
Co(II) complexes loaded into metal–organic frameworks as efficient heterogeneous catalysts for aerobic epoxidation of olefins†
Jingjing
Wang
a,
Mu
Yang
a,
Wenjun
Dong
a,
Zhaokui
Jin
a,
Jia
Tang
a,
Shuang
Fan
a,
Yunfeng
Lu
b and
Ge
Wang
*a
aBeijing Key Laboratory of Function Materials for Molecule & Structure Construction, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, PR China. E-mail: gewang@mater.ustb.edu.cn; Tel: +86 10 62333765
bDepartment of Materials Science and Engineering, University of California, Los Angeles, CA 90034, USA
First published on 8th October 2015
Abstract
A series of efficient cobalt(II)-anchored Cr-MOF (Cr-MIL-101-NH2) catalysts, such as Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I, have been successfully synthesized by one-pot modification of the terminal amino group with salicylaldehyde, pyridine-2-aldehyde or pyridine-3-aldehyde and anchoring of Co(II) ions into the mesoporous Cr-MOF supports. The Co(II)@Cr-MIL-101-P2I catalyst exhibited high catalytic performance for epoxidation of olefins with air as an oxidant due to the nitrogen atom in the pyridine ring as a strong electron-withdrawing substituent, high dispersion of Co(II) species and high surface area for sufficient contact between the substrate and active sites. The strong coordination interaction between the Co(II) ions and chelating groups in the Co(II)@Cr-MIL-101-P2I catalyst guaranteed the excellent recycling performance. Furthermore, the synthesized Co(II)@Cr-MIL-101-P2I catalyst realized its general applicability towards various olefins, such as cyclic olefins, tri-substituted olefins, aliphatic olefins and aromatic olefins.
Introduction
Metal–organic frameworks (MOFs) are crystalline hybrid inorganic/organic solids with extended structures which can be utilized in many fields, such as gas separation and storage, sensors, and drug delivery.1–3 Especially, MOFs used for heterogeneous catalysis materials have sparked immense interest due to their high crystallinity with well-defined pore properties, high specific surface area and tailorable structures.4–7 However, established MOFs have relatively low catalytic activity due to their limited selections of metals and organic ligands.8 In order to further enhance the catalytic activity, post-synthetic modification (PSM), defined as the chemical derivatization of MOFs after their formation, becomes a practical and powerful tool to generate MOF-based heterogeneous catalysts with improved catalytic performance.9,10MIL-101 discovered by Férey et al. shows a rigid zeotype cubic structure, which defines nanocages of about 2.9 and 3.4 nm with windows of 1.2 and 1.6 nm, respectively.11 MIL-101-NH2 retains the features of MIL-101, in which amino groups are introduced into the organic frames of the MOF lattice.12 Cr-MIL-101-NH2 can be a good candidate for PSM due to its large pores, good thermal stability as well as excellent chemical stability to water and common organic solvents.13 Moreover, the reactive amino groups existing on the surface of the Cr-MIL-101-NH2 pores can provide great opportunities to covalently attach functional species. Tremendous efforts for the PSM strategy of various Cr-MIL-101-NH2, such as urea-derived MIL-101 by introducing ethyl isocyanate14,15 and photo-switchable MIL-101 by installing an azo functionality,12 have been achieved successfully. However, the PSM strategy of Cr-MIL-101-NH2 in the catalytic oxidation reaction has received much less attention.
Cobalt(II)-based organometallics, as efficient and selective homogeneous catalysts, have been applied to the epoxidation of olefins.16,17 However, the main drawbacks of these catalysts are catalyst recycling and product separation. To overcome these problems, various supports, such as polymers,18,19 zeolites,20 and silica,21,22 have been used to load the homogeneous complexes. Nevertheless, these heterogeneous catalysts typically involve some problems, such as trivial preparation, low chemical stability, non-uniform structure and low efficiency. Therefore, how to obtain an accessible, reusable and novel heterogeneous cobalt(II)-based catalyst exhibiting the high activity of a homogeneous catalyst and the good reusability of a heterogeneous catalyst remains a challenge.
In this paper, a novel system for the aerobic oxidation of olefins under air atmosphere employing metal–organic framework-anchored cobalt(II) catalysts has been developed. Co(II)-based complexes were immobilized into mesoporous Cr-MIL-101-NH2 by a one-pot method using salicylaldehyde, pyridine-2-aldehyde or pyridine-3-aldehyde as linker agents (Scheme 1). The amino group of Cr-MIL-101-NH2 upon condensation with salicylaldehyde, pyridine-2-aldehyde or pyridine-3-aldehyde afforded a Schiff base moiety in the porous matrix. The Schiff base moieties as chelating groups can anchor the Co(II) ions. The Cr-MIL-101-NH2 MOFs with high surface area were used as supports to improve the dispersion of Co(II) ions. The uniform and big pores of the MOFs guarantee sufficient contact between the catalytic active sites and the substrate. The strong coordination interaction between the Co(II) ions and chelating groups in the Co(II)@Cr-MIL-101-P2I catalyst enhances its reusability after the reaction. The developed cobalt(II)-based solid catalysts showed high catalytic activity, easy recovery and excellent recycling stability for the epoxidation of olefins under mild reaction conditions.
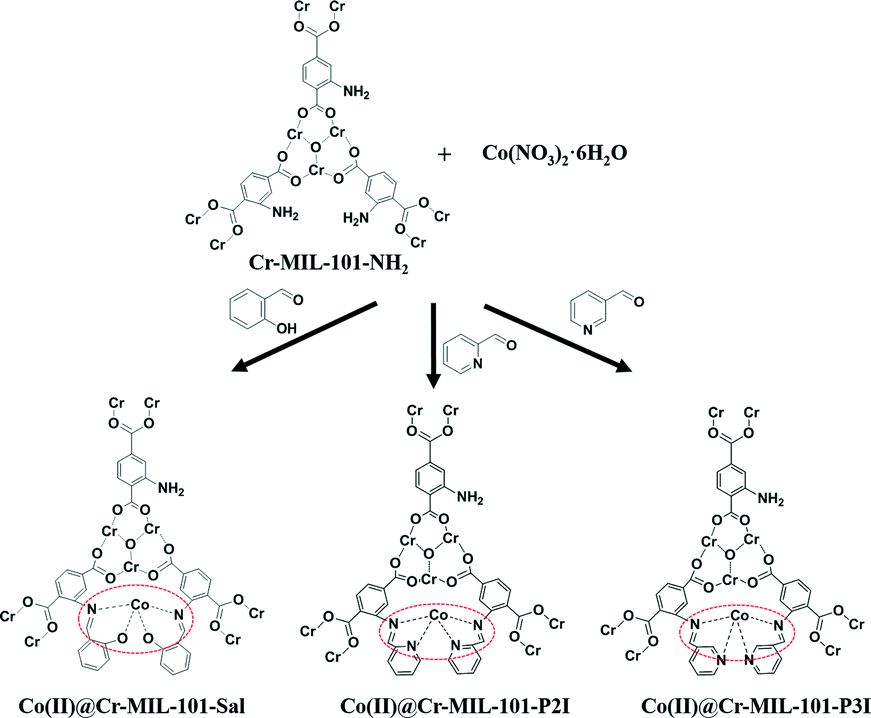 | ||
| Scheme 1 Schematic illustration of the preparation of Co(II) complexes loaded into Cr-MIL-101-NH2. | ||
Experimental
Synthesis of Cr-MIL-101-NH2
Cr-MIL-101-NH2 was prepared by a hydrothermal synthesis method according to the literature.23 In a typical process, chromic nitrate hydrate (6.40 g, 16 mmol), 2-aminoterephthalic acid (2.88 g, 16 mmol) and sodium hydroxide (1.60 g, 40 mmol) were dissolved in deionized water (120 mL) under stirring, then the solution was transferred into a 200 mL Teflon-lined stainless steel autoclave and kept at 150 °C for 12 h. The autoclave was left to cool down to room temperature. The green precipitate was collected by centrifugation, and washed with deionized water, DMF and methanol. Finally, the products were dried under vacuum at 40 °C overnight.Preparation of cobalt(II)-based solid catalysts
In order to ensure that sufficient Co(II) moieties could be immobilized in the MOF pores, the molar ratio of the terminal amino group of MOFs, the modifying aldehyde group and Co(II) was set at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 during the synthesis of the three kinds of catalysts.
:4:1 during the synthesis of the three kinds of catalysts.
To modify Cr-MIL-101-NH2 with salicylaldehyde and load Co(II) ions, 0.40 g of Cr-MIL-101-NH2 was first dispersed into 30 mL of methanol in a flask under ultrasound, and 0.62 mL of salicylaldehyde and 0.43 g of cobalt nitrate hexahydrate were added to the mixture, then the solution was refluxed at 85 °C for 10 h. Afterwards, the precipitate was washed with abundant ethanol three times and dried under vacuum at 40 °C overnight. The obtained product was labeled as Co(II)@Cr-MIL-101-Sal.
The synthesis procedure of Cr-MIL-101-NH2 modified with pyridine-2-aldehyde or pyridine-3-aldehyde and loaded with cobalt(II) was the same as that of Co(II)@Cr-MIL-101-Sal but replacing salicylaldehyde with pyridine-2-aldehyde or pyridine-3-aldehyde, respectively. The obtained products were accordingly named as Co(II)@Cr-MIL-101-P2I or Co(II)@Cr-MIL-101-P3I.
Catalytic epoxidation reactions
The catalytic epoxidation reaction was carried out in the liquid phase in a 25 mL two-neck flask. Olefins (1 mmol), catalyst (0.6 mol% cobalt) and isobutyraldehyde (2 mmol) were added to acetonitrile (5 mL). The reaction mixture was stirred at 35 °C for 5 h under air atmosphere. After the reaction, nitrobenzene (0.2 mmol) as an internal standard was added to the mixture for the determination of yield and selectivity. The filtrate was analyzed using an Agilent 7890/5975C-GC/MS.For catalyst recycling, the catalyst was filtered from the reaction solution, washed with acetonitrile and dried for the next run. The dried catalyst was reused for the epoxidation of cis-cyclooctene in acetonitrile under the same reaction conditions.
The hot filtration test was performed by separating the catalyst from the reaction mixture at 1 h, and the filtrate was stirred further for 4 h. The reaction progress was monitored by GC/MS.
Characterization
Field-emission scanning electron microscopy (FESEM) images were acquired using a ZEISS SUPRA55 operated at an accelerating voltage of 10 kV. Fourier transform infrared (FTIR) spectra were obtained with a Nicolet 6700 using the potassium bromide (KBr) pellet technique. Diffuse reflectance UV-vis spectra were recorded on a UV-2550 spectrometer equipped with a diffuse reflectance integrating sphere coated with BaSO4, which also served as a standard. Powder X-ray diffraction (XRD) patterns were recorded by using an M21X diffractometer (MAC Science Co. Ltd., Japan) using a Cu Kα radiation (k = 1.541 Å) source at 40 kV and 200 mA. The chemical compositions were analysed by inductively coupled plasma-atomic emission spectrometry (ICP-AES, Varian 715-ES) and X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi). The C 1s line at 284.8 eV from adventitious carbon was used as the reference for binding energy. Thermal gravimetric analysis (TGA) was used to investigate the thermal decomposition behavior of the samples using a Netzsch STA449F3 instrument at a heating rate of 10 °Cmin−1 under an air flow. Nitrogen adsorption–desorption isotherms were measured at 77 K with a Micromeritics ASAP 2420 adsorption analyzer. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method. The pore size distributions were derived from the adsorption branches of the isotherms by using the Barrett–Joyner–Halenda (BJH) model. The catalytic results were analyzed by means of gas chromatography-mass spectrometry (GC-MS, Agilent 7890/5975C-GC/MSD), and nitrobenzene was used as an internal standard.
Results and discussion
Catalyst characterization
Typical morphologies of the Cr-MIL-101-NH2, Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples are shown in Fig. 1. The SEM image of Cr-MIL-101-NH2 (Fig. 1a) showed that the as-prepared MOF was composed of particles and the particle size was around 50–300 nm, which was similar to previous reports.23 After modifying with salicylaldehyde, pyridine-2-aldehyde or pyridine-3-aldehyde followed by anchoring of Co(II) ions, the morphology and size of Co(II)@Cr-MIL-101-Sal (Fig. 1b), Co(II)@Cr-MIL-101-P2I (Fig. 1c) and Co(II)@Cr-MIL-101-P3I (Fig. 1d) were found to be similar to those of Cr-MIL-101-NH2, indicating the strong chemical stability of the Cr-MIL-101-NH2 support during the aldehyde group modification and Co(II) ion anchoring procedure. | ||
| Fig. 1 SEM images of (a) Cr-MIL-101-NH2, (b) Co(II)@Cr-MIL-101-Sal, (c) Co(II)@Cr-MIL-101-P2I and (d) Co(II)@Cr-MIL-101-P3I. | ||
The XRD patterns of the Cr-MIL-101-NH2, Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples are shown in Fig. 2. The XRD pattern of the Cr-MIL-101-NH2 sample corresponded well to the literature data, which indicated that the desired MOF support was obtained.14,23 The Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples showed diffraction patterns similar to that of Cr-MIL-101-NH2, which clearly suggested that the framework structure of Cr-MIL-101-NH2 remained intact during the modification and loading procedure. Moreover, compared with Cr-MIL-101-NH2, a decrease in the peak intensity at 2θ = 3.2° of the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples was observed, which was attributed to the modifying complexes inside the pores.24
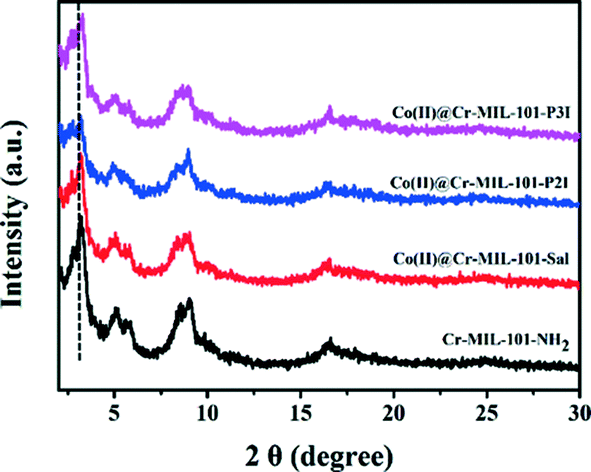 | ||
| Fig. 2 Powder XRD patterns of Cr-MIL-101-NH2, Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I. | ||
In order to evaluate the surface area, pore volume and pore structure of the samples, nitrogen adsorption–desorption was performed and the obtained isotherms are shown in Fig. 3. All the samples featured the same type IV isotherm, which further indicated that the pore structure was well retained after functionalization of the linker and anchoring of Co(II) ions. The surface area of the samples was calculated using the Brunauer–Emmett–Teller (BET) method. The total pore volume was obtained from the volume of N2 adsorption at P/P0 = 0.985. The BET surface area and the total pore volume of the Cr-MIL-101-NH2 sample were 1039.2 m2 g−1 and 1.56 cm3 g−1, respectively. An obvious decrease in the BET surface area as well as the total pore volume was observed in the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples, which demonstrated that the pores of Cr-MIL-101-NH2 were occupied by Co(II) complexes.21,24 FTIR (Fig. S1†), 1H NMR (Fig. S2†) and diffuse reflectance UV-vis spectra (Fig. S3a†) results also confirmed the successful modification of Cr-MIL-101-NH2. The Co(II)@Cr-MIL-101-P2I sample had the lowest BET surface area and total pore volume, which could be ascribed to its high content of the anchored Co(II) complex. The content of cobalt measured by ICP-AES in the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples (Table 1) was 0.12 mmol g−1, 0.29 mmol g−1 and 0.03 mmol g−1, respectively. Under the same synthesis procedure and the same molar ratio of raw materials, the varied loading content of cobalt in these samples should be due to the different coordination environments of Co(II) ions.
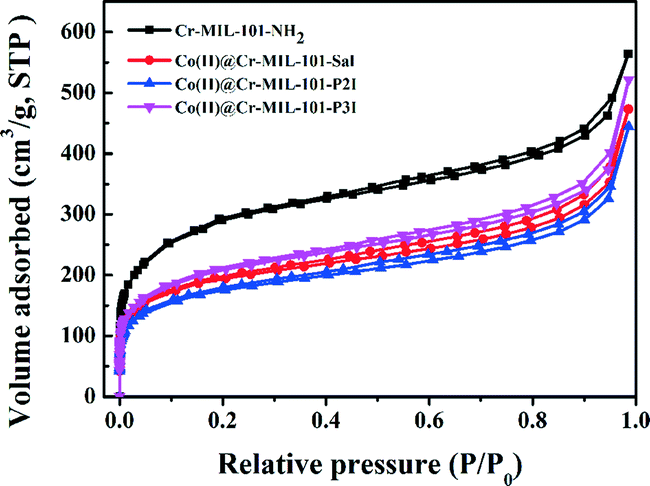 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of Cr-MIL-101-NH2, Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I. | ||
| Samples | S BET (m2 g−1) | Total pore volume (cm3 g−1) | ICP (Co, mmol g−1) |
|---|---|---|---|
| Cr-MIL-101-NH2 | 1039.2 | 1.56 | — |
| Co(II)@Cr-MIL-101-Sal | 702.5 | 0.73 | 0.12 |
| Co(II)@Cr-MIL-101-P2I | 632.2 | 0.69 | 0.29 |
| Co(II)@Cr-MIL-101-P3I | 757.1 | 0.81 | 0.03 |
In order to gain deeper insight into the surface composition of the catalysts, the XPS spectrum of the Co(II)@Cr-MIL-101-P2I sample was investigated. As shown in Fig. 4a, the sample contained C, O, Cr, N and Co elements. The Cr 2p1/2 and Cr 2p3/2 signals showed two peaks at 586.8 eV and 577.2 eV (Fig. 4b), respectively. Both peaks corresponded to the typical binding energies of Cr3+.25 No other oxidation states of chromium were observed, which indicated that the chromium trimers were chemically stable during functionalization of the linker and anchoring of the cobalt precursor. The peak-fitted N 1s core-line spectrum of Co(II)@Cr-MIL-101-P2I (Fig. 4c) showed three resolved peaks at 400.4 eV, 399.7 eV and 398.9 eV, corresponding to imine (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) groups,26 amino (–NH2) groups27 and pyridine N,28 respectively. Additionally, the peak at 406.7 eV was due to the existence of the N in the NO3− group.29 The Co 2p3/2 and Co 2p1/2 core level peaks were at the binding energies of 781.7 eV and 796.9 eV, respectively (Fig. 4d). The Co 2p1/2 and Co 2p3/2 separation of 15.2 eV for the tethered cobalt catalyst was identical to the value reported for Co(salen)·1/2H2O.30 Additionally, the presence of satellite lines at the binding energies of 803.4 eV and 786.4 eV was furthermore indicative of a high-spin Co2+ center in the Co(II)@Cr-MIL-101-P2I sample.31
N–) groups,26 amino (–NH2) groups27 and pyridine N,28 respectively. Additionally, the peak at 406.7 eV was due to the existence of the N in the NO3− group.29 The Co 2p3/2 and Co 2p1/2 core level peaks were at the binding energies of 781.7 eV and 796.9 eV, respectively (Fig. 4d). The Co 2p1/2 and Co 2p3/2 separation of 15.2 eV for the tethered cobalt catalyst was identical to the value reported for Co(salen)·1/2H2O.30 Additionally, the presence of satellite lines at the binding energies of 803.4 eV and 786.4 eV was furthermore indicative of a high-spin Co2+ center in the Co(II)@Cr-MIL-101-P2I sample.31
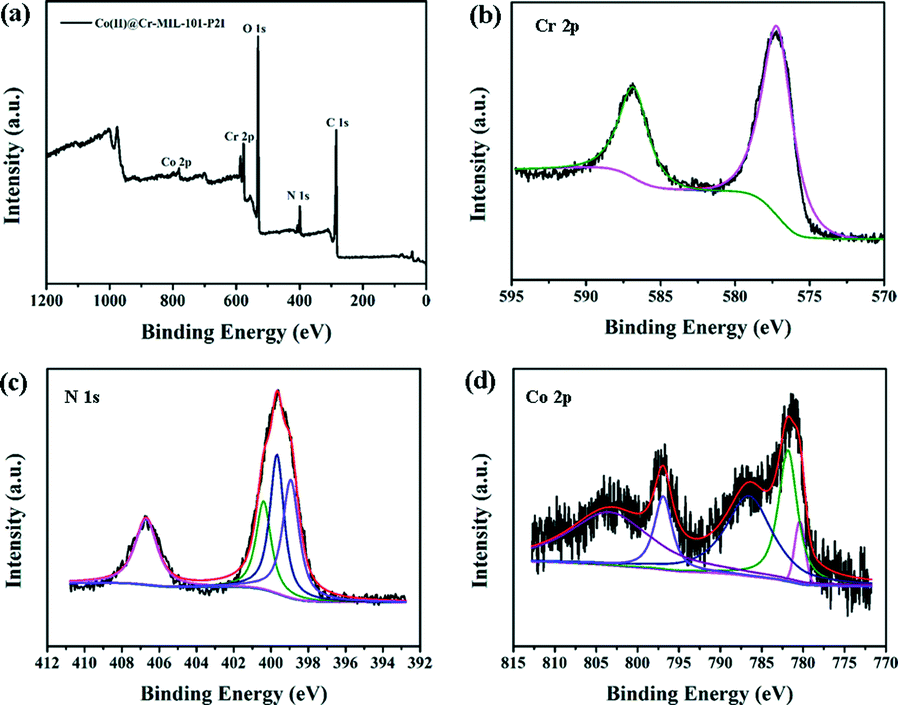 | ||
| Fig. 4 X-ray photoelectron spectra of (a) the Co(II)@Cr-MIL-101-P2I sample, (b) Cr 2p, (c) N 1s and (d) Co 2p. | ||
The TGA curves of the Cr-MIL-101-NH2, Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples are shown in Fig. S4.† The total weight loss of the Cr-MIL-101-NH2 sample occurred through two weight-loss steps. The initial weight loss of 9.8% in the range 40–230 °C was assigned to the loss of free guest water in the pores. The subsequent weight loss of 56.6% in the range 230–330 °C was due to the decomposition of the organic framework. It was noteworthy that the weight losses of the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples were similar to that of Cr-MIL-101-NH2. The cobalt(II)-based solid catalysts showed a higher decomposition temperature and were more stable than Cr-MIL-101-NH2 attributed to the rigid structure of the formed coordinated Co(II) complexes.24
Catalytic study
The catalytic activity of the cobalt(II)-based solid catalysts was studied by catalyzing olefin epoxidation with 1 atm air and isobutyraldehyde in acetonitrile. Epoxidation of cis-cyclooctene was carried out as a model reaction to identify the optimal reaction conditions. Initially, a blank control experiment was performed, and the conversion of cis-cyclooctene was found to be only 20% after 5 h (Table 2, entry 1), which agreed with previous reports.32 No change in the conversion of cis-cyclooctene was observed upon employing Cr-MIL-101-NH2 as the catalyst (Table 2, entry 2). Interestingly, when 0.6 mol% Co(NO3)2·6H2O was used as the catalyst, the conversion was about 99% (Table 2, entry 3). The conversion of cis-cyclooctene was only 47% when 0.3 mol% Co(NO3)2·6H2O was applied as the catalyst (Table 2, entry 4). The conversion of cis-cyclooctene was not improved when the mixture of Co(NO3)2·6H2O and diaminocyclohexane (Table 2, entry 5) or the mixture of Co(NO3)2·6H2O and 2-aminoterephthalic acid (Table 2, entry 6) was used as the homogeneous catalyst, which indicated that the homogeneous organic amino compounds did not contribute to the catalytic activity and Co(II) ions played a significant role in the epoxidation of cis-cyclooctene. However, the homogeneous system leads to difficulties in product separation and catalyst recycling. When Cr-MIL-101-NH2-anchored cobalt(II) complexes including the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I samples were used as catalysts, the conversion of cis-cyclooctene was 78% (Table 2, entry 7), 99% (Table 2, entry 8) and 92% (Table 2, entry 9), respectively. The TOFs for cis-cyclooctene epoxidation catalysed by the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I catalysts were calculated to be 25.74 h−1, 32.67 h−1 and 30.36 h−1, respectively, which were higher than that for the Co6-CP-catalysed cis-cyclooctene epoxidation with a value of 3.72 h−1 at 120 °C.33 Although the TOFs of the solid catalysts were lower than that of Co(NO3)2·6H2O with a value of 54.45 h−1, they could be recollected and reused. The enhanced catalytic efficiency for the epoxidation of cis-cyclooctene was due to the high dispersion of active catalytic sites at the pore walls of the MOFs. Importantly, the conversion of cis-cyclooctene for the Co(II)@Cr-MIL-101-Sal catalyst was lower than those for the Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I catalysts, which could be due to the nitrogen atom in the pyridine ring being a π-electron acceptor.34,35 It can be regarded as a strong electron-withdrawing substituent which could be conducive to electrophilic attack of the Co-peroxy species and then accelerate the rate of epoxidation (Fig. S5†). However, the conversion of cis-cyclooctene was significantly decreased after the re-collected Co(II)@Cr-MIL-101-P3I sample was used again as the catalyst (Table 2, entry 12), which can be ascribed to the relatively weak coordination ability of the chelating groups. The XRD patterns of the Co(II)@Cr-MIL-101-Sal, Co(II)@Cr-MIL-101-P2I and Co(II)@Cr-MIL-101-P3I catalysts after 2 cycles of reaction are shown in Fig. S6.† No obvious change can be observed upon comparing the powder XRD patterns of the fresh and reused catalysts. Furthermore, the cobalt content of the reused Co(II)@Cr-MIL-101-Sal and Co(II)@Cr-MIL-101-P2I catalysts determined by ICP-AES analysis also showed almost no obvious reduction. However, the cobalt content of the reused Co(II)@Cr-MIL-101-P3I catalyst was reduced to 0.018 mmol g−1 from 0.03 mmol g−1. ICP-AES analysis of the reaction solution after one cycle of epoxidation for the Co(II)@Cr-MIL-101-P3I catalyst showed the presence of 0.34 ppm cobalt in the reaction solution, which further indicated that the Co(II) ions were relatively weakly chelated in the Co(II)@Cr-MIL-101-P3I sample. Therefore, among the three kinds of heterogeneous cobalt(II)-based catalysts, Co(II)@Cr-MIL-101-P2I was chosen as the optimal catalyst for the epoxidation of cis-cyclooctene.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conv.b (%) | Sel.b (%) | TOFepo (h−1) |
| a Reaction conditions: cis-cyclooctene (1.0 mmol), catalyst, isobutyraldehyde (2 mmol), acetonitrile (5.0 mL), at 35 °C under air atmosphere with stirring for 5 h. b Conversion and selectivity were determined by GC/MS using nitrobenzene as the internal standard. c Reaction was performed using 20 mg of MOF. d Reaction was under stirring for 3 h. e 0.3 mol% Co(II), reaction was under stirring for 3 h. f 0.3 mol% Co(II) + diaminocyclohexane, reaction was under stirring for 3 h. g 0.3 mol% Co(II) + 2-aminoterephthalic acid, reaction was under stirring for 3 h. h Fresh catalyst, 0.6 mol% Co(II) based on the amount of cobalt in the sample. i Catalyst underwent 2 cycles. | ||||
| 1 | No | 20 | >99 | — |
| 2c | Cr-MIL-101-NH2 | 21 | >99 | — |
| 3d | Co(NO3)2·6H2O | >99 | >99 | 54.45 |
| 4e | Co(NO3)2·6H2O | 47 | >99 | — |
| 5f | Co(NO3)2·6H2O + diaminocyclohexane | 48 | >99 | — |
| 6g | Co(NO3)2·6H2O + 2-aminoterephthalic acid | 51 | >99 | — |
| 7h | Co(II)@Cr-MIL-101-Sal | 78 | >99 | 25.74 |
| 8h | Co(II)@Cr-MIL-101-P2I | >99 | >99 | 32.67 |
| 9h | Co(II)@Cr-MIL-101-P3I | 92 | >99 | 30.36 |
| 10i | Co(II)@Cr-MIL-101-Sal | 76 | >99 | — |
| 11i | Co(II)@Cr-MIL-101-P2I | >99 | >99 | — |
| 12i | Co(II)@Cr-MIL-101-P3I | 57 | >99 | — |
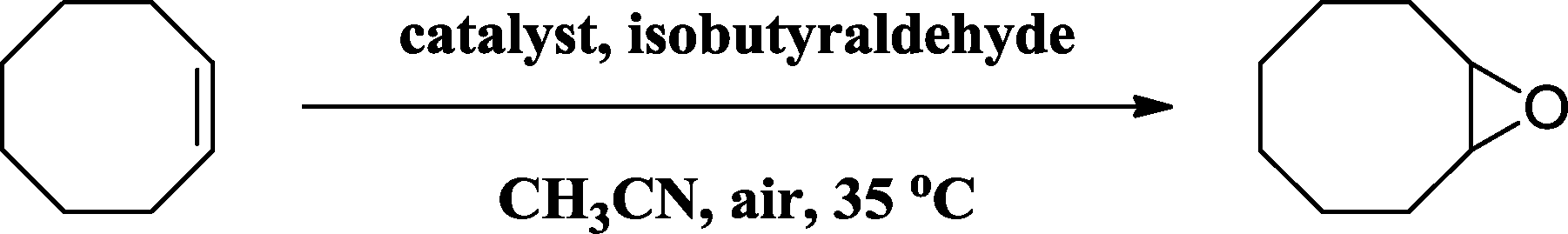
Aldehydes, acting as sacrificial co-reductants, could be transformed in situ to carboxylperoxo radicals which facilitated oxygen transfer to promote the aerobic epoxidation of olefins.36,37 Several aldehydes were evaluated as additives for the epoxidation of cis-cyclooctene in the presence of the Co(II)@Cr-MIL-101-P2I catalyst. The blank control experiment showed no reactivity for the formation of 1,2-epoxycyclooctane (Table 3, entry 1), which indicated that aldehydes were critical additives for aerobic epoxidation. The yield of 1,2-epoxycyclooctane reached up to 99% when isobutyraldehyde was used as the additive (Table 3, entry 2), which was higher than those obtained for the cyclic cyclohexanecarboxaldehyde (86%, Table 3, entry 3), aliphatic heptaldehyde (71%, Table 3, entry 4) and aromatic benzaldehyde (<5%, Table 3, entry 5). The distinctly different yields further suggested that the steric and electronic effects of aldehydes were vital for dioxygen activation in the epoxidation of olefins. Moreover, compromised yields of 1,2-epoxycyclooctane using lower amounts of isobutyraldehyde were obtained (Table 3, entries 6–7).
|
|
|||
|---|---|---|---|
| Entry | Aldehyde (mmol) | Conv.b (%) | Sel.b (%) |
| a Reaction conditions: cis-cyclooctene (1.0 mmol), Co(II)@Cr-MIL-101-P2I catalyst (0.6 mol% cobalt), aldehyde, acetonitrile (5.0 mL), at 35 °C under air atmosphere with stirring for 5 h. b Conversion and selectivity were determined by GC/MS using nitrobenzene as the internal standard. | |||
| 1 | — | — | — |
| 2 | Isobutyraldehyde (2 mmol) | >99 | >99 |
| 3 | Cyclohexanecarboxaldehyde (2 mmol) | 86 | >99 |
| 4 | Heptaldehyde (2 mmol) | 71 | >99 |
| 5 | Benzaldehyde (2 mmol) | <5 | >99 |
| 6 | Isobutyraldehyde (1 mmol) | 44 | >99 |
| 7 | Isobutyraldehyde (0.5 mmol) | 18 | >99 |
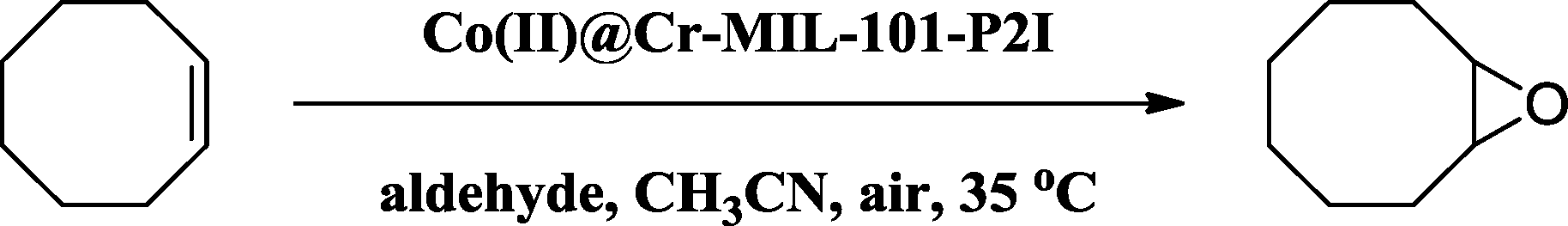
Solvents play a crucial role in epoxidation reactions. The selected model reaction was performed in different solvents and the results are summarized in Table 4. The yields of 1,2-epoxycyclooctane were 99%, 82%, 59%, 48% and 25% when acetonitrile, acetone, ethyl acetate, dichloromethane and n-hexane were employed as solvents, respectively (Table 4, entries 1–5), which revealed that the solvent with greater polarity was beneficial to the enhancement of the conversion of cis-cyclooctene. This can be due to the greater polarity of the solvent facilitating the dispersion and collision of the catalyst, the substrate and the oxygen source.38 However, no 1,2-epoxycyclooctane was observed when ethyl alcohol was used as the solvent due to the coordination with Co(II) ions and competing oxidation reaction.5,27
|
|
|||
|---|---|---|---|
| Entry | Solvent | Conv.b (%) | Sel.b (%) |
| a Reaction conditions: cis-cyclooctene (1.0 mmol), Co(II)@Cr-MIL-101-P2I catalyst (0.6 mol% cobalt), isobutyraldehyde (2.0 mmol), solvent (5.0 mL), at 35 °C under air atmosphere with stirring for 5 h. b Conversion and selectivity were determined by GC/MS using nitrobenzene as the internal standard. | |||
| 1 | Acetonitrile | >99 | >99 |
| 2 | Acetone | 82 | >99 |
| 3 | Ethyl acetate | 59 | >99 |
| 4 | Dichloromethane | 48 | >99 |
| 5 | n-Hexane | 25 | >99 |
| 6 | Ethyl alcohol | 0 | — |
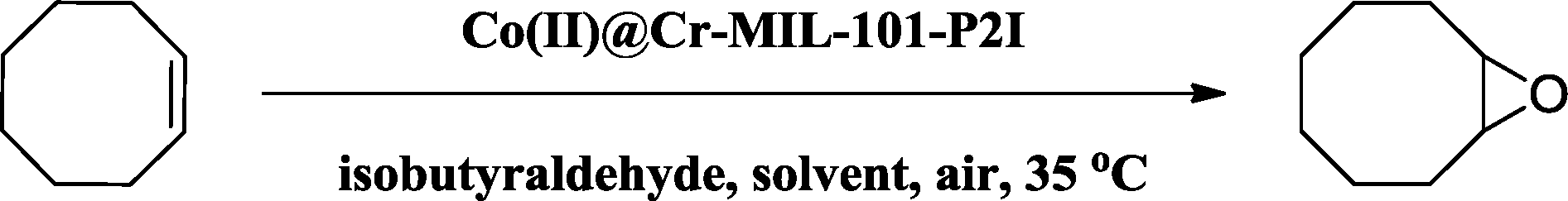
The evolution of the conversion of cis-cyclooctene as a function of reaction time using the fresh Co(II)@Cr-MIL-101-P2I catalyst was explored and is shown in Fig. 5 (curve a). The conversion increased linearly with reaction time and reached 77% at 2 h. Then, the conversion increased gradually and cis-cyclooctene was completely converted to 1,2-epoxycyclooctane at 5 h. Additionally, the selectivity to the desired product was maintained at 99% during the whole reaction process. Therefore, the optimized catalytic conditions involved 1 mmol of the substrate, 0.6 mol% Co(II)@Cr-MIL-101-P2I catalyst based on cobalt and 2 mmol of isobutyraldehyde as an additive under 1 atm air atmosphere at 35 °C with stirring for 5 hours. Such a catalytic system is more efficient and eco-friendly than the cobalt(II)-based zeolite X catalyst at 100 °C (ref. 39) and the cobalt(II)-based MCM-41 catalyst at 110 °C.21
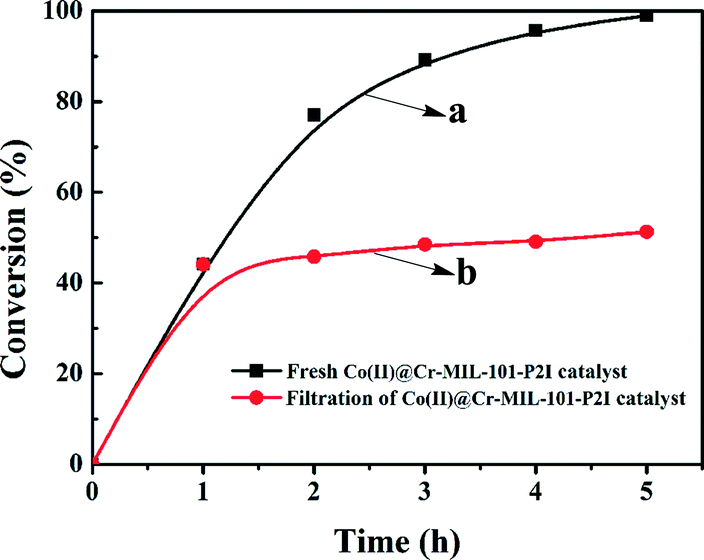 | ||
| Fig. 5 Curve (a): change of conversion over reaction time for the model reaction catalyzed by fresh Co(II)@Cr-MIL-101-P2I catalyst. Curve (b): leaching test. | ||
To confirm whether the Co(II)@Cr-MIL-101-P2I catalyst was heterogeneous for the epoxidation reaction, a hot filtration test was carried out. The solid catalyst was separated by centrifugation at 1 h in the first cycle, and the residual liquid phase was transferred into a clean reactor vessel and stirred for another 4 h under the same reaction conditions. In the absence of the catalyst (Fig. 5b), GC results showed that the conversion of cis-cyclooctene increased to 51% from 44%, which was caused by isobutyraldehyde (Fig. S7†). Furthermore, ICP-AES analysis of the filtrate showed almost no presence of cobalt in the solution.
In order to evaluate the reusability of the Co(II)@Cr-MIL-101-P2I catalyst, the epoxidation of cis-cyclooctene was performed using the recovered catalyst under the optimized catalytic conditions. After each reaction, the catalyst was recovered by simple centrifugation and washed with acetonitrile for the next reaction run. The catalyst remained active during each recycle and maintained 95% conversion and 93% selectivity even after 5 recycles (Fig. 6). The XRD and FTIR results (Fig. S8†) of the fresh and recovered Co(II)@Cr-MIL-101-P2I catalyst showed that the composition of the catalyst had no obvious change after reaction, while the SEM images (Fig. S8†) showed that the structure of the catalyst was slightly aggregated. ICP-AES analysis indicated that the cobalt content was reduced from 0.29 mmol g−1 to 0.26 mmol g−1. This may lead to the slight reduction in the conversion and selectivity of the epoxidation of cis-cyclooctene.
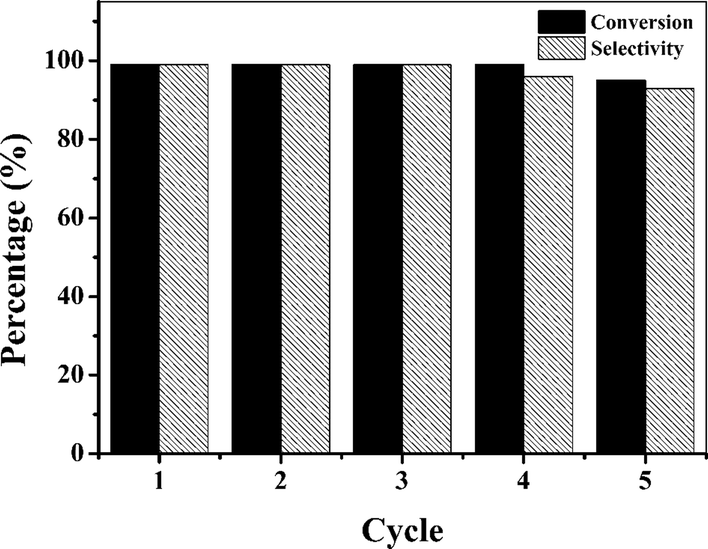 | ||
| Fig. 6 The reusability of the Co(II)@Cr-MIL-101-P2I catalyst for the epoxidation of cis-cyclooctene. | ||
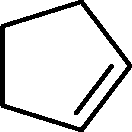
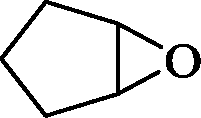
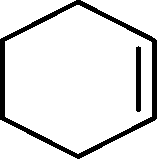
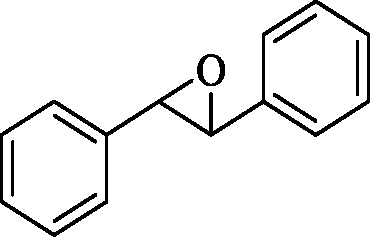
The general applicability of the epoxidation in the presence of air over the Co(II)@Cr-MIL-101-P2I catalyst is demonstrated in Table 5. Various cyclic olefins, such as cyclopentene, cyclohexene, cycloheptene and norbornene, were transformed into the corresponding epoxides with high conversion and selectivity (Table 5, entries 1–4). The relatively bulky cyclic olefin, cyclododecene, showed lower catalytic activity than cis-cyclooctene under the same reaction conditions because of more difficult electrophilic attack of the Co-peroxy species (Table 5, entry 5). The epoxidation of 1,5-cyclooctadiene occurred only at one double bond with a compromised yield and the main product was 9-oxabicyclo [6.1.0]non-4-ene (Table 5, entry 6). The tri-substituted olefin, α-pinene, was converted to the corresponding epoxide with excellent conversion (>99%) and selectivity (>99%) (Table 5, entry 7). Moreover, 1-decene, an aliphatic olefin, generally considered to be inactive towards epoxidation, was oxidized to 1,2-epoxydecane with decent conversion and high selectivity (Table 5, entry 8). The aromatic olefins, such as styrene, trans-stilbene and cis-stilbene, were also catalytically epoxidized. Interestingly, the epoxidation of trans-stilbene proceeded more smoothly than that of styrene and cis-stilbene due to the steric hindrance effect (Table 5, entries 9–11).40 The epoxidation of styrene proceeded in low yield along with the formation of the benzaldehyde by-product. Trans-stilbene showed remarkable conversion (over 91%) of the epoxide as the sole product while cis-stilbene only gave 45% yield of the epoxide.
| Entry | Substrate | Product | Time (h) | Conv.b (%) | Sel.b (%) | TOFepo (h−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: substrate (1.0 mmol), Co(II)@Cr-MIL-101-P2I catalyst (0.6 mol% cobalt), isobutyraldehyde (2.0 mmol), solvent (5.0 mL), at 35 °C under air atmosphere. b Conversion and selectivity were determined by GC/MS using nitrobenzene as the internal standard. | ||||||
| 1 |
|
|
5 | >99 | 82 | 27.06 |
| 2 |
|
|
5 | >99 | 74 | 24.42 |
| 3 |
|

|
5 | >99 | 94 | 31.02 |
| 4 |
|
|
5 | >99 | >99 | 33.00 |
| 5 |

|
|
5 | 69 | >99 | 22.77 |
| 6 |
|
|
5 | 46 | 97 | 14.87 |
| 7 |
|
|
5 | >99 | >99 | 33.00 |
| 8 |

|

|
12 | 62 | >99 | 8.53 |
| 9 |
|
|
24 | 80 | 59 | 3.28 |
| 10 |
|
|
24 | 91 | >99 | 6.26 |
| 11 |
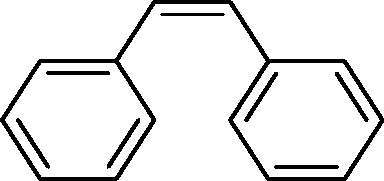
|
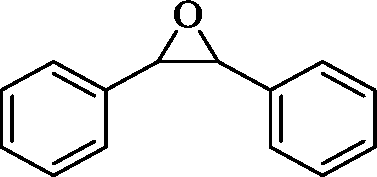
|
24 | 45 | >99 | 3.09 |
Conclusions
In summary, highly effective and recyclable cobalt(II)-based solid catalysts have been successfully synthesized and applied to the epoxidation of olefins with air as the oxidant and isobutyraldehyde as a sacrificial co-reductant. For the Co(II)@Cr-MIL-101-P2I catalyst, the nitrogen atom in the pyridine ring as a strong electron-withdrawing substituent ensured high activity, and the strong coordination interaction between the Co(II) ions and chelating groups guaranteed the excellent recycling performance. The high dispersion of Co(II) species promoted the sufficient contact between the substrate and the active sites. The catalyst showed good general applicability towards various olefins, such as cyclic olefins, tri-substituted olefins, aliphatic olefins and aromatic olefins. The Co(II) incorporation strategy provides a highly efficient, low-cost and simple approach for the synthesis of heterogeneous catalysts and can be extended to the structure design of other MOF-based nanohybrid catalysts.Acknowledgements
We thank the National High Technology Research and Development Program of China (863 Program, no. 2013AA031702) and the Co-building Special Project of Beijing Municipal Education.Notes and references
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734 CrossRef CAS PubMed.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés i Xamena and H. Garcia, Chem. – Eur. J., 2007, 13, 5106 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban and F. Taulelle, J. Am. Chem. Soc., 2008, 130, 6774 CrossRef CAS PubMed.
- C. D. Wu, A. G. Hu, L. Zhang and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS PubMed.
- Y. Qi, Y. Luan, J. Yu, X. Peng and G. Wang, Chem. – Eur. J., 2015, 21, 1589 CrossRef CAS PubMed.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Catal. Sci. Technol., 2011, 1, 856 CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275 RSC.
- Y. Luan, N. N. Zheng, Y. Qi, J. Tang and G. Wang, Catal. Sci. Technol., 2014, 4, 925 CAS.
- Z. Q. Wang and S. M. Cohen, Chem. Soc. Rev., 2009, 38, 1315 RSC.
- Y. Luan, N. N. Zheng, Y. Qi, J. Yu and G. Wang, Eur. J. Inorg. Chem., 2014, 26, 4268 CrossRef.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef PubMed.
- S. Bernt, V. Guillerm, C. Serre and N. Stock, Chem. Commun., 2011, 47, 2838 RSC.
- Y. B. Huang, S. J. Liu, Z. J. Lin, W. J. Li, X. F. Li and R. Cao, J. Catal., 2012, 292, 111 CrossRef CAS.
- S. Bernt, V. Guillerm, C. Serre and N. Stock, Chem. Commun., 2011, 47, 2838 RSC.
- X. W. Dong, T. Liu, Y. Z. Hu, X. Y. Liu and C. M. Che, Chem. Commun., 2013, 49, 7681 RSC.
- D. E. Hamilton, R. S. Drago and A. Zombeck, J. Am. Chem. Soc., 1987, 109, 374 CrossRef CAS.
- T. Punniyamurthy, B. Bhatia, M. M. Reddy, G. C. Maikap and J. Iqbal, Tetrahedron, 1997, 53, 7469 CrossRef.
- S. Jain and O. Reiser, ChemSusChem, 2008, 1, 534 CrossRef CAS PubMed.
- G. Kowalski, J. Pielichowski and M. Jasieniak, Appl. Catal., A, 2003, 247, 295 CrossRef CAS.
- C. Jin, W. B. Fan, Y. J. Jia, B. B. Fan, J. H. Ma and R. F. Li, J. Mol. Catal. A: Chem., 2006, 249, 23 CrossRef CAS.
- S. Bhunia, S. Jana, D. Saha, B. Dutta and S. Koner, Catal. Sci. Technol., 2014, 4, 1820 CAS.
- A. S. Amarasekara, A. R. Oki, I. McNeal and U. Uzoezie, Catal. Commun., 2007, 8, 1132 CrossRef CAS.
- Y. C. Lin, C. L. Kong and L. Chen, RSC Adv., 2012, 2, 6417 RSC.
- Z. G. Sun, G. Li, H. O. Liu and L. P. Liu, Appl. Catal., A, 2013, 466, 98 CrossRef CAS.
- H. Khajavi, H. A. Stil, H. P. C. E. Kuipers, J. Gascon and F. Kapteijn, ACS Catal., 2013, 3, 2617 CrossRef CAS.
- H. J. Kim, I. S. Bae, S. J. Cho, J. H. Boo, B. C. Lee, J. Heo, I. Chung and B. Hong, Nanoscale Res. Lett., 2012, 7, 1 CrossRef PubMed.
- J. Yu, Y. Luan, Y. Qi, J. Y. Hou, W. J. Dong, M. Yang and G. Wang, RSC Adv., 2014, 4, 55028 RSC.
- G. Liu, X. G. Li, J. W. Lee and B. N. Popov, Catal. Sci. Technol., 2011, 1, 207 CAS.
- V. I. Nefedov, Koord. Khim., 1978, 4, 1285 Search PubMed.
- E. F. Murphy, L. Schmid, T. Bürgi, M. Maciejewski and A. Baiker, Chem. Mater., 2001, 13, 1296 CrossRef CAS.
- Y. Yang, Y. Zhang, S. J. Shao and Q. B. Kan, Chem. Eng. J., 2011, 171, 1356 CrossRef CAS.
- A. P. Zhang, L. Q. Li, J. Li, Y. Zhang and S. Gao, Catal. Commun., 2011, 12, 1183 CrossRef CAS.
- J. K. Gao, L. L. Bai, Q. Zhang, Y. X. Li, G. Rakesh, J.-M. Lee, Y. H. Yang and Q. C. Zhang, Dalton Trans., 2014, 43, 2559 RSC.
- J. E. D. Bene, J. Am. Chem. Soc., 1979, 101, 6184 CrossRef.
- Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., 2011, 123, 7567 CrossRef.
- D. Saha, T. Maity, R. Bera and S. Koner, Polyhedron, 2013, 56, 230 CrossRef CAS.
- Z. F. Li, S. J. Wu, H. Ding, D. F. Zheng, J. Hu, X. Wang, Q. S. Huo, J. Q. Guan and Q. B. Kan, New J. Chem., 2013, 37, 1561 RSC.
- J. Tang, W. J. Dong, G. Wang, Y. Z. Yao, L. M. Cai, Y. Liu, X. Zhao, J. Q. Xu and L. Tan, RSC Adv., 2014, 4, 42977 RSC.
- K. M. Jinka, J. Sebastian and R. V. Jasra, J. Mol. Catal. A: Chem., 2007, 274, 33 CrossRef CAS.
- I. G. Bosch, X. Ribas and M. Costas, Adv. Synth. Catal., 2009, 351, 348 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR, 1H NMR, diffuse reflectance UV-vis spectra and TGA of the samples; proposed reaction mechanism; XRD, FTIR and SEM of the recovered catalyst; internal standard calibration. See DOI: 10.1039/c5cy01099c |
| This journal is © The Royal Society of Chemistry 2016 |