
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polynuclear alkoxy–zinc complexes of bowl-shaped macrocycles and their use in the copolymerisation of cyclohexene oxide and CO2†
James R.
Pankhurst
 a,
Shyeni
Paul
b,
Yunqing
Zhu
b,
Charlotte K.
Williams
*b and
Jason B.
Love
*a
a,
Shyeni
Paul
b,
Yunqing
Zhu
b,
Charlotte K.
Williams
*b and
Jason B.
Love
*a
aEaStCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: jason.love@ed.ac.uk
bChemistry Research Laboratory, 12 Mansfield Road, University of Oxford, Oxford, OX1 3TA, UK. E-mail: charlotte.williams@chem.ox.ac.uk
First published on 21st March 2019
Abstract
The reactions between alcohols and the tetranuclear ethyl–Zn complexes of an ortho-phenylene-bridged polypyrrole macrocycle, Zn4Et4(L1) 1 and the related anthracenyl-bridged macrocyclic complex, Zn4Et4(THF)4(L2) 2 have been studied. With long-chain alcohols such as n-hexanol, the clean formation of the tetranuclear hexoxide complex Zn4(OC6H13)4(L1) 3 occurs. In contrast, the use of shorter-chain alcohols such as i-propanol results in the trinuclear complex Zn3(μ2-OiPr)2(μ3-OiPr)(HL1) 4 that arises from demetalation; this complex was characterised by X-ray crystallography. The clean formation of these polynuclear zinc clusters allowed a study of their use as catalysts in the ring-opening copolymerisation (ROCOP) reaction between cyclohexene oxide and CO2. In situ reactions involving the pre-catalyst 1 and n-hexanol formed the desired polymer with the best selectivity for polycarbonate (90%) at 30 atm CO2, whilst the activity and performance of pre-catalyst 2 was poor in comparison.
Introduction
Multidentate macrocycles are attractive as ligands for di- and polynuclear complexes of transition- and f-block metals as they can control both the basic coordination chemistry and the relative spatial positioning of metals within the macrocyclic framework, so providing a pre-organised chemical environment.1–4 This ligand design strategy can deliver a diversity of physical and reaction properties in the resulting complexes leading to, for example, clustering and aggregation,5–14 catalytic activity,15–28 molecular magnetism,29 allosteric constructs,30,31 and molecular sensing.32–35We have been studying macrocycles in which two donor compartments comprising two dipyrromethane and two Schiff-base nitrogen donors (i.e. an N4-donor set) are separated by rigid aryl backbones (e.g. L3 and L4, Fig. 1).36,37 On metalation, the resulting dinuclear complexes adopt Pac-Man structures (e.g.A, Fig. 1) that promote a diversity of chemistry within the dinuclear molecular cleft, including dioxygen reduction catalysis,38–41 halide sensing,42 and uranyl reduction and oxo-group functionalisation.43–50 We have also exploited a steric variation of the meso-substituent (H instead of alkyl, L1 and L2, Fig. 1) which results in the adoption of bowl-shaped structures on metalation, hinging at the meso-carbon instead of the aryl groups.51 Importantly, using this latter ligand variant allows for the isolation of higher nuclearity complexes such as the tetranuclear zinc alkyl macrocyclic complexes 1 and 2 (Scheme 1); these complexes undergo subsequent protonolysis reactions with water to form tetranuclear Zn-oxo and hydroxo clusters.52
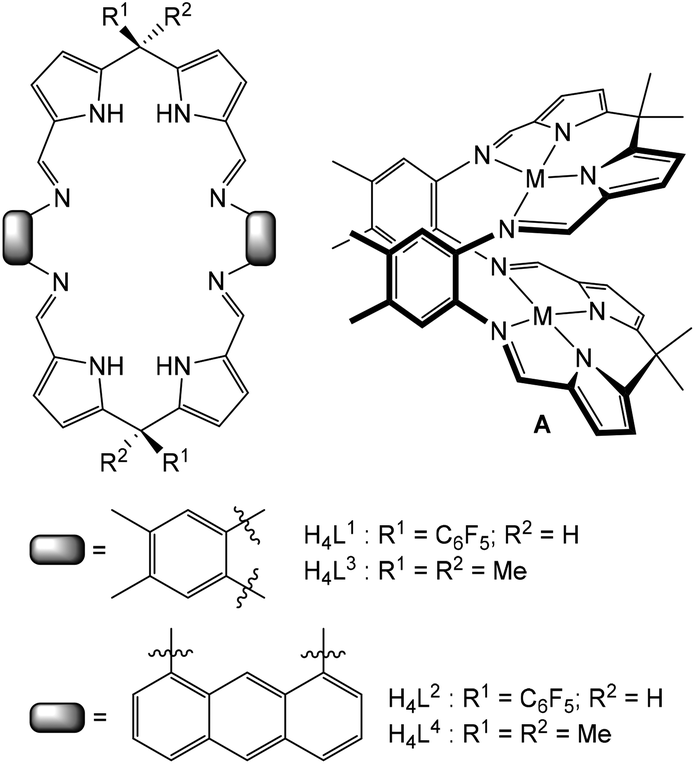 | ||
| Fig. 1 Schiff-base pyrrole macrocycles with varying meso-substituents and aryl linkers and the formation of generic dinuclear complexes of Pac-Man structures. | ||
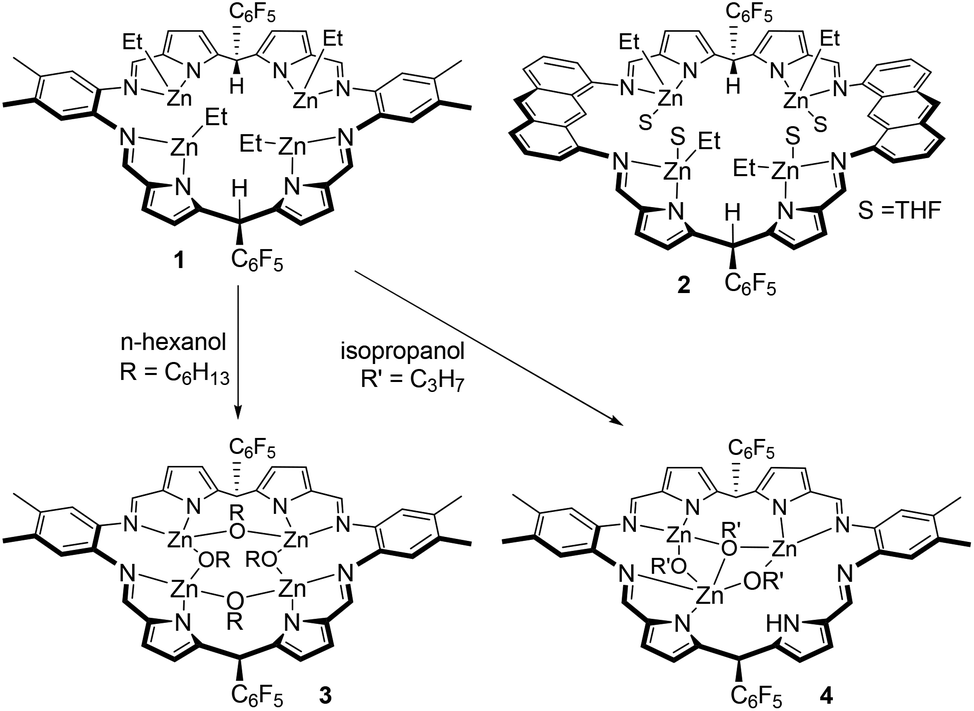 | ||
| Scheme 1 Tetranuclear ethyl–zinc complexes of the Schiff-base pyrrole macrocycles L1 and L2 and their reactions with alcohols; complexes 1 and 2 were reported previously.52 | ||
The straightforward syntheses of 1 and 2, and their facile hydrolysis, provides an opportunity to study the ring opening copolymerisation (ROCOP) of carbon dioxide and epoxides to produce aliphatic polycarbonates.53–57 ROCOP catalysts are often isolated Lewis-acidic metal–alkoxide complexes or are pre-catalysts that are activated by alcohols to form metal alkoxides in situ.58–75 Furthermore, homogeneous zinc catalysts are attractive for ROCOP as the metal is redox inert and sustainable. Zinc clusters formed by alcoholysis/hydrolysis of organo-zinc species act as ROCOP catalysts but have very slow rates.76 Highly active zinc β-diketiminate (BDI) catalysts were reported,77 with the best forming dimers under the polymerisation conditions.78–84 Dinuclear zinc macrocyclic complexes are also highly active and operate under low pressures of carbon dioxide.25,84–88 While higher nuclearity zinc catalysts have been reported, it is not yet understood if dinuclear catalysts are optimum.89–93 As such, we reasoned that the tetranuclear alkyl–zinc macrocyclic complexes 1 and 2 could be activated by alcoholysis and that the resulting zinc alkoxide complexes could act as catalysts for ROCOP of CO2 and epoxides.
Results and discussion
Multinuclear ZnII complexes considered for ROCOP catalysis
The two tetranuclear Zn–alkyl complexes [Zn4Et4(L)], where L is either the ortho-phenylene-bridged macrocycle L1 (1) or the anthracenyl-bridged macrocycle L2 (2), were prepared as previously described (Scheme 1).52 These complexes are inert towards insertion of CO2, but undergo protonolysis reactions with four equivalents of n-hexanol to generate Zn–alkoxide complexes. Specifically, the alkoxide complex, [Zn4(μ2-OC6H13)4(L1)] (3) was isolated, in 84% yield, from the reaction of 1 with four equivalents of n-hexanol in THF (Scheme 1). The reaction proceeds immediately as evident from the ethane gas evolution observed. The 1H NMR spectrum of 3 implies that it is fully symmetric with a single set of resonances for the macrocycle that are shifted in comparison with 1 (Fig. S1†); in C6D6, the imine protons appear as a single resonance at 8.07 ppm, and the meso-protons appear at 6.38 ppm. Importantly, the ethyl resonances, that appear at 1.32 and 0.42 ppm for 1 in C6D6, are absent from the spectrum of 3. Instead, there are a number of overlapping resonances between 1.89 and 0.57 ppm assigned to the new hexyl alkoxide ligands. Two triplet resonances, at 3.83 and 3.70 ppm, each showing integral values consistent with four protons are assigned to the methylene groups adjacent to the Zn–O bond. The distinct chemical shifts indicate that the alkoxide ligands bridge between two metals, with two alkoxides bridging between imine-donors and the other two bridging between pyrrole donors; the structurally characterised and analogous Zn–hydroxide complex, [Zn4(μ2-OH)4(L2)], also displayed similarly equivalent macrocycle resonances yet two distinct hydroxide environments.52 Furthermore, the 19F NMR resonance for the ortho-F groups is severely broadened due to restricted rotation of that group; such broadening is typically observed for bowl-shaped tetranuclear complexes.51 As such, the NMR data support the protonolysis of 1 to form 3 which is a bowl-shaped, tetranuclear Zn–(μ2-alkoxide) complex.Protonolysis reactions between 1 and alcohols other than n-hexanol are not straightforward. The reaction of 1 with iso-propanol occurs readily, evolving gas from the THF solution, to yield the new trinuclear complex, [Zn3(μ2-OiPr)2(μ3-OiPr)(HL1)] (4, Scheme 1). The 1H NMR spectrum of 4 in d8-THF at 300 K shows a number of broad resonances consistent with the formation of a symmetric product and with the successful loss of the ethyl groups from 1 (Fig. S3†). The broad NMR resonances suggests the complex has a fluxional solution structure and so a VT-NMR study was undertaken. At 213 K, the spectrum is sharper and consistent with an asymmetric macrocyclic ligand environment, with each of the four inequivalent imine proton resonances showing signals at 8.86, 8.68, 8.48 and 8.40 ppm (Fig. S4†). Notably, a resonance at 11.98 ppm is only observable at this temperature and is assigned to a single pyrrole N–H proton. In addition, only three iso-propoxyl ligands are seen, with the ipso-protons appearing as a single, broad resonance centred at 4.29 ppm, and the six individual methyl groups well resolved between 1.45 and 0.66 ppm. At 330 K, broad, thermally averaged resonances are seen, with the imine protons appearing as a single resonance at 8.36 ppm, whilst the three iso-propoxide ipso-protons resonate at 4.11 ppm; the associated methyl protons, with integral values of 18 protons per macrocycle, show a signal at 1.02 ppm.
Large, red, block crystals of 4 were grown from a benzene solution and the solid-state structure was determined by X-ray crystallography. Complex 4 is a trinuclear complex (Fig. 2) and adopts a highly distorted bowl-structure with a bite-angle of 102° between the two N4-donor compartments of the macrocycle. This bite-angle is small in comparison with other bowl-shaped complexes of the same ligand, for example, its CuII analogue (Cu2(py)4(L1), 152°).51 This small bite-angle is attributed to coordination of the ligand to an L-shaped, trinuclear Zn-iso-propoxide cluster, which resembles a cubane in which two vertices are removed.94–96 In this cluster, the Zn centres are bridged by two μ2-alkoxide ligands (O2 and O3) and one μ3-alkoxide ligand (O1). Each Zn centre is four-coordinate with highly distorted tetrahedral coordination geometries, with bond angles ranging from 81.58(8)° to 143.47(9)°; in order to accommodate this cluster an imine group (N5) from one of the imino-pyrrole chelates is non-coordinating. The inter-metallic distances between nearest neighbours in the cluster are 3.0071(5) Å (Zn1–Zn3) and 2.8213(6) Å (Zn2–Zn3). The Zn–O bond lengths that describe the edges of the cluster are regular and are in the range 1.920(2) Å to 2.143(2) Å. However, the cluster is distorted, with inequivalent bond angles in the hinge, of 121.4(1)° (Zn2–O1–Zn1) and 111.21(8)° (O3–Zn3–O2).
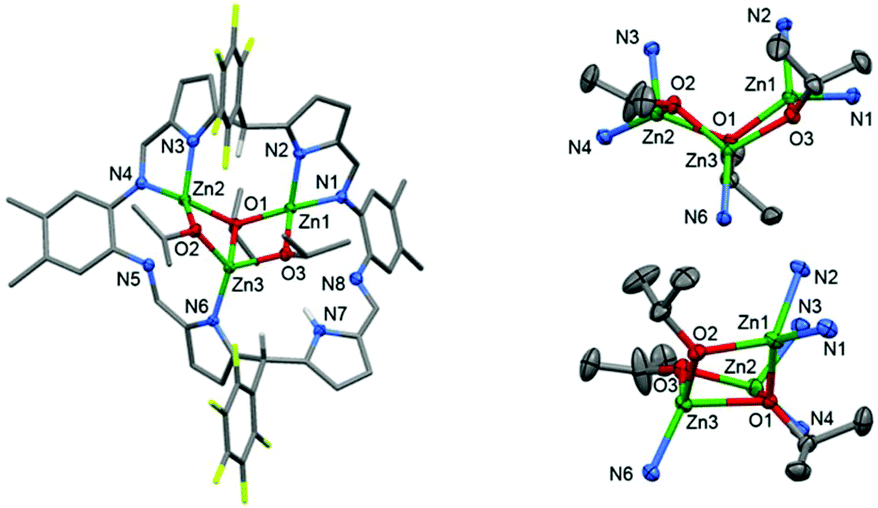 | ||
| Fig. 2 Solid-state structure of 4 (displacement ellipsoids drawn at 50% probability). For clarity, solvent molecules and all hydrogen atoms except the meso- and N–H hydrogen atoms are omitted. Right: Orthogonal views of the Zn3(OiPr)3 cluster. | ||
The reaction between 1 and four equivalents of phenol occurs readily and results in the formation of a complex that displays a similar 1H NMR spectrum to that of 4 (Fig. S5†). A single N–H proton resonance is seen at 11.76 ppm (at 300 K) which indicates that a similar demetalation reaction has occurred to form [Zn3(OPh)3(HL1)]. In an attempt to introduce a kinetic barrier towards demetalation, the reaction between 1 and 2,6-di-tert-butyl-phenol was investigated. No reaction is seen at room temperature, with the 1H NMR spectrum of 1 unchanged in the presence of the di-substituted phenol. However, after heating at 90 °C for 24 h, the 1H NMR spectrum shows that while partial protonolysis had occurred, no N–H proton is seen, consistent with the reaction avoiding demetalation side-processes (Fig. S6†). Nonetheless, the triplet resonance at 6.91 ppm, assigned to the para-proton of phenoxide co-ligands, shows an integral value consistent with there being only two phenoxides per macrocycle. There is also a quartet at 0.55 ppm and its integral is consistent with there being two ethyl ligands per macrocycle. Thus, the product is the tetranuclear complex [Zn4(OC6H3-tBu2-2,6)2Et2(L1)]. Although zinc–phenoxide complexes are able to initiate ROCOP,97 this heteroleptic complex was not investigated further as the mixture of co-ligands would likely complicate initiation processes. Overall, the attempted protonolysis reactions resulted in only 3 as an isolated catalyst suitable for the ROCOP and only n-hexanol was considered as an acceptable alcohol for the in situ generation of catalytic systems using 1 and 2.
Demetalation of a tetranuclear Zn-iso-propoxide complex, that presumably forms initially, would yield one equivalent of Zn(OiPr)2 per equivalent of 4. The relative instability of the tetranuclear zinc complex suggests that combining 1 or 2 with iso-propanol will not be an effective initiating system as the desired multinuclear zinc alkoxide complex will be contaminated by the homoleptic zinc alkoxide. Indeed, homoleptic zinc alkoxide complexes are known to catalyse the formation of ether linkages in ROCOP reactions.98 Demetalation was not observed during the reaction of 1 with n-hexanol, which may be a result of the longer-chain alkoxide ligands imparting kinetic stability. The pKa for n-hexanol is predicted at 16.6 in water99 and is essentially identical to that of iso-propanol (pKa = 16.5 in water).100 Therefore, whilst demetalation occurs through protonolysis of the Zn–pyrrolide bond, the formation of 4 is not attributed to a difference in acidity of the alcohol.
Polymerisation catalysis
Ring-opening copolymerisation reactions were conducted using complex 1 reacted in situ with four equivalents of n-hexanol, with a catalyst loading of 0.1 mol%, in neat cyclohexene oxide (CHO), under 1 bar pressure of CO2, at 80 °C for 24 h (Table 1, entry 1). Four equivalents of the alcohol (0.4 mol%) were added immediately before the mixture was exposed to carbon dioxide. The catalytic activity was low, with a TOF of 9 h−1. The polymer formed has a low molar mass (Mn = 4400 g mol−1) and broad dispersity (Đ = 1.67). Analysis of the polymer composition using 1H NMR spectroscopy showed that the majority of linkages are ether, with only 7% carbonate linkages. Complex 1 was also tested, under 1 bar CO2, using four equivalents of methanol as the alcohol (Table 1, entry 2). By analogy to the stoichiometric reactions with iso-propanol, it was proposed that a trinuclear Zn–methoxide complex would form and this species shows a low catalytic activity (TOF = 13 h−1). The resulting poly(ether-carbonate) shows a high proportion of ether linkages, moderate molar mass (Mn = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1) and broad dispersity (Đ = 2.69), the latter indicative of slow or multiple initiation reactions.
300 g mol−1) and broad dispersity (Đ = 2.69), the latter indicative of slow or multiple initiation reactions.
| Entry | Catalyst | [Epoxide]/M ([cat.]/mol%) | [CO2]/atm | TONb | TOFc/h−1 | Polycarbonate linkage selectivityd/% | M n, g mol−1 (Đ)e |
|---|---|---|---|---|---|---|---|
| Reactions were conducted for 24 h at 80 °C and when using 1 or 2, four equivalents of n-hexanol (except where stated otherwise) were added immediately prior to the addition of carbon dioxide. Reactions were either conducted in neat epoxide (i.e. [CHO] = 10 M) or in toluene ([CHO] = 5 M).a Methanol was added instead of n-hexanol.b TON = (moles epoxide consumed)/(moles catalyst), the conversion was determined by integration of the signals, in the 1H NMR spectrum for methine protons assigned to CHO (3.14 ppm) and polymer (4.65 ppm).c TOF = TON/time (h).d Selectivity for carbonate linkages was determined by comparison of the relative integrals in the 1H NMR spectrum for the signals of polycarbonate (4.65 ppm) and ether linkages (3.43 ppm).e The molar mass (Mn) and dispersity (Đ) values were determined using size-exclusion chromatography (SEC), in THF, which was calibrated with polystyrene standards. | |||||||
| 1 | 1 | 10 (0.1) | 1 | 220 | 9 | 7 | 4400 (1.67) |
| 2a | 1 | 10 (0.1) | 1 | 310 | 13 | 6 | 15300 (2.69) |
| 3 | 1 | 10 (0.1) | 30 | 510 | 21 | 56 | 18100 (3.03) |
| 4 | 1 | 10 (0.1) | 50 | 140 | 6 | 29 | 13700 (2.97) |
| 5 | 1 | 5 (0.2) | 30 | 360 | 15 | 90 | 11900 (14) |
| 6 | 1 | 5 (0.2) | 1 | 0 | 0 | 0 | — |
| 7 | 2 | 10 (0.1) | 1 | 70 | 3 | 0 | — |
| 8 | 2 | 10 (0.1) | 50 | 30 | 1 | 68 | — |
| 9 | 2 | 5 (0.2) | 30 | 10 | 0.5 | 88 | — |
| 10 | 3 | 5 (0.2) | 30 | 250 | 11 | 81 | 7700 (13) |
In order to increase the proportion of carbonate linkages for the catalyst system comprising 1/n-hexanol, the CO2 pressure was increased (Table 1, entries 3 and 4, Fig. S8†). Using 30 bar pressure of CO2, both the activity (TOF = 21 h−1) and the selectivity for carbonate linkages increased (carbonate linkages = 56%). In line with the greater conversion, the resulting polymer shows a higher molar mass (Mn = 18100 g mol−1) but the dispersity remains very broad (Đ = 3.03). When the reaction pressure is increased further to 50 bar, the catalyst activity, conversion of epoxide, and carbonate selectivity all decrease. This may be a result of gas expansion which is known to occur under such sub-critical conditions and which effectively dilutes the catalyst concentrations.101
As part of attempts to improve the polymerisation selectivity, polymerisations were conducted in toluene solutions to reduce the overall epoxide concentration and hence slow sequential enchainment reactions (Table 1, entries 5, 6, Fig. S9†). Overall, the absolute catalyst concentration was the same as in the previous reactions conducted in neat epoxide but its relative loading compared to epoxide is increased. Polymerisations conducted in toluene solution at 1 bar CO2 pressure were unsuccessful (Table 1, entry 6), but at 30 bar pressure polymerisation occurs to form a polymer with significantly increased carbonate linkages (Table 1, entry 5). However, the ROCOP activity is reduced compared to reactions in neat epoxide, for example the TOF decreased from 21 h−1 (10 M) to 15 h−1 (5 M) (Table 1, entries 3 and 5). The polymerisation control is very poor forming a polymer with an exceptionally broad dispersity (Mn = 11000 g mol−1; Đ = 14). To investigate further, the evolution of polycarbonate molar mass vs. conversion was analysed (Table S1, Fig. S11†). At low conversions, bimodal molar mass distributions are seen showing a characteristic very high molar mass peak (Mn = 194000 g mol−1; Đ = 1.89) and a lower molar mass peak (Mn = 2400; Đ = 3.00). The higher molar mass peak did not increase particularly as polymerisation progressed whereas the lower peak shows a clear increase in molar mass vs. conversion. Aliquots were taken and the 1H NMR spectra show the formation of both carbonate and ether linkages throughout the reaction. It is tentatively proposed that the higher molar mass peak is due to uncontrolled and rapid formation of polyether, whilst the lower molar mass peak arises from ROCOP to form predominantly polycarbonate. Nonetheless, a more detailed analysis is precluded by the very broad molar mass distributions that clearly signal problems with relative initiation rates and number of active sites.
Polymerisations were also conducted under a range of similar conditions using the catalyst system formed from 2 (Table 1, entries 7–9). Under all conditions, its activity is very low, although the carbonate selectivity could be somewhat increased at higher pressures. The isolated hexyl alkoxide complex 3 shows similar performance to the catalyst system formed using 1 and n-hexanol, and is consistent with 3 being the true initiating species formed during alcoholysis of 1. The polycarbonate product, formed using 3, shows a similar molecular weight (Mn = 7400 g mol−1) and very broad dispersity (Đ > 13) to that formed using the catalyst system of 1/hexyl alcohol. Finally, ROCOP reactions using propylene oxide and carbon dioxide (50 bar) were unsuccessful with all catalysts. Overall, the activity values for all catalysts are at the lower end in this field and cannot compete with leading catalysts, such as the di-zinc catalysts coordinated by β-diketiminate or macrocyclic ancillary ligands.77–88
Conclusions
The result of reactions between the tetranuclear ethyl zinc complex 1 and alcohols is highly dependent on the alcohol used. While reaction with n-hexanol provides the isolable tetranuclear Zn hexyl–alkoxide complex 3, use of isopropanol results in demetalation and the formation of the trinuclear Zn complex 4. Reactions between 1 and phenol similarly result in demetalation, while the use of the more sterically hindered alcohol HOC6H3-tBu-2,6 maintains the nuclearity of the complex but limits the protonolysis reaction, with two ethyl groups untouched. Complex 1 showed some activity and selectivity as a catalyst in ring-opening copolymerisation of cyclohexene oxide and carbon dioxide, with optimised conditions of 30 atm pressure of CO2, 0.1 mol% catalyst loading, 80 °C and in 5 M cyclohexene oxide (diluted in toluene). These conditions enabled the production of polycarbonates with 90% selectivity for carbonate linkages and with a TOF of 15 h−1. However, the polymers produced have very broad molar mass distributions suggesting that multiple catalytic sites are present which exhibit poor reaction control. The analogous anthracenyl-bridged complex 2 showed even lower activity and a similar lack of polymerisation control. While higher nuclearity macrocyclic zinc complexes have potential as catalysts in ROCOP reactions, the complexes used in this study appear too labile, with facile demetalation occurring under reaction conditions, making them unsuitable as catalysts. This highlights the need for improved ligand design and complex stability towards alcohols to prepare more active and selective catalysts for ROCOP reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Edinburgh, the Principal's Career Development Scholarship Scheme for funding (JRP), the EPSRC (EP/L017393/1; EP/K014668/1) and the Chinese Scholarship Council (studentship to YZ) for their support.Notes and references
- R. J. Hooley, Inorg. Chem., 2018, 57, 3497–3499 CrossRef CAS PubMed.
- Z. Liu, S. K. M. Nalluri and J. F. Stoddart, Chem. Soc. Rev., 2017, 46, 2459–2478 RSC.
- L. F. Lindoy, K.-M. Park and S. S. Lee, Chem. Soc. Rev., 2013, 42, 1713–1727 RSC.
- J. B. Love, Chem. Commun., 2009, 3154–3165 RSC.
- T. Ogoshi, T. Kakuta and T.-A. Yamagishi, Angew. Chem., Int. Ed., 2019, 58, 2197–2206 CrossRef CAS PubMed.
- M. T. Chaudhry, F. Lelj and M. J. MacLachlan, Chem. Commun., 2018, 54, 11869–11872 RSC.
- Z. Chen, S. Guieu, N. G. White, F. Lelj and M. J. MacLachlan, Chem. – Eur. J., 2016, 22, 17657–17672 CrossRef CAS PubMed.
- T. Hojo, R. Matsuoka and T. Nabeshima, Inorg. Chem., 2019, 58, 995–998 CrossRef CAS PubMed.
- J. Janczak, D. Prochowicz, J. Lewiński, D. Fairen-Jimenez, T. Bereta and J. Lisowski, Chem. – Eur. J., 2016, 22, 598–609 CrossRef CAS PubMed.
- M. Löffler, J. Gregoliński, M. Korabik, T. Lis and J. Lisowski, Dalton Trans., 2016, 45, 15586–15594 RSC.
- M. A. Minier and S. J. Lippard, Organometallics, 2014, 33, 1462–1466 CrossRef CAS PubMed.
- T. Nakamura, Y. Kaneko, E. Nishibori and T. Nabeshima, Nat. Commun., 2017, 8, 129 CrossRef PubMed.
- S. Zhang and L. Zhao, Acc. Chem. Res., 2018, 51, 2535–2545 CrossRef CAS PubMed.
- M. Fondo, A. M. García-Deibe, N. Ocampo, J. Sanmartín and M. R. Bermejo, Dalton Trans., 2004, 2135–2141 RSC.
- Y.-M. Zhao, G.-Q. Yu, F.-F. Wang, P.-J. Wei and J.-G. Liu, Chem. – Eur. J., 2019, 25, 3726–3739 CrossRef CAS PubMed.
- M. Biyikal, K. Löhnwitz, P. W. Roesky and S. Blechert, Synlett, 2008, 3106–3110 CAS.
- A. Gualandi, L. Cerisoli, H. Stoeckli-Evans and D. Savoia, J. Org. Chem., 2011, 76, 3399–3408 CrossRef CAS PubMed.
- R. W. Hogue, O. Schott, G. S. Hanan and S. Brooker, Chem. – Eur. J., 2018, 24, 9820–9832 CrossRef CAS PubMed.
- A. B. Kremer and P. Mehrkhodavandi, Coord. Chem. Rev., 2019, 380, 35–57 CrossRef CAS.
- P. Mahapatra, S. Ghosh, S. Giri, V. Rane, R. Kadam, M. G. B. Drew and A. Ghosh, Inorg. Chem., 2017, 56, 5105–5121 CrossRef CAS PubMed.
- K. Mase, S. Aoi, K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2016, 20, 935–949 CrossRef CAS.
- M. Quernheim, H. Liang, Q. Su, M. Baumgarten, N. Koshino, H. Higashimura and K. Müllen, Chem. – Eur. J., 2014, 20, 14178–14183 CrossRef CAS PubMed.
- C. Redshaw, Catalysts, 2017, 7, 165 CrossRef.
- X. Ribas and M. Devillard, Chem. – Eur. J., 2018, 24, 1222–1230 CrossRef CAS PubMed.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem., Int. Ed., 2016, 55, 8680–8685 CrossRef CAS PubMed.
- G. Tseberlidis, D. Intrieri and A. Caselli, Eur. J. Inorg. Chem., 2017, 3589–3603 CrossRef CAS.
- X.-X. Zheng and Z.-X. Wang, J. Organomet. Chem., 2016, 823, 14–22 CrossRef CAS.
- R. Liu, C. von Malotki, L. Arnold, N. Koshino, H. Higashimura, M. Baumgarten and K. Müllen, J. Am. Chem. Soc., 2011, 133, 10372–10375 CrossRef CAS PubMed.
- S. Dhers, H. L. C. Feltham, M. Rouzières, R. Clérac and S. Brooker, Dalton Trans., 2016, 45, 18089–18093 RSC.
- A. I. d'Aquino, H. F. Cheng, J. Barroso-Flores, Z. S. Kean, J. Mendez-Arroyo, C. M. McGuirk and C. A. Mirkin, Inorg. Chem., 2018, 57, 3568–3578 CrossRef PubMed.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- J. F. C. Mário and M. P. Sara, Curr. Org. Synth., 2017, 14, 704–714 Search PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342–4373 CrossRef CAS PubMed.
- B. M. Rambo and J. L. Sessler, Chem. – Eur. J., 2011, 17, 4946–4959 CrossRef CAS PubMed.
- G. Givaja, A. J. Blake, C. Wilson, M. Schroder and J. B. Love, Chem. Commun., 2003, 2508–2509 RSC.
- E. Askarizadeh, A. M. J. Devoille, D. M. Boghaei, A. M. Z. Slawin and J. B. Love, Inorg. Chem., 2009, 48, 7491–7500 CrossRef CAS PubMed.
- G. Givaja, M. Volpe, M. A. Edwards, A. J. Blake, C. Wilson, M. Schröder and J. B. Love, Angew. Chem., Int. Ed., 2007, 46, 584–586 CrossRef CAS PubMed.
- M. Volpe, H. Hartnett, J. W. Leeland, K. Wills, M. Ogunshun, B. J. Duncombe, C. Wilson, A. J. Blake, J. McMaster and J. B. Love, Inorg. Chem., 2009, 48, 5195–5207 CrossRef CAS PubMed.
- E. Askarizadeh, S. B. Yaghoob, D. M. Boghaei, A. M. Z. Slawin and J. B. Love, Chem. Commun., 2010, 46, 710–712 RSC.
- A. M. J. Devoille and J. B. Love, Dalton Trans., 2012, 41, 65–72 RSC.
- A. M. J. Devoille, P. Richardson, N. L. Bill, J. L. Sessler and J. B. Love, Inorg. Chem., 2011, 50, 3116–3126 CrossRef CAS PubMed.
- P. L. Arnold, M. S. Dutkiewicz, M. Zegke, O. Walter, C. Apostolidis, E. Hollis, A.-F. Pécharman, N. Magnani, J.-C. Griveau, E. Colineau, R. Caciuffo, X. Zhang, G. Schreckenbach and J. B. Love, Angew. Chem., Int. Ed., 2016, 55, 12797–12801 CrossRef CAS PubMed.
- P. L. Arnold, E. Hollis, G. S. Nichol, J. B. Love, J.-C. Griveau, R. Caciuffo, N. Magnani, L. Maron, L. Castro, A. Yahia, S. O. Odoh and G. Schreckenbach, J. Am. Chem. Soc., 2013, 135, 3841–3854 CrossRef CAS PubMed.
- P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, A. J. Blake, C. Wilson and J. B. Love, J. Am. Chem. Soc., 2006, 128, 9610–9611 CrossRef CAS PubMed.
- P. L. Arnold, D. Patel, C. Wilson and J. B. Love, Nature, 2008, 451, 315 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056 CrossRef CAS PubMed.
- B. E. Cowie, G. S. Nichol, J. B. Love and P. L. Arnold, Chem. Commun., 2018, 54, 3839–3842 RSC.
- P. L. Arnold, A.-F. Pécharman, R. M. Lord, G. M. Jones, E. Hollis, G. S. Nichol, L. Maron, J. Fang, T. Davin and J. B. Love, Inorg. Chem., 2015, 54, 3702–3710 CrossRef CAS PubMed.
- J. R. Pankhurst, T. Cadenbach, D. Betz, C. Finn and J. B. Love, Dalton Trans., 2015, 44, 2066–2070 RSC.
- T. Cadenbach, J. R. Pankhurst, T. A. Hofmann, M. Curcio, P. L. Arnold and J. B. Love, Organometallics, 2015, 34, 2608–2613 CrossRef CAS.
- Z. Qin, C. M. Thomas, S. Lee and G. W. Coates, Angew. Chem., Int. Ed., 2003, 42, 5484–5487 CrossRef CAS PubMed.
- D. J. Darensbourg, P. Rainey and J. Yarbrough, Inorg. Chem., 2001, 40, 986–993 CrossRef CAS.
- C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS.
- A. M. Chapman, C. Keyworth, M. R. Kember, A. J. J. Lennox and C. K. Williams, ACS Catal., 2015, 5, 1581–1588 CrossRef CAS.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- A. Thevenon, A. Cyriac, D. Myers, A. J. P. White, C. B. Durr and C. K. Williams, J. Am. Chem. Soc., 2018, 140, 6893–6903 CrossRef CAS PubMed.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- D. J. Darensbourg, R. M. Mackiewicz, A. L. Phelps and D. R. Billodeaux, Acc. Chem. Res., 2004, 37, 836–844 CrossRef CAS PubMed.
- D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335–6342 CrossRef CAS PubMed.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831–1833 CrossRef CAS PubMed.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212–214 RSC.
- E. K. Noh, S. J. Na, S. S, S.-W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082–8083 CrossRef CAS PubMed.
- X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574–3577 CrossRef CAS PubMed.
- X.-B. Lu, L. Shi, Y.-M. Wang, R. Zhang, Y.-J. Zhang, X.-J. Peng, Z.-C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664–1674 CrossRef CAS PubMed.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312–317 CrossRef CAS.
- C.-H. Li, H.-J. Chuang, C.-Y. Li, B.-T. Ko and C.-H. Lin, Polym. Chem., 2014, 5, 4875–4878 RSC.
- C.-Y. Tsai, F.-Y. Cheng, K.-Y. Lu, J.-T. Wu, B.-H. Huang, W.-A. Chen, C.-C. Lin and B.-T. Ko, Inorg. Chem., 2016, 55, 7843–7851 CrossRef CAS PubMed.
- C.-Y. Tsai, B.-H. Huang, M.-W. Hsiao, C.-C. Lin and B.-T. Ko, Inorg. Chem., 2014, 53, 5109–5116 CrossRef CAS PubMed.
- R. Eberhardt, M. Allmendinger, G. A. Luinstra and B. Rieger, Organometallics, 2003, 22, 211–214 CrossRef CAS.
- D. J. Darensbourg and M. W. Holtcamp, Macromolecules, 1995, 28, 7577–7579 CrossRef CAS.
- D. J. Darensbourg, M. W. Holtcamp, G. E. Struck, M. S. Zimmer, S. A. Niezgoda, P. Rainey, J. B. Robertson, J. D. Draper and J. H. Reibenspies, J. Am. Chem. Soc., 1999, 121, 107–116 CrossRef CAS.
- H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS.
- T. Aida, M. Ishikawa and S. Inoue, Macromolecules, 1986, 19, 8–13 CrossRef CAS.
- S. Inoue, H. Koinuma and T. Tsuruta, J. Polym. Sci., Part B: Polym. Lett., 1969, 7, 287–292 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599 CrossRef CAS PubMed.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911–11924 CrossRef CAS PubMed.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S.-i. Han, H. Yun, H. Lee and Y.-W. Park, J. Am. Chem. Soc., 2005, 127, 3031–3037 CrossRef CAS PubMed.
- S. Kissling, P. T. Altenbuchner, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, U. B. Seemann and B. Rieger, Chem. – Eur. J., 2015, 21, 8148–8157 CrossRef CAS PubMed.
- M. W. Lehenmeier, S. Kissling, P. T. Altenbuchner, C. Bruckmeier, P. Deglmann, A.-K. Brym and B. Rieger, Angew. Chem., Int. Ed., 2013, 52, 9821–9826 CrossRef CAS PubMed.
- S. Klaus, S. I. Vagin, M. W. Lehenmeier, P. Deglmann, A. K. Brym and B. Rieger, Macromolecules, 2011, 44, 9508–9516 CrossRef CAS.
- K. Nakano, S. Hashimoto and K. Nozaki, Chem. Sci., 2010, 1, 369–373 RSC.
- C. Romain, M. S. Bennington, A. J. P. White, C. K. Williams and S. Brooker, Inorg. Chem., 2015, 54, 11842–11851 CrossRef CAS PubMed.
- M. R. Kember, P. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931–933 CrossRef CAS PubMed.
- J. A. Garden, P. K. Saini and C. K. Williams, J. Am. Chem. Soc., 2015, 137, 15078–15081 CrossRef CAS PubMed.
- F. Jutz, A. Buchard, M. R. Kember, S. B. Fredriksen and C. K. Williams, J. Am. Chem. Soc., 2011, 133, 17395–17405 CrossRef CAS PubMed.
- C. Romain, J. A. Garden, G. Trott, A. Buchard, A. J. P. White and C. K. Williams, Chem. – Eur. J., 2017, 23, 7367–7376 CrossRef CAS PubMed.
- D. J. Darensbourg, J. R. Wildeson and J. C. Yarbrough, Inorg. Chem., 2002, 41, 973–980 CrossRef CAS PubMed.
- M. B. Dinger and M. J. Scott, Inorg. Chem., 2001, 40, 1029–1036 CrossRef CAS.
- K. L. Orchard, J. E. Harris, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2011, 30, 2223–2229 CrossRef CAS.
- R. Duchateau, W. J. van Meerendonk, S. Huijser, B. B. P. Staal, M. A. van Schilt, G. Gerritsen, A. Meetsma, C. E. Koning, M. F. Kemmere and J. T. F. Keurentjes, Organometallics, 2007, 26, 4204–4211 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535–9542 CrossRef CAS PubMed.
- J. W. Leeland, F. J. White and J. B. Love, J. Am. Chem. Soc., 2011, 133, 7320–7323 CrossRef CAS PubMed.
- P. E. Eaton and T. W. Cole, J. Am. Chem. Soc., 1964, 86, 3157–3158 CrossRef CAS.
- D. Schroder, H. Schwarz, S. Polarz and M. Driess, Phys. Chem. Chem. Phys., 2005, 7, 1049–1053 RSC.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487–12496 CrossRef CAS.
- M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 222–224 RSC.
- M. J. Citra, Chemosphere, 1999, 38, 191–206 CrossRef CAS PubMed.
- W. Reeve, C. M. Erikson and P. F. Aluotto, Can. J. Chem., 1979, 57, 2747–2754 CrossRef CAS.
- S. Mang, A. I. Cooper, M. E. Colclough, N. Chauhan and A. B. Holmes, Macromolecules, 2000, 33, 303–308 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterising data, detailed catalysis results. CCDC 1509316. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00595a |
| This journal is © The Royal Society of Chemistry 2019 |