
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nitrosonium ion catalysis: aerobic, metal-free cross-dehydrogenative carbon–heteroatom bond formation†
Luis
Bering
 ab,
Laura
D’Ottavio
ac,
Giedre
Sirvinskaite
a and
Andrey P.
Antonchick
*ab
ab,
Laura
D’Ottavio
ac,
Giedre
Sirvinskaite
a and
Andrey P.
Antonchick
*ab
aDepartment of Chemical Biology, Max-Planck-Institute of Molecular Physiology, Otto-Hahn-Straße 11, 44227 Dortmund, Germany. E-mail: Andrey.Antonchick@mpi-dortmund.mpg.de
bFaculty of Chemistry and Chemical Biology, TU Dortmund University, Otto-Hahn-Straße 4a, 44227 Dortmund, Germany
cUniversity of Bologna, Department of Pharmacy and Biotechnology, Via Belmeloro 6, 40126 Bologna, Italy
First published on 2nd November 2018
Abstract
Catalytic cross-dehydrogenative coupling of heteroarenes with thiophenols and phenothiazines has been developed under mild and environmentally benign reaction conditions. For the first time, NOx+ was applied for catalytic C–S and C–N bond formation. A comprehensive scope for the C–H/S–H and C–H/N–H cross-dehydrogenative coupling was demonstrated with >60 examples. The sustainable cross-coupling conditions utilize ambient oxygen as the terminal oxidant, while water is the sole by-product.
The formation of carbon–heteroatom bonds is fundamental for the synthesis of natural products, pharmaceuticals and materials science.1 To overcome the requirement for pre-functionalized starting materials, cross-dehydrogenative coupling (CDC) has emerged as a highly efficient strategy.2 Transition-metal-catalyzed C–S and C–N bond formation has been widely reported.3 Cost, toxicity and oxygen sensitivity of catalysts limit the general applicability.4 Consequently, metal-free synthesis has gained increasing interest.5 Different metal-free approaches for the C–H/S–H CDC have been reported.6 Additionally, the unique dehydrogenative amination with phenothiazines has received significant attention.7 High temperatures, excess of oxidants and harmful solvents are common limitations.
Nitronium and nitrosonium salts are inexpensive, stable and non-toxic single-electron oxidants.8 Radner's group reported the synthesis of biaryls using NOBF4 as catalyst (Fig. 1a).9 Ambient oxygen was identified as the terminal oxidant and water as the by-product.10 Later, Wang's group reported the catalytic intramolecular C–C bond formation (Fig. 1b).11 Under acidic reaction conditions, NO+ is generated in situ from NaNO2. The oxidative coupling of phenols is well studied.12 Recently, our group reported the NO+ catalyzed coupling for the construction of C–C bonds.13 Despite the impact of NO+ as catalyst for oxidative C–C bond formation, the application in carbon–heteroatom bond formation via C–H bond functionalization is unprecedented. Herein, we demonstrate the first NOx+ catalyzed C–H/S–H and C–H/N–H CDC under mild and environmentally benign reaction conditions (Fig. 1d).
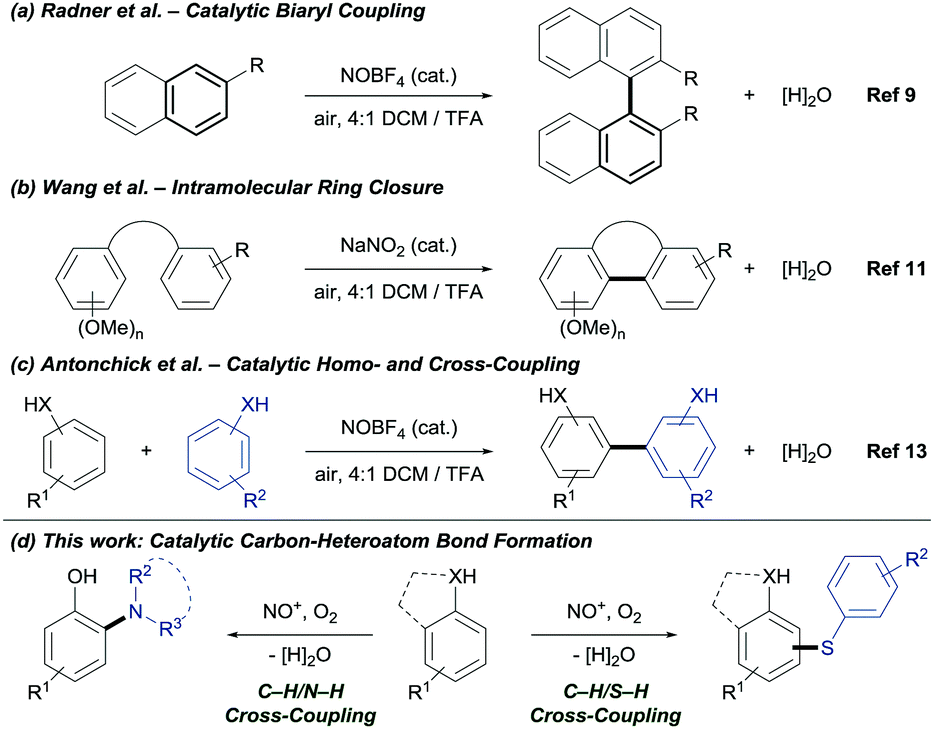 | ||
| Fig. 1 Prior work on the oxidative carbon–carbon bond formation via C–H bond functionalization and newly developed transformation catalyzed by NOx+. | ||
Nitrosonium salts are capable to convert thiols to disulfides.14 Oxidation of thiols proceeds via transient S-nitrosation and recombination of S-centred radicals. Due to the low bond dissociation energy (BDE) of phenols and thiophenols, the possibility for a radical–radical recombination reaction of phenoxy and sulfur radicals was hypothesized.15 A multi parameter optimization for the cross-coupling of p-cresol (1a) and 4-chlorothiophenol (2a) was performed (Table S1, ESI†). To our delight, 3a was isolated in excellent yield, by using NO2BF4 as the catalyst. Hexafluoroisopropanol (HFIP) was identified as the best solvent, due to its acidic character and the unique ability to stabilize radical intermediates.16
Initially, the scope for the cross-coupling of phenols and thiophenols was studied (Scheme 1). The reaction was scaled to 1 mmol, which did not alter the outcome of the reaction. Functional groups at the para-position of phenols were well tolerated (3b–3d). 2,4-Substituted phenols yielded products 3e–3h in good to excellent yields, covering electron-rich and sterically demanding functional groups. Product 3g was isolated in quantitative yield and product 3l was synthesized with high para-selectivity. Electron-rich product 3j revealed selectivity for the meta-position of phenol. The same outcome was observed for product 3k by blocking the ortho- and para-positions. Dearomatization and subsequent 1,4-addition appeared to be an alternative pathway. 3l was isolated in moderate yield, using a 3,5-subsituted phenol. Polysubstituted phenols allowed the isolation of products 3m–o in 76–98% yields. Naphthol derivatives were compatible, affording products 3p, 3q. Next, substituted thiophenols were tested. Different functional groups on the para-position were well tolerated (3s–v). Alkyl or chloro substituents at the ortho- and meta-position afforded products 3w–3y in good yields. Double thioarylation was not observed under the developed conditions. Alkyl and benzyl thiols did not yield the desired products either. To stress the utility of the obtained products, 3a was transformed into benzothiophene 3z by applying a dual C–H bond activation strategy.17
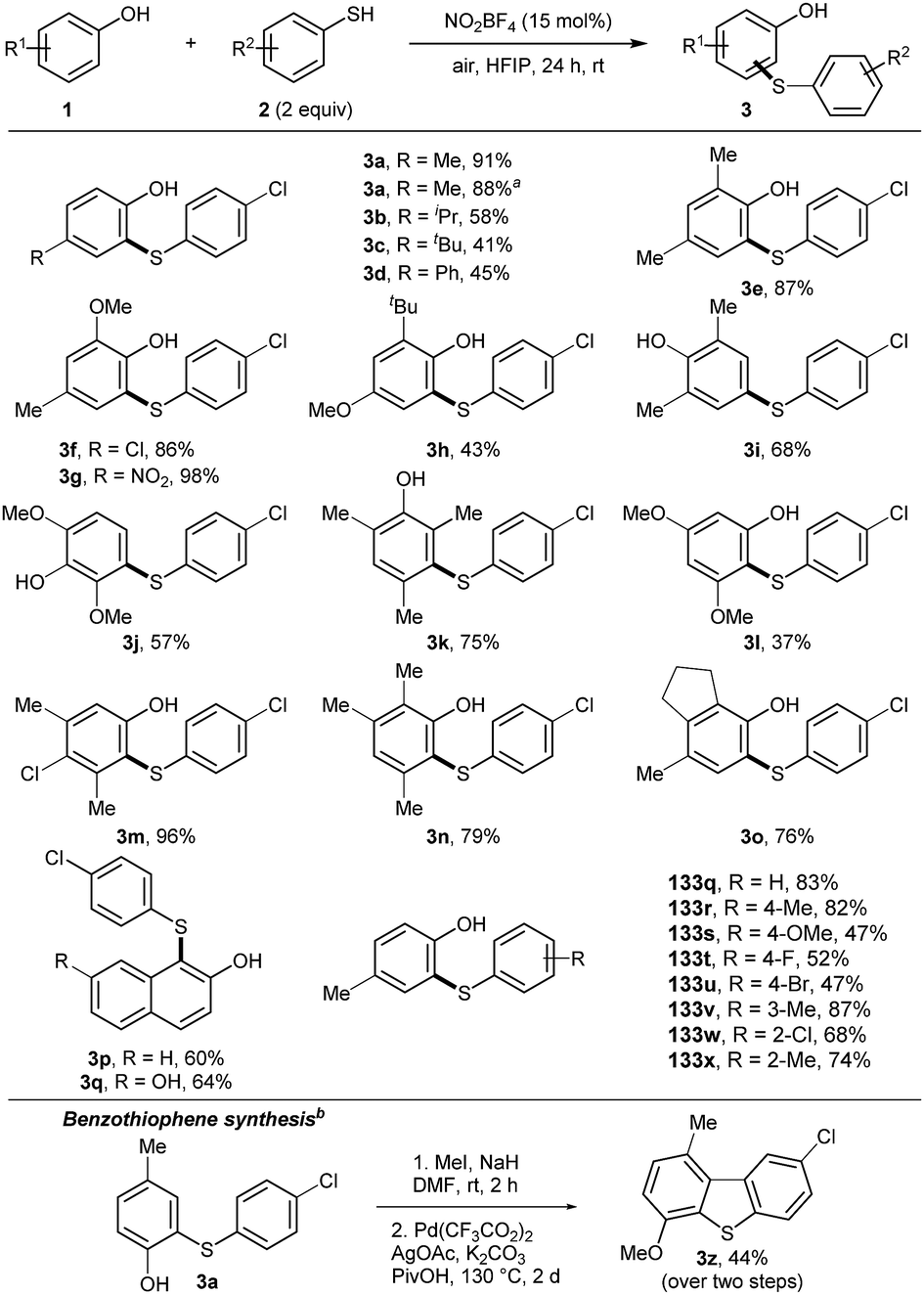 | ||
Scheme 1 Scope with respect to phenols (1) and thiophenols (2). Reaction conditions: 1 (0.1 mmol, 1 equiv.), 2 (2 equiv.), HFIP (0.05 M), at room temperature under air atmosphere. Yields are given for isolated products after column chromatography. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Reaction carried out with 1 mmol of phenol 1a. b1. 3a (0.3 mmol, 1 equiv.), MeI (1.5 equiv.), NaH (1.2 equiv.) in DMF (0.1 M) at room temperature; 2. (CF3CO2)2Pd (20 mol%), AgOAc (5 equiv.), K2CO3 (1.5 equiv.) in PivOH (0.3 M) at 130 °C for 2 d. Reaction carried out with 1 mmol of phenol 1a. b1. 3a (0.3 mmol, 1 equiv.), MeI (1.5 equiv.), NaH (1.2 equiv.) in DMF (0.1 M) at room temperature; 2. (CF3CO2)2Pd (20 mol%), AgOAc (5 equiv.), K2CO3 (1.5 equiv.) in PivOH (0.3 M) at 130 °C for 2 d. | ||
Next, the thioarylation of indoles was studied (Scheme 2). Unprotected indoles gave better results than N-protected analogues. This result makes the reaction conditions more attractive for other applications. The cross-coupling of indole 4a and thiophenol 2a yielded 5a in 77% yield. Scaling the reaction to 1 mmol gave 5a unaffectedly. Functional groups with different electronic properties at the indole skeleton were well tolerated (5b–f). Further, thiophenols were decorated with functional groups at the para (5h–j), ortho (5k–l) and meta (5m) position. Product 5n was isolated in 51% yield bearing a napthyl moiety. Polysubstituted products 5o–5s were synthesized in good yields, covering combinations of electron-rich and electron-deficient functional groups. Next, substituted pyrroles were tested. Product 5t was isolated in 64% yield. 2-Substituted pyrrol yielded the double functionalized product 5v in good yield. To further stress the applicability, 5l was transformed into the indol-fused benzothiophenes 5v.18
 | ||
| Scheme 2 Scope with respect to indoles (4) and thiophenols (2). Reaction conditions: 4 (0.1 mmol, 1 equiv.), 2 (2 equiv.), HFIP (0.05 M), at room temperature under air atmosphere. Yields are given for isolated products after column chromatography. a Reaction carried out with 1 mmol of indol 4a. b5l (0.08 mmol, 1 equiv.), Pd(PPh3)2Cl2 (5 mol%), CsPiv (2 equiv.) in N,N-dimethylacetamide (0.1 M). | ||
Next, the time course of the cross-coupling reactions was analysed by GC-MS-FID (Fig. 2). Interestingly, thiophenol 2a was fully converted to disulfide 6a, prior to the coupling step with phenol 1a (Fig. 2A). In contrast, indole 4a and thiophenol 2a underwent synchronous cross-coupling without initial formation of 6a (Fig. 2B). Conducting the coupling reaction with disulfide 6a confirmed the discrete recombination selectivities (Schemes S2 and S5, ESI†). Consequently, disulfide 6a is an intermediate for the coupling with phenol, but indoles undergo direct recombination with thiophenols. Further control experiments were conducted (ESI† for the details). No product was formed in the presence of radical trap butylated hydroxytoluene (BHT) and the product formation was inhibited under inert gas atmosphere (Schemes S2 and S4, ESI†). Ambient oxygen was crucial to maintain the catalytic activity. Initiation of the coupling reaction through oxidation of weak heteroatom–hydrogen bonds was studied by methylating the starting materials. Drastically reduced conversion was observed, which excludes a direct single-electron-transfer (Schemes S2 and S4, ESI†). Thiophenol 2a was quantitatively oxidized to disulfide 6a in the presence of NOBF4 (Scheme S3, ESI†). Conversion to disulfide 6a was tied to the presence of air and did not take place in the presence of BHT (Scheme S3, ESI†).
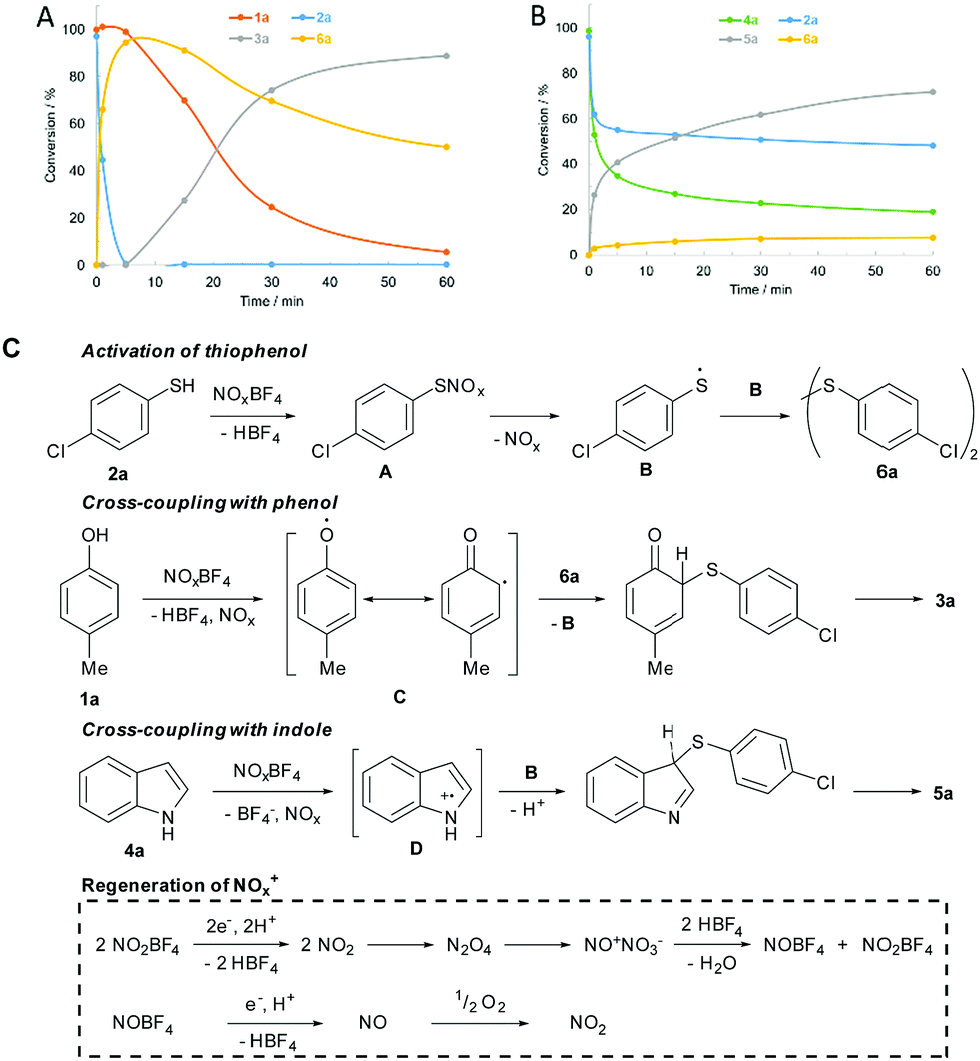 | ||
| Fig. 2 Reaction profiles and proposed mechanism. (A) Reaction profiles for the thioarylation of phenol 1a with thiophenol 2a. (B) Reaction profiles for the thioarylation of indole 4a with thiophenol 2a. (C) Reaction mechanism. | ||
Based on the control experiments, a mechanism for the cross-coupling of phenol 1a and indole 4a with thiophenol 2a was proposed (Fig. 2C). Initially, intermediate A is formed by S-nitrosylation of 2a. Homolytic cleavage releases radical B, which forms disulfide 6a by recombination with itself. Phenol 1a is oxidized to form a phenoxy radical. Delocalization of the phenoxy radical (C) and attack at the disulfide bond leads to release of radical B. Rearomatization furnishes product 3a. Oxidation of indole 4a gives intermediate D, which undergoes recombination with B. Oxidation of indole 4a and thiophenol 2a occurs synchronously, leading to the formation of 5a before disulfide 6a begins to form. Further, NO2BF4 oxidizes the starting materials to form NO2. NO2 dimerizes to form N2O4, which undergoes disproportionation to NONO3.11a Water is released upon protonation by HBF4 and nitrosonium and nitronium tetrafluoroborate are regenerated. NOBF4 is capable of oxidizing the substrates in the same way as the nitronium salt. Oxidation of substrates results in the formation of nitrogen monoxide, which has to be oxidized by ambient oxygen, in order to maintain the catalytic cycle.
Based on the revealed radical mechanism, we hypothesized that phenothiazines represent suitable substrates for the radical recombination with phenols, due to the low N–H BDE and the unique ability to stabilize radical intermediates.19 Multi parameter optimization was performed (Table S2, ESI†). Sustainable C–H bond amination of phenols with phenothiazines was achieved using NaNO2 as catalyst, omitting halogenated reagents and solvents. The developed conditions overcome environmental issues of known methods.7
Next, the scope of the catalytic C–H/N–H cross-dehydrogenative coupling was studied (Scheme 3). Initially, the cross-coupling of 4-methoxyphenol (7a) and different phenothiazines was performed. Products 9a–d were isolated in good yields. Different 4-substituted phenols yielded the desired cross-coupling products in moderate to excellent yields (9e–j). Cross-coupling with differently substituted phenothiazines was achieved for several phenols. Product 9k was isolated in good yield as single regioisomer. Polyfunctionalized products 9l–q were isolated in moderate to good yields. Phenoxazine proved to be compatible for the cross-coupling reaction (9r). 2-Naphthol yielded the desired product 9s in 73% yield. Finally, the cross-coupling of 2-phenylindole and phenoxazine was successfully performed. However, synthesis of 9t worked superior if NOBF4 was used as catalyst. The underlying mechanism for the aerobic C–H bond amination proceeds analogously as described before via direct radical–radical recombination under aerobic conditions (Scheme S8, ESI†).
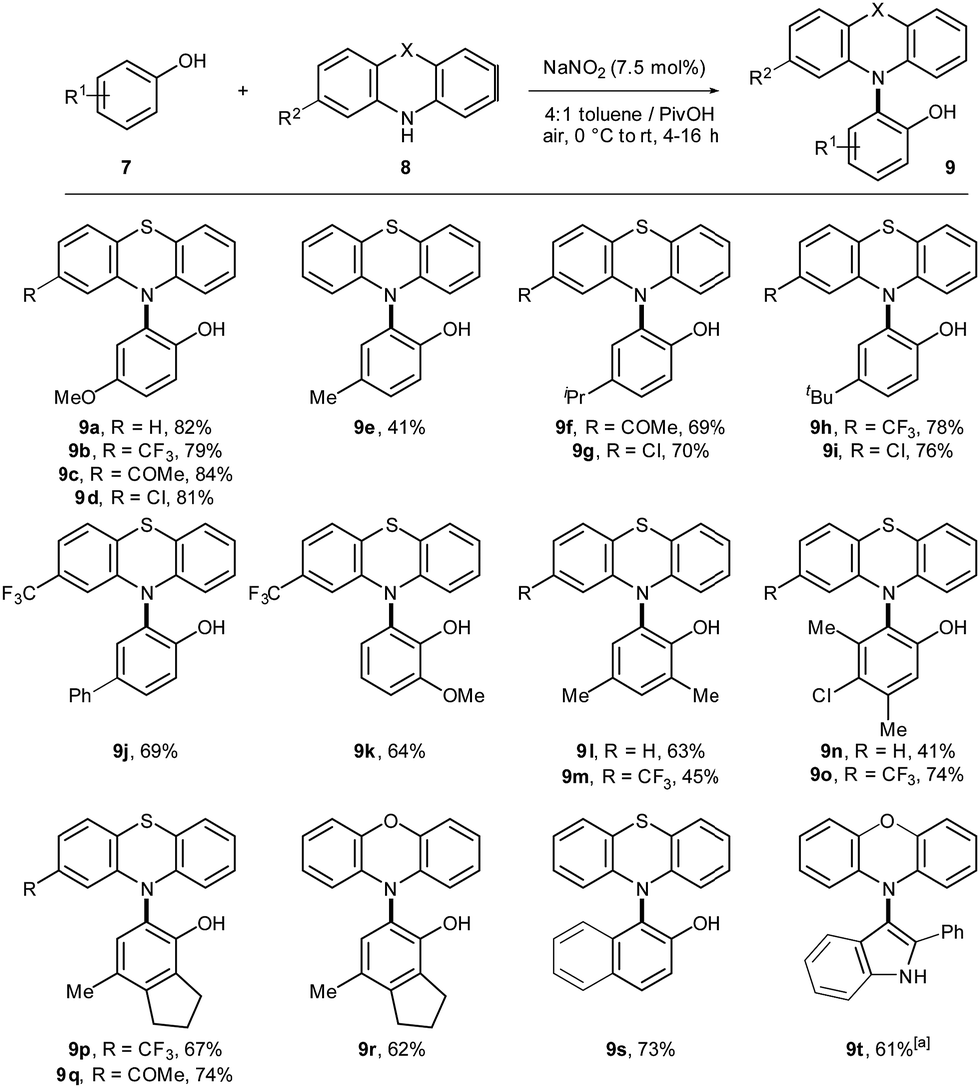 | ||
Scheme 3 Scope for the cross-dehydrogenative C–H bond amination. Reaction conditions: 8 (0.2 mmol, 1 equiv.), 9 (3 equiv.), 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene/PivOH (0.05 M), 0 °C to rt under air atmosphere. Yields are given for isolated products after column chromatography. aNOBF4 (0.02 mmol, 10 mol%) was used as catalyst. :1 toluene/PivOH (0.05 M), 0 °C to rt under air atmosphere. Yields are given for isolated products after column chromatography. aNOBF4 (0.02 mmol, 10 mol%) was used as catalyst. | ||
In summary, we have reported the first application of NOx+ as efficient and environmentally friendly catalyst for carbon–heteroatom bond formation. The operationally simple and sustainable protocol enables the C–H/S–H and C–H/N–H CDC. Ambient oxygen serves as stoichiometric oxidant and water is generated as by-product. A broad scope was demonstrated in good yields and regioselectivities. The reported methodology offers mild reaction conditions and does not require an excess of reagents or any specialized equipment.
A. P. A. acknowledges the support of the DFG (AN 1064/4-1) and the Boehringer Ingelheim Foundation (Plus 3). L. B. is supported by the Verband der Chemischen Industrie e.V. We gratefully acknowledge Ms R. Perinbarajah for assistance. Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS PubMed; (b) A. K. Yudin and J. F. Hartwig, Catalyzed Carbon-Heteroatom Bond Formation, Wiley-VCH, Weinheim, 2010 CrossRef.
- (a) S. A. Girard, T. Knauber and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS PubMed; (b) J. F. Hartwig and M. A. Larsen, ACS Cent. Sci., 2016, 2, 281 CrossRef CAS PubMed.
- (a) I. P. Beletskaya and V. P. Ananikov, Chem. Rev., 2011, 111, 1596 CrossRef CAS PubMed; (b) J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283 RSC; (c) L. Chin-Fa, L. Yi-Chen and B. S. Singh, Chem. – Asian J., 2014, 9, 706 CrossRef PubMed; (d) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901 RSC; (e) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed.
- (a) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed; (b) R. A. D. Arancon, C. Sze Ki Lin, C. Vargas and R. Luque, Org. Biomol. Chem., 2014, 12, 10 RSC; (c) V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957 RSC.
- (a) R. Samanta, K. Matcha and A. P. Antonchick, Eur. J. Org. Chem., 2013, 5769 CrossRef CAS; (b) R. Narayan, K. Matcha and A. P. Antonchick, Chem. – Eur. J., 2015, 21, 14678 CrossRef CAS PubMed; (c) L. Bering, S. Manna and A. P. Antonchick, Chem. – Eur. J., 2017, 23, 10936 CrossRef CAS PubMed.
- (a) Y. Liu, Y. Zhang, C. Hu, J.-P. Wan and C. Wen, RSC Adv., 2014, 4, 35528 RSC; (b) S. K. R. Parumala and R. K. Peddinti, Green Chem., 2015, 17, 4068 RSC; (c) Z. Huang, D. Zhang, X. Qi, Z. Yan, M. Wang, H. Yan and A. Lei, Org. Lett., 2016, 18, 2351 CrossRef CAS PubMed; (d) S. Song, Y. Zhang, A. Yeerlan, B. Zhu, J. Liu and N. Jiao, Angew. Chem., Int. Ed., 2017, 56, 2487 CrossRef CAS PubMed; (e) L. T. Silva, J. B. Azeredo, S. Saba, J. Rafique, A. J. Bortoluzzi and A. L. Braga, Eur. J. Org. Chem., 2017, 4740 CrossRef CAS; (f) R. Rahaman, S. Das and P. Barman, Green Chem., 2018, 20, 141 RSC; (g) H.-H. Wang, T. Shi, W.-W. Gao, Y.-Q. Wang, J.-F. Li, Y. Jiang, Y. S. Hou, C. Chen, X. Peng and Z. Wang, Chem. – Asian J., 2017, 12, 2675 CrossRef CAS PubMed; (h) P. Wang, S. Tang, P. Huang and A. Lei, Angew. Chem., Int. Ed., 2017, 56, 3009 CrossRef CAS PubMed; (i) R. Ohkado, T. Ishikawa and H. Iida, Green Chem., 2018, 20, 984 RSC; (j) H. Iida, R. Demizu and R. Ohkado, J. Org. Chem., 2018, 83, 12291 CrossRef CAS PubMed.
- (a) M.-L. Louillat-Habermeyer, R. Jin and F. W. Patureau, Angew. Chem., Int. Ed., 2015, 54, 4102 CrossRef CAS PubMed; (b) R. Jin and F. W. Patureau, Org. Lett., 2016, 18, 4491 CrossRef CAS PubMed; (c) Y. Zhao, B. Huang, C. Yang and W. Xia, Org. Lett., 2016, 18, 3326 CrossRef CAS PubMed; (d) M. Goswami, A. Konkel, M. Rahimi, M.-L. Louillat-Habermeyer, H. Kelm, R. Jin, B. de Bruin and F. W. Patureau, Chem. – Eur. J., 2018, 24, 11936 CrossRef CAS PubMed; (e) R. Jin, C. L. Bub and F. W. Patureau, Org. Lett., 2018, 20, 2884 CrossRef CAS PubMed; (f) S. Tang, S. Wang, Y. Liu, H. Cong and A. Lei, Angew. Chem., Int. Ed., 2018, 57, 4737 CrossRef CAS PubMed.
- (a) K. Y. Lee, D. J. Kuchynka and J. K. Kochi, Inorg. Chem., 1990, 29, 4196 CrossRef CAS; (b) G. A. Olah, G. K. S. Prakash, Q. Wang and X.-Y. Li, Nitrosonium Tetrafluoroborate, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, New Jersey, 2001 Search PubMed; (c) G. I. Borodkin and V. G. Shubin, Russ. Chem. Rev., 2001, 70, 211 CrossRef CAS; (d) I. B. Gennady and G. S. Vyacheslav, Russ. Chem. Rev., 2017, 86, 18 CrossRef.
- F. Radner, J. Org. Chem., 1988, 53, 702 CrossRef CAS.
- (a) M. Tanaka, H. Nakashima, M. Fujiwara, H. Ando and Y. Souma, J. Org. Chem., 1996, 61, 788 CrossRef CAS PubMed; (b) M. Tanaka, E. Muro, H. Ando, Q. Xu, M. Fujiwara, Y. Souma and Y. Yamaguchi, J. Org. Chem., 2000, 65, 2972 CrossRef CAS PubMed.
- (a) B. Su, L. Li, Y. Hu, Y. Liu and Q. Wang, Adv. Synth. Catal., 2012, 354, 383 CrossRef CAS; (b) B. Su, M. Deng and Q. Wang, Org. Lett., 2013, 15, 1606 CrossRef CAS PubMed.
- (a) Y. E. Lee, T. Cao, C. Torruellas and M. C. Kozlowski, J. Am. Chem. Soc., 2014, 136, 6782 CrossRef CAS PubMed; (b) A. Libman, H. Shalit, Y. Vainer, S. Narute, S. Kozuch and D. Pappo, J. Am. Chem. Soc., 2015, 137, 11453 CrossRef CAS PubMed; (c) M. Koji, S. Kazuma, O. Takao, D. Toshifumi and K. Yasuyuki, Angew. Chem., Int. Ed., 2016, 55, 3652 CrossRef PubMed; (d) H. Shalit, A. Libman and D. Pappo, J. Am. Chem. Soc., 2017, 139, 13404 CrossRef CAS PubMed; (e) A. Wiebe, B. Riehl, S. Lips, R. Franke and S. R. Waldvogel, Sci. Adv., 2017, 3, eaao3920 CrossRef PubMed; (f) H. Kang, Y. E. Lee, P. V. G. Reddy, S. Dey, S. E. Allen, K. A. Niederer, P. Sung, K. Hewitt, C. Torruellas, M. R. Herling and M. C. Kozlowski, Org. Lett., 2017, 19, 5505 CrossRef CAS PubMed; (g) S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706 CrossRef CAS PubMed.
- (a) L. Bering, F. M. Paulussen and A. P. Antonchick, Org. Lett., 2018, 20, 1978 CrossRef CAS PubMed; (b) L. Bering, M. Vogt, F. M. Paulussen and A. P. Antonchick, Org. Lett., 2018, 20, 4077 CrossRef CAS PubMed.
- (a) T. Itoh, N. Tsutsumi and A. Ohsawa, Bioorg. Med. Chem. Lett., 1999, 9, 2161 CrossRef CAS PubMed; (b) A. S. Demir, I. A. Cigdem and A. S. Mahasneh, Tetrahedron, 1999, 55, 12399 CrossRef CAS; (c) N. Tsutsumi, T. Itoh and A. Ohsawa, Chem. Pharm. Bull., 2000, 48, 1524 CrossRef CAS PubMed; (d) J.-M. Lü, J. M. Wittbrodt, K. Wang, Z. Wen, H. B. Schlegel, P. G. Wang and J.-P. Cheng, J. Am. Chem. Soc., 2001, 123, 2903 CrossRef.
- (a) M. I. de Heer, H.-G. Korth and P. Mulder, J. Org. Chem., 1999, 64, 6969 CrossRef CAS; (b) Y.-D. Wu and D. K. W. Lai, J. Org. Chem., 1996, 61, 7904 CrossRef CAS PubMed.
- (a) L. Eberson, M. P. Hartshorn and O. Persson, J. Chem. Soc., Perkin Trans. 2, 1995, 1735, 10.1039/P29950001735; (b) M. Lucarini, V. Mugnaini, G. F. Pedulli and M. Guerra, J. Am. Chem. Soc., 2003, 125, 8318 CrossRef CAS PubMed; (c) J.-P. Bégué, D. Bonnet-Delpon and B. Crousse, Synlett, 2004, 18 Search PubMed.
- C. Rui, W. Zhiqing, L. Zhengkai, X. Haifeng and Z. Xiangge, Chem. – Eur. J., 2014, 20, 7258 CrossRef PubMed.
- P. Saravanan and P. Anbarasan, Org. Lett., 2014, 16, 848 CrossRef CAS PubMed.
- (a) M. Lucarini, P. Pedrielli, G. F. Pedulli, L. Valgimigli, D. Gigmes and P. Tordo, J. Am. Chem. Soc., 1999, 121, 11546 CrossRef CAS; (b) L. A. Farmer, E. A. Haidasz, M. Griesser and D. A. Pratt, J. Org. Chem., 2017, 82, 10523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08328b |
| This journal is © The Royal Society of Chemistry 2018 |