
Synthesis of poly(styrene-b-4-(tert-butyldimethylsiloxy)styrene) block copolymers and characterization of their self-assembled patterns†
Yoon Hyung
Hur‡
a,
Seung Won
Song‡
a,
Jimmy
Mays
b,
YongJoo
Kim
*c,
Beom-Goo
Kang
*b and
Yeon Sik
Jung
 *a
*a
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 302-701, Republic of Korea. E-mail: ysjung@kaist.ac.kr (Y.S. Jung)
bDepartment of Chemistry, University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: beomgook@princeton.edu (B.-G. Kang)
cKI for NanoCentury, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Yuseong-gu, Daejeon 302-701, Republic of Korea. E-mail: cjyjee@kaist.ac.kr (Y.J. Kim)
First published on 16th October 2017
Abstract
Directed self-assembly (DSA) of block copolymers (BCPs) is regarded as one of the alternative methods to traditional top-down lithography approaches due to its ability to form well-aligned nanostructures such as spheres, cylinders, and lamellae with controlled domain size. Despite extensive research activities in this field, the question of how the Flory–Huggins interaction parameter (χ) and polydispersity of chains affect self-assembled pattern formation remains. Here, we report the self-assembly behavior of poly(styrene-b-4-(tert-butyldimethylsiloxy)styrene) (PS-b-P4BDSS) BCP, which has an intermediate χ between those of poly(styrene-b-methyl methacrylate) (PS-b-PMMA) and poly(styrene-b-dimethylsiloxane) (PS-b-PDMS), which allows pattern formation of the BCP in a wide range of length scales induced by simple thermal annealing. We also demonstrate the effect of polydispersity on self-assembled pattern quality by comparing BCPs synthesized via anionic polymerization with those synthesized via reversible addition fragmentation chain transfer (RAFT) polymerization. The self-assembled pattern of BCPs with a narrow PDI (PDI < 1.05 via anionic polymerization) shows 38% lower line edge roughness than that of BCPs with a broad PDI (PDI > 1.15 via RAFT polymerization).
Design, System, ApplicationThe self-assembly behaviors and pattern quality of block copolymers are highly dependent on their material parameters. For example, the line edge roughness of directed self-assembly (DSA) patterns can be significantly improved by increasing the Flory–Huggins interaction parameter (χ) of block copolymers, while this approach can seriously slow down the kinetics of pattern alignment of DSA. In this study, we designed and synthesized poly(styrene-b-4-(tert-butyldimethylsiloxy)styrene) block copolymers with an intermediate χ and a controlled polydispersity index (PDI). To figure out the effect of the PDI on the pattern quality, two different synthetic routes – anionic polymerization and reversible addition fragmentation chain transfer (RAFT) polymerization – are used. This work shows how the pattern quality and pattern formation kinetics of the DSA process can be influenced by different synthetic methods and the χ parameter, respectively. |
Introduction
Directed self-assembly (DSA) of block copolymers (BCPs) is considered a promising route to form a variety of nanostructured patterns with high resolution, high throughput, and low cost.1–7 The self-assembly behavior of BCPs is governed by the segregation strength χN and the volume fraction f of each block, where χ is the Flory–Huggins interaction parameter and N is the degree of polymerization of BCPs. BCPs self-assemble into various morphologies such as spheres, cylinders, gyroids and lamellae as a function of f for χN above the order–disorder transition (ODT),8 and their domain size d is determined by d ∼ N2/3χ1/6. In this respect, we can decrease the domain size of the self-assembled pattern of BCPs simply by reducing the degree of polymerization N while maintaining χN above the ODT.Poly(styrene-b-methyl methacrylate) (PS-b-PMMA) is regarded as a promising material for DSA application because of its relatively higher etching selectivity and ability to form thermally self-assembled vertical lamellar patterns owing to a negligible difference of surface energies between two blocks.9 However, the low χ value of PS-b-PMMA prevents formation of sub-15 nm lamellar patterns because a relatively longer BCP chain length is required to obtain χN above the ODT (∼10.5) of the lamellar morphology.10 In addition, the relatively poor pattern quality of low-χ BCPs due to large line edge roughness (LER) and line width roughness (LWR)11 can affect the reliability of device performances.12,13 Therefore, BCPs with higher χ values have been extensively studied such as poly(styrene-b-ethylene oxide) (PS-b-PEO),14 poly(cyclohexylethylene-b-methyl methacrylate) (PCHE-b-PMMA),15 poly(lactide-b-trimethylsilylstyrene) (PLA-b-PTMSS),16 poly(styrene-b-dimethylsiloxane) (PS-b-PDMS),17–19 poly(methyl methacrylate-b-dimethylsiloxane) (PMMA-b-PDMS),20 poly(lactide-b-dimethylsiloxane) (PLA-b-PDMS),21 poly(2vinylpyridine-b-dimethylsiloxane) (P2VP-b-PDMS), and poly((styrene-r-vinylnaphthalene)-b-methyl methacrylate) (PSVN-b-PMMA).22 Among high-χ BCPs, Si-containing BCPs have been widely studied due to their extremely high etch selectivity from the formation of robust silica films under an oxygen plasma process.23,24 However, for the rapid and reliable self-assembly of high-χ BCPs, a solvent vapor annealing process is required to increase the chain mobility to overcome a large defect kinetic barrier and generate well-ordered patterns of high-χ BCPs,17–19,25,29 which results in process complexity. Although some research groups have shown that the blending of BCPs and homopolymers can substantially increase the self-assembly kinetics at the expense of pattern quality compared to pure BCPs, it would be desirable to develop a BCP with an intermediate χ, which can be long-range ordered by thermal annealing. Also, some BCPs with extremely high χ values demonstrated a good pattern quality despite having a high polydispersity index (PDI), which is the molecular weight distribution (Mw/Mn) of polymers.22,26,27 Although the PDI may significantly affect the LER and LWR of self-assembled BCP patterns for low-χ BCPs from previous theoretical and experimental studies,28–32 it is not certain whether an intermediate or high χ can compromise the effect of some degree of chain size distribution. Therefore, it would be worthwhile to explore other Si-containing BCPs with reduced χ and controlled PDI and with the ability to be assembled via thermal annealing to form sub-15 nm patterns having high etching selectivity and reasonable pattern quality.
