
A desymmetrization-based approach to morphinans: application in the total synthesis of oxycodone†‡
Kun Ho (Kenny)
Park
and
David Y.-K.
Chen
 *
*
Department of Chemistry, Seoul National University, Gwanak-1 Gwanak-ro, Gwanak-gu, Seoul 08826, South Korea. E-mail: davidchen@snu.ac.kr
First published on 5th November 2018
Abstract
Here we report a total synthesis of the pharmacologically significant morphinan alkaloid, oxycodone. The centerpiece of the developed strategy features the first application of the Rovis desymmetrization of peroxyquinol in target-oriented total synthesis to access an optically active phenanthrene framework shared by the morphinans. A Stork–Ueno radical cyclization under photoredox conditions installed the all-carbon quaternary stereocenter, and a late-stage reductive detosylation with concomitant piperidine formation secured the core structure of the target molecule.
The ready availability of symmetrical compounds, either naturally occurring or synthetic, has had a profound impact on synthetic organic chemistry.1 In the retrosynthetic analysis of molecules which do not possess obvious symmetry element(s), unveiling a symmetrical sub-structure or a potential synthetic precursor is often non-trivial. Even if a potential synthetic precursor can be identified, the choice of desymmetrization method in the forward synthesis remains highly challenging especially if the process is also intended to deliver enantioselectivity. Our laboratory has a long-standing interest in desymmetrization-based synthetic approaches to access architecturally complex natural products, and during the course of our studies has successfully demonstrated conceptually novel solutions to challenging synthetic problems.2
Naturally occurring and synthetic morphinans are well-recognized for their therapeutic value as well as illicit usage, and continue to be the center of chemical3 and biological investigations4 more than 90 years after Robinson's landmark structural elucidation of morphine.5 In accordance to the proposed desymmetrization approach to access the generic morphinan core structure shown in Scheme 1, our synthesis commenced with the preparation of biaryl phenol intermediate 1. As shown in Scheme 2, a Wittig-olefination6 between benzaldehyde derivative 37 and phosphonium salt 48 followed by a controlled hydrogenation (Pd/C, H2) readily delivered biaryl phenol 1 on a multi-gram scale, provided that hydrogenation was performed under carefully monitored reaction conditions to avoid any undesired reductive debromination. Oxidative dearomatization of phenol 1 under hypervalent iodine conditions (PIDA) afforded hydroxy dienone 5 uneventfully,9 which underwent an intramolecular Heck cyclization [Pd(OAc)2, PPh3] followed by a chemoselective hydrogenation [Rh(PPh3)3Cl, H2] to furnish hydroxy enone 7. Hydroxy enone 7 could also be prepared through a Rh(PPh3)3Cl catalyzed hydrosilylation of dienone 5 followed by an intramolecular Heck reaction of the intermediate hydroxy enone 8. Interestingly, the intramolecular Heck condition developed for hydroxy dienone 5 was totally ineffective for hydroxy enone 8, therefore, after extensive studies, an alternative protocol involving PdCl2(PPh3)2, X-Phos, and tBu3PH·BF4 was implemented to realize the desired transformation.10 With phenanthrene system 7 in hand, installation of the all-carbon quaternary stereocenter in the morphinans was accomplished using both conventional (nBu3SnH, Et3B) and a photoredox variant11 of the Stork–Ueno radical cyclization12 through the intermediacy of iodoacetal 9, to deliver tetracyclic acetal 11 as an inconsequential mixture of diastereoisomers in a 57–62% overall yield. Evidently, the operationally benign photoredox protocol circumvented extensive chromatographic removal of organotin residues which proved synthetically more attractive. Treatment of 11 with mCPBA in the presence of BF3·OEt213 smoothly oxidized its acetal moiety to the corresponding lactone, which also fortuitously removed the MOM ether to afford phenol 12. Advancing tetracyclic ketone 12 next called for the formation of the five-membered oxacycle that resides in several flagship morphinans.4 To this end, application of the phenol-directed bromination (PyH·Br3) followed by a thermally induced cyclization developed by the Fukuyama laboratory14 completed the synthesis of pentacyclic key intermediate 13 in nine-steps from 3 and 4.
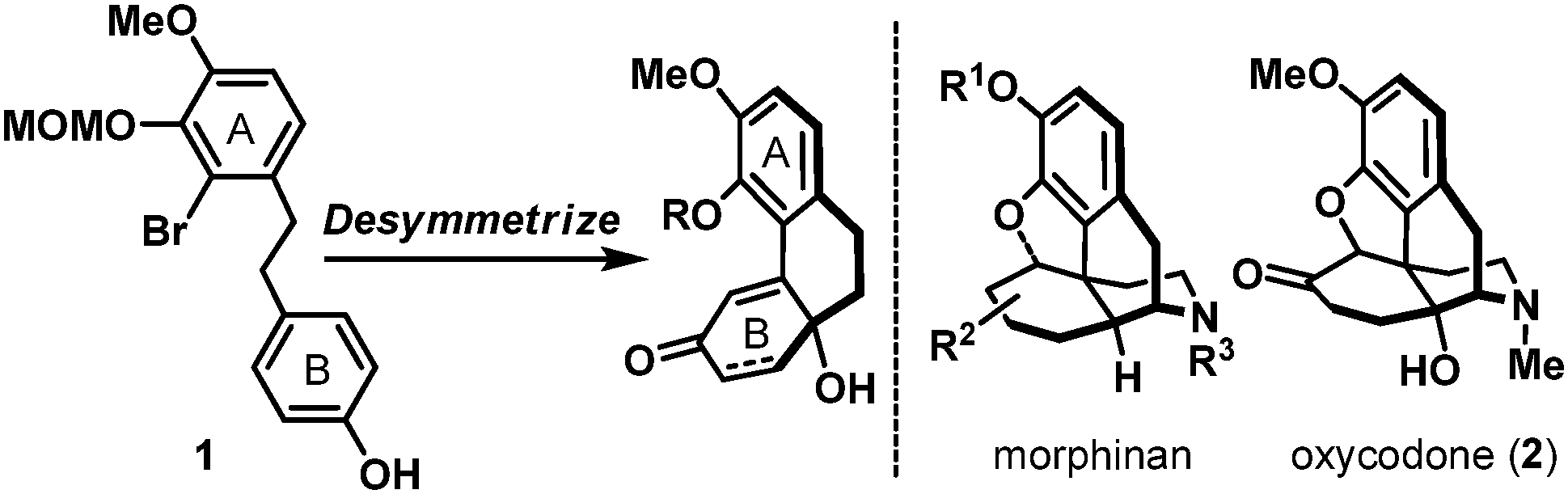 | ||
| Scheme 1 Proposed desymmetrization approach to morphinans and oxycodone (2). MOM = methoxymethyl. | ||
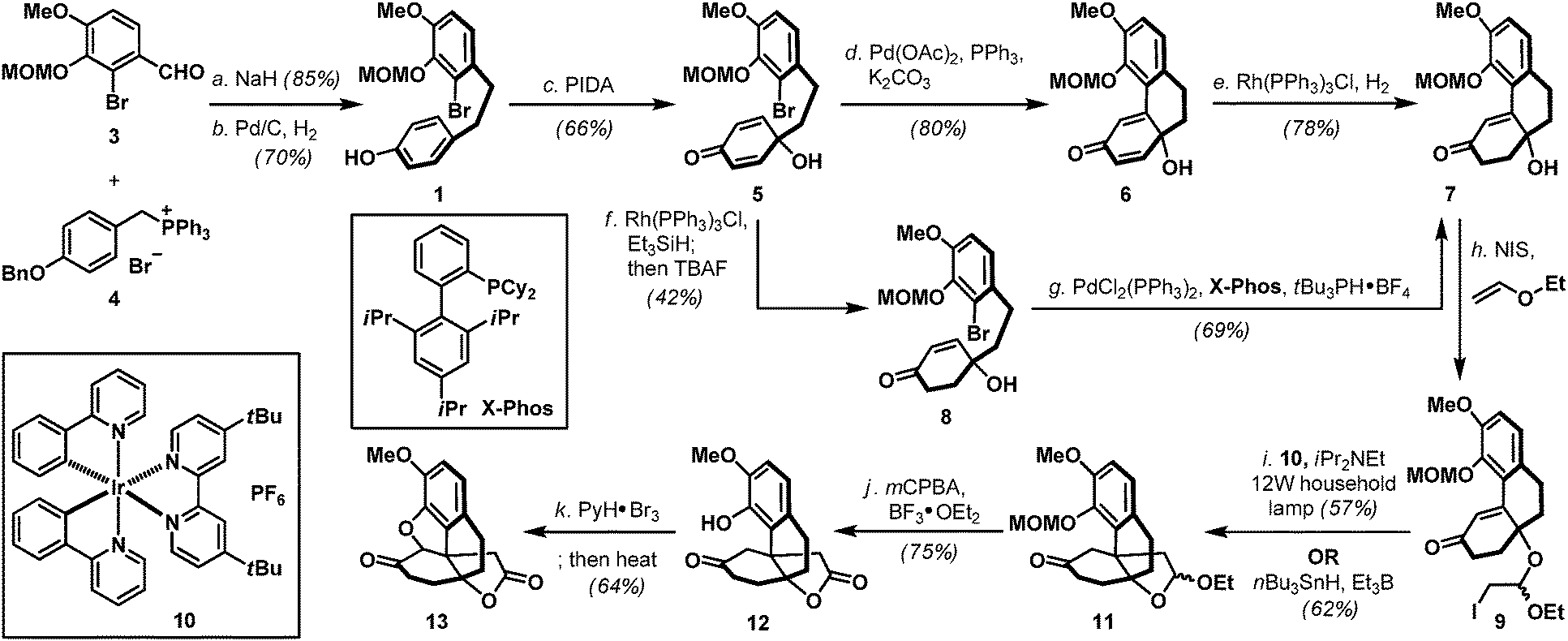 | ||
Scheme 2 Synthesis of pentacyclic ketolactone 13. Reagents and conditions: (a) NaH (3.5 equiv.), THF, 0 °C to 23 °C, 2 h, 85%; (b) Pd/C (10% wt/wt, 0.05 equiv.), H2, EtOAc/MeOH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 23 °C, 1 h, 70%; (c) PIDA (1.2 equiv.), MeCN/H2O (1:1), 0 °C, 2 h, 66%; (d) Pd(OAc)2 (0.15 equiv.), K2CO3 (3.0 equiv.), PPh3 (0.3 equiv.), toluene, 110 °C, 24 h, 80%; (e) Rh(PPh3)3Cl (0.05 equiv.), H2, benzene, 23 °C, 12 h, 78%; (f) Rh(PPh3)3Cl (0.05 equiv.), Et3SiH (2.0 equiv.), benzene, 50 °C, 10 h, recovered 5: 16%; then TBAF (1.0 M in THF, 1.8 equiv.), THF, 0 °C to 23 °C, 4 h, 8: 42% for two steps; (g) PdCl2(PPh3)2 (0.15 equiv.), Na2CO3 (3.0 equiv.), X-Phos (0.4 equiv.), tBu3PH·BF4 (0.8 equiv.), DMSO, 100 °C, 40 h, 69%; (h) NIS (6.0 equiv.), ethyl vinyl ether (8.0 equiv.), CH2Cl2, −20 °C to 23 °C, 12 h; (i) [Ir(ppy)2(dtbbpy)]PF6 (0.03 equiv.), iPr2NEt (10 equiv.), 12 W lamp, MeCN, 36 h, 57% for two steps; or Et3B (1.3 equiv.), nBu3SnH (1.3 equiv.), air, benzene, 23 °C, 12 h, 62% for two steps; (j) mCPBA (1.8 equiv.), BF3·OEt2 (2.0 equiv.), CH2Cl2, 0 °C, 1 h, 75%; (k) PyH·Br3 (1.1 equiv.), CH2Cl2/AcOH (1:3.4), 23 °C, 30 min; then LiBr (6.1 equiv.), Et3N (1.6 equiv.), CH3CN, 60 °C, 10 min, 64%. Bn = benzyl; mCPBA = meta-chloroperoxy benzoic acid; Cy = cyclohexyl; [Ir(ppy)2(dtbbpy)]PF6 = [4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[2-(2-pyridinyl-N)phenyl-C]iridium(III) hexafluorophosphate; NIS = N-iodosuccinimide; PIDA = [bis(acetoxy)iodo]benzene; Pd(OAc)2 = palladium(II) acetate; PyH·Br3 = pyridinium tribromide; TBAF = tetra-n-butylammonium fluoride; and X-Phos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl. :1), 23 °C, 1 h, 70%; (c) PIDA (1.2 equiv.), MeCN/H2O (1:1), 0 °C, 2 h, 66%; (d) Pd(OAc)2 (0.15 equiv.), K2CO3 (3.0 equiv.), PPh3 (0.3 equiv.), toluene, 110 °C, 24 h, 80%; (e) Rh(PPh3)3Cl (0.05 equiv.), H2, benzene, 23 °C, 12 h, 78%; (f) Rh(PPh3)3Cl (0.05 equiv.), Et3SiH (2.0 equiv.), benzene, 50 °C, 10 h, recovered 5: 16%; then TBAF (1.0 M in THF, 1.8 equiv.), THF, 0 °C to 23 °C, 4 h, 8: 42% for two steps; (g) PdCl2(PPh3)2 (0.15 equiv.), Na2CO3 (3.0 equiv.), X-Phos (0.4 equiv.), tBu3PH·BF4 (0.8 equiv.), DMSO, 100 °C, 40 h, 69%; (h) NIS (6.0 equiv.), ethyl vinyl ether (8.0 equiv.), CH2Cl2, −20 °C to 23 °C, 12 h; (i) [Ir(ppy)2(dtbbpy)]PF6 (0.03 equiv.), iPr2NEt (10 equiv.), 12 W lamp, MeCN, 36 h, 57% for two steps; or Et3B (1.3 equiv.), nBu3SnH (1.3 equiv.), air, benzene, 23 °C, 12 h, 62% for two steps; (j) mCPBA (1.8 equiv.), BF3·OEt2 (2.0 equiv.), CH2Cl2, 0 °C, 1 h, 75%; (k) PyH·Br3 (1.1 equiv.), CH2Cl2/AcOH (1:3.4), 23 °C, 30 min; then LiBr (6.1 equiv.), Et3N (1.6 equiv.), CH3CN, 60 °C, 10 min, 64%. Bn = benzyl; mCPBA = meta-chloroperoxy benzoic acid; Cy = cyclohexyl; [Ir(ppy)2(dtbbpy)]PF6 = [4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis[2-(2-pyridinyl-N)phenyl-C]iridium(III) hexafluorophosphate; NIS = N-iodosuccinimide; PIDA = [bis(acetoxy)iodo]benzene; Pd(OAc)2 = palladium(II) acetate; PyH·Br3 = pyridinium tribromide; TBAF = tetra-n-butylammonium fluoride; and X-Phos = 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl. | ||
As shown in Scheme 3a, pentacyclic ketone 13 was next protected as its dioxolane derivative followed by treatment of the resulting lactone 14 with methylamine to provide hydroxy amide 15. Exhaustive reduction of amide 15 and subsequent tosylation of the resulting amine furnished sulfonamide 16, which further underwent a benzylic bromination (NBS) and HBr elimination to afford styrene derivative 17. Interestingly, switching the order of exhaustive amide reduction and styrene olefin introduction led to the formation of saturated tosylamide 16 (Scheme 3b), whereas reduction of styrene containing lactone 18/18a afforded diol 21 uneventfully with complete retention of the olefin. These findings clearly suggested that the proximal nitrogen played an assisting role in the unexpected saturation of the styrene olefin in 19. Furthermore, introduction of the morphinan nitrogen atom via amidation of lactone 14 proved uniquely effective, as conventional substitution reactions engaging diol 21 and reductive amination protocols on acetal or hemiacetal 24 were both unsuccessful in our studies (Scheme 3c).
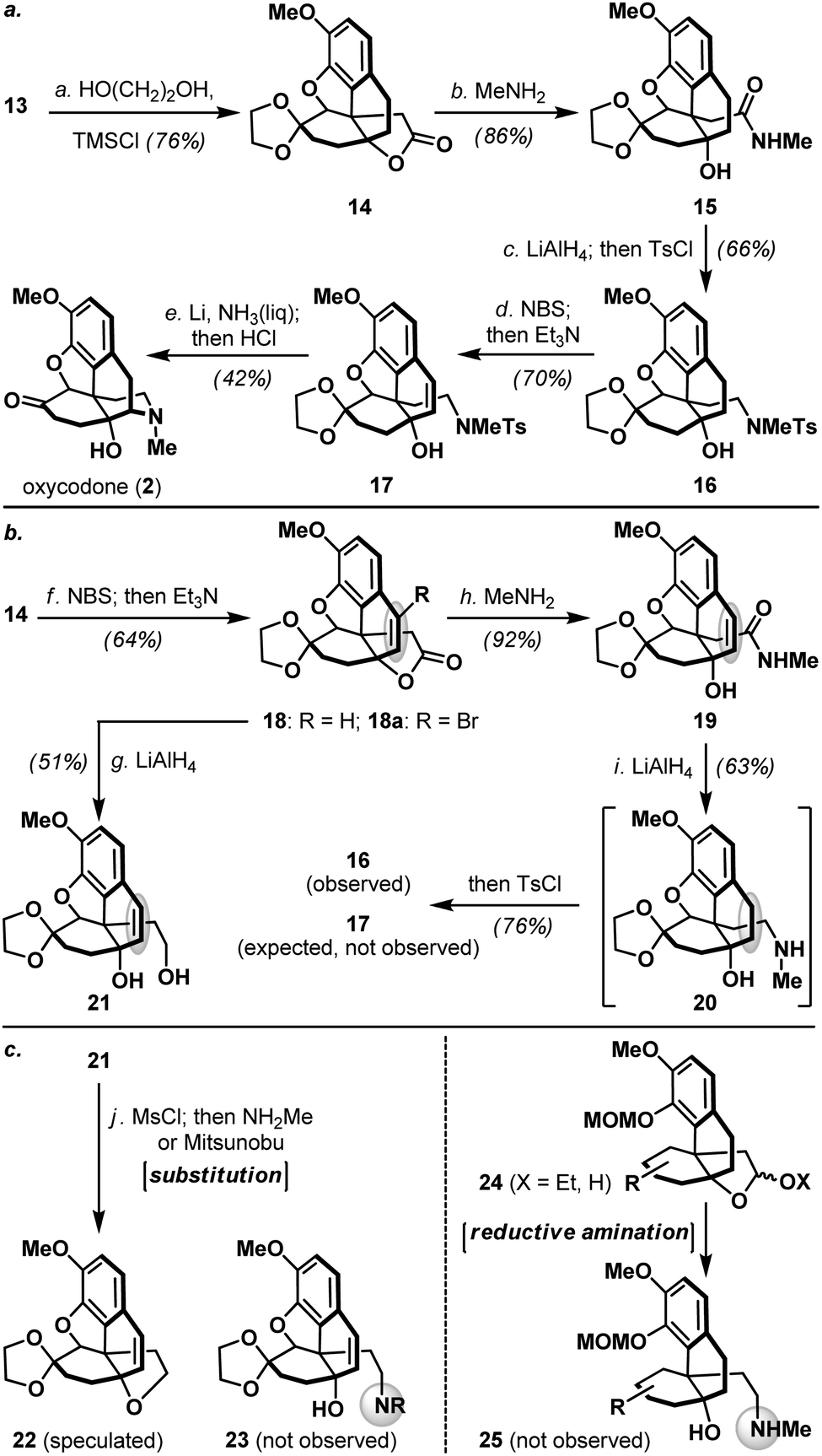 | ||
| Scheme 3 (a) Completion of total synthesis of oxycodone (2); (b) contrasting results in the lithium aluminium hydride mediated reduction of styrene-containing lactone 18 and amide 19; (c) attempted introduction of morphinan nitrogen via substitution and reductive amination conditions. Reagents and conditions: (a) TMSCl (4.8 equiv.), CH2Cl2/ethylene glycol (1:1), 23 °C, 7 h, 76%; (b) MeNH2 (10 equiv.), MeOH/CH2Cl2 (6:1), 23 °C, 12 h, 86%; (c) LiAlH4 (10 equiv.), THF, reflux, 12 h; then TsCl (1.4 equiv.), Et3N (2.6 equiv.), CH2Cl2, 23 °C, 4 h, 66% for two steps; (d) NBS (1.1 equiv.), benzoyl peroxide (0.1 equiv.), CCl4, reflux, 45 min; then Et3N (16.5 equiv.), reflux, 10 min, 70%; (e) Li (excess), NH3, tBuOH/THF (1:10), −78 °C, 15 min, 70%; then HCl (2.0 N aq.)/THF (1:10), 80 °C, 12 h, 61%; (f) NBS (1.1 equiv.), benzoyl peroxide (0.2 equiv.), CCl4, reflux, 45 min; then Et3N (5.6 equiv.), reflux, 10 min, 18:18aca. 5:1, 64%; (g) LiAlH4 (8.3 equiv.), THF, 50 °C, 5 h, 51%; (h) MeNH2 (10 equiv.), MeOH/CH2Cl2 (6:1), 23 °C, 12 h, 92%; (i) LiAlH4 (10 equiv.), THF, reflux, 12 h, 63%; then TsCl (1.2 equiv.), Et3N (10.8 equiv.), CH2Cl2, 23 °C, 3 h, 76%. MsCl = methanesulfonyl chloride; NBS = N-bromosuccinimide; TMSCl = trimethylsilyl chloride; Ts = para-toluenesulfonyl; TsCl = para-toluenesulfonyl chloride. | ||
Tosylamide 17 bearing a tertiary alcohol was poised for the completion of oxycodone (2, Scheme 3a), a pharmacologically important semi-synthetic morphinan currently used in clinics.15 Accordingly, detosylation of 17 under Birch-type conditions took place with concomitant piperidine formation, and subsequently furnished oxycodone (2) upon dioxolane removal (HCl aq.). Related Birch-type reductive cyclization was featured in several previous syntheses of morphinans,3 however, the feasibility of allylic alcohol system 17 has never been demonstrated. All physical properties of synthetic oxycodone (2) were in perfect agreement with the reported data,14 and this developed sequence represented a significant reduction in step-count compared to the first and only reported total synthesis of oxycodone (2) to date by the Fukuyama laboratory.14
Finally, by accessing optically active tricyclic tertiary alcohol 7, an asymmetric version of the aforementioned synthetic pathway is outlined in Scheme 4. Inspired by the work of Feringa,16 our initial efforts toward an optically active dienone 6 were based on the asymmetric Heck reaction of hydroxy dienone 5 in the presence of a chiral ligand. However, after screening numerous conditions only a minuscule level of asymmetric induction was observed (Scheme 4a). After contemplating several cyclohexadienone-based desymmetrization protocols17 including the enantioselective conjugate reduction of prochiral 2,5-cyclohexadione recently reported by Corey and co-workers (Scheme 4b),18 we were attracted to the effectiveness and novelty of the enantioselective desymmetrization of peroxyquinols developed by the Rovis laboratory (Scheme 4c).19 In this context, application of the Rovis technology took advantage of the earlier described phenol intermediate 1 and its oxidized peroxyquinol derivative 26, which on treatment under the conditions described by Rovis and co-workers in the presence of chiral phosphoric acid (R)-TRIP afforded peroxyacetal 27 in 66% yield as a single diastereoisomer. Interestingly, a racemic version of the same process mediated by diphenylphosphinic acid resulted in a mixture of diastereoisomers (ca. 3:1), suggesting the influential role of chiral phosphoric acid in both enantio- and diastereocontrol (see the ESI‡). Optically active peroxyacetal 27 further underwent a chemoselective hydrogenation (Rh/Al, PtO2, and H2) followed by a Kornblum–DeLamare type fragmentation20 to furnish hydroxy enone 8 (95:5 er, determined by chiral HPLC analysis). Optically active hydroxy enone 8 also participated in an intramolecular Heck reaction under the previously described conditions for racemic 8 to afford optically active enone 7 with no erosion of optical purity, thereby establishing an asymmetric pathway to oxycodone (2).
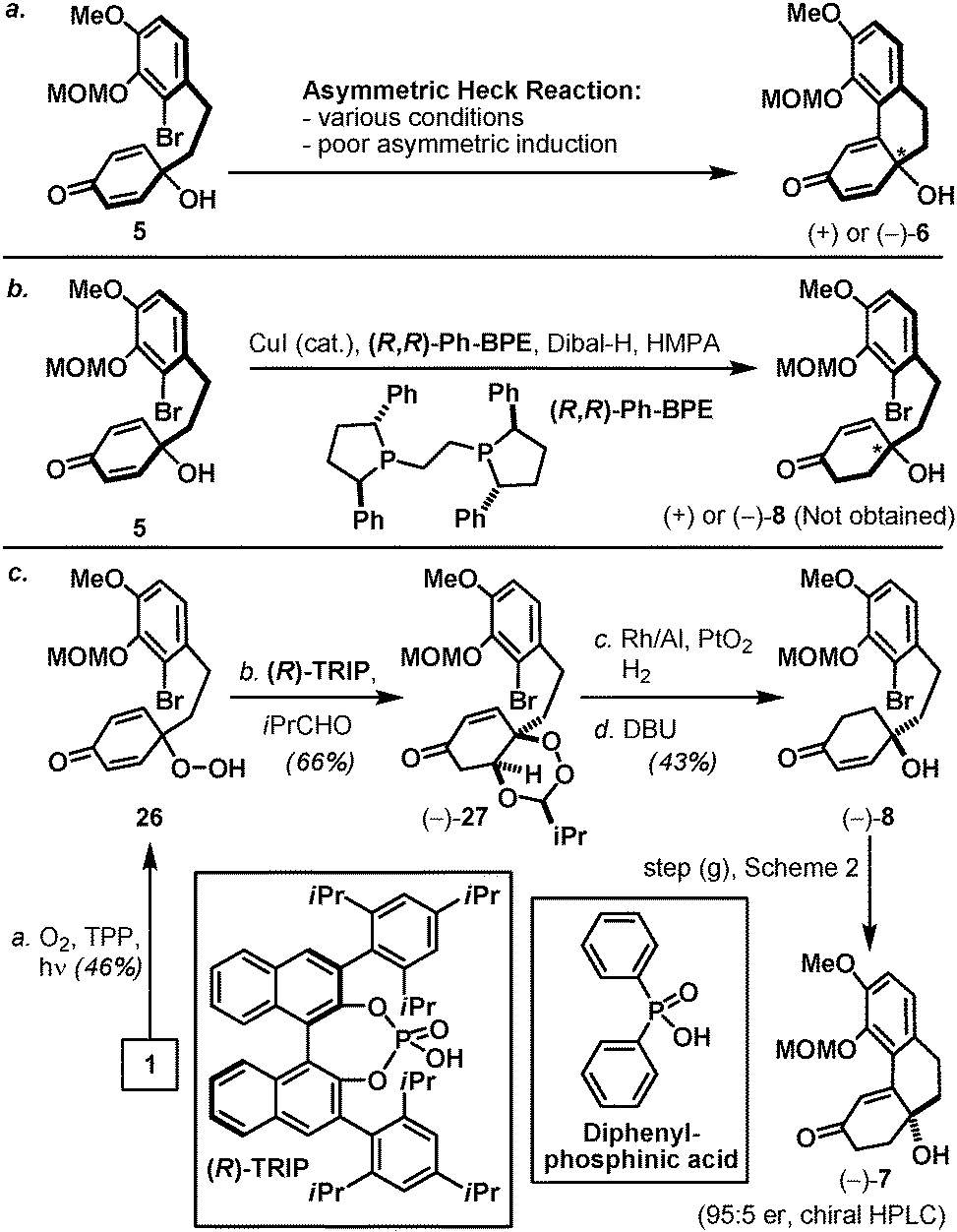 | ||
| Scheme 4 Preparation of optically active intermediate 7. (a) Initial exploration of an asymmetric intramolecular Heck reaction of prochiral hydroxy dienone 5; (b) attempted enantioselective desymmetrizing reduction of prochiral hydroxy dienone 5; (c) synthesis of optically active hydroxy enone 7 using Rovis asymmetric peroxyquinol chemistry. Reagents and conditions: (a) TPP (0.05 equiv.), O2, 27 W lamp, CHCl3, 23 °C, 5 d, 46%; (b) (R)-TRIP (0.05 equiv.), isobutyraldehyde (1.2 equiv.), 4 Å MS, 1,2-dichloroethane, 45 °C, 21 h, 66%; (c) Rh/Al, PtO2 (0.3 equiv.), H2, EtOAc, 3 h; then DBU (2.6 equiv.), CH2Cl2, 45 °C, 4 h, 43% for two steps. MS = molecular sieves; TPP = 5,10,15,20-tetraphenyl-21H,23H-porphine; (R)-TRIP = (R)-3,3′-bis(2,4,6-triisopropylphenyl)-1,1′-binaphthyl-2,2′-diylhydrogenphosphate; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene. | ||
In summary, a desymmetrization-based synthetic strategy has enabled a concise and asymmetric entry to oxycodone (2) from a readily accessible, reduced stilbene system 1. A salient feature of the developed synthesis featured a Rovis desymmetrization of peroxyquinol 26 through chiral Bronsted acid catalysis, an intramolecular photoredox radical cyclization to forge the congested quaternary stereocenter, and a late-stage reductive detosylation with concomitant piperidine formation. Further improvements of the developed synthetic sequence and application of the synthetic technologies discussed herein to access other naturally occurring and designed morphinans are currently under investigation in our laboratory.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (No. 2013R1A1A2057837; and 2014R1A5A1011165, Center for New Directions in Organic Synthesis), and Novartis. Kenny Park was supported by the BK21Plus Program, Ministry of Education. We thank Hyeonuk Woo and Shohee Park for preliminary synthetic studies.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews and selected recent examples of the use of desymmetrization in total synthesis, see: (a) S. R. Magnuson, Tetrahedron, 1995, 51, 2167 CrossRef CAS; (b) M. C. Willis, J. Chem. Soc., Perkin Trans. 1, 1999, 1765 RSC; (c) M. Wang, M. Feng, B. Tang and X. Jiang, Tetrahedron Lett., 2014, 55, 7147 CrossRef CAS.
- Y. Yoshii, H. Tokuyama and D. Y.-K. Chen, Angew. Chem., Int. Ed., 2017, 56, 12277 CrossRef CAS PubMed.
- For an extensive list of morphinan syntheses, see ESI‡.
- For a recent book and review chapter, see: (a) R. B. Herbert, H. Venter and S. Pos, Nat. Prod. Rep., 2000, 17, 317 RSC; (b) H. Nagase, Chemistry of Opioids; Top. Curr. Chem., Springer-Verlag, Berlin Heidelberg, 2011, vol. 299, pp. 1–312 CrossRef.
- J. M. Gulland and R. Robinson, Mem. Proc. – Manchester Lit. Philos. Soc., 1925, 69, 79 CAS.
- For a related Wittig reaction followed by chemoselective hydrogenation, see: J. C. C. Morales, A. G. Torres and E. Gonazlez-Zamora, Eur. J. Org. Chem., 2011, 3165 CrossRef.
- J. Duchek, G. Piercy, J. Gilmet and T. Hudlicky, Can. J. Chem., 2011, 89, 709 CrossRef CAS.
- F. Colobert, R. Des Mazery, G. Solladie and M. C. Carreno, Org. Lett., 2002, 4, 1723 CrossRef CAS PubMed.
- For a review, see: R. M. Moriaty and O. Prakash, Org. React., 2001, 57, 327 CrossRef.
- Heck reaction of substrate 8 under the conditions developed for substrate 5 only afforded recovered starting material (8). For an example of the modified conditions, see: T. N. Gowala and J. Pabba, Tetrahedron Lett., 2015, 56, 1801 CrossRef CAS.
- H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303 CrossRef CAS PubMed.
- For a review, see: X. J. Salom-Roig, F. Denes and P. Renaud, Synthesis, 2004, 1903 CAS.
- M. Inoue, T. Sato and M. Hirama, Angew. Chem., Int. Ed., 2006, 45, 4843 CrossRef CAS PubMed.
- A. Kimishima, H. Umihara, A. Mizoguchi, S. Yokoshima and T. Fukuyama, Org. Lett., 2014, 16, 6244 CrossRef CAS PubMed.
- For chemical, biological and clinical studies, see: (a) T. Heiskanen and E. Kalso, Pain, 1997, 73, 37 CrossRef CAS PubMed; (b) S. Mercadante and E. Arcuri, Cancer Treat. Rev., 1998, 24, 425 CrossRef CAS PubMed; (c) C. P. Watson and N. Babul, Neurology, 1998, 50, 1837 CrossRef CAS PubMed; (d) B. Silvestri, E. Bandieri, S. Del Prete, G. P. Ianniello, G. Micheletto, M. Dambrosio, G. Sabbatini, L. Endrizzi, A. Marra, E. Aitini, A. Calorio, F. Garetto, G. Nastasi, F. Piantedosi, V. Sidoti and P. Spanu, Clin. Drug Invest., 2008, 28, 399 CrossRef CAS PubMed.
- R. Imbos, A. J. Minnard and B. L. Feringa, J. Chem. Soc., Dalton Trans., 2003, 2017 RSC.
- For a review of desymmetrization methods of cyclohexadienones, see: K. A. Kalstabakken and A. M. Harned, Tetrahedron, 2014, 70, 9571 CrossRef CAS PubMed.
- Y. Han, S. Breitler, S.-L. Zheng and E. J. Corey, Org. Lett., 2016, 18, 6172 CrossRef CAS PubMed.
- D. M. Rubush, M. A. Morges, B. J. Rose, D. H. Thamm and T. Rovis, J. Am. Chem. Soc., 2012, 134, 13554 CrossRef CAS PubMed . Absolute stereochemical assignments of optically active intermediates depicted in Scheme 4c were inferred based on the work of Rovis and co-workers.
- For a related process, see: C. W. Jefford, J.-C. Rossier and J. Boukouvalas, Chem. Commun., 1987, 1593 RSC . Furthermore, a E1cb-type mechanism cannot be excluded to account for this transformation.
Footnotes |
| † This paper is dedicated to Professor Chi-Huey Wong on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc07667g |
| This journal is © The Royal Society of Chemistry 2018 |