuel, and only correlates with NO and CO emissions during hard acceleration indicating different mechanisms of production.33
In addition to laboratory measurements for HNCO directly from exhaust, ambient measurements have also been used to attribute HNCO emission to the combustion of gasoline and diesel. In ambient measurements, HNCO correlated with markers of vehicle exhaust including benzene, toluene, and black carbon (BC).30,72 Further, from the diurnal cycle observed in HNCO measurements in Toronto, two separate sources for HNCO were identified based on time of day.30 First, concentrations of HNCO generally increased each morning due to morning rush hour traffic. Concentration increases correlated well with both benzene and toluene, common tracers for vehicle emissions. Later in the day when maximum concentrations of HNCO are generally observed, there are high correlations between HNCO and the photochemically produced species (Section 3.3).30,55,57
Diesel oxidation catalysts function in similar ways to gasoline catalytic converters and aim to reduce NO, CO, and unburned hydrocarbons. These diesel oxidation catalysts however are not efficient at reducing NOx and thus, diesel emissions require an additional clean-up step. Often, the method used to reduce NOx in diesel emissions is selective catalytic reduction with urea.73
Today, diesel emissions of NOx are reduced through urea-based selective catalytic reduction systems.74 In these systems, urea thermally decomposes into NH3 and HNCO.35,49,75 The reaction proceeds as follows (R7):35
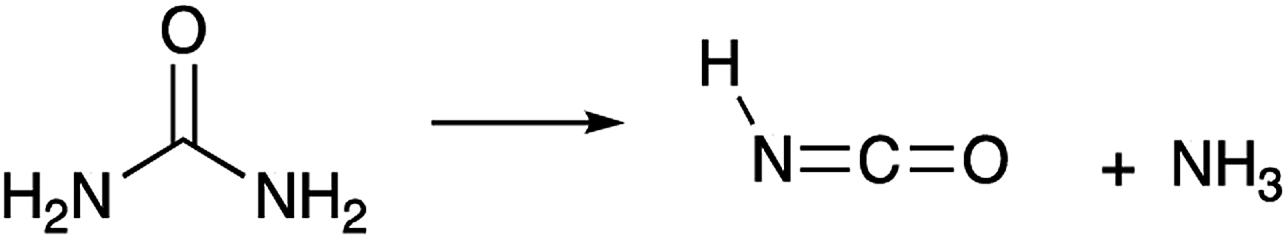 | (R7) |
HNCO is then hydrolysed to produce NH3 and CO2 (R9). NH3 acts as the reducing agent for NO and NO2 yielding N2 and H2O.35,76 However it is possible for some of the HNCO to be emitted through the exhaust.
Several studies have investigated the emissions of HNCO from light duty diesel vehicles with some conflicting results (Table 3).30,34–37 In some cases, HNCO production was found to increase significantly with the selective catalytic reduction system on and with increasing urea injection levels, from 0.028 g HNCO per h for a low urea dosing to 0.26 g HNCO per h for a high urea dosing.36,76 However, in two other studies, the engines equipped with selective catalytic reduction systems, when compared to non-selective catalytic reduction systems, had lower emissions of HNCO.35,37 One possible explanation is that selective catalytic reduction systems are not sources of HNCO to diesel exhaust, but rather that HNCO is produced within the engine cylinder during combustion.35 These contrasting results demonstrate the uncertainties present in the quantification of HNCO emissions from diesel engines. Differences could also arise from the different measurement techniques employed in the studies. For instance, Heeb et al. (2011, 2012)36,76 monitored the HNCO concentrations using an offline system where HNCO was hydrolysed in the aqueous phase and detected as NH3. This technique makes the quantification of HNCO sensitive to any changes in the concentrations of NH3 emitted during testing.
In an investigation by Link et al., diesel engine emissions were monitored for HNCO under both idle and 50% load conditions with both conventional diesel and biodiesel.34 Emission factors were found to be 3 times larger for engines during idle compared to those at 50% load with values of 54 mg kg−1 fuel and 17 mg kg−1 fuel, respectively. Biodiesel did not have a significant difference in emission factors when compared to conventional diesel. These values are in good agreement with those obtained by Jathar et al. (2017), who studied the emission factors on the same engine.35 These values are much larger than those obtained in Wentzell et al. (2013), who calculated emission factors between 0.21 and 3.96 mg kg−1 fuel for measurements during city driving and aggressive driving, respectively.30 The range in reported emission factors could result from the differences in measurement techniques, for example between on-road (Wentzell et al. (2013)) and laboratory (Link et al. & Jathar et al.) measurements34 as well as differences in engine size.35
The emission factors of diesel engines ranged from 0.21 ± 0.14 mg HNCO per kg fuel for a consumer diesel vehicle undergoing a driving procedure (FTP75) that simulates city driving,30 to 282 ± 7 mg HNCO per kg fuel for a commercial diesel engine running the ISO 8178/4 cycle with selective catalytic reduction equipped (Table 3).36,76
Furthermore, two mobile studies have quantified HNCO emissions from traffic. Wren et al. (2018) conducted a mobile study using a CIMS operated in a mobile lab to measure real time HNCO and HCN mixing ratios of on-road vehicles.56 They found that on-road vehicles were clear emitters of HNCO to the atmosphere, with higher emissions observed in winter than in summer. Algorithms were used to calculate both plume-based and timed-based emission factors from the combined diesel and gasoline emissions. The winter and summer time-based emission factors (mean) were 2.6 and 3.1 mg kg−1 fuel, respectively. These values agree closely to the values obtained for diesel vehicles in Wentzell et al. (2013)30 and gasoline vehicles in Brady et al.,33 but much lower than dynamometer measurements by Suarez-Bertoa et al.,37 Link et al.,34 and Jathar et al.35 Further evidence of summer and winter emission differences are provided by Suarez-Bertoa et al. who found that on average in their fleet (both gasoline and diesel vehicles) that vehicles operated at −7 °C had emission ratios more than four times greater than when operated at 23 °C.37
In the second mobile study, Liggio et al. conducted a regional aircraft study in the Athabasca oil sands in northern Alberta, Canada, where bitumen deposits are extracted and refined to crude oil resulting in heavy industrial activity.72 In the study, HNCO was measured using a time-of-flight CIMS over 22 flights. Elevated HNCO concentrations were attributed to diesel exhaust through correlation with black carbon levels. Their study found that off-road oil sands diesel activities had emissions factors of 9.2 mg HNCO per kg fuel (mean) contributing approximately 0.15 tons HNCO per day to the atmosphere. In the Alberta Oil Sands region, the diesel emissions are dominated by large industrial engines compared with relatively smaller commercial and freight trucks on Toronto highways, which may explain the higher emission factors compared to Wentzell et al. (2013)30 and Wren et al. (2018).56 Additionally, because the plane intercepted the vehicle exhaust much further downwind of the source that the on-road mobile lab, there was more time for secondary production of HNCO to occur.
3.2. Biomass burning
Biomass burning, which includes wildfires and agricultural burning, can occur on a large scale with important local to regional impacts on air quality and health. Furthermore, the occurrence and intensity of wildfires are expected to increase in the future due to the effects of climate change.80Several laboratory studies have looked at the constituents of biomass burning emissions from different types of materials. Shea nut meal, yellow peas, soya beans, and whey protein have been used as model compounds to study the combustion emission of these N-containing biomass material.27 Indeed, all these crops emit HNCO during pyrolysis. The whey and soya beans emissions of HNCO are temperature dependent with higher temperatures corresponding to lower HNCO production. Similar to the pyrolysis of coal, the cracking of cyclic amides likely explains the production of HNCO from biomass burning.
During the burning of California sage brush, HNCO/CO emission ratios can be 5–10 times larger during the flaming state of combustion than during the smoldering stage.19 This result is consistent with measurements made during a nationwide bonfire event in the UK where HNCO/CO ratios were observed to be 3 times higher during a period when emissions were thought to be dominated by flaming combustion compared to smouldering.58 In addition, this Guy Fawkes Night study found that plumes associated with bonfires had maximum mixing ratios of 1.64 ppbv HNCO, a two-order of magnitude enhancement of the mean non-bonfire mixing ratio of 0.012 ppbv.58
Different parameters such as protein content and temperature determine how much HNCO is released during biomass burning.27 HNCO emission ratios can vary from 0.80 ± 0.57 mmol per mole of CO for fuels commonly found in southwestern USA28 to 4.6 mmol mol−1 of CO for fuels commonly found in northern USA.81 Similar results with emission ratios reaching 0.76 mmol mol−1 of CO for fuels common to the southwestern USA burned in laboratory studies.21 Further, emissions from 30 laboratory biomass burning fires during the 2016 Fire Influence on Regional Global Environment Experiment (FIREX 2016) were found to contain on average 1% HNCO, in terms of total ion count measured by HR-TOF-CIMS using iodide reagent ion.82 In these same experiments, the high- and low-temperature pyrolysis profiles were compared.83 High-temperature pyrolysis resulted in larger emissions of HNCO versus low-temperature pyrolysis experiments and had mole fractions of HNCO over double that of HCN.83
During the Fourmile Canyon fire in 2010, enhancements in HNCO and CO were measured in nearby Boulder, CO. HNCO was measured at mixing ratios up to 0.220 ppbv; about 20 times the ambient background concentrations in that region.19 Similarly, in areas affected by local agricultural burning in Mohali, India mixing ratios were increased by approximately 0.500 ppbv, 30%, versus times when there was no agricultural burning.53 Regional scale wildfires are known to have important consequences on local and regional air quality, yet whether HNCO is causing part of adverse health effects related to biomass burning events remains unclear.
3.3. Secondary production
HNCO might have important secondary photochemical sources in the atmosphere. In fact, we know that amines, such as monoethanolamine, oxidize to amides and are subsequently oxidized to isocyanates.43–45,65 In laboratory studies, HNCO is the main oxidation product from the oxidation of formamide with OH, NO3, or Cl (common atmospheric oxidants).40–42 Urea also produces HNCO as the sole C-containing oxidation product upon Cl-initiated urea oxidation.40 Other amides including N-methylformamide, acetamide, and propanamide produce HNCO as either a first or second generation product of oxidation.41Secondary production of HNCO from diesel exhaust was observed by photochemically aging the exhaust in a potential aerosol mass reactor.34 The enhancement factor for HNCO increased by a factor of four when compared to the emission factor of fresh diesel exhaust. When aged for 1.5 days, HNCO enhancement factors reached 230 mg HNCO per kg fuel during idle conditions. In addition, the aging of diesel exhaust was studied in the Athabasca oil sands.72 Plumes downwind of oil sands sites were examined using a top down emission rate retrieval algorithm to filter out primary emissions. Indeed, photooxidation of diesel exhaust accounted for the production of 116–186 kg HNCO per h or approximately 1.5 tons HNCO per day. Compared to the value measured for direct (primary) HNCO emissions of 6.2 kg HNCO per h, photooxidation of diesel exhaust accounts for over 20 times the HNCO production than what is directly emitted from the tailpipe. This process of exhaust aging should be further investigated to determine which components of exhaust are contributing to the large HNCO increase and whether this increase in HNCO holds true for exhaust from other combustion sources.
Correlations of HNCO diurnal cycles with respect to several photochemically produced species including HNO3, O3, and formic acid, have been observed.30,55,57 Early afternoon peaks in mixing ratios along with the aforementioned correlations have provided additional evidence for secondary production.
Therefore, the combined contributions of ambient HNCO from photochemical enhancements of vehicle exhaust and from formation by amine and amide oxidation are likely to be more relevant to ambient HNCO concentrations than primary emissions. However, the ratio of primary emissions to secondary emissions would be dependent on geographic location and fire activity.
3.4. Cigarette smoke
There are three potential sources of HNCO from cigarettes: (1) pyrolysis of the protein-enriched tobacco and other N-containing organics, (2) the pyrolysis of urea, a common ingredient added to many cigarettes, and (3) the oxidation of nicotine.38 In an approximation made by Roberts et al.19 based on parameters developed by Baker and Bishop,39 an estimated 36 μg and 108 μg of HNCO is inhaled from just one filtered and unfiltered cigarette, respectively. This concentration translates to a mixing ratio of 40000 to 140000 ppbv being inhaled by the smoker, well above the estimated 1 ppbv threshold for human carbamoylation risk.19
In a laboratory study, nicotine was found to produce formamide and HNCO upon photo-oxidation, which could occur in both indoor and outdoor environments.38 The photo-oxidation of nicotine therefore, leads to an important exposure pathways of HNCO through first, second, third hand smoke.38,84
3.5. Material combustion from the built environment
The combustion of materials from the built environment presents a diverse and potentially large yet sporadic source of HNCO to air. HNCO and other isocyanates have been measured to be a combustion product of materials commonly found in buildings, such as glass, rubber, wool, foam, optical cables, carpet, mattresses and wood.85–89 Glass wool, nitrile rubber and melamine emitted isocyanates between 2000 and 4000 ppbv in laboratory test fires.86 HNCO was consistently the major product quantified in test fires with manufactured polyurethane materials.86 In laboratory test fires of both flexible polyurethane foam and viscoelastic memory foam mattresses, the total yields of isocyanates ranged from 1.43–11.95 mg m−3 and 0.05–6.13 mg m−3, respectively.89 Further, it was found that HNCO made up 34–99% and 2–98% of total NCO emissions from flexible polyurethane foam and viscoelastic memory foam mattresses, respectively.89 These results depended on the fire temperature, with the highest yield percentage corresponding to the highest burn temperature.89Fire tests involving vehicles and their interior, exterior, and engine components have also been investigated for their emissions of isocyanates.90,91 Significant amounts of isocyanates were found to be emitted from vehicular fires ranging from 0.0059–0.0668 mg m−3.85,90 Of those emissions, all of the isocyanates originated from the combustion of the cabin materials and not from the engine compartment. The implications of the combustion of building materials for exposure to HNCO remains constrained to a building fire, and likely contributes negligibly to the global budget of atmospheric HNCO.
4. HNCO loss processes
4.1. Gas phase photochemistry
The atmospheric removal of HNCO though gas phase reactions is thought to be slow. By extrapolating available data at high temperatures,92,93 Tsang et al.47 recommends 1.06 × 10−18T2exp(−1290/T) cm3 per molecule per s for the temperature-dependent rate coefficient for reaction with OH radicals. This value corresponds to a rate coefficient of 1.24 × 10−15 cm3 per molecule per s at 298 K, and to a lifetime of several decades.
Moreover, HNCO is stable against photolysis from sunlight. The lowest energy absorption band of HNCO is below 280 nm and thus the molecule cannot absorb in the actinic region of radiation in the troposphere.94 Consequently, the lifetime of HNCO with respect to gas phase photolysis is on the order of months.19 Most loss of HNCO in the atmosphere is therefore thought to be through dry deposition or uptake to the aqueous phase followed by hydrolysis or wet deposition.19,95
4.2. Heterogeneous uptake and hydrolysis
The lifetime of HNCO with respect to heterogenous uptake and hydrolysis depends strongly on atmospheric parameters such as the liquid water content, temperature, and pH of cloud droplets.2,4 The air–water phase partitioning of HNCO is thus an important parameter to investigate. Roberts et al. reported an intrinsic Henry's law coefficient of 21 M atm−1, inferred from measurements made at pH = 3.19 This value was later confirmed by Borduas et al. who determined the Henry's law coefficient and enthalpy of dissolution to be 26 ± 2 M atm−1 and −34 kJ mol−1, respectively.2 The Ka for HNCO is 2.1 × 10−4 M.2,3 The enthalpy of dissociation for HNCO is 5.4 kJ mol−1.95 With these values, the partitioning of HNCO from the gas phase to the aqueous phase can be predicted at varying temperatures and pH values. Additionally, HNCO exhibits a salting out effect—a decrease in the solubility of a solute with the increase in ionic strength of the solution – at increasing NaCl concentrations at pH 3.4 Roberts and Liu reported the Setschenow constant, ks, a measure of a solute's solubility as a function of salt content, of HNCO to be 0.097 ± 0.0011 M−1 from a series of solubility experiments at varying ionic strengths.4 The Setschenow constant for HNCO is also well predicted with its octanol–water constant, Kow,96 which was recently measured to be 4.4.4Once HNCO is dissolved into an aqueous solution, it can undergo hydrolysis, through three pH dependent reactions, to irreversibly produce NH3 and CO2. These two products may then form ammonium and bicarbonate depending on the pH (R8–R10).2,97
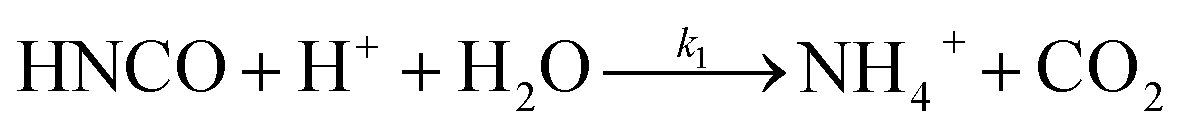 | (R8) |
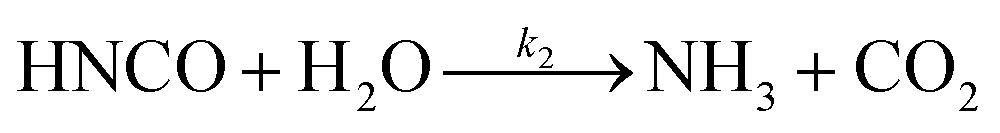 | (R9) |
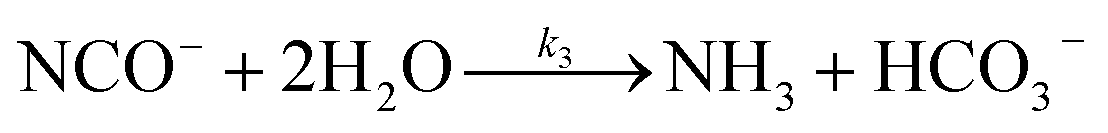 | (R10) |
The overall pseudo first order rate constant for the hydrolysis of the total amount of isocyanate in solution [HNCO + NCO−] can be written as eqn (1).2,97 Because each hydrolysis rate is pH dependent, each term (k1, k2, or k3) in eqn (1) changes in importance with respect to the overall rate as pH changes (Fig. 3). The values for each of the individual rate constants k1, k2, and k3 were calculated using k1 = (4.4 ± 0.2) × 107exp(−6000 ± 240/T) M s−1, k2 = (8.9 ± 0.9) × 106exp(−6770 ± 450/T) s−1, and k3 = (7.2 ± 1.5) × 108exp(−10900 ± 1400/T) s−1 as recommend in Borduas et al. 2016.2 We note that similar values were obtained by Roberts et al. for atmospherically relevant temperatures and pH.4 The Ka value used for this calculation was 2.1 × 10−4 M. At this temperature, the k1 term dominates the overall rate constant equation at pH values between 0 and 1; while at pH values above 9 the k3 term dominates. The k2 term dominates the overall rate constant in the pH range of 3–7.
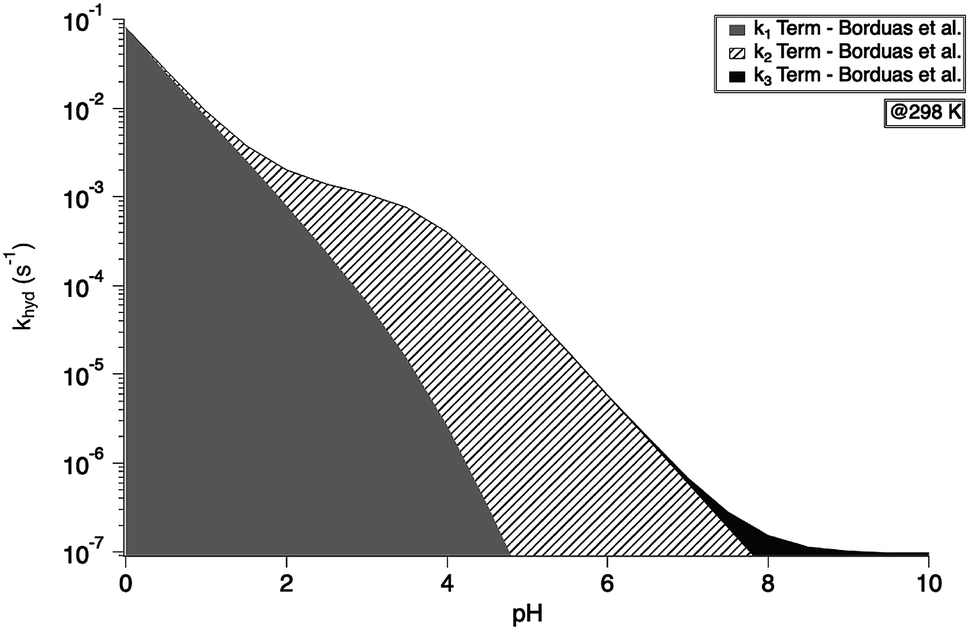 | ||
| Fig. 3 Overall hydrolysis rate constant khyd for varying pH values at 298 K. Graph is coloured with respect to the contributions of each term of the overall rate constant equation (eqn (1)). Values of k1, k2, and k3 were calculated using the activation energies reported in Borduas et al. 2016.2 The Ka value used was 2.1 × 10−4. | ||
Overall rate constant for the hydrolysis of HNCO:
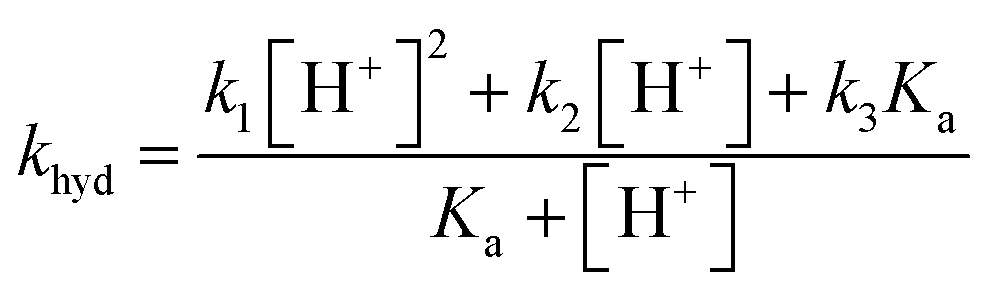 | (1) |
The extent to which aqueous phase hydrolysis can impact the lifetime of HNCO in the atmosphere depends on the liquid water content of the atmosphere, and the temperature and pH of the liquid water droplets.2 At atmospherically relevant cloud water pH values between 4 and 6, and temperatures between 273 K and 298 K, the lifetime of HNCO in solution can range from five hours to over a month. At a given liquid water content of 1 g m−3, the pH-dependence of the solubility and the pH-dependence of the hydrolysis rate (between pH 4 and pH 8) counteract each other, yielding a lifetime of HNCO against uptake and hydrolysis relatively insensitive to pH.2 In this pH range, the temperature dependence of the overall loss rate is driven by the temperature dependence of k2, which increases by an order of magnitude between 273 K and 298 K. The atmospheric fate of HNCO relies heavily on an accurate measurement of k2.
In addition to hydrolysis, it has recently been reported that HNCO reacts with ammonia in solution to form urea (R11).4 The reaction has a rate constant of 1.2 (±0.1) × 10−3 M−1 s−1 and proceeds at almost twice the rate of hydrolysis at pH 3.4 Note that ammonium has a pKa of 9.24 and thus, at pH 3 virtually all of the ammonia will be protonated to ammonium.
| HNCO + NH3 → H2NC(O)NH2 | (R11) |
In regards to the heterogeneous uptake of HNCO, Zhao et al. 201455 investigated the ability of HNCO to be scavenged by cloud droplets. To do so, atmospheric concentrations of HNCO were measured pre-cloud, in the gas-phase, and then during a cloud event, in the aqueous phase, while excluding interstitial air. The fraction of the HNCO in the water ranged from 7–17% of the original gas phase amount depending on the pH of the cloud droplet; with the higher percentage of aqueous HNCO corresponding to a higher pH value. These values far exceeded the theoretical values calculated using the pH and effective Henry's law constant which ranged between 0.02% and 0.7%. These discrepancies cannot be explained solely by partitioning even when considering the updated information available regarding temperature dependence of HNCO's Henry's law coefficient and suggests the existence of an aqueous source of HNCO.2,4 A recent study proposed the increase in HNCO measured in the cloud droplets55 could be due to NCO− present in seawater from biological activity.4 The resulting NCO− enriched sea spray and subsequent acidification could be a source of gaseous HNCO to the atmosphere.4
4.3. Dry deposition
Given the absence of other gas-phase loss processes, it seems likely that dry deposition may be an important sink for HNCO. Prior to the calculation of the deposition velocity of HNCO, its deposition has been implemented into a chemical transport model (discussed further in Section 5) by assuming the same for formic acid.95 The resulting lifetimes against deposition are currently estimated to be 1–3 days over the ocean and, 1–2 weeks over vegetation.95 Roberts et al. recently revisited these numbers, and estimate that the lifetime with respect to dry deposition is 1–2 days and the lifetime with respect to aerosol deposition is 6–12 days.45. Fate modelling
5.1. Chemical transport modelling
Chemical transport models are a necessary tool in atmospheric chemistry to evaluate our current understanding of chemical and physical processes, to estimate exposure risks where measurements have yet to be made and to help identify missing and/or inaccurate sources and sinks of pollutants of interest. Young et al., used the chemical transport model, MOZART-4 (Model for Ozone and Related Chemical Tracers), to simulate the global tropospheric budget and spatial distribution of HNCO.95 The model inputs included biomass burning sources along with anthropogenic emissions, but did not include secondary photochemical sources. Emissions were determined by scaling the emissions of HCN by a factor of 0.3 for biomass burning, and of 0.3 for anthropogenic emissions. However, based on recent biomass combustion studies, we now know that the scaling factor of 0.3 is likely low for many fuels.81,83 The authors identified biofuel combustion as the major anthropogenic source of HNCO within their model.95 The result of this scaling exercise generated global HNCO emissions of 1490 Gg per year.95The loss processes within the MOZART-4 simulation included OH radical oxidation, using a temperature-independent rate coefficient of 10−15 cm3 per molecule per s which itself was extrapolated to 298 K from Tsang et al. (1992),47 and dry deposition, using deposition velocities for formaldehyde, a previously used surrogate for formic and acetic acid in models.95 Heterogeneous uptake was included as an irreversible loss process and was calculated using an intrinsic Henry's law coefficient of 21 M atm−1, taking into account both pH and liquid water content dependence of solubility. The pH and temperature dependences of the hydrolysis reactions were not considered however; hydrolysis was assumed to occur with unit probability following dissolution. As a result of considering the pH-dependence solely of the solubility, the importance of the heterogeneous loss was strongly pH-dependent.
In the simulations, the lifetime of HNCO reached a minimum of 5 days, which seems somewhat inconsistent with the diurnal trends observed for HNCO in the atmosphere. The model output was compared to observations from Erie, CO, where it simulated mixing ratios of a reasonable magnitude (0.030–0.080 ppbv) but did not match the patterns of variability in the observations. The model simulations identified several regions across the globe where mixing ratios were predicted to exceed the 1 ppbv threshold for potential carbamoylation and health effects.19,95 Regions of potentially high HNCO exposure were those affected by wildfires or high anthropogenic emissions. Note that these simulation results predict ppbv levels of HNCO without the inclusion of photochemically produced HNCO, thereby likely underestimating ambient concentrations near amine and amide emissions. Since the publication of the modelling study, measurements in India and Nepal have confirmed that mixing ratios can reach values in excess of 1 ppbv, especially in regions with agricultural burning.51,52
Regional HNCO emissions have also been modelled with respect to areas downwind from the Athabasca oil sands in Alberta, Canada.72 Using the Canadian regional air quality model (GEM-MACH v2.0), mean HNCO concentrations of 0.025 ppbv and 0.052 ppbv were predicted for Fort McMurray, 70 km away from oil sands industrial activity, and Edmonton, an urban city 575 km from the oil sands, respectively.72 Emissions of HNCO were separated into two categories: emissions from primary sources and emissions from secondary production. Contributions from primary oil sands emissions were determined through source specific emission factors of 0.0028, 2.3, and 2.9 mmol HNCO per mol of CO for gasoline, on-road diesel, and off-road diesel emissions, respectively. Secondary production was also taken into account and was calculated for the oxidation of VOCs and enhancement of diesel exhaust. Both primary and secondary oil sands activity accounted on average for 55% of HNCO mixing ratio in Fort McMurray and increased to values greater than 80% when directly impacted by oil sands plumes.72 Indeed, modelled concentrations reached levels of 0.600 ppbv in Fort McMurray during oil sands plume events, 60 times the mixing ratio of non-plume times. These studies are helping us understand the potential exposure to HNCO on global and local scales.
5.2. Cloud modelling
A gas-aqueous zero-dimensional model study was conducted to investigate the fate of HNCO in various cloud chemistry conditions.98 The model included 25 chemical species participating in 34 gas-phase reactions and 31 aqueous-phase reactions. The pH of the clouds could be held constant or could vary depending on the chemical constituents in the system. Unlike the Young et al. study, this model treated the kinetics of the aqueous phase hydrolysis reactions explicitly. Dissolution of HNCO into cloud water was reversible and as the liquid water content decreased or the pH decreased, HNCO could return to the gas phase. The conditions associated with cumulus clouds were found to cause the largest reduction of HNCO in the atmosphere with a corresponding lifetime of around 2 h for a cloud-containing airmass. It should be noted that the droplets in the cumulus clouds corresponding to the shortest lifetimes had a pH of 3.7 due to a SO2 concentration of 2 ppbv. This short lifetime assumes that a given airmass has constant contact with clouds and thus an overall lifetime of HNCO in an air mass is more likely closer to 3–5 days.98 The cloud properties were shown to have an effect on HNCO loss, with larger reductions corresponding to lower pH and higher temperature. Thus, the most efficient removal of HNCO is suggested to occur in warm low-lying clouds near urban regions.5.3. HNCO as a source of NH3
The hydrolysis of HNCO in an aqueous droplet leads to the production of NH3, and here we consider whether this production could be a secondary source of NH3. Young et al. estimated the global emissions of HNCO to be 1.5 Tg per year,95 and since N represents a third of the mass of HNCO, it corresponds to a nitrogen emission of around 0.5 Tg N per year. If we assume that the oxidation of amines yields HNCO, then we can use the global amine production estimated by Schade and Crutzen99 at 0.3 Tg N per year in combination with the estimate that oxidation accounts for 25–75% of the sinks for amines.100 This calculation yields, at maximum, 0.2 Tg N per year of HNCO from amines. Combining the primary emissions estimates to the secondary production and allowing every HNCO to hydrolyze to NH3 suggests that the maximum amount of NH3 produced through HNCO hydrolysis would be on the order of 0.7 Tg N per year. Comparing this value to the global estimate of NH3 emissions of 60 Tg N per year,101 HNCO seems to be an insignificant source globally, contributing only around 1%. However, there could be local environments where NH3 sources are low and the contribution of HNCO hydrolysis may not be negligible for a particular ecosystem.6. Exposure and risks
Much of the motivation for the study of HNCO is rooted in the risk associated with human exposure. HNCO is in equilibrium with its anion, cyanate (NCO−). When in the presence of molecules with amino groups, NCO− will react to form carbamoyl derivatives.16 Further, the enzyme activity of ribonuclease can be lost when exposed to cyanate due to protein function alteration through reaction with cyanate by carbamoylation.13,16 Changes in amino acid structure by carbamoylation alters the folding and functional ability of proteins leading to major complications such as renal failure,15,102 cardiovascular disease,10,14,103 cataracts,12 and rheumatoid arthritis.13Based on an effective Henry's law constant of 105 M atm−1, Roberts et al. estimated that with an ambient gas phase concentration of HNCO of just 1 ppbv, inhalation could result in blood cyanate concentrations high enough to participate in carbamoylation reactions.19 Furthermore, revised values to include the temperature dependences of the solubility and the acid dissociation constants, produces only a 15% difference (1.15 ppbv) in the inhaled concentration required to cause blood concentrations of 100 μM. Yet, the connection between inhalation of HNCO and its presence in the blood stream is to the best of our knowledge unknown. Another possible fate of cyanate in the blood is hydrolysis; using a temperature of 310 K, a pH of 7.4 and the Arrhenius expressions, the lifetime against hydrolysis is calculated to be 291 h,2 implying that the hydrolysis pathway is likely not fast enough to destroy the cyanate before it enters cells to further undergo carbamoylation. The transfer of HNCO from the blood stream into cells should be an efficient process with a logKOW value of 0.64 at 298 K.4
However, there has yet to be any direct epidemiological or toxicological studies relating HNCO exposure via inhalation to adverse health effects. Such studies would help uncover if HNCO in fact does cause in vivo carbamoylation and at which ambient mixing ratios. Until then, there are several options to reduce the rate of carbamoylation including amino acid supplementation, modified dialysis prescriptions and anti-inflammatory therapies.15
7. Future research
Since the atmospheric chemistry community has identified HNCO in the atmosphere, efforts have been focused on identifying and quantifying its sources and sinks. Importantly, there is potential for this molecule to have adverse health impacts and so understanding its fate to better predict its location and its concentrations is an important problem. Furthermore, HNCO can serve as a tracer of photochemically processed organo-nitrogen as well as wet partitioning. Ambient measurements remain scarce and geographically limited (Fig. 2) though additional unpublished datasets may exist due to the increasing deployment of time-of-flight CIMS and PTR-MS instruments in field campaigns. In fact, there is the opportunity for datamining existing acetate- and iodide-CIMS measurements to better quantify HNCO temporally and spatially. Chemical transport models will continue to help fill in data gaps, but require updated information on emissions, secondary sources, and heterogeneous loss processes. We foresee the need for further ambient measurements particularly through biomass burning and residential cooking, with a focus on developing countries. Understanding seasonal differences in mixing ratios and diurnal cycles could also be prioritized.The main sinks of HNCO are wet and dry deposition. Yet these sinks are typically slow, giving significant time for HNCO to react through other processes. For example, organic molecules with nucleophilic moieties present in the atmosphere could be a sink for HNCO. Moreover, the amplitude of the HNCO diurnal profile could be suggesting a relatively fast sink in the atmosphere.
Development of emission factors and enhancement ratios, hydrolysis rate constants, and amine and amide oxidation pathways might warrant a revisit of a chemical transport model study. Model predictions could help identify geographic locations at risk of high exposure to HNCO and assist in informing policy in regard to its regulation.
Finally, the pre-eminent gap in the understanding of HNCO is its impact on human health. The study of the fate of this weak acid has been led by atmospheric chemists speculating through the use of Henry's law coefficient that HNCO could be present in the blood and cause carbamoylation reactions, leading to cardiovascular problems. However, no direct study has linked ambient air HNCO exposure to carbamoylation and thus to health effects. These toxicological and even epidemiological studies could help motivate our understanding of the fate of HNCO. If this compound is truly toxic through inhalation and/or dermal uptake, then more resources could then be allocated to mitigating HNCO exposure.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
NBD acknowledges funds from NSERC PDF and from Ambizione SNSF grant project SNF_179703.References
- J. Liebig and F. Wöhler, Untersuchungen über die Cyansäure, Ann. Phys., 1830, 96, 369–400 CrossRef.
- N. Borduas, B. Place, G. R. Wentworth, J. P. D. Abbatt and J. G. Murphy, Solubility and reactivity of HNCO in water: insights into HNCO's fate in the atmosphere, Atmos. Chem. Phys., 2016, 16, 703–714 CrossRef CAS.
- A. R. Amell, Kinetics of the hydrolysis of cyanic acid, J. Am. Chem. Soc., 1956, 78, 6234–6238 CrossRef CAS.
- J. M. Roberts and Y. Liu, Solubility and Solution-phase Chemistry of Isocyanic Acid, Methyl Isocyanate, and Cyanogen Halides, Atmos. Chem. Phys. Discuss., 2018, 1–30 Search PubMed.
- W. H. Hocking, M. C. L. Gerry and G. Winnewisser, The Microwave and Millimetre Wave Spectrum, Molecular Constants, Dipole Moment, and Structure of Isocyanic Acid, HNCO, Can. J. Phys., 1975, 53, 1869–1901 CrossRef CAS.
- L. H. Jones, J. N. Shoolery, R. G. Shulman and D. M. Yost, The molecular structure of isocyanic acid from microwave and infra-red absorption spectra, J. Chem. Phys., 1950, 18, 990–991 CrossRef CAS.
- M. Mladenović, M. Elhiyani and M. Lewerenz, Electric and magnetic properties of the four most stable CHNO isomers from ab initio CCSD(T) studies, J. Chem. Phys., 2009, 131, 034302 CrossRef PubMed.
- M. Mladenović and M. Lewerenz, Equilibrium structure and energetics of CHNO isomers: steps towards ab initio rovibrational spectra of quasi-linear molecules, J. Chem. Phys., 2008, 343, 129–140 Search PubMed.
- C. K. Lee and J. M. Manning, Kinetics of the carbamylation of the amino groups of sickle cell hemoglobin by cyanate, J. Biol. Chem., 1973, 248, 5861–5865 CAS.
- F. H. Verbrugge, W. H. W. Tang and S. L. Hazen, Protein carbamylation and cardiovascular disease, Kidney Int., 2015, 88, 474–478 CrossRef CAS PubMed.
- R. A. Koeth, K. Kalantar-Zadeh, Z. Wang, X. Fu, W. H. W. Tang and S. L. Hazen, Protein Carbamylation Predicts Mortality in ESRD, J. Am. Soc. Nephrol., 2013, 24, 853–861 CrossRef CAS PubMed.
- H. T. Beswick and J. J. Harding, Conformational changes induced in bovine lens a-crystallin by carbamylation, Biochem. J., 1984, 223, 221–227 CrossRef CAS PubMed.
- P. Mydel, Z. Wang, M. Brisslert, A. Hellvard, L. E. Dahlberg, S. L. Hazen and M. Bokarewa, Carbamylation-Dependent Activation of T Cells: A Novel Mechanism in the Pathogenesis of Autoimmune Arthritis, J. Immunol., 2010, 184, 6882–6890 CrossRef CAS PubMed.
- Z. Wang, S. J. Nicholls, E. R. Rodriguez, O. Kummu, S. Hörkkö, J. Barnard, W. F. Reynolds, E. J. Topol, J. A. DiDonato and S. L. Hazen, Protein carbamylation links inflammation, smoking, uremia and atherogenesis, Nat. Med., 2007, 13, 1176–1184 CrossRef CAS PubMed.
- S. Delanghe, J. R. Delanghe, R. Speeckaert, W. Van Biesen and M. M. Speeckaert, Mechanisms and consequences of carbamoylation, Nat. Rev. Nephrol., 2017, 13, 580–593 CrossRef CAS PubMed.
- G. R. Stark and W. H. Stein, Reactions of the Cyanate Present in Aqueous Urea with Amino Acids and Proteins, J. Biol. Chem., 1960, 235, 3177–3181 CAS.
- D. J. Belson and A. N. Strachan, Preparation and properties of isocyanic acid, Chem. Soc. Rev., 1982, 11, 41–56 RSC.
- W. Jelkmann, ‘O’, erythropoietin carbamoylation versus carbamylation, Nephrol., Dial., Transplant., 2008, 23, 3033 CrossRef PubMed.
- J. M. Roberts, P. R. Veres, A. K. Cochran, C. Warneke, I. R. Burling, R. J. Yokelson, B. Lerner, J. B. Gilman, W. C. Kuster, R. Fall and J. de Gouw, Isocyanic acid in the atmosphere and its possible link to smoke-related health effects, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8966–8971 CrossRef CAS PubMed.
- J. M. Roberts, P. Veres, C. Warneke, J. A. Neuman, R. A. Washenfelder, S. S. Brown, M. Baasandorj, J. B. Burkholder, I. R. Burling, T. J. Johnson, R. J. Yokelson and J. de Gouw, Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions, Atmos. Meas. Tech., 2010, 3, 981–990 CrossRef CAS.
- P. Veres, J. M. Roberts, I. R. Burling, C. Warneke, J. de Gouw and R. J. Yokelson, Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry, J. Geophys. Res.: Atmos., 2010, 115, 1–14 CrossRef.
- P. Veres, J. M. Roberts, C. Warneke, D. Welsh-Bon, M. Zahniser, S. Herndon, R. Fall and J. de Gouw, Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere, Int. J. Mass Spectrom., 2008, 274, 48–55 CrossRef CAS.
- D. Gylestam, D. Karlsson, M. Dalene and G. Skarping, Determination of gas phase isocyanates using proton transfer reaction mass spectrometry, Anal. Chem. Lett., 2011, 1, 261–271 CrossRef CAS.
- M. J. Jankowski, R. Olsen, C. J. Nielsen, Y. Thomassen and P. Molander, The applicability of proton transfer reaction-mass spectrometry (PTR-MS) for determination of isocyanic acid (ICA) in work room atmospheres, Environ. Sci.: Processes Impacts, 2014, 16, 2423–2431 RSC.
- M. J. Jankowski, R. Olsen, Y. Thomassen and P. Molander, The stability and generation pattern of thermally formed isocyanic acid (ICA) in air – potential and limitations of proton transfer reaction-mass spectrometry (PTR-MS) for real-time workroom atmosphere measurements, Environ. Sci.: Processes Impacts, 2016, 18, 810–818 RSC.
- J. M. Roberts, P. R. Veres, T. C. VandenBoer, C. Warneke, M. Graus, E. J. Williams, B. Lefer, C. A. Brock, R. Bahreini, F. Öztürk, A. M. Middlebrook, N. L. Wagner, W. P. Dubé and J. A. de Gouw, New insights into atmospheric sources and sinks of isocyanic acid, HNCO, from recent urban and regional observations: Atmospheric sources and sinks of HNCO, J. Geophys. Res.: Atmos., 2014, 119, 1060–1072 CAS.
- K.-M. Hansson, J. Samuelsson, C. Tullin and L.-E. Amand, Formation of HNCO, HCN, and NH3 from the Pyrolysis of Bark and Nitrogen-Containing Model Compounds, Combust. Flame, 2004, 137, 265–277 CrossRef CAS.
- J. B. Gilman, B. M. Lerner, W. C. Kuster, P. D. Goldan, C. Warneke, P. R. Veres, J. M. Roberts, J. A. de Gouw, I. R. Burling and R. J. Yokelson, Biomass burning emissions and potential air quality impacts of volatile organic compounds and other trace gases from fuels common in the US, Atmos. Chem. Phys., 2015, 15, 13915–13938 CrossRef CAS.
- M. M. Coggon, P. R. Veres, B. Yuan, A. Koss, C. Warneke, J. B. Gilman, B. M. Lerner, J. Peischl, K. C. Aikin, C. E. Stockwell, L. E. Hatch, T. B. Ryerson, J. M. Roberts, R. J. Yokelson and J. A. de Gouw, Emissions of nitrogen-containing organic compounds from the burning of herbaceous and arboraceous biomass: Fuel composition dependence and the variability of commonly used nitrile tracers: Organic nitrogen in smoke, Geophys. Res. Lett., 2016, 43, 9903–9912 CrossRef CAS.
- J. J. B. Wentzell, J. Liggio, S.-M. Li, A. Vlasenko, R. Staebler, G. Lu, M.-J. Poitras, T. Chan and J. R. Brook, Measurements of Gas phase Acids in Diesel Exhaust: A Relevant Source of HNCO?, Environ. Sci. Technol., 2013, 47, 7663–7671 CrossRef CAS PubMed.
- P. F. Nelson, C.-Z. Li and E. Ledesma, Formation of HNCO from the Rapid Pyrolysis of Coals, Energy Fuels, 1996, 10, 264–265 CrossRef CAS.
- P. M. Nicholls and P. F. Nelson, Detection of HNCO during the Low-Temperature Combustion of Coal Chars, Energy Fuels, 2000, 14, 943–944 CrossRef CAS.
- J. M. Brady, T. A. Crisp, S. Collier, T. Kuwayama, S. D. Forestieri, V. Perraud, Q. Zhang, M. J. Kleeman, C. D. Cappa and T. H. Bertram, Real-Time Emission Factor Measurements of Isocyanic Acid from Light Duty Gasoline Vehicles, Environ. Sci. Technol., 2014, 48, 11405–11412 CrossRef CAS PubMed.
- M. F. Link, B. Friedman, R. Fulgham, P. Brophy, A. Galang, S. H. Jathar, P. Veres, J. M. Roberts and D. K. Farmer, Photochemical processing of diesel fuel emissions as a large secondary source of isocyanic acid (HNCO): Photochemical Source of Isocyanic Acid, Geophys. Res. Lett., 2016, 43, 4033–4041 CrossRef CAS.
- S. H. Jathar, C. Heppding, M. F. Link, D. K. Farmer, A. Akherati, M. J. Kleeman, J. A. de Gouw, P. R. Veres and J. M. Roberts, Investigating diesel engines as an atmospheric source of isocyanic acid in urban areas, Atmos. Chem. Phys., 2017, 17, 8959–8970 CrossRef CAS.
- N. V. Heeb, R. Haag, C. Seiler, P. Schmid, M. Zennegg, A. Wichser, A. Ulrich, P. Honegger, K. Zeyer, L. Emmenegger, Y. Zimmerli, J. Czerwinski, M. Kasper and A. Mayer, Effects of a combined diesel particle filter-deNOx system (DPN) on reactive nitrogen compounds emissions: A parameter study, Environ. Sci. Technol., 2012, 46, 13317–13325 CrossRef CAS PubMed.
- R. Suarez-Bertoa and C. Astorga, Isocyanic acid and ammonia in vehicle emissions, Transport. Res. Transport Environ., 2016, 49, 259–270 CrossRef.
- N. Borduas, J. G. Murphy, C. Wang, G. da Silva and J. P. D. Abbatt, Gas Phase Oxidation of Nicotine by OH Radicals: Kinetics, Mechanisms, and Formation of HNCO, Environ. Sci. Technol. Lett., 2016, 3, 327–331 CrossRef CAS.
- R. R. Baker and L. J. Bishop, The pyrolysis of tobacco ingredients, J. Anal. Appl. Pyrolysis, 2004, 71, 223–311 CrossRef CAS.
- I. Barnes, G. Solignac, A. Mellouki and K. H. Becker, Aspects of the Atmospheric Chemistry of Amides, ChemPhysChem, 2010, 11, 3844–3857 CrossRef CAS PubMed.
- N. Borduas, G. da Silva, J. G. Murphy and J. P. D. Abbatt, Experimental and Theoretical Understanding of the Gas Phase Oxidation of Atmospheric Amides with OH Radicals: Kinetics, Products, and Mechanisms, J. Phys. Chem. A, 2015, 119, 4298–4308 CrossRef CAS PubMed.
- A. J. C. Bunkan, T. Mikoviny, C. J. Nielsen, A. Wisthaler and L. Zhu, Experimental and Theoretical Study of the OH-Initiated Photo-oxidation of Formamide, J. Phys. Chem. A, 2016, 120, 1222–1230 CrossRef CAS PubMed.
- N. Borduas, J. P. D. Abbatt and J. G. Murphy, Gas Phase Oxidation of Monoethanolamine (MEA) with OH Radical and Ozone: Kinetics, Products, and Particles, Environ. Sci. Technol., 2013, 47, 6377–6383 CrossRef CAS PubMed.
- H.-B. Xie, C. Li, N. He, C. Wang, S. Zhang and J. Chen, Atmospheric Chemical Reactions of Monoethanolamine Initiated by OH Radical: Mechanistic and Kinetic Study, Environ. Sci. Technol., 2014, 48, 1700–1706 CrossRef CAS PubMed.
- H.-B. Xie, F. Ma, Y. Wang, N. He, Q. Yu and J. Chen, Quantum Chemical Study on ·Cl-Initiated Atmospheric Degradation of Monoethanolamine, Environ. Sci. Technol., 2015, 49, 13246–13255 CrossRef CAS PubMed.
- S. S. Brown, H. L. Berghout and F. F. Crim, The HNCO heat of formation and the N–H and C–N bond enthalpies from initial state selected photodissociation, J. Chem. Phys., 1996, 105, 8103–8110 CrossRef CAS.
- W. Tsang, Chemical kinetic data base for propellant combustion. II. Reactions involving CN, NCO, and HNCO, J. Phys. Chem. Ref. Data, 1992, 21, 753–791 CrossRef CAS.
- D. Karlsson, M. Dalene, G. Skarping and Å. Marand, Determination of isocyanic acid in air, J. Environ. Monit., 2001, 3, 432–436 RSC.
- O. Kroecher, M. Elsener and M. Koebel, An ammonia and isocyanic acid measuring method for soot containing exhaust gases, Anal. Chim. Acta, 2005, 537, 393–400 CrossRef CAS.
- R. Woodward-Massey, Y. M. Taha, S. G. Moussa and H. D. Osthoff, Comparison of negative-ion proton-transfer with iodide ion chemical ionization mass spectrometry for quantification of isocyanic acid in ambient air, Atmos. Environ., 2014, 98, 693–703 CrossRef CAS.
- B. P. Chandra and V. Sinha, Contribution of post-harvest agricultural paddy residue fires in the N.W. Indo-Gangetic Plain to ambient carcinogenic benzenoids, toxic isocyanic acid and carbon monoxide, Environ. Int., 2016, 88, 187–197 CrossRef CAS PubMed.
- C. Sarkar, V. Sinha, V. Kumar, M. Rupakheti, A. Panday, K. S. Mahata, D. Rupakheti, B. Kathayat and M. G. Lawrence, Overview of VOC emissions and chemistry from PTR-TOF-MS measurements during the SusKat-ABC campaign: high acetaldehyde, isoprene and isocyanic acid in wintertime air of the Kathmandu Valley, Atmos. Chem. Phys., 2016, 16, 3979–4003 CrossRef CAS.
- V. Kumar, B. P. Chandra and V. Sinha, Large unexplained suite of chemically reactive compounds present in ambient air due to biomass fires, Sci. Rep., 2018, 8, 626 CrossRef CAS PubMed.
- J. M. Mattila, P. Brophy, J. Kirkland, S. Hall, K. Ullmann, E. V. Fischer, S. Brown, E. McDuffie, A. Tevlin and D. K. Farmer, Tropospheric sources and sinks of gas-phase acids in the Colorado Front Range, Atmos. Chem. Phys., 2018, 18, 12315–12327 CrossRef CAS.
- R. Zhao, A. K. Y. Lee, J. J. B. Wentzell, A. M. Mcdonald, D. Toom-Sauntry, W. R. Leaitch, R. L. Modini, A. L. Corrigan, L. M. Russell, K. J. Noone, J. C. Schroder, A. K. Bertram, L. N. Hawkins, J. P. D. Abbatt and J. Liggio, Cloud partitioning of isocyanic acid (HNCO) and evidence of secondary source of HNCO in ambient air, Geophys. Res. Lett., 2014, 41, 6962–6969 CrossRef CAS.
- S. N. Wren, J. Liggio, Y. Han, K. Hayden, G. Lu, C. M. Mihele, R. L. Mittermeier, C. Stroud, J. J. B. Wentzell and J. R. Brook, Elucidating real-world vehicle emission factors from mobile measurements over a large metropolitan region: a focus on isocyanic acid, hydrogen cyanide, and black carbon, Atmos. Chem. Phys., 2018, 18, 16979–17001 CrossRef CAS.
- E. L. Mungall, J. P. D. Abbatt, J. J. B. Wentzell, A. K. Y. Lee, J. L. Thomas, M. Blais, M. Gosselin, L. A. Miller, T. Papakyriakou, M. D. Willis and J. Liggio, Microlayer source of oxygenated volatile organic compounds in the summertime marine Arctic boundary layer, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6203–6208 CrossRef CAS PubMed.
- M. Priestley, M. Le Breton, T. J. Bannan, K. E. Leather, A. Bacak, E. Reyes-Villegas, F. De Vocht, B. M. A. Shallcross, T. Brazier, M. Anwar Khan, J. Allan, D. E. Shallcross, H. Coe and C. J. Percival, Observations of Isocyanate, Amide, Nitrate, and Nitro Compounds From an Anthropogenic Biomass Burning Event Using a ToF-CIMS, J. Geophys. Res.: Atmos., 2018, 123, 7687–7704 CAS.
- J. de Gouw and C. Warneke, Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 2007, 26, 223–257 CrossRef CAS PubMed.
- P. R. Veres, J. M. Roberts, A. K. Cochran, J. B. Gilman, W. C. Kuster, J. S. Holloway, M. Graus, J. Flynn, B. Lefer, C. Warneke and J. de Gouw, Evidence of rapid production of organic acids in an urban air mass, Geophys. Res. Lett., 2011, 38, 1–5 CrossRef.
- R. D. Daniels, Occupational asthma risk from exposures to toluene diisocyanate: A review and risk assessment, Am. J. Ind. Med., 2018, 61, 282–292 CrossRef CAS.
- P. A. Smith, J. Lodwick, J. Dartt, J. R. Amani and K. M. Fagan, Methemoglobinemia resulting from exposure in a confined space: Exothermic self-polymerization of 4,4′-methylene diphenyl diisocyanate (MDI) material, J. Occup. Environ. Hyg., 2017, 14, D13–D21 CrossRef CAS PubMed.
- R. L. Prueitt, H. N. Lynch, K. Zu, L. Shi and J. E. Goodman, Dermal exposure to toluene diisocyanate and respiratory cancer risk, Environ. Int., 2017, 109, 181–192 CrossRef CAS PubMed.
- M. J. Jankowski, R. Olsen, Y. Thomassen and P. Molander, Comparison of air samplers for determination of isocyanic acid and applicability for work environment exposure assessment, Environ. Sci.: Processes Impacts, 2017, 19, 1075–1085 RSC.
- N. Borduas, J. P. D. Abbatt, J. G. Murphy, S. So and G. da Silva, Gas-Phase Mechanisms of the Reactions of Reduced Organic Nitrogen Compounds with OH Radicals, Environ. Sci. Technol., 2016, 50, 11723–11734 CrossRef CAS.
- S. Wang, W. Wei, D. Li, K. Aunan and J. Hao, Air pollutants in rural homes in Guizhou, China – Concentrations, speciation, and size distribution, Atmos. Environ., 2010, 44, 4575–4581 CrossRef CAS.
- S. Kambara, T. Takarada and M. Toyoshima, Relation between functional forms of coal nitrogen and NOx emissions from pulverized coal combustion, Fuel, 1995, 74, 1247–1253 CrossRef CAS.
- K.-M. Hansson, J. Samuelsson, L.-E. Åmand and C. Tullin, The temperature's influence on the selectivity between HNCO and HCN from pyrolysis of 2,5-diketopiperazine and 2-pyridone, Fuel, 2003, 82, 2163–2172 CrossRef CAS.
- O. C. Mullins, S. Mitra-Kirtley, J. Van Elp and S. P. Cramer, Molecular Structure of Nitrogen in Coal from XANES Spectroscopy, Appl. Spectrosc., 1993, 47, 1268–1275 CrossRef CAS.
- R. Dumpelmann, N. Cant and D. Trimm, Formation of isocyanic acid during the reaction of mixtures of NO, CO and H2 over supported platinum catalysts, Appl. Catal., B, 1995, 6, L291–L296 CrossRef.
- N. W. Cant, D. C. Chambers and I. O. Y. Liu, The formation of isocyanic acid and ammonia during the reduction of NO over supported platinum group metals, Catal. Today, 2004, 93–95, 761–768 CrossRef CAS.
- J. Liggio, C. A. Stroud, J. J. B. Wentzell, J. Zhang, J. Sommers, A. Darlington, P. S. K. Liu, S. G. Moussa, A. Leithead, K. Hayden, R. L. Mittermeier, R. Staebler, M. Wolde and S.-M. Li, Quantifying the Primary Emissions and Photochemical Formation of Isocyanic Acid Downwind of Oil Sands Operations, Environ. Sci. Technol., 2017, 51, 14462–14471 CrossRef CAS PubMed.
- A. Russell and W. S. Epling, Diesel Oxidation Catalysts, Catal. Rev., 2011, 53, 337–423 CrossRef CAS.
- M. Koebel and M. Elsener, Oxidation of Diesel-Generated Volatile Organic Compounds in the Selective Catalytic Reduction Process, Ind. Eng. Chem. Res., 1998, 37, 3864–3868 CrossRef CAS.
- Z. Chen, W. Yang, J. Zhou, H. Lv, J. Liu and K. Cen, HNCO hydrolysis performance in urea-water solution thermohydrolysis process with and without catalysts, J. Zhejiang Univ., Sci., A, 2010, 11, 849–856 CrossRef CAS.
- N. V. Heeb, Y. Zimmerli, J. Czerwinski, P. Schmid, M. Zennegg, R. Haag, C. Seiler, A. Wichser, A. Ulrich, P. Honegger, K. Zeyer, L. Emmenegger, T. Mosimann, M. Kasper and A. Mayer, Reactive nitrogen compounds (RNCs) in exhaust of advanced PM-NOx abatement technologies for future diesel applications, Atmos. Environ., 2011, 45, 3203–3209 CrossRef CAS.
- Reciprocating internal combustion engines—Exhaust emission measurement—Part 4: Steady-state and transient test cycles for different engine applications, International Organization for Standardization, 2007 Search PubMed.
- M. Tutuianu, P. Bonnel, B. Ciuffo, T. Haniu, N. Ichikawa, A. Marotta, J. Pavlovic and H. Steven, Development of the World-wide harmonized Light duty Test Cycle (WLTC) and a possible pathway for its introduction in the European legislation, Transport. Res. Transport Environ., 2015, 40, 61–75 CrossRef.
- M. Nesbit, M. Fergusson, A. Colsa, J. Ohlendorf, C. Hayes, K. Paquel and J.-P. Schweitzer, Comparative study on the differences between the EU and US legislation on emissions in the automotive sector, European Parliament: Policy Department A: Economic and Scientific Policy, 2016 Search PubMed.
- M. D. Flannigan and C. E. Wagner, Climate change and wildfire in Canada, Can. J. For. Res., 1991, 21, 66–72 CrossRef.
- A. R. Koss, K. Sekimoto, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, J. L. Jimenez, J. Krechmer, J. M. Roberts, C. Warneke, R. J. Yokelson and J. de Gouw, Non-methane organic gas emissions from biomass burning: identification, quantification, and emission factors from PTR-ToF during the FIREX 2016 laboratory experiment, Atmos. Chem. Phys., 2018, 18, 3299–3319 CrossRef CAS.
- S. Tomaz, T. Cui, Y. Chen, K. G. Sexton, J. M. Roberts, C. Warneke, R. J. Yokelson, J. D. Surratt and B. J. Turpin, Photochemical Cloud Processing of Primary Wildfire Emissions as a Potential Source of Secondary Organic Aerosol, Environ. Sci. Technol., 2018, 52, 11027–11037 CrossRef CAS PubMed.
- K. Sekimoto, A. R. Koss, J. B. Gilman, V. Selimovic, M. M. Coggon, K. J. Zarzana, B. Yuan, B. M. Lerner, S. S. Brown, C. Warneke, R. J. Yokelson, J. M. Roberts and J. de Gouw, High- and low-temperature pyrolysis profiles describe volatile organic compound emissions from western US wildfire fuels, Atmos. Chem. Phys., 2018, 18, 9263–9281 CrossRef CAS.
- P. F. DeCarlo, A. M. Avery and M. S. Waring, Thirdhand smoke uptake to aerosol particles in the indoor environment, Sci. Adv., 2018, 4, 1–8 CrossRef PubMed.
- L. Bengtström, M. Salden and A. A. Stec, The role of isocyanates in fire toxicity, Fire Sci. Rev., 2016, 5, 1–23 CrossRef.
- P. Blomqvist, T. Hertzberg, M. Dalene and G. Skarping, Isocyanates, aminoisocyanates and amines from fires - a screening of common materials found in buildings, Fire Mater., 2003, 27, 275–294 CrossRef CAS.
- P. Blomqvist, M. S. McNamee, A. A. Stec, D. Gylestam and D. Karlsson, Detailed study of distribution patterns of polycyclic aromatic hydrocarbons and isocyanates under different fire conditions: Distribution patterns of PAHs and isocyanates under different fire conditions, Fire Mater., 2014, 38, 125–144 CrossRef CAS.
- P. Blomqvist, T. Hertzberg, H. Tuovinen, K. Arrhenius and L. Rosell, Detailed determination of smoke gas contents using a small-scale controlled equivalence ratio tube furnace method, Fire Mater., 2007, 31, 495–521 CrossRef CAS.
- M. A. Garrido, A. C. Gerecke, N. Heeb, R. Font and J. A. Conesa, Isocyanate emissions from pyrolysis of mattresses containing polyurethane foam, Chemosphere, 2017, 168, 667–675 CrossRef CAS PubMed.
- K. W. Fent and D. E. Evans, Assessing the risk to firefighters from chemical vapors and gases during vehicle fire suppression, J. Environ. Monit., 2011, 13, 536 RSC.
- A. Lönnermark and P. Blomqvist, Emissions from an automobile fire, Chemosphere, 2006, 62, 1043–1056 CrossRef PubMed.
- J. D. Mertens, A. Y. Chang, R. K. Hanson and C. T. Bowman, A shock tube study of reactions of atomic oxygen with isocyanic acid, Int. J. Chem. Kinet., 1992, 24, 279–295 CrossRef CAS.
- F. P. Tully, R. A. Perry, L. R. Thorne and M. D. Allendorf, Free-radical oxidation of isocyanic acid, Proc. Combust. Inst., 1989, 22, 1101–1106 CrossRef.
- R. A. Brownsword, T. Laurent, R. K. Vatsa, H.-R. Volpp and J. Wolfrum, Photodissociation dynamics of HNCO at 248 nm, Chem. Phys. Lett., 1996, 258, 164–170 CrossRef CAS.
- P. J. Young, L. K. Emmons, J. M. Roberts, J.-F. Lamarque, C. Wiedinmyer, P. Veres and T. C. VandenBoer, Isocyanic acid in a global chemistry transport model: Tropospheric distribution, budget, and identification of regions with potential health impacts: Isocyanic Acid in a Global Transport Model, J. Geophys. Res.: Atmos., 2012, 117, 1–14 Search PubMed.
- N. Ni and S. H. Yalkowsky, Prediction of Setschenow constants, Int. J. Pharm., 2003, 254, 167–172 CrossRef CAS PubMed.
- M. B. Jensen, On the Kinetics of the Decomposition of Cyanic Acid, Acta Chem. Scand., 1959, 13, 659–664 CrossRef CAS.
- M. C. Barth, A. K. Cochran, M. N. Fiddler, J. M. Roberts and S. Bililign, Numerical modeling of cloud chemistry effects on isocyanic acid (HNCO), J. Geophys. Res.: Atmos., 2013, 118, 8688–8701 CAS.
- G. W. Schade and P. J. Crutzen, Emission of Aliphatic Amines from Animal Husbandry and their Reactions: Potential Source of N20 and HCN, J. Atmos. Chem., 1995, 22, 319–346 CrossRef CAS.
- F. Yu and G. Luo, Modeling of gaseous methylamines in the global atmosphere: impacts of oxidation and aerosol uptake, Atmos. Chem. Phys., 2014, 14, 12455–12464 CrossRef.
- A. F. Bouwman, D. S. Lee, W. A. H. Asman, F. J. Dentener, K. W. Van Der Hoek and J. G. J. Olivier, A global high-resolution emission inventory for ammonia, Global Biogeochem. Cycles, 1997, 11, 561–587 CrossRef CAS.
- C. M. Balion, T. F. Draisey and R. J. Thibert, Carbamylated hemoglobin and carbamylated plasma protein in hemodialyzed patients, Kidney Int., 1998, 53, 488–495 CrossRef CAS PubMed.
- G. Asci, A. Basci, S. V. Shah, A. Basnakian, H. Toz, M. Ozkahya, S. Duman and E. Ok, Carbamylated low-density lipoprotein induces proliferation and increases adhesion molecule expression of human coronary artery smooth muscle cells, Nephrology, 2008, 13, 480–486 CrossRef CAS PubMed.
- R. F. Hems, C. Wang, D. B. Collins, S. Zhou, N. Borduas-Dedekind, J. A. Siegel and J. P. D. Abbatt, Sources of isocyanic acid (HNCO) indoors: A focus on cigarette smoke, Environ. Sci.: Processes Impacts, 2019 10.1039/C9EM00107G.
| This journal is © The Royal Society of Chemistry 2019 |