
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Essential role of hydride ion in ruthenium-based ammonia synthesis catalysts†
Masaaki
Kitano
a,
Yasunori
Inoue
b,
Hiroki
Ishikawa
b,
Kyosuke
Yamagata
b,
Takuya
Nakao
b,
Tomofumi
Tada
a,
Satoru
Matsuishi
a,
Toshiharu
Yokoyama
ac,
Michikazu
Hara
*bcd and
Hideo
Hosono
*abcd
aMaterials Research Center for Element Strategy, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: hosono@msl.titech.ac.jp
bLaboratory for Materials and Structures, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: mhara@msl.titech.ac.jp
cACCEL, Japan Science and Technology Agency, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dFrontier Research Center, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
First published on 21st April 2016
Abstract
The efficient reduction of atmospheric nitrogen to ammonia under low pressure and temperature conditions has been a challenge in meeting the rapidly increasing demand for fertilizers and hydrogen storage. Here, we report that Ca2N:e−, a two-dimensional electride, combined with ruthenium nanoparticles (Ru/Ca2N:e−) exhibits efficient and stable catalytic activity down to 200 °C. This catalytic performance is due to [Ca2N]+·e1−x−Hx− formed by a reversible reaction of an anionic electron with hydrogen (Ca2N:e− + xH ↔ [Ca2N]+·e1−x−Hx−) during ammonia synthesis. The simplest hydride, CaH2, with Ru also exhibits catalytic performance comparable to Ru/Ca2N:e−. The resultant electrons in these hydrides have a low work function of 2.3 eV, which facilitates the cleavage of N2 molecules. The smooth reversible exchangeability between anionic electrons and H− ions in hydrides at low temperatures suppresses hydrogen poisoning of the Ru surfaces. The present work demonstrates the high potential of metal hydrides as efficient promoters for low-temperature ammonia synthesis.
Introduction
Catalytic ammonia synthesis is vital for the production of synthetic fertilizers and serves as an active nitrogen source for important chemicals. Dinitrogen (N2) is a typically inert molecule because of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond (945 kJ mol−1).1 Therefore, high temperature (400–600 °C) and high pressure (20–40 MPa) are required for industrial ammonia synthesis (Haber–Bosch process),2 which results in high energy consumption. Recently, ammonia has also attracted much attention as a hydrogen storage material due to its high capacity for hydrogen storage (17.6 wt%) and facile liquefaction under mild conditions.3 Ammonia synthesis at low temperature is thermodynamically favorable but still presents a major challenge. Extensive studies on N2 activation with organometallic complexes have been conducted over the last decade.4–7 Although NH3 has been successfully produced under ambient conditions, the rate of formation is still far from appropriate for practical application, and strong reducing agents and extra proton sources are required to afford NH3.
N bond (945 kJ mol−1).1 Therefore, high temperature (400–600 °C) and high pressure (20–40 MPa) are required for industrial ammonia synthesis (Haber–Bosch process),2 which results in high energy consumption. Recently, ammonia has also attracted much attention as a hydrogen storage material due to its high capacity for hydrogen storage (17.6 wt%) and facile liquefaction under mild conditions.3 Ammonia synthesis at low temperature is thermodynamically favorable but still presents a major challenge. Extensive studies on N2 activation with organometallic complexes have been conducted over the last decade.4–7 Although NH3 has been successfully produced under ambient conditions, the rate of formation is still far from appropriate for practical application, and strong reducing agents and extra proton sources are required to afford NH3.
In heterogeneous catalysts, it is widely recognized that ruthenium (Ru) catalysts work under milder conditions than iron-based catalysts for the Haber–Bosch process.8,9 The activity of Ru catalysts is substantially enhanced by electron injection from alkali or alkali earth metal oxide promoters.8,10 Although these electronic promoters lower the energy barrier for N2 dissociation, the enthalpy of hydrogen adsorption on the Ru catalyst is also increased, leading to high surface coverage by H atoms (hydrogen poisoning).11 Accordingly, the electronic promotion effect for N2 dissociation is retarded by the competitive adsorption of H2. It is therefore highly desirable to develop a new Ru catalyst that can promote N2 dissociation and prevent hydrogen poisoning. It was demonstrated that the 12CaO·7Al2O3 electride (C12A7:e−)12-supported Ru catalyst exhibits much higher activity for ammonia synthesis than alkali-promoted Ru catalysts.13,14 The intrinsically low work function (ca. 2.4 eV) of C12A7:e−15 in this catalyst promotes N2 dissociation on Ru, which leads to a reduction in the activation energy to half (ca. 55 kJ mol−1) of that for conventional Ru catalysts. A recent kinetic analysis revealed that the bottleneck for ammonia synthesis is shifted from N2 dissociation to the formation of N–Hn species.16 In addition, this catalyst has reversible exchangeability of electrons and hydride ions, and is almost immune to hydrogen poisoning of the Ru surface, which is a serious drawback for conventional Ru catalysts. These results imply that both electrons and hydride ions play a crucial role in effective ammonia synthesis. However, the outstanding activity of C12A7:e− is diminished at low temperatures (<320 °C), which is strongly correlated with the weak H2 desorption properties at low temperatures. The electron–hydride ion exchange reaction in C12A7:e− is accomplished by H desorption through a cage wall composed of a rigid monolayer of Ca–Al–O; therefore, the exchange reaction in C12A7:e− requires a relatively high temperature that is sufficient to excite thermal vibration and allow H to escape from the cage. Therefore, our design concept for a highly active low-temperature ammonia synthesis catalyst is embodied by inorganic electride materials with hydride ions exposed to the surface, i.e., metal hydrides.
Dicalcium nitride, [Ca2N]+·e− (denoted as Ca2N:e−), was confirmed as a two-dimensional (2D) electride with a low work function (2.6 eV), in which anionic electrons confined between the [Ca2N]+ layers as counter anions17–19 can be partly exposed to the surfaces. In addition, this material can be converted into Ca2NH ([Ca2N]+·H−) by the reaction between an anionic electron and a hydrogen, which is analogous to that for C12A7:e−. Therefore, Ca2NH was selected as the first test bed material to verify our hypothetical design concept of metal hydrides for low-temperature ammonia synthesis.
Here we report that metal hydride materials such as Ca2NH and CaH2, which are not intrinsically low work function materials, strongly promote the cleavage of N2 to form NH3 on Ru nanoparticles under low pressure and temperature conditions. The low work function (2.3 eV) is immediately realized by the formation of hydrogen vacancies in these Ru-loaded hydride materials during ammonia synthesis, which in turn facilitates N2 dissociation and prevents hydrogen poisoning of the Ru surface.
Results and discussion
Catalytic performance of Ru-loaded Ca2N:e−
Fig. 1 shows the temperature dependence for ammonia synthesis over various Ru catalysts and Table 1 summarizes the catalytic properties of various Ru catalysts. The Ru dispersion of Ru/Ca2N:e− (3.1%) is much smaller than that of Ru(2%)–Cs/MgO (50.4%) because of its low surface area (1.5 m2 g−1), which is similar to the results for Ru/C12A7:e−. However, Ru/Ca2N:e− exhibits high catalytic activity (at 340 °C) comparable to Ru(2%)–Cs/MgO, which is one of the most active catalysts for ammonia synthesis reported to date.20,21 Accordingly, the turnover frequency (TOF) with Ru/Ca2N:e− is higher than that with Ru–Cs/MgO by an order of magnitude. It is noteworthy that Ru/Ca2N:e− exhibits higher catalytic activity than Ru/C12A7:e− or Ru(2%)–Cs/MgO, especially below 300 °C, and ammonia formation can be distinctly observed even at 200 °C. The apparent activation energy of Ru/Ca2N:e− for ammonia synthesis is 60 kJ mol−1, which is one-half that of Ru–Cs/MgO (120 kJ mol−1). We have previously reported16 that a change in the activation energy is observed for Ru/C12A7:e−, i.e., the activation energy at 400–320 °C is 50 kJ mol−1 but exceeds 90 kJ mol−1 at lower reaction temperatures (320–250 °C) and no reaction is observed at 200 °C. These results indicate that Ru/Ca2N:e− is superior to Ru/C12A7:e− for low-temperature ammonia synthesis. Furthermore, we note that Ru/Ca2N:e− functions as a stable catalyst for ammonia synthesis over long periods without degradation in activity. Fig. 1b shows that the initial ammonia synthesis rate was maintained even after 54 h under high pressure conditions (1.0 MPa), and the total amount of produced ammonia reached 27 mmol, which is more than 25 times the total nitrogen content in Ca2N:e− (1.06 mmol). This result indicates that the ammonia produced is not derived from the decomposition of the Ca2N:e− support.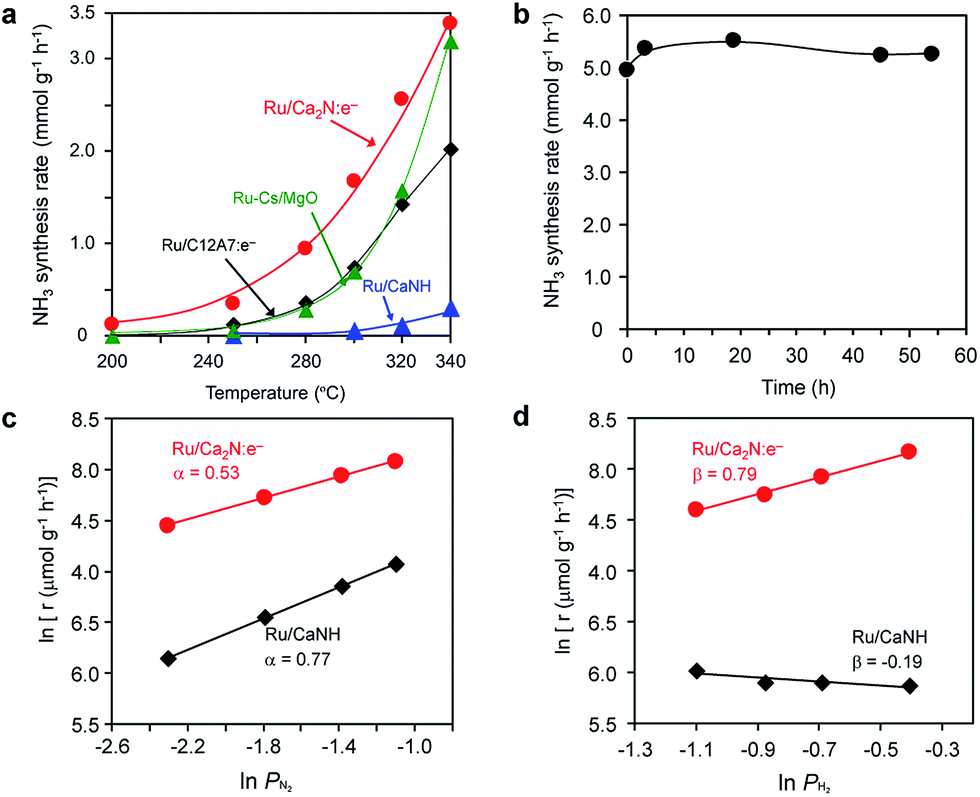 | ||
Fig. 1 (a) Catalytic activity for ammonia synthesis over various Ru catalysts (2 wt%) as a function of reaction temperature (reaction conditions: catalyst, 0.1 g; WHSV, 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mL gcat−1 h−1; reaction pressure, 0.1 MPa). (b) Reaction time profile for ammonia synthesis over Ru (5 wt%)/Ca2N at 340 °C (reaction conditions: catalyst, 0.1 g; WHSV, 36000 mL gcat−1 h−1; reaction pressure, 1.0 MPa). (c, d) Dependence of ammonia synthesis on the partial pressure of (c) N2 and (d) H2 using various Ru catalysts (2 wt%) at 340 °C under atmospheric pressure. 000 mL gcat−1 h−1; reaction pressure, 0.1 MPa). (b) Reaction time profile for ammonia synthesis over Ru (5 wt%)/Ca2N at 340 °C (reaction conditions: catalyst, 0.1 g; WHSV, 36000 mL gcat−1 h−1; reaction pressure, 1.0 MPa). (c, d) Dependence of ammonia synthesis on the partial pressure of (c) N2 and (d) H2 using various Ru catalysts (2 wt%) at 340 °C under atmospheric pressure. | ||
| Catalyst | Ru loading (wt%) | S BET (m2 g−1) | D m (%) | d (nm) | N S (mmol g−1) | r NH3 (mmol g−1 h−1) | TOFc (×103) (s−1) | E a (kJ mol−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 340 °C | 300 °C | 340 °C | 300 °C | |||||||
|
a Dispersion (Dm), particle size (d), and the number of surface Ru atoms (NS) were calculated on the basis of CO chemisorption values, assuming spherical metal particles and a stoichiometry of Ru/CO = 1.37
b NH3 synthesis rate (rNH3); conditions: catalyst (0.1 g), synthesis gas (H2/N2 = 3, 60 mL min−1), weight hourly space velocity (WHSV) = 36000 mL gcat−1 h−1, pressure (0.1 MPa).
c TOF was calculated from the rate of ammonia synthesis divided by NS.
d
E
a is the apparent activation energy calculated from Arrhenius plots for the reaction rate in the temperature range of 340–250 °C.
|
||||||||||
| Ru/Ca2N:e− | 1.8 | 1.5 | 3.1 | 42.8 | 5.1 | 3386 | 1674 | 185.1 | 91.5 | 60 |
| Ru/CaNH | 1.8 | 1.0 | 4.5 | 30.2 | 7.2 | 308 | 53 | 11.9 | 2.0 | 110 |
| Ru/C12A7:e− | 1.8 | 1.0 | 4.7 | 28.7 | 8.3 | 2021 | 745 | 67.5 | 24.9 | 51 (400–320 °C) |
| 91 (320–200 °C) | ||||||||||
| Ru/CaH2 | 2.0 | 3.8 | 12.3 | 10.9 | 22.3 | 4002 | 2549 | 153.9 | 98.0 | 51 |
| Ru–Cs/MgO | 2.0 | 12.0 | 50.4 | 2.5 | 45.7 | 3200 | 697 | 19.5 | 4.2 | 120 |
Fig. 1c and d show the dependence of the ammonia synthesis rate on the partial pressure of N2 and H2, respectively. The reaction orders with respect to N2, H2, and NH3 over various Ru catalysts are also summarized in Table S1.† The reaction order for N2 with conventional heterogeneous catalysts is 0.8–1.0,11,20,21 where N2 dissociation is the rate-determining step for ammonia synthesis. In contrast, the reaction order for N2 with Ru/Ca2N:e− is almost one-half, which is attributed to a more dense population of N adatoms on Ru/Ca2N:e− than on the other catalysts. Two results were noted; one is that the Ru/Ca2N:e− catalyst also facilitates the N2 isotopic exchange reaction (15N2 + 14N2 = 215N14N) with a lower activation energy (59 kJ mol−1) than conventional Ru catalysts (>130 kJ mol−1)16 (Fig. S1†), indicating that the energy barrier for N2 dissociation is significantly lowered by Ru/Ca2N:e−. The other result is that Ru/Ca2N:e− has a positive reaction order with respect to H2, in contrast to the case for Ru–Cs/MgO (β < 0), which indicates that ammonia synthesis over Ru/Ca2N:e− is not inhibited by hydrogen adsorption, i.e., hydrogen poisoning.11,20 Thus, Ru/Ca2N:e− maintains the key kinetics observed with Ru/C12A7:e−, in that N2 cleavage is not the rate-determining step for ammonia synthesis16 and the reactions are free from hydrogen poisoning on the Ru surface. Generally, the promotion effect of alkali compounds such as Cs-oxide in a Ru catalyst is a trade-off between lowering the activation barrier for N2 dissociation and increasing the competitive adsorption of H2.11 However, this serious drawback can be overcome by using Ca2N:e− as a support material. Furthermore, the catalytic activity of Ru/Ca2N:e− increased with an increase in the reaction pressure at 320 °C (Fig. S2†). On the other hand, the increment in the catalytic activity of Ru/C12A7:e− is moderate at this temperature, which is due to the poisoning effect of H atoms on Ru/C12A7:e− at low reaction temperature (≤320 °C).16 These results clearly indicate that Ru/Ca2N:e− exhibits improved performance for ammonia synthesis compared with Ru/C12A7:e−, even at lower temperatures and elevated pressures.
Structural properties of Ca-nitride catalysts
X-ray diffraction (XRD) and Raman spectroscopy measurements confirmed that Ca2NH, an inorganic hydride, is formed in the Ru/Ca2N:e− catalyst during the ammonia synthesis reaction. Fig. 2a shows that Ru/Ca2N:e− after ammonia synthesis has no diffraction peaks attributable to the Ca2N phase (Fig. 2b),19,22,23 whereas new peaks due to Ca2NH with cubic structure (Fig. 2c) appear.23–25 There is no CaNH phase (cubic structure, Fig. 2d), which consists of Ca2+ and NH2− ions (the formal charge of hydrogen is +1), in Ru/Ca2N:e− after the reaction.26–28 The formation of Ca2NH in the catalyst was also elucidated from in situ Raman spectroscopy measurements (Fig. 2e). Two intense bands at 173 and 299 cm−1 for Ca2N:e− are red-shifted to 180 and 322 cm−1, respectively, under the reaction conditions. The Raman spectrum for CaNH has broad bands in the range from 100 to 1000 cm−1 and is completely different from that of Ca2NH (Fig. S3†). In addition, CaNH has an intense band centered at 3122 cm−1, which is attributed to the N–H stretching mode in imide ions.29,30 Although Ru/Ca2N:e− also showed weak bands in the range of 3100–3300 cm−1 after the reaction, the intensity is much smaller than that for CaNH, which indicates that the catalyst consists mainly of Ca2NH. In addition, Ru/CaNH has much lower catalytic activity and a higher activation energy (110 kJ mol−1) than Ru/Ca2N:e− (Fig. 1, S2† and Table 1). The kinetic analysis (Fig. 1 and Table S1†) revealed that N2 cleavage is the rate-determining step for Ru/CaNH and the catalyst is subject to hydrogen poisoning like the conventional Ru catalysts.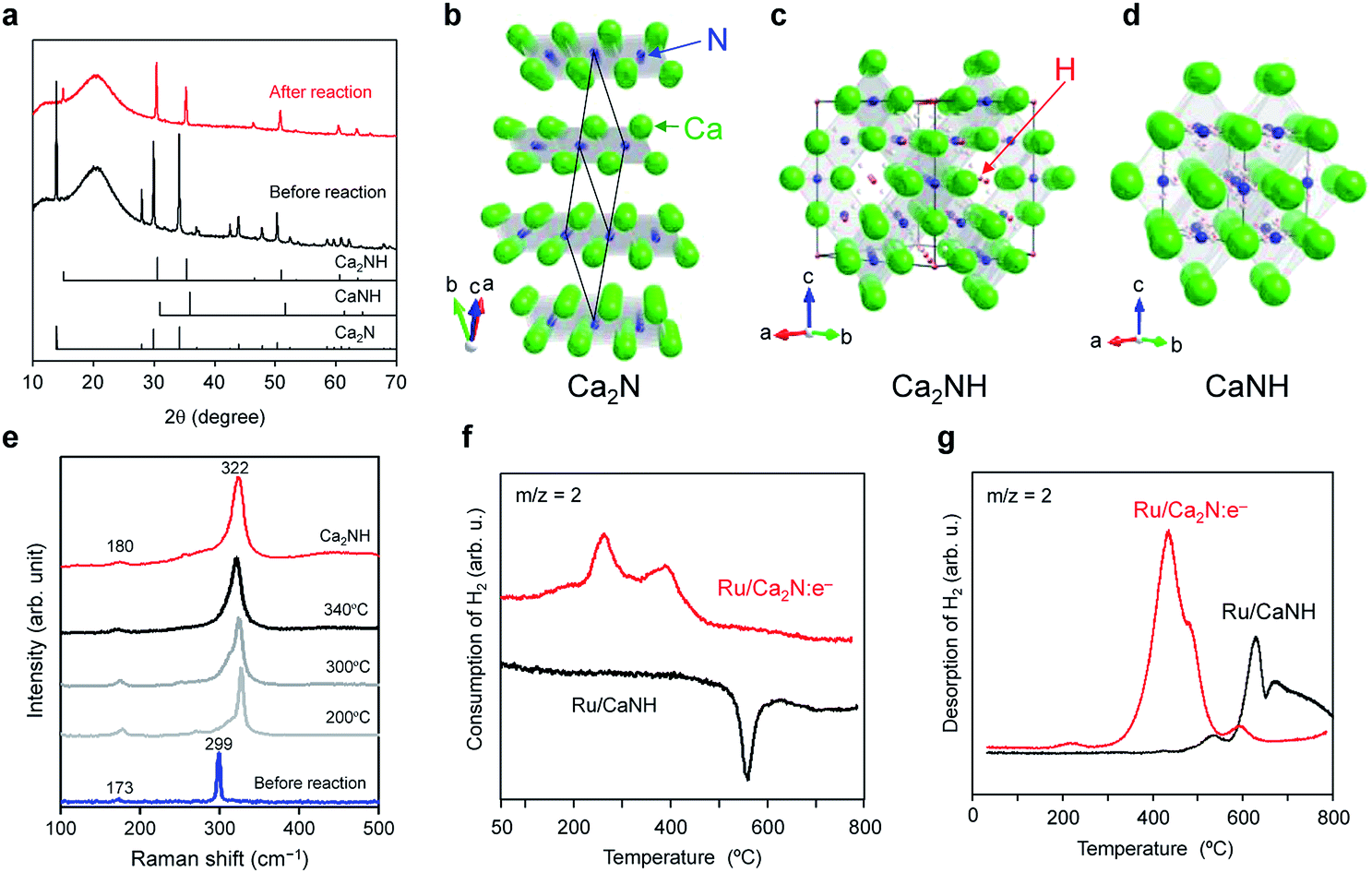 | ||
Fig. 2 (a) XRD patterns for Ru/Ca2N:e− before and after ammonia synthesis reaction at 340 °C for 20 h. Standard JCPDS diffraction patterns for Ca2N (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, PDF: 70-4196), CaNH (space group Fmm, PDF: 75-0430), and Ca2NH (space group Fdm, PDF: 76-608) are provided for reference. (b–d) Crystal structures of Ca2N (b), Ca2NH (c), and CaNH (d) were visualized using the VESTA program.38 (b) Ca2N:e− has a hexagonal layered structure with anionic electron layers between the cationic framework layers ([Ca2N]+) composed of edge-sharing NCa6 octahedra. (c) Ca2NH is composed of Ca2+, N3−, and H− ions, where Ca atoms form a slightly distorted cubic close packed structure, and N and H are ordered in each anion layer. (d) CaNH, an inorganic imide compound with a cubic structure, consists of Ca2+ and NH2− ions. (e) In situ Raman spectra for Ru/Ca2N:e− measured under ammonia synthesis conditions (N2:H2 = 1:3, 0.1 MPa, 60 mL min−1). The Raman spectrum for Ca2NH is also shown as a reference. (f) H2 TPA profiles for Ru/Ca2N:e− and Ru/CaNH catalysts. The TPA measurements were performed (1 °C min−1) with a dilute mixture of H2 (5%) in Ar. (g) H2 TPD profiles for Ru/Ca2N:e− and Ru/CaNH after ammonia synthesis reaction at 340 °C for 10 h. The TPD measurements were performed (1 °C min−1) under Ar flow. m, PDF: 70-4196), CaNH (space group Fmm, PDF: 75-0430), and Ca2NH (space group Fdm, PDF: 76-608) are provided for reference. (b–d) Crystal structures of Ca2N (b), Ca2NH (c), and CaNH (d) were visualized using the VESTA program.38 (b) Ca2N:e− has a hexagonal layered structure with anionic electron layers between the cationic framework layers ([Ca2N]+) composed of edge-sharing NCa6 octahedra. (c) Ca2NH is composed of Ca2+, N3−, and H− ions, where Ca atoms form a slightly distorted cubic close packed structure, and N and H are ordered in each anion layer. (d) CaNH, an inorganic imide compound with a cubic structure, consists of Ca2+ and NH2− ions. (e) In situ Raman spectra for Ru/Ca2N:e− measured under ammonia synthesis conditions (N2:H2 = 1:3, 0.1 MPa, 60 mL min−1). The Raman spectrum for Ca2NH is also shown as a reference. (f) H2 TPA profiles for Ru/Ca2N:e− and Ru/CaNH catalysts. The TPA measurements were performed (1 °C min−1) with a dilute mixture of H2 (5%) in Ar. (g) H2 TPD profiles for Ru/Ca2N:e− and Ru/CaNH after ammonia synthesis reaction at 340 °C for 10 h. The TPD measurements were performed (1 °C min−1) under Ar flow. | ||
To understand the reactivity of these materials with hydrogen, temperature-programmed absorption (TPA) and desorption (TPD) of H2 on the catalysts were examined. Ru/Ca2N:e− can absorb hydrogen above 150 °C, which means that the hydrogen storage reaction (H0 + e− → H−) takes place to form Ca2NH (Fig. 2f). In contrast, no H2 absorption peak was observed for Ru/CaNH and a negative peak appeared at 500–600 °C, which corresponds to hydrogen desorption from the sample, i.e., decomposition. Fig. 2g shows that H2 is released (H− → H0 + e−) from Ru/Ca2N:e− after reaction above 200 °C (the actual material is Ru/Ca2NH) and the H− ion content was estimated to be 10.6 mmol g−1, which is in good agreement with the theoretical amount (10.5 mmol g−1) of H− ions in Ca2NH. Furthermore, the onset temperature for H2 desorption from Ru/Ca2N:e− after the reaction is much lower than that for Ru/CaNH. The TPA and TPD results show that hydrogen incorporation and desorption reactions proceed above 200 °C over Ru/Ca2N:e−. Therefore, a nonstoichiometric phase represented by [Ca2N]+·e1−x−Hx−, rather than stoichiometric Ca2NH, is formed by the reaction between anionic electrons and H− ions (Ca2N:e− + H ↔ [Ca2N]+·H−) during ammonia synthesis. This exchangeability is a key reaction confirmed in Ru/C12A7:e−, but the significant difference between Ru/Ca2N:e− and Ru/C12A7:e− is the onset temperature for H2 desorption, i.e., 200 °C for Ru/Ca2N:e− and 350 °C for Ru/C12A7:e−.
DFT calculations
To shed more light on the electronic state of Ca2NH, density functional theory (DFT) calculations were conducted (detailed conditions are described in the ESI†). Fig. 3a shows a computational model of the Ca2NH(100) surface, and Fig. 3b–d show the calculated electronic states of Ca2NH(100), Ca2NH1−x(100), and Ru-loaded Ca2NH(100), respectively. The calculations were executed under the condition of an electrically neutral unit cell. The calculated work functions (WFs)31 for Ca2N (3.3, 2.5 eV) and Ru (4.8 eV) are close to the measured values19,32,33 (Table 2), which indicates the reliability of the DFT calculations. The calculated WF for Ca2NH is significantly smaller than that for CaNH, which indicates the higher electron-donating ability of Ca2NH than CaNH. Notably, Ca2NH(100) with a hydrogen vacancy (VH) (i.e., Ca2NH1−x(100)) shows a much lower WF (2.3 eV) than both Ca2NH (2.8 eV) and Ca2N:e− (2.5 eV). When an anionic electron is confined at the VH position in Ca2NH (see the inset of Fig. 3c), the anionic electron state is located above the valence band maximum (VBM) of Ca2NH, as shown in Fig. 3c. The confinement of the anionic electron in Ca2NH1−x(100) is stronger than that of the 2D layered space in Ca2N, and thus Ca2NH(100) with VH has a small WF, which reflects the strong electron-donation ability of Ca2NH1−x.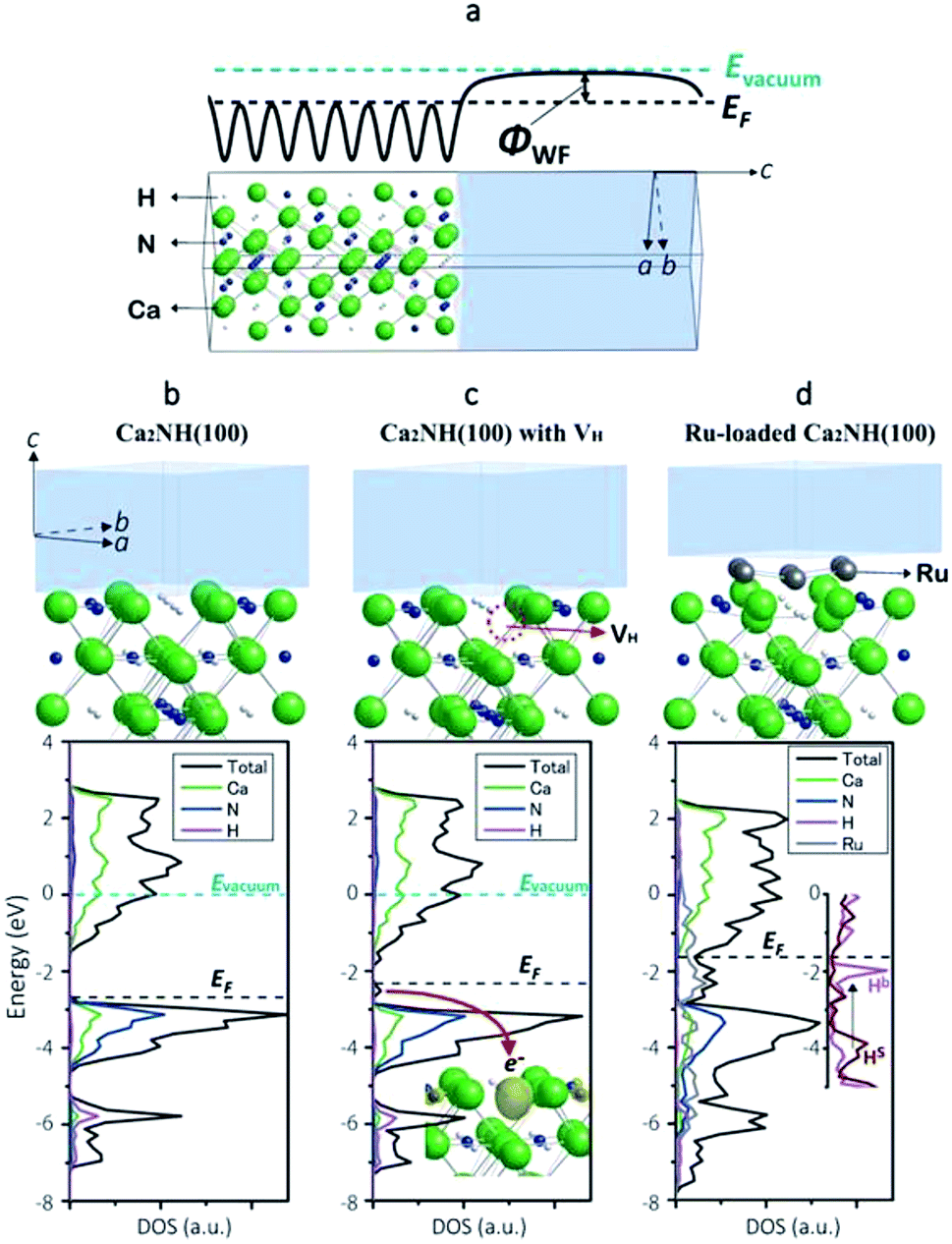 | ||
| Fig. 3 (a) Computational model used in the calculation of the work function of Ca2NH(100). The vacuum region (transparent gray) is included in the model to determine the vacuum level from the electrostatic potential profile (solid thick line) in the region. (b) Surface structure and spin-averaged DOS of Ca2NH(100), where an energy of 0.0 eV corresponds to the vacuum level. (c) Surface structure and spin-averaged DOS of Ca2NH(100) with a hydrogen vacancy (VH), indicated with a red dotted circle. The energy of 0.0 eV corresponds to the vacuum level. Inset: the local density (yellow) of anionic electron states just below EF (a red arrow) depicted with an isosurface value of 0.015 e− bohr−3. The anionic electron states are purely spin polarized states. (d) Surface structure and spin-averaged DOS of Ru-loaded Ca2NH(100). The vacuum level was not determined in this model; therefore, the DOS were represented to match the VBM of nitrogen with those of Ca2NH(100) with/without VH. Inset: DOS of H bonded with Ru(Hb) and H on the free surface (Hs). Green, blue, white, and gray atoms in the atomistic models correspond to Ca, N, H, and Ru, respectively. These crystal structures and charge distributions were visualized using the VESTA program.38 | ||
| Compound | Space group | Surface index | WFcalc (eV) | WFexp (eV) | ΔE(VH) (eV per atom) |
|---|---|---|---|---|---|
| a ΔE(VH) for compound AH1−x is the total energy difference, defined as [E(AH1−x) + xE(H2)/2] − E(AH), where E(AH), E(AH1−x), and E(H2) are, respectively, the total energies of the stoichiometric AH, H-deficient AH1−x, and hydrogen molecule. b Ref. 13. c Ca2NH and CaNH are not metallic compounds, and thus the values of WFcalc correspond to the positions of the valence band maximum with respect to the vacuum level. d The H atoms in CaNH show partial occupation, so that determination of the hydrogen positions for DFT calculations is a difficult task; therefore, the following assumptions were adopted only for CaNH: that (1) the lattice parameters are fixed to the experimental values, (2) the positions of hydrogen were determined to maintain the local stoichiometry at the topmost layer of the CaNH surface, and (3) the ionic positions were all fixed to those in the bulk to avoid an artificial surface reconstruction caused by an ordered configuration of H atoms. If the surface structure of CaNH with the ordered configuration of hydrogen is relaxed, then the vacuum level required for the WF calculation cannot be determined. In a realistic situation of CaNH, ionic relaxations lead to a more stable electronic structure, and thereby the position of the valence band maximum becomes deeper. Therefore, the calculated value based on the above assumptions can be recognized as a lower limit. | |||||
| Ca2N:e− |
Rm |
(111) | 3.3 | 3.5b | — |
| Ca2N:e− |
Rm |
(112) | 2.5 | 2.6b | — |
| Ca2NH |
Fdm |
(100) | 2.8c | — | — |
| Ca2NH1–1/16 |
Fdm |
(100) | 2.3 | — | 1.00 |
| Ca2NH1–2/16 |
Fdm |
(100) | 2.3 | — | 0.88–1.02 |
| Ru/Ca2NH1–1/16 | — | (100) | — | — | 0.43–0.67 |
| Ru/N2/Ca2NH1–1/16 | — | (100) | — | — | 0.49–0.91 |
| CaH2 | Pnma | (010) | 4.0 | — | — |
| CaH2–1/24 | Pnma | (010) | 2.3 | — | 1.08–1.11 |
| CaNH |
Fmm |
(100) | ≥3.6c,d | — | — |
| CaNH1–1/32 |
Fmm |
(100) | — | — | 1.71 |
Ca2NH1−x can be readily formed by the combination of Ca2NH with Ru nanoparticles. Table 2 lists the calculated formation energies of VH on the surfaces of the catalysts in the presence or absence of Ru. Relatively large VH formation energies (0.88–1.02 eV) were obtained for Ru-free Ca2NH(100), which indicates the difficulty of hydrogen vacancy formation. The density of states (DOS) for an H− ion in Ca2NH(100) is found at a relatively deep level, as shown in Fig. 3c and the inset of Fig. 3d (−6 eV below the Fermi level (EF) for bulk H− and −4 eV below EF for surface H−); therefore, the formation of anionic electrons by H desorption requires a relatively large amount of energy. However, the situation is significantly changed when a Ru cluster is loaded on the Ca2NH(100) surface; the VH formation energy for Ru6-loaded Ca2NH(100) is decreased to 0.43–0.67 eV. Fig. 3d shows that the DOS for an H− ion bonded with Ru (Hb in the inset of Fig. 3d) are energetically shifted up from a relatively deep level to a shallow level (−0.5 eV below EF). Thus, an H− ion bonded with Ru is almost ready to form an anionic electron by the desorption of H0. The lift-up of the H− states is caused by the fraction of the anti-bonding level with Ru, and thus originates from the presence of surface H−. In other words, the orbital hybridization between Ru and surface H− leads to an electron donation from surface H− to Ru, which makes the desorption as H0 easier. This situation is maintained even when an N2 molecule is adsorbed on Ru; the formation energy for VH on Ru6N2–Ca2NH(100) is 0.49 eV. In contrast, a large VH formation energy is observed for CaNH (1.71 eV), which corresponds well to the experimental evidence presented in Fig. 2g. These computational results support the idea that anionic electrons with a low WF are formed in nonstoichiometric hydrides, [Ca2N]+·e1−x−Hx−, by Ru catalysts during the reaction, which facilitates N2 cleavage on the hydride via electron donation from the anionic electrons to the loaded Ru nanoparticles.
Reaction mechanism over Ru/[Ca2N]+·e1−x−Hx−
Ammonia synthesis from N2 and D2 was examined to elucidate the reaction mechanism over Ru/[Ca2N]+·e1−x−Hx−. The initial gas (reaction time = 0 h) consists mainly of N2 (m/z = 28) and D2 (m/z = 4), whereas the amounts of other species such as H2 (m/z = 2) and HD (m/z = 3) are negligibly small (Fig. S4†). As the reaction time increased, the N2 and D2 peaks decreased and the signals with m/z = 19, 18, 17, and 16 increased (Fig. 4a and S4 and S5†). These signals are attributed to ND2H, NDH2, ND2 (a fragment of ND3), NH3, and NH2 (a fragment of NH3). The signals with m/z = 17 and 16 are much larger than those with m/z = 18 and 19 in the early stage (0–3 h), which indicates that the dominant product from the reaction of N2 and D2 is NH3, rather than NH2D, NHD2, or ND3. Therefore, N adatoms react with H atoms derived from [Ca2N]+·e1−x−Hx− to form NH3, whereas D adatoms produced by the dissociative adsorption of D2 on Ru surfaces prefer to be incorporated into [Ca2N]+·e1−x−Hx− as D− ions rather than directly react with N adatoms. TPD measurements (Fig. 4b) show that H2, HD, and D2 species were desorbed from the catalyst after the reaction. It is thus evident that some H− ions in Ru/[Ca2N]+·e1−x−Hx− are replaced by D− ions during the ammonia synthesis reaction. The incorporation of D− ions proceeds simultaneously with NH3 formation, so that the exchange rate of H− ions in Ru/[Ca2N]+·e1−x−Hx− with D− ions can be estimated to be 1.66 mmol g−1 h−1 from the initial NH3 formation rate (0–3 h). On the other hand, when Ru/[Ca2N]+·e1−x−Hx− is heated at 340 °C under a D2 atmosphere without N2 (Fig. S6†), the exchange rate of H− with D− ions remains at only 0.18 mmol g−1 h−1, which is lower by an order of magnitude than that (1.66 mmol g−1 h−1) shown in Fig. 4a. These results reveal that N adatoms on Ru/[Ca2N]+·e1−x−Hx− preferentially react with H atoms derived from H− ions in [Ca2N]+·e1−x−Hx− rather than H atoms produced by the dissociative adsorption of H2 on Ru surfaces because H atoms on Ru are readily incorporated into the support material to form H− ions by reaction with an anionic electron.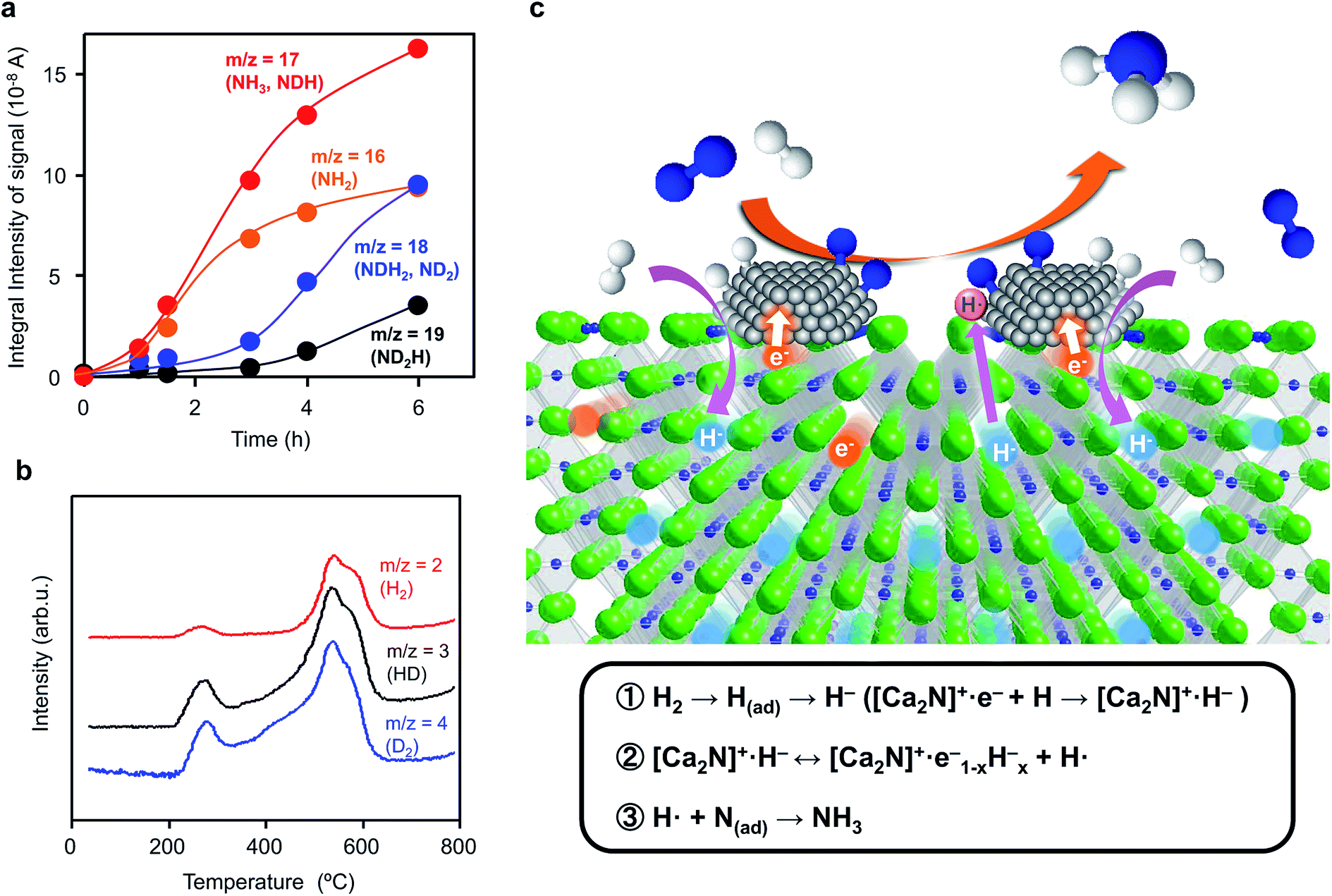 | ||
| Fig. 4 (a) Reaction time profiles for ammonia synthesis from N2 and D2 over Ru/Ca2N:e− at 340 °C (reaction conditions: catalyst, 0.2 g; reaction gas, N2:D2 = 1:3; reaction pressure, 60 kPa). Prior to reaction, Ru/Ca2N:e− was heated under N2 + H2 flow (N2:H2 = 1:3) at 340 °C for 10 h to form Ru/Ca2NH. (b) TPD profiles of Ru/Ca2N:e− after the reaction (a). TPD measurements were performed (10 °C min−1) with Ar flow. (c) Schematic illustration of ammonia synthesis over Ru/Ca2N:e−. During ammonia synthesis over Ru/Ca2N:e−, H2 is incorporated into Ca2N:e− as H− ions to form Ca2NH (reaction 1). The H− ions are released from Ca2NH, which leaves electrons to form a hydrogen vacancy near the Ru-support interface (reaction 2). The cleavage of N2 proceeds effectively on Ru surfaces due to electron injection from [Ca2N]+·e1−x−Hx− and the nitrogen adatoms react with H radicals to form ammonia (reaction 3). | ||
To summarize these results, we propose the reaction mechanism illustrated in Fig. 4c. The dissociative adsorption of H2 forms H adatoms on Ru surfaces under the reaction conditions, and Ca2N:e− is readily transformed into Ca2NH by the reaction of anionic electrons in Ca2N:e− with spillover H adatoms ([Ca2N]+·e− + H → [Ca2N]+·H−, reaction 1 in Fig. 4c). Ca2N:e− and Ca2NH are in an equilibrium through the reversible hydrogen storage reaction; therefore, nonstoichiometric [Ca2N]+·e1−x−Hx− rather than stoichiometric Ca2NH is expected to result from the transformation (reaction 2 in Fig. 4c). [Ca2N]+·e1−x−Hx− with small work functions strongly donate electrons into Ru, which facilitates the cleavage of N2 molecules on Ru surfaces. N adatoms on Ru surfaces prefer to react with H atoms derived from the hydride to form ammonia and anionic electrons (hydrogen vacancies) in [Ca2N]+·e1−x−Hx− (reaction 3 in Fig. 4c), as demonstrated in the D2 experiments. This mechanism is distinct from ammonia synthesis on conventional heterogeneous catalysts, where the reaction between N and H adatoms on transition metal surfaces produces ammonia.34 The incorporation of hydrogen adatoms formed on Ru surfaces into the 2D layer of Ca2N is driven by reaction with anionic electrons and results in the suppression of active site saturation by hydrogen, i.e., hydrogen poisoning of Ru. Thus, the reaction mechanism is very similar to that for Ru/C12A7:e−, as elucidated by kinetic analyses.16 Therefore, the rate-determining step for ammonia synthesis over Ru/Ca2N:e− is not N2 cleavage, but subsequent processes, possibly the formation of NH species. However, Ru/Ca2N:e− is far superior to Ru/C12A7:e− in terms of catalytic performance for ammonia synthesis below 300 °C; Ru/C12A7:e− has moderate catalytic activity and high activation energy (90 kJ mol−1) for ammonia synthesis below 300 °C, where the reaction mechanism is analogous to that for conventional catalysts. The H2 absorption–desorption characteristics of Ru/C12A7:e− are observed above ca. 350 °C,16 which is due mainly to the stabilization of H− ions in the positively charged sub-nanometer sized cages, so that a larger thermal energy is necessary to release hydrogen through the cage wall.35 On the other hand, the facile hydrogen exchange reaction on Ru/Ca2N:e− proceeds at lower temperatures (from 200 °C) than that on Ru/C12A7:e− because H− ions are located in the open spaces between two cationic slabs [Ca2N]+. This facile hydrogen exchange at lower temperatures thus makes it possible for noticeable ammonia synthesis to occur, even at ca. 200 °C.
The experimental results described above demonstrate the validity of our proposed design concept for the Ru-support for low-temperature NH3 synthesis, i.e., the reversible exchangeability between H− ions and electrons plays an important role for effective ammonia synthesis at low temperatures. The effectiveness of this idea is further demonstrated by the use of CaH2, the simplest hydride, as a support for a Ru catalyst. Although CaH2 itself has a WF of 4.0 eV, the WF of CaH2 with VH (i.e., CaH2−x) has a small value (2.3 eV) similar to that for nonstoichiometric Ca2NH (Table 2). Ru/CaH2 exhibits high catalytic activity with a low activation energy (51 kJ mol−1), as expected from its low work function (Table 1), and has reaction orders for N2 and H2 similar to those for Ru/Ca2N:e− (Table S1†). The formation of hydrogen vacancies in CaH2 is difficult because hydrogen desorption from CaH2 occurs above 600 °C.36 However, hydrogen can easily desorb from the surface of CaH2 above 200 °C in the presence of a Ru catalyst (Fig. S7†). Thus, anionic electrons are formed in Ru/CaH2 during the reaction, which results in efficient ammonia synthesis at lower reaction temperatures.
Conclusions
Ru/Ca2N:e− exhibits much higher catalytic performance for ammonia synthesis at low temperatures than heterogeneous catalysts reported to date, including Ru/C12A7:e−. This is not attributed to the Ca2N:e− electride, but to a hydrogen-deficient Ca2NH hydride ([Ca2N]+·e1−x−Hx−) formed during the ammonia synthesis reaction. Both Ru/CaH2 and Ru/Ca2NH hydrides exhibit high catalytic activity for ammonia synthesis. The formation of anionic electrons in these hydrides results in a small work function (2.3 eV), which accounts for the strong electron donation ability that facilitates efficient N2 cleavage on Ru. N adatoms on Ru preferentially react with H atoms derived from the hydride to form NH species. These reactions proceed even at ca. 200 °C, so that ammonia synthesis is catalyzed above 200 °C. In addition, the hydrides suppress H2 poisoning of the Ru surface due to their hydrogen storage properties. The present results demonstrate that the strong electron-donating ability and the reversible exchangeability between H− ions in the lattice and anionic electrons at low temperatures are requisite for the Ru catalyst support in low-temperature ammonia synthesis.Acknowledgements
The authors appreciate M. Okunaka, S. Fujimoto, E. Sano, and Y. Takasaki for technical assistance and S. Kanbara for conducting catalytic tests. This work was supported by funds from the Accelerated Innovation Research Initiative Turning Top Science and Ideas into High-Impact Values (ACCEL) program of the Japan Science and Technology Agency (JST). A portion of this work was supported by a Kakenhi Grant-in-Aid (No. 15H04183) from the Japan Society for the Promotion of Science (JSPS).Notes and references
- S. Gambarotta and J. Scott, Angew. Chem., Int. Ed., 2004, 43, 5298–5308 CrossRef CAS PubMed.
- A. Mittasch, Adv. Catal., 1950, 2, 81–104 Search PubMed.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- J. A. Pool, E. Lobkovsky and P. J. Chirik, Nature, 2004, 427, 527–530 CrossRef CAS PubMed.
- K. Arashiba, Y. Miyake and Y. Nishibayashi, Nat. Chem., 2011, 3, 120–125 CrossRef CAS PubMed.
- B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed.
- J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84–88 CrossRef CAS PubMed.
- K. Aika, A. Ozaki and H. Hori, J. Catal., 1972, 27, 424–431 CrossRef CAS.
- A. Ozaki, Acc. Chem. Res., 1981, 14, 16–21 CrossRef CAS.
- H. Bielawa, O. Hinrichsen, A. Birkner and M. Muhler, Angew. Chem., Int. Ed., 2001, 40, 1061–1063 CrossRef CAS.
- S. E. Siporin and R. J. Davis, J. Catal., 2004, 225, 359–368 CrossRef CAS.
- S. Matsuishi, Y. Toda, M. Miyakawa, K. Hayashi, T. Kamiya, M. Hirano, I. Tanaka and H. Hosono, Science, 2003, 301, 626–629 CrossRef CAS PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- S. Kanbara, M. Kitano, Y. Inoue, T. Yokoyarna, M. Hara and H. Hosono, J. Am. Chem. Soc., 2015, 137, 14517–14524 CrossRef CAS PubMed.
- Y. Toda, H. Yanagi, E. Ikenaga, J. J. Kim, M. Kobata, S. Ueda, T. Kamiya, M. Hirano, K. Kobayashi and H. Hosono, Adv. Mater., 2007, 19, 3564–3569 CrossRef CAS.
- M. Kitano, S. Kanbara, Y. Inoue, N. Kuganathan, P. V. Sushko, T. Yokoyama, M. Hara and H. Hosono, Nat. Commun., 2015, 6, 6731 CrossRef CAS PubMed.
- Y. J. Kim, S. M. Kim, H. Hosono, J. W. Yang and S. W. Kim, Chem. Commun., 2014, 50, 4791–4794 RSC.
- Y. J. Kim, S. M. Kim, E. J. cho, H. Hosono, J. W. Yang and S. W. Kim, Chem. Sci., 2015, 6, 3577–3581 RSC.
- K. Lee, S. W. Kim, Y. Toda, S. Matsuishi and H. Hosono, Nature, 2013, 494, 336–340 CrossRef CAS PubMed.
- K. Aika, M. Kumasaka, T. Oma, O. Kato, H. Matsuda, N. Watanabe, K. Yamazaki, A. Ozaki and T. Onishi, Appl. Catal., 1986, 28, 57–68 CrossRef CAS.
- F. Rosowski, A. Hornung, O. Hinrichsen, D. Herein, M. Muhler and G. Ertl, Appl. Catal., A, 1997, 151, 443–460 CrossRef CAS.
- D. H. Gregory, A. Bowman, C. F. Baker and D. P. Weston, J. Mater. Chem., 2000, 10, 1635–1641 RSC.
- O. Reckeweg and F. J. DiSalvo, Solid State Sci., 2002, 4, 575–584 CrossRef CAS.
- Y. Kojima and Y. Kawai, Chem. Commun., 2004, 2210–2211 RSC.
- J. F. Brice, J. P. Motte, A. Courtois, J. Protas and J. Aubry, J. Solid State Chem., 1976, 17, 135–142 CrossRef CAS.
- H. H. Franck, M. A. Bredig and G. Hoffmann, Naturwissenschaften, 1933, 21, 330–331 CrossRef CAS.
- S. Hino, T. Ichikawa, H. Y. Leng and H. Fujii, J. Alloys Compd., 2005, 398, 62–66 CrossRef CAS.
- Y. F. Liu, T. Liu, Z. T. Xiong, J. J. Hu, G. T. Wu, P. Chen, A. T. S. Wee, P. Yang, K. Murata and K. Sakata, Eur. J. Inorg. Chem., 2006, 4368–4373 CrossRef CAS.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, J. Phys. Chem. B, 2003, 107, 10967–10970 CrossRef CAS.
- G. T. Wu, Z. T. Xiong, T. Liu, Y. F. Liu, J. J. Hu, P. Chen, Y. P. Feng and A. T. S. Wee, Inorg. Chem., 2007, 46, 517–521 CrossRef PubMed.
- N. D. Lang and W. Kohn, Phys. Rev. B: Solid State, 1971, 3, 1215–1223 CrossRef.
- H. B. Michaelson, J. Appl. Phys., 1977, 48, 4729–4733 CrossRef CAS.
- H. L. Skriver and N. M. Rosengaard, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 7157–7168 CrossRef CAS.
- A. Vojvodic, A. J. Medford, F. Studt, F. Abild-Pedersen, T. S. Khan, T. Bligaard and J. K. Nørskov, Chem. Phys. Lett., 2014, 598, 108–112 CrossRef CAS.
- K. Hayashi, P. V. Sushko, Y. Hashimoto, A. L. Shluger and H. Hosono, Nat. Commun., 2014, 5, 3515 Search PubMed.
- L. George and S. K. Saxena, Int. J. Hydrogen Energy, 2010, 35, 5454–5470 CrossRef CAS.
- Y. V. Larichev, B. L. Moroz, V. I. Zaikovskii, S. M. Yunusov, E. S. Kalyuzhnaya, V. B. Shur and V. I. Bukhtiyarov, J. Phys. Chem. C, 2007, 111, 9427–9436 CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, kinetic analyses, catalytic performance, detailed characterization, and DFT calculations. See DOI: 10.1039/c6sc00767h |
| This journal is © The Royal Society of Chemistry 2016 |