
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid fabrication of functionalised poly(dimethylsiloxane) microwells for cell aggregate formation†
A.
Forget‡
 ab,
A. L. S.
Burzava‡
bc,
B.
Delalat
c,
K.
Vasilev
bde,
F. J.
Harding
*bef,
A.
Blencowe
*befg and
N. H.
Voelcker
*c
ab,
A. L. S.
Burzava‡
bc,
B.
Delalat
c,
K.
Vasilev
bde,
F. J.
Harding
*bef,
A.
Blencowe
*befg and
N. H.
Voelcker
*c
aScience and Engineering Faculty, Queensland University of Technology, Brisbane, QLD 4001, Australia
bCooperative Research Centre for Cell Therapy Manufacturing (CRC-CTM), Adelaide 5000, Australia
cMonash Institute of Pharmaceutical Sciences, Monash University, Parkville, VIC 3052, Australia. E-mail: fnicolas.voelcker@monash.edu
dSchool of Engineering, University of South Australia, Mawson Lakes 5095, Australia
eFuture Industries Institute, University of South Australia, Mawson Lakes 5095, Australia
fCell Therapies Pty Ltd, Victorian Comprehensive Cancer Centre (VCCC), Melbourne 3000, Australia. E-mail: franharding@celltherapies.com.au
gSchool of Pharmacy and Medical Science, University of South Australia, Adelaide 5000, Australia. E-mail: anton.blencowe@unisa.edu.au
First published on 9th March 2017
Abstract
Cell aggregates reproduce many features of the natural architecture of functional tissues, and have therefore become an important in vitro model of tissue function. In this study, we present an efficient and rapid method for the fabrication of site specific functionalised poly(dimethylsiloxane) (PDMS) microwell arrays that promote the formation of insulin-producing beta cell (MIN6) aggregates. Microwells were prepared using an ice templating technique whereby aqueous droplets were frozen on a surface and PDMS was cast on top to form a replica. By employing an aqueous alkali hydroxide solution, we demonstrate exclusive etching and functionalisation of the microwell inner surface, thereby allowing the selective absorption of biological factors within the microwells. Additionally, by manipulating surface wettability of the substrate through plasma polymer coating, the shape and profile of the microwells could be tailored. Microwells coated with antifouling Pluronic 123, bovine serum albumin, collagen type IV or insulin growth factor 2 were employed to investigate the formation and stability of MIN6 aggregates in microwells of different shapes. MIN6 aggregates formed with this technique retained insulin expression. These results demonstrate the potential of this platform for the rapid screening of biological factors influencing the formation and response of insulin-producing cell aggregates without the need for expensive micromachining techniques.
Introduction
Diabetes affects more than 380 million people worldwide, and this number is alarmingly set to increase by 20% in the next years. To date, there is no available cure.1 Therefore, diabetes biology, preventative measures and cures are the subject of intense research. Diabetes results from the death or dysfunction of pancreatic islets, which are aggregates of endocrine cells responsible for regulating glucose levels. In vitro models for studying islet biology rely on animal and human donor sourced islets, cell monolayers and a few scaffold based culture models.2,3 As islets are globular communities of cells, models based upon cell monolayers do not accurately represent the native tissue. It is well accepted that accurate modelling of natural tissues in a laboratory setting requires cells to be organised into a three-dimensional (3D) geometry that mimics their native environment.4A common approach to form natural structures similar to mammalian tissue is to force cells to organise into spheroids.5 These cellular aggregates are of particular importance for studying biological processes such as angiogenesis,6 or the action of pharmaceutical agents (e.g., anti-cancer drugs, toxicology screening).7 In islet biology, the aggregation of insulin-producing cells offers a method to recapture the 3D organisation of islet tissues without resorting to animal isolated islets.8 It has been demonstrated that cell–cell interaction is key to improving insulin production of islet-derived cells.9–11 In order to create cell aggregates, different techniques that force the cells to associate with each other while restricting the size of the cluster have been employed. As well as the hanging-drop technique12 and non-adherent surface culture,13 microwell culture has proven to be particularly efficient for aggregate formation while being compatible with standard biological characterisation assays.14–19
Several techniques have been investigated for manufacturing microwells for the formation of MIN6 cell aggregates, which are used as a model to study islet biology. Frequently, microwell arrays are cast from low-fouling materials such as agarose, poly(ethylene glycol) or poly(dimethylsiloxane) (PDMS) using moulds manufactured through intensive micromachining, laser ablation or lithography techniques.14,17,20–25 In addition, microwell array platforms have been studied for the aggregation of dissociated pancreatic mouse islet single cells26 and insulin-producing cells,8 although both platforms require specialised and expensive manufacturing techniques, which are not amenable to rapid prototyping of novel microwell designs.
An alternative approach for the fabrication of microwell arrays that avoids the use of intensive and time-consuming manufacturing techniques involves the application of moulds prepared from patterned ice templates. Ice templates can be rapidly prepared in various sizes and shapes, and have been used to imprint microwell patterns into collagen,27,28 and PDMS.29
Interestingly, previous studies have demonstrated that PDMS surface chemistries can be altered through aqueous sodium hydroxide (NaOH) etching, which in turn modifies the surface chemistry and allows protein adsorption.30 In contrast to conventional moulds, an appealing feature of ice templates is the ability to load the template (i.e. the ice droplets) with biofactors or biomacromolecules (e.g., growth factors) that can be then used for the selective functionalisation of the microwells. This was demonstrated by the selective functionalisation of collagen replica microwells that can absorb the biological signals from the ice template onto the collagen fibers.27 This unique feature allows the internal surface of the microwell to be selectively functionalised without altering the interstitial surfaces between the microwells. Fabrication of site-specific functionality or site-specific biological patterning generally requires the use of complex masks to selectively decorate the substrate with biological factors.31 Such platforms have the potential to be used for the high-throughput screening of cell interactions with biofactors. Whilst these techniques are applicable to protein adherent materials such as collagen, common casting materials such as PDMS generally display limit adhesion of proteins.32 Because the PDMS surface is not strongly protein adherent, simply loading the ice template with proteins is not applicable for the site-specific immobilisation of factors within the PDMS replica microwells.32
We hypothesised that PDMS microwells could be rapidly prototyped with tailorable microwell shapes and site-specific surface functionality by using frozen concentrated aqueous NaOH droplets as the templating agent. Furthermore, the size and shape of the microwells could be tailored by modifying the surface chemistry of the substrate onto which the NaOH droplets are deposited. Subsequently, the sodium hydroxide activated PDMS surfaces allowed for the facile and site-specific immobilisation of biofactors within the microwells by incubating the complete array in a biofactor solution. These surfaces provide a cost-effective and rapid approach to manufacture microwell arrays with different dimensions and isolated functionalities. The resulting microwell arrays could then be used to study and screen the formation of cell aggregates in the presence of different geometries and biofactors. To demonstrate the potential of this approach, we chose to utilise insulin-producing mouse MIN6 cells, a pancreatic beta cell line, investigating their formation into insulin-producing cell aggregates resembling the natural morphology of islets. The aggregate stability and cell viability was screened against various (bio)macromolecules, including extracellular matrix (ECM) protein collagen IV (Col-IV), insulin-like growth factor 2 (IGF-2) that have been reported to support islet survival,33 and an anti-fouling synthetic polymer. This work presents the potential of rapid microwell platform prototyping for the screening of cellular environments to facilitate functional cell aggregate formation.
Results and discussion
Tailoring the microwell shape
A simple process was devised to prepare the microwell arrays with different well shapes via an ice templating approach (Fig. 1). Aqueous water droplets were deposited on tissue culture polystyrene (TCPS) Petri dishes coated with different plasma polymers and frozen to afford the template. A PDMS prepolymer solution was then poured onto template and cured to afford the microwell arrays.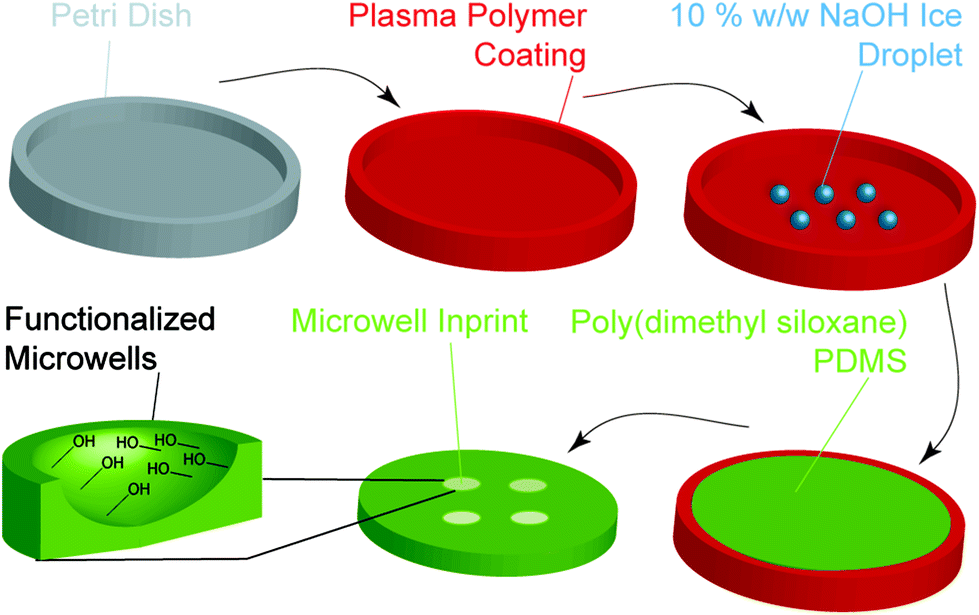 | ||
| Fig. 1 Schematic showing the steps involved in the fabrication of functionalised PDMS microwells using the ice templating technique. Initially, an uncoated tissue culture polystyrene (TCPS) Petri dish was used or Petri dishes coated with a plasma polymer based from ethanol, octadiene or allylamine monomers. Subsequently, water droplets or 10% w/w NaOH droplets were deposited on the surface and snap-frozen in liquid nitrogen. A PDMS prepolymer solution was then poured on top of the frozen droplets and allowed to cure for 16 h at 60 °C. The resulting PDMS microwell array was then separated from the Petri dish. | ||
The initial step in this process was to generate surfaces of different hydrophobicity by coating TCPS Petri dishes with a thin layer of octadiene, ethanol or allylamine plasma polymers (ESI, Fig. S1A†), as reported previously.34–37 To measure coating thickness via ellipsometry, control silicon wafer substrates were plasma polymer coated alongside the Petri dishes. For the octadiene, ethanol and allylamine plasma polymer coatings, the thicknesses were determined to be 87.1 ± 0.2, 16.3 ± 0.02 and 30.7 ± 0.2 nm, respectively (ESI, Fig. S1B†).38,39 Attenuated total reflectance FTIR spectroscopy was used to characterise the surface functionalisation of the Petri dishes. Each plasma polymer coating exhibited the expected characteristic functional groups, with ethanol, allylamine and octadiene coatings displaying alcohol (νO–H = 3350 cm−1), amine (νN–H = 3240 cm−1), and alkene (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C = 1680 cm−1) vibrations, respectively (ESI, Fig. S1C and D†).
C = 1680 cm−1) vibrations, respectively (ESI, Fig. S1C and D†).
The hydrophobicity of the surface coatings was determined by sessile drop water contact angle measurements. The contact angle formed between a droplet of deionised water and the freshly prepared plasma polymer coated surfaces was compared to bare TCPS (Fig. 2A). For deionised water, the contact angle of the droplet was found to decrease from 99 ± 2° for TCPS, 80 ± 8° for octadiene, 61 ± 3° for ethanol to 51 ± 3° for allylamine plasma polymer coatings (Fig. 2C) and these results were consistent with published studies.40,41
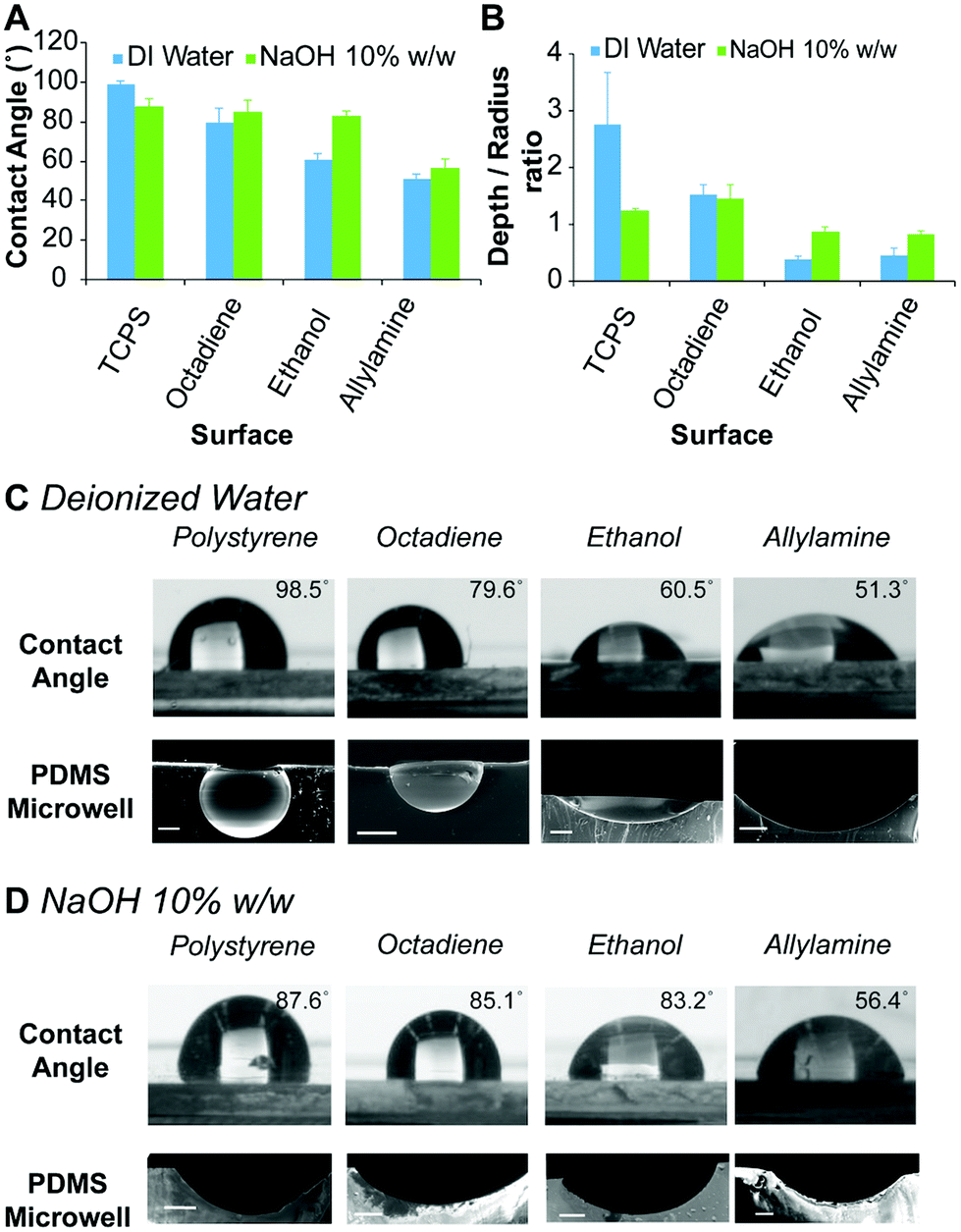 | ||
| Fig. 2 (A) Contact angle measurements of deionised water and 10% w/w aqueous NaOH solution measured by the sessile drop approach for TCPS and different plasma polymer coated surfaces. Error bars represent standard deviation for n = 4. (B) Shape profile of PDMS microwells obtained from different surfaces and ice templates of deionised water and 10% w/w aqueous NaOH solution (1 μL). Error bars represent standard deviation for n = 3 microwells. Digital images of (C) water and (D) 10% w/w aqueous NaOH droplets (top) on various surfaces, and scanning electron microscopy (SEM) images (bottom) showing the cross-section of PDMS microwells prepared using (C) deionised water and (D) 10% w/w aqueous NaOH ice templates. Scale bars 200 μm. | ||
To prepare microwells with selectively functionalised inner surfaces, a 10% w/w aqueous NaOH solution was used to prepare the ice templates. Initially, the contact angle made by the alkali solution on the different surfaces was measured. In contrast to the results obtained using deionised water, the contact angle of the aqueous NaOH solution was similar on TCPS, octadiene and ethanol plasma polymer surfaces (∼85°), but lower on allylamine plasma polymer surfaces (∼56°) (Fig. 2D). Allylamine plasma polymer coatings contain amine groups that can be protonated, which accounts for the higher surface hydrophilicity (i.e., lower contact angles) with water. In the presence of a strong base, the allylamine plasma polymer surface become deprotonated and slightly less hydrophilic, hence the slight increase in the contact angle for aqueous NaOH droplets versus water droplets.34
Microwell arrays were initially prepared by curing PDMS on top of the ice templates composed of water and aqueous NaOH solutions (1 μL) on the different plasma polymer coated substrates, followed by washing and drying. The dimensions of the resulting microwells were measured via analysis of scanning electron microscopy (SEM) images of the microwell cross-sections, and the shape profiles were calculated (Fig. 2B). The shape profile is described by dividing the measured depth by the radius (at the surface), with 1 being hemispherical and values <1 being shallower sub-hemispherical shapes. We observed that the PDMS microwells obtained from water ice templates on TCPS substrates have a super-hemispherical shape while templates on octadiene, ethanol and allylamine plasma polymer coated substrates gave hemispherical or sub-hemispherical microwells. For microwells prepared using NaOH ice templates on TCPS and octadiene plasma polymer substrates, the shape profiles were approximately hemispherical, whereas for ethanol and allylamine plasma polymer substrates the shape profiles were sub-hemispherical, but deeper than the respective microwells prepared from water ice templates on the same surface (Fig. 2B). In general, the trends observed for the contact angle measurements (Fig. 2A) and the shape of the droplets accurately reflected the respective PDMS microwell shapes (Fig. 2C and D) showing an increase of the microwell diameter as the contact angle decreased.
To demonstrate that the ice templating approach can also be used to prepare microwells with different dimensions and volumes, ice templates of deionised water were prepared on all of the substrates using different droplet volumes, ranging from 25 nL to 1 μL (ESI, Fig. S5†). In all cases, the shape profile of the microwells was effectively conserved across the different microwell sizes, i.e. across the different droplets volumes used to imprint the microwells into the PDMS (ESI, Fig. S3†).
These results demonstrate that the dimensions and shape of the PDMS microwells can be tailored by utilising surfaces of different hydrophobicity, different aqueous templating solutions, and different template volumes. While microwells obtained with water templates span a broad range of shape profiles (0.38 ± 0.05 to 2.76 ± 0.9), the range of microwell shapes obtained with NaOH templates was somewhat more restricted (0.83 ± 0.04 to 1.46 ± 0.24), albeit appropriate for the current study. Conceivably, other shape profiles could be prepared by using other coatings and surfaces as the template, including superhydrophobic or superhydrophilic surfaces.29
Functionalisation of microwell arrays
Previously it has been reported that PDMS can be etched with aqueous NaOH solutions30 to produce surfaces exhibiting silanol groups (Fig. 3A). The etching of the microwell surfaces by the NaOH droplets was assessed via ATR-FTIR spectroscopy and compared to freshly cured PDMS imprinted with water droplets (Fig. 3B and ESI, Fig. S2†). The spectrum of the etched surface revealed a strong OH stretch (νOH = 3500 cm−1) (Fig. 3B) and a reduction in the intensity of the Si–O–Si band (νSi–O–Si = 1010 cm−1) (ESI, Fig. S2†) consistent with hydrolysis of the PDMS surface and the formation of silanols. Further confirmation of the etching was obtained with energy-dispersive X-ray (EDX) spectroscopy (Fig. 3C and ESI, Fig. S3†), which revealed a 20% increase in the ratio of oxygen-to-silicon atoms at the surface of the etched PDMS compared to unmodified PDMS. These results confirm that the microwell surface is etched to produce silanol groups (Fig. 3A).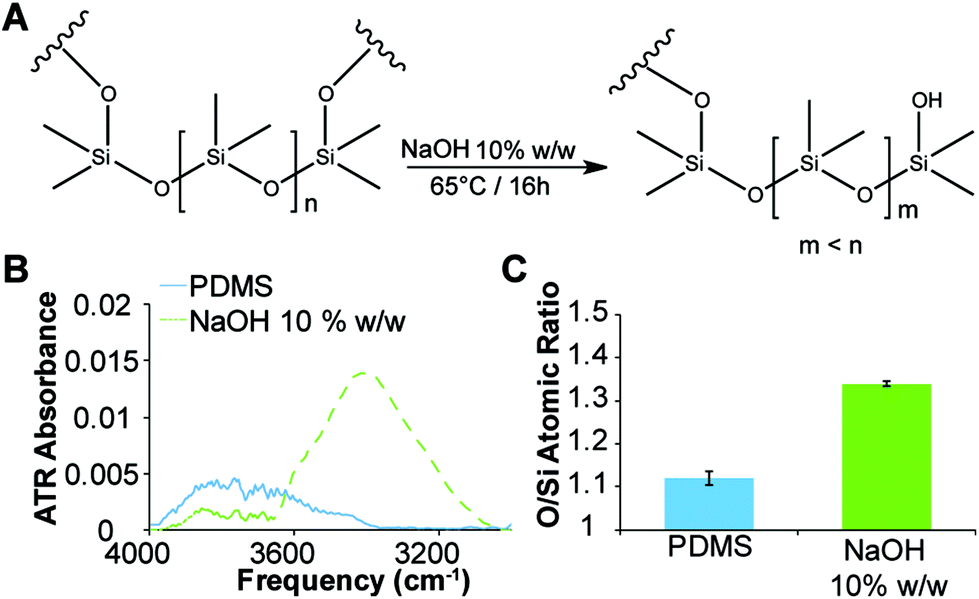 | ||
| Fig. 3 (A) Scheme showing etching of PDMS with concentrated aqueous NaOH solution. (B) Attenuated total reflectance FTIR spectra of the etched PDMS microwells (dashed) and unmodified microwells. (C) Ratio of oxygen to silicon atoms for etched PDMS microwells and unmodified microwells, as determined by EDX spectroscopy. Error bars represent standard deviation for n = 3. | ||
Immobilisation of biological factors
The unique ability to fabricate PDMS microwells with site-isolated silanol groups using 10% w/w NaOH ice templates provides facile access to microwells that can be selectively functionalised or coated. In order to demonstrate this, a fluorescently labelled protein was selectively physisorbed on to the etched microwells. The complete microwell arrays were covered in fluorescein isocyanate labelled bovine serum albumin (FITC-BSA) solution and incubated for 16 h at 4 °C in the dark. After rinsing thoroughly with Dulbecco's phosphate buffered saline (dPBS), the microwell arrays were imaged (Fig. 4). Confocal laser scanning microscopy revealed that the FITC-BSA was only adsorbed within the etched microwells, and importantly, did not coat the non-etched PDMS surface between the microwells (Fig. 4B). Validation of the uniform immobilisation of the FITC-BSA within the microwells was obtained by performing z-stack imaging on a confocal microscope (Fig. 4C and D). The reconstructed 3D images show that only the etched surface in the microwells is covered with FITC-BSA, whereas the cross-section of the 3D reconstructed image shows that the etched microwell is uniformly covered with FITC-BSA. This facile fabrication and coating approach provides a considerable advantage when compared to alternative microfabrication techniques such as micromachining or lithography.14,17,20–22 Indeed, our approach allows selective functionalisation of the inner wall of the microwells without the recourse of mask fabrication, optimisation of the mask alignment, and mask contact with the PDMS surface. Therefore, the etchant droplet technique presented herein, offers an affordable and rapid means to fabricate functionalised PDMS microwell arrays.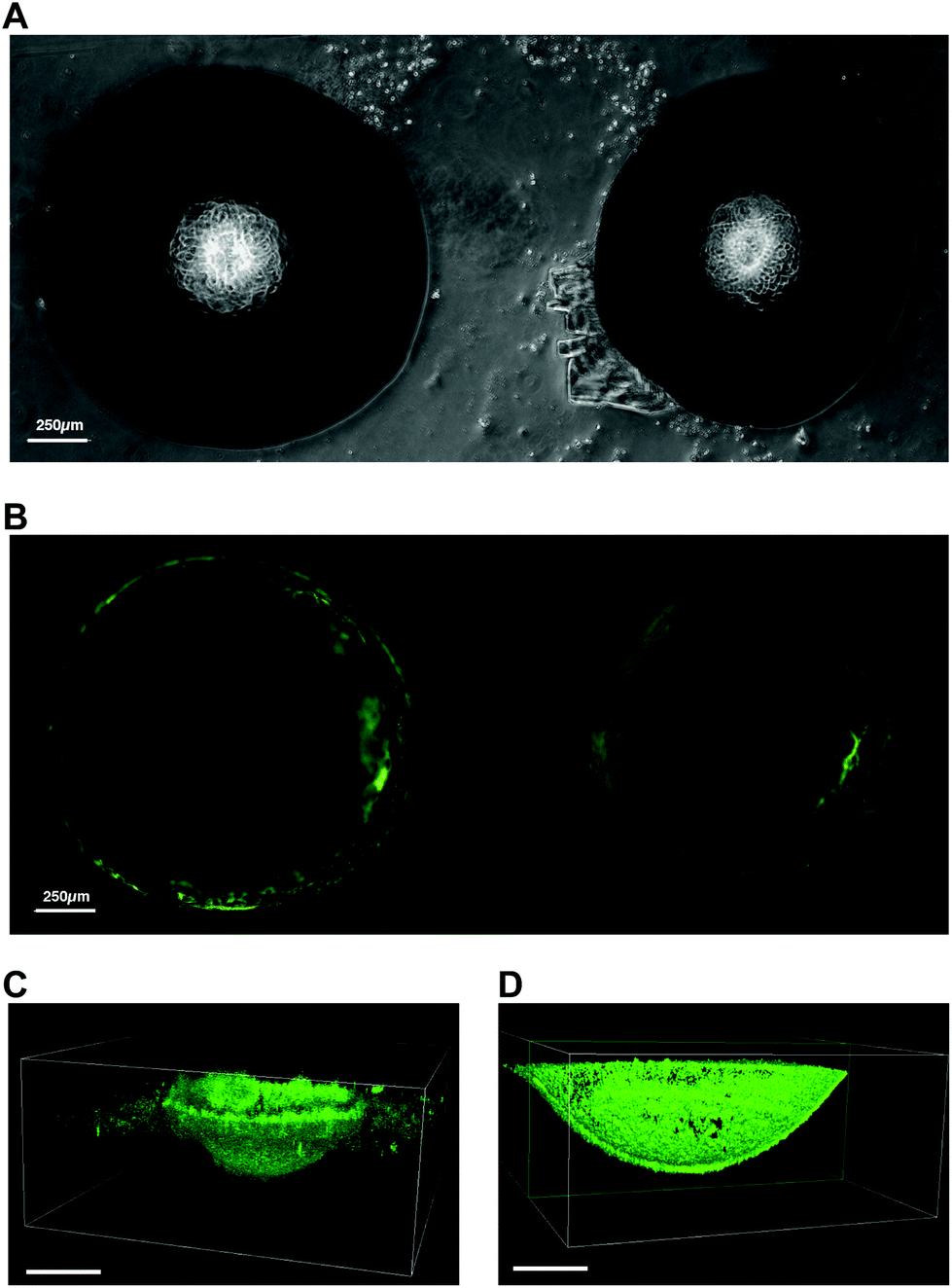 | ||
| Fig. 4 (A) Brightfield microscopy images of aqueous NaOH etched PDMS microwells. (B) Fluorescence microscopy images of NaOH etched PDMS microwells coated with FITC-BSA. (C) Confocal microscopy 3D reconstructed z-stack of aqueous NaOH etched PDMS microwell coated with FITC-BSA and (D) cross-section of the same coated microwell. Scale bars 250 μm. | ||
Screening microwell shape profile for MIN6 cell aggregate formation
Current laboratory models for the in vitro study of insulin producing cells often necessitate expensive isolated animal islets or employ physiologically inaccurate monolayer cell cultures. Therefore, the development of in vitro models that enable the formation of insulin-producing cell aggregates that mimic pancreatic islets is of particular benefit for the study of islet biology.8,26 Furthermore, because islet function is highly related to their size, a versatile platform that allow for a rapid fabrication of size and shape specified microwells would be favorable.15 Forced aggregation is known to aid spheroid formation.42 This was successfully demonstrated for the formation of embryoid bodies induced pluripotent stem cells,43,44 but also for the formation of tumour cells spheroid.45 In this study, we used the immortalised insulin-producing beta cell line MIN6 as a model for dissociated islets, to demonstrate the ability of the functionalised PDMS microwells to facilitate cell aggregate formation. To study the effect of the microwell shape profile on the formation of cell aggregates, MIN6 cells were cultured on deep and shallow microwells coated with Pluronic 123 and compared to a flat PDMS surface coated with Pluronic 123. The deep microwell arrays with shape profiles of 1.46 were prepared using NaOH droplets on octadiene plasma polymer coated Petri dishes. The shallow microwell arrays with shape profiles of 0.88 were prepared using NaOH droplet on ethanol plasma polymer coated Petri dishes.The behaviour of the cells on the microwells and flat surfaces was monitored by recording brightfield microscopy images over 72 h (Fig. 5A), and the projected surface area occupied by the cells or cell aggregates was measured directly from the microscopic images (Fig. 5B). On flat surfaces, no apparent aggregate formation was noted and the surface area covered by the MIN6 cells was measured. This value slightly decreased from 78 to 59 mm2 over 24 h, but remained stable thereafter. In contrast, both the shallow and deep microwells were found to support cell aggregate formation with similar aggregate surface areas (∼28 mm2) from 24 h to 72 h. The cell viability of the MIN6 cells after 72 h as determined by Trypan blue staining on the dissociated cell aggregate revealed that all cells cultured on flat surfaces were dead (Fig. 5C).
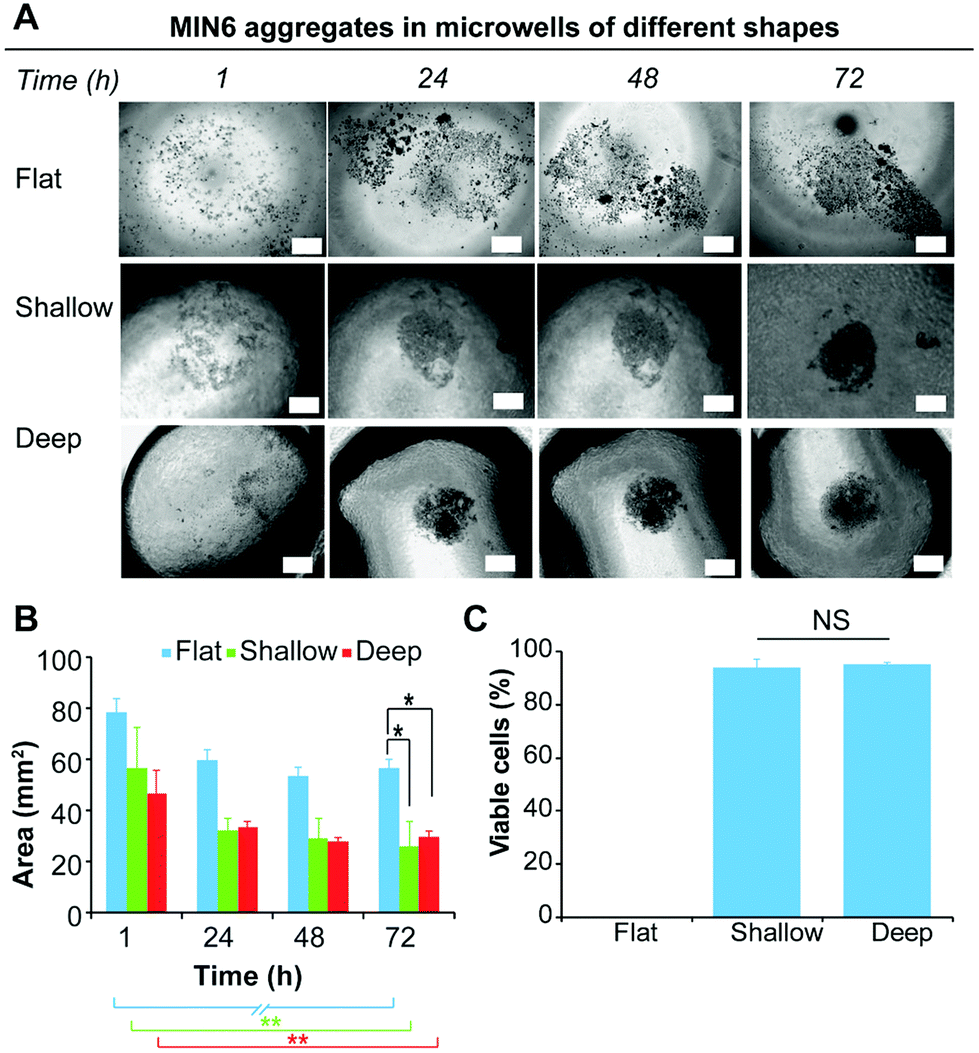 | ||
| Fig. 5 (A) Brightfield microscopy images over 72 h showing the morphology of MIN6 cells on flat PDMS, and shallow and deep microwells coated with Pluronic 123; scale bars 250 μm. (B) Area of cells or cell aggregates as a function of time. (C) Concentration of dead and live cells determined by Trypan blue staining of dissociated MIN6 cell aggregates after 72 h of culture. Error bars represent standard deviation for n = 5. T-Test one tail with *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001. | ||
On the flat surface the cells cannot adhere to the Pluronic 123 coated PDMS substrate. Under these conditions, MIN6 cells are unable to attach to the substrate, but poorly adhere to each other and thus do not form spheroids, inducing apoptosis in the cell population.46 In contrast, MIN6 cells cultured on shallow and deep microwells displayed excellent viability (Fig. 5C). While deep and shallow microwells exhibited similar cell viability within aggregates, aggregates formed in the deep microwells were more difficult to dissociate, and thus this microwell shape profile was chosen for further experiments.
Impact of biological factors on MIN6 cell aggregate formation
In their natural environment, cells and cell aggregates are in constant interaction with the surrounding extracellular matrix (ECM), which consists of a complex mixture of structural proteins and polysaccharides, and signalling factors. The presented microwell arrays provide an excellent platform to investigate and screen these natural interactions and determine if the presence of certain macromolecules or signalling molecules can promote or disturb aggregate formation. MIN6 cells were cultured in deep PDMS microwells (shape profile 1.46) coated with various biological molecules and compared to PDMS microwells coated with Pluronic 123 as a positive control. PDMS microwells functionalised with BSA were utilised as a comparison of a non-adherent biological surface. PDMS microwells coated with Col-IV, a molecule of the ECM found in pancreatic tissue,47 and IGF-2, a growth factor that has been shown to improve islet viability,33 were used to study the effect of biological factors on aggregate formation. This microwell approach represents the converse approach to the utilisation of microparticles coated with biological factors to force cell aggregation.48 The behaviour of MIN6 cells on the differently functionalised microwell surfaces was monitored over 72 h via brightfield microscopy (Fig. 6A), and the area covered by the cells or cell aggregates was determined via measuring the projected area of the cell aggregate over time (Fig. 6B).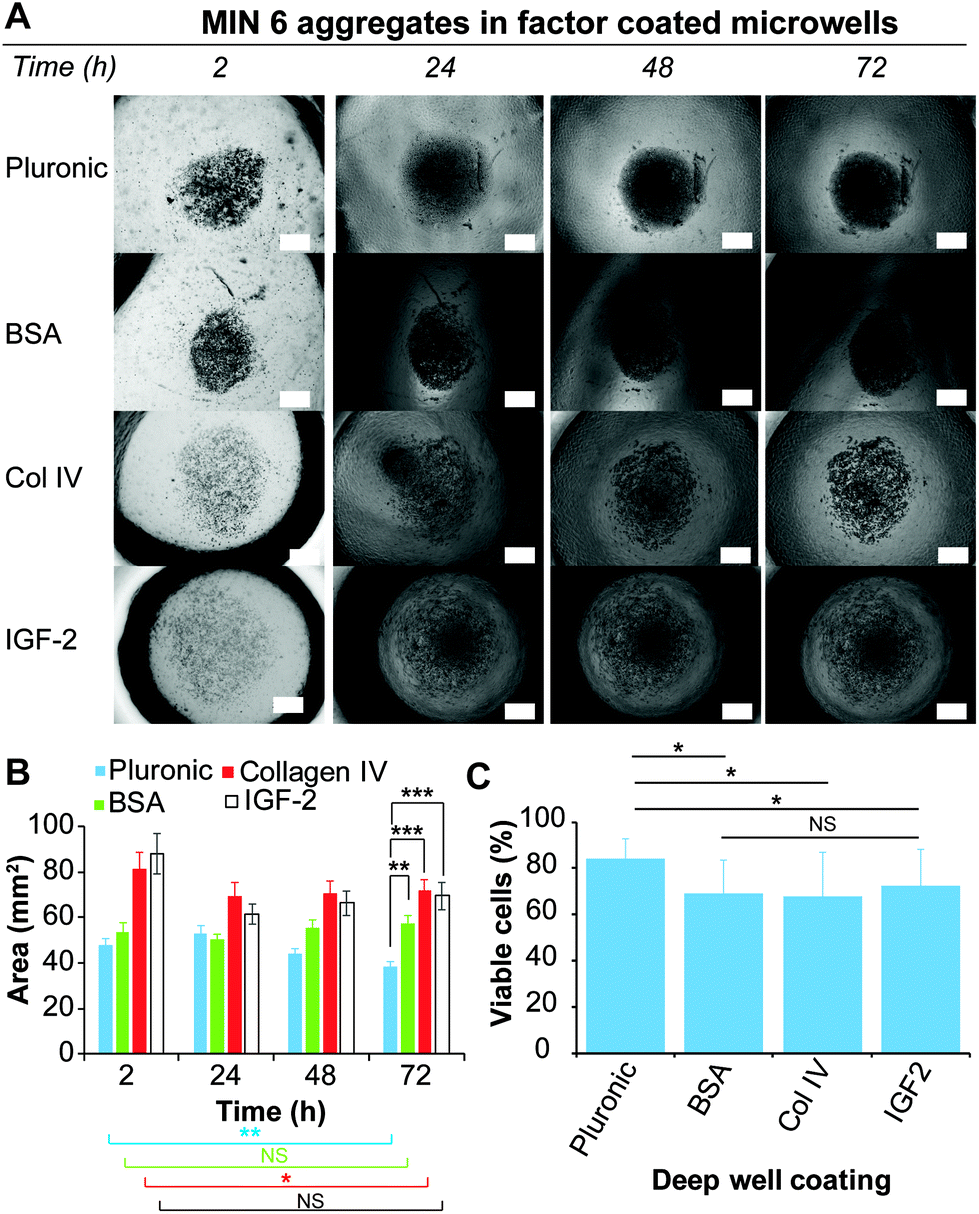 | ||
| Fig. 6 (A) Brightfield microscopy images over 72 h showing the morphology of MIN6 cells on deep PDMS microwells coated with Pluronic 123, BSA, Col-IV and IGF-2; scale bar 250 μm. (B) Area of cells or cell aggregates as a function of time. (C) Concentration of dead and live cells determined by Trypan blue staining of dissociated MIN6 aggregates after 72 h of culture. Error bars represent standard deviation for n = 10. T-Test one tail with *p ≤ 0.05, **p ≤ 0.01 and ***p ≤ 0.001. | ||
For the Pluronic 123 coated microwells, the cells were observed to form dense aggregates after 24 h, which continued to decrease in size over 72 h. Similarly, the Col-IV and IGF-2 coated microwells showed a size decrease after the first 24 h. However, this size remained constant until 72 h. In contrast, cells loaded onto the BSA coated microwells formed dense cell aggregates similarly to the one formed onto Pluronic 123 coated microwells. The size of the spheroids on BSA remained approximately constant from 2 h to 72 h. Since IGF-2 and Col-IV are molecules that support cell adhesion, it is possible that the adhesion to the coated microwell surface competed with the cell–cell interactions required from spheroid formation.49,50
The viability of the cells after 72 h was determined by a Trypan blue staining on dissociated cell aggregates for each of the coated microwells (Fig. 6C). Interestingly, while only Pluronic 123 and BSA coated microwells supported the formation of dense cell aggregates, the Pluronic 123 coated microwells retained the highest cell viability, while having no statistical differences with the previous experiment depicted in Fig. 5C. Across the three other protein coatings, cells derived from spheroids cultured in Col-IV, BSA and IGF-2 coated microwells did not show any significant differences in cell viability. However, between these three biological coatings only the BSA coating resulted into a dense cell aggregate similar to Pluronic 123 coating after 24 h of culture. In contrast to BSA, Col-IV and IGF-2 can bind to cell surface receptors,51,52 which may compete with cell–cell aggregation necessary for aggregate formation.
These results demonstrate that the functionalised microwells provide a platform to rapidly screen biological molecules or combinations thereof for the optimisation of the formation of cell aggregates.
Insulin expression of MIN6 cell aggregates
MIN6 is an insulin-producing pancreatic beta cell line, and therefore, MIN6 cell aggregates should express insulin if viable. It has been demonstrated that cell–cell interactions through tight junctions improve MIN6 stimulus response and proliferation.46,53 For validation of cell aggregate function, an insulin immunostaining assay was performed with dense MIN6 cell aggregates generated on the Pluronic 123 coated microwells. The cell aggregates were fixed and stained for insulin, along with MIN6 cells cultured as a monolayer on TCPS (Fig. 7). Fluorescence imaging confirmed that the dense MIN6 cell aggregate produced insulin, demonstrating that functional aggregates can be formed in the microwells.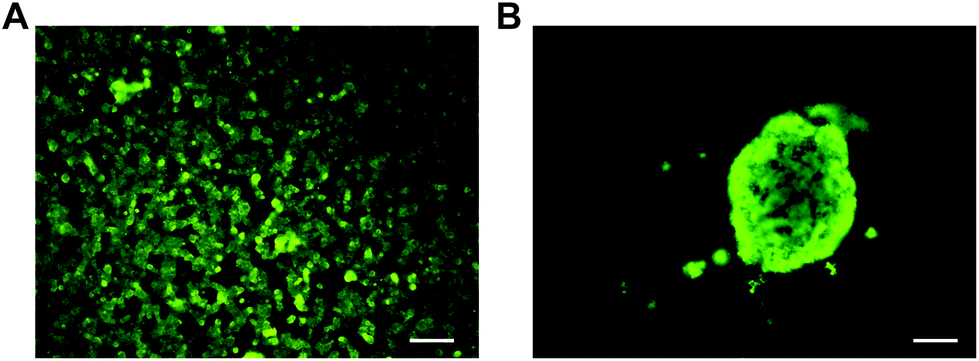 | ||
| Fig. 7 Insulin immunostaining of MIN6 cells (A) cultured as a monolayer and (B) as an aggregate in PDMS deep microwells coated with Pluronic 123. Scale bars 100 μm. | ||
Experimental
Plasma coating
Plasma deposition was performed in a purpose-built capacitively coupled bell-chamber reactor equipped with a 13.56 MHz plasma generator (Advance Energy, USA).39 Clean tissue culture polystyrene Petri dishes from Thermo Fisher (Waltham, Massachusetts, USA) were added to the plasma reactor and the chamber brought to a vacuum. To further clean and prime the Petri dish surfaces, atmospheric air was introduced into the chamber until a constant pressure of 1.1 × 10−1 mbar was achieved. The plasma was ignited with a 50 W continuous wave (CW) radio frequency (RF) and left on for 5 min. This process ensures that any unwanted organic material on the surface are removed prior to polymer deposition and activation. Subsequently, the pressure in the chamber was lowered to 3.5 × 10−2 mbar and the monomer – 1,7 octadiene (98%), ethanol (technical grade) or allylamine (98%) from Sigma Aldrich (St Louis, Missouri) – was introduced via a needle valve until steady working flow rates were achieved. The plasma was ignited with a 50 W CW RF for 5 min. Along with the Petri dishes, clean silicon wafers were also exposed to the same treatment for further characterisation of the plasma polymer coatings.Microwell array fabrication
The functionalised PDMS microwell arrays were fabricated through a simple and rapid casting process. Initially, droplets of water from a Merck MilliQ water dispenser (Darmstadt, Germany), or 10% w/w NaOH solution freshly prepared from NaOH pellets (AR grade; Chem-Supply, South Australia), were deposited with a micropipette onto either tissue culture polystyrene (TCPS) Petri dishes or plasma-polymer-coated Petri dishes. The droplets were then snap frozen by immersion in a liquid nitrogen bath to afford the ice templates. PDMS (Sylgard 184; Dow; Midland, Michigan) was mixed with a catalyst solution in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (PDMS: catalyst solution) as specified by the manufacturer, and degassed under vacuum to remove any trapped air bubbles prior to being slowly poured on top of the frozen droplets in the Petri dishes. The Petri dishes were heated at 60 °C for 16 h to cure the PDMS and thaw the ice template, allowing etching of the PDMS microwell surface (NaOH solutions only). The cured PDMS microwell arrays were then removed from the Petri dish and thoroughly washed with water (to remove the NaOH solution), rinsed with ethanol and dried under vacuum for 16 h (0.1 mbar). The resulting microwell arrays were then used for aggregate formation. Alternatively, for small volumes, the droplet patterns of water or 10% w/w NaOH solution were made with an automated non-contact spotting system S3 sciFLEXARRAYER (Scienion AG, Germany) using a piezoelectric printing technology. The instrument was equipped with a glass nozzle and single drops ejected from the nozzle had a volume of 25 to 1000 nL. The distance between printed spots was selected based on the diameter of the water droplets. The water droplets were deposited on the substrates, which were pre-chilled in a 100% humidified environment, and then frozen in liquid nitrogen to afford the ice templates.
:1 ratio (PDMS: catalyst solution) as specified by the manufacturer, and degassed under vacuum to remove any trapped air bubbles prior to being slowly poured on top of the frozen droplets in the Petri dishes. The Petri dishes were heated at 60 °C for 16 h to cure the PDMS and thaw the ice template, allowing etching of the PDMS microwell surface (NaOH solutions only). The cured PDMS microwell arrays were then removed from the Petri dish and thoroughly washed with water (to remove the NaOH solution), rinsed with ethanol and dried under vacuum for 16 h (0.1 mbar). The resulting microwell arrays were then used for aggregate formation. Alternatively, for small volumes, the droplet patterns of water or 10% w/w NaOH solution were made with an automated non-contact spotting system S3 sciFLEXARRAYER (Scienion AG, Germany) using a piezoelectric printing technology. The instrument was equipped with a glass nozzle and single drops ejected from the nozzle had a volume of 25 to 1000 nL. The distance between printed spots was selected based on the diameter of the water droplets. The water droplets were deposited on the substrates, which were pre-chilled in a 100% humidified environment, and then frozen in liquid nitrogen to afford the ice templates.
Ellipsometry
Coating thickness on the silicon wafers was determined via ellipsometry using a J. A. Woolam Co. (Lincoln, Nebraska, USA) variable angle spectroscopic ellipsometer (VASE). All measurements and data were analysed using WVASE32 software provided with the instrument. Three silicon wafers from independent plasma depositions were used for thickness measurements. Polymer thickness values were estimated by applying a Cauchy model.Fourier-transform infrared spectroscopy
Fourier transform infrared spectroscopy was performed using a Hyperion 1000 FTIR Microscope from Bruker (Ettlingen, Germany). An attenuated total reflectance (ATR) accessory with a germanium crystal and a liquid nitrogen-cooled mercury cadium telluride (MCT) detector was used to record the spectra. All spectra were collected as an average of 64 scans, with a resolution of 4 cm−1, and over the range of 650–4000 cm−1. A background of air was subtracted from the raw spectra. Spectra were analysed using Opus software from Bruker (Ettlingen, Germany).Contact angle measurements
A custom-built sessile drop apparatus with an Olympus SZ-PT microscope and lens system connected to a Sony CCD camera was employed to measure the wettability of the plasma coated surfaces. A 10 μL syringe (Hamilton, Reno, USA) was used to dispense droplets of MilliQ water or 10% w/w NaOH solutions of approximately 1 μL on the coated Petri dish. A minimum of three contact angle measurements were taken for each surface. Angle analysis of captured droplets was performed with ImageJ software v1.50 with the DropSnake plugin.54Energy-dispersive X-ray spectroscopy (EDX)
Microwells made using water or 10% w/w NaOH ice templates were characterised via EDX spectroscopy on a scanning electron microscope (Quanta 450 FEG ESEM FEI; Hillsboro, Oregon, USA) equipped with an Apollo X SDD EDX detector. The ratio of O/Si was then calculated for each sample.Confocal microscopy
Freshly made microwells were incubated for 16 h with BSA-FITC and then washed three times with dPBS from Sigma Aldrich (St Louis, Missouri). The microwells were then dried at room temperature and imaged upside down on a confocal fluorescence microscope (Nikon A1; Tokyo, Japan) equipped with appropriate filters.Scanning electronic microscopy (SEM)
Microscopic images were obtained using a field emission SEM (Merlin®, Zeiss, USA), fitted with a GEMENI II® column (Zeiss, USA) and a secondary electron detector, operating at 1 kV in high vacuum mode. Measurements of microwell dimensions were performed using the software ImageJ 1.50.MIN6 cell culture
MIN6 were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) from Sigma (St Louis, Missouri) supplemented with 15% foetal bovine serum, 2.5% HEPES 1 M (Gibco) glutamax (Gibco) and 1% v/v penicillin/streptomycin from Thermo Fisher (Waltham, Massachusetts, USA), and 1% v/v β-mercaptoethanol (5 μL L−1) from Sigma (St Louis, Missouri, USA). Once confluent, cells were trypsinised with a solution of 0.05% trypsin/EDTA obtained from Sigma Aldrich.Aggregate formation
PDMS microwell arrays were initially sterilised in a 0.4 mg mL−1 penicillin/streptomycin solution from Thermo Fischer (Waltham, Massachusetts, USA) for 16 h at 37 °C in an incubator. The arrays were then coated with either a 2% w/v solution of Pluronic 123 (BASF, Ludwighafen, Germany), 100 μg mL−1 BSA (Sigma Aldrich, St Louis, USA), 100 μg mL−1 collagen IV from Stemcell Technologies (Vancouver, Canada) or 100 μg mL−1 IGF-2 (GroPep, Adelaide, Australia). The macromolecules were allowed to absorb on the microwell surface over 16 h. The arrays were then washed thoroughly washed with dPBS prior to aggregate formation. The first step of aggregate formation involved producing MIN6 cell hanging droplets. MIN6 cells were seeded at 106 cells mL−1 arranged in 10 μL drops on the inside of a 60 mm Petri dish lid. The lid was quickly inverted and placed onto a dPBS-filled bottom chamber to avoid evaporation of the media. MIN6 hanging droplets were incubated at 37 °C, 5% CO2 for 3 d and then transferred into the coated microwells, and cultured for a further 3 d. Cell behaviour and aggregate formation was followed over time by taking pictures every day on an Eclipse TiS microscope from Nikon (Tokyo, Japan).Cell viability
The viability of the MIN6 aggregates was assessed using a Trypan blue count. Cells were initially transferred from the different microwells, centrifuged, and re-suspended in trypsin with 2% chicken serum from Thermo Fisher (Waltham, Massachusetts, USA). Then, a more refined dissociation was obtained by using an 18G needle from Braun (Kronberg im Taunus, Germany) to re-suspend the cells. The trypsin was then inactivated with complete media, and the cells were centrifuged and re-suspended in complete media and Trypan blue solution dye from Sigma Aldrich (St Louis, Missouri) (1:4). Viable cells were counted using a hematocytometer, blue cells were counted as dead and unstained cells were counted as alive. Three counts were performed for each condition. Viable cells were counted as a percentage of the total amount of cells counted (dead and alive). Data were analysed using Prism 7 software from Graphpad software (La Jolla, California) and the results were expressed as mean ± SEM.
Insulin immunostaining
After 6 d of culture, MIN6 cell aggregates were carefully removed from the microwells. To stain the insulin expressing cells, aggregates were washed with dPBS, fixed with 4% paraformaldehyde solution for 20 min, permeabilised with 0.3% Triton X-100 in dPBS for 20 min, and then blocked with 3% goat serum in dPBS for 30 min. Primary antibody (guinea pig anti-insulin, EMD Millipore, City, Country Billerica, MA, USA) diluted in dPBS was incubated with the cells for 16 h at 4 °C. The spheroids were washed with dPBS, followed by incubation with a goat anti-guinea pig rhodamine conjugated antibody (Jackson ImmunoResearch, City, Country West Grove, PA, USA) diluted in dPBS incubated for 2 h at room temperature. Aggregates were imaged using a Nikon TiS Eclipse inverted fluorescence microscope (Tokyo, Japan).Conclusions
Rapid manufacturing of shape tailorable and site-specific functionalised microwell arrays was achieved through patterned ice templates prepared from frozen aqueous NaOH droplets. Compared to techniques utilising micromachining or lithography, the method described in this study allows for the cost-effective fabrication of PDMS functionalised microwells that can be made with limited resources and without specialised equipment. The novel site-specific etching approach introduced in this study affords PDMS microwell arrays onto which proteins and biofactors can be selectively immobilised within the microwells. This strategy enables the interaction of cells with libraries of physisorbed polymers, proteins and other biological molecules to be easily screened, with particular reference to the formation of cell aggregates. The application of the microwell arrays for the formation of physiologically relevant MIN6 cell aggregates that exhibit insulin production was demonstrated. This work paves the way for further site-specific functionalisation of PDMS microwells by utilisation of different templating surfaces and silane chemistries, as well as providing a versatile platform for the extension of aggregate formation techniques to other cell types.Acknowledgements
The authors would like to thank Drs Cavallaro and McGregor for their help with the plasma polymer coatings, Dr Sweetman for his help with FTIR acquisition and A/Prof. Claudine Bonder for providing the MIN6 cells.Notes and references
- J. E. Shaw, R. A. Sicree and P. Z. Zimmet, Diabetes Res. Clin. Pract., 2010, 87, 4–14 CrossRef CAS PubMed.
- M. Skelin, M. Rupnik and A. Cencic, ALTEX, 2010, 27, 105–113 CrossRef PubMed.
- J. T. Daoud, M. S. Petropavlovskaia, J. M. Patapas, C. E. Degrandpré, R. W. Diraddo, L. Rosenberg and M. Tabrizian, Biomaterials, 2011, 32, 1536–1542 CrossRef CAS PubMed.
- M. Ravi, V. Paramesh, S. R. Kaviya, E. Anuradha and F. D. Paul Solomon, J. Cell. Physiol., 2015, 230, 16–26 CrossRef CAS PubMed.
- E. Fennema, N. Rivron, J. Rouwkema, C. van Blitterswijk and J. De Boer, Trends Biotechnol., 2013, 31, 108–115 CrossRef CAS PubMed.
- A. Stahl, X. Wu, A. Wenger, M. Klagsbrun and P. Kurschat, FEBS Lett., 2005, 579, 5338–5342 CrossRef CAS PubMed.
- F. Pampaloni and E. H. Stelzer, Biotechnol. Genet. Eng. Rev., 2010, 26, 117–138 CrossRef CAS PubMed.
- J. Hilderink, S. Spijker, F. Carlotti, L. Lange, M. Engelse, C. van Blitterswijk, E. de Koning, M. Karperien and A. van Apeldoorn, J. Cell. Mol. Med., 2015, XX, 1–11 Search PubMed.
- D. Pipeleers, P. I. in't Veld, E. Maes and M. Van De Winkel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 7322–7325 CrossRef CAS.
- M. J. Luther, A. Hauge-Evans, K. L. A. Souza, A. Jörns, S. Lenzen, S. J. Persaud and P. M. Jones, Biochem. Biophys. Res. Commun., 2006, 343, 99–104 CrossRef CAS PubMed.
- H. C. Brereton, M. J. Carvell, H. Asare-Anane, G. Roberts, M. R. Christie, S. J. Persaud and P. M. Jones, Biochem. Biophys. Res. Commun., 2006, 344, 995–1000 CrossRef CAS PubMed.
- R. Foty, J. Visualized Exp., 2011, 20, 4–7 Search PubMed.
- J. Z. Tong, P. De Lagausie, V. Furlan, T. Cresteil, O. Bernard and F. Alvarez, Exp. Cell Res., 1992, 200, 326–332 CrossRef CAS PubMed.
- T. Liu, C.-C. C. Chien, L. Parkinson and B. Thierry, ACS Appl. Mater. Interfaces, 2014, 6, 8090–8097 CAS.
- Y. Ichihara, R. Utoh, M. Yamada, T. Shimizu and Y. Uchigata, Heliyon, 2016, 2, e00129 CrossRef PubMed.
- M. Shinohara, H. Kimura, K. Montagne, K. Komori, T. Fujii and Y. Sakai, Biotechnol. Prog., 2014, 30, 178–187 CrossRef CAS PubMed.
- A. B. Bernard, C.-C. Lin and K. S. Anseth, Tissue Eng., Part C, 2012, 18, 583–592 CrossRef CAS PubMed.
- A. Forget, M. Waibel, D. M. Rojas-Canales, S. Chen, N. Kawazoe, F. J. Harding, T. Loudovaris, P. T. H. Coates, A. Blencowe, G. Chen and N. H. Voelcker, J. Mater. Chem. B, 2017, 5, 220–225 RSC.
- R. El Assal, U. A. Gurkan, P. Chen, F. Juillard, A. Tocchio, T. Chinnasamy, C. Beauchemin, S. Unluisler, S. Canikyan, A. Holman, S. Srivatsa, K. M. Kaye and U. Demirci, Nat. Publ. Gr., 2016, 1–10 Search PubMed.
- S.-E. Yeon, D. Y. No, S.-H. Lee, S. W. Nam, I.-H. Oh, J. Lee and H.-J. Kuh, PLoS One, 2013, 8, e73345 CAS.
- J. Dahlmann, G. Kensah, H. Kempf, D. Skvorc, A. Gawol, D. A. Elliott, G. Dräger, R. Zweigerdt, U. Martin and I. Gruh, Biomaterials, 2013, 34, 2463–2471 CrossRef CAS PubMed.
- B. K. Babur, P. Ghanavi, P. Levett, W. B. Lott, T. Klein, J. J. Cooper-White, R. Crawford and M. R. Doran, PLoS One, 2013, 8, e58865 CAS.
- H. Rashidi, J. Yang and K. M. Shakesheff, Biomater. Sci., 2014, 2, 1318 RSC.
- Y. Morimoto and S. Takeuchi, Biomater. Sci., 2013, 1, 257–264 RSC.
- J. L. Albritton, J. D. Roybal, S. J. Paulsen, N. Calafat, J. A. Flores-Zaher, M. C. Farach-Carson, D. L. Gibbons and J. S. Miller, RSC Adv., 2016, 6, 8980–8991 RSC.
- J. W. Hwang, B. R. Lee, M. J. Jung, H. S. Jung, Y. H. Hwang, M. J. Kim, S. H. Lee and D. Y. Lee, Macromol. Res., 2011, 19, 1320–1326 CrossRef CAS.
- H. H. Oh, Y.-G. Ko, H. Lu, N. Kawazoe and G. Chen, Adv. Mater., 2012, 24, 4311–4316 CrossRef CAS PubMed.
- A. Forget, M. Waibel, D. Rojas-Canales, S. Chen, N. Kawazoe, F. J. Harding, T. Loudovaris, P. T. H. Coates, A. Blencowe, G. Chen and N. H. Voelcker, J. Mater. Chem. B, 2017, 5, 220–225 RSC.
- T. Liu, M. Winter and B. Thierry, Biomaterials, 2014, 35, 6060–6068 CrossRef CAS PubMed.
- I. Hoek, F. Tho and W. M. Arnold, Lab Chip, 2010, 10, 2283–2285 RSC.
- M. Singh, C. Berkland and M. S. Detamore, Tissue Eng., Part B, 2008, 14, 341–366 CrossRef CAS PubMed.
- A. Backovic, D. Wolfram, B. Del-Frari, H. Piza, L. a Huber and G. Wick, J. Immunol. Methods, 2007, 328, 118–127 CrossRef CAS PubMed.
- a. Hughes, D. Rojas-Canales, C. Drogemuller, N. H. Voelcker, S. T. Grey and P. T. H. Coates, J. Endocrinol., 2014, 221, R41–R48 CrossRef CAS PubMed.
- A. Mierczynska, A. Michelmore, A. Tripathi, R. V. Goreham, R. Sedev and K. Vasilev, Soft Matter, 2012, 8, 8399 RSC.
- K. Vasilev, V. Sah, K. Anselme, C. Ndi, M. Mateescu, B. Dollmann, P. Martinek, H. Ys, L. Ploux and H. J. Griesser, Nano Lett., 2010, 10, 202–207 CrossRef CAS PubMed.
- F. J. Harding, L. R. Clements, R. D. Short, H. Thissen and N. H. Voelcker, Acta Biomater., 2012, 8, 1739–1748 CrossRef CAS PubMed.
- P. Y. Wang, L. R. Clements, H. Thissend, W. B. Tsaia and N. H. Voelckere, Acta Biomater., 2015, 11, 58–67 CrossRef CAS PubMed.
- K. Vasilev, A. Michelmore, P. Martinek, J. Chan, V. Sah, H. J. Griesser and R. D. Short, Plasma Processes Polym., 2010, 7, 824–835 CrossRef CAS.
- K. Vasilev, A. Michelmore, H. J. Griesser and R. D. Short, Chem. Commun., 2009, 3600–3602 RSC.
- X. Liu, Q. Feng, A. Bachhuka and K. Vasilev, Appl. Surf. Sci., 2013, 270, 473–479 CrossRef CAS.
- M. Ramiasa-MacGregor, A. Mierczynska, R. Sedev and K. Vasilev, Nanoscale, 2016, 8, 4635–4642 RSC.
- E. S. Ng, R. Davis, E. G. Stanley and A. G. Elefanty, Nat. Protoc., 2008, 3, 768–776 CrossRef CAS PubMed.
- E. S. Ng, R. P. Davis, L. Azzola, E. G. Stanley and A. G. Elefanty, Blood, 2005, 106, 1601–1603 CrossRef CAS PubMed.
- M. Pesl, I. Acimovic, J. Pribyl, R. Hezova, A. Vilotic, J. Fauconnier, J. Vrbsky, P. Kruzliak, P. Skladal, T. Kara, V. Rotrekl, A. Lacampagne, P. Dvorak and A. C. Meli, Heart Vessels, 2013, 29, 834–846 CrossRef PubMed.
- J. M. Kelm, N. E. Timmins, C. J. Brown, M. Fussenegger and L. K. Nielsen, Biotechnol. Bioeng., 2003, 83, 173–180 CrossRef CAS PubMed.
- M. J. Luther, E. Davies, D. Muller, M. Harrison, A. J. Bone, S. J. Persaud and P. M. Jones, Am. J. Physiol.: Endocrinol. Metab., 2005, 288, E502–E509 CrossRef CAS PubMed.
- J. C. Stendahl, D. B. Kaufman and S. I. Stupp, Cell Transplant., 2009, 18, 1–12 Search PubMed.
- A. B. Bernard, R. Z. Chapman and K. S. Anseth, Biotechnol. Bioeng., 2014, 111, 1028–1037 CrossRef CAS PubMed.
- T. Kaido, M. Yebra, V. Cirulli and A. M. Montgomery, J. Biol. Chem., 2004, 279, 53762–53769 CrossRef CAS PubMed.
- T. W. Van Haeften and T. B. Twickler, Eur. J. Clin. Invest., 2004, 34, 249–255 CrossRef CAS PubMed.
- L. M. Weber, K. N. Hayda and K. S. Anseth, Tissue Eng., Part A, 2008, 14, 1959–1968 CrossRef CAS PubMed.
- M. Cornu, J. Yang, E. Jaccard, C. Poussin, C. Widmann and B. Thorens, Diabetes, 2009, 58, 1816–1825 CrossRef CAS PubMed.
- A. C. Hauge-Evans, P. E. Squires, S. J. Persaud and P. M. Jones, Diabetes, 1999, 48, 1402–1408 CrossRef CAS PubMed.
- A. F. Stalder, G. Kulik, D. Sage, L. Barbieri and P. Hoffmann, Colloids Surf., A, 2006, 286, 92–103 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The chemical structure of the monomers used for the plasma polymer coating, FTIR spectra and thickness (as determined by ellipsometry) of the plasma polymer coating, the full ATR-FTIR and EDX spectra of the unmodified and etched PDMS, the depth and diameter of microwells for the different surfaces and different volume and the respective microwells aspect ratio. See DOI: 10.1039/c6bm00916f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |