
Antibiotic gold: tethering of antimicrobial peptides to gold nanoparticles maintains conformational flexibility of peptides and improves trypsin susceptibility†
Parvesh
Wadhwani‡
a,
Nico
Heidenreich‡
b,
Benjamin
Podeyn
b,
Jochen
Bürck
a and
Anne S.
Ulrich
 *ab
*ab
aKarlsruhe Institute of Technology (KIT), 1Institute of Biological Interfaces (IBG-2) P.O.B. 3640, D 76021 Karlsruhe, Germany. E-mail: Anne.Ulrich@kit.edu
bKIT, 2Institute of Organic Chemistry & CFN, Fritz-Haber-Weg 6, D-76131 Karlsruhe, Germany
First published on 9th March 2017
Abstract
Peptide-coated nanoparticles are valuable tools for diverse biological applications, such as drug delivery, molecular recognition, and antimicrobial action. The functionalization of pre-fabricated nanoparticles with free peptides in solution is inefficient either due to aggregation of the particles or due to the poor ligand exchange reaction. Here, we present a one-pot synthesis for preparing gold nanoparticles with a homogeneous distribution that are covered in situ with cationic peptides in a site-selective manner via Cys-residue at the N-terminus. Five representative peptides were selected, which are known to perturb cellular membranes and exert their antimicrobial and/or cell penetrating activity by folding into amphiphilic α-helical structures. When tethered to the nanoparticles at a single site, all peptides were found to switch their conformation from unordered state (in aqueous buffers) to their functionally relevant α-helical conformation in the presence of model membranes, as shown by circular dichroism spectroscopy. The conjugated peptides also maintained the same antibacterial activity as in the free form. Most importantly, when tethered to the gold nanoparticles the peptides showed an enormous increase in stability against trypsin digestion compared to the free forms, leading to a dramatic improvement of their lifetimes and activities. These findings suggest that site-selective surface tethering of peptides to gold nanoparticles has several advantages: (i) it does not prevent the peptides from folding into their biologically active conformation, (ii) such conjugation protects the peptides against protease digestion, and (iii) this way it is possible to prepare stable, water soluble antimicrobial nanoparticles as promising antibacterial agents.
Introduction
The increasing prevalence of multidrug resistant bacterial strains calls for the development of new antibiotic agents and antimicrobial surfaces.1–3 Cationic amphiphilic peptides have gained much attention as drug candidates, as polypeptides can nowadays be economically prepared on a large scale, either by chemical synthesis or by fermentation.4 It is well documented that antimicrobial peptides fold into an amphiphilic structure upon binding to the membrane and when acting on bacterial membranes, these agents can induce instantaneous permeabilization, such that the bacteria are unlikely to develop resistance.5 Much effort has been spent to optimize the antimicrobial activity of various natural peptides and designer sequences, and to minimize their toxic side effects on the host. One of the remaining concerns of peptide-based drugs, however, is their intrinsic instability towards proteolytic enzymes.6,7 Most antimicrobial sequences contain basic Arg or Lys side chains,8–10 so they are particularly susceptible to serine proteases such as trypsin, which can cause rapid degradation even before the bacterial targets are reached.7,11 Therefore, in order to exploit the full potential of cationic amphiphilic peptides it is important to stabilize these peptides against proteases such as trypsin. Chemical attempts to avoid protease degradation include, for example, modification of sites prone to protease attack,12–16 the use of unnatural homologues like ornithine17,18 or N-methylated amino acids,12,19 cyclization of the backbone,20,21 or inversion of the stereochemistry into all-D-enantiomers.21,22Peptide-covered gold nanoparticles have found increasing use in biomedical applications such as molecular recognition,23 nuclear targeting,24 and as protein mimicking agents.25 Only few approaches are available to obtain gold nanoparticles that are decorated with the desired cationic peptides26–29 and there are only handful of reports where antimicrobial peptides have been directly attached to gold nanoparticles.30–32 The two most common modes of attaching a peptide to gold involve either a labile Au–N bond (as in Lys, for easy release of the drug) or a relatively stable covalent Au–S bond (for surface functionalization).33,34 Most often the peptide–gold conjugates are invariably obtained either by ligand exchange reaction lasting over weeks, or by hydrophobic interactions or by unspecific interaction of the peptides bearing free Lys (and other) side chains to the gold surface. In other words, in all these methods the peptides are first synthesized, cleaved from the solid support which removes their side chain protection and are later conjugated to the gold nanoparticles. Thus, any of the above described synthetic approaches (ligand exchange or direct conjugation) where the Lys or Arg side chains are free, may allow these side chains to also interact favorably with gold surface thereby leading to an uncontrolled mode of attachment especially if more than one Lys is present in the peptide as is the case for most antimicrobial peptides used here. The reaction is usually performed in water where most of the antimicrobial peptides are usually unstructured. This situation leads to attachment of a randomly folded peptide to the gold core as evidenced previously.32,35,36 This implies if the peptide in question does not need any particular structural folding for the function (unlike here: amphiphilic structure for antimicrobial activity) these methods are still well suited but they cannot be generalized for most of the membrane active peptides where folding into an amphiphilic structure is a pre-requirement for their function (antimicrobial activity).8,37–40 As a result most previous reports have either focused on synthesis strategies to produce peptide–gold conjugates,26,28,36,41 or they present biophysical data on the functionalized systems.27,42,43 Most biological and biophysical assays require peptide quantification and there are no straightforward methods available for quantification of the peptide bound to nanoparticles especially when the peptides are devoid of Tyr or Trp residues. Thus, to overcome all these hurdles, here, we demonstrate a strategy for synthesis (Fig. 1) and quantification of antimicrobial peptides tethered to gold nanoparticles in a site selective manner.
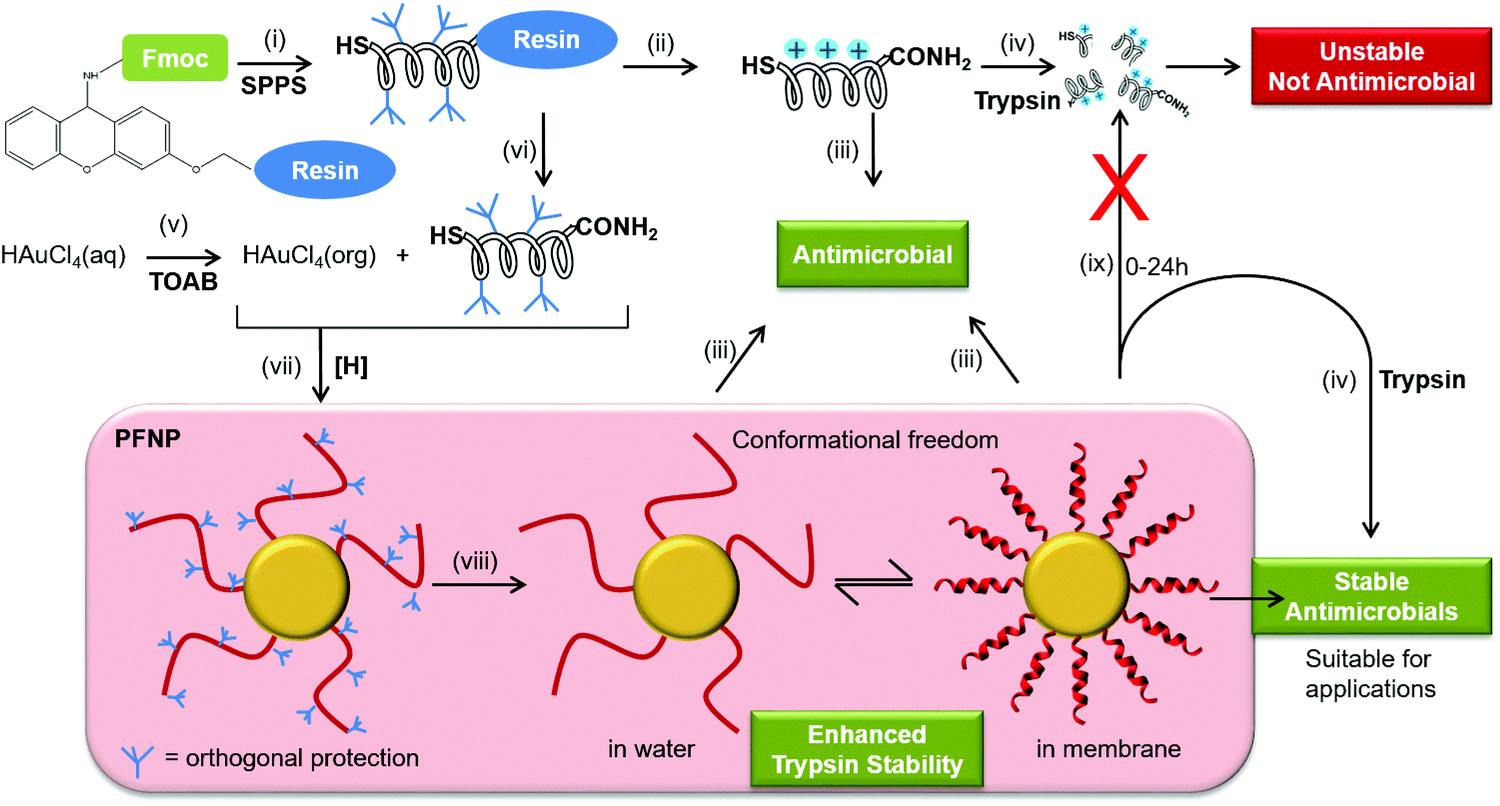 | ||
Fig. 1 Schematic representation to obtain antimicrobial peptides tethered to gold nanoparticles where peptides show conformational freedom and enhanced stability towards trypsin degradation upon attachment to a gold surface. (i) Fmoc-solid phase peptide synthesis (SPPS) was carried out on Sieber amide resin using HBTU/HOBt/DIPEA to obtain antimicrobial peptides with cysteine at the N-terminus. (ii) Regular cleavage of peptides from solid support was done using trifluoroacetic acid/triisopropylsilane/water (92.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2.5%, v/v) followed by HPLC purification. (iii) Purified free peptides were used to perform CD and antimicrobial activity (minimal inhibitory concentration (MIC) determination). (iv) Trypsin treatment of free peptide resulted in fast degradation of peptides (15 min) making them unsuitable for any biological application. (v) Tetraoctylammonium bromide (TOAB) was used as a phase-transfer catalyst to transfer gold ions into toluene solution. (vi) Mild cleavage reaction was performed in presence of trifluoroacetic acid (1%, v/v) in dichloromethane to detach fully side chain protected peptides from the solid support having only one free cysteine at the N-terminus to react with gold. (vii) Gold ions in the organic phase were reduced by NaBH4 in presence of side chain protected peptides, in order to attach the peptides exclusively via their N-terminus to obtain peptide functionalized gold nanoparticles (PFNP). (viii) Cleavage of the orthogonal protection groups present on peptides was performed after attaching the protected peptides as described in step (ii) followed by solvent extraction to remove any free or unbound peptides before performing CD spectroscopy and antimicrobial assay (MIC determination) on the immobilized peptides as described in step (iii) where the peptides bound to PFNP should be able to fold into amphiphilic helices and remain as functional in bacterial killing as the free peptides in solution. (ix) Peptides tethered to the gold nanoparticles remain stable up to 24 h. :5:2.5%, v/v) followed by HPLC purification. (iii) Purified free peptides were used to perform CD and antimicrobial activity (minimal inhibitory concentration (MIC) determination). (iv) Trypsin treatment of free peptide resulted in fast degradation of peptides (15 min) making them unsuitable for any biological application. (v) Tetraoctylammonium bromide (TOAB) was used as a phase-transfer catalyst to transfer gold ions into toluene solution. (vi) Mild cleavage reaction was performed in presence of trifluoroacetic acid (1%, v/v) in dichloromethane to detach fully side chain protected peptides from the solid support having only one free cysteine at the N-terminus to react with gold. (vii) Gold ions in the organic phase were reduced by NaBH4 in presence of side chain protected peptides, in order to attach the peptides exclusively via their N-terminus to obtain peptide functionalized gold nanoparticles (PFNP). (viii) Cleavage of the orthogonal protection groups present on peptides was performed after attaching the protected peptides as described in step (ii) followed by solvent extraction to remove any free or unbound peptides before performing CD spectroscopy and antimicrobial assay (MIC determination) on the immobilized peptides as described in step (iii) where the peptides bound to PFNP should be able to fold into amphiphilic helices and remain as functional in bacterial killing as the free peptides in solution. (ix) Peptides tethered to the gold nanoparticles remain stable up to 24 h. | ||
Using this approach, the peptides should be able to retain their backbone conformational freedom allowing them to fold into their native amphiphilic α-helical structure in lipid environment where they should fully retain their antimicrobial activity and might gain enhanced stability towards trypsin digestion. To examine and compare the behavior of several different membrane-active peptides when tethered to gold nanoparticles, we selected five well-known amphiphilic α-helical representatives (Table 1).9,10,38,44–46 These include (i) the naturally occurring 21-residue antimicrobial PGLa from the skin of an African frog,44 (ii) another 21-mer sequence MSI-103 that was derived from PGLa with a higher therapeutic index,45 (iii) a related 18-mer cell penetrating peptide MAP (model amphiphilic peptide) that also has a high antimicrobial activity,9 (iv) the 11-mer cecropin–melittin hybrid BP100 that exhibits both antimicrobial and cell penetrating properties,10,46 and (v) a designated cell penetrating peptide TP10 (transportan-10) that is a 21-mer chimera derived from the neuropeptide galanin and wasp venom mastoparan.38 All these peptides are known to exhibit a pronounced antimicrobial activity against Gram-positive and Gram-negative bacteria, irrespective of their origin (naturally occurring or designer made) or classification (antimicrobial or cell penetrating).10,47
| Peptide | Amino acid sequence | Peptide amounta [mass%] |
|---|---|---|
| a Determined using quantitative amino acid analysis, x = CGGGGG. | ||
| x-PGLa | x-GMASKAGAIAGKIAKVALKAL-NH2 | 26 ± 2 |
| x-MSI103 | x-KIAGKIAKIAGKIAKIAGKIA-NH2 | 28 ± 4 |
| x-MAP | x-KLALKLALKALKAALKLA-NH2 | 17 ± 1 |
| x-BP100 | x-KKLFKKILKYL-NH2 | 24 ± 3 |
| x-TP10 | x-AGYLLGKINLKALAALAKKIL-NH2 | 24 ± 2 |
Experimental
Materials
Fmoc-protected amino acids were purchased either from Novabiochem or from Iris Biotech, Germany. Cl-HOBt (1-hydroxybenzotriazol), chlorotritylchlorid-resin, diisopropylethylene-diamine and HBTU (1H-benzotriazol-1-yl-1,1,3,3-tetramethyluronium-hexafluorophosphat) were purchased from Iris Biotech GmbH, Germany. Dichloromethane and trifluoroacetic acid were purchased from Biosolve, Netherlands. Diethylether, dimethylene formamide, ethanol, methanol, sodium borohydride (NaBH4), N-methyl-pyrrolidone and toluene were purchased from VWR, Germany. Tetrachloroaurate(III)–trihydrate (HAuCl4·3H2O), triisopropyl silane (TIS) and tetraoctyl-ammonium bromide (TOAB) were purchased from Sigma-Aldrich, Germany. Piperidine and Müller Hinton broth were purchased by Carl Roth, Germany. Trypsin was purchased from VWR, Germany. Water was purified with Milli-Q-Guard 2 (Biocel Millipore).Peptide synthesis and purification
All peptides were synthesized on an automated peptide synthesizer (Multipep XP Syro II from Multisyntech GmbH). Peptides with an amidated C-terminus were synthesized using a Sieber-amide resin, following standard Fmoc-protocols as described previously.48 100 μmol resin was placed into a reaction vessel and was allowed to swell using a mixture of DMF and DCM (50%, v/v) for 1 h. The Fmoc group was removed by using piperidine (20% v/v in DMF), and each amino acid was coupled using a 4-fold excess of Fmoc-protected acid and HOBt/HBTU in the presence of DIPEA for 1 h. To test the quality of the synthesized peptides and to perform CD and antimicrobial test, a sufficient fraction of the resin was cleaved using TFA/triisoproplysilane/water as described previously.49,50 The bulk of the synthesized peptides were cleaved from the resin with TFA (1%, v/v) in DCM for 60 min, such that the side chain protections remained unaffected. Crude peptides were obtained by lyophilisation in acetonitrile/water (1:1, v/v). For CD and antimicrobial tests, the peptides were purified on a reverse phase preparative C18 column using water acetonitrile gradients containing 5 mM HCl as previously reported.10
Synthesis of peptide functionalized gold nanoparticles
An aqueous solution (5 mL) containing HAuCl4 (15 mg) was added to TOAB (100 mg) in 20 ml toluene. The fully protected x-peptide carrying an N-terminal cysteine (20 mg) was added to this mixture. An additional 15 ml of toluene was added and the mixture was slowly stirred until all gold was transferred to the organic phase. The aqueous phase was discarded. After further stirring for 15 min, a freshly prepared solution of sodium borohydride (NaBH4, 10 mg in water) was added dropwise. The colour of the solution gradually changed to red and a dark red precipitate appeared. The conjugates were separated from the reaction solution by centrifugation (14000 rpm for 10 min), and were washed thrice with water, ethanol and methanol to remove excess gold ions and free peptides. The precipitate was treated with TFA to cleave the side chain protections as previously described to obtain the PFNP.10
Transmission electron microscopy
The size distribution of the AuNP-conjugates was obtained by transmission electron microscopy (TEM) with a Zeiss T-109 microscope. Samples were prepared by suspending the conjugates in water and subsequent evaporation on a TEM copper grid coated with carbon. The images were analysed with Image-J software.Dynamic light scattering
The average size of the PFNP was determined by dynamic light scattering (DLS) using Zetasizer Nano Series (Malvern Instruments Ltd). Series of measurements were performed to obtain the size distribution. Each measurement was repeated thrice and the data was analysed via the software provided by the Malvern Instruments.Circular dichroism spectroscopy
The secondary structure of the peptides tethered to the gold nanoparticles was investigated by circular dichroism spectroscopy (CD) using a Jasco J-810 spectropolarimeter. As a membrane-mimicking environment we used either a 1:1 mixture of 50% trifluoroethanol in 10 mM phosphate buffer, or small unilamelar vesicles (SUVs) composed of a mixture of 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC) and 1,2-dimyristoyl-sn-glycero-3-phosphatidylglycerol (DMPG) in the molar ratio of 3:1. SUVs were produced by 3 × 5 minutes sonification. The measurements were done in quartz glass cuvettes with 0.1 cm optical path length. The CD spectra were smoothed with the Jasco spectra analysis software.
Amino acid analysis
Amino acid analysis was performed to quantify the amount of peptides present in the PFNP, by hydrolysing the peptide in the presence of 6 N HCl at 110 °C for 48 h. This was followed by post-column derivatization with ninhydrin and the complex was detected at 570 nm, using an amino acid analyser (model S 433, Sykam GmbH, Germany). Amino acid standards were purchased from Sykam. Briefly, 0.5 mg PFNP were hydrolysed with 6 N HCl, and excess HCl was removed under vacuum. The resulting solid was dissolved in 1.5 ml sample dilution buffer, and 150 μl was injected over an ion exchange column to separate the constituent amino acids, which were subsequently derivatized by ninhydrin. Each quantification step was repeated thrice, and the statistical error in determination of peptide amount is indicated in Table 1.Antimicrobial activity
Minimum Inhibitory Concentration (MIC) assay was performed to access the antimicrobial activity of the free and bound peptides. MIC values were determined in a sterile 96 well plate against bacterial strains E. coli DSM 1103, B. subtilis DSM 346, S. aureus DSM 1104 and M. luteus DSM 1790 in a 96-well microtiter plate, as previously described.10 The peptide stock solutions were prepared in water and successively diluted by using Müller-Hinton broth (pH = 7.3) to obtain the desired concentration. The assay was validated using three standard antibiotics as a reference. The microtiter plates were incubated overnight and stained with Alamar Blue™, where a pink colour indicates living cells while a blue colour represents dead bacteria.Trypsin stability tests
Trypsin stability tests were performed as previously described, with slight modifications.11 Briefly, a stock solution of the peptides or PFNP was dissolved in NH4HCO3 buffer (5 mM, pH 7.8) to a final concentration of 1 mg ml−1 for free peptides and 3 mg ml−1 for the PFNP, respectively. 100 μl of this solution was added to 900 μl NH4HCO3 buffer (5 mM, pH = 7.8), followed by addition of 10 μl of trypsin (50 μg ml−1) in NH4HCO3 buffer. The reaction mixture was incubated at 37 °C using a heating block. At regular intervals an aliquot was withdrawn to determine the residual intact peptide. To inhibit the activity of trypsin, 150 μl of the reaction solution were mixed with 10 μl of a benzamidine hydrochloride solution (25 mM in MeOH) at any given point of time, vortexed thoroughly and analysed using LC-MS.Results and discussion
Synthesis and characterization of peptide functionalized gold nanoparticles (PFNP)
Peptides were synthesized using standard Fmoc solid phase protocols, using Sieber amide resin as previously described.9,10,38 Each sequence was extended at the N-terminus with a cysteine-pentaglycyl linker (CG5, denoted by “x” in the peptide names) to serve as a spacer between the active peptide and the gold surface. In standard antimicrobial activity assays, we found that this modification slightly diminished the antimicrobial activity of the free x-peptides compared to the original forms (Table S1, ESI†). Interestingly, this slight loss of activity was regained upon tethering them to the nanoparticles (see below). After synthesis, peptides were cleaved from the resin under mild conditions to retain the side-chain protections such that the resulting peptides contain only one reactive site to couple with gold nanoparticles. In addition, small aliquots were also cleaved with the usual harsh procedure40 to control the product quality via analytical HPLC coupled to a mass spectrometer. All crude peptides were pure to >85% and could thus be used without further purification to prepare the functionalized gold nanoparticles in a one-pot synthesis. For circular dichroism spectroscopy and for antimicrobial test the peptides were purified as described in the Experimental section.The peptide functionalized gold nanoparticles (PFNP) were characterized using UV-VIS spectroscopy and transmission electron microscopy (TEM). The gold conjugates had a characteristic red-violet color. The absorption bands around 525 to 555 nm are indicative of the formation of peptide–gold nanoparticle conjugates, as previously reported (Fig. 2A).51 The TEM images in Fig. 2B show a homogeneous size distribution with a mean diameter of around 5–7 nm (Fig. S1, ESI†), as supported by dynamic light scattering (DLS) data (Fig. 2C and Table S2, ESI†).
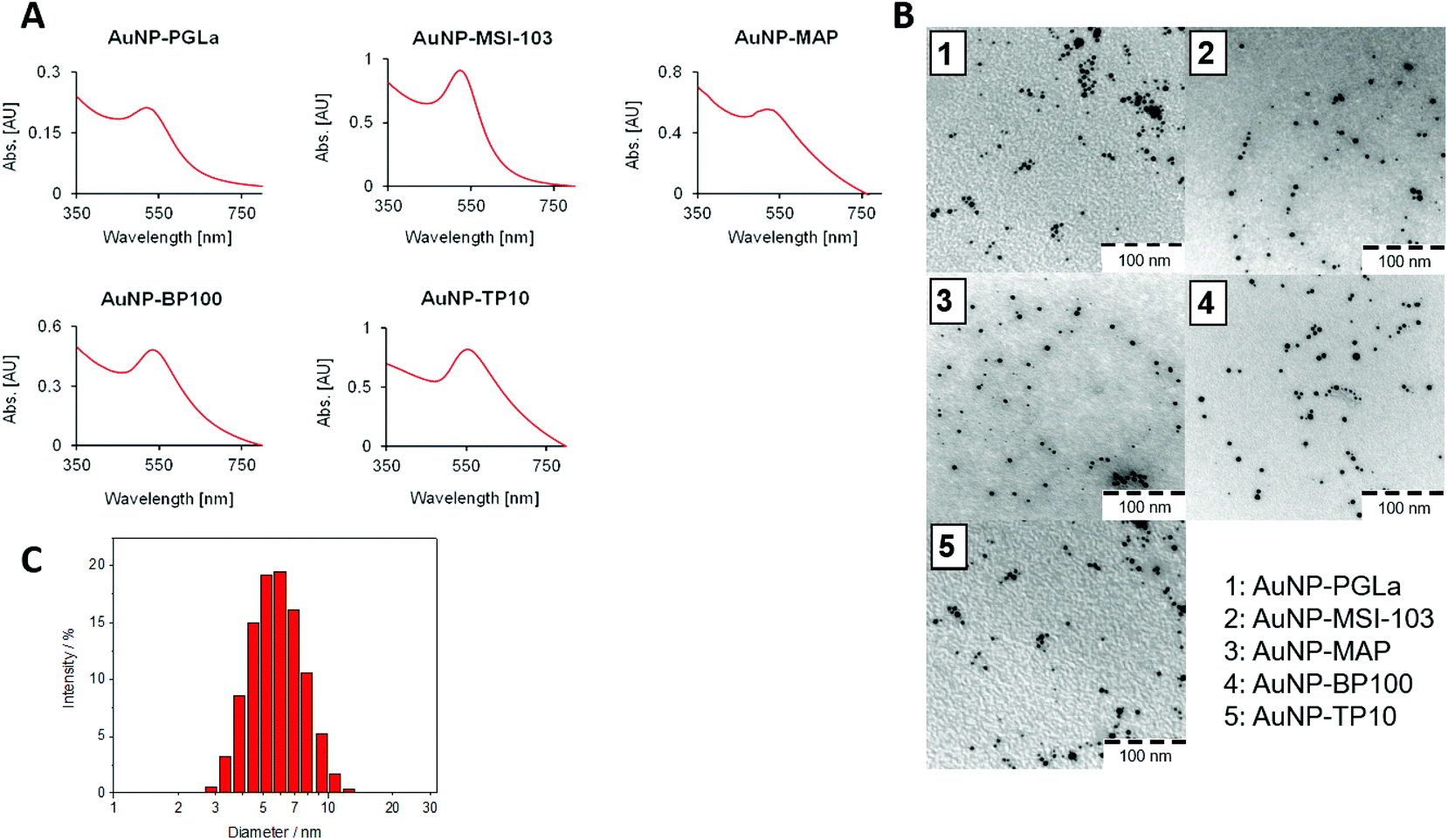 | ||
| Fig. 2 (A) UV-VIS spectra of PFNP. All conjugates show a characteristic red color, and plasmon resonance with a maximum around 525–555 nm. (B) TEM images of PFNP, showing a homogeneous size distribution (scale bar = 100 nm) and (C) DLS data showing a series of measurements illustrating a narrow size distribution of functionalized gold nanoparticles (here shown for PGLa) with a mean diameter of around 5–7 nm measured as described previously. | ||
It is important to note that all of the peptides used here are rich in basic amino acid Lys which is charged and crucial for the antimicrobial action. Amino groups are known to interact favorably with free gold surfaces, and cationic peptides have been reported to cause precipitation of nanoparticles.25,34,43 In traditional synthesis routes based on thiol-capping agent exchange reactions, which often results in either poor ligand exchange or in an inhomogeneous mixture where the peptides are bound to the gold core either via Lys side chains or via the N-terminal amino group as illustrated in Fig. 3A and C.51,52 Although there are several reports where peptides are coated,26 integrated,31 self-assembled,30 or even directly bonded onto gold nanoparticles,27,28,32,36 but to the best of our knowledge there is no synthetic report where peptides are site specifically conjugated to gold nanoparticles as shown in Fig. 3B, such that they have the necessary conformational freedom (Fig. 1) to fold into their amphipathic helical and functional structure (Fig. 3C) and to avoid an inactive state (Fig. 3A) where the charged Lys residues necessary for bacterial cell death are instead bonded to the gold core and cannot contribute to the peptide conformation necessary for bacterial cell death. Most previous reports have either focused on synthesis strategies to produce peptide–gold conjugates utilizing above described methods, or by using peptides with no side chain protection, or they present biophysical data on the functionalized systems.27,36,42,43 We thus developed the present protocol (Fig. 1) to overcome the above mentioned problems with the aim to keep the cationic side chains freely available (Fig. 3C) for interactions with water and/or bacterial membranes such that the resulting conjugates could be optimized as antimicrobial agents.
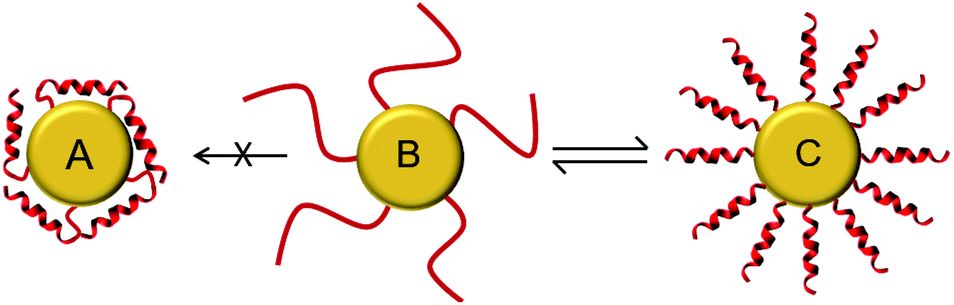 | ||
| Fig. 3 Schematic illustration of peptides tethered to the gold nanoparticle (B). They should be able to fold into functional amphiphilic helices upon interaction with bacterial membranes (C). Any interaction of the side chain amino groups of Lys with the gold surface is not desired (A), as this would interfere with the biological activity (figure not drawn to scale). | ||
For the one-pot synthesis, we optimized the Brust–Schiffrin method28,53 to prepare gold nanoparticles in the presence of the side-chain protected x-peptides that carry a free Cys residue at the extended N-terminus. Details of the procedure are described in the Experimental section. Briefly, an aqueous solution of HAu(III)Cl4 is stirred with TOAB in the presence of toluene, followed by reduction of Au(III) in the presence of the side-chain protected x-peptides using NaBH4 (Fig. 1). The protection groups are subsequently removed with TFA (92.5%) in the presence of triisopropylsilane (5%) and water (2.5%),49 and the product is washed by centrifugation in water/ethanol to remove unbound peptides and salts. This method allows coating the freshly formed gold nanoparticles in situ with the thiol groups present in the x-peptides. There is no need to use any passivating agents such as organic thiols or citrate-ions as are often employed in synthesis protocols.25,53 Using side chain protected peptides our strategy was designed to (i) assure attachment of the x-peptides to the gold core exclusively via the N-terminal Cys, (ii) achieve a high surface coverage, (iii) avoid unspecific interactions of the Lys side chains with the gold core, and (iv) thereby prevent inactivation of the functional peptide sequences. This way, all peptides should be able to fold into the amphipathic structure once their protection groups are removed as illustrated in Fig. 3C, such that the desired biological function would be preserved.
CD spectroscopy
The free antimicrobial peptides investigated here are unfolded in aqueous buffers as shown in Fig. 4A and in presence of bacterial membrane they fold as amphiphilic α-helices to be biologically active, i.e. to bind and lyse bacterial membranes.10,39 To find out whether the peptides can still assume this conformation when they are tethered to the gold surface, we performed circular dichroism (CD) spectroscopy on the PFNP. There are only few reports so far where CD has been used to characterize peptides conjugated to gold nanoparticles.27,36,54Fig. 4A and C respectively show a disordered state of the free and AuNP-bound peptides in water (except for TP10 when bound to gold nanoparticles). Fig. 4C directly confirms the state of the bound peptides as shown in Fig. 3B. To model bacterial membranes, we used a well-known lipid mixture composed of DMPC and DMPG in the molar ratio of 3:1, prepared as small unilamellar vesicles as previously described.40 This corresponds very well to the behavior of the free x-peptides (Fig. S2, ESI†) as well as the native peptides which have been comprehensively described in the past.10,39Fig. 4B and D show the bound peptides are able to fold into a distinct α-helical conformation in the presence of model membranes similar to the free peptides. Given our synthetic methodology using single site of attachment of peptides to the gold and having evidenced bound but yet unordered peptides as shown in Fig. 3B, Fig. 4D strongly supports the state of the peptides described in Fig. 3C. The same α-helical conformation was observed for free peptides, free-x-peptides and for the peptides bound to the gold nanoparticles in the membrane-mimicking solvent 2,2,2-trifluoroethanol (Fig. S3, ESI†). This conformational behavior suggests that our new coupling strategy was successful in reaching the aim outlined in Fig. 3.
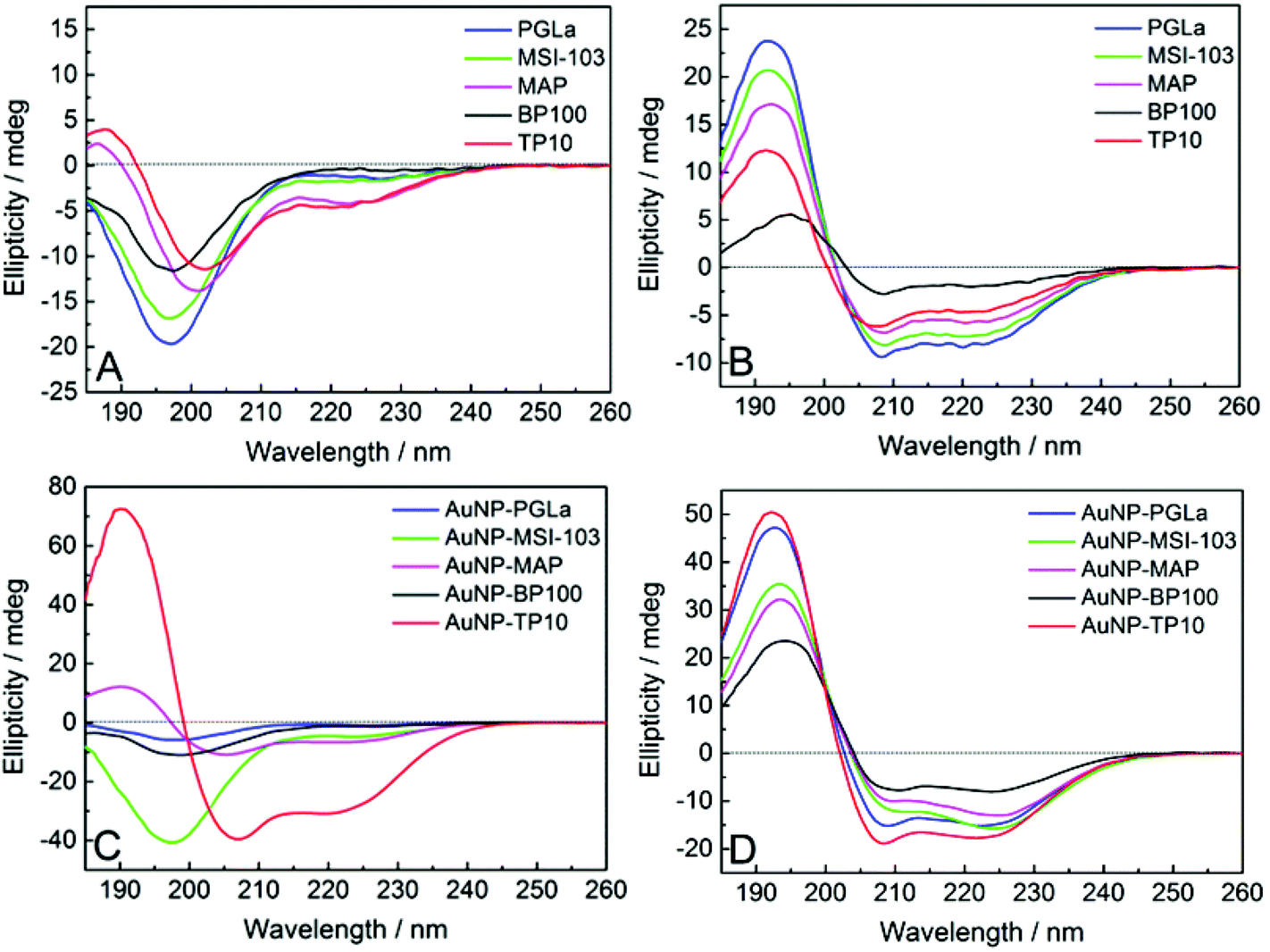 | ||
| Fig. 4 CD spectra of the free peptides (A, B), and when tethered to gold nanoparticles (C, D). In aqueous buffer (A, C) the peptides are largely disordered (except for AuNP-TP10), but in the presence of lipid vesicles composed of DMPC/DMPG (B, D) they become distinctly α-helical (Fig. S2 and S3, ESI†). | ||
Peptide quantification
To compare the biological activity of the tethered peptides with that of the free molecules in a quantitative manner, we needed to find out the amount of peptide present in each PFNP preparation. Although quantification of gold present in the nanoparticles is well documented55,56 there are no straightforward protocols for this purpose in the literature especially for the peptides which are devoid of Tyr and Trp residues as is the case here. This may explain the lack of studies (and hence applications) that require quantification of peptides bound to nanoparticles. To overcome this limitation, we performed quantitative amino acid analysis to determine the mass percentage (see Table 1). Briefly, the conjugates were first treated with 6 N HCl for 24–48 h at 110 °C to hydrolyze the peptides into the constituent amino acids (see Experimental section). The free amino acids were separated over an ion exchange column, followed by post-column derivatization using ninhydrin.57 Interestingly, we did not find any sign of Cys in the amino acid analysis, which must have remained coupled to the gold surface, if not via a covalent bond then via coordinated bond as previously reported.58 Given our synthetic procedure which involves centrifugation of the PFNP twice and thereby removing the uncoupled peptides in supernatant solution, it is safe to assume that all peptide was covalently bound to the nanoparticle and none had remained free in solution. The obtained mass percentages are summarized in Table 1 which can be now used to determine biological activity of the PFNP.Antimicrobial activity
To examine their antimicrobial activities, the minimum inhibitory concentrations (MIC) of the tethered peptides were compared with those of the same amount of free peptide. Four bacterial strains were examined (Gram-negative E. coli DSM 1103, and Gram-positive B. subtilis DSM 346, M. luteus DSM 1790, S. aureus DSM 1104) in standard 2-fold dilution assays that have been previously described.8,10 The results are shown in Fig. 5. Note that a low concentration implies that a peptide is highly active, whereas MIC values ≥256 μg ml−1 indicate a lack of antimicrobial activity. Small differences in the MIC value by a factor of 2 are within the intrinsic error range of this serial dilution assay. With an exception of three cases namely PGLa (for E. coli) and MAP (for S. aureus and M. luteus) all values are either identical or within an acceptable error range of one dilution factor. Thus, all our gold tethered peptides have clearly retained essentially the same activity as the free molecules in solution, when compared on an unbiased weight-to-weight basis of the pure peptidic material. As a negative control we used thioalkane capped gold nanoparticles, which were found to be inactive (data not shown). Therefore, we may conclude that the antimicrobial behavior of the functionalized gold nanoparticles is exclusively due to the presence of the membrane-active peptides tethered onto their surface.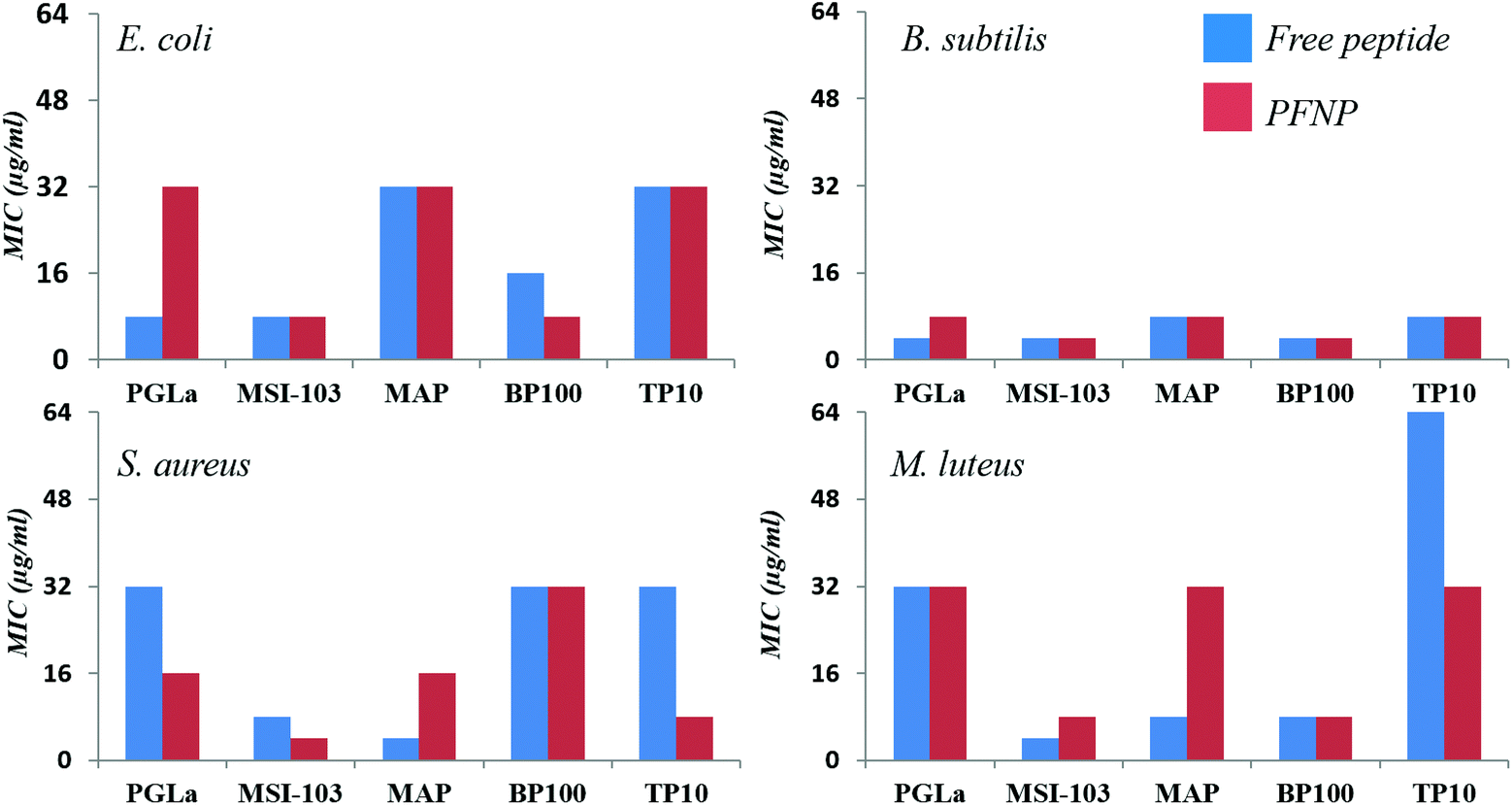 | ||
| Fig. 5 Antimicrobial activity of the free peptides (blue bars on left) in comparison to the peptides tethered to gold nanoparticles (PFNP, red bars on right) for four different bacterial strains showing that free and bound peptides have similar antimicrobial activities. | ||
Trypsin stability
The peptide quantification and the fully preserved antimicrobial activity of the peptide-functionalized nanoparticles promise new applications in vivo as soluble antibacterial agents. However, in any biological context the peptides would need to be protected from protease degradation, which is a major challenge in the development and application of peptidic drugs.59 To examine the protease stability of the tethered peptides, the nanoparticle conjugates were tested against enzymatic degradation by trypsin, using established procedures.11 Both, the PFNP as well as the free peptides for comparison, were dissolved in NH4HCO3 buffer and treated with 500 μg l−1 trypsin. This concentration is slightly above the reported occurrence of trypsin in blood serum (150 and 400 μg l−1).60,61 The actual peptide concentration was set to 50 μM, which is above their minimally required MIC values, and the concentration of the PFNP was adjusted accordingly (i.e. to provide the same mass of peptide). Trypsin is known to cleave proteins and peptides behind Lys or Arg residues, resulting in small fragments of variable length, and invariably resulting in a total loss of activity of our Lys-rich sequences. The resulting peptide fragments are readily detected and characterized by liquid chromatography-mass spectrometry (LC-MS), which was used to monitor the time-dependent degradation of the various preparations. The rapid degradation of the free peptides by trypsin is shown in Fig. 6 which clearly demonstrates the short half-life of the peptides.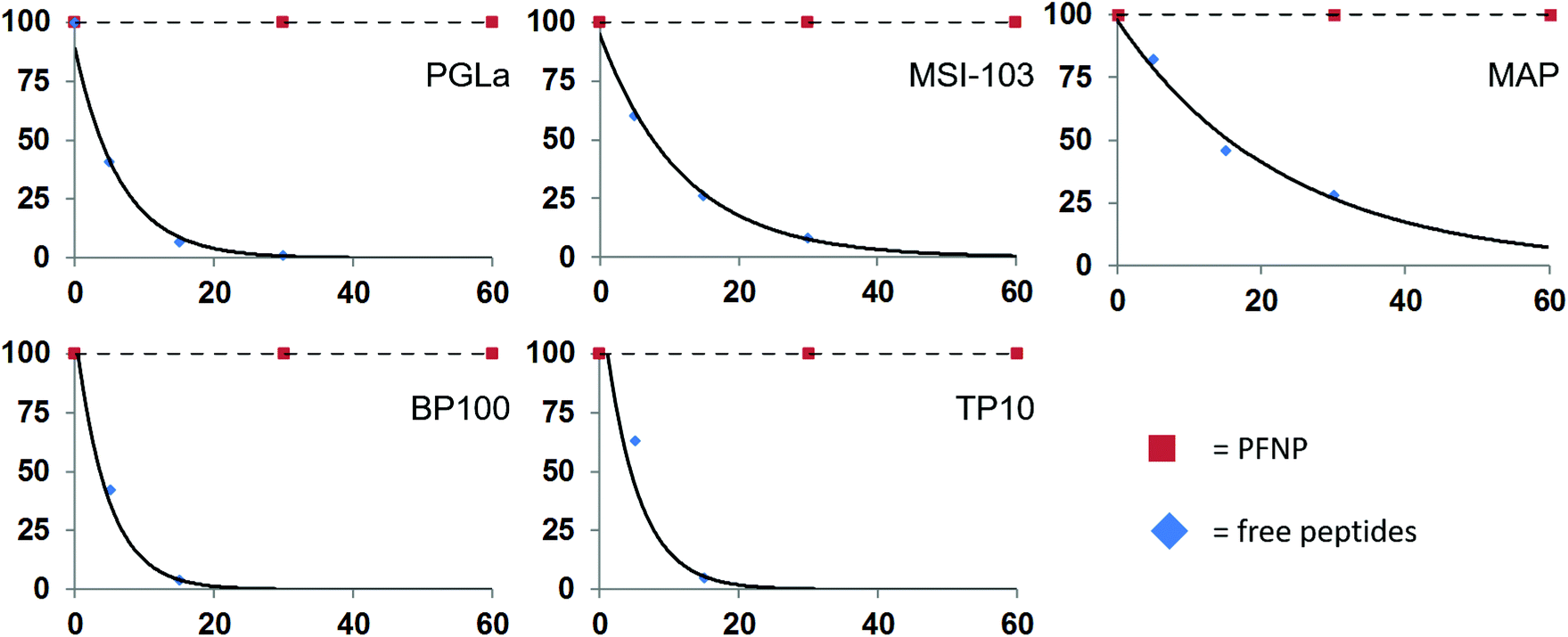 | ||
| Fig. 6 Degradation of the free peptides in the presence of trypsin occurs rapidly within 30 min while the peptide bound to the gold surface (PFNP) are completely stable during this time. | ||
As expected, the free peptides are highly susceptible and showed significant losses (∼50%) already in the first 5–10 minutes.59,61 Almost 95% of the free peptides were degraded within 30 min of trypsin treatment, a time frame which is often claimed to be necessary for killing the bacteria.62–64 We note that at each time point some different fragments of the parent peptide appeared in the LC-MS chromatogram (Fig. S4, ESI†), indicating cleavage at successive sites in each given peptide sequence. Some of the observed fragment masses are indicated in Fig. S5 in the ESI.† In contrast, we found no such peptide fragments when the PFNP were incubated with trypsin. Neither by liquid chromatography nor by sensitive mass spectrometry could we detect any traces of degraded peptides even after 24 h of protease treatment (Fig. S6, ESI†). This observation indicates an enormous stability of the tethered peptides towards trypsin as compared to the free peptides, suggesting that the gold nanoparticles maintain the active peptides firmly on their surface making it difficult for the trypsin to act.
Antimicrobial activity after trypsin incubation
To ultimately prove the stability of the peptides against proteolytic degradation, we therefore incubated the PFNP with trypsin for 0, 2, 6, and 24 h, and we then tested the antimicrobial activity of these trypsin treated conjugates. If the tethered peptides are unstable, they would have been degraded into smaller fragments and PFNP would have lost their antimicrobial activity. On the other hand, if the tethered peptides are protected from protease degradation on the nanoparticle surface, PFNP should still exhibit a high antimicrobial activity (lower MIC values) afterwards.Fig. 7 shows the decline in antimicrobial activity (increasing MIC values) for all five peptide systems, both free and when tethered to the gold nanoparticles, as a function of incubation time with 500 μg l−1 trypsin for B. subtilis DSM 346. It is clear from Fig. 5 and 7A that in absence of any trypsin both free peptides and PFNP are highly active showing MIC in the range of 4–8 μg ml−1. A lower MIC value indicates a lower peptide concentration is required for the inhibition of microbial growth. As expected with the increase in trypsin incubation time to 2 h, the free peptides begin to degrade and Fig. 7B shows that PGLa and TP10 are completely inactive after 2 h with MIC values of over 256 μg ml−1. PFNP, on the other hand are fully active after two hours as seen by the lower MIC values represented by red bars. As the trypsin incubation time is increased to 6 or 24 h, the free peptides were completely degraded and lose their activity. The PFNP, on the other hand, remain much more stable and maintained a high antimicrobial activity even after 6 h and sometimes even after 24 h (MAP and TP10) of incubation with trypsin. Similar data is observed for other Gram-positive and Gram-negative bacterial strains (Fig. S7–S9, ESI†). It appears that given the dense coverage of peptides on the gold nanoparticles there is significant steric hindrance for trypsin to bind into the cleavage site and the reaction is therefore slowed down and less effective as compared to the trypsin cleavage of the free peptides. This is supported by comparing the Fig. 7A and D where the MIC values at 0 min and that after 24 h of trypsin treatment do show a slight increase in the values suggesting that degradation by trypsin is sluggish for the peptides bound to gold nanoparticles. We further note the rapid degradation of the free peptides as shown in Fig. 6 where the trypsin cleavage was performed in NH4HCO3 buffer at an optimal pH of 7.8 as compared to the slow trypsin cleavage performed under the conditions of the MIC assay (i.e. in Muller-Hinton broth at a pH of 7.3). Trypsin is known to have pH dependent activity as previously demonstrated65 and given the composition of Muller Hinton broth it is not surprising that the degradation of the free peptides under these latter conditions might be compromised which increases their lifetime from 30 min to about 2 h. Nevertheless, between 2 and 6 h, all free peptides were completely degraded indicating that trypsin might be slow under these condition but continues to cleave the free peptides which makes them inactive as indicated by the high values of the blue bars in Fig. 7B and C. On the other hand, the red bars representing PFNP (Fig. 7D), continue to show high antimicrobial activity even after 24 h of trypsin exposure and clearly demonstrate the advantage of the peptide when bound to the gold core. Thus, the intrinsic problem of the low bioavailability of peptidic drugs can be overcome by tethering them to a gold core provided the peptides do not lose their biological function (here antimicrobial activity). These data demonstrate that the peptides tethered to the gold nanoparticle have gained a strongly enhanced stability against trypsin degradation and can now be further optimized for several biomedical applications requiring disinfection or equipment which needs sterile conditions.
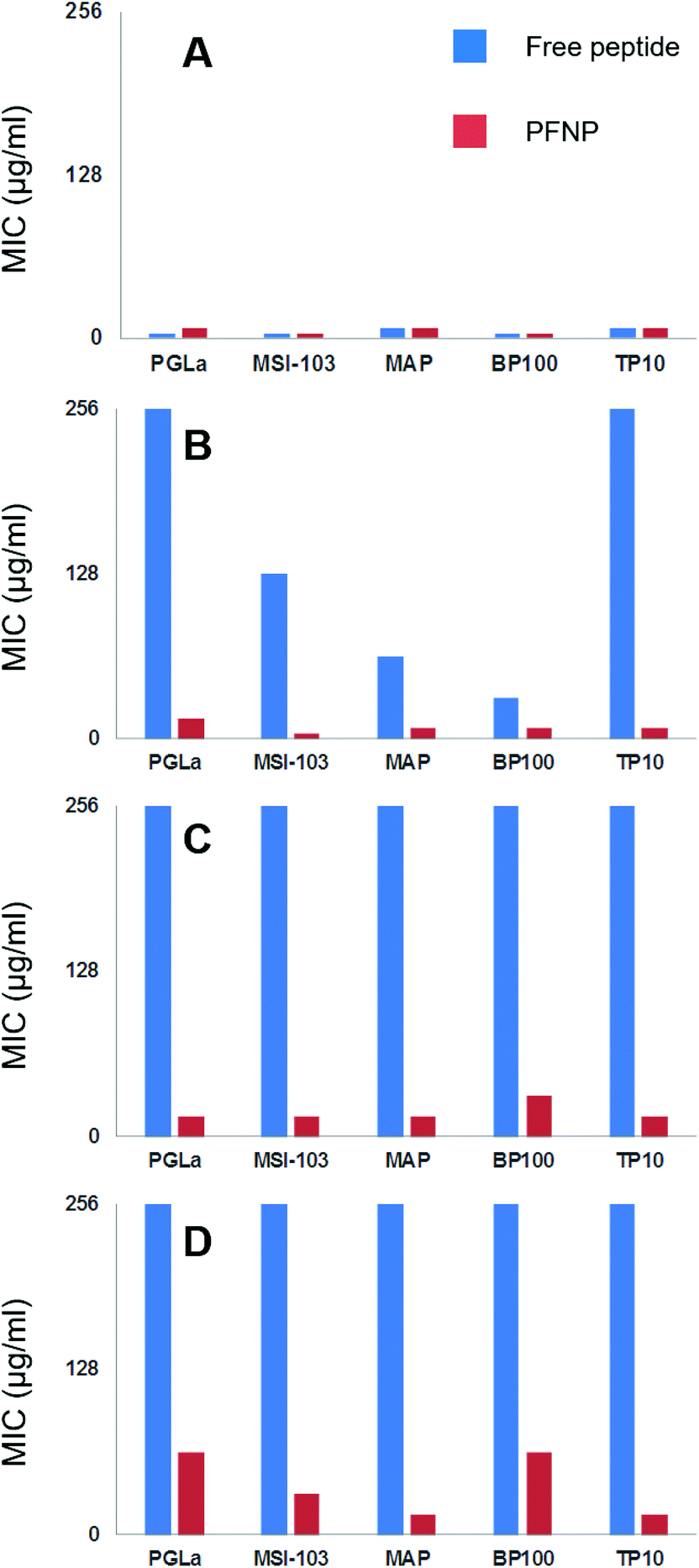 | ||
| Fig. 7 Decline in antimicrobial action (seen as the increase in the MIC values) as a function of trypsin incubation time (A = 0 min, B = 2 h, C = 6 h and D = 24 h) investigated on five free peptides (blue bars, left) and on PFNP (red bars on right). Free peptides are fully inactive within 2–6 h as seen by higher bars (blue) indicating loss of activity whereas PFNP retain their antimicrobial action even up to 24 h and show only slight decline in antimicrobial action. Each panel shows MIC values (μg ml−1) for B. subtilis DSM 346. For similar data on other bacterial strains see Fig. S7–S9 in ESI.† | ||
Conclusions
In summary, we have prepared and characterized some novel peptide functionalized gold nanoparticles that (i) are water soluble, (ii) bear conformationally flexible peptides, (iii) are antimicrobially active, and (iv) are proteolytically stable towards trypsin. For synthesis, the Brust–Schiffrin method was modified to avoid the generation of side products, and to use a one-pot reaction without the need for capping agents. Two main advantages of this new procedure are (i) tethering exclusively at one terminus and (ii) its universal applicability to virtually any peptide sequence. The possibility to prepare such mono-conjugated water soluble gold nanoparticles gives the tethered peptides enough conformational freedom to switch their secondary structure depending on the environment, as illustrated here by CD spectroscopy (Fig. 4). Being able to fold into their functionally relevant amphiphilic α-helical structure in a membraneous environment, the peptides also retained their full antimicrobial activity, as documented here against various bacterial strains (Fig. 5). Amino acid analysis allows determination of peptide concentration especially for the peptides which contain no Tyr or Trp. Finally, our results have highlighted for the first time a vast increase in the stability of the tethered peptides, as they are protected from protease degradation by trypsin for several hours or a day, as compared to a few minutes in the case of the free peptides (Fig. 7). It thus appears that a high surface coverage, and possibly the high charge density of the peptides on the nanoparticles is sufficient to prevent them being digested by trypsin. Although there are several methods to tether antimicrobial peptides to various surfaces,2 it is not clear yet as to how the nanoparticle-tethered peptides actually permeabilize the inner bacterial membranes, as it has been the case for the free peptides but it seems likely that membrane is the target as previously evidenced.66 Given that the functionalized nanoparticles have a comparatively small diameter of <10 nm, it is possible that they might still reach the plasma membrane. There, the high positive charge density may either lead to lipid clustering which causes depolarization of the membrane thereby influencing membrane integrity as seen for some of the peptides used here,10,29,37 or the membrane could become remodeled around the nanoparticle which could result in local leakage. On the other hand it is highly likely that certain PFNP could penetrate the cells as previously reported56,67 and interfere with the intracellular machinery of the bacteria particularly given the significant overlap in properties of antimicrobial and cell-penetrating peptides used here.68 By selecting the right cell penetrating sequence, these conjugates can be optimized for nanoparticle based drug delivery systems or as biosensors for the detection of pathogens at lower infection level before it is too late.69 The exact mechanism of bacterial killing is beyond the scope of the present study, the advantage of our gold nanoparticles is the fact that they are water soluble, they offer enhanced protease stability, they can be detected in a biological tissue by electron microscopy and possibly also by the intrinsic fluorescence of the gold core. While the peptide stability towards other proteases must be considered before a suitable application is conceived,70 the present results show a promising way to overcome the trypsin susceptibility of peptides, while retaining their biological activity not only in solution but also on surfaces. The self-assembly,71,72 conjugation of peptides and proteins to silica,73 silver74 and gold nanoparticles is known, the selective single-point, direct attachment of peptides on to gold nanoparticles presented here allows the peptides to switch conformations in response to their external environment into their functionally active state. Simultaneous enhancement of peptide half-life from 15 min to 24 h further opens new possibilities for application of peptide conjugated gold nanoparticles in the field of biomedical nanoscience.Acknowledgements
This work was supported by the DFG Centre for Functional Nanostructures (TP E1.2) and the HGF-programme BioInterfaces. We thank Andrea Eisele and Kerstin Scheubeck for their assistance in peptide synthesis and Bianca Posselt for help with the CD measurements.Notes and references
- M. Hensher, Br. Med. J., 2013, 346, f2135 CrossRef PubMed.
- S. A. Onaizi and S. S. Leong, Biotechnol. Adv., 2011, 29, 67–74 CrossRef CAS PubMed.
- L. J. Sherrard, M. M. Tunney and J. S. Elborn, Lancet, 2014, 384, 703–713 CrossRef CAS.
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CAS.
- W. C. Wimley and K. Hristova, J. Membr. Biol., 2011, 239, 27–34 CrossRef CAS PubMed.
- S. M. Howell, S. V. Fiacco, T. T. Takahashi, F. Jalali-Yazdi, S. W. Millward, B. Hu, P. Wang and R. W. Roberts, Sci. Rep., 2014, 4, 6008 CrossRef CAS PubMed.
- H. Kim, J. H. Jang, S. C. Kim and J. H. Cho, J. Antimicrob. Chemother., 2014, 69, 121–132 CrossRef CAS PubMed.
- G. Manzo, M. A. Scorciapino, P. Wadhwani, J. Burck, N. P. Montaldo, M. Pintus, R. Sanna, M. Casu, A. Giuliani, G. Pirri, V. Luca, A. S. Ulrich and A. C. Rinaldi, PLoS One, 2015, 10, e0116379 Search PubMed.
- P. Wadhwani, J. Burck, E. Strandberg, C. Mink, S. Afonin and A. S. Ulrich, J. Am. Chem. Soc., 2008, 130, 16515–16517 CrossRef CAS PubMed.
- P. Wadhwani, R. F. Epand, N. Heidenreich, J. Burck, A. S. Ulrich and R. M. Epand, Biophys. J., 2012, 103, 265–274 CrossRef CAS PubMed.
- J. Svenson, W. Stensen, B.-O. Brandsdal, B. E. Haug, J. Monrad and J. S. Svendsen, Biochemistry, 2008, 47, 3777–3788 CrossRef CAS PubMed.
- C. Adessi and C. Soto, Curr. Med. Chem., 2002, 9, 963–978 CrossRef CAS PubMed.
- P. Henklein and T. Bruckdorfer, Biopolymers, 2009, 92, 365–366 Search PubMed.
- S. L. Johnson and M. Pellecchia, Curr. Top. Med. Chem., 2006, 6, 317–329 CrossRef CAS PubMed.
- R. Karstad, G. Isaksen, E. Wynendaele, Y. Guttormsen, B. De Spiegeleer, B. O. Brandsdal, J. S. Svendsen and J. Svenson, J. Med. Chem., 2012, 55, 6294–6305 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowski, J. Martinez and M. Khrestchatisky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
- N. Berthold, P. Czihal, S. Fritsche, U. Sauer, G. Schiffer, D. Knappe, G. Alber and R. Hoffmann, Antimicrob. Agents Chemother., 2013, 57, 402–409 CrossRef CAS PubMed.
- D. Knappe, N. Kabankov and R. Hoffmann, Int. J. Antimicrob. Agents, 2011, 37, 166–170 CrossRef CAS PubMed.
- O. Ovadia, S. Greenberg, B. Laufer, C. Gilon, A. Hoffman and H. Kessler, Expert Opin. Drug Discovery, 2010, 5, 655–671 CrossRef CAS PubMed.
- N. L. Daly, L. Thorstholm, K. P. Greenwood, G. J. King, K. J. Rosengren, B. Heras, J. L. Martin and D. J. Craik, J. Biol. Chem., 2013, 288, 36141–36148 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- E. M. Molhoek, A. van Dijk, E. J. Veldhuizen, H. P. Haagsman and F. J. Bikker, Peptides, 2011, 32, 875–880 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS PubMed.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS PubMed.
- R. Levy, N. T. Thanh, R. C. Doty, I. Hussain, R. J. Nichols, D. J. Schiffrin, M. Brust and D. G. Fernig, J. Am. Chem. Soc., 2004, 126, 10076–10084 CrossRef CAS PubMed.
- D. Bartczak, O. L. Muskens, T. Sanchez-Elsner, A. G. Kanaras and T. M. Millar, ACS Nano, 2013, 7, 5628–5636 CrossRef CAS PubMed.
- M. Schade, A. Moretto, P. M. Donaldson, C. Toniolo and P. Hamm, Nano Lett., 2010, 10, 3057–3061 CrossRef CAS PubMed.
- T. Serizawa, Y. Hirai and M. Aizawa, Langmuir, 2009, 25, 12229–12234 CrossRef CAS PubMed.
- G. T. Qin, A. Lopez, C. Santos, A. M. McDermott and C. Z. Cai, Biomater. Sci., 2015, 3, 771–778 RSC.
- W. Y. Chen, H. Y. Chang, J. K. Lu, Y. C. Huang, S. G. Harroun, Y. T. Tseng, Y. J. Li, C. C. Huang and H. T. Chang, Adv. Funct. Mater., 2015, 25, 7189–7199 CrossRef CAS.
- L. H. Peng, Y. F. Huang, C. Z. Zhang, J. Niu, Y. Chen, Y. Chu, Z. H. Jiang, J. Q. Gao and Z. W. Mao, Biomaterials, 2016, 103, 137–149 CrossRef CAS PubMed.
- A. Rai, S. Pinto, T. R. Velho, A. F. Ferreira, C. Moita, U. Trivedi, M. Evangelista, M. Comune, K. P. Rumbaugh, P. N. Simoes, L. Moita and L. Ferreira, Biomaterials, 2016, 85, 99–110 CrossRef CAS PubMed.
- Y. Cheng, A. C. Samia, J. Li, M. E. Kenney, A. Resnick and C. Burda, Langmuir, 2010, 26, 2248–2255 CrossRef CAS PubMed.
- I. Fratoddi, I. Venditti, C. Cametti and M. V. Russo, J. Mater. Chem. B, 2014, 2, 4204–4220 RSC.
- A. A. Shemetov, I. Nabiev and A. Sukhanova, ACS Nano, 2012, 6, 4585–4602 CrossRef CAS PubMed.
- J. M. Slocik, A. O. Govorov and R. R. Naik, Nano Lett., 2011, 11, 701–705 CrossRef CAS PubMed.
- R. F. Epand, W. L. Maloy, A. Ramamoorthy and R. M. Epand, Biochemistry, 2010, 49, 4076–4084 CrossRef CAS PubMed.
- S. Fanghanel, P. Wadhwani, E. Strandberg, W. P. Verdurmen, J. Burck, S. Ehni, P. K. Mykhailiuk, S. Afonin, D. Gerthsen, I. V. Komarov, R. Brock and A. S. Ulrich, PLoS One, 2014, 9, e99653 Search PubMed.
- P. Wadhwani, J. Reichert, J. Burck and A. S. Ulrich, Eur. Biophys. J., 2012, 41, 177–187 CrossRef CAS PubMed.
- P. Wadhwani, E. Strandberg, N. Heidenreich, J. Burck, S. Fanghanel and A. S. Ulrich, J. Am. Chem. Soc., 2012, 134, 6512–6515 CrossRef CAS PubMed.
- K. Hilpert, M. Elliott, H. Jenssen, J. Kindrachuk, C. D. Fjell, J. Korner, D. F. Winkler, L. L. Weaver, P. Henklein, A. S. Ulrich, S. H. Chiang, S. W. Farmer, N. Pante, R. Volkmer and R. E. Hancock, Chem. Biol., 2009, 16, 58–69 CrossRef CAS PubMed.
- J. Kim, M. J. Sadowsky and H. G. Hur, Biomacromolecules, 2011, 12, 2518–2523 CrossRef CAS PubMed.
- G. Scari, F. Porta, U. Fascio, S. Avvakumova, V. Dal Santo, M. De Simone, M. Saviano, M. Leone, A. Del Gatto, C. Pedone and L. Zaccaro, Bioconjugate Chem., 2012, 23, 340–349 CrossRef CAS PubMed.
- M. Ieronimo, S. Afonin, K. Koch, M. Berditsch, P. Wadhwani and A. S. Ulrich, J. Am. Chem. Soc., 2010, 132, 8822–8824 CrossRef CAS PubMed.
- E. Strandberg, N. Kanithasen, D. Tiltak, J. Burck, P. Wadhwani, O. Zwernemann and A. S. Ulrich, Biochemistry, 2008, 47, 2601–2616 CrossRef CAS PubMed.
- P. Wadhwani, E. Strandberg, J. van den Berg, C. Mink, J. Burck, R. A. Ciriello and A. S. Ulrich, Biochim. Biophys. Acta, 2014, 1838, 940–949 CrossRef CAS PubMed.
- R. B. Arrighi, C. Ebikeme, Y. Jiang, L. Ranford-Cartwright, M. P. Barrett, U. Langel and I. Faye, Antimicrob. Agents Chemother., 2008, 52, 3414–3417 CrossRef CAS PubMed.
- G. B. Fields and R. L. Noble, Int. J. Pept. Protein Res., 1990, 35, 161–214 CrossRef CAS PubMed.
- A. Isidro-Llobet, M. Alvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- P. Wadhwani, S. Afonin, M. Ieronimo, J. Buerck and A. S. Ulrich, J. Org. Chem., 2006, 71, 55–61 CrossRef CAS PubMed.
- F. Porta, G. Speranza, Z. Krpetic, V. Dal Santo, P. Francescato and G. Scari, Mater. Sci. Eng., B, 2007, 140, 187–194 CrossRef CAS.
- I. M. Rio-Echevarria, R. Tavano, V. Causin, E. Papini, F. Mancin and A. Moretto, J. Am. Chem. Soc., 2011, 133, 8–11 CrossRef CAS PubMed.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656, 10.1039/C39950001655.
- E. Longo, A. Orlandin, F. Mancin, P. Scrimin and A. Moretto, ACS Nano, 2013, 7, 9933–9939 CrossRef CAS PubMed.
- Z. Krpetic, S. Saleemi, I. A. Prior, V. See, R. Qureshi and M. Brust, ACS Nano, 2011, 5, 5195–5201 CrossRef CAS PubMed.
- P. Nativo, I. A. Prior and M. Brust, ACS Nano, 2008, 2, 1639–1644 CrossRef CAS PubMed.
- S. S. Wasko, G. Z. Tay, A. Schwaighofer, C. Nowak, J. H. Waite and A. Miserez, Biomacromolecules, 2014, 15, 30–42 CrossRef CAS PubMed.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2014, 5, 4348 CAS.
- S. C. Penchala, M. R. Miller, A. Pal, J. Dong, N. R. Madadi, J. Xie, H. Joo, J. Tsai, P. Batoon, V. Samoshin, A. Franz, T. Cox, J. Miles, W. K. Chan, M. S. Park and M. M. Alhamadsheh, Nat. Chem. Biol., 2015, 11, 793–798 CrossRef CAS PubMed.
- J. M. Artigas, M. E. Garcia, M. R. Faure and A. M. Gimeno, Postgrad. Med. J., 1981, 57, 219–222 CrossRef CAS PubMed.
- W. S. Ruddell, C. J. Mitchell, I. Hamilton, J. P. Leek and J. Kelleher, Br. Med. J., 1981, 283, 1429–1432 CrossRef CAS PubMed.
- Y. R. Bommineni, H. E. Dai, Y. X. Gong, J. L. Soulages, S. C. Fernando, U. DeSilva, O. Prakash and G. L. Zhang, FEBS J., 2007, 274, 418–428 CrossRef CAS PubMed.
- J. Noore, A. Noore and B. Li, Antimicrob. Agents Chemother., 2013, 57, 1283–1290 CrossRef CAS PubMed.
- Y. Xiao, Y. Cai, Y. R. Bommineni, S. C. Fernando, O. Prakash, S. E. Gilliland and G. Zhang, J. Biol. Chem., 2006, 281, 2858–2867 CrossRef CAS PubMed.
- H. P. Kasserra and K. J. Laidler, Can. J. Chem., 1969, 47, 4021–4029 CrossRef CAS.
- Z. V. Feng, I. L. Gunsolus, T. A. Qiu, K. R. Hurley, L. H. Nyberg, H. Frew, K. P. Johnson, A. M. Vartanian, L. M. Jacob, S. E. Lohse, M. D. Torelli, R. J. Hamers, C. J. Murphy and C. L. Haynes, Chem. Sci., 2015, 6, 5186–5196 RSC.
- Y. Zhou, Y. Kong, S. Kundu, J. D. Cirillo and H. Liang, J Nanobiotechnology, 2012, 10, 19 CrossRef CAS PubMed.
- S. T. Henriques, M. N. Melo and M. A. Castanho, Biochem. J., 2006, 399, 1–7 CrossRef CAS PubMed.
- P. B. Lillehoj, C. W. Kaplan, J. He, W. Shi and C. M. Ho, J. Lab. Autom., 2014, 19, 42–49 CrossRef CAS PubMed.
- A. Sood and R. Panchagnula, Chem. Rev., 2001, 101, 3275–3303 CrossRef CAS PubMed.
- Z. Yu, Z. Cai, Q. Chen, M. Liu, L. Ye, J. Ren, W. Liao and S. Liu, Biomater. Sci., 2016, 4, 365–374 RSC.
- M. Hughes, S. Debnath, C. W. Knapp and R. V. Ulijn, Biomater. Sci., 2013, 1, 1138–1142 RSC.
- C. C. Lechner and C. F. W. Becker, Biomater. Sci., 2015, 3, 288–297 RSC.
- S. Ramesh, M. Grijalva, A. Debut, B. G. de la Torre, F. Albericio and L. H. Cumbal, Biomater. Sci., 2016, 4, 1713–1725 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Antimicrobial activity of peptides, DLS data, TEM image analysis, CD data under different conditions, LCMS profile showing trypsin degradation of peptides, and stable antimicrobial activity of bound peptides for E. coli, M. luteus, and S. aureus. See DOI: 10.1039/c7bm00069c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2017 |