
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplications of boroxide ligands in supporting small molecule activation by U(III) and U(IV) complexes†
Polly L.
Arnold
 *a,
Laura
Puig-Urrea
a,
Jordann A. L.
Wells‡
a,
Dan
Yuan
b,
Faye L.
Cruickshank
a and
Rowan D.
Young
a
*a,
Laura
Puig-Urrea
a,
Jordann A. L.
Wells‡
a,
Dan
Yuan
b,
Faye L.
Cruickshank
a and
Rowan D.
Young
a
aEaStCHEM School of Chemistry, Joseph Black Building, The King's Buildings, The University of Edinburgh, Edinburgh, EH9 3FJ, UK
bKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Dushu Lake Campus, Suzhou 215123, People's Republic of China
First published on 29th March 2019
Abstract
The boroxide ligand [OBAr2]− (Ar = Mes, Trip) is shown to be able to support both UIII and UIV centres for the first time. The synthesis and structures of homoleptic and heteroleptic UIII and UIV complexes are reported. The UX3 complex with larger substituents, [U(OBTrip2)3]2, exhibits greater thermal stability compared to less encumbered [U(OBMes2)3]2 but reacts with a smaller range of the small molecules tested to date. Initial studies on their capacity to participate in small molecule chemistry show that dark purple [U(OBMes2)3]2 binds and/or reductively activates a variety of small molecules such as pyridine-oxide, triphenylphosphineoxide, sulfur, and dicyclohexylcarbodiimide. While [U(OBMes2)3]2 shows no reaction with CO or CO2, [U(OBTrip2)3]2 is oxidised by both, in the former case forming [U(OBTrip2)4], and in the latter case forming a small quantity of the structurally characterised μ-carbonate product [(μ-CO3){U(OBTrip2)3}2].
Introduction
Complexes of the form UX3 where X is a large, monoanionic ligand such as an amide or aryloxide have shown great utility in the binding and reductive activation of small molecules. The choice of X provides great control over the space at the Lewis acidic metal, and the strength of the reducing capacity of the UIII centre. For example, the common silylamide and aryloxide complexes [UN′′3] and [U(ODtbp)3] (N′′ = N(SiMe3)2, ODtbp = O-2,6-tBu2C6H3) reductively couple carbon monoxide gas at room temperature and pressure to form the [OCCO]2− fragment as the sole product in [X2UOCCOUX2].1,2 The dinuclear tren derivative [{U(tren′)}2(μ-η2:η2-N2)] (tren′ = N(NCH2CH2NSitBuMe2)3) and [U(ODtbp)3] show ready but reversible side-on N2 coordination,2 but the OTtbp analogue (OTtbp = O-2,4,6-tBu3C6H2) [{U(OTtbp)3}2(μ-η2:η2-N2)] releases N2 only upon heating a solution to 80 °C, even in the presence of small O-containing reagents such as CO2 and CO.2We showed that bulky trimesitylsiloxide complexes bind N2 even more strongly in [{U(OSiMes3)3}2(μ-η2:η2-N2)],3 and others have shown that further functionalisation of the trapped and reduced N2 in polymetallic nitrido complexes supported by tertbutylsiloxides is possible.4 Flexible uranium siloxides have also allowed reactions such as CO2 deoxygenation and carbonate formation in [{U(OSi(OtBu)3)3}2(μ-η1:η2CO3)].5
The capacity to transfer one X ligand has also provided significant advantages for new reactivity. For example, storage of arene solutions of [UX3] (X = N′′, ODtbp) results in the spontaneous reductive activation of the arene (over 90 h at 90 °C, or 30 days at 25 °C) through the formation of [{X2U}2(μ-η6:η6-C6H6)] and elimination of two equivalents of UX4 by-product.6 The trapped arenes are also sufficiently reduced that a mild, homogeneous borylation of one C–H group is possible.
Boroxides provide an interesting alternative to alkoxides and aryloxides as these can provide greater delocalisation of the electron density across the OBM bond, and even a distant Lewis acidity via an empty orbital on the boron atom, thus allowing the electron density to be tuned at the metal centre.7 The aryl functionalisation can also provide a larger protected pocket at the metal as the bulk of the aryl groups is maintained at a greater distance from the metal binding site.8
We have studied the capacity of the boroxide ligand [OBAr2]− (Ar = Mes, Trip) to bind UIII for the first time. Here we show the synthesis and structures of new UIII and UIV complexes and initial studies on their capacity to participate in small molecule chemistry.
Results and discussion
Syntheses
The formation of dark purple [U(OBMes2)3]21m from HOBMes2 and UN′′3 occurs in less than 3 hours at room temperature in toluene solution, Scheme 1. Work-up from hexanes affords dark crystals in 46% yield, suitable for a single crystal X-ray diffraction study, Fig. 1. At room temperature the 1H NMR spectrum of 1 shows the retention of the dimeric structure in solution with two different sets of ligand resonances assignable to terminal and bridging ligands. The bulkier aryl groups on the boroxide still lead to similar reaction chemistry to afford [U(OBTrip2)3]21t, although the major by-product in the synthesis is the tetrakis(ligand) complex 2t, and both UX3 and UX4 are isolated by fractional crystallisation in similar (ca. 30%) yields. The disproportionation of uranium(III) to uranium(IV) and uranium metal is a known oxidative pathway.6,9,10 Dark red crystals of the mononuclear diethylether adduct [(Et2O)U(OBTrip2)3] 1t-Et2O are formed from reactions in diethyl ether and have also been structurally characterised by a single crystal X-ray diffraction study, Fig. 1. The 1H NMR spectrum of 1m suggests exchange between unbound and bound Ar groups on bridging OBMes2 at ambient temperature and support the dimeric conformation. The resonances assigned to the bridging ligands are strongly paramagnetically shifted out of the diamagnetic region (from −3.71 to −15.13 ppm) due to proximity to the uranium centres, whereas the resonances corresponding to the terminal ligands (double the relative intensity) are between 8.29 to 2.53 ppm. The 11B NMR spectrum contains a resonance at 73.7 ppm that is tentatively assigned to one of what should be two different boron environments, but only one other signal attributed to trace free ligand (50.8 ppm) is observed over an extended frequency range. | ||
| Scheme 1 Synthesis of complexes 1 showing common by-products and decomposition products. | ||
 | ||
| Fig. 1 Displacement ellipsoid drawing of the molecular structure of (a) 1m and (b) 1t-Et2O. The hydrogen atoms are omitted and the mesityl and triisopropylphenyl groups are depicted as wireframe and capped sticks for clarity. The thermal ellipsoids are displayed at 50% probability. | ||
Clean starting materials and precise control of the stoichiometry is key to the formation of pure samples, as both derivatives of 1 are very air sensitive. The homoleptic UIV complexes [U(OBAr2)4] 2 are significantly more stable, and can be formed as minor side products in syntheses of 1, and from independent metathetical routes from the reaction between uranium tetraiodide and the salt NaOBAr2, Scheme 1. A high-yielding route to 2m is available from the reaction of NaOBMes2 with [UI4(1,4-dioxane)2] in toluene, affording [U(OBMes2)4(dioxane)0.5]2 (2m·diox). Colourless crystals of 2m·diox and 2t·C7H8 suitable for XRD analysis were obtained from slow diffusion of hexanes into the reaction mixture, and standing of a concentrated toluene solution, respectively, see below. Complex 2m·diox was characterised by 1H NMR spectroscopy, mass spectrometry (APPI-MS) and single crystal XRD analysis. The 1H NMR spectrum show the corresponding signals of one boroxide ligand environment. Mass spectrometric analysis showed the molecular ion at m/z = 1298.76 that corresponds to [M]˙+ fragment [U(OBMes2)4]˙+. The 1H NMR spectrum of 2t contains many more resonances than that of 1t due to the congestion around metal centre; twelve magnetically different methyl group resonances are measured in the range of 4.13–18.40 ppm and five different methine groups are discernible in the range 8.86–4.78 ppm.
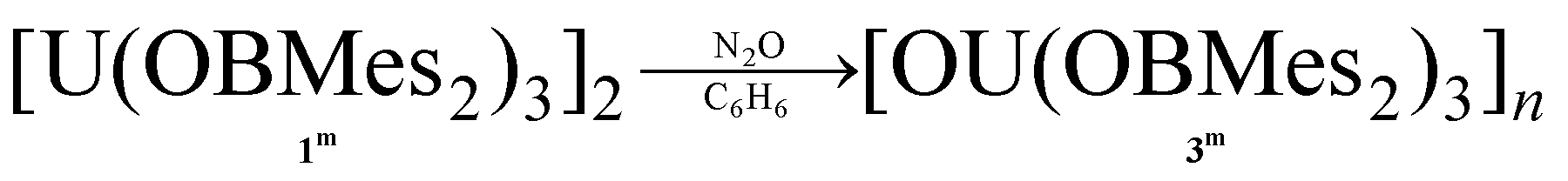 | (1) |
Additionally, complex 1m is readily oxidised to the oxo UIV product [OU(OBMes2)3]n3m, due to the presence of traces of oxygen, which has been fully characterised from an independent synthesis. Samples also decompose when heated to 100 °C in toluene or in the solid state (10−3 mbar, 115 °C, 6 h). Upon exposure of a benzene solution of complex 1m to one atmosphere of dry N2O, a rapid colour change from dark purple to light brown is observed due to the formation of the UIV oxo complex 3m, which is assigned as containing bridging oxo ligands by comparison with the literature and in consideration of the size of the OBMes2 ligands, eqn (1). The 1H and 13C NMR spectra show resonances corresponding to one mesityl environment, which is slightly shifted due to paramagnetic contributions. The 11B NMR spectrum contains one resonance at 89.29 ppm, which confirms the presence of only one boron environment. The elemental analysis result is in agreement with the formation of 3m. Reactions designed to reduce complex 3m on a bulk scale have so far been unsuccessful; a small quantity of a UIII product K2[{(OBMes2)3U}2(μ-O)2] (10m) that was isolated from a reaction with KC8 has been structurally characterised (see ESI† for details).
Any traces of the metallacycle [UN′′2{κ2-N′(SiMe2CH2)}] (N′ = N(SiMe3), a common UIV contaminant in UN′′3, forms [UN′′3(OBMes2)] and [UN′′3(OBTrip2)] (vide infra).
Crystallography
The solid-state structure of 1m confirms the formation of a centrosymmetric binuclear UIII complex in which the two uranium centres are bridged by two boroxide ligands (Fig. 1a) in a distorted trigonal bipyramid geometry due to the interaction of the uranium centres with the bridging mesityl groups. The U⋯C distances for these interactions (U⋯Cavg 2.845 Å) are slightly longer than other η1 interactions with uranium, such as the complexes bearing the ligand {1,3[2,5-(iPr)2PhNC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)]C6H4} reported by Gambarotta and co-workers (2.678(12) Å).11 The U–O bond lengths for the boroxide ligands are 2.548(10) Å and 2.372(10) Å for the bridging boroxides, and an average U–Oavg bond length of 2.196 Å for the terminal boroxides. Both are in the same range as the U–O distances for the siloxide-bridged UIII dimer reported by Mazzanti and co-workers, [U(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2, which features two UIII centres with two bridging and two terminal siloxide ligands with U–O distances of 2.549(3) Å and 2.396(3) Å for the bridging siloxides, and a U–Oavg of 2.193(4) Å for the terminal ligands. Moreover, the U⋯U distance for complex 1m is 3.966 Å, which is similar to the UIII siloxide complex (U⋯U 3.9862(2) Å).5 The U–Oavg distance for the terminal ligands is similar to that of [(Et2O)U(OBTrip2)3] 1t-Et2O (mean 2.183(7) Å) and slightly longer than that of the uranium tris(aryloxide) complex, [U(ODtbp)3], with U–Oavg = 2.159 Å. The average B–O bond length of complex 1m is 1.351 Å. For both complexes the B–Oavg fall within range for Ar2BO–M complexes reported in the literature (1.295–1.405 Å, mean 1.350(1) Å for a fragment search for Mes2BO–M in the Cambridge Structural Database).12 In the solid-state structure of 1t-Et2O the average U–Oboroxide distance is 2.183 Å, slightly shorter in 1m but significantly longer than in [U(ODtbp)3] (2.159 Å). The U–Oether is 2.530(7) Å and is much shorter than in the seven-coordinate complex [{(NeopArO)3tacn}U(OEt2)] (2.669(2) Å, NeopArO = 2-neopentyl-4-methylphenoxide, tacn = triazacyclononane), which is the only other crystallographically characterised UIII diethyletherate.13 This is consistent with both the more electron-deficient uranium centre engendered by the poor π donor properties of the boroxide ligand and the lower coordination number in 1t-Et2O.
CH2)]C6H4} reported by Gambarotta and co-workers (2.678(12) Å).11 The U–O bond lengths for the boroxide ligands are 2.548(10) Å and 2.372(10) Å for the bridging boroxides, and an average U–Oavg bond length of 2.196 Å for the terminal boroxides. Both are in the same range as the U–O distances for the siloxide-bridged UIII dimer reported by Mazzanti and co-workers, [U(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2, which features two UIII centres with two bridging and two terminal siloxide ligands with U–O distances of 2.549(3) Å and 2.396(3) Å for the bridging siloxides, and a U–Oavg of 2.193(4) Å for the terminal ligands. Moreover, the U⋯U distance for complex 1m is 3.966 Å, which is similar to the UIII siloxide complex (U⋯U 3.9862(2) Å).5 The U–Oavg distance for the terminal ligands is similar to that of [(Et2O)U(OBTrip2)3] 1t-Et2O (mean 2.183(7) Å) and slightly longer than that of the uranium tris(aryloxide) complex, [U(ODtbp)3], with U–Oavg = 2.159 Å. The average B–O bond length of complex 1m is 1.351 Å. For both complexes the B–Oavg fall within range for Ar2BO–M complexes reported in the literature (1.295–1.405 Å, mean 1.350(1) Å for a fragment search for Mes2BO–M in the Cambridge Structural Database).12 In the solid-state structure of 1t-Et2O the average U–Oboroxide distance is 2.183 Å, slightly shorter in 1m but significantly longer than in [U(ODtbp)3] (2.159 Å). The U–Oether is 2.530(7) Å and is much shorter than in the seven-coordinate complex [{(NeopArO)3tacn}U(OEt2)] (2.669(2) Å, NeopArO = 2-neopentyl-4-methylphenoxide, tacn = triazacyclononane), which is the only other crystallographically characterised UIII diethyletherate.13 This is consistent with both the more electron-deficient uranium centre engendered by the poor π donor properties of the boroxide ligand and the lower coordination number in 1t-Et2O.
Together, the nearly linear U–O–B angle for the terminal boroxide ligands (mean 171.6° in 1m 171.0° in 1t-Et2O) and the elongation of the U–O bonds are a direct consequence of the reduced π-donor ability of the ligand compared with a carbocyclic aryloxide, which is decreased by having a boron substituent on the ligand. Electron density from the oxygen is accepted by the boron, leading to a lower degree of π-donation from the boroxide to the uranium centre.
The solid-state structure of 2m·diox reveals two UIV ions both in a distorted trigonal bipyramidal coordination environment (Fig. 2a). Each uranium centre is ligated by one end of a bridging dioxane molecule in an axial position with a U–O distance of 2.577(3) Å, which is considerably longer than in [UI4(1,4-dioxane)2] or the [UI2(dioxane)2(aryloxide)] previously reported by our group (mean = 2.333(6) Å and 2.087 Å respectively).14–16 The boroxide ligands in the equatorial positions have a U–Oavg distance of 2.144(3) Å, which is shorter than in the uranium(III) complex 1m, consistent with a UIV centre, and similar to the U–O bond length for [U(OBTrip)4] with a U–O distance of 2.159(5) Å. The boroxide ligands in the axial positions have a U–O distance of 2.095(3) Å, which is much shorter than the equatorial boroxides. The B–Oavg distance is 1.356 Å, which is similar to complex 1m and falls within range for reported metal boroxide complexes. The U–O–Bavg angles are 167.1°, which is comparable to the U–O–B angles of the terminal boroxide ligands in 1m (mean 171.6°).
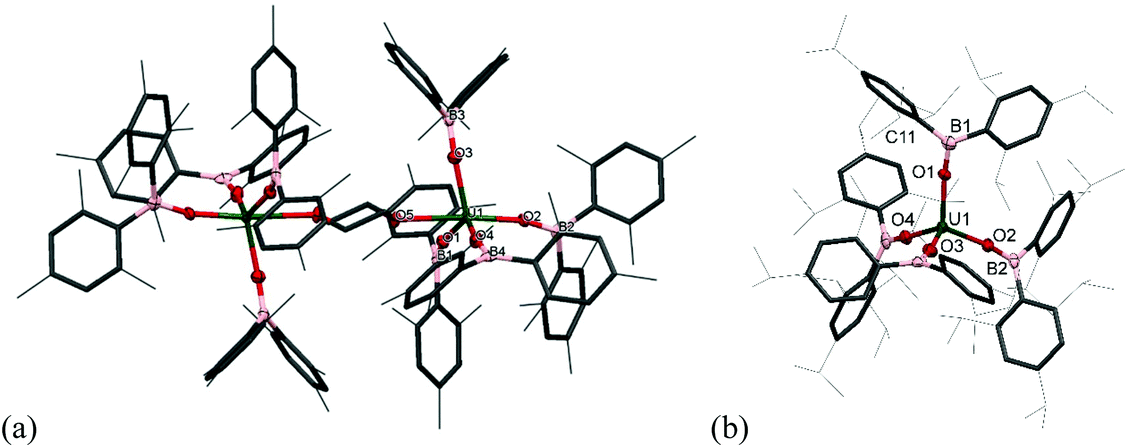 | ||
| Fig. 2 Displacement ellipsoid drawing of the molecular structure of (a) 2m·diox and (b) 2t·C7H8. Thermal ellipsoids are displayed at 50% probability. Lattice solvent and hydrogen atoms omitted, and aryl substituents are depicted as wireframe and capped sticks for clarity. | ||
Single crystals of 2t·C7H8 suitable for X-ray diffraction analysis were obtained from concentrated toluene solution, and its solid-state structure is depicted in Fig. 2b. The solid-state structure shows an unsolvated four-coordinate uranium centre in tetrahedral geometry. The mean boroxide ligand U–O distance of 2.159(5) Å is shorter than in the uranium(III) complex 1t-Et2O, consistent with a uranium(IV) centre. The U–O bond lengths in 2t are longer than those in homoleptic uranium(IV) aryloxide [U(ODtbp)4] (2.135(4) Å), due to the greater steric demand of the boroxide ligand and poorer metal to ligand backbonding. The U–O–B angles are comparable to those in 1t-Et2O (mean 171.0(5)°), despite the considerable bulk about the uranium centre. The average B–O bond length of 1.364(7) Å falls within range for reported metal boroxide complexes and is slightly longer than in 1t-Et2O. The longer B–O bond may be due to the more Lewis acidic uranium(IV) centre in 2t.
Reactions of 1m and 1t
Compound 1m has been treated with a wide range of small molecules, Scheme 2.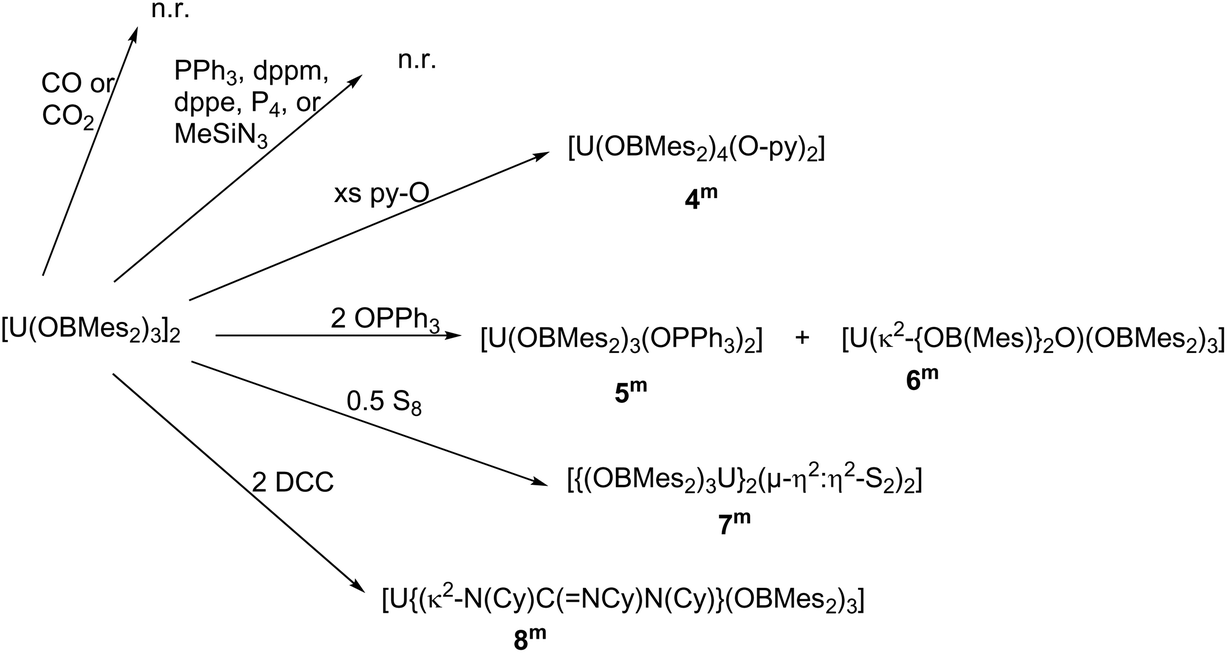 | ||
| Scheme 2 Reactions of complex 1m investigated. | ||
Reactions with small, neutral N- or P-donor reagents that are readily visible by NMR spectroscopy were designed to identify whether the boron atoms in the ligands retain sufficient Lewis acidic character to participate in reactions. However, arene solutions of 1m showed no reaction with added Me3SiN3, PPh3, CH2(PPh2)2 (dppm), C2H4(PPh2)2 (dppe) or white phosphorus, P4. Normal, simple UX3 complexes with large monoanionic X ligands would be anticipated to react with these, readily forming oxidised uranium complexes or adducts, for example, [Me3SiNUN′′3],17,18 [Me3SiNU{OSi(OtBu)3}3],19 [U(BH4)3(PEt3)2].20
Pyridine N-oxide
The formation of oxo complexes with structures such as ULnO,21–23 LnU![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O13,24 or U–O–U25 have been reported from reactions with pyridine N-oxide. In this case, addition of benzene to a mixture of [U(OBMes2)3]2 and an excess of pyridine N-oxide provides a brown solution from which [U(OBMes2)4(py-O)2] (4m) can be isolated as dark orange solids after 72 hours, Scheme 2. The reaction proceeds through oxidation of complex 1m to mononuclear UIV complex 4m with ligand redistribution. Coordination of pyridine N-oxide to a metal is not unique, although relatively rare.13,26–29 The identity of complex 4m was confirmed by 1H NMR spectroscopy, APPI mass spectrometry and single crystal XRD analysis. Mass spectrometric data showed the expected molecular ion at m/z = 1223.65, which corresponds to the [M]˙+ fragment, [U(OBMes2)4(Py–O)2]˙+. Dark orange crystals of 4m suitable for XRD analysis were grown from a concentrated benzene solution. The data are of sufficient quality to discuss connectivity information but not for bond metrics. The solid-state structure reveals a UIV complex with a distorted octahedral geometry with the boroxide ligands in the equatorial positions and the pyridine N-oxide ligands in the axial positions (Fig. 3).
O13,24 or U–O–U25 have been reported from reactions with pyridine N-oxide. In this case, addition of benzene to a mixture of [U(OBMes2)3]2 and an excess of pyridine N-oxide provides a brown solution from which [U(OBMes2)4(py-O)2] (4m) can be isolated as dark orange solids after 72 hours, Scheme 2. The reaction proceeds through oxidation of complex 1m to mononuclear UIV complex 4m with ligand redistribution. Coordination of pyridine N-oxide to a metal is not unique, although relatively rare.13,26–29 The identity of complex 4m was confirmed by 1H NMR spectroscopy, APPI mass spectrometry and single crystal XRD analysis. Mass spectrometric data showed the expected molecular ion at m/z = 1223.65, which corresponds to the [M]˙+ fragment, [U(OBMes2)4(Py–O)2]˙+. Dark orange crystals of 4m suitable for XRD analysis were grown from a concentrated benzene solution. The data are of sufficient quality to discuss connectivity information but not for bond metrics. The solid-state structure reveals a UIV complex with a distorted octahedral geometry with the boroxide ligands in the equatorial positions and the pyridine N-oxide ligands in the axial positions (Fig. 3).
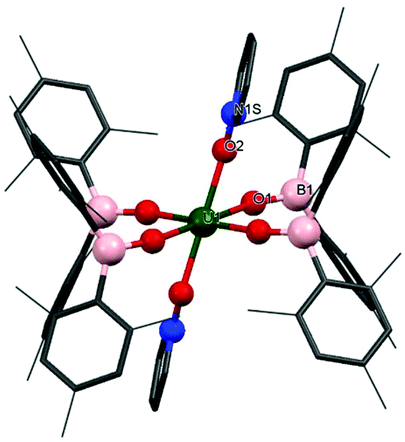 | ||
| Fig. 3 Solid-state structure of 4m. Hydrogens atoms are omitted and mesityl and pyridine oxide ligands are depicted as wireframe and capped sticks for clarity. | ||
Triphenylphosphine oxide
Phosphine oxides are widely used ligands in f-element chemistry. As a result of the oxophilic and Lewis acidic character of lanthanides and actinides, phosphine oxides coordinate readily, and are able to stabilise a variety of complexes in low and high oxidation states.30–35 Moreover, applications in catalysis and liquid–liquid extraction processes have been found.36 Therefore, the reaction of a benzene solution of the uranium boroxide complex 1m with two equivalents of OPPh3 was carried out, Scheme 3. This leads to the formation of [U(OBMes2)3(OPPh3)2] (5m), which co-crystallised together with a minor by-product [U(κ2-{OB(Mes)}2O)(OBMes2)3] (6m).7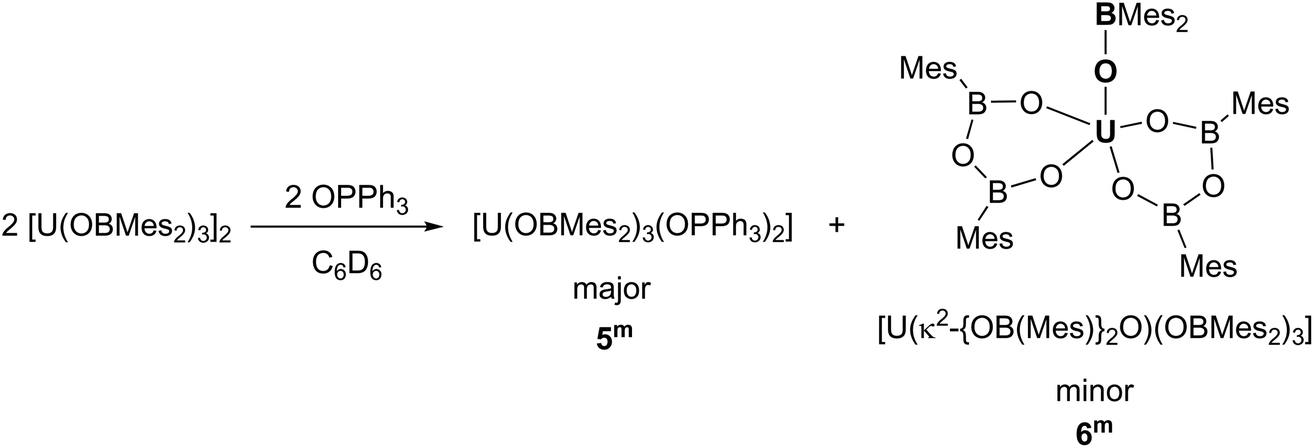 | ||
| Scheme 3 Synthesis of compounds 5m and 6m. | ||
The 1H NMR spectrum of 5m in C6D6 contains six resonances which range from 6.9 to 1.87 ppm. The 31P NMR spectrum shows a single broad resonance at −17.83 ppm, and this is significantly shifted from the free OPPh3 resonance (∼25 ppm) as a result of its proximity to the metal centre. Mass spectrometric analysis (APPI-MS) showed a molecular ion peak at m/z = 1589.77 that corresponds to fragment [5m]˙+. By-product [U(κ2-{OB(Mes)}2O)(OBMes2)3] (6m) contains a coordinated boroxinato ligand, a cyclic anhydride of borinic acid,37 which is formally a product of ligand condensation, but probably formed from a contaminant in the ligand. Boroxines are rigid bidentate ligands which have drawn attention recently, mostly by forming complexes of main group elements, e.g. for Al;38–40 Sn, Sb and Bi;41–47 but also for transition metals such as Mn,48 Au37,49 and Pt.50
Single crystals of 5m and 6m were obtained by slow diffusion of hexanes into a benzene solution. Complex 5m has an approximately trigonal bipyramidal coordination geometry (Fig. 4). The U–OPPh3 bond lengths are 2.273(4) Å and 2.270(4) Å, which are ∼0.1 Å shorter than other UIII–OPPh3 distances previously reported.36,51 The U–O–P angles for the phosphine oxide ligands are 170.4(3)° and 174.4(2)° respectively, and the O4–U–O5 angle is 176.62(13)°. These angles are similar to the angles in the known complex, [U(NPhArF)3(OPPh3)2].24 The U–Oavg distance for the boroxide ligands is 2.124 Å, which is ∼0.07 Å shorter than the U–O bond lengths for the terminal boroxides in 1m (mean 2.196 Å). Similarly, the average B–O distance is 1.345 Å which is slightly shorter than complex 1m (mean 1.351 Å). Moreover, the mean U–O–B angle is 176.0°, and this is less bent than the terminal boroxide in 1m (mean 171.6°). Complex 6m has a nearly square pyramidal coordination geometry. The metallacycle formed from the [OBOBO]2− unit has similarities with early metal complexes such as (η2-catechol-μ-catecholborate)3ThCl(C4H8O)3·C4H8O that are coordinated by alternating catechol and catecholborate groups in a hexaoxo trianionic macrocyclic structure, observed for Zr, Nd, Sm, U and Th centres.52,53 For the boroxine ligand, the U–O bond distances are 2.193(4) Å and 2.191(4) Å. The B–O distances range from 1.342(9)–1.370(9) Å, which are in the same range as previously reported for main group boroxines. The six-membered ring unit has internal angles close to 120°, except for the O9–U–O11 bond which is much smaller, being only 77.40(16)°. The boroxide ligands have similar U–O and B–O bond lengths (U–Oavg 2.172 Å, B–Oavg 1.316 Å) to the terminal boroxides in 1m (mean U–O 2.196 Å, B–O 1.351 Å). Finally, the U–O–B angles for the boroxides range from 155.2(4)° to 168.7(4)°, which are significantly more bent than for the terminal boroxides in 1m (mean 171.6°).
 | ||
| Fig. 4 Solid-state structure of (a) 5m and (b) 6m. Mesityl and phenyl ligands are depicted as capped sticks and wireframe for clarity. Hydrogen atoms are omitted. The thermal ellipsoids are displayed at 50% probability. | ||
Elemental sulfur
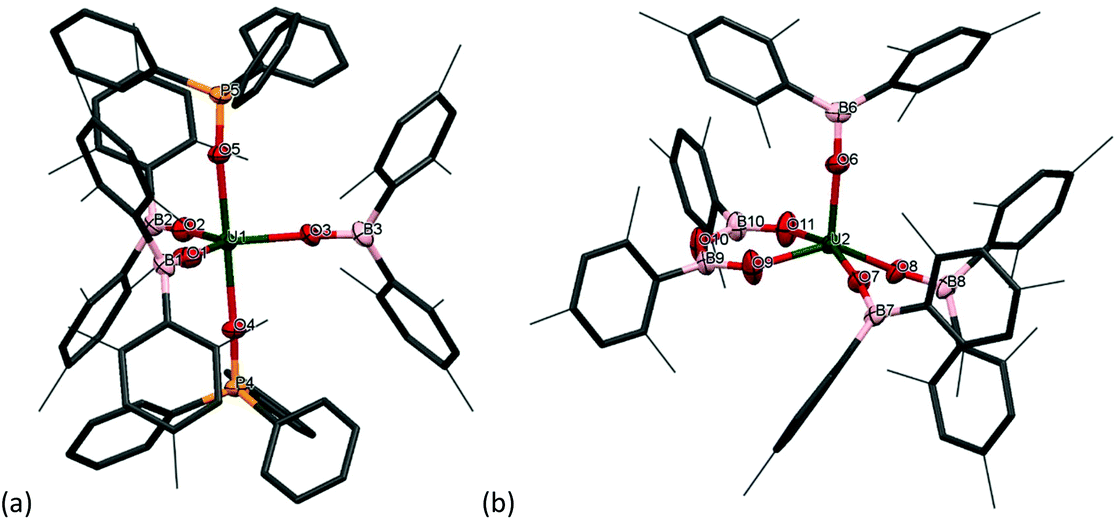
The reactivity of chalcogens and chalcogenides with actinide complexes has gained attention recently. Of particular interest is the potential of the metal–ligand bonding in these more polarisable systems to advance our understanding of the degree of covalency in uranium–ligand bonds, as this can provide insights into better separation of actinides from lanthanides in spent nuclear fuel.54–58 Addition of 0.5 equivalents of S8 to a stirring solution of [U(OBMes2)3]21m leads to the formation of [{(OBMes2)3U}2(μ–η2:η2–S2)2] (7m), which contains two independent bridging S2 units. There is only one other example in the literature of a uranium complex featuring two bridging S2 units, [{([AdArO]3N)U}2(μ-η2:η2-S2)2].59 The 1H NMR spectrum contains a set of resonances corresponding to the OBMes2 ligand, indicating one ligand environment. Mass spectrometric analysis shows the molecular ion at m/z = 1033.56 that corresponds to the [M]˙+ fragment, [{(OBMes2)3U}(μ-η2:η2-S2)]˙+. Translucent red crystals of the sulfur-bridged complex 7m suitable for XRD analysis were grown from a concentrated toluene solution.Fig. 5 shows the molecular structure of 7m, confirming the formation of a uranium dimer with two S2 units. Each uranium ion has a distorted pentagonal bipyramidal coordination geometry. The S1B–S2B bond length is 2.050(2) Å, which is a typical value for a S–S single bond, and comparable to H2S2 with a S–S bond length of 2.055 Å.44 The S⋯S distance between the two units is 3.491 Å, excluding the possibility of a S42− unit.
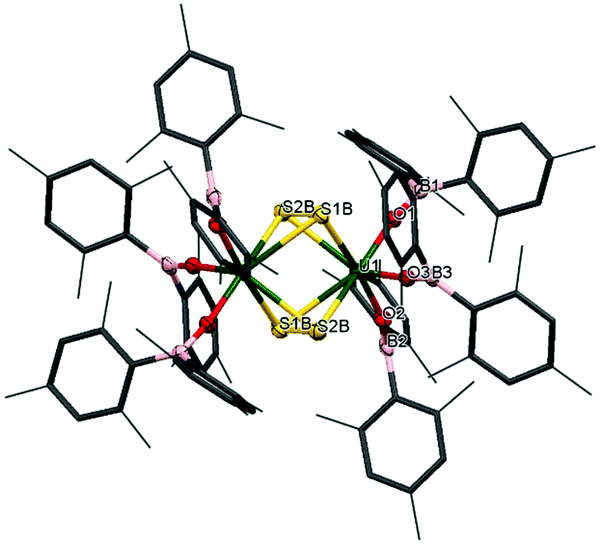 | ||
| Fig. 5 Solid-state structure of complex 7m. Mesityl ligands are depicted as capped sticks and wireframe for clarity. Hydrogen atoms are omitted. The thermal ellipsoids are displayed at 50% probability. | ||
Thus, although one would normally anticipate a UIV formal oxidation state product to form from the reduction of sulfur, here the UV assignment appears more appropriate. The ligands are both identified as bridging persulfido (S22−), giving a formal UV oxidation state for 7m by comparison of the U–E bonds. Previously reported uranium persulfido S22− complexes show S–S distances from 2.050 Å to 2.103 Å.60–68 Previously reported transition metal supersulfido S2− complexes show S–S distances from 1.944 to 2.023 Å.69–72 Furthermore, the U–Oavg bond length is 2.078 Å, which is significantly shorter than the U–O bond lengths for the terminal boroxides in UIII1m (mean 2.196 Å), and in 2m (mean 2.119 Å), also suggesting a formal oxidation state of +5 is most likely. Moreover, the B–O average distance is 1.359 Å, which is slightly longer than for complexes 1m and 2m (mean 1.351 Å and 1.356 Å respectively). The U–O–B angle of 165(3)° is much more bent than for the terminal boroxide ligands in 1m (mean 171.6°), which is reasonable if more electron density from the O atoms are being distributed into a backdonation from the uranium centres to the sulfido ligands.
DCC, N,N′–dicyclohexylcarbodiimide
The direct reaction of carbodiimides such as (DCC = N,N′–dicyclohexylcarbodiimide) with uranium was first reported in 2009.73 The aim of these reactions was to insert a carbodiimide into a U–C ligand bond in order to increase the steric crowding in organouranium complexes. Since then, several examples of insertion of carbodiimides into metal–ligand σ-bonds have been reported,74–77 as well as further reactivity towards small molecules with these ligands.78–80 Moreover, reactions of carbodiimides have also led to the formation of metallacycles that have unique structural properties and possible applications in catalysis.81–84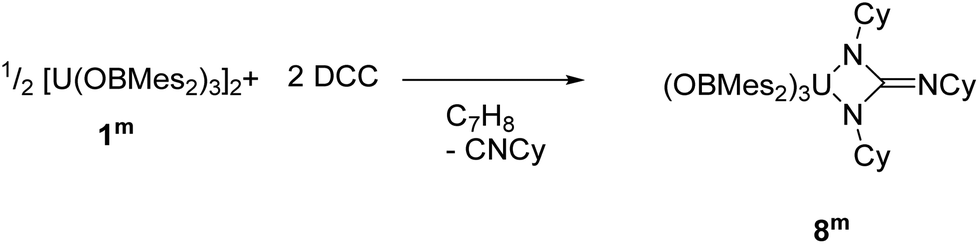 | (2) |
Dropwise addition of a toluene solution of DCC to a dark purple toluene solution of 1m produced a green solution from which [U{η2-N(Cy)C(NCy)N(Cy)}(OBMes2)3] (8m, Cy = cyclo-C6H11), eqn (2), was isolated in close to quantitative yield after work-up. Mass spectroscopic analysis confirms the formulation of 8m as the molecular ion at m/z = 1337.85 that corresponds to [M + H]+ was observed. Single crystals for XRD characterisation could not be obtained. The 1H NMR spectrum of 8m has resonances from 58.36 to −10.62 ppm, widely shifted out of the diamagnetic region by proximity to the paramagnetic uranium centre. Taken together, the characterising data show that the carbodiimide has been coupled by the reducing U centre to form the rearranged cyclohexyl-substituted guanidinate ligand via loss of cyclo-hexylisonitrile. This presumably occurs similarly to the procedure reported for the reaction that converts [U(η5–C5Me5)2(η2-bipy)] to [U(η5–C5Me5)2{η2–N(Cy)C(NCy)N(Cy)}] via extrusion of [CyNC] from a coordinated DCC ligand then a [2 + 2] addition to the transiently formed UVNCy group.81 The fact that this forms from the UIV product again supports the stabilising effect of this ligand on the higher formal oxidation states of uranium.
Reactions of 1t with CO and CO2
In contrast to 1m, the bulkier 1t reduces both CO and CO2; reaction with the former affording the known oxidation product 2t, and the latter affording a bridging carbonate 9t, although the yields are moderate and minimal respectively, Scheme 4.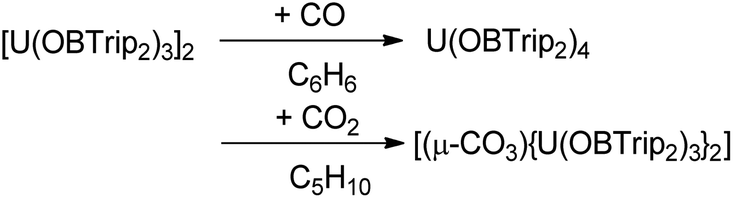 | ||
| Scheme 4 Reactions of complex 1t with CO and CO2. | ||
Exposure of a degassed purple-brown solution of 1t in cyclopentane to a 1 atm pressure of CO2 results in the immediate formation of a green solution. Colourless crystals can be obtained upon allowing the solution to stand at room temperature overnight. The single crystals collected were of sufficient quality to obtain connectivity information by single crystal XRD analysis to identify the product as [(μ-CO3){U(OBTrip2)3}2] (9t). The solid-state structure is reminiscent of other bimetallic carbonate-bridged uranium(IV) complexes,85,86 with a disordered bridging carbonate linking the uranium centres with distorted trigonal bipyramidal geometries. The boroxide ligands are staggered along the U1–C1–U1A axis to minimise the steric interactions of the ligands. The formation of carbonate bridged bimetallic uranium(IV) complexes has been postulated to proceed via a terminal uranium(V) oxo.87,88
Comparatively, exposure of 1t solutions to a CO atmosphere only results in decomposition to the homoleptic U(IV) boroxide complex 2t and no CO containing products could be identified.
Formation of complexes [U(OBMes2)N′′3] 11m and [U(OBTrip2)N′′3] 11t
As mentioned previously, any traces of the metallacycle [UN′′2{κ2-N′(SiMe2CH2)}] in the synthesis of complexes 1m and 1t leads to the formation of complexes [UN′′3(OBMes2)] (11m) and [UN′′3(OBTrip2)] (11t). Moreover, an alternative synthesis for complex 11m is achieved by addition of just one equivalent of HOBMes2 to an orange-brown solution of the uranium metallacycle [UN′′2{κ2-N′(SiMe2CH2)}], which provides a brown solution from which [UN′′3(OBMes2)] (11m) can be isolated as brown solids (Scheme 5). Formation of complex 11m was confirmed by 1H and 29Si spectroscopies, elemental analysis and single crystal XRD analysis.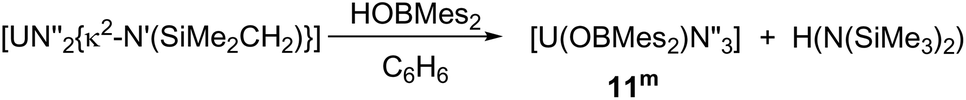 | ||
| Scheme 5 Synthesis of heteroleptic complex 11m. | ||
The 1H NMR spectrum shows a paramagnetically shifted resonance at −6.56 ppm, which corresponds to the methyl groups in the tris(silylamide) ligand, and three smaller sets of resonances for the boroxide ligand. Moreover, the 29Si NMR resonances shift from −74.46 and −81.84 ppm for the uranium metallacycle to −131.74 ppm for complex 11m.
Single crystal XRD analysis revealed that the solid-state structure of complexes 11m and 11t have a distorted tetrahedral coordination environment where the uranium centre is coordinated by one terminal boroxide ligand and three silylamide groups (Fig. 6). Crystals of complex 11m suitable for XRD analysis are obtained from a concentrated toluene solution. The average U–Navg bond length is 2.268 Å, which is ∼0.03 Å shorter than that of the previously reported complex [U(N(SiMe3)4],89 which is reasonable if more electron density is being distributed to the boroxide ligand and more electron density is being pulled from the tris(silylamide) ligands, thus decreasing the U–N bond length. The B–Oavg is 1.365(4) Å, which is longer than the B–Oavg bond length in complex 1m (mean 1.351 Å). Moreover, the N–U–N angles are 105.00(8)°, 113.72(8)° and 116.96(9)°, which are in the same range as for [U(N(SiMe3)4]. The U–O distance is 2.1559(18) Å, which is slightly shorter than the U–O distance in the terminal boroxides for complex 1m (U–Oavg is 2.196 Å) but longer than the U–Oavg for 2m (2.144 Å). The U–O–B angle is 167.23(19)°, which is in the same range as U–O–B angles for the terminal boroxides in complex 1 (mean 171.6°). Colourless crystals of complex 11t suitable for X-ray diffraction analysis were obtained from a concentrated benzene solution upon standing at room temperature. The U–O distance amounts to 2.167(3) Å, which is longer than that of OB(Mes)2-coordinating analogue 11m. The mean U–N bond length is 2.272(3) Å, which is longer than complex 11m and the previously reported [U(N(SiMe3)4].89 Bond length of B–O (1.365(5) Å) is essentially the same as that of U(IV) complex 2t (1.364(7) Å). Bond angle of U–O–B is 169.8(3)°, which is in the same range as the U–O–B angle in complex 11m.
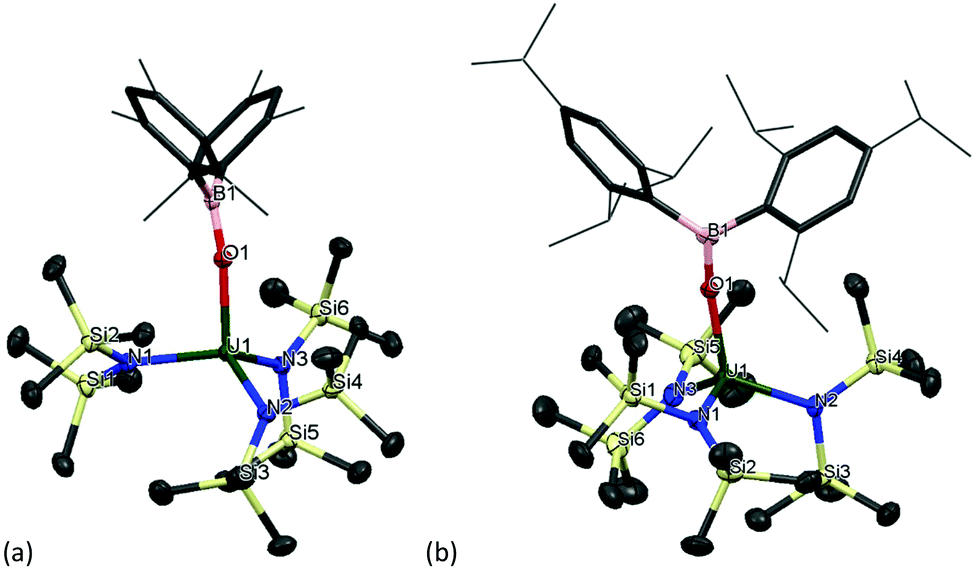 | ||
| Fig. 6 Solid-state structure of (a) 11m and (b) 11t. Mesityl and triisopropylphenyl ligands are depicted as capped sticks and wireframe for clarity. Hydrogen atoms are omitted. The thermal ellipsoids are displayed at 50% probability. | ||
Conclusions
Diaryl boroxide ligands are capable ligands for both UIII and UIV cations but the UIII complexes are particularly air-sensitive and decompose readily to different UIV complexes depending on the conditions. We suggest that rather than working in tandem as a second Lewis acidic reaction centre, the role of the boron is simply to remove electron density from the reducing UIII centre in 1 to such an extent that it does not efficiently reduce an incoming substrate, and neither the U or B centres in these complexes can bind a phosphine. No reactions were observed for 1m with carbon monoxide or carbon dioxide at ambient pressure and temperature, underlining these observations. Reactions at elevated temperatures simply result in the decomposition of 1m. Meanwhile, with larger boroxide aryl substituents, complex 1t exhibits greater thermal stability compared to less encumbered 1m and greater reactivity with the less reducible, smaller carbon oxides, but lower reactivity with the less reactive, but slightly larger small molecules tested so far. In general, the reactions of 1m with small molecules and other substrates give low yields due to the extensive formation of 3m from traces of oxygen, or perhaps also reactions with vacuum grease and/or glassware. Qualitatively, the sum of these reactions suggest that the OBAr2 ligands make the UX3 centre less strongly reducing than the analogous aryloxide ligands ODtbp, which is in line with expectation, but we caution that due to the additional modes of reactivity available to the boron centre, a direct and more precise estimate of the relative oxidation potentials that the ligands impose on the U(III) centre in each complex is more difficult to make. For example, the aryloxide complexes U(OAr)3 reductively activate N2 but this has not been observed in the boroxide system, although the increased capacity for both OBAr2 ligands to bridge to a second U centre hampers the comparison. Work on finding a more quantitive measure for this is in progress. The isolation of small quantity of a μ-carbonate product [(μ-CO3){U(OBTrip2)3}2] suggests there are opportunities however, and work is in progress to identify new substrates that could be usefully activated by the combination of the two potentially Lewis acidic centres in this system.Experimental section
[U(OBMes2)3]2 (1m)
A Schlenk flask was charged with U[N(SiMe3)2]3 (1.08 g, 1.5 mmol) and HOBMes2 (1.20 g, 4.5 mmol, 3 eq.) and a stir bar. The reaction mixture was dissolved in toluene (40 mL) to yield a purple solution, which was allowed to stir at room temperature for 4 hours. Volatiles were removed by evaporation to dryness. The residue was washed with cold hexane (−78 °C, 4 mL) and recrystallized from hexane providing dark purple block crystals suitable for single-crystal X-ray crystallography. Yield: 634 mg, 41%.1H NMR (500 MHz, C6D6): δH 8.29 (Ar-H terminal Mes, 16H); 7.20 (o-CH3 terminal Mes, 48H); 2.53 (p-CH3 terminal Mes, 24H); −3.71 (o-CH3 bridging Mes, 12H); −9.20 (Ar-H and p-CH3 bridging Mes, 20H); −15.13 (o-CH3 bridging Mes, 12H).
11B NMR (160.46 MHz, C6D6): δB 73.65 (OBMes). The latter is a tentative assignment. The sample analysed was dissolved single crystals, but this resonance is close to that of hydrolysed ligand so it may be the other resonance is not observed in the window.
MS (APPI): m/z = 1033 ([M]˙+ [U(OBMes2)3]) C54H66B3O3U˙+ [M]˙+ requires 1033.5799, found 1033.5675 (+12.4 ppm).
FTIR (cm−1): 3612, 2728, 1721, 1608, 1552, 1280, 1228, 1176, 1151, 1081, 1029, 960, 928, 847, 745, 672.
Anal. calc for C104H132B6O6U2: C, 62.75 H, 6.44 N, 0.00%. Found: C, 62.63 H, 6.47 N, 0.00%.
[U(OBMes2)4] (2m)
A toluene solution (1.5 mL) of NaOBMes2 (115 mg, 0.4 mmol, 4 eq.) was added dropwise over 10 min to a solution of UI4(dioxane)2 (92 mg, 0.1 mmol) in toluene (1.5 mL) in a 7 cm3 vial with stirring for 18 hours. The reaction was centrifuged and filtered. Colourless plates suitable for single-crystal X-ray crystallography were grown from slow diffusion of hexanes into the reaction mixture. Yield: 16.7 mg, 12%.1H NMR (500 MHz, C6D6): δH 6.71 (Ar-H Mes, 16H); 2.15 (o-CH3 Mes, 48H); 1.99 (p-CH3 Mes, 24H). 11B NMR (160.46 MHz, C6D6): not observed in the window +130–−130 ppm.
MS (APPI): m/z = 1299 ([M]˙+ [U(OBMes2)4]). C72H88B4O4U [M]˙+ requires 1298.7563, found 1298.7595 (−3.2 ppm).
[U(OBTrip2)3]2 (1t) [U(OBTrip2)4] (2t)
HOBTrip2 (104 mg, 0.24 mmol) and UN′′3 (72 mg, 0.1 mmol) were dissolved in benzene (0.8 mL) in a 7 mL vial. The resulting dark red solution was stored at room temperature for 60 h during which time U(OBTrip2)42t crystallised as dark red crystals which can be isolated and washed with benzene (0.3 mL × 3). A second crop of 2t was isolated from the mother liquor to give a combined yield of 25% of 2t (30 mg, 0.02 mmol). Complex 1t (39 mg, 0.013 mmol, 32% yield) was isolated after removal of volatiles from the mother liquor.Characterization of complex 1t:
1H NMR (500 MHz, C6D6): δ 8.10 (br-s, 12H, Ar–H), 2.90 (br-s, 18H, CH), 1.45 (br-s, 108H, Me). 11B NMR (160.46 MHz, C6D6): 110.19 (O–B–Ar). Repeated submissions of samples for elemental analysis did not yield reasonable data.
Characterization of complex 2t:
1H NMR (500 MHz, C6D6): δ 44.14 (s, 3H, Ar–H), 17.87 (s, 3H, Ar–H), 10.66 (s, 4H, Ar–H), 8.86 (s, 6 Ar–H + 4 CH), 7.41 (s, 8H, CH), 6.74 (s, 4H, CH), 5.46 (s, 4H, CH), 4.78 (s, 4H, CH), 4.13 (s, 12H, CH3), 1.49 (m, 24H, CH3), 1.24 (s, 24H, CH3), 0.52 (d, J = 6 Hz, 12H, CH3), 0.25 (d, J = 6 Hz, 12H, CH3), 0.02 (d, J = 6 Hz, 9H, CH3), −1.34 (s, 3H, CH3), −3.43 (s, 12H, CH3), −4.29 (s, 12H, CH3), −13.75 (s, 12H, CH3), −16.19 (s, 9H, CH3), −18.40 (s, 3H, CH3). 11B NMR (160.46 MHz, C6D6): no obvious resonance in the range of 230–40 ppm. Repeated submissions of samples for elemental analysis did not yield reasonable data.
[U(OBTrip2)3(OEt2)] (1t-OEt2)
A solution of HOBTrip2 (326 mg, 0.75 mmol) in Et2O (1 mL) was added dropwise to solution of UN′′3 (216 mg, 0.3 mmol) in Et2O (1 mL) in a 7 mL vial. The resulting dark red solution was stirred at room temperature for 0.5 h, which was left at −30 °C for a further ca. 60 h. A brown precipitate was separated and washed with Et2O (0.5 mL × 3) as complex 11t (see below). The dark red mother liquid was concentrated, and left for standing, which gave complex 1t-OEt2 (46 mg, 0.029 mmol) as dark-red crystals. The isolated yield is low due to the very high solubility of the product.1H NMR (500 MHz, C6D6): δ 8.94 (br, 12H, Ar–H), 3.56 (br-s, 18H, CH), 1.73 (br-s, 108H, Me). 11B NMR (160.46 MHz, C6D6): not observed in the window +130–−130 ppm. No reasonable elemental analysis data were obtained despite repeated trials.
[OU(OBMes2)3]n (3m)
Method A. This product is observed when the complex [U(OBMes2)2(μ-OBMes2)]2 is stored as a solution or when solutions are heated, due to reaction with traces of oxygen.Method B. A purple solution of [U(OBMes2)2(μ-OBMes2)]2 (25 mg, 0.012 mmol) in C6D6 (0.4 mL) was prepared in situ in a Young's NMR tube. The solution was then degassed by three freeze pump thaw cycles and exposed to 1 bar pressure of N2O. The mixture was agitated to give an immediate colour change to light brown. Formation of complex [OU(OBMes2)3]n was confirmed by NMR spectroscopy and elemental analysis.
1H NMR (500 MHz, C6D6): δH 5.90 (Ar-H Mes); 1.89 (p-CH3 Mes); −0.52 (o-CH3 Mes).
11B NMR (160.46 MHz, C6D6): δB 89.29 (OBMes).
13C NMR (126 MHz, C6D6): δH 184.55 (free CO); 166.10 (o-C Mes); 137.10 (p-C Mes); 133.59 (C-B Mes); 126.94 (Ar-CH Mes); 21.29 (p-CH3 Mes); 5.05 (o-CH3 Mes).
Elemental analysis: C 61.80%, H 6.34% calculated. C 61.75%, H 6.37% found.
FTIR (cm−1): 3617, 2728, 1721, 1608, 1552, 1280, 1228, 1175, 1152, 1081, 1029, 960, 928, 840, 745, 670.
[U(OBMes2)4(OPy)2]n (4m)
A Young's NMR tube was charged with [U(OBMes2)2(μ-OBMes2)]2 (25 mg, 0.012 mmol), C5H5NO (5 mg, 0.048 mmol, 4 eq.) and C6D6 (0.4 mL). The reaction mixture turned dark brown immediately and was allowed to react for 72 hours, during which time dark orange crystals of [U(OBMes2)4(Py-O)2] deposited on the reaction vessel walls. Yield: 19 mg, 65%.1H NMR (500 MHz, C6D6): δH 6.76 (Ar-H Mes, 12H); 4.42 (Py–O H, 6H); 2.31 (o-CH3 Mes, 36H); 2.16 (p-CH3 Mes, 18H) 1.96 (Py–O H, 4H).
11B NMR (160.46 MHz, C6D6): not observed in the window +130–−130 ppm.
MS (APPI): m/z = 1223 ([M]˙+ [U(OBMes2)3(Py-O)2]). C64H76B3N2O5U˙+ [M]˙+ requires 1223.6541, found 1223.6552 (−1.1 ppm).
Dark orange crystals suitable for SCXRD analysis were grown from concentrated benzene solutions, however, the data are only of sufficient quality to discuss connectivity and not detailed metrics.
[U(OBMes2)3(OPPh3)2] (5m and 6m)
[U(OBMes2)2(μ-OBMes2)]2 (25 mg, 0.012 mmol) and OPPh3 (7 mg, 0.024 mmol, 2 eq.) were placed in a Young's NMR tube. The tube's contents were dissolved in C6D6 (0.4 mL). 1H NMR confirmed the formation of [U(OBMes2)3(OPPh3)2] (5m). Co-crystals of 5m and the side product [U(OBMes2)3(B2O3Mes2)] (6m) suitable for X-ray diffraction studies were grown by slow diffusion of hexane into a concentrated benzene solution of [U(OBMes2)3(OPPh3)2]. Yield: 23 mg, 60%.1H NMR (500 MHz, C6D6): δH 6.90 (Ar-H Mes, 12H); 6.46 (p-Ph H, 6H); 5.92 (o-Ph H, 12H); 3.64 (o-CH3 Mes, 36H); 2.85 (m-Ph H, 12H); 1.87 (p-CH3 Mes, 18H).
11B NMR (160.46 MHz, C6D6) (5m): δB not observed in the window +130–−130 ppm.
31P NMR (202 MHz, C6D6): δP −17.83(OPPh3).
MS (APPI): m/z = 1589 ([M]˙+ [U(OBMes2)3(OPPh3)2]) C90H96B3O5P2U˙+ [M]˙+ requires 1589.7520, found 1589.7790 (−27.0 ppm).
[{U(OBMes2)3}2(μ-η2:η2-S2)2] (7m)
To a magnetically stirred, dark purple solution of [U(OBMes2)2(μ-OBMes2)]2 (165 mg, 0.08 mmol) in 2 mL of toluene in a 7 cm3 vial was added elemental sulfur (10 mg, 0.04 mmol, 0.5 eq.) with stirring and the reaction was allowed to stir for 2 hours. Dark red crystals of [{(OBMes2)3U}2(μ-η2:η2-S2)2] suitable for X-ray diffraction can be obtained from storage of a concentrated benzene solution at room temperature. Yield: 32 mg, 18%.1H NMR (500 MHz, C6D6): δH 6.88 (Ar-H Mes, 24H); 2.38 (p-CH3 Mes, 36H); 2.23 (o-CH3 Mes, 72H).
11B NMR (160.46 MHz, C6D6): not observed in the window +130–−130 ppm. MS (APPI): m/z = 1097 ([M]˙+. [U(OBMes2)3(μ-S2)]) C54H66B3O3S2U˙+ [M]˙+ requires 1097.5240, found 1097.5259 (−1.9 ppm).
[U(η2-DCC)(OBMes2)3] (8m)
A solution of N,N′-dicyclohexylcarbodiimide [(DCC = η2-N(Cy)C(NCy)N(Cy)]; Cy = C6H11) (10 mg, 0.048 mmol, 2 eq.) in benzene (1 mL) was added dropwise to a stirring benzene solution of [U(OBMes2)2(μ-OBMes2)]2 (50 mg, 0.024 mmol) in a 7 cm3 vial. The solution turned green immediately and was allowed to stir for 18 hours. The solution was then filtered and evaporated to dryness. 1H NMR spectroscopy confirmed the formation of the title compound. Yield: 22 mg, 69%.
1H NMR (500 MHz, C6D6): δH 58.36 (Cy H, 1H); 43.14 (Cy H, 1H); 28.62 (Cy H, 3H); 13.73 (Cy H, 2H); 12.48 (Cy H, 1H); 11.15 (Cy H, 2H); 9.66 (Cy H, 1H); 9.06 (Cy H, 3H); 7.64 (Cy H, 2H); 7.32 (Cy H, 1H); 6.75 (Ar-H Mes, 12H); 6.48 (Cy H, 2H); 3.89 (Cy H, 1H); 3.08 (p-CH3 Mes, 18H); 2.92 (Cy H, 4H); 2.55 (Cy H, 1H); 2.16 (Cy H, 1H); 1.76 (Cy H, 1H); −4.75 (Cy H, 1H); −6.63 (Cy H, 1H); −10.39 (o-CH3 Mes, 36H); −10.62 (Cy H, 4H).
MS (APPI): m/z = 1338 ([M + H] (OBMes2)3U[η2-N(C6H11)C(NC6H11)N-(C6H11)]) C73H100B3N3O3U+ [M + H]+ requires 1337.8552, found 1337.8577 (−2.5 ppm).
[{U(OBTrip2)3}2(μ-CO3)] (9t)
A purple-brown solution of [U(OBTripp2)3]2 (75 mg, 0.048 mmol, 1 eq.) in C5H10 prepared in situ in a Young's NMR tube was degassed by three freeze pump thaw cycles and placed under a 1 bar pressure of CO2 at room temperature. The mixture was agitated to give an immediate colour change to light green-brown. The reaction mixture was allowed to stand overnight at room temperature. Crystalline [U(OBTrip2)3]2(μ-CO3) was obtained by slow diffusion of hexanes into the reaction mixture. Yield: 4 mg, 2%. Due to the extremely low yield of isolated material, no further characterising data were obtainable.[U(OBMes2)N′′3] (11m)
Method A: UIN′′3 (225 mg) and KOBMes2 (100 mg) were placed into a Schlenk flask with a stirrer bar. Toluene (15 mL) was introduced into the flask and the contents were stirred overnight. The solution was then cannula filtered and the solvent removed. The resulting residue was washed with cold hexanes (2 × 5 mL) to yield the title compound 11m (205 mg, 78%).Method B: (N′′)2U{κ2-(N,C)-N(SiMe3)SiMe2CH2} (10 mg) and dimesitylborinic acid (3.5 mg) were placed in an NMR tube and dissolved in C6D6 (0.5 mL). 1H NMR showed that quantitative formation of U(OBMes2)N′′3 had occurred. Crystals suitable for X-ray diffraction studies were grown by slow evaporation of a toluene solution of U(OBMes2)N′′3. Local code P18053_tri.
1H NMR (C6D6): δH 7.52 (s, 4 H), 4.89 (s, 12 H), 2.48 (s, 6 H), −6.68 (s, 54 H); anal. Calc for C36H76B1N3O1Si6U1: C, 52.58 H, 8.01 N, 3.41%. Found: C, 52.39 H, 7.34 N, 3.53%; EI-MS(m/z): 823.2 [M − N′′]+.
[U(OBTrip2)N′′3] (11t)
Complex U(OBTrip2)N′′311t was also isolated as a side product in 5% yield from the synthesis of 1t·OEt2 as a brown precipitate, and washed with Et2O (0.5 mL × 3) in low yield (46 mg, 0.04 mmol, 5% yield based on HOBTrip2). Colourless crystals of 11t suitable for X-ray diffraction analysis were obtained from concentrated benzene solution upon standing at room temperature. Local code P18083.1H NMR (500 MHz, C6D6): δ 41.93 (s, 1H, Ar–H), 16.81 (s, 1H, Ar–H), 11.70 (s, 2H, Ar–H), 10.47 (s, 1H, Ar–H), 9.29 (s, 2H, CH), 8.01 (s, 1H, CH), 7.48 (s, 2H, CH), 3.25 (s, 12H, CH3), 1.98 (s, 3H, CH3), 1.21 (s, 3H, CH3), 1.10 (s, 3H, CH3), 0.52 (s, 3H, CH3), 1.48 (s, 9H, CH3), −4.63 (s, 3H, CH3), −7.36 (s, 12H, CH3), −10.03 (s, 24H, CH3), −14.56 (s, 18H, CH3). There is restricted rotation of the CH3 groups at room temperature on the 1H NMR timescale. 11B NMR (160.46 MHz, C6D6): 109.74 (O–B–Ar).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Edinburgh and the EPSRC for funding through EP/H004823/1 and EP/M010554/1, EP/J018139/1 and the UK Catalysis Hub EP/K014714/1. This project has also received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 740311). The support provided by China Scholarship Council (CSC) during a visit of D. Y. to the University of Edinburgh is acknowledged. We would also like to thank Dr Colin Logan Mackay of the University of Edinburgh for mass spectrometry and the University of Edinburgh SIRCAMS facility for use of the FTICRMS.References
- P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC.
- S. M. Mansell, N. Kaltsoyannis and P. L. Arnold, J. Am. Chem. Soc., 2011, 133, 9036–9051 CrossRef CAS PubMed.
- S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS.
- M. Falcone, L. Chatelain, R. Scopelliti, I. Živković and M. Mazzanti, Nature, 2017, 547, 332 CrossRef CAS PubMed.
- V. Mougel, C. Camp, J. Pécaut, C. Copéret, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed., 2012, 51, 12280–12284 CrossRef CAS PubMed.
- P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed.
- S. C. Cole, M. P. Coles and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 2002, 4168–4174 RSC.
- M. P. Coles, Coord. Chem. Rev., 2016, 323, 52–59 CrossRef CAS.
- O. J. Cooper, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 5074–5076 RSC.
- D. M. King, W. Lewis and S. T. Liddle, Inorg. Chim. Acta, 2012, 380, 167–173 CrossRef CAS.
- I. Korobkov, S. Gorelsky and S. Gambarotta, J. Am. Chem. Soc., 2009, 131, 10406–10420 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- M. J. Monreal, R. K. Thomson, T. Cantat, N. E. Travia, B. L. Scott and J. L. Kiplinger, Organometallics, 2011, 30, 2031–2038 CrossRef CAS.
- J. A. L. Wells, M. L. Seymour, M. Suvova and P. L. Arnold, Dalton Trans., 2016, 45, 16026–16032 RSC.
- J. A. L. Wells, PhD thesis, University of Edinburgh, 2018.
- A. Zalkin, J. G. Brennan and R. A. Andersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1553–1554 CrossRef.
- C. J. Burns, W. H. Smith, J. C. Huffman and A. P. Sattelberger, J. Am. Chem. Soc., 1990, 112, 3237–3239 CrossRef CAS.
- C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed.
- H. J. Wasserman, D. C. Moody and R. R. Ryan, J. Chem. Soc., Chem. Commun., 1984, 532–533 RSC.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS.
- D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1993, 115, 9840–9841 CrossRef CAS.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed.
- L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245–4254 CrossRef CAS.
- A. J. Lewis, U. J. Williams, J. M. Kikkawa, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2012, 51, 37–39 CrossRef CAS PubMed.
- N. L. Bell, P. L. Arnold and J. B. Love, Dalton Trans., 2016, 45, 15902–15909 RSC.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2005, 127, 1338–1339 CrossRef CAS PubMed.
- N. A. Siladke, J. LeDuc, J. W. Ziller and W. J. Evans, Chem. – Eur. J., 2012, 18, 14820–14827 CrossRef CAS PubMed.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Dalton Trans., 2004, 2814–2821 RSC.
- S. Kannan, M. A. Moody, C. L. Barnes and P. B. Duval, Inorg. Chem., 2006, 45, 9206–9212 CrossRef CAS PubMed.
- J. J. Kiernicki, J. S. Harwood, P. E. Fanwick and S. C. Bart, Dalton Trans., 2016, 45, 3111–3119 RSC.
- T. W. Hayton, J. M. Boncella, B. L. Scott, E. R. Batista and P. J. Hay, J. Am. Chem. Soc., 2006, 128, 10549–10559 CrossRef CAS PubMed.
- R. E. Jilek, L. P. Spencer, R. A. Lewis, B. L. Scott, T. W. Hayton and J. M. Boncella, J. Am. Chem. Soc., 2012, 134, 9876–9878 CrossRef CAS PubMed.
- R. E. Jilek, N. C. Tomson, B. L. Scott and J. M. Boncella, Inorg. Chim. Acta, 2014, 422, 78–85 CrossRef CAS.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Polyhedron, 2003, 22, 3475–3482 CrossRef CAS.
- N. A. Ayoub, A. R. Browne, B. L. Anderson and T. G. Gray, Dalton Trans., 2016, 45, 3820–3830 RSC.
- Z. Yang, X. Ma, R. B. Oswald, H. W. Roesky and M. Noltemeyer, J. Am. Chem. Soc., 2006, 128, 12406–12407 CrossRef CAS PubMed.
- X. Ma, Z. Yang, X. Wang, H. W. Roesky, F. Wu and H. Zhu, Inorg. Chem., 2011, 50, 2010–2014 CrossRef CAS PubMed.
- Z. Yang, P. Hao, Z. Liu, X. Ma, H. W. Roesky and J. Li, J. Organomet. Chem., 2014, 751, 788–791 CrossRef CAS.
- M. Kořenková, B. Mairychová, R. Jambor, Z. Růžičková and L. Dostál, Inorg. Chem. Commun., 2014, 47, 128–130 CrossRef.
- M. Kořenková, M. Erben, R. Jambor, A. Růžička and L. Dostál, J. Organomet. Chem., 2014, 772–773, 287–291 CrossRef.
- B. Mairychová, T. Svoboda, P. Štěpnička, A. Růžička, R. W. A. Havenith, M. Alonso, F. D. Proft, R. Jambor and L. Dostál, Inorg. Chem., 2013, 52, 1424–1431 CrossRef PubMed.
- B. Mairychová, P. Štěpnička, A. R
![[u with combining dot above]](https://www.rsc.org/images/entities/char_0075_0307.gif) žička, L. Dostál and R. Jambor, Organometallics, 2014, 33, 3021–3029 CrossRef.
žička, L. Dostál and R. Jambor, Organometallics, 2014, 33, 3021–3029 CrossRef. - B. Mairychová, I. V. Kityk, A. Maciag, F. Bureš, M. Klikar, A. Růžička, L. Dostál and R. Jambor, Inorg. Chem., 2016, 55, 1587–1594 CrossRef PubMed.
- L. Dostál, R. Jambor, A. Růžička, R. Jirásko, A. Lyčka, J. Beckmann and S. Ketkov, Inorg. Chem., 2015, 54, 6010–6019 CrossRef PubMed.
- M. Kořenková, B. Mairychová, A. Růžička, R. Jambor and L. Dostál, Dalton Trans., 2014, 43, 7096–7108 RSC.
- U. Bossek, H. Hummel, T. Weyhermüller, K. Wieghardt, S. Russell, L. van der Wolf and U. Kolb, Angew. Chem., Int. Ed., 1996, 35, 1552–1554 CrossRef CAS.
- A. R. Browne, N. Deligonul, B. L. Anderson, M. Zeller, A. D. Hunter and T. G. Gray, Chem. Commun., 2015, 51, 15800–15803 RSC.
- I. Pantcheva and K. Osakada, Organometallics, 2006, 25, 1735–1741 CrossRef CAS.
- H. Yin, A. J. Lewis, U. J. Williams, P. J. Carroll and E. J. Schelter, Chem. Sci., 2013, 4, 798–805 RSC.
- E. Barnea, T. Andrea, M. Kapon and M. S. Eisen, J. Am. Chem. Soc., 2004, 126, 5066–5067 CrossRef CAS PubMed.
- R. K. Das, E. Barnea, T. Andrea, M. Kapon, N. Fridman, M. Botoshansky and M. S. Eisen, Organometallics, 2015, 34, 742–752 CrossRef CAS.
- K. I. M. Ingram, N. Kaltsoyannis, A. J. Gaunt and M. P. Neu, J. Alloys Compd., 2007, 444–445, 369–375 CrossRef CAS.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- S. R. Daly, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor and R. L. Martin, J. Am. Chem. Soc., 2012, 134, 14408–14422 CrossRef CAS PubMed.
- D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 814–825 CrossRef CAS PubMed.
- J. K. Pagano, D. S. J. Arney, B. L. Scott, D. E. Morris, J. L. Kiplinger and C. J. Burns, Dalton Trans., 2019, 48, 50–57 RSC.
- S. M. Franke, F. W. Heinemann and K. Meyer, Chem. Sci., 2014, 5, 942–950 RSC.
- B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207–6217 RSC.
- D. E. Smiles, G. Wu and T. W. Hayton, New J. Chem., 2015, 39, 7563–7566 RSC.
- D. L. Perry, A. Zalkin, H. Ruben and D. H. Templeton, Inorg. Chem., 1982, 21, 237–240 CrossRef CAS.
- D. J. Grant, Z. Weng, L. J. Jouffret, P. C. Burns and L. Gagliardi, Inorg. Chem., 2012, 51, 7801–7809 CrossRef CAS PubMed.
- J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS.
- E. M. Matson, M. D. Goshert, J. J. Kiernicki, B. S. Newell, P. E. Fanwick, M. P. Shores, J. R. Walensky and S. C. Bart, Chem. – Eur. J., 2013, 19, 16176–16180 CrossRef CAS PubMed.
- A. C. Sutorik and M. G. Kanatzidis, Polyhedron, 1997, 16, 3921–3927 CrossRef CAS.
- J.-E. Kwak, D. L. Gray, H. Yun and J. A. Ibers, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, i86–i87 CrossRef CAS.
- C. Camp, M. A. Antunes, G. García, I. Ciofini, I. C. Santos, J. Pécaut, M. Almeida, J. Marçalo and M. Mazzanti, Chem. Sci., 2014, 5, 841–846 RSC.
- A. Terzis and R. Rivest, Inorg. Chem., 1973, 12, 2132–2136 CrossRef CAS.
- R. C. Elder and M. Trkula, Inorg. Chem., 1977, 16, 1048–1051 CrossRef CAS.
- J. T. York, E. C. Brown and W. B. Tolman, Angew. Chem., Int. Ed., 2005, 44, 7745–7748 CrossRef CAS PubMed.
- S. Yao, C. Milsmann, E. Bill, K. Wieghardt and M. Driess, J. Am. Chem. Soc., 2008, 130, 13536–13537 CrossRef CAS PubMed.
- W. J. Evans, J. R. Walensky, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 3350–3357 CrossRef CAS.
- W. J. Evans, M. K. Takase, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 5802–5808 CrossRef CAS.
- M. K. Takase, N. A. Siladke, J. W. Ziller and W. J. Evans, Organometallics, 2011, 30, 458–465 CrossRef CAS.
- O. J. Cooper, D. P. Mills, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2014, 43, 14275–14283 RSC.
- M. A. Boreen, B. F. Parker, T. D. Lohrey and J. Arnold, J. Am. Chem. Soc., 2016, 138, 15865–15868 CrossRef CAS PubMed.
- W. J. Evans, J. R. Walensky and J. W. Ziller, Organometallics, 2010, 29, 101–107 CrossRef CAS.
- W. J. Evans, J. R. Walensky and J. W. Ziller, Inorg. Chem., 2010, 49, 1743–1749 CrossRef CAS PubMed.
- N. A. Siladke, J. W. Ziller and W. J. Evans, J. Am. Chem. Soc., 2011, 133, 3507–3516 CrossRef CAS PubMed.
- L. Zhang, C. Zhang, G. Hou, G. Zi and M. D. Walter, Organometallics, 2017, 36, 1179–1187 CrossRef CAS.
- L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2016, 138, 5130–5142 CrossRef CAS PubMed.
- L. Zhang, B. Fang, G. Hou, G. Zi, W. Ding and M. D. Walter, Organometallics, 2017, 36, 898–910 CrossRef CAS.
- G. Zi, Chem. Commun., 2018, 54, 7412–7430 RSC.
- O. T. Summerscales, A. S. P. Frey, F. G. N. Cloke and P. B. Hitchcock, Chem. Commun., 2008, 198–200 RSC.
- C. J. Inman, A. S. P. Frey, A. F. R. Kilpatrick, F. G. N. Cloke and S. M. Roe, Organometallics, 2017, 36, 4539–4545 CrossRef CAS.
- N. Tsoureas, L. Castro, A. F. R. Kilpatrick, F. G. N. Cloke and L. Maron, Chem. Sci., 2014, 5, 3777–3788 RSC.
- L. Castro, O. P. Lam, S. C. Bart, K. Meyer and L. Maron, Organometallics, 2010, 29, 5504–5510 CrossRef CAS.
- A. J. Lewis, U. J. Williams, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2013, 52, 7326–7328 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental considerations, preparation of reagents and additional experimental details. CCDC 1886802–1886811, 1886182 and 1886183 (complex 11t and 2t). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt05051a |
| ‡ Current address. Department of Chemistry, Ångström Laboratory, Uppsala University, Uppsala, Sweden. |
| This journal is © The Royal Society of Chemistry 2019 |