Here, we introduce Si-containing BCPs consisting of a polystyrene (PS) block and a poly(4-(tert-butyldimethylsiloxy)styrene) (P4BDSS) block with an intermediate χ parameter. Such an intermediate χ enables simple thermal annealing to promote the pattern formation in the width range approximately from 16 to 55 nm. Also, different synthesis routes – anionic polymerization and reversible addition fragmentation chain transfer (RAFT) polymerization – result in a relatively low (<1.05) and high (>1.15) PDI, respectively, which provides an opportunity to investigate the effect of the PDI on pattern quality.
Experimental
Materials
Styrene (Aldrich, ≥99%) and 4-(tert-butyldimethylsiloxy)styrene (Gelest) were distilled over CaH2in vacuo and subsequently distilled from di-n-butylmagnesium on a vacuum line. Commercially available sec-butyllithium (s-BuLi, 1.4 M in cyclohexane, Aldrich) was used as received. The monomers and initiators were diluted with THF and n-hexane, respectively, and stored at −30 °C in ampules equipped with break-seals.Measurements
Molecular weight and molecular weight distribution were determined using a Polymer Laboratories GPC-120 size exclusion chromatograph (SEC) equipped with a Precision Detector PD2040 (two angle static light scattering detector), Precision Detector PD2000DLS (dynamic light scattering detector), Viscotek 220 differential viscometer, and Polymer Laboratories refractometer. THF was used as the mobile phase at a flow rate of 1.0 mL min−1 at 40 °C. The installed column set consists of the following four columns: Polymer Laboratories PLgel; 7.5 × 300 mm; 10 μm; 500, 1 × 104, 1 × 106, and 1 × 107 Å. The 1H NMR spectrum of the block copolymer was obtained using CDCl3 as a solvent at 25 °C (JEOL JNM-ECX400). The chemical shifts are referenced to tetramethylsilane (TMS) at 0 ppm. A field emission scanning electron microscope (FE-SEM: Hitachi S-4800) with an accelerating voltage of 12 kV and a working distance of 7.0 mm was used to confirm self-assembled BCP patterns. Commercial image analysis software (SuMMIT) was used for quantitative analysis of self-assembled BCP patterns: the critical dimension (CD), pitch, line width roughness (LWR), and line edge roughness (LER). Small-angle X-ray scattering (SAXS) measurement was performed at Pohang Accelerator Laboratory (PAL). The operating conditions were chosen to be a wavelength of 1.18 Å and a sample-to-detector distance (SDD) of 2.51 m. Grazing-incidence small-angle X-ray scattering (GISAXS) analysis was also carried out at PAL. The operating conditions were chosen to be a wavelength of 1.18 Å and a SDD of 3.98 m. To probe the surface and internal structures of BCP films, the incidence angle (αi) was set from 0.08° to 0.16°. 2D patterns were recorded by using a Mar-CCD detector (Mar-165) positioned at the end of a vacuum guide tube.Synthesis of PS-b-P4BDSS via anionic polymerization
The polymerization of styrene (6.20 mmol) was carried out using s-BuLi (0.015 mmol) in THF at −78 °C for 0.5 h in an all-glass apparatus equipped with break-seals under high vacuum. After a portion of the living PS was sampled for SEC characterization, 4-(tert-butyldimethylsiloxy)styrene (1.27 mmol) was added to the living PS solution, and the polymerization was continued for 0.5 h. The polymerization was terminated with methanol and the solution was poured into a large excess of methanol to obtain PS and PS-b-P4BDSS. The block copolymer was characterized by SEC and 1H NMR. PS-b-P4BDSS (Mn(obsd) = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1, Mw/Mn = 1.01 (A-25)). 1H NMR (400 MHz, CDCl3): δ = 0.05–0.21 (methyl group of P4BDSS), 0.91–1.03 (tert-butyl group of P4BDSS), 1.17–2.24 (main chain), 6.18–7.24 (phenyl group of PS and P4BDSS).
300 g mol−1, Mw/Mn = 1.01 (A-25)). 1H NMR (400 MHz, CDCl3): δ = 0.05–0.21 (methyl group of P4BDSS), 0.91–1.03 (tert-butyl group of P4BDSS), 1.17–2.24 (main chain), 6.18–7.24 (phenyl group of PS and P4BDSS).
Synthesis of PS-b-P4BDSS via RAFT polymerization
The polymerization of styrene was carried out using a RAFT CTA agent, 2-{[(butylsulfanyl)carbonothioyl]sulfanyl}propanoic acid (0.0210 mmol), distilled styrene (1.64 mmol), AIBN (0.0105 mmol), and toluene (2 mL). The reactants were put in a 3-necked flask purged with argon gas and were degassed by freeze-pump-thaw cycles. Subsequently, the solution was stirred at 83 °C for 12 h and the polymerization was terminated by the addition of tetrahydrofuran (THF). The polymer solution was precipitated, put in a 3-necked flask and purged with argon gas. Subsequently, the solution was degassed by three freeze-pump-thaw cycles and then the solution was stirred at 83 °C for 12 h. After 12 h, the polymerization was terminated by the addition of THF and the polymer solution was precipitated in hexane (350 mL). The block copolymer was characterized by SEC and 1H NMR. PS-b-P4BDSS (Mn(obsd) = 25700 g mol−1, Mw/Mn = 1.21 (R-25)). 1H NMR (400 MHz, CDCl3): δ = 0.05–0.21 (methyl group of P4BDSS), 0.91–1.03 (tert-butyl group of P4BDSS), 1.17–2.24 (main chain), 6.18–7.24 (phenyl group of PS and P4BDSS).
Thin film preparation and characterization
Silicon wafers were washed via three steps in acetone, isopropanol, and DI water for 30 min, respectively. Thin films of each BCP were generated by spin-casting from 0.5 wt% or 1 wt% toluene solution at 4000 rpm for 30 s onto a wafer (1 × 1 inch2). BCP films were self-assembled by the thermal annealing process at 200 °C for 1 h under vacuum. Reactive ion etching (RIE) was carried out with CF4 plasma (50 W, 30 sccm, 15 mTorr) and O2 plasma (60 W, 30 sccm, 15 mTorr) consecutively.Results and discussion
Synthesis of block copolymers
First, BCPs were synthesized by anionic polymerization. The block copolymerization was carried out by sequential addition of styrene as the first monomer and 4-(tert-butyldimethylsiloxy)styrene as the second monomer using s-BuLi in tetrahydrofuran (THF) at −78 °C, as shown in Scheme 1a. After 4-(tert-butyldimethylsiloxy)styrene was added to the living PS solution, the distinct deep orange color of living PS-b-P4BDSS was maintained during polymerization, indicating the occurrence of a rapid crossover reaction from the active chain end of PS to 4-(tert-butyldimethylsiloxy)styrene. It was confirmed from the 1H NMR spectrum that the sequential block copolymerization proceeded exclusively. As shown in Fig. S1,† the characteristic peaks of each block were clearly observed. In addition, Table S1† shows that well-defined PS-b-P4BDSS samples with controlled Mn (12500–106600 g mol−1) and narrow Mw/Mn (1.01–1.10) were synthesized. For example, the observed Mn (54200 g mol−1) of the synthesized block copolymer was in good agreement with the calculated one (53200 g mol−1) based on the monomer-to-initiator ratio (Table 1, A-54), and the narrow SEC curve of PS-b-P4BDSS (Mw/Mn = 1.05) completely shifted from the starting PS toward the higher Mn region (see Scheme 1a). These results demonstrate that well-controlled PS-b-P4BDSS can be synthesized under the polymerization conditions employed in this work.
 | ||
| Scheme 1 Synthetic scheme and SEC results of BCPs synthesized via (a) anionic polymerization and (b) RAFT polymerization. BCPs synthesized by anionic polymerization had a relatively narrow molecular weight distribution compared to BCPs synthesized by RAFT polymerization. | ||
| Block copolymer (homopolymer) | |||||
|---|---|---|---|---|---|
| Samplea | M n × 10−3 | M w/Mnc | f B | Domain sizee | Morphologyf |
| obsdb | % | nm | |||
| a Sample name refers to the components, where A = anionic polymerized block copolymer, R = RAFT polymerized block copolymer. b M n of the block copolymer was determined by using Mn of poly(styrene) and the molar ratio of each block estimated by 1H NMR. c M w/Mn was determined by using SEC calibration with poly(styrene) standards in a THF solution containing 2% triethylamine as the eluent at 40 °C. d Volume fraction of poly(4-(tert-butyldimethylsiloxy)styrene). e Domain size of BCPs was measured by bulk SAXS. f The morphology of BCPs was confirmed by bulk SAXS, where DIS = disorder, CYL = cylinder, LAM = lamellae, and HPL = hexagonally perforated lamellae. | |||||
| A-13 | 12.5 (7.5) | 1.01 (1.02) | 36.4 | — | DIS |
| A-54 | 54.2 (37.1) | 1.05 (1.06) | 28.4 | 38.3 | CYL |
| A-25 | 25.3 (16.3) | 1.01 (1.05) | 32.2 | 16.4 | CYL |
| R-55 | 55.1 (36.8) | 1.14 (1.21) | 29.0 | 39.5 | CYL |
| R-54 | 54.3 (36.8) | 1.32 (1.21) | 28.8 | 39.2 | CYL |
| R-25 | 25.2 (16.2) | 1.15 (1.17) | 32.7 | 16.5 | CYL |
| R-26 | 25.7 (16.2) | 1.21 (1.17) | 33.5 | 16.8 | CYL |
To compare the self-assembled pattern quality of the same BCPs with different PDIs, BCPs were synthesized by RAFT polymerization as shown in Scheme 1b. The polymerization of styrene was carried out using a RAFT CTA agent (2-{[(butylsulfanyl)carbonothioyl]sulfanyl}propanoic acid), distilled styrene, AIBN, and toluene. The reactants were put in a 3-necked flask, purged with argon gas and degassed three times by freeze-pump-thaw cycles. Subsequently, the solution was stirred at 83 °C for 12 h and the polymerization was terminated by the addition of THF. The polymer solution was precipitated in hexane. PS-b-P4BDSS was polymerized by using PS as the macro-RAFT CTA. The PS macro-RAFT CTA agent, distilled 4-(tert-butyldimethylsiloxy)styrene, AIBN, and toluene were put in a 3-necked flask purged with argon gas. Subsequently, the solution was degassed by three freeze-pump-thaw cycles and then the solution was stirred at 83 °C for 12 h. After 12 h, the polymerization was terminated by the addition of THF and the polymer solution was precipitated in hexane. Table S2† shows that PS-b-P4BDSS with controlled Mn (25200–55100 g mol−1) and Mw/Mn (1.14–1.32) was synthesized by RAFT polymerization. For example, the Mn (36800 g mol−1) of macro-RAFT CTA completely shifted toward the higher Mn (54300 g mol−1) of PS-b-P4BDSS (Table 1, R-54) as shown in Scheme 1b. The Mw/Mn of PS-b-P4BDSS was controlled by the termination rate.
Calculation of the Flory–Huggins interaction parameter of block copolymers
The anionic polymerized PS-b-P4BDSS was characterized by small-angle X-ray scattering (SAXS) measurement after thermal annealing at 200 °C for 24 h. Typically, the domain size of the BCP d is defined as d = 2π/q*, where q* is the first-order peak in Bragg peaks. The SAXS results of PS-b-P4BDSS are described in Fig. 1. The weaker and broader SAXS peaks of A-13 (Mn: 12500 g mol−1, Mw/Mn: 1.01) compared to other PS-b-P4BDSS peaks show the disordered structure of A-13 at 200 °C. However, A-54 (Mn: 54.2 kg mol−1, Mw/Mn: 1.05) indicated clear Bragg peaks of 1q*, √3q*, 2q*, √7q*, and 3q* with q* = 0.1369 nm−1. The calculated value of d was 38.3 nm with a well-ordered cylindrical morphology. The d of other BCPs was calculated by the same procedure and the results are summarized in Tables S1 and S2.† The BCPs were able to generate different morphologies with superior ordering and diversity in domain size from 16.4 to 54.8 nm.
 | ||
| Fig. 1 SAXS profiles of bulk BCPs synthesized by anionic polymerization. The detailed information on BCPs is summarized in Table S1.† The diffraction peaks of A-54 and A-25 at 1q*, √3q*, 2q* and √7q* indicate a cylindrical morphology. The diffraction peaks of A-33, A-117 and A-30 at 1q*, 2q*, 3q* and 4q* indicate a lamellar morphology. The broad peak of A-27 shows a mixed phase of at least two distinct morphologies. | ||
To investigate the χ value of PS-b-P4BDSS, the SAXS of A-30 (Mn: 29800 g mol−1, Mw/Mn: 1.03) was measured at various temperatures, as shown in Fig. 2a. The intensity of the SAXS profiles significantly decreased at temperatures between 250 °C and 255 °C and this discontinuous change exhibited an order–disorder transition temperature (TODT) of PS-b-P4BDSS.20,33Fig. 2c shows the change in the SAXS profiles of A-13 as a function of temperature. The continuous profiles with broader peak width indicate that A-13 is in a disordered state in the temperature range from 25 °C to 255 °C. To quantitatively determine the TODT of PS-b-P4BDSS, first-order peaks at various temperatures were investigated in terms of the maximum intensity (Im) and the half-width at half-maximum (σq). Fig. 2b and d are the Im−1 and σq2 plots as a function of T−1 for A-30 and A-13. A remarkable discontinuity shows that the TODT of A-30 is between 250 °C and 255 °C. However, the continuous Im−1 and σq2 curve of A-13 shows that the TODT of A-13 stays below 25 °C.
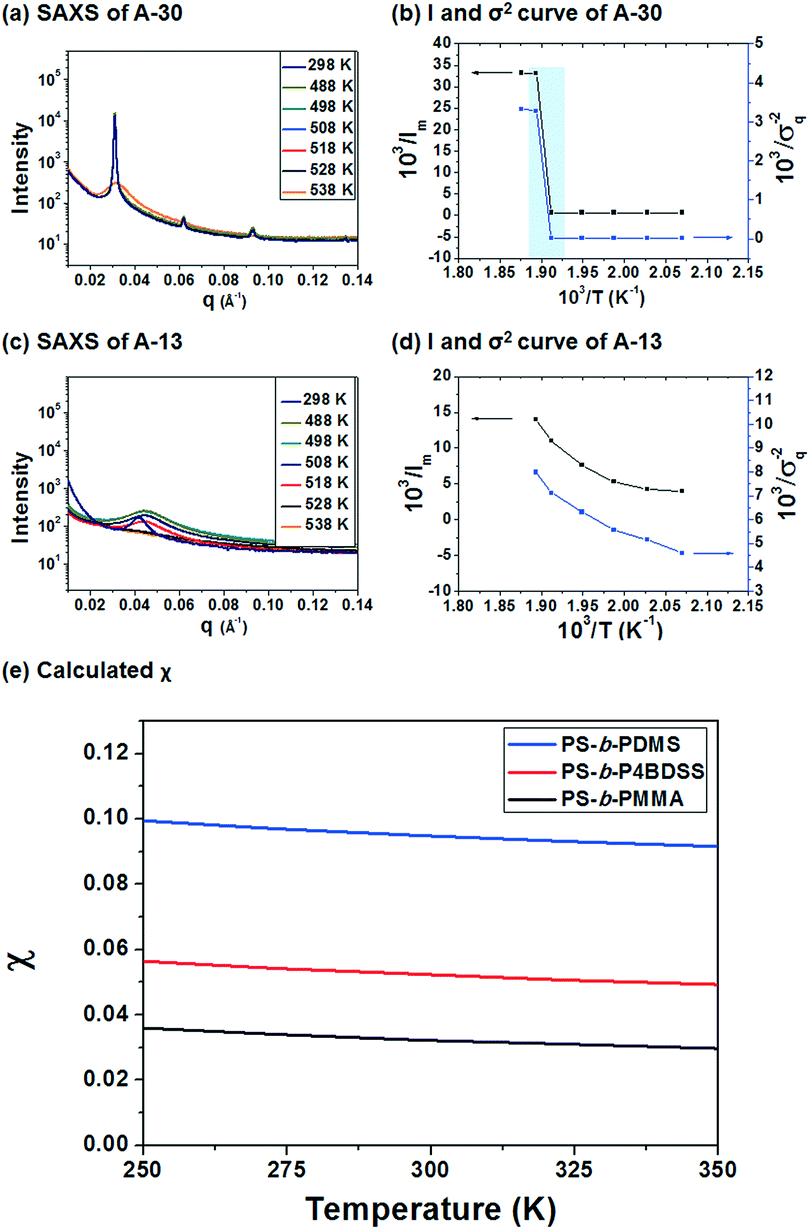 | ||
| Fig. 2 (a and c) SAXS profiles of A-30 and A-13 in the temperature range from 25 to 255 °C. (b and d) Plots of inverse intensity (Im−1) of q* and square of half-width at half-maximum (σq) of A-30 and A-13 as a function of inverse temperature (T−1). (e) Flory–Huggins interaction parameter (χ) of PS-b-P4BDSS calculated using the absolute intensity of SAXS. The χ values of BCP are described by χ(T) = α/T + β for PS-b-P4BDSS (α = 6.19 K, β = 0.0317), PDMS-b-PS (α = 15.3 K, β = 0.0401) and PS-b-PMMA (α = 5.20 K, β = 0.0150). | ||
The χ value of PS-b-P4BDSS was calculated by analyzing the SAXS profiles from disordered A-13 using Leibler's mean-field theory, which was modified considering the effect of polydispersity and segmental volume asymmetry.30,33–35 The χ value is defined in terms of the enthalpic component (α) and the entropic component (β), as described by the following equation:
| χ(T) = α/T + β | (1) |
The detailed fitting information for calculation of the χ value is provided in Fig. S3 and S4† and the obtained χ value of PS-b-P4BDSS as a function of temperature is described by the following equation:
| χ(T) = 6.19/T + 0.0317 | (2) |
The χ value of PS-b-P4BDSS was 0.0525 at 25 °C, which was higher than that of PS-b-PMMA (0.0324 at 25 °C, see Fig. S4†) and was lower than that of commonly studied Si-containing high-χ BCPs such as PS-b-PDMS (0.0950 at 25 °C, see Fig. S5†) calculated with the same procedure. This suggests that PS-b-P4BDSS can be classified as an intermediate-χ BCP. If the kinetic energy barrier is effectively lowered by the reduced χ parameter, it can generate well-ordered nanostructures with diverse domain sizes via thermal annealing.
Effect of polydispersity on self-assembled pattern quality
The spin-casted PS-b-P4BDSS films were self-assembled by thermal annealing at 200 °C for 1 h on a graphoepitaxy template (1 μm) and characterized by field emission scanning electron microscopy (FE-SEM) and grazing incidence small-angle X-ray scattering (GISAXS) measurements after RIE treatment. As shown in Fig. S6,† the self-assembled PS-b-P4BDSS films generated well-defined morphologies except A-13 due to its low segregation strength. A-54, A-33, and A-25 with volume fractions of P4BDSS fB ranging from 28.4% to 32.2% form a cylindrical morphology while A-117 and A-30 with fB ranging from 42.2% to 42.9% form a lamellar morphology. These morphologies were confirmed by GISAXS measurement, where the incident angle (αi) was set as 0.120°, which is below the critical angle, 0.140°. As shown in Fig. 3a, A-54 had strong Bragg peaks along a line at qx, 1q*, 2q*, 3q*, 4q*, 5q*, and 6q*, indicating a well-ordered cylindrical morphology with an orientation parallel to the template (see Fig. S6†). A-117, however, had Bragg peaks along a line at qy, which demonstrates a lamellar morphology (see Fig. S7†). | ||
| Fig. 3 Comparison of self-assembled patterns of BCPs with different PDIs. (a) GISAXS profiles collected at 25 °C and (b) SEM images with the CD, LWR and LER of BCP thin films (A-54, R-54, A-25 and R-26) after thermal annealing at 200 °C for 1 h. (c) SAXS profiles collected at 25 °C for bulk BCPs (A-54, R-54, A-25 and R-26) after thermal annealing at 200 °C for 24 h. | ||
To investigate the effect of the PDI on the pattern quality, we investigated the self-assembled patterns of PS-b-P4BDSS samples synthesized by anionic polymerization (A-54 and A-25) and RAFT polymerization (R-54 and R-26), which have similar molecular weights but different PDIs, as shown in Table 1. Fig. 3b shows SEM images of self-assembled cylindrical patterns of A-54 and R-54. Despite having similar Mn values, the pitch of A-54 was smaller than that of R-54 from the GISAXS profiles shown in Fig. 3a. The Bragg peaks of A-54 and R-54 are q* = 0.1645 nm−1 and 0.1603 nm−1 and the domain sizes are calculated to be 38.2 nm and 39.2 nm, respectively, from the Bragg peaks. The smaller domain size of PS-b-P4BDSS samples synthesized by anionic polymerization compared to BCPs synthesized by RAFT polymerization was also observed in the case of A-25 and R-26 (Bragg peaks of q* = 0.4347 nm−1 and 0.4098 nm−1 and domain sizes are calculated to be 14.5 nm and 15.3 nm, respectively, from Bragg peaks). Also, the GISAXS profiles of A-54 and A-25 show clearer higher-order peaks than those of R-54 and R-26. In addition, the bulk SAXS profiles of A-54 and A-25 had sharper and higher-order peaks than those of R-54 and R-26, as shown in Fig. 3c. This can be explained as follows. The domain size of self-assembled BCPs is governed by the competition between the interfacial energy between two blocks and the entropy penalty of chain stretching. Because BCPs with a broad PDI effectively release chain stretching energy, the domain size of BCPs is increased but the increased fluctuation of the interface due to the broad PDI results in lower pattern quality.29,36,37
To quantitatively analyze the effect of the PDI on the self-assembled pattern quality, A-54, A-26, R-55, and R-25 BCPs with various molecular weights and PDIs were investigated (see Table 1 for detailed information). The SEM images of the self-assembled patterns are presented in Fig. 4 and the detailed pattern quality factors such as CD, LER, and LWR were analysed via image analysis. Fig. 4 shows that, regardless of the molecular weights, PDI dominantly affected LER and LWR.
 | ||
| Fig. 4 Comparison of the effect of molecular weight and PDI on the pattern quality of the self-assembled patterns of BCPs. | ||
For a quantitative comparison of the correlation between PDI and pattern quality, the power spectral density (PSD) of LER and LWR is analyzed in Fig. 5. More detailed comparison results for line edge and line width deviations are summarized in Fig. S11 and S12,† respectively. In the PSD curve for LER and LWR, the self-assembled pattern of A-54 shows lower intensity values than the self-assembled patterns of R-54 and R-55 in the whole frequency region which demonstrates that A-54 is able to form patterns with better quality. This tendency originated from a stress field induced by different domain sizes of polymers with different chain lengths within thin films of PS-b-P4BDSS with a broad PDI. In this respect, PS-b-P4BDSS with a low PDI can self-assemble into patterns with lower roughness values. To confirm the consistent effect of the PDI on the pattern quality, we further investigated the PSD curve of the self-assembled patterns of BCPs with a lower Mn near 25500 g mol−1 (A-25, R-25, and R-26) in Fig. S13† and revealed that the pattern of A-25 with a narrow PDI shows better quality compared to the patterns of R-25 and R-26. These results suggest that, at least for intermediate-χ BCPs, the control of the PDI is also very important for controlling the χ parameters.
 | ||
| Fig. 5 Power spectral density (PSD) analysis of the (a) line edge roughness (LER) and (b) line width roughness (LWR) of A-54 (PDI = 1.05), R-55 (PDI = 1.14) and R-54 (PDI = 1.32). | ||
A quantitative comparison of pattern quality between PS-b-P4BDSS and other BCPs was conducted. We chose PS-b-PDMS and PS-b-PMMA as high-χ and low-χ BCPs, respectively. First, the LER of the patterns derived from PS-b-P4BDSS was relatively higher than that of PS-b-PDMS (LER = 2.85 ± 0.12 for PS-b-PDMS, LER = 3.41 ± 0.85 for PS-b-P4BDSS). Also, the LER of self-assembled PS-b-PMMA was 4.85 ± 0.97.38 A more detailed comparison of other different BCPs is listed in Table S3.† Therefore, we can conclude that the self-assembled pattern of PS-b-P4BDSS with a narrow PDI shows decreased domain size and roughness, which can be a huge benefit for the DSA application of BCPs.
Conclusions
A poly(styrene-b-4-(tert-butyldimethylsiloxy)styrene) BCP was synthesized by anionic polymerization and RAFT polymerization for nanoscale patterning applications. Thermally annealed morphologies (cylinders, HPLs and lamellae) with domain sizes ranging from 16.4 to 54.8 nm were obtained by controlling the molecular weight of the BCP. The χ value of the BCP (0.0525 at 25 °C) was calculated using Leibler's mean-field theory and absolute intensity SAXS analysis above TODT. In particular, the BCP can form cylindrical patterns with domain sizes from 16.4 nm to 39.5 nm by thermal annealing, with superior etch selectivity. Furthermore, the effect of the PDI of BCPs on the self-assembled pattern quality was studied. The domain size and roughness of the self-assembled patterns significantly increased when the BCPs had a broad PDI due to the decreased entropic penalty for chain stretching. As a result, the LER and LWR of self-assembled patterns, generated from the BCP with a smaller PDI of 1.05, were reduced by 38% and 19%, respectively, compared to the BCP with a PDI of 1.32.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge Pohang Accelerator Laboratory for small-angle X-ray scattering measurements. JM and BGK acknowledge support from the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division.References
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525–557 CrossRef CAS PubMed.
- F. S. Bates and G. H. Fredrickson, Phys. Today, 1999, 52, 32–38 CrossRef CAS.
- C. M. Bates, T. Seshimo, M. J. Maher, W. J. Durand, J. D. Cushen, L. M. Dean, G. Blachut, C. J. Ellison and C. G. Willson, Science, 2012, 338, 775–779 CrossRef CAS PubMed.
- J. D. Cushen, C. M. Bates, E. L. Rausch, L. M. Dean, S. X. Zhou, C. G. Willson and C. J. Ellison, Macromolecules, 2012, 45, 8722–8728 CrossRef CAS.
- S. Hosaka, T. Akahane, M. Huda, H. Zhang and Y. Yin, ACS Appl. Mater. Interfaces, 2014, 6, 6208–6211 CAS.
- K. Aissou, M. Mumtaz, G. Fleury, G. Portale, C. Navarro, E. Cloutet, C. Brochon, C. A. Ross and G. Hadziioannou, Adv. Mater., 2015, 27, 261–265 CrossRef CAS PubMed.
- C. Sinturel, F. S. Bates and M. A. Hillmyer, ACS Macro Lett., 2015, 4, 1044–1050 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- S. O. Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef CAS PubMed.
- C. B. Tang, E. M. Lennon, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Science, 2008, 322, 429–432 CrossRef CAS PubMed.
- D. Broseta, G. H. Fredrickson, E. Helfand and L. Leibler, Macromolecules, 1990, 23, 132–139 CrossRef CAS.
- J. L. Sturtevant, J.-Y. Lee, J. Shin, H.-W. Kim, S.-G. Woo, H.-K. Cho, W.-S. Han and J.-T. Moon, Proc. SPIE, 2004, 5376, 426–433 CrossRef.
- J. A. Allgair, V. Constantoudis, C. J. Raymond and E. Gogolides, Proc. SPIE, 2008, 6922, 692223 CrossRef.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- J. G. Kennemur, L. Yao, F. S. Bates and M. A. Hillmyer, Macromolecules, 2014, 47, 1411–1418 CrossRef CAS.
- J. D. Cushen, L. Wan, G. Pandav, I. Mitra, G. E. Stein, V. Ganesan, R. Ruiz, C. G. Willson and C. J. Ellison, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 36–45 CrossRef CAS.
- Y. S. Jung and C. A. Ross, Nano Lett., 2007, 7, 2046–2050 CrossRef CAS PubMed.
- Y. S. Jung and C. A. Ross, Adv. Mater., 2009, 21, 2540–2545 CrossRef CAS.
- Y. S. Jung and C. A. Ross, Small, 2009, 5, 1654–1659 CrossRef CAS PubMed.
- Y. D. Luo, D. Montarnal, S. Kirn, W. C. Shi, K. P. Barteau, C. W. Pester, P. D. Hustad, M. D. Christianson, G. H. Fredrickson, E. J. Kramer and C. J. Hawker, Macromolecules, 2015, 48, 3422–3430 CrossRef CAS.
- M. D. Rodwogin, C. S. Spanjers, C. Leighton and M. A. Hillmyer, ACS Nano, 2010, 4, 725–732 CrossRef CAS PubMed.
- S. X. Zhou, D. W. Janes, C. B. Kim, C. G. Willson and C. J. Ellison, Macromolecules, 2016, 49, 8332–8340 CrossRef CAS.
- J. W. Jeong, W. I. Park, M. J. Kim, C. A. Ross and Y. S. Jung, Nano Lett., 2011, 11, 4095–4101 CrossRef CAS PubMed.
- W. I. Park, K. Kim, H. I. Jang, J. W. Jeong, J. M. Kim, J. Choi, J. H. Park and Y. S. Jung, Small, 2012, 8, 3762–3768 CrossRef CAS PubMed.
- Y. S. Jung, J. B. Chang, E. Verploegen, K. K. Berggren and C. A. Ross, Nano Lett., 2010, 10, 1000–1005 CrossRef CAS PubMed.
- J. M. Kim, Y. Kim, W. I. Park, Y. H. Hur, J. W. Jeong, D. M. Sim, K. M. Baek, J. H. Lee, M. J. Kim and Y. S. Jung, Adv. Funct. Mater., 2015, 25, 306–315 CrossRef CAS.
- J. M. Kim, Y. H. Hur, J. W. Jeong, T. W. Nam, J. H. Lee, K. Jeon, Y. Kim and Y. S. Jung, Chem. Mater., 2016, 28, 5680–5688 CrossRef CAS.
- N. Torikai, A. Noro, M. Okuda, F. Odamaki, D. Kawaguchi, A. Takano and Y. Matsushita, Phys. B, 2006, 385, 709–712 CrossRef.
- N. A. Lynd and M. A. Hillmyer, Macromolecules, 2005, 38, 8803–8810 CrossRef CAS.
- L. Leibler, Macromolecules, 1980, 13, 1602–1617 CrossRef CAS.
- S. S. Rane and P. Choi, Chem. Mater., 2005, 17, 926 CrossRef CAS.
- P. C. Hiemenz and T. P. Lodge, Polymer chemistry, CRC Press, New York, 2007 Search PubMed.
- Y. Zhao, E. Sivaniah and T. Hashimoto, Macromolecules, 2008, 41, 9948–9951 CrossRef CAS.
- T. P. Russell, J. S. Lin, S. Spooner and G. D. Wignall, J. Appl. Crystallogr., 1988, 21, 629–638 CrossRef CAS.
- S. Sakurai, K. Mori, A. Okawara, K. Kimishima and T. Hashimoto, Macromolecules, 1992, 25, 2679–2691 CrossRef CAS.
- N. A. Lynda, A. J. Meulerb and M. A. Hillmyer, Prog. Polym. Sci., 2008, 33, 875–893 CrossRef.
- D. C. Hur, A. Noro, A. Takano and Y. Matsushita, Macromolecules, 2005, 38, 4371–4376 CrossRef.
- K. S. Lee, J. Lee, J. Kwak, H. C. Moon and J. K. Kim, ACS Appl. Mater. Interfaces, 2017, 9, 31245–31251 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00085e |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |