
Spatially-controllable and uniform photochemical transfer printing of block copolymer nanopatterns†
Dustin W.
Janes
 *a,
Takejiro
Inoue
bc,
Nathan D.
Prisco
d,
Michael J.
Maher
e,
Paul F.
Nealey
b and
Christopher J.
Ellison
*f
*a,
Takejiro
Inoue
bc,
Nathan D.
Prisco
d,
Michael J.
Maher
e,
Paul F.
Nealey
b and
Christopher J.
Ellison
*f
aJoris Helleputteplein 5, 3000 Leuven, Belgium. E-mail: djanes1@gmail.com
bInstitute for Molecular Engineering, The University of Chicago, 5801 South Ellis Avenue, Chicago, IL 60637, USA
cElectronic and Imaging Research Laboratories, Toray Industries, Inc., 1-2, Sonoyama 3-chome, Otsu, Shiga 520-0842, Japan
dMcKetta Department of Chemical Engineering, The University of Texas at Austin, 200 East Dean Keaton Stop C0400, Austin, TX 78712, USA
eDepartment of Chemistry, The University of Texas at Austin, 105 E. 24th St. Stop A5300, Austin, TX 78712, USA
fDepartment of Chemical Engineering and Materials Science, University of Minnesota, 421 Washington Ave. SE, Minneapolis, MN 55455, USA. E-mail: cellison@umn.edu
First published on 1st November 2017
Abstract
Transfer printing processes with resolution approaching 10 nm are not very common because pattern fidelity is often lost due to translational motion of the patterning molecules. To overcome this challenge, we describe here a photochemical transfer printing (PTP) process in which covalent bonds are formed between a nanopatterned master film formed by self-assembled poly(styrene)-block-poly(methyl methacrylate) (PS-b-PMMA) and solution-deposited conformal layer poly(styrenesulfonylazide-alt-maleic anhydride) in the solid state, where the pattern is fully stable. The monolayer of grafted PS-b-PMMA is then transferred to an initially blank replica film using a photopolymerizable liquid conformal layer possessing very low viscosity (hexanedioldiacrylate, 9 cP), which is known to promote dimensional uniformity in UV nanoimprint technology. The chemical nanopatterns transferred to the replica substrate is continuous and robust enough to successfully direct the self-assembly of new PS-b-PMMA films cast upon it after thermal annealing. The experiments in this work demonstrate patterning resolution down to 14 nm half-periodicity. The PTP process is mechanistically controlled by light and therefore can be spatially controlled by photomasks.
Design, System, ApplicationWhen the transfer printing of block copolymer nanopatterns was first described by Ji and Nealey in 2010, they and their coworkers utilized rigid silicon substrates, limiting the area of transfer to ca. 1 mm2. To expand the area of transfer, new conformal materials need to be developed which allow two substrates to contact one another continuously over large areas (>100 mm2). It must be possible to process these conformal materials into uniform and ultrathin layers such that subsequent plasma etch processes are capable of transferring the pattern into the replica substrate. Furthermore the conformal materials need to chemically react to components dissolved in the block copolymer nanopattern, or the block copolymer itself, and be thermally stable during all subsequent processing steps. These design criteria led us to synthesize a new polymer, poly(styrenesulfonylazide-alt-maleic anhydride), that can be spin coated orthogonally atop block copolymer nanopatterns and photochemically activated to form covalent bonds with neighboring molecules. This enables very low-viscosity liquid conformal layers to be utilized in transferring the pattern to other substrates over large, continuous areas, which is practically desirable in nanopatterning applications. |
1. Introduction
Transfer printing processes are possibly the most essential method of replicating information. From Gutenberg's printing press to its modern equivalent, roll-to-roll offset printing, conformal contact between two surfaces over large areas enables mass production of tangible printed objects on a nearly instantaneous basis, with patterning resolution at the millimeter scale.1 Biological processes like DNA replication and RNA transcription can even be considered transfer printing processes conducted in cellular solution. In those cases, freely dissolved base-pairs are drawn in contact to their complements via hydrogen bonding interactions as their order is preserved by polymerization. Conceptually, atomistic scale biological transfer printing processes are reliant on precise self-assembly while visual-scale printing replicates consciously invented designs. Researchers designing intermediate-scale transfer printing processes, hoping to combine the throughput of offset printing with the molecular resolution and fidelity of biology, might therefore be most successful when they merge the “top-down” strategy with “bottom-up” self-assembly paradigms.2Transfer printing processes capable of spatial control on nanometer length scales are not very common.3–6 For example, microcontact printing relies on conformal contact between an elastomeric relief structure and a blank replica surface, but is resolution-limited for features <100 nm in width due to translational motion of liquid inks.7 In principle, this limitation could be circumvented by performing transfer printing in the solid state.8–10 To meet this need, we have developed a new process called photochemical transfer printing (PTP). The PTP introduced here utilizes a highly reactive polymer, poly(styrenesulfonyl azide-alt-maleic anhydride) (PSSMA),11,12 to facilitate transfer printing of patterns from a nanostructured solid film to a blank, unpatterned substrate via interfacial grafting reactions. Because the covalent bonds are formed in response to light stimuli, PTP is spatially controllable and compatible with existing lithographic tools.13,14
As a model nanostructured solid we have chosen poly(styrene-block-methyl methacrylate) (PS-b-PMMA) thin films possessing perpendicularly oriented microdomains.15 The domain periodicity, and hence the patterning resolution, was rationally varied from 28 to 87 nm by choosing an appropriate PS-b-PMMA molecular weight.16 We show conclusively that a critical exposure dose of ultraviolet light must be applied to PSSMA to replicate fingerprint patterns and that the critical dose is increased for the finest nanostructures. Furthermore, a set of methodologies exists to direct the long-range alignment of the microdomains into useful, device oriented structures over macroscopically large areas using current-generation lithographic tools and process steps.17,18 We replicated the patterns formed by lithographically-directed templates to show that PTP is fully capable of using simple and inexpensive equipment to copy nanoscale features fabricated within high-capital cost facilities inaccessible to most materials researchers.
2. Experimental
Materials
The synthesis and characterization of the PSSMA sample used in this study was described previously.12 The L0 = 42 nm and L0 = 87 nm PS-b-PMMA (37 kDa-b-37 kDa and 105 kDa-b-106 kDa,19 respectively) were purchased from Polymer Source, Inc. A solution of L0 = 28 nm PS-b-PMMA in propylene glycol methyl ether acetate (PME-312) was provided by EMD Performance Materials. The synthesis of poly(styrene-stat-methyl methacrylate-stat-gycidyl methacrylate) used as a surface neutral treatment (SNT) on silicon wafers was described previously.20,21Solutions of the solid conformal layer, PSSMA, were made by dissolving ca. 2 mg PSSMA in a couple of drops of trimethylamine/water solution (50 vol%) and then further dissolving in methanol until a film of the desired 20 nm thickness could be obtained by spin coating.11,12 Hexane diol diacrylate (HDDA) was purchased from Sigma-Aldrich and ethoxylated pentaerythritol tetraacrylate was provided by Sartomer. Irgacure 2100 photoinitiator was provided by BASF. Liquid conformal layer was prepared by dissolving 0.5 wt% photoinitiator in HDDA or ethoxylated pentaerythritol tetraacrylate.
Master patterns used in PTP
Chemically nanopatterned substrates that are capable of aligning L0 = 28 nm PS-b-PMMA microdomains over large areas were prepared by methods described previously.22,23 The samples were rinsed of soluble material by toluene and a 100 nm thick layer of PS-b-PMMA was spin-coated upon them. To obtain concentrated solutions of L0 = 28 nm PS-b-PMMA suitable for coating a 100 nm thick layer, the polymer was precipitated from the provided solution in methanol, dried under vacuum, and redissolved in cyclopentanone. After spin coating, the samples were annealed for 4 h at 230 °C under vacuum.Master films of L0 = 87 nm PS-b-PMMA fingerprint patterns were prepared by a procedure similar to that described previously.24 A 100 nm thick layer of PS-b-PMMA was spin-coated atop SNT-treated silicon wafer. The sample was solvent annealed in a sealed 100 mL glass vial containing 10 mL tetrahydrofuran for 45 min to lower the effective segregation strength between the blocks during the initial stages of self-assembly. Afterwards it was quickly removed and thermally annealed on a hot plate at 250 °C for 1 h. A representative SEM image of this film is provided as ESI† (see Fig. S1).
Master films of L0 = 42 nm PS-b-PMMA fingerprint patterns possessing characteristically large grain size were prepared by procedures similar to that described previously.5 Relatively concentrated solutions (5–10 wt%) of PS-b-PMMA in toluene or cyclopentanone were spin coated atop SNT-treated silicon wafers to obtain block copolymer layers >5 L0 thick, and they were annealed under vacuum on a 250 °C hotplate for at least one hour. The fingerprint morphologies obtained are visually distinct from those obtained from 1.0L0 thick films.5,25 42 nm thick master films of L0 = 28 nm PS-b-PMMA fingerprint patterns were prepared by spin coating the as-received BCP solution at 1000 rpm on SNT-treated silicon wafers. They were annealed at 250 °C in air for 5 min to obtain vertically oriented lamella.
Photochemical transfer printing (PTP)
PTP was performed by first spin-coating PSSMA atop master films. The master films were placed in a quartz-topped chamber that was continuously purged by N2 gas for at least 30 min prior to exposure. This procedure removes 85% of the ambient oxygen.12 Then collimated broadband UV light was applied from a fiber guide connected to a spot-cure system (Novacure 2100, EXFO Inc.) at a collimating lens-to-sample distance of 7 cm. The intensity at this distance is 25 mW cm−2 as measured by a radiometer (Fieldmax II TO, Coherent Inc.). Photographs of this PTP step are provided as ESI† (see Fig. S2).The second step of PTP was performed by placing a drop of liquid conformal layer atop a methacrylate-silane treated glass slide.5 The glass slide was then inverted and pressed gently to the master film and irradiated for 30 s under ambient conditions. The duration, dose, and wavelengths of light used in this exposure step are only what are necessary to fully photopolymerize the liquid conformal layer, since the covalent bonds between PSSMA and master pattern were formed during the previous step. Photographs of this PTP step are provided as ESI† (see Fig. S3). These steps of PTP were performed in the absence of ambient UV light (i.e. a “yellow lab”) to preserve the light-sensitive conformal materials and also while wearing orange UV filter safety glasses.
Portions of photopolymerized conformal layer stuck to the edges of the sample stack (i.e. the “flash”) were trimmed using a razor blade. The sample stack was soaked in 80 °C toluene until the master and replica substrates could be separated with gentle force applied to the stack corner by a razor blade. The master and replica substrates were rinsed 5× with toluene while they rotated on a spin coater; they were then bath sonicated for 5 minutes in toluene and rinsed 5 additional times with toluene. A 1.0L0 reassembly layer of PS-b-PMMA was spin coated atop them and thermally annealed. For example, a 42 nm thick film of L0 = 42 nm PS-b-PMMA was obtained by spin-coating a 1.6 wt% solution in toluene at 1500 rpm atop replica samples. A 28 nm thick layer of L0 = 28 nm PS-b-PMMA was obtained on replica substrates by spin coating the as-received BCP solution at 2300 rpm.
Film characterization
10×, 50×, and 100× optical micrographs of samples were obtained with an Olympus BX60 optical microscope. Top-down scanning electron microscope (SEM) images were taken using a secondary in-lens detector on a Zeiss Supra 40 VP SEM. The typical accelerating voltage was 1 keV and the typical working distance was 2–5 mm. The thickness of spin-coated polymer films was determined from fitting of optical parameters to the data measured by spectroscopic ellipsometry (J.A. Woollam Co. Inc. M-2000 V, λ = 382–984 nm, 65° angle).3. Results and discussion
A schematic for PTP is shown in Fig. 1. The input to this replication process is a supported thin block copolymer film in which the microdomains are oriented perpendicularly to the free surface. After coating it with PSSMA the sample stack is irradiated from above by UV light in a N2-purged, quartz-topped chamber (Fig. 1a). UV irradiation photochemically generates highly reactive nitrene intermediates from the azide precursor that covalently bond to nearby C–H groups via insertion and free-radical recombination mechanisms.12 The PSSMA layer crosslinks and affixes the adjacent block copolymer molecules to it, thereby preserving the chemical nanopattern. Next, a glass slide coated with photopolymerizable liquid conformal layer is pressed against the master stack and irradiated under ambient conditions (Fig. 1b). This step causes the PSSMA and affixed chemical nanopattern to adhere to the now-solidified conformal layer and glass slide. The sample stack is separated by soaking in warm solvent, which dissolves ungrafted block copolymer molecules (Fig. 1c). This produces a coated glass slide with the transferred chemical nanopattern at its top surface, as well as recovers the substrate of the original master nanopattern. These substrates are cleaned of untethered polymers by a brief sonication/rinsing procedure. Coating a new layer of block copolymer atop these substrates and annealing provides mobility and enables self-assembly into structures directed by the chemical nanopattern(s). This generates a replica with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 domain registration (Fig. 1d) and recovers the original master pattern (Fig. 1e).
:1 domain registration (Fig. 1d) and recovers the original master pattern (Fig. 1e).
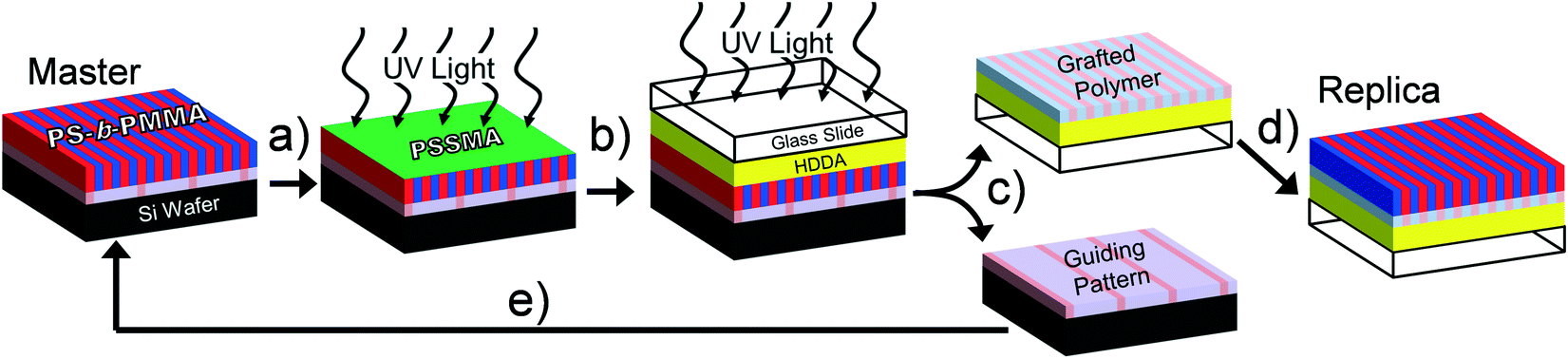 | ||
| Fig. 1 Schematic of PTP. a) A block copolymer thin film is coated by PSSMA. b) After exposure to UV light in a N2-purged, quartz topped chamber, a glass slide coated in HDDA is placed atop it. c) After a second irradiation step performed in ambient conditions, the sample stack is separated in 80 °C toluene and the substrates are cleaned by sonication and rinsing in toluene. d) The chemically nanopatterned glass substrate and e) recovered guiding pattern are coated by a new block copolymer layers and thermally annealed. | ||
A demonstration of the PTP process described above using a lithographically-directed master pattern is shown in Fig. 2. While PTP is in principle capable of utilizing any organic chemical nanopattern as input, we used symmetric PS-b-PMMA aligned over large areas into dense line-and-space patterns with 14 nm half-pitch because this system has served as a model for scalable production on 300 mm diameter substrates.23,26 The characteristic patterns atop the master pattern are indeed transferred to the glass substrate and can be inspected with clarity after the steps indicated by Fig. 1c and d as observed in the micrographs representing the “Grafted Polymer” and “Replica” samples in Fig. 2. Equally important is the demonstrated recovery of the original guiding pattern as shown in the micrographs representing the “Guiding Pattern” and “Recovered Master”. Please note that the thickness of the PS-b-PMMA reassembly layer atop the replica and recovered master is 28 nm (≈1.0L0) while that used atop the original master was 100 nm (≈3.6L0). As a consequence, in the recovered master micrograph the underlying crosslinked PS guiding lines provide additional SEM imaging contrast at every third line in the self-assembled PS-b-PMMA film, and it appears somewhat brighter. Each PS microdomain appears equally bright in the micrograph of the master sample because the PS guidestripes are far below the inspection depth of SEM microscopy. While the PS regions also appear equally bright in the replica sample, it is because it was aligned with 1:1 registration between the microdomains and chemical pattern. A full, unedited micrograph of the replica sample is provided as ESI† (see Fig. S4).
 | ||
| Fig. 2 Representative SEM micrographs of PTP process using a lithographically-directed master pattern. | ||
Wan et al. have recently demonstrated that even sparse and discontinuous 1:1 chemical guiding patterns may improve the quality of nanopatterns formed by the directed self-assembly (DSA) of block copolymers.27 Ultimately even higher-fidelity, more uniform, and continuous 1:1 chemical nanopatterns are desirable to reduce defects in lithographically-fabricated DSA patterns.3 In this context, it is worthwhile to consider the thickness of grafted PS-b-PMMA that is transferred to the replica substrate. A robust thickness of grafted polymer is highly desirable because this would minimize the influence of non-uniformities and block interpenetration artifacts. Our previous characterization11 of thicknesses of poly(styrene) monolayers photochemically grafted to PSSMA revealed close agreement with that theoretically expected for physically adsorbed monolayers (i.e. a “Guiselin brush”28). L0 = 28 nm PS-b-PMMA can be anticipated to possess a total Mw = 45–50 kDa based on the archived trend16 and assuming the polydispersity index is between 1.0 and 1.1. The relationship between Mw and Guiselin brush thickness irreversibly adsorbed to an interface is experimentally established for poly(styrene) and poly(methyl methacrylate) homopolymers.29,30 Therefore we can expect the grafted thicknesses of L0 = 28 nm PS-b-PMMA monolayers to be 3.1–3.3 nm.
Having demonstrated effective transfer printing of chemical nanopatterns, we now seek to demonstrate that the PTP process is photochemically triggered and occurs over macroscopically large areas (>1 cm2) using PSSMA as a light-sensitive conformal layer. In this and subsequent examples we are utilizing self-assembled PS-b-PMMA fingerprint patterns as master films that possess characteristically large grain-size due to very thick BCP layers and long annealing times.5,25 A master substrate where PSSMA is not present in a “longhorn” shaped region was prepared by placing a lapel pin face-down on top of the sample during the photoexposure depicted in Fig. 1a (see Fig. S5†). Subsequently the unexposed PSSMA was rinsed from the sample by methanol (as shown in Fig. 3a). Due to the film thickness variations between areas coated and uncoated by PSSMA these regions are visually distinct. Since only the area outside of the “longhorn” contains PSSMA, the area inside the “longhorn” will not transfer print chemical nanopatterns via the PTP process.
We then conducted the transfer of the chemical nanopattern to a glass slide using ethoxylated pentaerythritol tetraacrylate as a liquid, photopolymerizable conformal layer. To promote uniform squeezing pressure across the sample, a custom clamp was fabricated from machined flange caps, a quartz disc window, and a silicon rubber press pad. Hex nuts were turned to center the clamping pressure and bubbles were removed by vacuum prior to photopolymerization of the liquid conformal layer. Photographs of these steps are provided as ESI† (See Fig. S5). A photograph of the solidified sample stack is shown in Fig. 3b.
 | ||
| Fig. 3 Spatial control of PTP over large areas. a) Photo of master substrate where a “longhorn” shaped region is not coated by PSSMA. b) Photo of sample stack after a glass slide coated by ethoxylated pentaerythritol tetraacrylate was clamped to the master film and irradiated. c) Photograph (upper) of replica sample after separation, cleaning, and coating by PS-b-PMMA reassembly layer. The optical micrograph (lower) was taken at the edge of the “longhorn” shaped region. d) Photograph (upper) and optical micrograph (lower) of replica sample after annealing. e) and f) Show representative SEM images taken inside and outside, respectively, of the longhorn shaped region. | ||
After separating the sample stack in warm toluene, and removing untethered materials with rinsing and bath sonication steps, a 42 nm thick PS-b-PMMA was spin coated on top of the substrate. Photographs and optical micrographs of the glass slide prior to annealing are shown in Fig. 3c. The “longhorn” shaped region is not yet visually distinct (upper panel), although the optical micrograph indicates both regions are macroscopically flat (lower panel). Fig. 3d shows the same sample after thermal annealing for 5 min at 250 °C. The “longhorn” shaped region appears cloudy when illuminated at an oblique angle (upper panel). Microscopic inspection (lower panel) reveals the presence of discrete film thickness variations inside the “longhorn” region. These macroscopic features represent an “islands and holes” topology that scatters visible light,15,31 confirming that the PS-b-PMMA microdomains are oriented parallel to the substrate due to the lack of an underlying chemical nanopattern. Outside of the “longhorn” shaped region the film remains smooth, and does not scatter visible light, suggesting perpendicular orientation of PS-b-PMMA microdomains.
SEM inspection of the sample agrees with these observations. Fig. 3e and f show representative micrographs located inside and outside the “longhorn” shaped region, respectively. Outside of the “longhorn” shaped region, where PTP occurred, perpendicularly-oriented lamellae possessing a grain structure matching the master film are present. Inside the “longhorn” shaped region, where PTP was blocked by the lapel pin, perpendicularly oriented lamellae are not present.
To demonstrate that photochemical reactions between the PSSMA and block copolymer pattern underlie the success of PTP, we designed mechanistic control experiments. Shown in Fig. 4 are top-down SEM images of replica samples after various stages of PTP. The PTP shown in the upper row was performed with the PSSMA layer. The center row shows the control where each step was performed identically but in the absence of the PSSMA layer. Immediately after separating and rinsing the master and replica samples (Fig. 1c) a block copolymer nanopattern characteristic of the master is present on the replica sample surface for both the PTP and control sample. After briefly bath sonicating these samples in toluene, PS-b-PMMA that is not bound by covalent bonds is removed from the surface. In the control without PSSMA, no evidence of residual PS-b-PMMA is present on the surface, which suggests no grafting occurred. The pattern present prior to bath sonication on the control sample was evidently made of weakly adsorbed and/or loosely entangled PS-b-PMMA molecules. Since PTP is driven by the formation of covalent bonds between PSSMA and the BCP, the pattern is still present in the sample made with PSSMA after sonication. This monolayer of bound polymer serves to direct the self-assembly of the new block copolymer spin coated atop it and annealed.
 | ||
| Fig. 4 Representative 1 μm2 SEM images of replica substrates fabricated by the PTP process with a PSSMA layer (upper row), without a PSSMA layer (middle row), and with an inert analog of PSSMA (bottom row). SEM micrographs were taken at different stages in the PTP process. HDDA was used as liquid conformal layer. | ||
In the control without PSSMA, the subsequent coating and annealing of a new BCP yields a distorted fingerprint pattern possessing very small grain size because there is no underlying pattern to guide those domains. We also performed a similar control experiment substituting an inert analogue of PSSMA, poly(4-methylstyrene-alt-maleic anhydride), for PSSMA. The results are shown in the lower row of Fig. 4. Indeed, without the presence of the photochemically active sulfonyl azide group, no PTP takes place and the pattern is not transferred.
Having demonstrated that photochemical grafting reactions between the block copolymer and PSSMA are necessary for PTP, next we show that the resolution of patterning is related to the applied dose of UV light in the step depicted by Fig. 1a. Master films were prepared from PS-b-PMMA possessing L0 = 87 nm, 42 nm, and 28 nm. SEM images representing replica films formed by PTP with varying dose are shown in Fig. 5–7. The photochemical conversion of azide groups at given exposure doses was determined from transmission Fourier transform infrared spectroscopy measurements described in detail in our previous work.12 Note that in these experiments we utilized a λ = 365 nm bandpass filter in the exposure step depicted by Fig. 1b. Since this wavelength of light is not absorbed by PSSMA, the success or failure of PTP can solely be attributed to the formation of covalent bonds between PSSMA and the master pattern during the primary exposure step depicted by Fig. 1a, or lack thereof. The color of the micrograph's border indicates our assessment of the quality of PTP; green indicates a defect-free, continuous chemical nanopattern was obtained, orange indicates the presence of defects due to insufficient dose, and red indicates a total failure of PTP.
 | ||
| Fig. 5 Representative 1 μm2 SEM images of grafted L0 = 87 nm PS-b-PMMA monolayers formed atop replica samples after separation, sonication, and rinsing by toluene. The broadband dose, duration of exposure, and photochemical conversion of azide groups was varied in the step Fig. 1a and is indicated above the images. | ||
Intuitively, one may expect that a greater areal density of grafting reactions would be necessary to transfer print a pattern formed by lower molar mass BCP. Indeed, this relationship is present in the obtained data. As shown in Fig. 5, a continuous chemical nanopattern of grafted L0 = 87 nm BCP is formed atop the replica sample after an exposure dose of 400 mJ cm−2. This dose corresponds to only 16% of azide groups being photochemically converted to their reactive nitrene intermediates. For the L0 = 42 nm, a dose of 500 mJ cm−2 is necessary to obtain successful PTP, corresponding to 21% conversion of azide groups (Fig. 6). Finally for the smallest periodicity BCP, L0 = 28 nm, a full 98.9% of azide groups must be converted, using a dose of 10.5 J cm−2 (Fig. 7). For each of these cases where a minimum dose has been determined, larger doses also obtain successful PTP results. The full set of critical dose experiments are summarized in the ESI,† Fig. S6–S9.
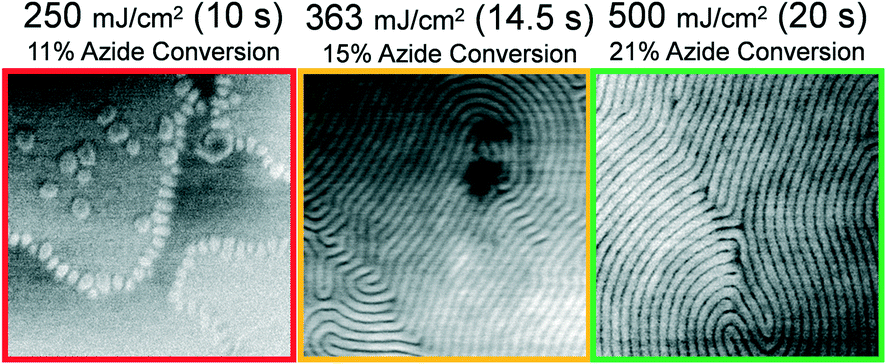 | ||
| Fig. 6 Representative 1 μm2 SEM images of L0 = 42 nm PS-b-PMMA patterns formed atop replica samples after spin-coating and annealing of a 1.0L0 thick reassembly layer atop them. The broadband dose, duration of exposure, and photochemical conversion of azide groups was varied in the step Fig. 1a and is indicated above the images. | ||
 | ||
| Fig. 7 Representative 1 μm2 SEM images of L0 = 28 nm PS-b-PMMA patterns formed atop replica samples after spin-coating and annealing of a 1.0L0 thick reassembly layer atop them. The broadband dose, duration of exposure, and photochemical conversion of azide groups was varied in the step Fig. 1a and is indicated above the images. | ||
The reader should note that the critical azide conversion necessary for PTP to occur increases slightly (400 mJ cm−2 to 500 mJ cm−2) as L0 reduces from 87 nm to 42 nm, but increases sharply (500 mJ cm−2 to 10.5 J cm−2) as L0 reduces from 42 nm to 28 nm. We hypothesize that this relationship is likely highly non-linear, especially when very high azide conversions are necessary to perform PTP. In this limit, nearly all photoactive species in the solid conformal layer are exhausted, it becomes highly crosslinked, and its material properties may change in a way that reduces the stoichiometric efficiency of each converted azide towards the interfacial PS-b-PMMA. Beyond the scope of the present study, we suggest that additional experiments using a significantly larger set of test L0 values could provide additional insight on how these parameters are linked.
Finally, to provide evidence of the outstanding conformal properties of the PTP process and materials described here, we have provided representative optical micrographs of master and replica samples in Fig. 8. In these samples all solutions and liquid conformal materials were syringe-filtered prior to coating, so the resulting films are free of particulate defects which would create non-uniformities in the film thickness. The color present in the image of the master film is related to its overall film thickness. Therefore the uniform change in color before and after coating with PSSMA is representative of a uniform increase in film thickness after coating it with a very thin layer of PSSMA.
 | ||
| Fig. 8 Optical micrographs and photos of thick PS-b-PMMA master film, before (left) and after (center) coating with PSSMA, and of the replica fabricated from it atop an HDDA coated glass slide (right). | ||
Accordingly, the replica sample also has a uniform appearance, with no presence of thickness variations visible in the optical micrograph. The ability to obtain a relatively uniform thickness of photopolymerized conformal layer is closely related to the very low viscosity of HDDA which allows it to distribute evenly between the top of the master film and bottom of the glass slide in Fig. 1b. HDDA possesses a viscosity of only 9 cP, which is roughly compatible with UV nanoimprint technologies. It would not have been possible to obtain this result with a previous materials set,5,6 because they possessed very high viscosities. The residual layer thickness obtained in UV nanoimprint lithography is typically ca. 10 nm,32 which represents the minimum HDDA thickness that would be practically achievable over large areas if the PTP technique were formally integrated with UV nanoimprint lithography tools.
Another advantage of using both a solid and photopolymerizable conformal layer we wish to emphasize is that the solid conformal layer, here PSSMA, acts as a barrier and inhibits infiltration of the block copolymer nanopattern by low-viscosity monomers. It is suggested by the results from the center row of Fig. 4 that there is some infiltration of HDDA into the BCP when there is not a barrier material such as PSSMA protecting the master film. We note that vapor deposited silicon nitride also works exceptionally well as a solid conformal layer, both in terms of reactivity to hydroxyl-terminated homopolymer “inks” and as a barrier to monomer infiltration.3
4. Conclusions
In this work we demonstrated a pattern replication procedure based on transfer printing that utilizes an ultrathin, solution-cast, and highly reactive conformal layer. Irradiation of the sample stack with light induces the formation of covalent bonds between the solid conformal layer and chemical nanopattern, and allows a monolayer of the chemical nanopattern material to be transferred to a previously unpatterned substrate. The experiments described here successfully transferred chemical nanopatterns with resolution as small as 14 nm. Since the stimuli used in transfer printing is light, it can be spatially controlled such that the pattern is replicated only in specific areas. The material selection of a light-reactive, solid conformal layer and low-viscosity, photopolymerizable conformal layer is practically desirable because it promotes uniform and thin conformal layer stacks to be transferred to the replica substrate.We presently see no fundamental reason that photochemical transfer printing (PTP) procedures could not be utilized or otherwise adapted to replicate chemical nanopatterns different than the PS-b-PMMA thin films utilized here. However, some practical challenges will remain present if PTP were to be extended to patterns formed by other block copolymers or nanomaterials. The photoreactive solid conformal layer must form covalent bonds with the master pattern at sufficiently high areal density for the pattern to be transferred with acceptable fidelity. The very high azide conversions necessary to achieve successful PTP at L0 = 28 nm shown in Fig. 7 suggest that higher azide content in the PSSMA and/or the use of grafting chemistries that are more stoichiometrically efficient would be necessary to achieve PTP at even higher resolution (i.e. smaller L0). A solution to this obstacle could involve synthesizing new block copolymers which contain functional groups that react efficiently to complementary chemistry present in the conformal materials.33
Additionally, the conformal layers must be deposited in a way that does not distort or erase the master pattern. The methods described here become more challenging to apply to PS-b-PMMA with smaller L0 in part because low molar mass poly(methyl methacrylate) is soluble in the methanol/water mixtures from which the PSSMA is applied.34 Solubilizing one block in a block copolymer thin film is known to structurally reorganize the phase-separated microdomains, which is presumably undesirable for PTP.35 We circumvented this obstacle here by using relatively thick films as master substrates, such as the 100 nm thick film prepared for the experiment in Fig. 2. Another known solution in the case of block copolymer nanopatterns is to utilize all-styrenic structures which are fully insoluble in methanol/water mixtures.36
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Guanyang Lin and Ralph Dammel of EMD Performance Materials, a subsidiary of Merck KGaA Darmstadt, for providing a PS-b-PMMA used in this study. The authors thank Roel Gronheid, Paulina Rincon, Hari Pathangi, Nadia Vandenbroeck, and Boon Teik Chan of Imec vzw for providing chemically nanopatterned substrates for directed self-assembly. The authors thank Butch Cunningham for machining large-area clamps and suggesting the use of silicone rubber press pads. SEM was performed at the Microscopy and Imaging Facility of the Institute for Cellular and Molecular Biology at the University of Texas at Austin. Partial financial support for this work was provided by the Robert A. Welch Foundation (No. F-1709), the Norman Hackerman ARP, Nissan Chemical Industries, Ltd., a 3M Nontenured Faculty Grant, the Lam Research Corporation, the University of Minnesota, and a DuPont Young Professor Award.Notes and references
- K. R. Carter, ACS Nano, 2010, 4, 595–598 CrossRef CAS PubMed.
- C. J. Hawker and T. P. Russell, MRS Bull., 2005, 30, 952–966 CrossRef CAS.
- T. Inoue, D. W. Janes, J. Ren, H. S. Suh, X. Chen, C. J. Ellison and P. F. Nealey, Adv. Mater. Interfaces, 2015, 2, 1500133 CrossRef.
- S. Ji, C.-C. Liu, G. Liu and P. F. Nealey, ACS Nano, 2010, 4, 599–609 CrossRef CAS PubMed.
- D. W. Janes, C. J. Thode, C. G. Willson, P. F. Nealey and C. J. Ellison, Macromolecules, 2013, 46, 4510–4519 CrossRef CAS.
- D. W. Janes, T. Inoue, B. D. McCoy, I. Madan, P. F. Nealey, C. G. Willson and C. J. Ellison, J. Photopolym. Sci. Technol., 2014, 27, 435–440 CrossRef CAS.
- D. Qin, Y. Xia and G. M. Whitesides, Nat. Protoc., 2010, 5, 491–502 CrossRef CAS PubMed.
- J. Y. Kim, H. Kim, B. H. Kim, T. Chang, J. Lim, H. M. Jin, J. H. Mun, Y. J. Choi, K. Chung, J. Shin, S. Fan and S. O. Kim, Nat. Commun., 2016, 7, 12911 CrossRef PubMed.
- J. Y. Kim, J. Lim, H. M. Jin, B. H. Kim, S.-J. Jeong, D. S. Choi, D. J. Li and S. O. Kim, Adv. Mater., 2016, 28, 1591–1596 CrossRef CAS PubMed.
- J. Y. Kim, B. H. Kim, J. O. Hwang, S.-J. Jeong, D. O. Shin, J. H. Mun, Y. J. Choi, H. M. Jin and S. O. Kim, Adv. Mater., 2013, 25, 1331–1335 CrossRef CAS PubMed.
- D. W. Janes, C. B. Kim, M. J. Maher and C. J. Ellison, Langmuir, 2016, 32, 6940–6947 CrossRef CAS PubMed.
- D. W. Janes, M. J. Maher, G. T. Carroll, D. M. Saylor and C. J. Ellison, Macromolecules, 2015, 48, 8361–8368 CrossRef CAS.
- A. P. Lane, M. J. Maher, C. G. Willson and C. J. Ellison, ACS Macro Lett., 2016, 5, 460–465 CrossRef CAS.
- M. J. Maher, C. M. Bates, G. Blachut, M. C. Carlson, J. L. Self, D. W. Janes, W. J. Durand, A. P. Lane, C. J. Ellison and C. G. Willson, ACS Macro Lett., 2014, 3, 824–828 CrossRef CAS.
- R. D. Peters, X. M. Yang, T. K. Kim, B. H. Sohn and P. F. Nealey, Langmuir, 2000, 16, 4625–4631 CrossRef CAS.
- A. M. Mayes, T. P. Russell, V. R. Deline, S. K. Satija and C. F. Majkrzak, Macromolecules, 1994, 27, 7447–7453 CrossRef CAS.
- C.-C. Liu, A. Ramírez-Hernández, E. Han, G. S. W. Craig, Y. Tada, H. Yoshida, H. Kang, S. Ji, P. Gopalan, J. J. de Pablo and P. F. Nealey, Macromolecules, 2013, 46, 1415–1424 CrossRef CAS.
- C.-C. Liu, C. J. Thode, P. A. Rincon-Delgadillo, G. S. W. Craig, P. F. Nealey and R. Gronheid, J. Vac. Sci. Technol., B, 2011, 29, 06F203 Search PubMed.
- R. P. Kingsborough, R. B. Goodman, K. Krohn and T. H. Fedynyshyn, Proc. SPIE, 2009, 7271, 72712D CrossRef.
- J. H. Cho, R. Katsumata, S. X. Zhou, C. B. Kim, A. R. Dulaney, D. W. Janes and C. J. Ellison, ACS Appl. Mater. Interfaces, 2016, 8, 7456–7463 CAS.
- E. Han, K. O. Stuen, Y.-H. La, P. F. Nealey and P. Gopalan, Macromolecules, 2008, 41, 9090–9097 CrossRef CAS.
- C.-C. Liu, E. Han, M. S. Onses, C. J. Thode, S. Ji, P. Gopalan and P. F. Nealey, Macromolecules, 2011, 44, 1876–1885 CrossRef CAS.
- R. Gronheid, P. Rincon Delgadillo, H. Pathangi, D. Van den Heuvel, D. Parnell, B. T. Chan, Y.-T. Lee, L. Van Look, Y. Cao, Y. Her, G. Lin, R. Harukawa, V. Nagaswami, L. D'Urzo, M. Somervell and P. Nealey, Proc. SPIE, 2014, 9049, 904905 CrossRef.
- E. Kim, H. Ahn, S. Park, H. Lee, M. Lee, S. Lee, T. Kim, E.-A. Kwak, J. H. Lee, X. Lei, J. Huh, J. Bang, B. Lee and D. Y. Ryu, ACS Nano, 2013, 7, 1952–1960 CrossRef CAS PubMed.
- S. Ji, C.-C. Liu, W. Liao, A. L. Fenske, G. S. W. Craig and P. F. Nealey, Macromolecules, 2011, 44, 4291–4300 CrossRef CAS.
- H. Pathangi, V. Vaid, B. T. Chan, N. Vandenbroeck, J. Li, S. E. Hong, Y. Cao, B. Durairaj, G. Lin, M. Somervell, T. Kitano, R. Harukawa, K. Sah, A. Cross, H. Bayana, L. D'Urzo and R. Gronheid, Proc. SPIE, 2016, 9777, 97770G CrossRef.
- L. Wan, R. Ruiz, H. Gao and T. R. Albrecht, ACS Nano, 2017, 11, 7666–7673 CrossRef CAS PubMed.
- O. Guiselin, Europhys. Lett., 1992, 17, 225–230 CrossRef CAS.
- Y. Fujii, Z. Yang, J. Leach, H. Atarashi, K. Tanaka and O. K. C. Tsui, Macromolecules, 2009, 42, 7418–7422 CrossRef CAS.
- C. J. Durning, B. O'Shaughnessy, U. Sawhney, D. Nguyen, J. Majewski and G. S. Smith, Macromolecules, 1999, 32, 6772–6781 CrossRef CAS.
- T. P. Russell, G. Coulon, V. R. Deline and D. C. Miller, Macromolecules, 1989, 22, 4600–4606 CrossRef CAS.
- A. P. Lane, X. Yang, M. J. Maher, G. Blachut, Y. Asano, Y. Someya, A. Mallavarapu, S. M. Sirard, C. J. Ellison and C. G. Willson, ACS Nano, 2017, 11, 7656–7665 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- J. M. G. Cowie, M. A. Mohsin and I. J. McEwen, Polymer, 1987, 28, 1569–1572 CrossRef CAS.
- H. Cho, H. Park, T. P. Russell and S. Park, J. Mater. Chem., 2010, 20, 5047–5051 RSC.
- M. J. Maher, C. M. Bates, G. Blachut, S. Sirard, J. L. Self, M. C. Carlson, L. M. Dean, J. D. Cushen, W. J. Durand, C. O. Hayes, C. J. Ellison and C. G. Willson, Chem. Mater., 2014, 26, 1471–1479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7me00106a |
| This journal is © The Royal Society of Chemistry 2017 |