
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of chemical warfare agent simulants and hydrolysis products in biological samples by paper spray mass spectrometry†
Josiah
McKenna‡
a,
Elizabeth S.
Dhummakupt‡
b,
Theresa
Connell
c,
Paul S.
Demond
c,
Dennis B.
Miller
b,
J.
Michael Nilles
c,
Nicholas E.
Manicke
*a and
Trevor
Glaros
 *b
*b
aDepartment of Chemistry and Chemical Biology, Indiana University-Purdue University Indianapolis, Indianapolis, IN 46202, USA. E-mail: nmanicke@iupui.edu
bBioSciences Division, BioDefense Branch, US Army Edgewood Chemical Biological Center, Aberdeen Proving Ground, MD 21010, USA. E-mail: trevor.g.glaros.civ@mail.mil
cExcet, Inc., 6225 Brandon Ave, Suite 360, Springfield, VA 22150, USA
First published on 17th March 2017
Abstract
Paper spray ionization coupled to a high resolution tandem mass spectrometer (a quadrupole orbitrap) was used to identify and quantitate chemical warfare agent (CWA) simulants and their hydrolysis products in blood and urine. Three CWA simulants, dimethyl methylphosphonate (DMMP), trimethyl phosphate (TMP), and diisopropyl methylphosphonate (DIMP), and their isotopically labeled standards were analyzed in human whole blood and urine. Calibration curves were generated and tested with continuing calibration verification standards. Limits of detection for these three compounds were in the low ng mL−1 range for the direct analysis of both blood and urine samples. Five CWA hydrolysis products, ethyl methylphosphonic acid (EMPA), isopropyl methylphosphonic acid (IMPA), isobutyl methylphosphonic acid (iBuMPA), cyclohexyl methylphosphonic acid (CHMPA), and pinacolyl methylphosphonic acid (PinMPA), were also analyzed. Calibration curves were generated in both positive and negative ion modes. Limits of detection in the negative ion mode ranged from 0.36 ng mL−1 to 1.25 ng mL−1 in both blood and urine for the hydrolysis products. These levels were well below those found in victims of the Tokyo subway attack of 2 to 135 ng mL−1.1 Improved stability and robustness of the paper spray technique in the negative ion mode was achieved by the addition of chlorinated solvents. These applications demonstrate that paper spray mass spectrometry (PS-MS) can be used for rapid, sample preparation-free detection of chemical warfare agents and their hydrolysis products at physiologically relevant concentrations in biological samples.
Introduction
Modern use of chemical warfare agents (CWAs) dates back to World War I with the use of chlorine gas and other agents.2 More recent CWA usage on human populations occurred during the Iran-Iraq War in the 1980s3 and the Tokyo subway sarin attack in 1995.4 While efforts to ban the stockpiling and use of these weapons have been made, CWAs continue to be a threat to the warfighters, first responders, and civilian populations, especially when possessed by rogue states and terrorist groups. For example, in the last few years Syrian populations have been attacked with sarin.5–7 Additionally, a recent news report indicates terrorist groups have been using sulfur mustard.8Analysis of CWAs by mass spectrometry (MS) has grown substantially in the last 10 years.9–13 Many of the fielded MS methods require the use of gas chromatography (GC) and/or liquid chromatography (LC), which pose difficulties for rapid analysis due to sample preparation needs14 and lengthy run times. Several rapid, on-site methods have been proposed, such as atmospheric pressure chemical ionization (APCI) MS,15,16 selected ion flow tube (SIFT) MS,17,18 and proton transfer reaction (PTR) MS.19 However, these methods can only measure a limited range of volatile small molecules and have strong carry-over between runs, which decreases the rate of sample analysis. Direct analysis in real time (DART) MS, an ambient ionization method, has been shown to successfully analyze (and quantitate) CWAs20 and explosives.21 However, DART ionization has the logistical burden of using a heated purified gas flow.
Paper spray ionization (PS) MS is an alternative approach to these fieldable techniques. PS-MS is an ambient ionization technique that requires little to no sample preparation,22 analysis can be performed in seconds, and can directly sample complex biological and environmental samples.23–27 Numerous papers have utilized paper spray coupled to low resolution tandem mass spectrometers such as ion traps and triple quadrupole MS.25 Combining paper spray with mass spectrometers capable of both high resolution and tandem mass spectrometry further improves selectivity.28,29 To date, PS-MS has been used to analyze pesticides and herbicides in food and environmental samples,30,31 which have chemical similarities to CWAs. However, this type/class of chemicals has not been analyzed via paper spray MS in complex biological matrices (i.e. whole blood and urine). Paper spray has the potential to improve upon the robustness and fieldability of other low/no prep ambient ionization methods because it does not require purified ionizing gas. Improvements in fieldable mass spectrometers are still needed to fully realize the advantages of ambient ionization techniques. Progress toward miniaturization has been made by several vendors including the BaySpec Portability, MassTech Explorer, Inficon Hapsite, Bruker E2 M, and Griffon 824.32 Additionally, an effort to integrate paper spray cartridges in a plug-and-play format with a miniaturized MS has been reported.33 In this format, paper spray would be useful in a field-forward lab to rapidly analyze unknown samples collected by the warfighter and feedback the relevant information regarding CWAs.
Previous analysis of CWAs has focused not only on the parent compound but also on the breakdown products or hydrolysis products.34 This is due to the short half-life and low stability of parent compounds in environments containing water.35 An example of the short half-life was shown in Noort et al. analysis of serum samples from the 1995 Tokyo subway sarin attack victims. In blood samples collected 1.5–2.5 hours after the attack, sarin was not detectable but the hydrolysis product O-isopropyl methylphosphonic acid (IMPA) was detected and ranged from 2 to 135 ng mL−1.1 As a result of the rapid hydrolysis of the CWA parent compounds in biofluids, detection of the hydrolysis products is essential. However, the CWA hydrolysis products in the field are traditionally analyzed via GC-MS and thus require extensive derivatization prior to analysis34 which pose difficulties for rapid analysis. In this study, PS-MS analyses of CWA simulants of G-series nerve agents (i.e., sarin, soman, tabun) as well as major hydrolysis products of sarin, VX, soman, Russian VX, and cyclosarin were conducted in blood and urine matrices.
Materials and methods
Chemicals and materials
High-performance liquid chromatography (HPLC) grade solutions of methanol, acetonitrile, formic acid, and water were purchased from Sigma Aldrich (St Louis, MO, USA); anhydrous carbon tetrachloride was also purchased from Sigma Aldrich. Optima grade ammonium hydroxide and ultra-high-performance liquid chromatography (UHPLC) grade methanol were purchased from Fisher Scientific (Fair Lawn, NJ, USA). Raw human urine and human whole blood with K2EDTA anticoagulant were purchased from Innovative Research (Novi, MI, USA). Trimethyl phosphate (TMP), diisopropyl methylphosphonate (DIMP), and dimethyl methylphosphonate (DMMP) were purchased from Sigma. The d9TMP was purchased from Cerilliant (Round Rock, TX, USA), and the 13Cd3DIMP was synthesized by Dr. Bob Williams and Mark Alverez at the Los Alamos National Laboratory (Los Alamos, NM, USA). Ethyl methylphosphonic acid (EMPA), isopropyl methylphosphonic acid (IMPA), isobutyl methylphosphonic acid (iBuMPA), cyclohexyl methylphosphonic acid (CHMPA), and pinacolyl methylphosphonic acid (PinMPA) dissolved in water were from Cerilliant (product number NAx8-CAL). A mixture of stable isotope labeled (SIL) analogs of the hydrolysis products—d5EMPA, 13C3IMPA, 13Cd3iBuMPA, 13C6CHMPA, and 13C6PinMPA dissolved in water—was also from Cerilliant (product number NAx8-IS). Velox sample cartridges were purchased from Prosolia (Indianapolis, IN, USA).Sample preparation
The CWA simulant stock solutions—TMP, DIMP, and DMMP—were diluted from neat material to a stock concentration of 10 mg mL−1 in high purity methanol for DIMP and HPLC grade water for all others. These were then diluted to 100 μg mL−1 in HPLC grade water. The calibration curve standards were then prepared in blood or urine at concentrations of 1000, 500, 200, 100, 50, and 0 ng mL−1 by performing dilutions of the aqueous working solution in the biological matrix. The internal standards were spiked into the biological matrix under study to a final concentration of approximately 1000 ng mL−1.Solutions containing the CWA hydrolysis products (EMPA, IMPA, iBuMPA CHMPA, and PinMPA) were purchased in concentrations of 5000, 2500, 1250, 625, 250, 125, 63, and 25 ng mL−1. Calibration standards were then prepared in blood or urine at concentrations of 250, 125, 62.5, 31.25, 12.5, 5, 2.5, and 1.25 ng mL−1 by performing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilutions of the aqueous working solutions in the biological matrix. A 5 μL aliquot of an aqueous internal standard (ISTD) solution was spiked into a 100 μL aliquot of each biological sample; the ISTD solution contained 525 ng mL−1 each of the five SIL CWA hydrolysis products.
:20 dilutions of the aqueous working solutions in the biological matrix. A 5 μL aliquot of an aqueous internal standard (ISTD) solution was spiked into a 100 μL aliquot of each biological sample; the ISTD solution contained 525 ng mL−1 each of the five SIL CWA hydrolysis products.
Paper spray ionization
The Velox360 autosampler and ionization source from Prosolia (Indianapolis, IN, USA) was used. Prior to MS analysis, a 12 μL aliquot of blood or urine sample was pipetted onto the PS cartridge, taking care to make sure the sample spot wicked to the edges of the triangular-shaped paper. Sample spots were dried at 37 °C for 30 minutes immediately prior to analysis. All cartridges were loaded into the Velox360 for analysis. For positive ion mode, the spray solvent was 95:5 methanol:water with 0.01% formic acid. During the solvent addition step, pump A was programmed to dispense 3 μL of solvent five times for a total of 15 μL solvent directly onto the dried sample spot. This was immediately followed by pump B dispensing 10 μL of solvent eight times for a total of 80 μL at the rear of the cartridge. The addition of this solvent was spread out over 60 seconds.
In the negative ion mode, a spray solvent of 9:1 methanol:carbon tetrachloride with 0.01% ammonium hydroxide was used to reduce the propensity for discharge and encourage ion formation. Pump A was programmed to dispense 3 μL four times and pump B was programmed to dispense 10 μL thirteen times, using 142 μL of solvent total. When directly comparing both positive and negative ion polarities, the solvent pump programming was spread out over the course of 1.4 minutes, with the negative ion mode program utilizing smaller delays between subsequent pumps to prevent excessive solvent evaporation.
Mass spectrometry data acquisition
MS analyses of CWA simulants were performed using a Thermo Fisher Orbitrap Elite mass spectrometer (Thermo Scientific Inc., San Jose, CA, USA). The temperature of the MS capillary inlet was set at 325 °C. The tube lens voltage was set at 60 V. The MS method run time was 1.0 minute, broken down into 4 time segments with varying spray voltages: 0–0.1 min, 0 kV; 0.1–0.8 min, +5 kV; 0.8–0.9 min, 0 kV; and 0.9–1.0 min, −5 kV. Tandem mass spectra were recorded using collision-induced dissociation (CID). The observed precursor ions and primary fragment ions for the five simulant compounds, as well as the optimized collision energy (CE) for each, can be found in ESI Table 1.†Detection of CWA hydrolysis products was performed on a Thermo Q-Exactive Focus mass spectrometer with the S-lens set to 50 and capillary temperature set to either 325 °C or 320 °C for positive or negative ion modes, respectively. The instrument methods were both 1.4 minutes long with the spray voltage on at +4 kV or −4 kV (depending on the method-specified polarity) from 0–1.1 min, the voltage then being set to 0 kV from 1.1–1.4 min. The spray voltage was turned off to give zero-intensity scans, a requirement for automatic peak integration. The mass spectrometer was operated in tandem mass spectrometry (MS/MS) mode using an inclusion list. The precursor and fragment ions for the five hydrolysis products and their SIL analogs can be found in ESI Table 1.†
Data processing and statistics
For all CWA simulants, raw data were manually extracted and processed using XCalibur v. 2.2 (Thermo Scientific Inc., San Jose, CA, USA). Briefly, base peaks for each transition were extracted from the raw total ion chromatogram (TIC) within a 10 ppm window and the total area was determined. The total area was then used to determine the ratio relative to the fixed ISTD. MS analysis was performed on each of the calibration standards in triplicate. To generate the calibration curves, average ratios were graphed with the known concentrations. Linear lines of best fit were calculated by using 1/x weighted least squares. The limit of detection (LOD) was determined by multiplying the error in the y-intercept by 3.3 and dividing by the slope.All data for the CWA hydrolysis products were automatically processed using TraceFinder v. 3.3 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Peaks within a 5 ppm window of the target compound's fragment ion were integrated. The analyte peak area was divided by the area of the corresponding fragment ion of the appropriate ISTD. Each calibration point was run in triplicate and the ratios of analyte signal to ISTD signal were plotted against their known concentrations to generate the calibration curve, which was linearly fit using 1/x weighted least squares. LODs for the CWA hydrolysis products were determined by multiplying the standard error of the y-intercept by 3.3 and dividing by the slope of the curve; for positive ion mode, some of these calculated LODs were lower than the lowest-detected calibration samples, in which case the concentration of the first reliably detected calibration level was reported as the LOD.
Results and discussion
Chemical warfare agent simulants
Full-scan paper spray mass spectra were collected for DMMP, TMP, and DIMP and the isotopically labeled internal standards d9TMP and 13Cd3DIMP spiked into human blood and urine (Fig. 1). TMP and DMMP were detected primarily as protonated ions whereas DIMP was sodiated in both urine and blood. Additionally, it demonstrates that 10 μg mL−1 (DMMP, TMP, d9TMP) and 2 μg mL−1 (DIMP and 13Cd3DIMP) concentrations were easily distinguishable even among the complex biological background. For all subsequent CWA simulant experiments, tandem mass spectra generated by CID for each of the five compounds were used (ESI Fig. 1†).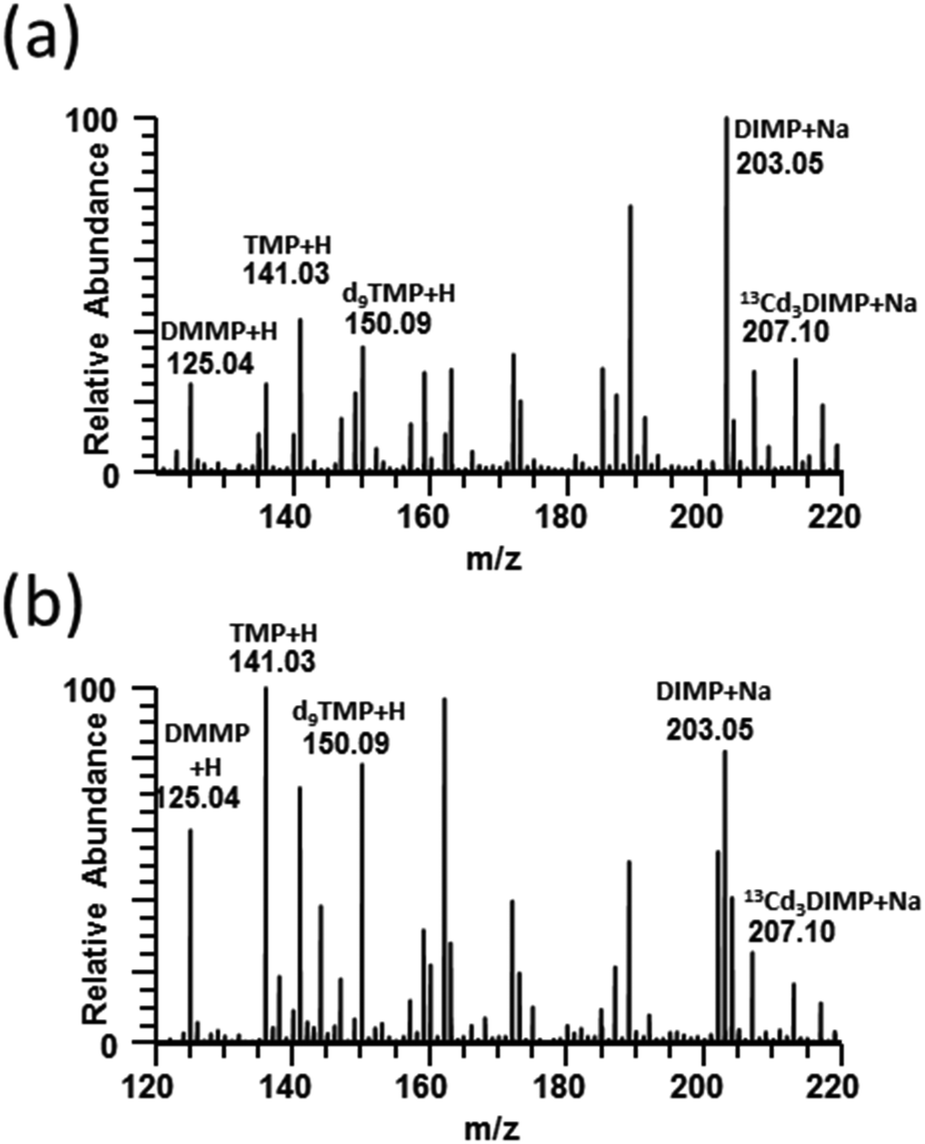 | ||
| Fig. 1 PS-MS full scan mass spectra for 10 ppm TMP, d9TMP, & DMMP; 2 ppm DIMP & 13Cd3DIMP spiked into blood (a) and urine (b). Data were collected in positive ion mode using 95/5 methanol/water with 0.01% formic acid. | ||
When developing a paper spray method, optimization of spray solvent composition for PS ionization is important because the solvent needs to sufficiently extract the analyte(s) from the paper while also being compatible with ionization. Solvent considerations even for the same compound can vary widely based upon its source or matrix background. Therefore, for the CWA simulants in blood and urine, we optimized the solvent system by varying ratios of acetonitrile (ESI Fig. 2a and d†) and methanol (ESI Fig. 2b and e†) in water. A spray solvent of 95:5 methanol:water was found to produce the highest signal intensity universally in both blood and urine. Next, formic acid was used as a solvent modifier to determine the ideal concentration of free protons. As is evident in blood (ESI Fig. 2c†) and urine (ESI Fig. 2f†), the addition of formic acid did not seem to have an effect on the overall signal intensity except when the concentration exceeded 0.1% for blood. In the case of urine, formic acid did not suppress the total signal even at 1.0%. For all subsequent experiments, we used 95:5 methanol:water with 0.01% formic acid. A small amount of formic acid was included in the final spray solvent because prior experience suggested that it improves spray stability.
Quantitative analyses of the three CWA simulants were performed by spiking internal standards (ISTDs) and analyte(s) into blood or urine. ISTD selection is crucial for quantitative analysis, with stable isotope labeled (SIL) analogs of the analytes being the optimal choice. However, isotopically labeled standards are sometimes difficult to obtain. In this study, d9TMP and 13Cd3DIMP were available, but a SIL analog of DMMP was not available. Calibration curves of each CWA simulant were generated using both d9TMP and 13Cd3DIMP to evaluate the quantitative performance of PS-MS. Table 1 shows the average LOD, standard deviation of the LOD, relative error in the slope, and range of R2 values of the two calibration curves generated for each of the 3 analytes from repeats of the same urine donor. As expected, the SIL analogs resulted in better calculated LODs for TMP and DIMP. In the case of DMMP, d9TMP yielded the more reproducible results than when using 13Cd3DIMP as the ISTD. These differences are likely because both DMMP and d9TMP are protonated ions, whereas 13Cd3DIMP is a sodiated ion. [13Cd3DIMP + Na]+ consistently produced significantly higher signal intensities than the [DMMP + H]+, typically on the order of 10–30× (ESI Fig. 2†). These higher intensities resulted in proportionally higher absolute variances. These translated, through the relative ratios to ISTD, to greater variation in the y-intercept and therefore the calculated LOD. Calibration curves for the three CWA simulants using the best performing ISTD in both blood and urine are shown in Fig. 2. Two continuing calibration verification (CCV) standards were analyzed 24–48 hours following the generation of the calibration curve to verify the accuracy of the calibration. All CCVs fell on the calibration curve, demonstrating excellent recovery. In all cases, the percent error was less than ±20% (ESI Table 2†).
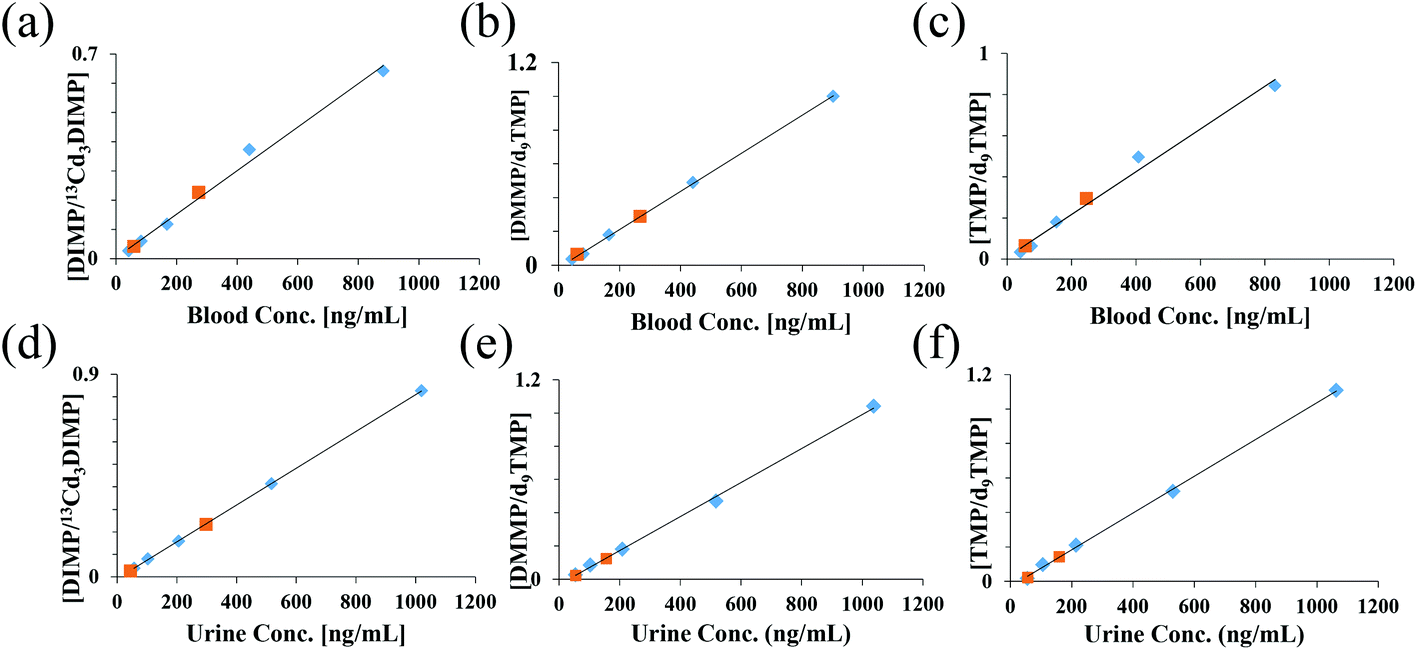 | ||
| Fig. 2 Calibration curves for each compound from 10 μL of blood (a–c) or urine (d–f) using an isotopically labeled internal standard were generated by PS-MS. Average ratios (N = 3) were plotted against the known concentration. Data were collected in positive ion mode using 95/5 methanol/water w 0.01% formic acid. Blue points – calibration curve levels; orange points – continuing calibration verification; refer to ESI Table 2.† | ||
| Analyte | ISTD | Avg. LOD [ng mL−1] | St. dev. LOD [ng mL−1] | Rel. error in slope [%] | R 2 range |
|---|---|---|---|---|---|
| T dry = 30 minutes | |||||
| DIMP | d9TMP | 29.0 | 5.0 | 8.0 | 0.986–0.998 |
| 13Cd3DIMP | 7.0 | 2.0 | 2.0 | 0.999–1.000 | |
| DMMP | d9TMP | 28.6 | 0.9 | 8.0 | 0.999–1.000 |
| 13Cd3DIMP | 31.0 | 14.0 | 9.0 | 0.986–0.997 | |
| TMP | d9TMP | 7.4 | 0.7 | 2.0 | 0.986–0.997 |
| 13Cd3DIMP | 37.0 | 6.0 | 10.0 | 0.976–0.993 | |
Ion suppression due to different matrix sources was evaluated by preparing calibration curves in blood and urine from three different donors and are presented in Tables 2 and 3, respectively. The LOD, standard line slopes, and R2 values showed minimal variation across the three donors, indicating that the matrix did not affect the assay performance across these three lots of biofluid. DIMP, which was detected as the [M + Na]+ ion, consistently had a tenfold lower detection limit in urine compared to blood.
| Donor # | LOD [ng mL−1] | Slope (×104) | R 2 | |
|---|---|---|---|---|
| DIMP/13Cd9DIMP | 1 | 28.0 | 7.7 | 0.992 |
| 2 | 28.0 | 7.6 | 0.994 | |
| 3 | 33.0 | 6.4 | 0.990 | |
| Avg | 30.0 | 7.2 | ||
| Std dev | 3.0 | 0.7 | ||
| DMMP/d9TMP | 1 | 12.0 | 11.4 | >0.999 |
| 2 | 16.0 | 14.0 | 0.999 | |
| 3 | 13.0 | 11.3 | 0.999 | |
| Avg | 14.0 | 12.0 | ||
| Std dev | 2.0 | 2.0 | ||
| TMP/d9TMP | 1 | 37.0 | 11.0 | 0.987 |
| 2 | 12.0 | 12.4 | >0.999 | |
| 3 | 10.0 | 14.0 | >0.999 | |
| Avg | 20.0 | 12.0 | ||
| Std dev | 15.0 | 2.0 |
| Donor # | LOD [ng mL−1] | Slope (×104) | R 2 | |
|---|---|---|---|---|
| DIMP/13Cd3DIMP | 1 | 4.1 | 8.2 | >0.999 |
| 2 | 2.0 | 8.1 | >0.999 | |
| 3 | 3.1 | 7.7 | >0.999 | |
| Avg | 3.1 | 8.0 | >0.999 | |
| Std dev | 1.1 | 0.2 | ||
| DMMP/d9TMP | 1 | 17.6 | 10.1 | 0.998 |
| 2 | 12.7 | 12.1 | >0.999 | |
| 3 | 7.4 | 10.7 | >0.999 | |
| Avg | 12.6 | 11.0 | ||
| Std dev | 5.1 | 1.0 | ||
| TMP/d9TMP | 1 | 24.8 | 10.8 | 0.999 |
| 2 | 6.6 | 11.1 | >0.999 | |
| 3 | 12.6 | 11.1 | >0.999 | |
| Avg | 14.7 | 11.0 | ||
| Std dev | 9.3 | 0.2 |
For the measurements described above, ISTD was mixed into the liquid samples prior to spotting. One advantage of paper spray MS is the possibility to store samples as dried matrix spots, which could improve analyte stability36 and, in the case of a dry blood spot (DBS), make it easier to ship since DBSs are not considered a biological hazard.37 However, to fully take advantage of PS-MS, biofluids would need to be applied to the paper spray cartridges in the field, necessitating the addition of the ISTD to the dried sample spot after transporting the cartridge to the laboratory for analysis. To evaluate the feasibility of this approach, blood and urine standards were dried on the paper spray cartridge followed by addition of an ISTD solution containing d9TMP and 13Cd3DIMP. Previous studies have demonstrated that the spot location and size are critical factors for PS-MS analysis, especially reproducibility. To improve robustness of the technique, an apparatus was developed and adapted to the PS cartridge to aid in reproducibly applying both the samples and especially the ISTD solution (Fig. 3). This device was 3D printed with autoclavable polylactic acid plastic filament on a Pegasus Touch (Full Spectrum Laser, Las Vegas, NV). It was designed to improve user reproducibility when samples are prepared in restricted spaces such as a biosafety cabinet. Table 4 shows the average LOD, standard deviation in the LOD, relative error in the slope, and range of R2 values for calibration curves prepared in blood and urine by applying the ISTD solution to the dried samples. Performances of the non-ideal ISTDs in the same experiment are highlighted in red. The quality of the curves in this experiment appear to be more sensitive to the selection of the internal standard. The difference in the quality of the curves between the ideal and non-ideal internal standard is more pronounced when applying the ISTD to the dried spot (Table 4) as compared to mixing the ISTD into the liquid samples (Table 1). Another important variable, not investigated here, is the amount of time between sample deposition and ISTD deposition. These results do show, however, that as long as the proper internal standard is utilized, application of the ISTD to the dried sample has the potential for real-world application with acceptable quantitative performance.
 | ||
| Fig. 3 A spotting apparatus was designed (a) and used to spot 12 μL of blood onto the Prosolia paper spray cartridges (b, c). | ||
| Analyte Tdry = 30 min | ISTD Tdry = 30 min | Avg. LOD [ng mL−1] | St. dev. LOD [ng mL−1] | Rel. error in slope [%] | R 2 range |
|---|---|---|---|---|---|
| Blood | |||||
| DIMP | 13Cd3DIMP | 38 | 13 | 6 | 0.993–0.998 |
| d9TMP | 112 | 41 | 17 | 0.920–0.977 | |
| DMMP | d9TMP | 27 | 3 | 5 | 0.991–0.998 |
| 13Cd3DIMP | 225 | 212 | 40 | 0.930–0.972 | |
| TMP | d9TMP | 35 | 6 | 6 | 0.988–0.998 |
| 13Cd3DIMP | 125 | 34 | 15 | 0.922–0.959 | |
| Urine | |||||
| DIMP | 13Cd3DIMP | 11 | 10 | 2 | 0.998–1.000 |
| d9TMP | 55 | 25 | 8 | 0.977–0.998 | |
| DMMP | d9TMP | 20 | 9 | 3 | 0.997–0.999 |
| 13Cd3DIMP | 52 | 14 | 8 | 0.982–0.991 | |
| TMP | d9TMP | 12 | 10 | 2 | 0.999–1.000 |
| 13Cd3DIMP | 47 | 27 | 7 | 0.968–0.998 | |
The CWA simulants are representative of organophosphate nerve agents such as tabun, sarin, and soman. These chemical warfare agents are highly lethal and volatile with an inhalation LC50 of 1 ppm for 10 minutes of exposure and a percutaneous LD50 of 300 mg per person.35 The data presented here show PS-MS is capable of detecting these simulants below lethal concentrations in clinical matrices.
Chemical warfare agent hydrolysis products
The CWA hydrolysis products can be detected in the positive ion mode as both protonated and sodiated ions, but they also form intense [M − H]− ions in the negative ion mode because they contain an acidic phosphonic acid moiety. We compared the quantitative performance in both positive and negative ion modes for five organophosphonate compounds. Using the optimized solvent system as determined with the parent CWA simulants—95:5 methanol:water with 0.01% formic acid—calibration curves could be generated in both blood and urine (ESI Fig. 3 and 4†). Detection limits, the relative error of the slopes, and the correlation coefficients obtained from the five calibration curves are shown in Table 5. Overall, detection of the CWA hydrolysis products as positive ions was complicated by their poor fragmentation in MS/MS mode. Despite good intensities for the positive molecular ions, the signal strength of their corresponding fragment ions was typically 10–100× lower than their negative ion counterparts, making quantitative analysis for positive ionization generally less sensitive. Furthermore, fragmentation for some of the hydrolysis products in positive ion mode was found to be unreliable, resulting in poor precision of analyte/ISTD measurements, as demonstrated for IMPA in ESI Fig. 5.† When operating in negative ion mode, [M − H]− molecular ions formed readily, and fragmentation by HCD in the q-orbitrap was efficient. The difficulty with pursuing negative ionization, however, was poor Taylor cone stability and irreproducibility of the MS signal. Greater spray instability in the negative ion mode is known to occur in conventional electrospray due to the lower onset potential for corona discharge compared to positive ion electrospray.38–41 This problem appears to be more pronounced in paper spray.26 Formation of corona discharge significantly interferes with Taylor cone formation and typically eliminates any ion signal from analytes or endogenous blood/urine compounds generated via ESI.† The tendency to discharge is sensitive to several experimental parameters, such as the tip-to-inlet distance, absolute applied voltage, solvent composition, and, in the case of paper spray, even the amount of solvent used. There are several potential ways to eliminate or minimize corona discharge, some of which involve the incorporation of electron-scavenging gases such as SF639,42 or O2.43 However, the use of electron-scavenging gases is complicated, particularly for fieldable or field-forward applications. Another option is to utilize different types of porous substrates; polyethylene and polyester were shown to have better sensitivity and lower onset voltages compared to wood, a cellulosic material.44 Polyethylene and polyester are both hydrophobic materials, however; blood and urine samples will bead up on the surface of these materials rather than wick into the pores.
| ISTD | Positive ion mode | Negative ion mode | |||||
|---|---|---|---|---|---|---|---|
| LOD [ng mL−1] | Rel. error in slope [%] | R 2 | LOD [ng mL−1] | Rel. error in slope [%] | R 2 | ||
| Blood | |||||||
| EMPA | d5EMPA | 3.0 | 2.0 | 0.99 | 1.2 | 2.0 | 0.994 |
| IMPA | 13C3IMPA | 10.0 | 26.0 | 0.46 | 0.9 | 2.0 | 0.997 |
| iBuMPA | 13Cd3iBuMPA | 10.0 | 12.0 | 0.90 | 0.9 | 1.0 | 0.996 |
| CHMPA | 13C6CHMPA | 10.0 | 7.0 | 0.97 | 0.8 | 1.0 | 0.998 |
| PinMPA | 13C6PinMPA | 25.0 | 3.0 | 0.98 | 0.5 | 1.0 | 0.995 |
| Urine | |||||||
| EMPA | d5EMPA | 0.7 | 1.0 | 0.99 | 1.2 | 3.0 | 0.982 |
| IMPA | 13C3IMPA | 6.0 | 10.0 | 0.72 | 1.2 | 2.0 | 0.994 |
| iBuMPA | 13Cd3iBuMPA | 3.0 | 8.0 | 0.83 | 1.1 | 2.0 | 0.996 |
| CHMPA | 13C6CHMPA | 3.0 | 5.0 | 0.98 | 0.6 | 1.0 | 0.999 |
| PinMPA | 13C6PinMPA | 6.0 | 4.0 | 0.97 | 0.4 | 1.0 | 0.998 |
We explored alternative solvents to improve paper spray stability in the negative ion mode. Because of the high surface tension of water, it was eliminated from the solvent and a purely organic solvent was used in order to reduce the electrospray onset potential.39,45,46 Studies have also shown that the use of halogenated solvents in electrospray—particularly chlorinated solvents with a higher percent weight of chlorine—helps increase the onset potential for corona discharge, allowing for a larger working range of electrospray voltages.45,47,48 Chloroform was tested in the solvent base, but it failed to significantly inhibit discharge, even when comprising 10% of the solvent. When testing carbon tetrachloride (CCl4), we found that a 9:1 (v:v) mixture of methanol:CCl4 significantly decreased the propensity for corona discharge. Lower amounts of CCl4 were also tested, but they were not able to reduce the current to low (∼1 μA) or stable enough values for both blood and urine to be considered free of discharge consistently from sample to sample. To this 9:1 base solvent, ammonium hydroxide was added at a concentration of 0.01% to facilitate the production of negative ions. Fig. 4A shows a photograph of the paper tip and a typical mass spectrum obtained when using methanol with 0.01% NH4OH as the spray solvent for a blood sample. The expected [M − H]− molecular ions of the hydrolysis products as well as ions from endogenous blood compounds such as amino acids and other metabolites are entirely absent. The spectrum is instead dominated by ions formed via corona discharge such as CO3˙− (m/z 59.98), HCO3− (m/z 60.99), and HCO4− (m/z 76.99).49 The glowing tip, lack of a Taylor cone, and the relatively high unstable spray current (7–10 μA) are all further indications of corona discharge. Fig. 4B shows a typical result for the 9:1 methanol:CCl4 solvent under identical conditions. A stable Taylor cone can be observed, and a relatively low spray current of around 0.5 μA is obtained. The ions detected by the mass spectrometer indicate electrospray ionization, including endogenous compounds from the blood as well as the [M − H]− ions of the hydrolysis products. The CO3˙− ion at m/z 59.98 is present but at an intensity ∼1000× lower than in Fig. 4A, indicating the success of the CCl4 in suppressing discharge and the formation of ions that accompany it.
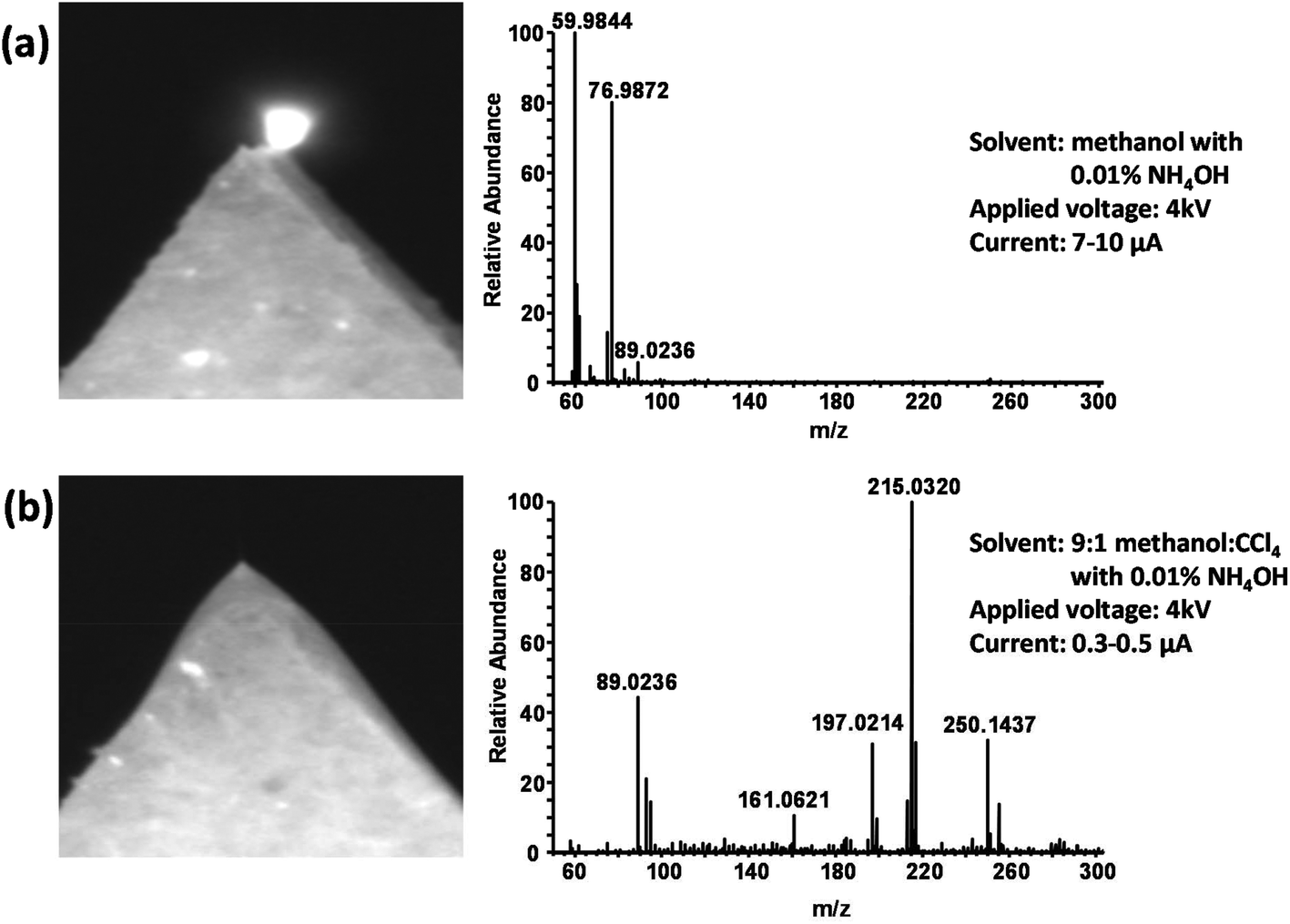 | ||
| Fig. 4 (a) Photograph of the paper tip after application of high voltage and a typical mass spectrum when using methanol as a spray solvent. Corona discharge is indicated by the glowing spot in the photograph, the high spray current, and the ions detected that overwhelm the full-MS spectrum. (b) Photograph and mass spectrum obtained under identical conditions using 9:1 methanol:CCl4 as the spray solvent. The lower spray current as well as the visible Taylor cone indicate a stable electrospray. | ||
Using this new solvent—optimized for paper spray in negative ion mode—calibration curves were generated for each of the CWA hydrolysis products. Calibration curves for IMPA, EMPA, PinMPA, iBuMPA, and CHMPA in both blood and urine are shown in Fig. 5. These compounds are major hydrolysis products of sarin (IMPA), VX (EMPA), soman (PinMPA), Russian VX (iBuMPA), and cyclosarin (CHMPA), respectively. The quantitative measures of these calibration curves are summarized in Table 5, comparing their LODs, relative errors in slope, and R2 values with the curves generated using positive and negative ionization paper spray. MS/MS was solely utilized for the quantitation of each analyte and ISTD—the specific fragmentations generating the ions monitored in negative ion mode are shown in ESI Fig. 1.†
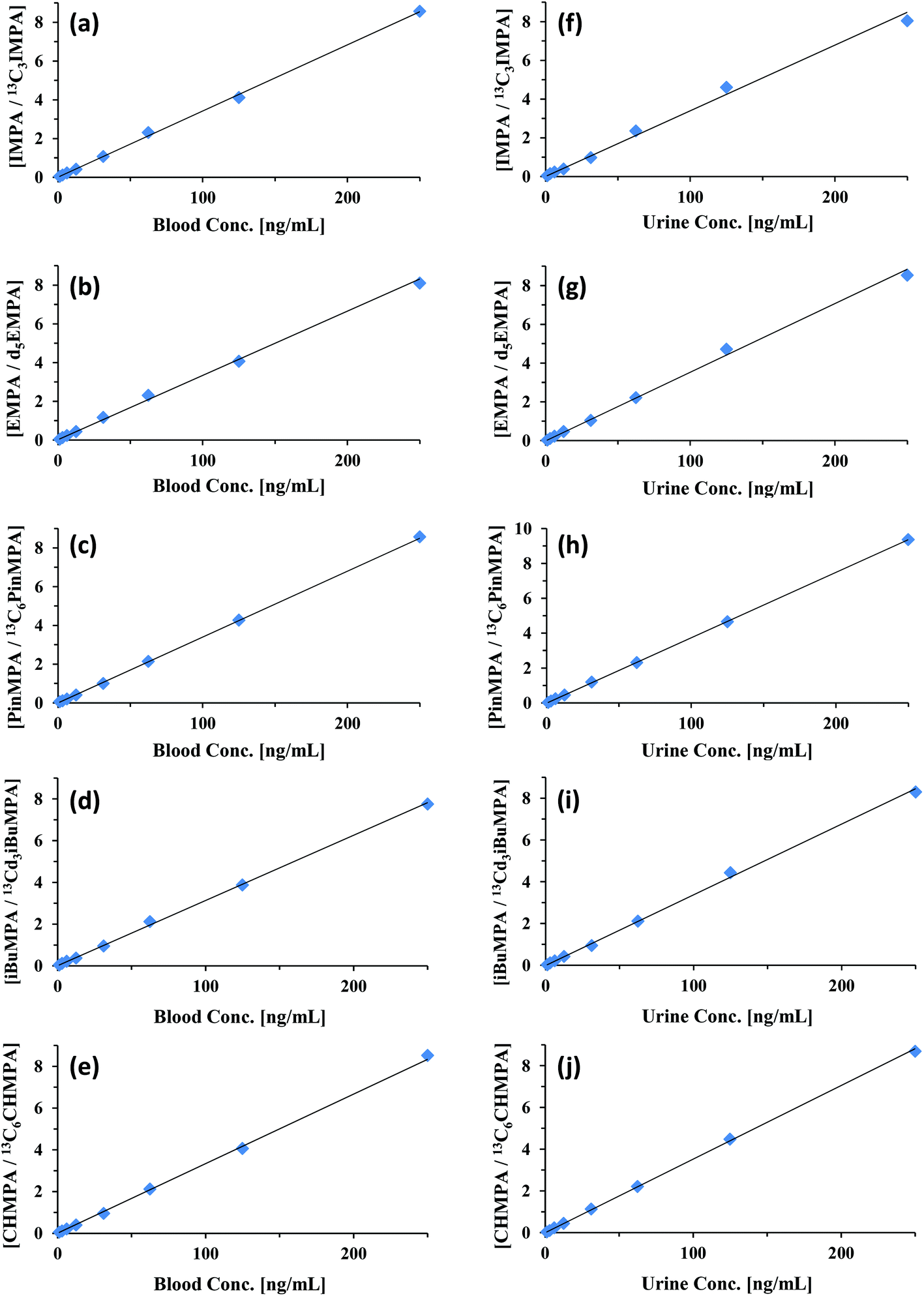 | ||
| Fig. 5 Paper spray MS/MS negative ion calibration curves for IMPA, EMPA, PinMPA, iBuMPA, and CHMPA from blood (a–e) and urine (f–j) using their corresponding SIL analogs as the internal standard. Average ratios (N = 3) were plotted against the prepared concentration. Data were collected in negative ion mode using 90/10 methanol/CCl4 w 0.01% ammonium hydroxide. | ||
In general, detection limits in negative ion mode improved with increasing analyte weight, achieving sub-ng mL−1 LODs for most compounds—down to 0.55 ng mL−1 in blood and 0.36 ng mL−1 in urine for PinMPA, respectively 50- and 15-fold improvements over its detection in positive ion mode. The hydrolysis products demonstrated better quantitation when moving from positive ionization to negative ionization. IMPA and iBuMPA especially showed marked improvement in linearity of the calibration curve and sample-to-sample precision. EMPA showed comparable quantitation between positive and negative ion modes for blood and urine (Table 5). As demonstrated with the parent CWA simulant data, the resolving power of orbitrap mass analyzers was capable of significantly reducing matrix interference as a potential problem for these analytes. Blank biofluid showed no signal within the 5 ppm m/z window for the fragment ions.
In summary, hydrolysis products can be detected as both positive and negative ions using PS-MS. Positive ion mode detection of hydrolysis products is useful because the parent CWAs generally ionize better in the positive ion mode. Negative ion mode, however, affords higher sensitivity, which is important for detecting CWA exposure in biological samples. To date, the levels of hydrolyzed CWA products found in exposure victims ranges from 2 to 135 ng mL−1.1
Conclusions
The objective of this work was to demonstrate paper spray's ability to analyze chemical warfare agent simulants as well as CWA hydrolysis products from biological matrices. PS-MS analysis requires no sample preparation, even when the CWA simulants are in complex matrices such as urine and whole blood. The three CWA parent simulants tested showed good linearity when isotopically labeled standards were spiked into the samples. Additionally, it was shown that the LOD and calibration curves are reproducible across different lots of biofluid.The evaluation of the CWA hydrolysis products was also demonstrated. Paper spray MS is capable of analyzing these hydrolysis products in both positive and negative ionization modes; however, higher sensitivity was found in negative ion mode. Utilizing a chlorinated spray solvent helped reduce the corona discharge potential and stabilize the spray, which are two problems that have plagued negative mode PS-MS analysis. The five CWA hydrolysis products showed good linearity when isotopically labeled standards were spiked into the samples. Additionally, the calculated LODs in negative mode are lower than the concentrations reported in real CWA exposure victims, which were between 2 and 135 ng mL−1. This is crucial, as the parent CWA compound is not likely to be seen in biofluids as a result of their rapid hydrolysis. While the 95/5 methanol/water with 0.01% formic acid is capable of analyzing the hydrolysis products, their LOD values are higher than some of the concentrations found in the Noort et al. study.
This work demonstrates that paper spray mass spectrometry is capable of rapidly detecting CWA and hydrolysis products in clinical biofluids. This distinction is important because MS-based methods are highly regarded in terms of specificity and sensitivity, but traditionally MS techniques require significant sample handling and processing procedures typically in a ‘brick and mortar’ laboratory. Furthermore, most MS detection techniques, especially if they required extensive processing, could take as long as 24 hours to get interpretable results. Using PS-MS, we have been able to get quantitative results in as little as one minute. This technology and approach could have immediate utility since analytical grade mass spectrometers such as the orbitrap are currently used in field-forward portable laboratories such as the US Army's JUPITR program.
Acknowledgements
Funding for this project is provided by the Defense Threat Reduction Agency – Joint Science and Technology Office for Chemical and Biological Defense to TG. The authors would like to thank Donna Hollinshead and Dr. Justin Wiseman at Prosolia for the use of the Velox360 paper spray source. The authors would like to thank Dr. Bob Williams and Mark Alverez the Los Alamos National Lab for generating 13Cd3DIMP. Special thanks to Dr. Bao Q. Tran and Dr. Phillip Mach for their critical review of this manuscript. This research was performed while the author (ESD) held an NRC Research Associateship Award at Edgewood Chemical Biological Center. Conclusions and opinions presented here are those of the authors and are not the official policy of the US Army, ECBC, or the US Government. Information in this report is cleared for public release and distribution is unlimited.Notes and references
- D. Noort, A. G. Hulst, D. H. J. M. Platenburg, M. Polhuijs and H. P. Benschop, Arch. Toxicol., 1998, 72, 671–675 CrossRef CAS PubMed.
- N. M. Elsayed and H. Salem, in Inhalation Toxicology, ed. H. Salem and S. A. Katz, CRC Press, 2nd edn, 2005, ch. 22, pp. 521–542 Search PubMed.
- Report of the Mission Dispatched by the Secretary-General to Investigate Allegations of the use of Chemical Weapons in the Conflict between the Islamic Republic of Iran and Iraq, United Nations Security Council, New York City, New York, 1986.
- A. T. Tu, Forensic Sci. Rev., 2014, 26, 115–120 CAS.
- A. King, Chem. Ind., 2013, 77, 8–8 CAS.
- D. A. Jett, Ann. N. Y. Acad. Sci., 2016, 1374, 5–9 CrossRef PubMed.
- O. Hakeem and S. Jabri, Lancet Glob. Health, 2015, 3, E196–E196 CrossRef PubMed.
- A. Deutsch, Samples Confirm Islamic State used Mustard Gas in Iraq, Published by Reuters on 23 February 2016 Search PubMed.
- H. John, F. Worek and H. Thiermann, Anal. Bioanal. Chem., 2008, 391, 97–116 CrossRef CAS PubMed.
- A. Gapeev and J. Yinon, J. Forensic Sci., 2004, 49, 227–237 CrossRef CAS PubMed.
- C. L. Crawford and H. H. Hill, Jr., in Mass Spectrometry Handbook, ed. M. S. Lee, John Wiley and Sons, New York, 2012, pp. 441–476 Search PubMed.
- P. D'Agostino, J. Hancock and L. Provost, J. Chromatogr., A, 1999, 840, 289–294 CrossRef.
- A. Weissberg, N. Tzanani and S. Dagan, J. Mass Spectrom., 2013, 48, 1340–1348 CrossRef CAS PubMed.
- H. Yoon, Arch. Pharmacal Res., 2007, 30, 387–395 CrossRef CAS.
- Y. Seto, M. Kanamori-Kataoka, K. Tsuge, I. Ohsawa, K. Iura, T. Itoi, H. Sekiguchi, K. Matsushita, S. Yamashiro, Y. Sano, H. Maruko, Y. Takayama, R. Sekioka, A. Okumura, Y. Takada, H. Nagano, I. Waki, N. Ezawa, H. Tanimoto, S. Honjo, M. Fukano and H. Okada, Anal. Chem., 2013, 85, 2659–2666 CrossRef CAS PubMed.
- S. N. Ketkar, J. G. Dulak, W. L. Fite, J. D. Buchner and S. Dheandhanoo, Anal. Chem., 1989, 61, 260–264 CrossRef CAS.
- D. Smith and P. Spanel, Mass Spectrom. Rev., 2005, 24, 661–700 CrossRef CAS PubMed.
- D. B. Milligan, G. J. Francis, B. J. Prince and M. J. McEwan, Anal. Chem., 2007, 79, 2537–2540 CrossRef CAS PubMed.
- R. S. Blake, C. Whyte, C. O. Hughes, A. M. Ellis and P. S. Monks, Anal. Chem., 2004, 76, 3841–3845 CrossRef CAS PubMed.
- J. Nilles, T. Connell and H. Durst, Anal. Chem., 2009, 81, 6744–6749 CrossRef CAS PubMed.
- J. M. Nilles, T. R. Connell, S. T. Stokes and H. D. Durst, Propellants, Explos., Pyrotech., 2010, 35, 446–451 CrossRef CAS.
- J. J. Liu, H. Wang, N. E. Manicke, J. M. Lin, R. G. Cooks and Z. Ouyang, Anal. Chem., 2010, 82, 2463–2471 CrossRef CAS PubMed.
- N. E. Manicke, Q. A. Yang, H. Wang, S. Oradu, Z. Ouyang and R. G. Cooks, Int. J. Mass Spectrom., 2011, 300, 123–129 CrossRef CAS.
- H. Evard, A. Kruve, R. Lohmus and I. Leito, J. Food Compos. Anal., 2015, 41, 221–225 CrossRef CAS.
- N. E. Manicke, B. J. Bills and C. Zhang, Bioanalysis, 2016, 8, 589–606 CrossRef CAS PubMed.
- N. E. Manicke, Q. Yang, H. Wang, S. Oradu, Z. Ouyang and R. G. Cooks, Int. J. Mass Spectrom., 2011, 300, 123–129 CrossRef CAS.
- Y. Su, H. Wang, J. Liu, P. Wei, R. G. Cooks and Z. Ouyang, Analyst, 2013, 138, 4443–4447 RSC.
- A. Y. Li, P. Wei, H. C. Hsu and R. G. Cooks, Analyst, 2013, 138, 4624–4630 RSC.
- F. P. M. Jjunju, S. Maher, D. E. Damon, R. M. Barrett, S. U. Syed, R. M. A. Heeren, S. Taylor and A. K. Badu-Tawiah, Anal. Chem., 2016, 88, 1391–1400 CrossRef CAS PubMed.
- Z. Zhang, R. G. Cooks and Z. Ouyang, Analyst, 2012, 137, 2556–2558 RSC.
- S. Reeber, S. Gadi, S. Huang and G. Glish, Anal. Methods, 2015, 7, 9808–9816 RSC.
- S. Giannoukos, B. Brkic, S. Taylor, A. Marshall and G. Verbeck, Chem. Rev., 2016, 116, 8146–8172 CrossRef CAS PubMed.
- L. Li, T.-C. Chen, Y. Ren, P. I. Hendricks, R. G. Cooks and Z. Ouyang, Anal. Chem., 2014, 86, 2909–2916 CrossRef CAS PubMed.
- R. M. Black and B. Muir, J. Chromatogr., A, 2003, 1000, 253–281 CrossRef CAS PubMed.
- N. B. Munro, S. S. Talmage, G. D. Griffin, L. C. Waters, A. P. Watson, J. F. King and V. Hauschild, Environ. Health Perspect., 1999, 107, 933–974 CrossRef CAS PubMed.
- N. E. Manicke, P. Abu-Rabie, N. Spooner, Z. Ouyang and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2011, 22, 1501–1507 CrossRef CAS PubMed.
- Shipping Guidelines for Dried Blood Spot Specimens, http://www.cdc.gov/labstandards/pdf/nsqap/Bloodspot_Transportation_Guidelines.pdf, (accessed May 11, 2015).
- A. P. Bruins, in Electrospray ionization mass spectrometry, ed. R. B. Cole, Wiley, New York, 1997, pp. 107–136 Search PubMed.
- M. G. Ikonomou, A. T. Blades and P. Kebarle, J. Am. Soc. Mass Spectrom., 1991, 2, 497–505 CrossRef CAS PubMed.
- G. J. Van Berkel, in Electrospray ionization mass spectrometry, ed. R. B. Cole, Wiley, New York, 1997, pp. 65–105 Search PubMed.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- F. M. Wampler, A. T. Blades and P. Kebarle, J. Am. Soc. Mass Spectrom., 1993, 4, 289–295 CrossRef CAS PubMed.
- M. Yamashita and J. B. Fenn, J. Phys. Chem., 1984, 88, 4671–4675 CrossRef CAS.
- M. Y.-M. Wong, S.-H. Man, C.-M. Che, K.-C. Lau and K.-M. Ng, Analyst, 2014, 139, 1482–1491 RSC.
- K. Hiraoka and I. Kudaka, Rapid Commun. Mass Spectrom., 1990, 4, 519–526 CrossRef CAS.
- H. Wang, J. Liu, R. G. Cooks and Z. Ouyang, Angew. Chem., 2010, 122, 889–892 CrossRef.
- R. B. Cole and A. K. Harrata, Rapid Commun. Mass Spectrom., 1992, 6, 536–539 CrossRef CAS.
- K. Hiraoka and I. Kudaka, Rapid Commun. Mass Spectrom., 1992, 6, 265–268 CrossRef CAS.
- J. D. Skalny, T. Mikoviny, S. Matejcik and N. J. Mason, Int. J. Mass Spectrom., 2004, 233, 317–324 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7an00144d |
| ‡ Joint first author/contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |