DOI:
10.1039/C8SC02547A
(Edge Article)
Chem. Sci., 2018,
9, 7115-7121
Metal-free alkene oxy- and amino-perfluoroalkylations via carbocation formation by using perfluoro acid anhydrides: unique reactivity between styrenes and perfluoro diacyl peroxides†
Received
10th June 2018
, Accepted 20th July 2018
First published on 1st August 2018
Abstract
We present a strategy for metal-free, alkene difunctionalization-type, oxy- and amino-perfluoroalkylations, using perfluoro acid anhydrides as practical and user-friendly perfluoroalkyl sources. This method provides efficient access to oxy-perfluoroalkylation products via carbocation formation due to the unique reactivity between styrenes and bis(perfluoroacyl) peroxides generated in situ from perfluoro acid anhydrides. This reaction is also applicable to metal-free intramolecular amino-perfluoroalkylation of styrenes bearing a pendant amino group. Synthetic utility of the oxy-trifluoromethylation products was confirmed by demonstrating derivatization via hydrolysis, elimination, and acid-catalyzed substitution with carbon nucleophiles. The mechanism of the carbocation formation was investigated experimentally and theoretically.
Introduction
Introduction of perfluoroalkyl groups is an important strategy for modifying the properties of bioactive compounds, agrochemicals and functional materials.1 Various methods are available for C–CF3 bond formation to construct functionalized CF3-containing compounds, and alkene difunctionalization-type trifluoromethylation has recently attracted particular interest.2–10 Styrene derivatives are often used as substrates in these reactions because of their unique reactivity and the utility of the products as CF3-containing synthetic building blocks.2–5 For example, transition-metal-catalyzed intermolecular oxy-trifluoromethylation to form C–O bonds has been well studied (Scheme 1a).3 As pioneering works, Szabó3a and we3b independently reported Cu-catalyzed intermolecular oxy-trifluoromethylation of styrenes with Togni reagent in 2012. In this reaction, the trifluoromethyl group is introduced into the β-position, and then 2-iodobenzoate group derived from the Togni reagent is introduced at the benzylic position via carbocation intermediate formation with the aid of copper-catalyst. In contrast to transition-metal-catalyzed reactions, metal-free oxy-trifluoromethylations generally proceed via the following steps: (1) formation of a CF3 radical, (2) formation of an alkyl radical intermediate by reaction of the CF3 radical and alkene, (3) trapping with an O-radical species.5 In 1993, Uneyama reported an electrochemical reaction of butyl acrylate with trifluoroacetic acid (TFA) and O2 as the trapping agent, affording CF3-containing alcohol products.5b In 2011, Xiao found that S-(trifluoromethyl)diphenylsulfonium salt could react with styrenes under aerobic conditions to afford ketone products.5c A similar transformation was also achieved by using CF3SO2Na in the presence of O2, obtaining a mixture of ketone and alcohol products.5e Lei developed the reaction with CF3SO2Na in the presence of O2 with the aid of K2S2O8 or the combination of NMP/PPh3, to obtain the ketone or alcohol product selectively.5f,h Fu reported oxazoline forming-trifluoromethylation of allylamide with CF3SO2Na by using oxidant.5g In 2012, Studer reported an efficient TEMPONa-promoted oxy-trifluoromethylation with Togni reagent,5d,j in which Togni reagent was decomposed via single electron transfer with TEMPONa as an electron donor, affording CF3 and TEMPO radicals and eventually providing the CF3-containing TEMPO adduct by addition to the alkene. In 2015, Tan and Liu reported the metal-free oxy-trifluoromethylation using hydroxamic acids, affording products containing an aminoxyl group.5i As regards metal-free reaction via a carbocation intermediate, Uneyama developed an electrochemical oxy-trifluoromethylation of butyl methacrylate with TFA and water as the oxygen nucleophile and solvent by careful tuning the current density to oxidize the radical intermediate, obtaining the alcohol product in up to 35% yield.5a Further, in 2016, Liu reported an amine-catalyzed intramolecular oxy-trifluoromethylation of alkenes bearing a 1,3-diaryl diketone group with Togni reagent, affording dihydrofuran products.5k,6
 |
| | Scheme 1 (a) Proposed mechanisms of previous oxy-trifluoromethylations, (b) our previous work on Cu-catalyzed amino-perfluoroalkylation with TFAA/urea·H2O2, and (c) metal-free difunctionalization-type perfluoroalkylation of styrenes by using perfluoro acid anhydrides (this work). | |
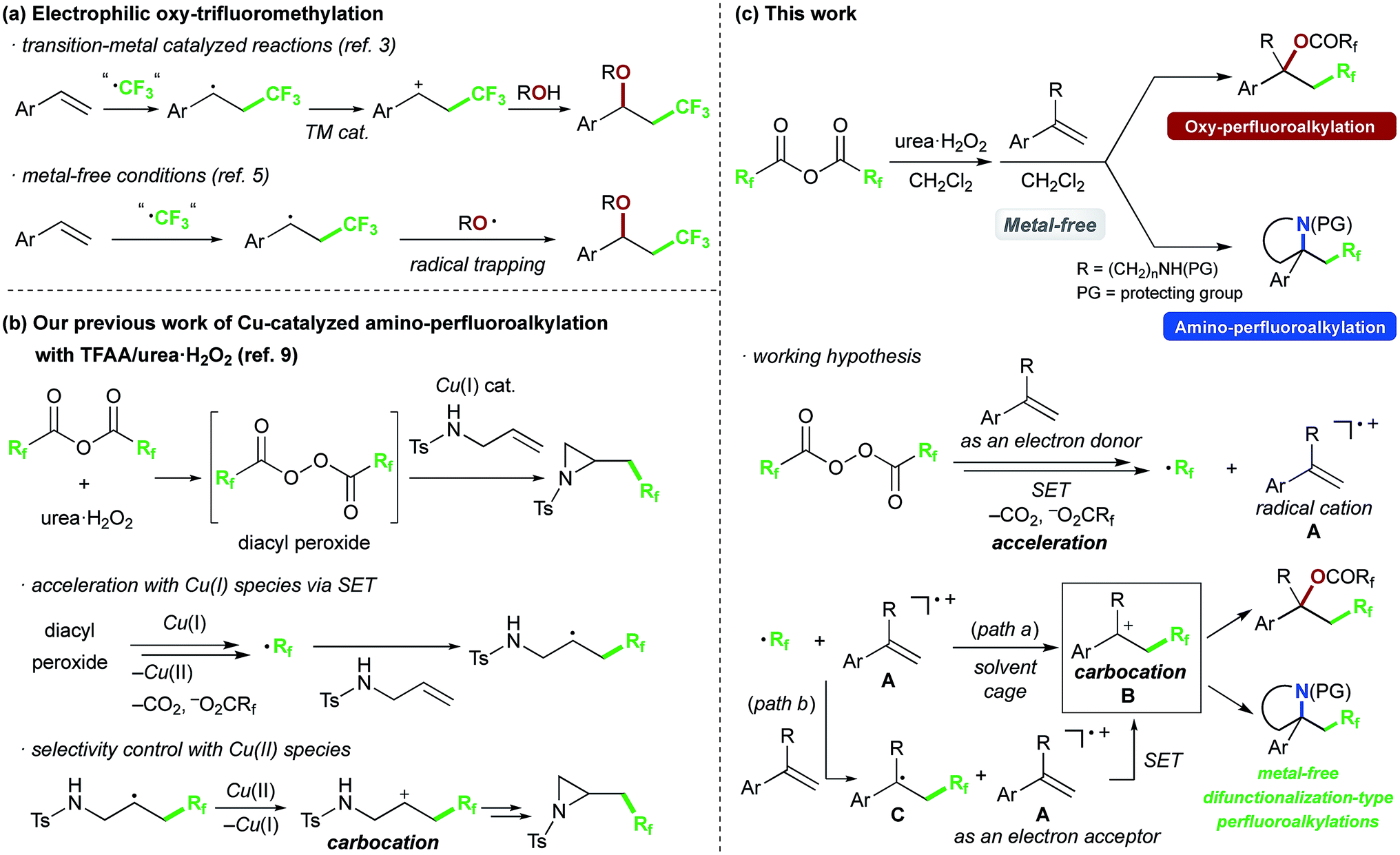
Recently, we have been interested in alkene perfluoroalkylation by using perfluoro acid anhydrides, which are convenient and practical perfluoroalkyl sources because of their low cost, ready availability and reasonable stability compared to conventional perfluoroalkylating reagents.9–12 We found that perfluoro diacyl peroxides prepared in situ from perfluoro acid anhydrides and urea·H2O2 showed excellent reactivity and selectivity in allylic perfluoroalkylation9a and in intramolecular amino-perfluoroalkylation9b of alkenes in the presence of Cu(I) salt as a catalyst. Our mechanistic studies of the amino-perfluoroalkylation indicated that it proceeds via (1) formation of a perfluoroalkyl radical (·Rf) and Cu(II) species from the peroxide and Cu(I) catalyst, (2) addition of the Rf radical to the double bond of the alkene, (3) oxidation of the resulting alkyl radical with Cu(II) species to afford a carbocation intermediate with recovery of the Cu(I) species, and (4) nucleophilic cyclization (Scheme 1b). In the absence of copper catalyst, the reaction of alkenes gave complex mixtures. Exceptionally, reaction of alkenes bearing an aromatic ring at an appropriate position selectively generated perfluoroalkyl group-containing carbocycles, because the aromatic ring acted as a scavenger of the alkyl radical. We were interested in the unique reactivity of styrenes and radical cation species in perfluoroalkylation with the perfluoro acid anhydride/urea·H2O2 system (Scheme 1c), and postulated that the styrene substrate serves to control the reactivity and selectivity in the formation of the carbocation intermediate without transition-metal-catalyst;9bi.e., styrene serves as an electron donor to accelerate generation of the perfluoroalkyl radical via decomposition of the diacyl peroxide by SET. Then, addition of the perfluoroalkyl radical to the resulting radical cation A affords the carbocation B (path a). Another possibility is that the perfluoroalkyl radical reacts with another styrene molecule (having higher electron density compared to the radical cation A), and the resulting benzyl radical intermediate C is oxidized by the radical cation A as an electron acceptor to afford the same benzyl cation intermediate B (path b). In this work, we focused on this carbocation formation, as a key process in difunctionalization-type perfluoroalkylation, and aimed to develop metal-free oxy- and amino-perfluoroalkylations of styrene derivatives by using perfluoro acid anhydrides. We also carried out various derivatizations to confirm the synthetic potential of the products.
Results and discussion
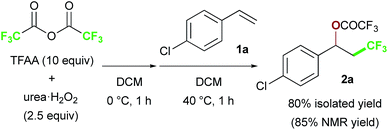
We chose commercially available 4-chlorostyrene 1a as a model substrate to explore the reaction. To our delight, after in situ generation of bis(trifluoroacetyl)peroxide (BTFAP) from trifluoroacetic anhydride (TFAA) with urea·H2O2 in DCM at 0 °C for 1 h, reaction with 1a at 40 °C for 1 h afforded the desired oxy-trifluoromethylation product 2a. Careful tuning of the ratio of the reagents and the reaction temperature improved the yield.13 Finally, the reaction with TFAA (10 equiv.) and urea·H2O2 (2.5 equiv.) provided the corresponding oxy-trifluoromethylated product 2a in 80% isolated yield (85% NMR yield) (Scheme 2).14 The scope of the optimized reaction conditions was then explored using a range of styrene-based substrates (Scheme 3). Various functional groups at the para position were tolerated and the corresponding oxy-trifluoromethylated products were formed efficiently (2a–i).15 The usefulness of the reaction was demonstrated in a gram-scale experiment with 4-fluorostyrene 1b, which was transformed into the desired compound 2b in 93% yield (4.7 g). meta- and ortho-substituted styrene substrates performed well in the oxy-trifluoromethylation reaction (2j–n), although higher temperatures were needed for meta-substituted substrate because of slow conversion compared to para- and ortho-substituted styrenes. A disubstituted styrene 1o afforded the target compound 2o in good yield. The generality of the reaction was also assessed with several internal alkenes, which afforded the corresponding difunctionalized products in moderate to good yields (2p–s). Quaternary carbon centres could be constructed successfully, and more complex compounds 2t and 2u were isolated in 59% and 80% yield, respectively. Finally, this metal-free procedure was applied to the oxy-perfluoroalkylation of styrene-based substrates with other perfluoro acid anhydrides, and the desired products 2b′ and 2d′′ were isolated in excellent yields.
 |
| | Scheme 2 Metal-free oxy-trifluoromethylation of styrene 1a with TFAA/urea·H2O2 under optimized conditions. | |
 |
| | Scheme 3 Substrate scope of the metal-free oxy-perfluoroalkylation reaction. aGram-scale synthesis using 2.0 g (16 mmol) of 1b. bRun at 0 °C for 10 min with Cs2CO3 (5 equiv.) as an additive. cRun at 60 °C in 1,2-dichloroethane. | |
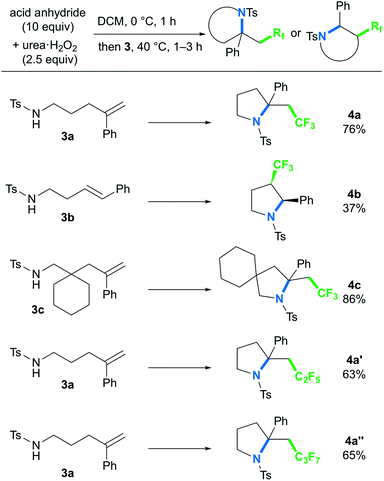
To further explore the ability of the styryl functionality to provide the carbocation intermediate, we next investigated whether pyrrolidines could be obtained by intramolecular amino-perfluoroalkylation of styrene derivatives bearing a pendant amino group via nucleophilic cyclization, based on our previous work.9b,16 In contrast to metal-free oxy-perfluoroalkylation, metal-free amino-perfluoroalkylation has rarely been reported,5a,8 probably because of the lack of appropriate N-radical trapping agents. Thus, we examined the reaction of styryl group-containing aminoalkene 3a with in situ-generated BTFAP under the optimal conditions for the oxy-trifluoromethylation (Scheme 4).17 As we had hoped, the amino-trifluoromethylation proceeded well to afford the corresponding CF3-containing pyrrolidine 4a in 76% yield. This styrene-driven amino-trifluoromethylation was also applicable to internal alkene 3b, which provided disubstituted pyrrolidine 4b as a syn-diastereomer.18 In this reaction, the oxy-trifluoromethylation product was obtained as a by-product in 40% yield, and it was not converted to the amino-trifluoromethylation product 4b even upon prolonged reaction. This observation suggested that this amino-trifluoromethylation does not proceed via nucleophilic substitution of the oxy-trifluoromethylation product under the conditions. Alkenyl amine 3c featuring a 6-membered ring as a tethering group in the carbon chain was tolerated, and the spirocyclic product 4c was formed in high yield. The use of acid anhydrides bearing longer perfluoroalkyl chains furnished C2F5- and C3F7-substituted pyrrolidines 4a′ and 4a′′ in good yields.
 |
| | Scheme 4 Metal-free amino-perfluoroalkylation reaction of alkenes. | |
To further expand the chemical space of the perfluoroalkylated compounds, we focused on the reactivity of the perfluoroacetoxy group, as a labile functional group, in the oxy-trifluoromethylation products (Scheme 5). First, we examined the reaction of 2b, as a representative substrate, with bases. When DBU was reacted with 2b in DME, the alcohol 5b was obtained in quantitative yield. On the other hand, KHMDS as the base was found to give the vinyl product 6bvia elimination reaction. Next, we attempted to construct attractive carbon skeletons and examined SN1-type nucleophilic substitution reactions with various carbon nucleophiles in the presence of acid catalysts. The trifluoroacetoxy group was readily dissociated with triflic acid, and trapping of the resulting carbocation with arenes resulted in C–C bond formation to give 7b and 8b.19 In the presence of a catalytic amount of B(C6F5)3 to promote formation of the carbocation, allylation with allylsilane proceeded to afford 9b in good yield.20 This approach was also applicable to the reaction with a ketene silyl acetal as a nucleophilic partner, providing 10b. The presented procedures for the substitution reactions of the benzylic trifluoroacetoxy group provide rapid access to various perfluoroalkyl-group-containing molecules, which should be useful building blocks in organic synthesis.
 |
| | Scheme 5 Examples of derivatization of benzyl trifluoroacetate 2b: (a) hydrolysis and elimination reaction (b) intermolecular C–C bond formation reactions. | |
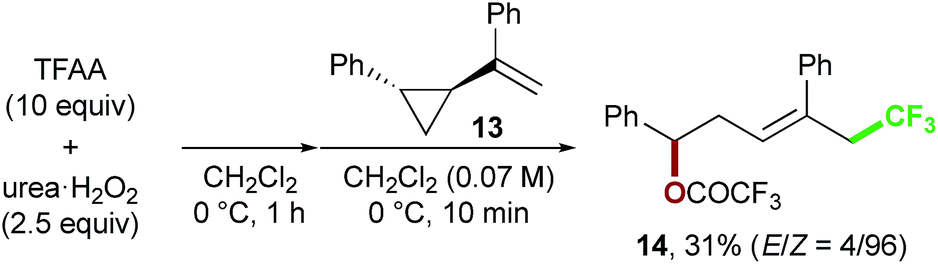
Finally, we focused on the reaction mechanism of the perfluoroalkylation (Scheme 1c). The reaction starts with the generation of an electrophilic CF3 radical via decomposition of the diacyl peroxide, i.e. BTFAP which is formed in situ from TFAA and urea·H2O2. Decomposition of BTFAP would be triggered by SET with styrene as the electron donor and/or by heating (Scheme 6). To trace the decomposition, TEMPO instead of styrene was reacted with in situ-generated BTFAP (Scheme 7a). TEMPO-CF3 adduct 11 was obtained in only 2% yield, which suggested that thermal decomposition would be very slow under these reaction conditions.21 Furthermore, the decomposition could not be observed by 19F NMR monitoring of the peroxide in CD2Cl2 at 40 °C without any substrate.13 Yoshida similarly found that aromatic compounds such as benzene accelerate the decomposition of BTFAP by SET.11b The HOMO level of 4-chlorostyrene 1a (−6.41 eV), used as the model substrate in this work, is higher than that of benzene (−7.09 eV).13 Accordingly, decomposition of BTFAP was concluded to be induced by SET with styrene under the present conditions at 40 °C, affording CF3 radical and radical cation A. Radical trapping with TEMPO under the optimized conditions afforded the CF3-containing TEMPO-benzyl adduct 12, generated via the benzyl radical intermediate C, in 7% yield (Scheme 7b). In addition to 12, TEMPO-CF311 was formed, together with a mixture of oxy-trifluoromethylation products 2a and its hydrolysis product 5a. Furthermore, a known radical probe alkene 13, 1-phenyl-1-(trans-2-phenylcyclopropyl)ethane,8b was subjected to the reaction (Scheme 8). The corresponding ring-opening product 14 was formed via the radical intermediate as the major product, along with a complex mixture of other products.13 These results proved that the CF3 radical reacts with styrene 1a (path b, Scheme 1c), although both path a and path b may be operated. Indeed, DFT calculation indicated that the activation energy of the reaction of the CF3 radical with 1a is low (ΔG‡ = +10.3 kcal mol−1, Scheme 9a). Next, we considered the oxidation step of the benzyl radical to the carbocation B. Comparison of the calculated LUMO levels of potential oxidants, BTFAP and radical cation A, indicated that radical cation A (−6.26 eV) has a lower LUMO level than that of BTFAP (−2.35 eV). In addition, the LUMO level of A was closer to that of Cu(II)(O2CCF3)2 (−5.09 eV) which was reported to oxidize the radical intermediate in the amino-perfluoroalkylation reaction (Scheme 1b).9b Thus, radical cation A was considered to act as the oxidant, affording carbocation B, which leads to the desired products. The DFT calculated activation energy of oxidation of benzyl radical C with A was ΔG‡ET = +6.2 kcal mol−1 (Scheme 9b), which is much lower than that of the addition of CF3 radical to 1a suggesting rapid conversion of the highly reactive benzyl radical C to the metastable benzyl cation intermediate B. These mechanistic studies supported our original hypothesis shown in Scheme 1c, in which substrate styrene itself acts as SET donor to trigger the perfluoroalkyl radical formation from the diacyl peroxide. The resulting perfluoroalkyl radical could react with styrene affording benzyl radical intermediate C, which is rapidly oxidized to the benzyl cation B by the radical cation A. The generated benzyl cation intermediate B is trapped by the perfluoro carboxylate anion or amine yielding the desired oxy- and amino-perfluoroalkylation products, 2 and 4.
 |
| | Scheme 6 Possible pathway of decomposition of BTFAP. | |
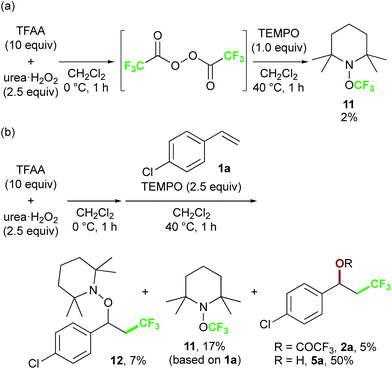 |
| | Scheme 7 TEMPO trapping test: (a) reaction of TEMPO with BTFAP and (b) oxy-trifluoromethylation in the presence of TEMPO. | |
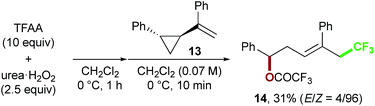 |
| | Scheme 8 Radical probe test using 13. | |
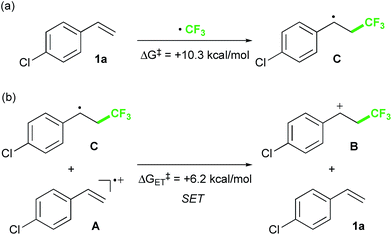 |
| | Scheme 9 Activation energies of (a) addition of CF3 radical to 1a and (b) SET between benzyl radical C and radical cation A. | |
Conclusions
We have developed a mild and efficient method for the metal-free oxy- and amino-perfluoroalkylation of styrenes via carbocation intermediates, using perfluoro acid anhydrides as inexpensive and practical perfluoroalkyl sources. The oxy-trifluoromethylation products were derivatized to a variety of CF3-containing unique molecules. We believe this method will prove useful in medicinal and agro-chemistry discovery programs. In addition, the unique reactivity between styrene and perfluoro diacyl peroxide may provide clues to design new reactions and catalysts in the future.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI (No. 15K17860) and Project Funding from RIKEN. E. V. is grateful for a JSPS Postdoctoral Fellowship (15F15340). The computational study was conducted on the HOKUSAI-GW (RIKEN).
Notes and references
- Selected reviews:
(a) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef PubMed;
(b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef PubMed;
(c) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef.
- Selected reviews:
(a) Y. Tian, S. Chen, Q.-S. Gu, J.-S. Lin and X.-Y. Liu, Tetrahedron Lett., 2018, 59, 203 CrossRef;
(b) X. Wang and A. Studer, Acc. Chem. Res., 2017, 50, 1712 CrossRef PubMed;
(c) A. Prieto, O. Baudoin, D. Bouyssi and N. Monteiro, Chem. Commun., 2016, 52, 869 RSC;
(d) T. Courant and G. Masson, J. Org. Chem., 2016, 81, 6945 CrossRef PubMed;
(e) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef PubMed;
(f) S.-M. Wang, J.-B. Han, C.-P. Zhang, H.-L. Qin and J.-C. Xiao, Tetrahedron, 2015, 71, 7949 CrossRef;
(g) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef PubMed;
(h) C. Alonso, E. M. de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef PubMed;
(i) T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2014, 20, 16830 CrossRef PubMed;
(j) Y. Shimizu and M. Kanai, Tetrahedron Lett., 2014, 55, 3727 CrossRef;
(k) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806 CrossRef PubMed;
(l) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef PubMed;
(m) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC;
(n) J. Xu, X. Liu and Y. Fu, Tetrahedron Lett., 2014, 55, 585 CrossRef;
(o) P. Chen and G. Liu, Synthesis, 2013, 45, 2919 CrossRef.
- Selected reports on transition-metal-catalyzed oxy-trifluoromethylations of styrenes:
(a) P. G. Janson, I. Ghoneim, N. O. Ilchenko and K. J. Szabó, Org. Lett., 2012, 14, 2882 CrossRef PubMed;
(b) H. Egami, R. Shimizu and M. Sodeoka, Tetrahedron Lett., 2012, 53, 5503 CrossRef;
(c) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2012, 134, 12462 CrossRef PubMed;
(d) Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef PubMed;
(e) R. Zhu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 12655 CrossRef PubMed;
(f) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747 CrossRef PubMed;
(g) X.-Y. Jiang and F.-L. Qing, Angew. Chem., Int. Ed., 2013, 52, 14177 CrossRef PubMed;
(h) N. O. Ilchenko, P. G. Janson and K. J. Szabó, J. Org. Chem., 2013, 78, 11087 CrossRef PubMed;
(i) A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240 CrossRef PubMed;
(j) H. Egami, R. Shimizu, Y. Usui and M. Sodeoka, J. Fluorine Chem., 2014, 167, 172 CrossRef;
(k) Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Org. Lett., 2014, 16, 780 CrossRef PubMed;
(l) R. Tomita, Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2014, 53, 7144 CrossRef PubMed;
(m) R. Zhu and S. L. Buchwald, J. Am. Chem. Soc., 2015, 137, 8069 CrossRef PubMed;
(n) Q.-H. Deng, J.-R. Chen, Q. Wei, Q.-Q. Zhao, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2015, 51, 3537 RSC;
(o) N. Noto, K. Miyazawa, T. Koike and M. Akita, Org. Lett., 2015, 17, 3710 CrossRef PubMed;
(p) Q. Wei, J.-R. Chen, X. Q. Hu, X. C. Yang, B. Lu and W. J. Xiao, Org. Lett., 2015, 17, 4464 CrossRef PubMed;
(q) S. Jana, A. Ashokan, S. Kumar, A. Verma and S. Kumar, Org. Biomol. Chem., 2015, 13, 8411 RSC;
(r) Y. Yang, Y. Liu, Y. Jiang, Y. Zhang and D. A. Vicic, J. Org. Chem., 2015, 80, 6639 CrossRef PubMed;
(s) L. Jarrige, A. Carboni, G. Dagousset, G. Levitre, E. Magnier and G. Masson, Org. Lett., 2016, 18, 2906 CrossRef PubMed;
(t) Y. Wu, G. Lu, T. Yuan, Z. Xu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 13668 RSC;
(u) X. Bai, L. Lv and Z. Li, Org. Chem. Front., 2016, 3, 804 RSC;
(v) N. Noto, T. Koike and M. Akita, J. Org. Chem., 2016, 81, 7064 CrossRef PubMed;
(w) H. Y. Zhang, C. Ge, J. Zhao and Y. Zhang, Org. Lett., 2017, 19, 5260 CrossRef PubMed;
(x) Y. F. Cheng, X. Y. Dong, Q. S. Gu, Z. L. Yu and X.-Y. Liu, Angew. Chem., Int. Ed., 2017, 56, 8883 CrossRef PubMed.
- Selected reports on transition-metal-catalysed amino-trifluoromethylations of styrenes:
(a) Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef PubMed;
(b) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881 CrossRef PubMed;
(c) G. Dagousset, A. Carboni, E. Magnier and G. Masson, Org. Lett., 2014, 16, 4340 CrossRef PubMed;
(d) J.-S. Lin, X.-Y. Dong, T.-T. Li, N.-C. Jiang, B. Tan and X.-Y. Liu, J. Am. Chem. Soc., 2016, 138, 9357 CrossRef PubMed;
(e) L.-Z. Yu, Y. Wei and M. Shi, Chem. Commun., 2016, 52, 13163 RSC;
(f) K. Shen and Q. Wang, Org. Chem. Front., 2016, 3, 222 RSC;
(g) H.-Y. Zhang, W. Huo, C. Ge, J. Zhao and Y. Zhang, Synlett, 2017, 28, 962 CrossRef.
- Metal-free oxy-trifluoromethylations:
(a) K. Uneyama, S. Watanabe, Y. Tokunaga, K. Kitagawa and Y. Sato, Bull. Chem. Soc. Jpn., 1992, 65, 1976 CrossRef;
(b) Y. Sato, S. Watanabe and K. Uneyama, Bull. Chem. Soc. Jpn., 1993, 66, 1840 CrossRef;
(c) C.-P. Zhang, Z.-L. Wang, Q.-Y. Chen, C.-T. Zhang, Y.-C. Gu and J.-C. Xiao, Chem. Commun., 2011, 47, 6632 RSC;
(d) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 CrossRef PubMed;
(e) H.-Q. Luo, Z.-P. Zhang, W. Dong and X.-Z. Luo, Synlett, 2014, 25, 1307 CrossRef;
(f) Q. Lu, C. Liu, Z. Huang, Y. Ma, J. Zhang and A. Lei, Chem. Commun., 2014, 50, 14101 RSC;
(g) J. Yu, H. Yang and H. Fu, Adv. Synth. Catal., 2014, 356, 3669 CrossRef;
(h) C. Liu, Q. Lu, Z. Huang, J. Zhang, F. Liao, P. Peng and A. Lei, Org. Lett., 2015, 17, 6034 CrossRef PubMed;
(i) L. Huang, S.-C. Zheng, B. Tan and X.-Y. Liu, Org. Lett., 2015, 17, 1589 CrossRef PubMed;
(j) M. Hartmann, Y. Li and A. Studer, Org. Biomol. Chem., 2016, 14, 206 RSC;
(k) N.-Y. Yang, Z.-L. Li, L. Ye, B. Tan and X.-Y. Liu, Chem. Commun., 2016, 52, 9052 RSC;
(l) J.-S. Lin, F.-L. Wang, X.-Y. Dong, W.-W. He, Y. Yuan, S. Chen and X.-Y. Liu, Nat. Commun., 2017, 8, 14841 CrossRef PubMed;
(m) X.-T. Li, Q.-S. Gu, X.-Y. Dong, X. Meng and X.-Y. Liu, Angew. Chem., Int. Ed., 2018, 57, 7668 CrossRef PubMed.
- Li's group reported a dihydrofuran-forming oxy-trifluoromethylation using 1,3-diaryl diketone-containing alkene in the presence of Cu-catalyst; they proposed a radical cyclization mechanism involving C–O bond formation between the radical carbon centre and O
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group: X. Bai, L. Lv and Z. Li, Org. Chem. Front., 2016, 3, 804 RSC.
C group: X. Bai, L. Lv and Z. Li, Org. Chem. Front., 2016, 3, 804 RSC.
- Nagano reported a metal-free hydroxy-trifluoromethylation reaction using CF3I. This reaction involved hydrolysis of the iodo-trifluoromethylation intermediate, and thus can be classified as iodo-trifluoromethylation:
(a) T. Yajima, C. Saito and H. Nagano, Tetrahedron, 2005, 61, 10203 CrossRef. Our group also reported KI-promoted, metal-free oxazoline-forming trifluoromethylation of allylamides with Togni reagent, in which an iodo-trifluoromethylation intermediate is also involved:
(b) S. Kawamura, D. Sekine and M. Sodeoka, J. Fluorine Chem., 2017, 203, 115 CrossRef.
-
(a) K. Arai, K. Watts and T. Wirth, ChemistryOpen, 2014, 3, 23 CrossRef PubMed;
(b) N. Noto, T. Koike and M. Akita, Chem. Sci., 2017, 8, 6375 RSC.
-
(a) S. Kawamura and M. Sodeoka, Angew. Chem., Int. Ed., 2016, 55, 8740 CrossRef PubMed;
(b) S. Kawamura, K. Dosei, E. Valverde, K. Ushida and M. Sodeoka, J. Org. Chem., 2017, 82, 12539 CrossRef PubMed.
- Zard reported thio-trifluoromethylation of alkenes by using S-trifluoromethyl xanthates prepared from TFAA: F. Bertrand, V. Pevere, B. Quiclet-Sire and S. Z. Zard, Org. Lett., 2001, 3, 1069 CrossRef PubMed.
- Trifluoromethylations of aromatic compounds with bis(trifluoroacetyl) peroxide (BTFAP):
(a) M. Yoshida, T. Yoshida, M. Kobayashi and N. Kamigata, J. Chem. Soc., Perkin Trans. 1, 1989, 909 RSC;
(b) H. Sawada, M. Nakayama, M. Yoshida, T. Yoshida and N. Kamigata, J. Fluorine Chem., 1990, 46, 423 CrossRef;
(c) M. Matsui, S. Kawamura, K. Shibata, M. Mitani, H. Sawada and M. Nakayama, J. Fluorine Chem., 1992, 57, 209 CrossRef;
(d) M. Nishida, S. Fujii, H. Kimoto, Y. Hayakawa, H. Sawada and L. A. Cohen, J. Fluorine Chem., 1993, 63, 43 CrossRef;
(e) M. Matsui, S. Kondoh, K. Shibata and H. Muramatsu, Bull. Chem. Soc. Jpn., 1995, 68, 1042 CrossRef;
(f) Y. Hayakawa, N. Terasawa and H. Sawada, Polymer, 2001, 42, 4081 CrossRef;
(g) S. Zhong, A. Hafner, C. Hussal, M. Nieger and S. Bräse, RSC Adv., 2015, 5, 6255 RSC.
- Stephenson and co-workers reported the photochemical perfluoroalkylation with pyridine N-oxides/TFAA adduct:
(a) J. W. Beatty, J. J. Douglas, K. P. Cole and C. R. J. Stephenson, Nat. Commun., 2015, 6, 7919 CrossRef PubMed;
(b) J. W. Beatty, J. J. Douglas, R. Miller, R. C. McAtee, K. P. Cole and C. R. J. Stephenson, Chem, 2016, 1, 456 CrossRef PubMed.
- See ESI† for details.
- Isolated yields were slightly lower than NMR yields because of hydrolysis of 2 during isolation by means of silica-gel column chromatography.
- Methoxy group-containing substrate showed very high reactivity, and only the moderate yield of the desired product was obtained due to undesired side reactions, even if the reaction was carried out at 0 °C.
-
(a) H. Egami, S. Kawamura, A. Miyazaki and M. Sodeoka, Angew. Chem., Int. Ed., 2013, 52, 7841 CrossRef PubMed;
(b) S. Kawamura, H. Egami and M. Sodeoka, J. Am. Chem. Soc., 2015, 137, 4865 CrossRef PubMed.
- The reaction of N-(2-vinylphenethyl)-p-toluenesulfonamide was examined. However, only 9% yield of tetrahydroisoquinoline was formed and 81% yield of an oxy-trifluoromethylation product (alcohol) was obtained. See ESI† for details.
- Liu reported Cu-catalyzed amino-trifluoromethylation of the same substrate with Togni reagent and obtained 4b with syn-selectivity: Y. Wang, M. Jiang and J.-T. Liu, Adv. Synth. Catal., 2016, 358, 1322 CrossRef.
- V. D. Vuković, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085 CrossRef PubMed.
- M. Rubin and V. Gevorgyan, Org. Lett., 2001, 3, 2705 CrossRef PubMed.
- As reported in ref. 9a, reaction of a simple alkene under the similar reaction conditions was very slow giving a small amount of complex mixture of products. A trace amount of CF3 radical generated via thermal decomposition could cause undesired reactions in the absence of an appropriate electron acceptor.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental procedures, experimental method, compound characterization, and NMR spectroscopic data. See DOI: 10.1039/c8sc02547a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Daisuke
Sekine
a and
Mikiko
Sodeoka
ab,
Daisuke
Sekine
a and
Mikiko
Sodeoka