
Facile solid-phase synthesis of PNA–peptide conjugates using pNZ-protected PNA monomers†
Yi-Chao
Huang
a,
Cheng
Cao
b,
Xiang-Long
Tan
a,
Xiaoyu
Li
*bc and
Lei
Liu
*a
aTsinghua-Peking Center for Life Sciences, Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: lliu@mail.tsinghua.edu.cn
bKey Laboratory of Bioorganic Chemistry and Molecular Engineering of the Ministry of Education, Beijing National Laboratory of Molecular Sciences, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: xiaoyuli@pku.edu.cn
cKey Laboratory of Chemical Genomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen, 518055, China
First published on 5th September 2014
Abstract
PNA–peptide conjugates are useful molecular tools in chemical biology and biotechnology. Although several approaches have been developed to synthesize PNA–peptide conjugates, more efficient methods are still needed. In this report a new pNZ (p-nitrobenzyloxycarbonyl)/bis-Boc strategy was developed as an alternative backbone/nucleobase protecting group method. The mild deprotection conditions of pNZ group and pNZ's full orthogonality with Fmoc solid-phase synthesis enable a new dimension of synthetic flexibility and practicality to generate versatile PNA–peptide conjugates.
Peptide nucleic acid (PNA) is an artificial nucleic acid analogue of DNA and RNA in which the phosphodiester backbone is replaced by a peptide moiety (Fig. 1A).1 Due to the absence of charge repulsion in PNA/DNA and PNA/RNA duplexes, PNA shows higher binding affinity and selectivity towards its complementary DNA or RNA sequence than its nucleic acid counterpart.2 Besides, the amide-based PNA backbone offers superior chemical, thermal and enzymatic stability in the presence of nucleases as well as proteases.3
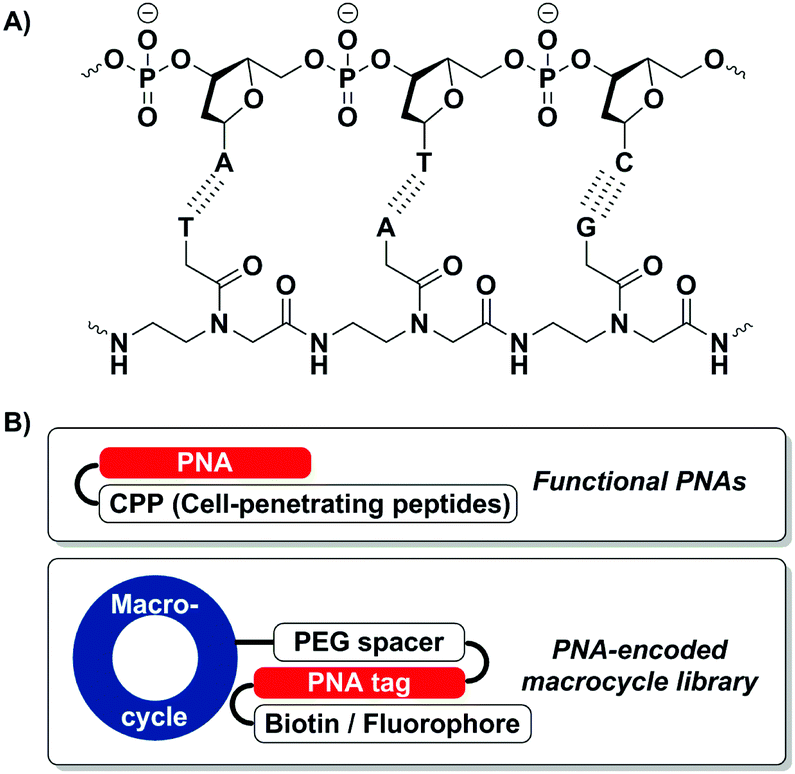 | ||
| Fig. 1 (A) Structure of a PNA–DNA duplex. (B) Applications of PNA–peptide conjugates. | ||
To expand the utility of PNA as diagnostic and therapeutic agents as well as molecular tools in chemical biology, many PNA–peptide conjugates have been explored (Fig. 1B).4 For instance, PNA–CPP (cell-permeable peptide) conjugates have been reported to enhance the cellular uptake of PNA.5 More recently, PNA-encoded chemical libraries emerged as an interesting platform for the rapid screening/selection of bioactive compounds.6 Ultimately, to realize the molecular and functional diversity of PNA–peptide conjugates, a robust and reliable synthetic method is in need to prepare these molecules with higher flexibility.
Up to now PNA oligomers have usually been synthesized in the solid phase using backbone (PG1) and side-chain nucleobase (PG2) protected monomers (PG1/PG2) (Fig. 2A). Three commercially available monomers employ the PG (protecting group) pairs of Boc/Z,7 Fmoc/Boc8 and Bts/Bhoc.9 Several drawbacks associated with these PG pairs include the harsh TFMSA deprotection conditions of the Z group, piperidine-induced acyl-shift side-reaction in Fmoc chemistry, and the malodorous thiol used to deprotect the Bts group.9 Other PG combinations have been developed which bear compatibility with the Fmoc/tBu chemistry, i.e. PG1 deprotection is orthogonal to piperidine-based Fmoc deprotection and PG2 should be cleaved off under TFA. However, these protecting strategies sometimes suffer from the limited orthogonality and the high-cost preparation of PNA monomers. For example, the starting materials for the synthesis of Dde,10f Azoc,10h and Mtt10i protected PNA backbones include Dde-OH, chloromethyl chloroformate (ClCH2OCOCl) and Mtt-Cl, which are expensive in scale-up synthesis. Other limitations involve the nucleophile lability of Dde and the weak acid lability of Mtt.10i These restrictions might limit the type of other operable chemical reactions in the solid phase.
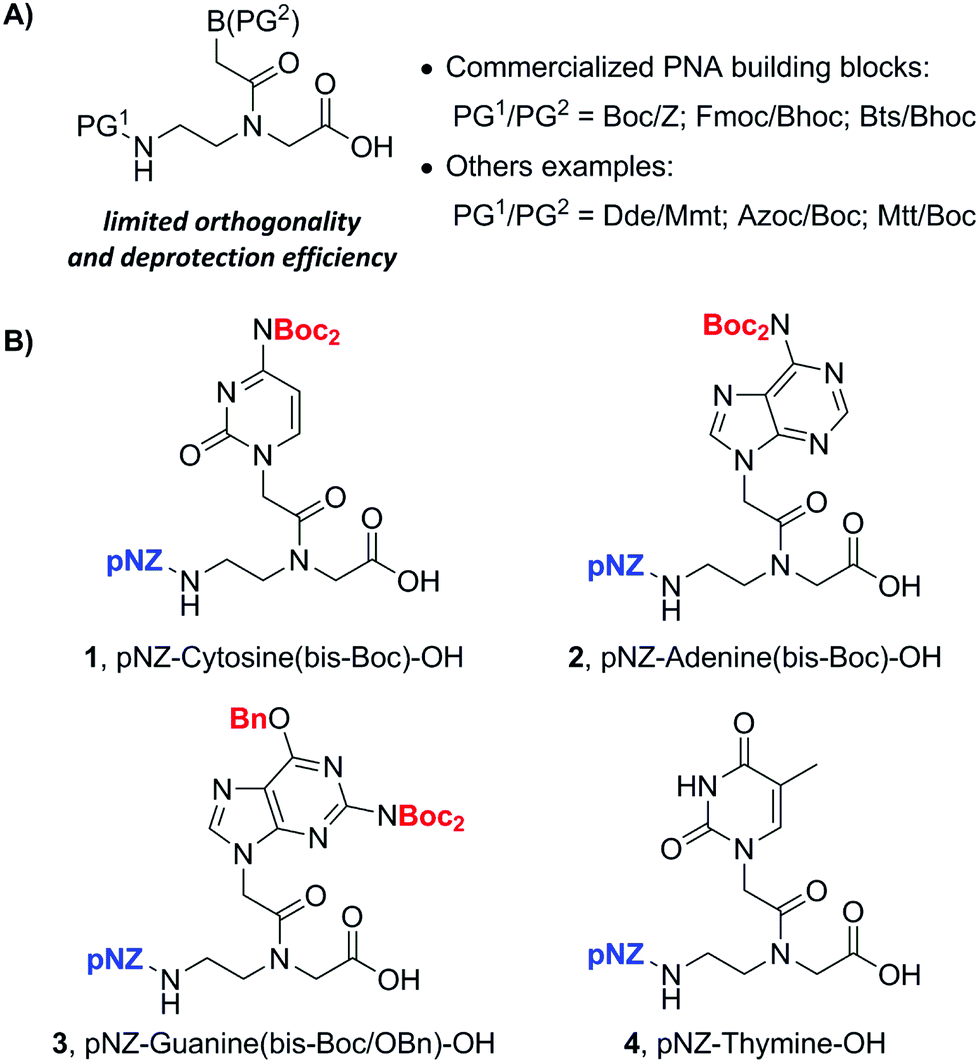 | ||
| Fig. 2 (A) Commercialized and tailor-made PNA monomers. (B) New pNZ/bis-Boc protected PNA monomers. | ||
We envisioned that an ideal PG1/PG2 removal condition for more flexible PNA–peptide conjugate synthesis should be compatible with commonly used protecting groups such as Lys(Alloc) and Cys(Mmt) in addition to Fmoc/tBu pairs. Herein, we report the design and synthesis of a novel type of pNZ (p-nitrobenzyloxycarbonyl)/bis-Boc protected PNA monomers. These monomers were produced cost-effectively from easily accessible pNZ chloride on a gram scale. This alternative building block family allows the efficient and robust synthesis of PNA oligomers. An important feature of our new strategy is that pNZ is fully compatible with common temporary side-chain protecting groups of amino acids such as Alloc and Mmt in addition to Fmoc. This advancement presents higher practical flexibility during the solid-phase synthesis of diversified PNA conjugates. As an example, a branched ester-linked PNA–peptide conjugate was synthesized using our pNZ-protected PNA monomers.
In our search for an ideal PG1/PG2 pair, we found pNZ/bis-Boc as an attractive option. pNZ is a carbamate-type protecting group which can be deblocked by many nitro-reducing methods.11 Among them SnCl2 has been used as a reliable reductant to deprotect pNZ-protected peptides in the solid phase.12 The reaction takes place under a catalytic amount of acid which can protonate the released free amino group. This acid treatment circumvents two side-reactions normally encountered in Fmoc SPPS, namely, the formation of aspartimides and DKP (diketopiperazines).12 We inferred that pNZ protection of the backbone amine of PNA monomer might also inhibit the piperidine-catalyzed formation of acyl-transfer byproducts13 as demonstrated in earlier literature. Moreover, the pNZ group was already identified to be orthogonal with Fmoc, Boc and Alloc protecting groups. This feature is important for the construction of branched or cyclic PNA–peptide conjugates. On the other hand, the bis-Boc group was previously reported by two teams in 2008 that blocked the aromatic amines of three nucleobases: adenine, cytosine and guanine.14 Hudson et al. stated that Fmoc/bis-Boc PNA monomers were more stable under weak acids than commercially available Fmoc/Bhoc PNA monomers.14a Compared to Boc, the hydrophobic bis-Boc group reduced one hydrogen-bond donor so that the monomer became more soluble in organic solvents.14a Based on the aforementioned reasons, we settled to prepare pNZ/bis-Boc protected PNA monomers.
The synthesis of the pNZ-protected backbone 5 and the bis-Boc protected nucleobases 1c, 2c, 3c, and 4a is shown in Scheme 1. Backbone 5 was synthesized from commercially available pNZ chloride in three steps (Scheme 1E). bis-Boc protected nucleobase derivatives were obtained through aromatic amine protection and alkylation with methyl bromoacetate. Note that O6-benzylguanine was used as a starting material in the preparation of 3c. This modification proved instrumental to improve the organic solubility of guanine derivatives presumably because of the removal of an aggregation-inducing amide N–H bond.14a With the nucleobases 1c, 2c, 3c, and 4a in hand (Scheme 1A–D), we carried out the amidation in the presence of DIC and HOAt and subsequent mild hydrolysis at 0 °C to afford pNZ/bis-Boc protected PNA monomers 1–4 on a gram scale (>96% HPLC purity) ready for the direct solid-phase synthesis (Fig. 3A). This modular synthetic design involves a universal backbone and independent side-chain nucleobases, which might be generally applicable to the synthesis of PNA monomers containing non-canonical nucleobases.
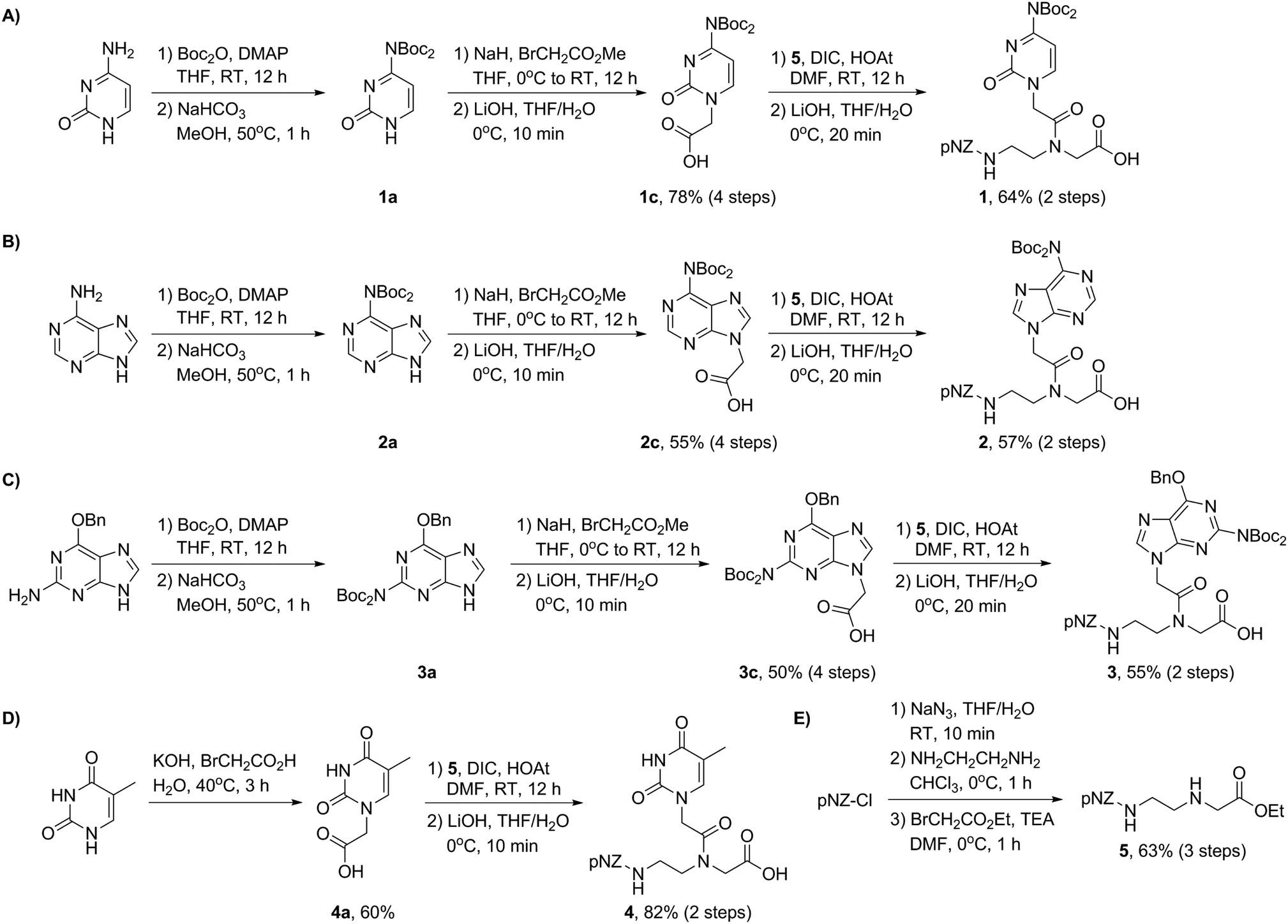 | ||
| Scheme 1 (A) Synthesis of the pNZ/bis-Boc protected cytosine monomer 1. (B) Synthesis of the pNZ/bis-Boc protected adenine monomer 2. (C) Synthesis of the pNZ/bis-Boc protected guanine monomer 3. (D) Synthesis of the pNZ/bis-Boc protected thymine monomer 4. (E) Synthesis of the pNZ backbone 5. | ||
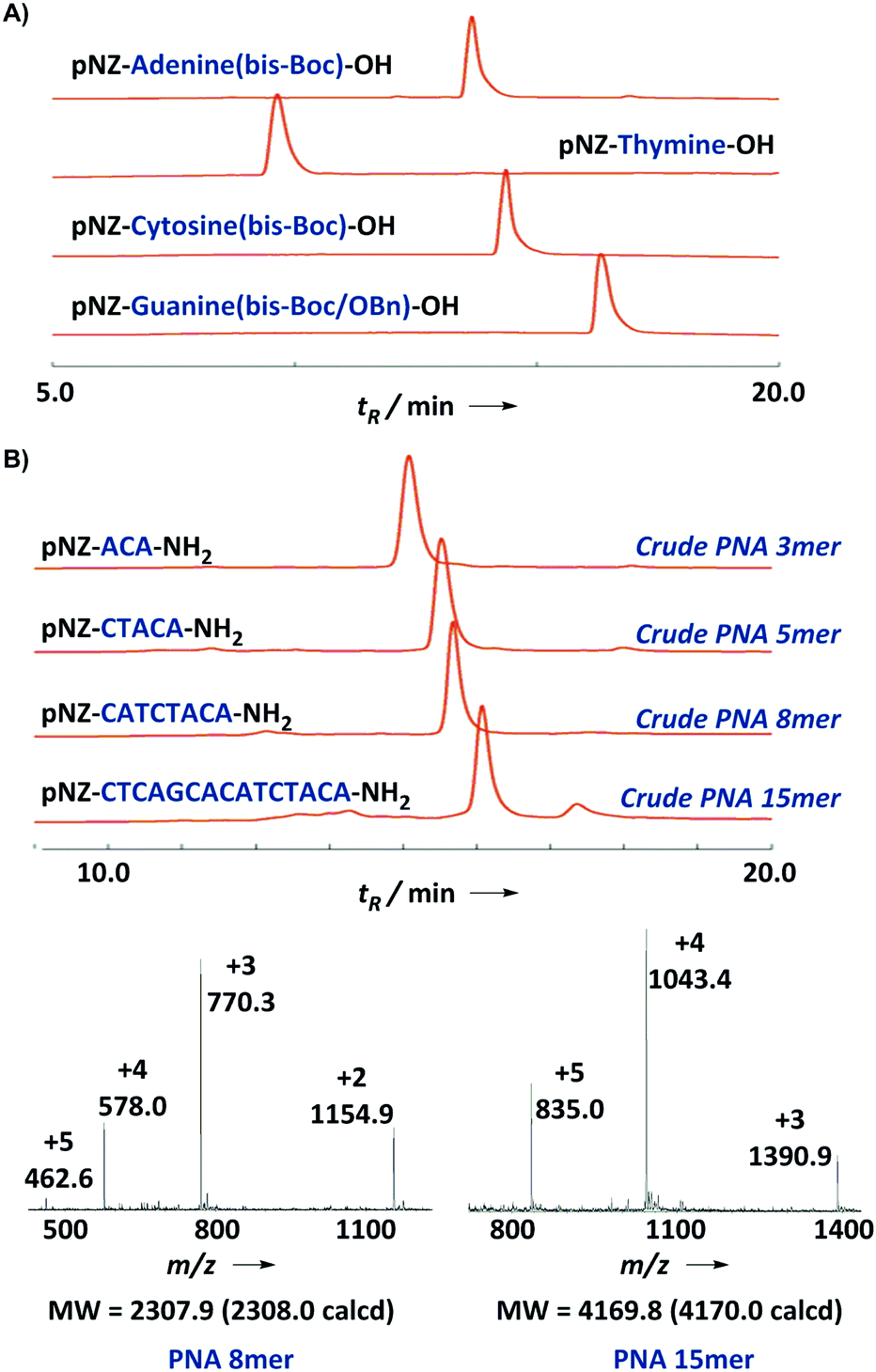 | ||
| Fig. 3 (A) LC characterization of four PNA building blocks. (B) HPLC trace (214 nm) and ESI-MS profile of a crude 15-mer model PNA sequence. | ||
Next we turned to the solid-phase oligomerization of our new PNA monomers (Scheme 2). The deprotected Rink amide resin was treated with 2.5 eq. PNA monomer, 2.5 eq. PyAOP, 5 eq. NMM in NMP (final monomer concentration 0.1 M) for 15 min twice. In the preliminary tests, we found that PyBOP/DMF could be used interchangeably with PyAOP/NMP for PNAs shorter than 8-mer. However, we preferred PyAOP/NMP in the synthesis of longer PNA oligomers. The coupling process was repeated and the resin was then capped with the Ac2O–lutidine mixture for 3 min to block unreacted amines. After that pNZ deprotection was conducted at 60 °C using the SnCl2 reducing solution12 slightly modified from the literature reported recipe. The resulting resin was cooled down to room temperature and ready for the next-round coupling. In order to assess the efficiency and efficacy of this protocol, we chose a 15-mer PNA9 previously studied in the literature as our model sequence. We monitored the on-resin coupling through analytical TFA cleavage and HPLC/ESI-MS characterization (Fig. 3B). Gratifyingly, this protocol worked quite well on the model sequence. The crude PNA 15-mer was obtained in high yield (90%) and good purity (82%).
 | ||
| Scheme 2 Typical coupling and deprotection procedures during PNA assembly in the solid phase. | ||
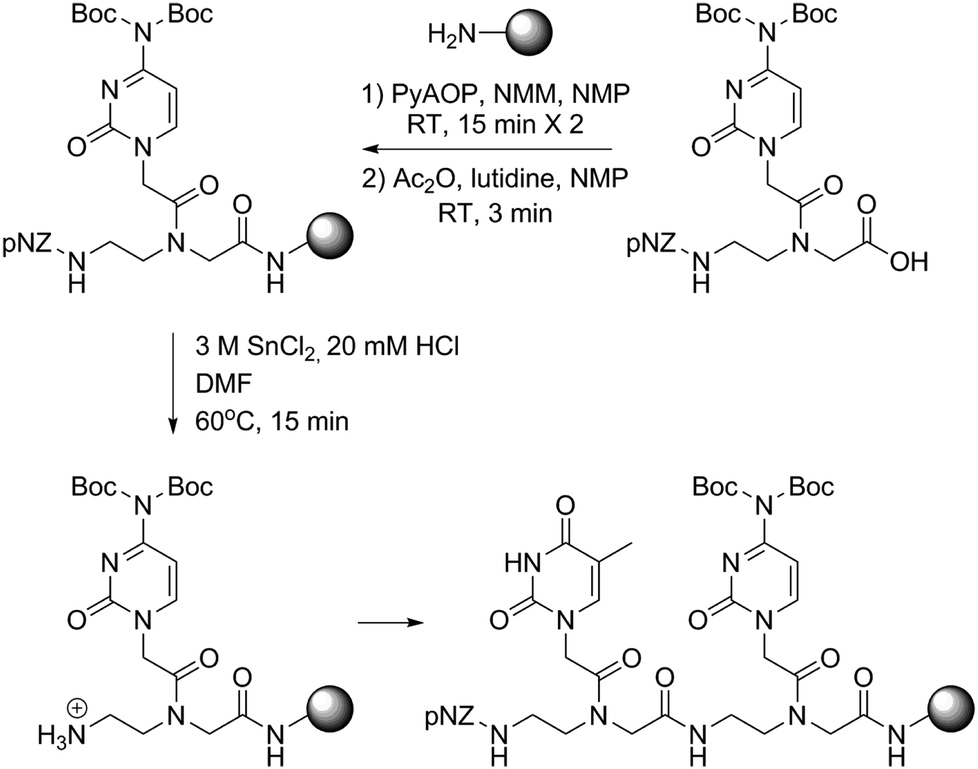
To further test the practicability and robustness of the new PNA monomers, we arbitrarily designed some extreme sequences. Homopentamers of adenine, thymine, cytosine and guanine were prepared using the standard protocol. All of the homopolymers gave satisfactory yields and purities according to analytical HPLC traces except polyguanine. The broad and asymmetrical peaks displayed on HPLC might be attributable to the poor solubility and aggregation-prone character of polyguanine. We noticed that this abnormal elution behavior was also observed in the case of certain hydrophobic peptides as reported earlier.15 Besides, a minor peak corresponding to the single ‘G’ deletion byproduct was found that eluted before the main peak. These data agreed with the perspective that the polyguanine sequence was generally considered difficult for both DNA and PNA solid-phase chemistry. In our next test, we chose a classical 17-mer PNA13 which contains all sixteen coupling combinations of the four PNA building blocks. The purine/pyrimidine ratio of the sequence was set to shift from a low to high level as PNA extends. These environmental changes thus made this sequence an ideal touchstone of our pNZ monomers and their coupling conditions. To our delight, the PNA 17-mer was successfully synthesized in high crude yield (82%) and purity (75%) (Fig. 4).
 | ||
| Fig. 4 HPLC trace (214 nm) of crude homo pentamer of adenine, thymine, cytosine and guanine; HPLC trace (214 nm) and ESI-MS profile of a crude 17-mer PNA sequence. | ||
Finally, we prepared a model branched PNA–peptide conjugate through alternate pNZ-based PNA coupling and Fmoc-based amino acid coupling (Fig. 5A). Starting from the Fmoc-Ser(TBDMS)-OH loaded Rink amide resin, we coupled a PNA oligomer (TTT) along the backbone and a peptide oligomer (KPK) along the side-chain. Amino acid couplings were carried out using PyBOP/DIEA in NMP for 20 min twice and PNA couplings were performed using PyAOP/NMM in NMP for 15 min twice. No premature Fmoc or pNZ cleavage was observed during the chain assembly according to a qualitative chloranil test. Fmoc-β-alanine (4 eq.) was coupled using 4 eq. DIC/0.1 eq. DMAP in CH2Cl2 overnight to form an ester bond on the side-chain of serine.16 This special amino acid was chosen to circumvent the notorious racemization and DKP issues normally encountered during the solid-phase ester formation. The ester-bond linked branched PNA–peptide conjugates might have potential to serve as a cleavable bifunctional molecules17 applied to intracellular PNA delivery. The final product was characterized by the use of HPLC and ESI-MS as shown in Fig. 5B, demonstrating the practicality of the new method for the synthesis of PNA–peptide conjugates.
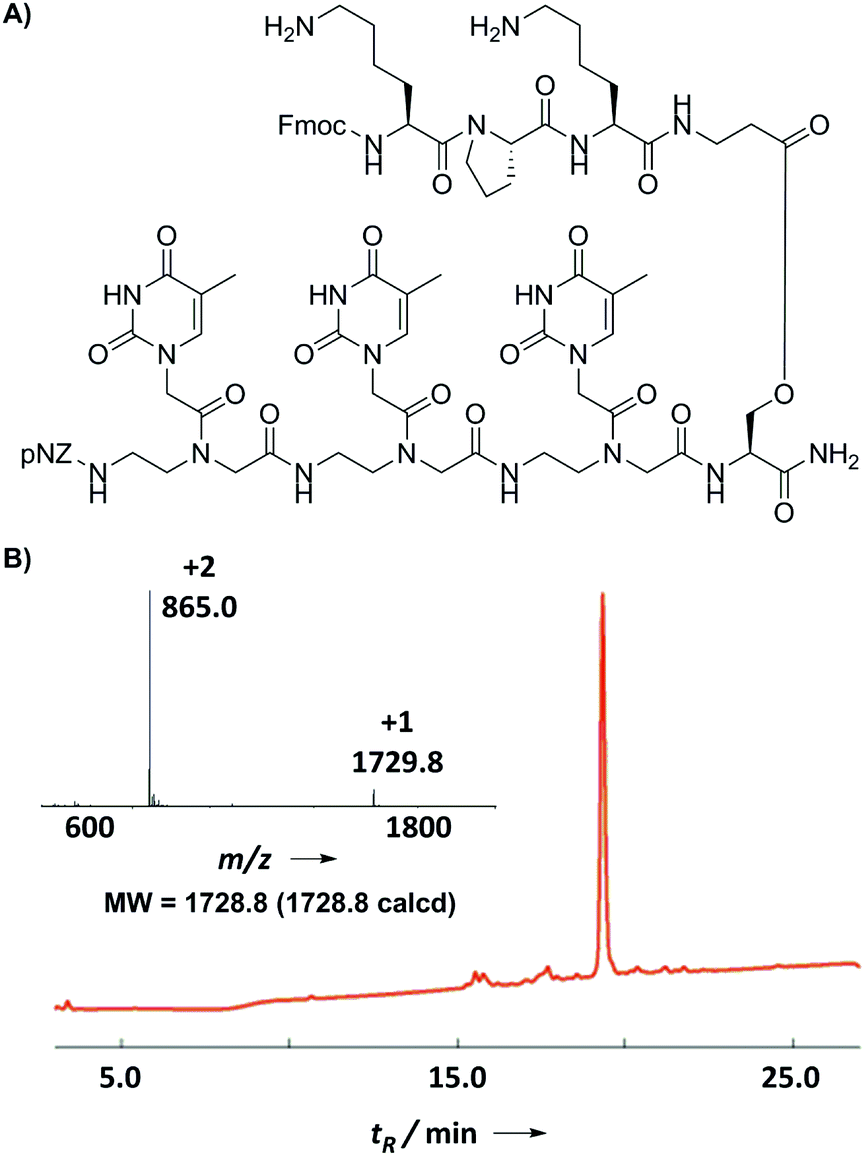 | ||
| Fig. 5 (A) Structure of the model branched PNA–peptide conjugate. (B) HPLC trace (214 nm) and ESI-MS profile of the crude PNA–peptide conjugate. | ||
To summarize, we have developed a new alternative pNZ/bis-Boc protecting group strategy for the solid-phase synthesis of PNA oligomers. The orthogonality of pNZ with Fmoc, Boc and Alloc protecting groups offers a new layer of flexibility in the synthesis of PNA–peptide conjugates. The feasibility of this strategy was demonstrated in the preparation of an ester-linked branched PNA–peptide conjugate. We considered that pNZ protected PNA monomers might find application in the generation of diversified PNA conjugates with higher-order complexity. These molecular toolkits should be useful to establish functional PNA probes and PNA-encoded chemical libraries.
Acknowledgements
This study was supported by the National Basic Research Program of China (973 program, no. 2013CB932800, 2011CB809100), NSFC grants (91313301, 21272016), and the Specialized Research Fund for the Doctoral Program of Higher Education (grant no. 20120002130004).Notes and references
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CAS
.
- M. Egholm, O. Buchardt, L. Christensen, C. Behrens, S. M. Freier, D. A. Driver, R. H. Berg, S. K. Kim, B. Nordern and P. E. Nielsen, Nature, 1993, 365, 566 CrossRef CAS PubMed
- V. Demidov, V. N. Potaman, M. D. Frank-Kamenetskii, O. Buchardt and P. E. Nielsen, Biochem. Pharmacol., 1994, 48, 1310 CrossRef CAS
- M. C. de Koning, G. A. van der Marel and M. Overhand, Curr. Opin. Chem. Biol., 2003, 7, 734 CrossRef CAS PubMed
-
(a) K. Braun, P. Peschke, R. Pipkom, S. Lampel, M. Wachsmuth, W. Waldeck, E. Friedrich and J. Debus, J. Mol. Biol., 2002, 318, 237 CrossRef CAS
-
(a) N. Winssinger, S. Ficarro, P. G. Schultz and J. L. Harris, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11139 CrossRef CAS PubMed
-
O. Buchardt, M. Egholm, P. E. Nielsen and R. H. Berg, PCT Int. ApplWO 92/20702, 1992 Search PubMed
-
J. M. Coull, M. Egholm, R. P. Hodge, M. Ismail and S. B. Rajur, PCT Int. ApplWO 96/40685, 1996 Search PubMed
- H. Lee, J. H. Jeon, J. C. Lim, H. Choi, Y. