
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Precious-metal free photoelectrochemical water splitting with immobilised molecular Ni and Fe redox catalysts†
Timothy E.
Rosser
,
Manuela A.
Gross
,
Yi-Hsuan
Lai
and
Erwin
Reisner
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB1 2EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 12th February 2016
Abstract
Splitting water into hydrogen and oxygen with molecular catalysts and light has been a long-established challenge. Approaches in homogeneous systems have been met with little success and the integration of molecular catalysts in photoelectrochemical cells is challenging due to inaccessibility and incompatibility of functional hybrid molecule/material electrodes with long-term stability in aqueous solution. Here, we present the first example of light-driven water splitting achieved with precious-metal-free molecular catalysts driving both oxygen and hydrogen evolution reactions. Mesoporous TiO2 was employed as a low-cost scaffold with long-term stability for anchoring a phosphonic acid-modified nickel(II) bis-diphosphine catalyst (NiP) for electrocatalytic proton reduction. A turnover number of 600 mol H2 per mol NiP was achieved after 8 h controlled-potential electrolysis at a modest overpotential of 250 mV. X-ray photoelectron, UV-vis and IR spectroscopies confirmed that the molecular structure of the Ni catalyst remains intact after prolonged hydrogen production, thereby reasserting the suitability of molecular catalysts in the development of effective, hydrogen-evolving materials. The relatively mild operating conditions of a pH 3 aqueous solution allowed this molecule-catalysed cathode to be combined with a molecular Fe(II) catalyst-modified WO3 photoanode in a photoelectrochemical cell. Water splitting into H2 and O2 was achieved under solar light illumination with an applied bias of >0.6 V, which is below the thermodynamic potential (1.23 V) for water splitting and therefore allowed the storage of solar energy in the fuel H2.
Introduction
Splitting water into hydrogen and oxygen using insolation, a process considered as ‘artificial photosynthesis’, is viewed as a promising sustainable solution to meeting the increasing global demand for transportable fuel and storable renewable energy. The reliance on the long excited state lifetimes1 and excellent catalytic properties2,3 characteristic of components based on low-abundance precious metal elements such as Ru and Pt remains a barrier to low-cost water splitting.4,5 Consequently, only a handful of systems that achieve full water splitting make use of catalysts made from only Earth-abundant elements.6–8 Of these, none rely on synthetic molecular catalysts driving both the H2 and O2 evolution half reactions, and as such the realisation of this goal remains a significant challenge.9,10The specific interest in molecular catalysts arises from the precise control afforded by modern synthetic chemistry over the individual catalytic centres, and therefore the opportunity to study the chemical and structural influences on catalysis.11–13 Indeed, there have been cases where only molecular catalysts, and not noble metal nanoparticles, are found to perform H2 evolution.14 Thus far, H2 evolution with molecular catalysts driving the reduction and oxidation reactions in homogeneous solution has only been achieved in the context of oxidation of an organic substrate.15,16 Thus, homogeneous approaches have failed to date in demonstrating full splitting of H2O into H2 and O2.
A photoelectrochemical (PEC) approach to water splitting utilising immobilised molecular catalysts has many advantages. It allows efficient use of highly active and selective catalytic centres (‘single-site-catalysis’), can overcome kinetic restrictions from diffusion limitations, separates the redox half-reactions to avoid quenching mechanisms and allows separation of the gaseous products, as well as providing a platform for the systematic study of molecular catalysts under aqueous conditions without the requirement of water-solubility.11,17 Recently, the first molecule-catalysed tandem PEC cells with an immobilised Co-based catalyst driving a dye-sensitised NiO photocathode and a Ru catalyst on a dye-sensitised photoanode have demonstrated full water splitting.18,19 However, this PEC cell relies on a precious metal (Ru) water oxidation catalyst and the lability of the axial pyridine on the cobalt H2 evolution catalyst limits the long-term applicability of the H2-evolving photocathode.
In this study, we present the first example of molecular catalyst-enabled water splitting using only Earth-abundant elements (Scheme 1). We have selected the Ni(II) bis-diphosphine class of H2 evolution catalyst due to its high activity in both aqueous20,21 and non-aqueous22 conditions and lack of a labile ligand that could undergo hydrolysis or displacement during catalysis, and immobilised a phosphonate-bearing example on a mesoporous TiO2 electrode. We have studied the activity and stability of this hybrid cathode, finding both to be excellent in mild aqueous conditions, which are essential for combination with a photoanode for solar water splitting. An Fe(II)-based molecular catalyst immobilised on WO3 has shown a well-characterised increase in activity and selectivity for O2 evolution in mildly acidic aqueous conditions compared to the unmodified electrodes,23 and as such was have combined the TiO2 hybrid cathode with a Fe-catalyst modified WO3 photoanode in a PEC water splitting cell.
 | ||
| Scheme 1 Schematic representation of PEC water splitting with the TiO2|NiP hydrogen evolution cathode wired to the WO3|FeP oxygen evolution photoanode in an aqueous electrolyte solution at pH 3. | ||
Results and discussion
Hybrid H2 evolution cathode
Ni(II) bis(diphosphine) complexes are a family of bio-inspired proton reduction catalysts,22,24,25 and the phosphonate-bearing catalyst NiP (Scheme 1) ranks among the most active precious-metal free molecular H2 evolution catalysts in aqueous conditions.14,20,26NiP was reported with turnover numbers (TONs) in excess of 700 mol H2 per mol NiP in photocatalytic schemes using a Ru(II)-based dye and ascorbic acid as an electron donor, and has been demonstrated as a homogeneous electrocatalyst in pH 4.5 aqueous solution.27NiP features phosphonic acid groups, which are well-established for effective binding to TiO2 under mildly acidic aqueous conditions,28–30 through a variety of modes such as P–O–Ti and hydrogen bonding, depending on the exposed TiO2 facet.31 These anchoring groups and the low ligand lability of NiP make it an ideal candidate for immobilisation on metal oxide electrodes for single-site heterogeneous proton reduction.Attachment of molecular H2-evolving catalysts bearing phosphonic acid groups to metal oxides in acidic and pH neutral conditions has been demonstrated,20,32 including the electrochemical reduction of aqueous protons with cobalt(III) catalysts immobilised onto mesoporous indium-doped tin oxide (ITO) electrodes.11,33 The performance of these electrodes, however, was low due to instability of ITO under reducing conditions, as well as the intrinsically low stability of the cobalt catalyst during turnover. NiP displays substantially higher activity than the previously employed Co-based catalysts and we replaced the electrodegrading ITO cathode with robust TiO2. Although TiO2 is often considered as a classical insulator and therefore unsuitable as electrode material, it has previously been used for electrocatalytic proton reduction with immobilised H2-cycling enzymes known as hydrogenases,34,35 and we aim here to establish its use as an electrode substrate for synthetic molecular catalysts such as NiP for long-term reductive electrocatalysis.
NiP was synthesised and characterised as described previously,20 and mesoporous TiO2 (mesoTiO2) electrodes were prepared by doctor blading a suspension of P25 TiO2 (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 anatase:rutile ratio, 25 nm average particle size) onto an FTO-coated glass substrates, followed by annealing at 450 °C. MesoTiO2 electrodes with a geometrical surface area of 1.0 cm2 were employed and scanning electron microscopy (SEM) revealed a film thickness of 4 μm (Fig. S1†). Modification with NiP was achieved by submersion of mesoTiO2 in a methanol solution of the Ni(II) compound (0.5 mM) for 18 h at room temperature, followed by rinsing with methanol and drying under a stream of N2. The amount of NiP per geometric surface area of the TiO2 electrode was determined as 14.6 ± 2.0 nmol cm−2 by spectrophotometry (at λabs = 257 and 300 nm) following desorption of the catalyst from TiO2 with aqueous NaOH (0.1 M). The loading of NiP is in agreement with previous results, where a surface coverage of 53 nmol cm−2 was observed for a phosphonic acid-modified ruthenium complex on 10 μm thick mesoporous TiO2 electrodes.36
:2 anatase:rutile ratio, 25 nm average particle size) onto an FTO-coated glass substrates, followed by annealing at 450 °C. MesoTiO2 electrodes with a geometrical surface area of 1.0 cm2 were employed and scanning electron microscopy (SEM) revealed a film thickness of 4 μm (Fig. S1†). Modification with NiP was achieved by submersion of mesoTiO2 in a methanol solution of the Ni(II) compound (0.5 mM) for 18 h at room temperature, followed by rinsing with methanol and drying under a stream of N2. The amount of NiP per geometric surface area of the TiO2 electrode was determined as 14.6 ± 2.0 nmol cm−2 by spectrophotometry (at λabs = 257 and 300 nm) following desorption of the catalyst from TiO2 with aqueous NaOH (0.1 M). The loading of NiP is in agreement with previous results, where a surface coverage of 53 nmol cm−2 was observed for a phosphonic acid-modified ruthenium complex on 10 μm thick mesoporous TiO2 electrodes.36
Cyclic voltammograms (CVs) of the resultant NiP-modified mesoTiO2 (TiO2|NiP) electrodes and bare mesoTiO2 in aqueous pH 3 electrolyte solution (0.1 M Na2SO4) are shown in Fig. 1a, and suggest H2 evolution activity of the immobilised Ni(II) catalyst. The CVs of the unmodified TiO2 electrode show a typical ‘trumpet plot’ response as expected for a semiconductor electrode.37 The reductive current observed upon the cathodic scan (Q = −0.6 mC at ν = 100 mV s−1), with an onset of approximately −0.1 V vs. the reversible hydrogen electrode (RHE), was followed by an oxidising current (Q = +0.4 mC) in the anodic scan. This observation was attributed to filling and emptying of the conduction band (CB) of TiO2. At pH 3, after modification with NiP, the oxidative current in the anodic scan is substantially diminished, with the charge ratio for the cathodic to anodic scans higher than 15:1, consistent with the consumption of the electrons from the CB of TiO2 for proton reduction catalysis.
 | ||
| Fig. 1 (a) CVs of NiP immobilised on mesoTiO2 (solid black line, TiO2|NiP) and unmodified mesoTiO2 (dashed red line) performed at ν = 100 mV s−1 (the arrow indicates the start of the experiment). (b) Spectroelectrochemistry of the same electrodes monitoring the absorbance change (ΔO.D.) at λ = 800 nm before (no applied potential), during CPE at Eappl = −0.43 V vs. RHE for 20 s (starts at 6 s), and following CPE (no applied potential). Conditions: a Ag/AgCl reference electrode and a Pt mesh counter electrode in aqueous Na2SO4 (0.1 M) solution at pH 3, N2 atmosphere and room temperature. | ||
Evidence that the electrons are transferred to the catalyst via the CB of TiO2 was obtained by spectroelectrochemistry.38 When an applied potential, Eappl, of −0.43 V vs. RHE was applied to TiO2 electrodes, a blue colour was observed, and the corresponding increase in absorbance between 600 and 900 nm in the UV-vis spectrum is shown in Fig. S2a.† We assigned this absorption to d–d transitions in Ti3+, which is formed by filling the CB of TiO2.39,40 When treated with NiP, the increase in absorbance between 600 and 900 nm was still observed at Eappl = −0.43 V vs. RHE (Fig. S2b†), but to a lesser extent, due to a lower steady-state concentration of electrons in the CB. To study the release of CB electrons to NiP, the time-resolved absorbance38 of an unmodified and NiP-modified mesoTiO2 electrode was monitored at λ = 800 nm with Eappl = −0.43 V vs. RHE for 20 s, followed by a return to no applied potential (Fig. 1b). In the absence of NiP, the CB electrons are only slowly released from TiO2 after charging at −0.43 V vs. RHE, which is consistent with the oxidising (discharging) current observed in the return scan of the CV in Fig. 1a. In the presence of NiP on mesoTiO2, the absorbance at λ = 800 nm decayed to its original value within a few seconds (τ1/2 = 2.3 s) of the potential being removed, supporting the efficient release of CB electrons to NiP.
Sustained electrocatalytic H2 production by the TiO2|NiP cathode was confirmed by controlled-potential electrolysis (CPE) in pH 3 aqueous electrolyte solution, and the amount of H2 produced alongside the theoretical maximum (assuming 100% faradaic efficiency) is shown in Fig. 2a. MesoTiO2 electrodes modified with NiP were held at Eappl = −0.25 V vs. RHE in a two-compartment electrolytic cell for 8 h, during which time a charge of 2.02 ± 0.17 C passed. The current decayed slowly during the first few hours, which is consistent with the good stability of the molecular catalyst in aqueous solution (Fig. S3a†).20,27 Analysis of the headspace of the electrochemical cell by gas chromatography revealed that 9.26 ± 2.1 μmol of H2 was produced after 8 h, which corresponds to a faradaic yield of 88 ± 17% and a Ni-based turnover number with a lower limit of 600 (assuming that all NiP remained electroactive on the electrode during CPE).
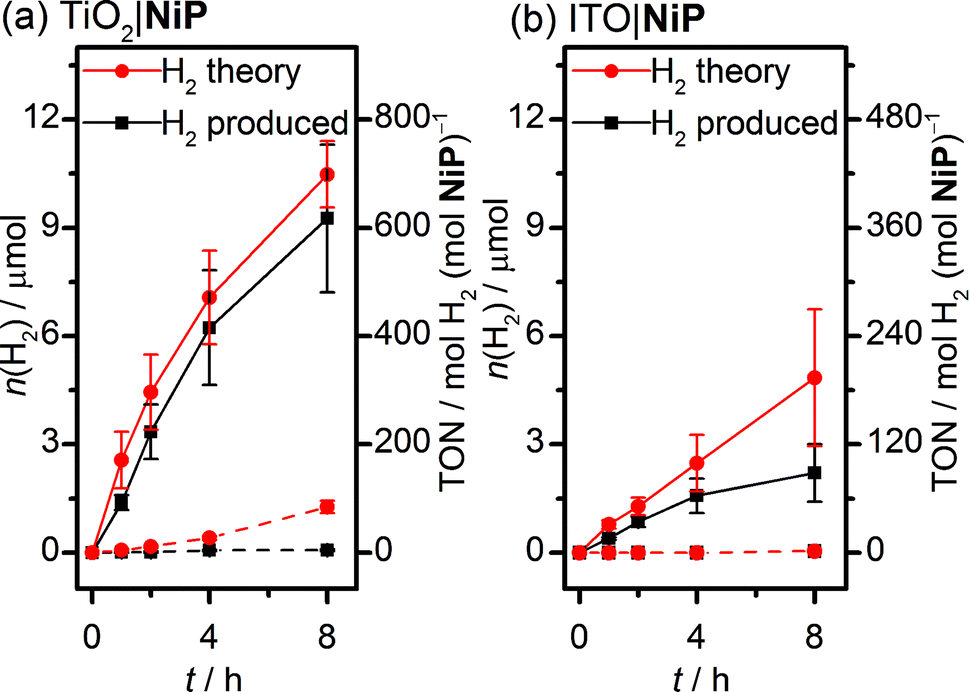 | ||
| Fig. 2 Amount of H2 generated over time during CPE of NiP-modified (solid line) and unmodified (dashed line) with (a) mesoTiO2 and (b) mesoITO electrodes at Eappl = −0.25 V vs. RHE. The theoretical amount of H2 was calculated based on 100% faradaic yield and the experimentally produced H2 was quantified by gas chromatography. Conditions: a Ag/AgCl reference electrode and a Pt mesh counter electrode in aqueous Na2SO4 (0.1 M) solution at pH 3, N2 atmosphere and room temperature. | ||
The fraction of adsorbed catalyst and molecular integrity were study by UV-vis absorption spectroscopy after electrolysis. NiP was desorbed from mesoTiO2 with NaOH (0.1 M) after 8 h CPE and the UV-vis spectrum of the resultant solution matched a reference spectrum recorded from freshly-modified electrodes treated in the same way, suggesting minimal degradation of the ligand framework of NiP (Fig. S4a†). The loading of NiP after CPE was 10.9 ± 2.5 nmol cm−2, which corresponds to 75% of that measured on the freshly-modified electrodes, thus demonstrating strong attachment between the phosphonate anchor and the TiO2 surface even under reducing conditions.
Further evidence for the retention of molecular NiP after CPE was obtained by attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy. Fig. S4b† shows the FTIR spectra of NiP powder, TiO2|NiP before and after 4 h CPE at −0.25 V vs. RHE, and TiO2 (treated with Na2SO4 electrolyte solution). Stretches at 1260 cm−1 (P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1440 and 1510 cm−1 (NiP ligand) were present in the reference NiP spectrum and TiO2|NiP before and after CPE, but not the TiO2 background, reasserting the molecular integrity of the catalyst surviving many turnovers on the electrode surface.
O), 1440 and 1510 cm−1 (NiP ligand) were present in the reference NiP spectrum and TiO2|NiP before and after CPE, but not the TiO2 background, reasserting the molecular integrity of the catalyst surviving many turnovers on the electrode surface.
MesoTiO2 displayed substantially improved performance to mesoITO, which has previously been used as a substrate for a phosphonate-bearing Co-based catalyst.33 We prepared mesoporous ITO electrodes (for synthetic details see the Experimental section) with a NiP loading per geometric surface area of 25 nmol cm−2. Although the CV supports a catalytic current for ITO|NiP in an aqueous pH 3 electrolyte solution (Fig. S5†), CPE at Eappl = −0.25 V vs. RHE (Fig. S3b†) shows the formation of only 2.2 ± 0.8 μmol of H2 after 8 h with a faradaic efficiency of 49 ± 14% (Fig. 2b). The low faradaic efficiency is attributed to competing reductive degradation of the ITO, thus demonstrating the fragility of the ITO electrodes under reducing conditions compared with TiO2.
Electrocatalytic activity of TiO2|NiP was highest at pH 3 and a lower performance was observed in electrochemical experiments in pH 4 and pH 2 electrolyte solutions. The CV of TiO2|NiP at pH 4 is shown in Fig. S6a† and it does not display the same loss of oxidative discharging of the CB as observed at pH 3 (Fig. 1a). This implies loss of performance, which is corroborated by a lower H2 production rate during CPE at Eappl = −0.33 V vs. RHE (Fig. S6b†). When electrolysis was performed at pH 2 under otherwise the same conditions, activity was again observed to be lower than at pH 3 (Fig. S5b†), establishing pH 3 aqueous solution as an optimum for TiO2|NiP. This optimum pH for TiO2|NiP is in agreement with the previously reported higher electrocatalytic activity of NiP in pH 3 compared to pH 4 in homogeneous solution.20 The pendant amines in NiP have a pKa of approximately 3,41 suggesting this to be the optimum pH for proton transfer to the Ni centre, which is a key mechanistic feature of this class of catalyst.22
We have also tested the influence of O2 on the activity of TiO2|NiP as O2 tolerance is an important property for a proton reduction catalyst in water splitting.27,42,43 The H2 production activity by the TiO2|NiP cathode was found to be less effective in a pH 3 electrolyte solution under an atmosphere of air, compared to when purged with N2 as presented above. When subjected to CPE at Eappl = −0.25 V vs. RHE for 4 h under air, the TiO2|NiP retained only some activity (Fig. S7†), achieving a TON of 39 ± 16 but with a low faradaic efficiency of 7%. This significant drop in activity in the presence of oxygen corroborates a similar deactivation observed for NiP in homogeneous aqueous solution.27 Thus, a membrane and a two-compartment electrochemical cell are required in PEC water splitting to protect NiP from O2 generated during simultaneous water oxidation at the photoanode (see below).
These results establish mesoTiO2 as an inexpensive, easily-prepared mesoporous cathode material for immobilisation of phosphonate-bearing molecular synthetic catalysts, particularly in terms of the stability of both the material itself and the interaction between the TiO2 and the molecular catalyst, enabling high turnover numbers to be reached and substantial amounts of H2 being generated at a modest overpotential. Our approach thereby complements previous work, where Ni(II) bis(diphosphine) complexes were attached onto electrode surfaces such as silicon44 and carbon-based materials.21,45,46 Compared to these previous approaches, TiO2|NiP is easier to assemble, requires a less expensive substrate, and can be studied spectroelectrochemically. Furthermore, the transparent nature of TiO2 to visible light and the demonstrated high activity in mild aqueous conditions make TiO2|NiP a suitable cathode material for combination with a photoanode to allow for full water splitting, which is difficult to achieve for cathodes operating under the less sustainable conditions used previously.21,44,46 The use of TiO2 as cathode material in this work is also of interest in the context of current work on its use as a protection layer for photocathode materials such as Cu2O,7,47 Si48–50 and CuInS2/CdS,51 all of which demonstrate photoelectrochemical H2 production in the presence of a Pt catalyst. Employment of phosphonated molecular catalysts such as NiP on such TiO2 layers appears as a promising approach to replace expensive noble metals.
Hybrid oxygen-evolving photoanode
Photoanodes composed of molecular hybrid materials developed thus far largely fall into the category of n-type TiO2 sensitised with molecular dyes, including those based on Ru(II),4,52,53 Zn(II)54 and organic molecules,19 and a co-immobilised molecular water oxidation catalyst. Alternatives have been developed utilising visible-light-harvesting semiconductors based on Earth-abundant metal oxides.23,55–57 Of these, WO3 is notable for good stability under the mildly acidic aqueous conditions required for operation with TiO2|NiP,58 whereas photoanodes made of α-Fe2O3 and BiVO4 are typically studied in alkaline57,59 and pH neutral conditions,56,60,61 respectively. It has recently been shown that an immobilised molecular Fe-based catalyst could improve the otherwise poor activity and selectivity62 for O2 evolution of WO3 in aqueous pH 3 Na2SO4 solution.23 We therefore assembled a photoanode using WO3,63 and an Fe(II) catalyst based on a phosphonic acid-modified tris(2-picolyl)amine (TPA) ligand, the unmodified triflate-coordinated analogue of which is known to perform water oxidation homogeneously at low pH in the presence of a chemical oxidant.12Incorporation of a phosphonic acid linker group into the TPA ligand was achieved via a multi-step chemical synthesis shown in Scheme 2 (see Experimental section for details). Reductive amination of 4-bromopyridine-2-carboxaldehyde with bis(2-picolyl)amine resulted in the bromine-derivatised TPA compound 1. The phosphonic acid was introduced first by Pd-catalysed cross coupling64 to yield the phosphonate ester 2 and subsequently hydrolysed in aqueous HCl to give TPAp1·3HCl. The phosphonic acid-modified TPA ligand was coordinated to FeCl2 in methanol in the presence of Et3N to precipitate FeP (Scheme 1). FeP was synthesised in an overall yield of 15% from 4-bromo-2-pyridinecarboxaldehyde and characterised by electrospray ionisation mass spectrometry (Fig. S8†), CHN elemental analysis and IR spectroscopy.
 | ||
| Scheme 2 Synthetic pathway to FeP. (i) Na(AcO)3BH (1.2 eq.), CH2Cl2, r.t., 2 d, 66%; (ii) Pd(OAc)2 (4 mol%), 1,1′-bis(diphenylphosphino)ferrocene (5 mol%), Et3N, HPO(OEt)2 (1.1 eq.), 80 °C, 2 d, 67%; (iii) HCl (18% in H2O), reflux, 18 h, 70%; (iv) FeCl2, Et3N, CH3OH, r.t., 47%. Full synthetic and characterisation details can be found in the Experimental section. | ||
The electrochemical response of immobilised FeP was first studied on a conducting mesoporous ITO electrode (synthesised as described in the Experimental section). Immobilisation was achieved by submersion in an FeP solution (2 mM in methanol) overnight at room temperature, and representative CVs are shown in Fig. S9.† The ITO|FeP electrodes displayed a reversible redox wave in pH 3 aqueous solution at E1/2 = 0.7 V vs. RHE and a linear dependence of the peak current with scan rate supports the immobilisation of FeP on the metal oxide electrode surface.
WO3 nanosheet (nanoWO3) electrodes were synthesised hydrothermally onto a WO3 seed layer deposited on FTO as previously described (SEM image shown in Fig. S1b†).63 Modification of nanoWO3 was achieved by submersion of the electrodes in a methanol solution of FeP (2 mM) overnight at room temperature in the dark, and the UV/vis spectrum shows a small increase in absorbance between 400 and 450 nm (Fig. S10a†), consistent with the presence of FeP (Fig. S10b†). Fig. 3a shows an increased photocurrent density (j/mA cm−2) from FeP-modified WO3 films in comparison to unmodified films and those treated with FeCl2 and the TPAp1 ligand was demonstrated by linear sweep voltammetry with chopped illumination (P = 0.2 W cm−2, air mass 1.5G). This result suggests an improved photoelectrocatalytic water oxidation by the WO3 film modified with the molecular catalyst, but not the constituent metal salt or ligand in isolation. This result was corroborated by CPE at Eappl = 1.0 V vs. RHE under illumination, with the charge equivalents and amount of O2 produced shown in Fig. 3b. After 2 h, the WO3|FeP was found to produce 3.7 ± 0.4 μmol O2 with 40 ± 4% faradaic efficiency, compared to 1.8 ± 0.3 μmol O2 with 21 ± 2.1% for the unmodified electrode. The low efficiencies can be explained by competing electrolyte oxidation and incomplete oxidation of water known to occur under acidic aqueous conditions,65 but the increase in selectivity in the presence of the iron(II) catalyst matched the precedent set previously.23
 | ||
| Fig. 3 (a) LSVs under chopped illumination (AM1.5G, 0.2 W cm−2) of WO3 (blue line), WO3|FeP (red line), WO3|FeCl2 (green line) and WO3|TPAp1 at ν = 5 mV s−1. (b) Amount of O2 generated after two h PEC water oxidation by nanoWO3 with and without FeP under solar illumination at Eappl = 1.0 V vs. RHE. The theoretical amount of O2 was calculated based on 100% faradaic yield and the experimentally measured O2 was quantified by a fluorescence probe. Conditions: a Ag/AgCl reference electrode and a Pt mesh counter electrode in aqueous Na2SO4 (0.1 M) solution at pH 3, N2 atmosphere and room temperature. | ||
The incident photon-to-current efficiency (IPCE) of WO3|FeP was found to vary with the wavelength of incident monochromatic light in accordance with the UV/vis spectrum of WO3 (Fig. S10a†), and made efficient use of the solar spectrum at wavelengths below 450 nm. The IPCE of WO3|FeP reached a peak value of 53% at a wavelength of 350 nm, and corresponded to previous reports for catalyst-modified WO3.66
Molecule-enabled PEC water splitting
The WO3|FeP photoanode displays good water oxidation activity and is compatible with the H2-evolving TiO2|NiP cathode in an aqueous pH 3 electrolyte solution. Fig. 4a shows a superposition of the three-electrode linear sweep voltammograms of the individual electrodes. The voltammograms of the individual electrodes imply that if the TiO2|NiP cathode and WO3|FeP photoanode are combined in a two-electrode PEC configuration, the onset of photocurrent should be at an applied voltage of approximately 0.6 V. This is lower than the thermodynamic potential requirement for water splitting (1.23 V) and the proposed device should have capacity for solar energy storage in the fuel H2.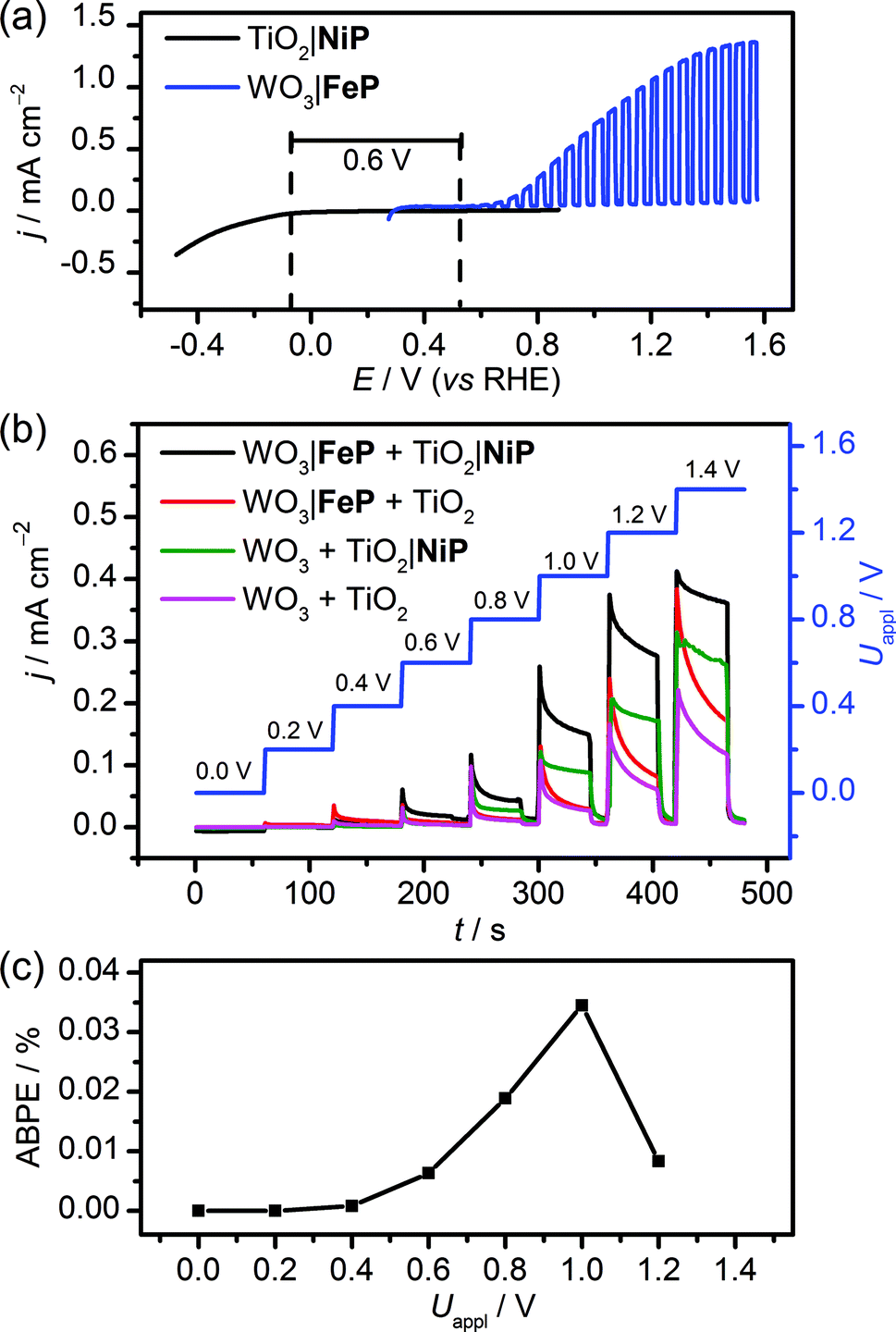 | ||
| Fig. 4 Comparison between (a) superimposed three-electrode LSVs with TiO2|NiP and WO3|FeP (ν = 5 mV s−1) and (b) two-electrode, stepped-potential PEC water splitting with TiO2|NiP directly wired to WO3|FeP (control experiments in the absence of one or both molecular catalysts are also shown). (c) Applied bias photon-to-current efficiency, ABPE for the cell comprising both catalyst-modified electrodes. Conditions: aqueous Na2SO4 (0.1 M) electrolyte solution at pH 3 and room temperature with solar light irradiation (100 mW cm−2 AM1.5G) using a TiO2|NiP cathode (geometrical surface area: 1.0 cm2) and a WO3|FeP photoanode (0.5 cm2). | ||
We therefore assembled a two-electrode PEC cell, with WO3|FeP as the photoanode under periodic simulated solar light irradiation and TiO2|NiP (shielded from the illumination to avoid UV band gap excitation of TiO2) as the cathode, with the electrodes separated by a proton-permeable Nafion membrane to prevent gas diffusion between the cathodic and anodic compartments (Scheme 1). As predicted from the voltammetric response of the individual electrodes, a notable photocurrent was observed at Uappl = 0.6 V in the two-electrode configuration linear sweep voltammogram shown in Fig. 4b. This demonstrates energy storage across the electrodes without the need for a precious-metal containing component (catalyst and electrode material). In the absence of NiP, the photocurrent decayed quickly within each 45 s illumination period, due to initial reductive charging of the TiO2 (Fig. 4b). The applied bias (Uappl) photon-to-current conversion efficiency (ABPE, Fig. 4c) for the PEC cell was calculated with eqn (1), with the photocurrent density (j/mA cm−2) taken after 45 s illumination at each Uappl under full simulated solar spectrum irradiation with a light intensity (P) of 100 mW cm−2.
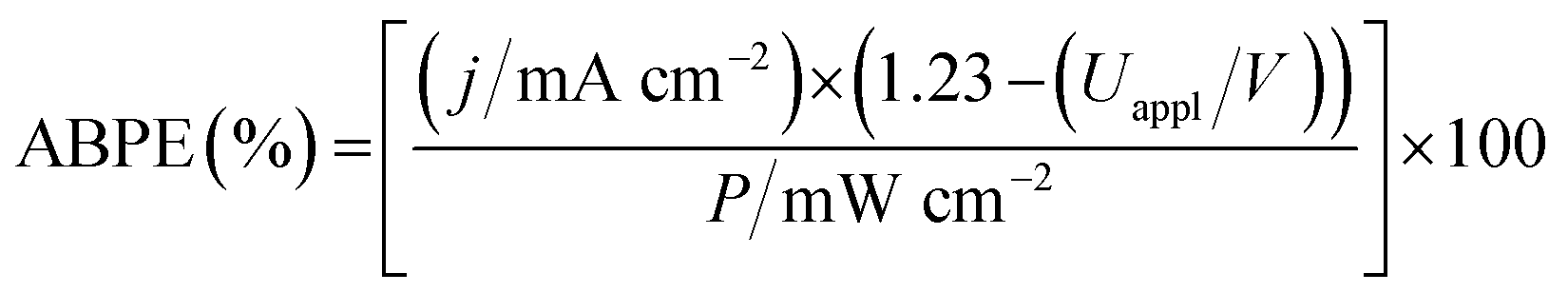 | (1) |
The ABPE was found to have a maximum at Uappl = 1.0 V with an ABPE = 0.035%. The low value for ABPE is due to incomplete use of the solar spectrum by WO3 shown in Fig. S10a† (note that the full simulated solar spectrum, not monochromatic light, was used to calculate the ABPE) and a relatively large applied bias required to achieve a significant photocurrent. Nevertheless, this efficiency is comparable to the only other efficiency reported for a fully molecular solar PEC water splitting cell with a solar-to-hydrogen (STH) efficiency of 0.05%, which required the use of precious metals. However it should be noted that STH accounts for the faradaic efficiency of H2 production, unlike the APBE presented here.19
It was essential to perform extended PEC water splitting under simulated solar light illumination for an in-depth assessment of the efficacy of the molecular catalysts. An electrochemical bias of 1.1 V, less than the thermodynamic potential for water splitting, was applied, and the generated charge, amounts of O2 and H2 are summarised in Table 1 and Fig. 5. After 1 h, 0.61 ± 0.06 μmol of O2 were detected by a fluorescence sensor (Fig. S11†) and 1.04 ± 0.29 μmol of H2 were analysed by gas chromatography with faradaic efficiencies of 61 ± 5% and 53 ± 17%, respectively. The O2-to-H2 ratio was close to the expected one-to-two ratio for full water splitting. Consistent with the three-electrode experiments was the observation that in the absence of FeP, no or only traces of oxygen were detected, again demonstrating the increased selectivity offered by the molecular FeP catalyst in these conditions. The lower faradaic efficiency for H2 evolution compared to the three-electrode CPE Table 1 may be due to initial competitive reduction of O2 or other contaminants trapped within the mesoporous TiO2 electrode in the early stages of electrolysis, as has been observed previously for other nanostructured electrodes.67 In agreement, we observed a faradaic efficiency of 58 ± 13% for H2 generation with TiO2|NiP after 1 h CPE in the experiments shown in Fig. 2a.
| Three-electrode configurationb | ||||||
|---|---|---|---|---|---|---|
| Description | E appl/V vs. RHE | Time/h | n(H2)/μmol | Faradaic yield (%) | n(O2)/μmol | Faradaic yield (%) |
| a Conditions: aqueous Na2SO4 (0.1 M) solution at pH 3 in a two compartment (photo)electrochemical cell at room temperature. Unless otherwise stated, the cell purged with N2 before each experiment and CH4 (2%) was present as internal standard for H2 quantification by GC. The WO3 photoanode was illuminated with solar light (AM1.5G) at 0.2 W cm−2 in the three-electrode experiments and 0.1 W cm−2 in two-electrode PEC water splitting. The TiO2 cathode was shielded from illumination. b A Pt counter electrode and a Ag/AgCl reference electrode were employed. c Performed under air with CH4 as external standard. d Below the limit of detection. | ||||||
| TiO2|NiP | −0.25 | 8 | 9.3 ± 2.1 | 88 ± 17 | ||
| TiO2 | −0.25 | 8 | 0.07 ± 0.03 | 5.6 ± 1.7 | ||
| TiO2|NiPc | −0.25 | 4 | 0.58 ± 0.24 | 7.4 ± 4.5 | ||
| WO3|FeP | 1.0 | 2 | 3.7 ± 0.4 | 40 ± 4 | ||
| WO3 | 1.0 | 2 | 1.8 ± 0.3 | 21 ± 2.1 | ||
| Two-electrode PEC water splitting | ||||||
|---|---|---|---|---|---|---|
| Description | U appl/V | Time/h | n(H2)/μmol | Faradaic yield (%) | n(O2)/μmol | Faradaic yield (%) |
| TiO2|NiP + WO3|FeP | 1.1 | 1 | 1.04 ± 0.29 | 53 ± 17 | 0.61 ± 0.06 | 61 ± 6 |
| TiO2|NiP + WO3 | 1.1 | 1 | 0.51 ± 0.19 | 47 ± 14 | 0.09 ± 0.02 | 18 ± 6 |
| TiO2 + WO3|FeP | 1.1 | 1 | 0.21 ± 0.1 | 38 ± 13 | 0.08 ± 0.08 | 26 ± 26 |
| TiO2 + WO3 | 1.1 | 1 | 0.41 ± 0.26 | 60 ± 18 | —d | — |
| TiO2|NiP + WO3|FeP | 1.23 | 1 | 2.3 ± 0.4 | 83 ± 16 | 0.82 ± 0.10 | 60 ± 20 |
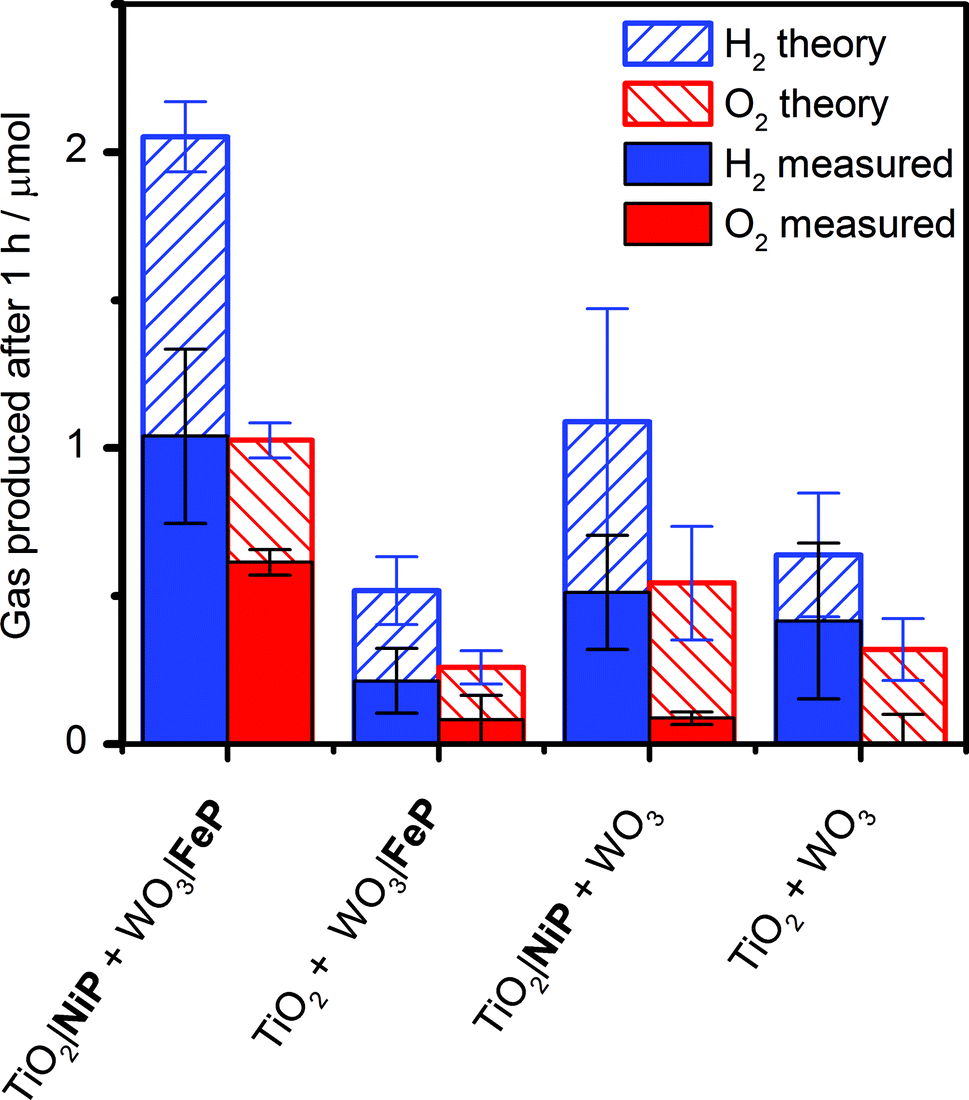 | ||
| Fig. 5 Summary of theoretical and practical H2 and O2 production by two-electrode PEC water splitting with TiO2|NiP cathode (geometrical surface area: 1.0 cm2) wired to WO3|FeP photoanode (0.5 cm2) and control experiments without one or both molecular catalysts. The theoretical amount of gaseous products were calculated based on 100% faradaic yield. Conditions: aqueous Na2SO4 (0.1 M) electrolyte solution at pH 3, N2 atmosphere and room temperature with an applied bias (Uappl) = 1.1 V for 1 h and solar light illumination (100 mW cm−2, AM1.5G). | ||
Without NiP, low photocurrents and consequently lower gaseous products were detected, showing that the molecular catalyst is required to perform proton reduction at a sufficiently low overpotential to be coupled to water oxidation in this system. We also performed two-electrode PEC water splitting of the molecular catalyst-modified electrodes at zero-energy storage (Uappl = 1.23 V vs. RHE) for 1 h, with the results summarised in Table 1. We observed a higher H2 (2.3 ± 0.4 μmol, 83 ± 16% faradaic yield) and O2 (0.82 ± 0.10 μmol, 60 ± 20% faradaic yield) evolution activity than at Uappl = 1.1 V. These results demonstrate that the molecular catalysts based on Earth-abundant transition metals are also capable of driving water splitting at a higher rate if better electrode materials become available. A tandem PEC device with a suitable photocathode modified with NiP wired to WO3|FeP would dramatically reduce the applied bias and result in higher photocurrents and ABPE in the future.7,68
Molecular integrity of catalysts in PEC water splitting
The study of molecular catalysts under strongly reductive and oxidative conditions requires an assessment of their integrity during catalysis.69 For instance, Fe-based catalysts related to FeP are known to remain molecular during water oxidation in homogeneous aqueous acidic conditions, but to oxidise to catalytically-active iron oxide nanoparticles under basic conditions.70 Although Ni-based molecular compounds have also been reported to decompose to catalytically-active Ni-containing nanoparticles under reductive conditions,71 this has not been observed for NiP in solution or in suspensions with semiconducting nanoparticles.14,20,26,27Fig. 6a shows diffuse reflectance UV-vis spectra of the NiP-modified mesoTiO2 electrode. Before electrolysis, the electrodes were purple in colour and showed a broad band at λmax = 520 nm in the UV-vis spectrum of NiP. The purple colour with the band at λmax = 520 nm were qualitatively retained following three-electrode CPE for 2 h and two-electrode solar water electrolysis coupled to WO3|FeP for 1 h, indicating that molecular NiP remained on mesoTiO2.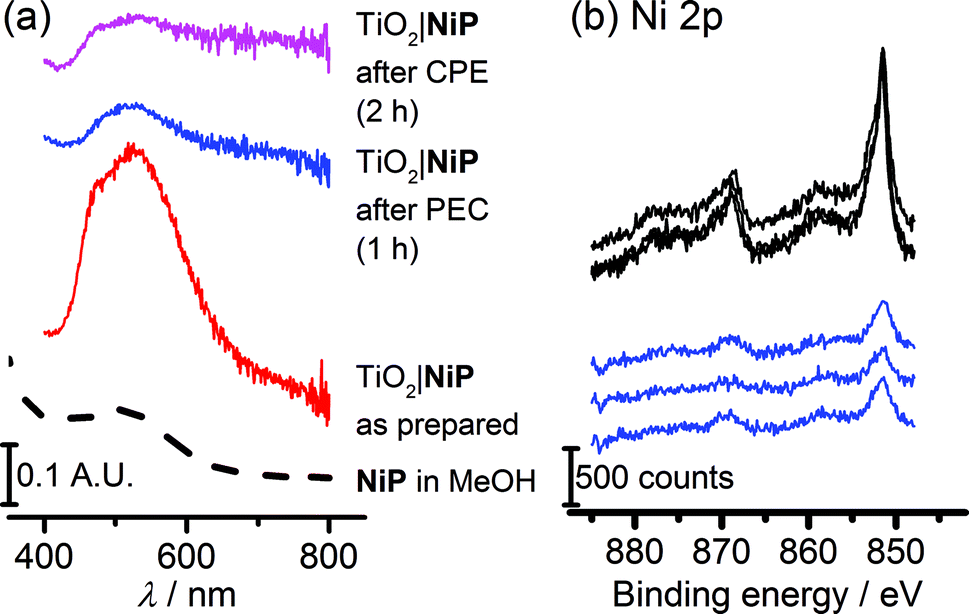 | ||
| Fig. 6 (a) Diffuse-reflectance UV-vis absorption spectra of NiP-modified TiO2 electrodes as prepared (red line), after 1 h PEC electrolysis at Uappl = 1.1 V, and after 2 h CPE at Eappl = −0.33 V vs. RHE, with a MeOH solution UV-vis spectrum as a reference (dashed line). (b) X-ray photoelectron spectra in the Ni 2p region of TiO2|NiP and electrodes before (black line) and after (blue line) 1 h PEC electrolysis at Uappl = 1.1 V with WO3|FeP. | ||
X-ray photoelectron spectroscopy (XPS) on both modified electrodes before and after running 1 h PEC water splitting at an applied bias of 1.1 V were performed to analyse the possible formation of metal-containing deposits. The Ni(2p) or Fe(2p):N(1s):P(2p) ratios, of approximately 1:4:8 for TiO2|NiP and 1:4:1 for WO3|FeP, before two-electrode PEC water splitting were as expected (Scheme 1 and Table S1†). In agreement with previous work, for WO3|FeP, no Fe peak was observed after PEC water splitting (Fig. S12†), suggesting that no iron-based deposit was formed on the electrode. Instead it is postulated that the iron(II) catalyst slowly desorbs from the electrode surface during photoelectrocatalysis,23 possibly due to the presence of only one phosphonate anchor group on FeP. Therefore, the limiting electrode in this PEC water splitting cell is the FeP-modified WO3 photoanode.
Peaks corresponding to Ni(2p), N(1s) and P(2p) were retained after catalysis (Fig. 6b and S13†), and remained in the same ratio before and after PEC water splitting for TiO2|NiP. Fig. 6b and the peak positions in Table S2† demonstrate that the Ni(2p) peak binding energies at 851.5 eV and 869 eV remain unchanged during PEC water splitting, and no new peaks were observed that could correspond to a new Ni-based species. The combination of the unchanged elemental ratios, unaltered peak positions in XPS and diffuse reflectance UV-vis data provides strong evidence that the molecular structure of NiP remains intact during electrolysis immobilised on mesoTiO2 electrodes. This result emphasises the suitability of molecular electrocatalyst design even when working on heterogeneous catalytic systems.
Conclusions
We have presented a hybrid proton reduction cathode based on a phosphonated Ni(II) bis(diphosphine) molecular catalyst and mesostructured TiO2, which is normally considered an insulator in the absence of UV irradiation and therefore of limited use in electrocatalysis. We observed, from complementary cyclic voltammetry and spectroelectrochemistry, that at mildly reducing potentials electrons are transferred first to the CB of TiO2, and then to NiP for proton reduction catalysis. During long term controlled-potential electrolysis, sustained H2 production was observed at an applied potential of Eappl = −0.25 V vs. RHE, with a NiP-based turnover number of 600 achieved after 8 hours. Characterisation and quantification of the catalyst after electrolysis revealed not only that the molecular structure was intact, but also 75% of the NiP initially present remained on the electrode surface. The modified TiO2 performed considerably better than an analogous mesoporous ITO electrode, which underwent degradation under reducing conditions. Therefore, the TiO2|NiP cathode exhibited excellent stability in terms of the material itself, the molecular structure of the catalyst, and the attachment between the two in mildly acidic aqueous conditions. These results establish TiO2|NiP as not only an effective H2 evolving electrode in its own right, but also suggest potential as a catalytically-active protection layer for photocathodes due to its ease of preparation, transparency to visible light, stability and high activity at a modest overpotential.We have demonstrated the utility of this cathode by constructing a precious-metal-free two-compartment PEC cell for full water splitting with molecular catalysts crucial to the performance of both the photoanode and the cathode for the first time. Under solar illumination and an applied bias of Uappl = 1.1 V, below the thermodynamic potential for water splitting, an approximately 2:1 ratio of H2:O2 was only obtained in the presence of FeP and NiP. Essential to this achievement was the development of hybrid electrodes operable under the same mild conditions. We have also presented a number of experiments showing consistent evidence that the molecular structure of NiP remained intact after the photoelectrolysis experiments, a result which emphasises the importance in molecular catalyst design in the development of molecule/material hybrids. These results demonstrate that if mild conditions are used, molecular catalysts can remain stable and effective under catalytic conditions. The immobilisation approach and characterisation techniques reported here provide a promising framework for the future systematic study of the activity and stability of a variety of molecular catalysts in environmentally-benign aqueous solution.
Experimental section
Materials and methods
All starting materials were obtained from commercial sources and used without further purification, unless otherwise stated. ITO nanopowder (<50 nm particle size) was obtained from Sigma-Aldrich and P25 TiO2 (8:2 anatase:rutile, 20 nm average particle size) from Evonik Industries. NiP20 and 4-bromo-2-pyridinecarboxaldehyde72 were synthesised following literature procedures. Dry solvents were dried and distilled under N2 prior to use, or in the case of methanol purchased as dry solvent and stored over molecular sieves. Solvent mixtures are reported as vol:vol ratios. Standard Schlenk line techniques were used where required. 1H, 13C and 31P NMR measurements were performed on a Bruker DPX400 spectrometer. Mass spectrometry measurements were performed on a Waters Micromass Quattro LS ESI or ThermoScientific Orbitrap Classic instrument (calculated and experimental isotope patterns were compared). SEM was performed on a FEI Phillips XL30 field emission gun SEM instrument. XPS was performed by the Nexus facility at the University of Newcastle on a K-Alpha (Thermo Scientific, East Grinstead, UK) spectrometer utilising a monochromatic Al-Kα X-ray source (1486.6 eV, 400 μm spot size, 36 W). Survey spectra were collected with a pass energy of 200 eV and 3 sweeps, while high resolution spectra were collected at a pass energy of 40 eV with 10 sweeps. Elemental analysis was carried out by the University of Cambridge Microanalysis Service using a Perkin-Elmer 240 Elemental Analyser. UV-vis absorption spectroscopy was performed using a Varian Cary 50 spectrophotometer. Where stated, a diffuse reflectance accessory for the spectrophotometer was used, with a Spectralon reference as a background. UV-vis spectra (reflectance mode) of WO3 were recorded on an Edinburgh Instruments FS5 spectrofluorometer equipped with an integrating sphere by running a synchronous scan (λex = λem) and subtracting a scan of the sphere background. ATR-FTIR measurements were performed on a Nicolet iS50 FTIR spectrometer.
Synthesis and characterisation
:Et3N (98:2) to yield 1 as a pale yellow oil (yield: 1.1 g, 66%). HR-MS calc. for [C18H18BrN4]+: 369.0709, found: 369.0724 (100% peak); 1H-NMR (400 MHz, CDCl3) δ/ppm: 8.58 (d, J = 4.1 Hz, 2H), 8.36 (d, J = 5.3 Hz, 1H), 7.80 (d, J = 1.5 Hz, 1H), 7.70 (td, J = 7.7, 1.6 Hz, 2H), 7.56 (d, J = 7.6 Hz, 2H), 7.34 (dd, J = 5.3, 1.8 Hz, 1H), 7.18 (dd, J = 6.6, 5.1 Hz, 2H), 3.93 (s, 4H, CH2), 3.91 (s, 2H, CH2); 13C NMR (101 MHz, CDCl3) δ/ppm 149.82, 148.46, 137.52, 136.93, 133.62, 126.67, 125.82, 123.75, 122.74, 120.88, 60.40, 59.33.
:Et3N:CH3OH (97.5:2:0.5) to yield 2 as a yellow oil that discoloured quickly and solidified on standing (yield: 0.78 g, 67%). HR-MS calc. for [C22H28N4O3P]+: 427.1894, found 427.1875 (100% peak); 1H-NMR (400 MHz, CDCl3) δ/ppm: 8.68 (t, J = 5.0 Hz, 1H), 8.53 (d, J = 4.0 Hz, 2H), 7.96 (d, J = 14.1 Hz, 1H), 7.67 (td, J = 7.5, 1.0 Hz, 2H), 7.58 (d, J = 8.0 Hz, 2H), 7.50 (dd, J = 13.1, 4.0 Hz, 1H), 7.15 (dd, J = 6.3, 5.3 Hz, 2H), 4.06–4.24 (m, 4H), 3.97 (s, 2H), 3.90 (s, 4H), 1.34 (t, J = 7.0 Hz, 6H); 13C (101 MHz, CDCl3) δ/ppm: 160.74 (d, J = 12.0 Hz), 159.45 (s), 149.78 (d, J = 12.8 Hz), 149.48 (s), 138.16 (d, J = 187.7 Hz), 136.86 (s), 124.92 (d, J = 8.8 Hz), 123.83 (d, J = 8.8 Hz), 123.39 (s), 122.47 (s), 63.07 (d, J = 5.6 Hz), 60.60 (s), 60.37 (s), 16.72 (d, J = 6.4 Hz) 31P-NMR (162 MHz, CDCl3) δ/ppm: −16.31; IR ν = 1250 cm−1 (PO).
O); EA calc. for C18H26Cl3N4O5P (TPAp1·3HCl·2H2O) C, 41.92; H, 5.08; N, 10.86; P, 6.01, found C 42.51, H 5.01, N 10.42, P 5.81.
O).
Preparation of nanostructured metal oxide electrodes
Prior to preparation of mesoporous electrodes, glass slides coated with indium-doped tin oxide (ITO) for mesoITO or fluorine-doped tin oxide (FTO) for mesoTiO2 of dimensions 3.0 cm × 1.0 cm were cleaned by heating at 70 °C in a 5:1:1 solution of H2O:H2O2 (30% aq.):NH4OH (conc. aq.) for 30 min, followed by rinsing with H2O and drying at 180 °C for 1 h. Suspensions of ITO (20% by weight of ITO in a 5 M acetic acid solution in ethanol) and TiO2 nanoparticles (100 mg TiO2 and 50 mg poly(ethylene glycol) in approximately 1 mL ethanol) were applied to the transparent conducting oxide-coated glass slides using the doctor blading method using a Scotch tape mask with aperture dimensions of either a 6 mm diameter circle (for cyclic voltammetry) or a 0.7 cm × 1.5 cm rectangle (for (photo)electrolysis). The slides were then annealed at 450 °C for 0.5 h (mesoTiO2) or at 400 °C for 1 h (mesoITO).
The mesoTiO2 and mesoITO electrodes were cleaned and dried with ammonia/hydrogen peroxide (see above) and/or using a BioForce UV/ozone cleaner. Immobilisation was achieved by submersion of the electrodes in a 0.5 mM solution of NiP in methanol for 18 h. The coverage of NiP was quantified by submersion of the modified electrodes in a NaOH(aq) solution (0.1 M) for 30 min, followed by UV-vis spectroscopy of the resultant solution and comparison with calibration data at λ = 257 and 300 nm.
WO3 electrodes were prepared as previously described by hydrothermal synthesis,63 and were characterised by SEM to reveal a nanosheet morphology (Fig. S1b†). Electrode areas were masked to 1.5 cm2 for three-electrode measurements and 0.5 cm2 for two-electrode measurements.
Electrochemical methods
All electrochemical measurements were performed on Ivium Technologies CompactStat or IviumStat, or PalmSens EmStat or MultiEmStat3+ potentiostats. All electrochemical measurements were performed in a Na2SO4 (0.1 M) aqueous solution, adjusted to the desired pH by titration with H2SO4 at room temperature and purged with N2 unless otherwise stated. All electrochemical experiments on the individual TiO2|NiP cathode or WO3|FeP photoanode were performed in a three-electrode configuration with a Pt counter electrode and a Ag/AgCl/KCl(sat) reference electrode, whereas full water splitting CPE experiments were performed in a two-electrode setup with TiO2|NiP directly (shielded from illumination) connected to WO3|FeP in a two-compartment airtight cell. Irradiation was provided through a quartz illumination window and the cathode was separated from the anode by a Nafion membrane. Simulated solar irradiation was provided by a Newport solar light simulator with an AM1.5G filter and a water IR filter. TiO2 was shielded from light to avoid UV band gap excitation. Light intensity was measured using an International Light Technologies 1400 photometer. IPCE measurements were performed using monochromatic light provided by a LOT 300 W Xe lamp equipped with a MSH300 monochromator (FWHM = 5 nm). IPCE was calculated using eqn (2). | (2) |
484 A s mol−1), λ is the wavelength of light (nm), W is the incident power of the monochromated light (W m−2), NA is Avogadro's number (6.022 × 1023 mol−1), h is Planck's constant (6.626 × 10−34 J s) and c is the speed of light (2.998 × 108 m s−1).
Resistance between electrodes in the two compartments was found to be <100 Ω by electrochemical impedance spectroscopy and the reported voltages are not corrected for cell resistance. The experiments were performed at room temperature, and the cell was purged with an inert gas before each experiment. All reduction potentials from the three-electrode experiments are reported against RHE using eqn (3).
| ERHE/V = EAg/AgCl/V + 0.197 + 0.059 × pH | (3) |
Spectroelectrochemical measurements were performed in a borosilicate glass one-compartment electrochemical cell purged with N2 in the beampath of a Varian Cary 50 spectrophotometer.
Quantification of gaseous products
Oxygen detection was performed using an Ocean Optics NeoFox phase fluorimeter equipped with a FOSPOR probe calibrated in the 0–3% O2 regime using a mass-flow controller. The chamber containing the oxygen probe (in the headspace, inserted through a rubber septum) was degassed by purging the solution with N2 for 30 min. Background O2 was then recorded for 30 min, before commencement of the photoelectrolysis experiment. Finally, a further 30 min background was run after ceasing illumination and the background levels of O2 were subtracted. The total amount of O2 produced was obtained using the ideal gas law for O2 in the headspace, and Henry's law for dissolved O2.For H2 quantification, the cell was purged with N2/CH4 (2%) for 10 min prior electrochemical experiments. The H2 produced was quantified by an Agilent 7890A gas chromatograph, with CH4 as an internal standard. For experiments performed under air, CH4 was used as an external standard. Gas detection experiments were repeated 3 times and values given as an average of the three ± standard deviation.
Acknowledgements
We acknowledge support by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research and Economy and National Foundation for Research, Technology and Development), the OMV Group and the EPSRC (DTA award and grant EP/H00338X/2). We thank Dr Katherine Orchard for SEM measurements of TiO2, the EPSRC NEXUS facility at the University of Newcastle for running XPS measurements, Mr Benjamin C. M. Martindale for assistance interpreting XPS data and Dr Bertrand Reuillard for valuable comments on the manuscript.References
- F. Lakadamyali, A. Reynal, M. Kato, J. R. Durrant and E. Reisner, Chem.–Eur. J., 2012, 18, 15464–15475 CrossRef CAS PubMed.
- M. Kirch, J.-M. Lehn and J.-P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- D. V. Esposito and J. G. Chen, Energy Environ. Sci., 2011, 4, 3900–3912 Search PubMed.
- Y. Gao, X. Ding, J. Liu, L. Wang, Z. Lu, L. Li and L. Sun, J. Am. Chem. Soc., 2013, 135, 4219–4222 CrossRef CAS PubMed.
- L. Alibabaei, M. K. Brennaman, M. R. Norris, B. Kalanyan, W. Song, M. D. Losego, J. J. Concepcion, R. A. Binstead, G. N. Parsons and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20008–20013 CrossRef CAS PubMed.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- C.-Y. Lin, Y.-H. Lai, D. Mersch and E. Reisner, Chem. Sci., 2012, 3, 3482–3487 RSC.
- D. Mersch, C.-Y. Lee, J. Z. Zhang, K. Brinkert, J. C. Fontecilla-Camps, A. W. Rutherford and E. Reisner, J. Am. Chem. Soc., 2015, 137, 8541–8549 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87, 946–964 CrossRef CAS PubMed.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 Search PubMed.
- J. Willkomm, N. M. Muresan and E. Reisner, Chem. Sci., 2015, 6, 2727–2736 RSC.
- J. L. Fillol, Z. Codolà, I. Garcia-Bosch, L. Gómez, J. J. Pla and M. Costas, Nat. Chem., 2011, 3, 807–813 CrossRef CAS PubMed.
- S. Roy, M. Bacchi, G. Berggren and V. Artero, ChemSusChem, 2015, 8, 3632–3638 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- W. M. Singh, D. Pegram, H. Duan, D. Kalita, P. Simone, G. L. Emmert and X. Zhao, Angew. Chem., Int. Ed., 2012, 51, 1653–1656 CrossRef CAS PubMed.
- P. Farràs, C. Di Giovanni, J. N. Clifford, E. Palomares and A. Llobet, Coord. Chem. Rev., 2015, 304–305, 202–208 CrossRef.
- A. Reynal, J. Willkomm, N. M. Muresan, F. Lakadamyali, M. Planells, E. Reisner and J. R. Durrant, Chem. Commun., 2014, 50, 12768–12771 RSC.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. Rakowski DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- B. M. Klepser and B. M. Bartlett, J. Am. Chem. Soc., 2014, 136, 1694–1697 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- A. Dutta, S. Lense, J. Hou, M. H. Engelhard, J. A. S. Roberts and W. J. Shaw, J. Am. Chem. Soc., 2013, 135, 18490–18496 CrossRef CAS PubMed.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 53, 11538–11542 CrossRef CAS PubMed.
- D. W. Wakerley, M. A. Gross and E. Reisner, Chem. Commun., 2014, 50, 15995–15998 RSC.
- E. Bae and W. Choi, J. Phys. Chem. B, 2006, 110, 14792–14799 CrossRef CAS PubMed.
- F. Lakadamyali, M. Kato and E. Reisner, Faraday Discuss., 2012, 155, 191–205 RSC.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC.
- C. Queffélec, M. Petit, P. Janvier, D. A. Knight and B. Bujoli, Chem. Rev., 2012, 112, 3777–3807 CrossRef PubMed.
- F. Lakadamyali and E. Reisner, Chem. Commun., 2011, 47, 1695–1697 RSC.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 12749–12753 CrossRef CAS PubMed.
- E. Reisner, J. C. Fontecilla-Camps and F. A. Armstrong, Chem. Commun., 2009, 550–552 RSC.
- S. Morra, F. Valetti, S. J. Sadeghi, P. W. King, T. Meyer and G. Gilardi, Chem. Commun., 2011, 47, 10566–10568 RSC.
- Z. Chen, J. J. Concepcion, J. W. Jurss and T. J. Meyer, J. Am. Chem. Soc., 2009, 131, 15580–15581 CrossRef CAS PubMed.
- G. Boschloo and D. Fitzmaurice, J. Electrochem. Soc., 2000, 147, 1117–1123 CrossRef CAS.
- B. O'Regan, M. Grätzel and D. Fitzmaurice, J. Phys. Chem., 1991, 95, 10525–10528 CrossRef.
- B. O'Regan, M. Grätzel and D. Fitzmaurice, Chem. Phys. Lett., 1991, 183, 89–93 CrossRef.
- G. Neri, J. J. Walsh, C. Wilson, A. Reynal, J. Y. C. Lim, X. Li, A. J. P. White, N. J. Long, J. R. Durrant and A. J. Cowan, Phys. Chem. Chem. Phys., 2015, 17, 1562–1566 RSC.
- A. Reynal, E. Pastor, M. A. Gross, S. Selim, E. Reisner and J. R. Durrant, Chem. Sci., 2015, 6, 4855–4859 RSC.
- F. Lakadamyali, M. Kato, N. M. Muresan and E. Reisner, Angew. Chem., Int. Ed., 2012, 51, 9381–9384 CrossRef CAS PubMed.
- D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2015, 8, 2283–2295 Search PubMed.
- J. Seo, R. T. Pekarek and M. J. Rose, Chem. Commun., 2015, 51, 13264–13267 RSC.
- A. K. Das, M. H. Engelhard, R. M. Bullock and J. A. S. Roberts, Inorg. Chem., 2014, 53, 6875–6885 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem., Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- A. Paracchino, V. Laporte, K. Sivula, M. Grätzel and E. Thimsen, Nat. Mater., 2011, 10, 456–461 CrossRef CAS PubMed.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef CAS PubMed.
- N. P. Dasgupta, C. Liu, S. Andrews, F. B. Prinz and P. Yang, J. Am. Chem. Soc., 2013, 135, 12932–12935 CrossRef CAS PubMed.
- Y.-H. Lai, H. S. Park, J. Z. Zhang, P. D. Matthews, D. S. Wright and E. Reisner, Chem.–Eur. J., 2015, 21, 3919–3923 CrossRef CAS PubMed.
- J. Zhao, T. Minegishi, L. Zhang, M. Zhong, Gunawan, M. Nakabayashi, G. Ma, T. Hisatomi, M. Katayama, S. Ikeda, N. Shibata, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2014, 53, 11808–11812 CrossRef CAS PubMed.
- R. Brimblecombe, A. Koo, G. C. Dismukes, G. F. Swiegers and L. Spiccia, J. Am. Chem. Soc., 2010, 132, 2892–2894 CrossRef CAS PubMed.
- D. L. Ashford, A. M. Lapides, A. K. Vannucci, K. Hanson, D. A. Torelli, D. P. Harrison, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6578–6581 CrossRef CAS PubMed.
- G. F. Moore, J. D. Blakemore, R. L. Milot, J. F. Hull, H. Song, L. Cai, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Energy Environ. Sci., 2011, 4, 2389–2392 Search PubMed.
- X. Chen, X. Ren, Z. Liu, L. Zhuang and J. Lu, Electrochem. Commun., 2013, 27, 148–151 CrossRef CAS.
- M. de Respinis, K. S. Joya, H. J. M. De Groot, F. D'Souza, W. A. Smith, R. van de Krol and B. Dam, J. Phys. Chem. C, 2015, 119, 7275–7281 CrossRef CAS.
- K. S. Joya, N. Morlanés, E. Maloney, V. Rodionov and K. Takanabe, Chem. Commun., 2015, 51, 13481–13484 RSC.
- W. A. Gerrard, J. Electroanal. Chem., 1978, 86, 421–424 CrossRef CAS.
- K. L. Hardee and A. J. Bard, J. Electrochem. Soc., 1976, 123, 1024–1026 CrossRef CAS.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- L. Zhang, C.-Y. Lin, V. K. Valev, E. Reisner, U. Steiner and J. J. Baumberg, Small, 2014, 10, 3970–3978 CrossRef CAS PubMed.
- Q. Mi, A. Zhanaidarova, B. S. Brunschwig, H. B. Gray and N. S. Lewis, Energy Environ. Sci., 2012, 5, 5694–5700 Search PubMed.
- Y.-H. Lai, T. C. King, D. S. Wright and E. Reisner, Chem.–Eur. J., 2013, 19, 12943–12947 CrossRef CAS PubMed.
- Y. Belabassi, S. Alzghari and J.-L. Montchamp, J. Organomet. Chem., 2008, 693, 3171–3178 CrossRef CAS PubMed.
- J. C. Hill and K.-S. Choi, J. Phys. Chem. C, 2012, 116, 7612–7620 CrossRef CAS.
- Y.-H. Lai, M. Kato, D. Mersch and E. Reisner, Faraday Discuss., 2014, 176, 199–211 RSC.
- N. Kaeffer, A. Morozan and V. Artero, J. Phys. Chem. B, 2015, 119, 13707–13713 CrossRef CAS PubMed.
- Y.-H. Lai, D. W. Palm and E. Reisner, Adv. Energy Mater. DOI:10.1002/aenm.201501668 Search PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
- D. Hong, S. Mandal, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, Inorg. Chem., 2013, 52, 9522–9531 CrossRef CAS PubMed.
- C. Wombwell and E. Reisner, Dalton Trans., 2014, 43, 4483–4493 RSC.
- N. Zaman, R. Guillot, K. Sénéchal-David and M.-L. Boillot, Tetrahedron Lett., 2008, 49, 7274–7275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04863j Additional data related to this publication are available at the University of Cambridge data repository (https://www.repository.cam.ac.uk/handle/1810/253744). |
| This journal is © The Royal Society of Chemistry 2016 |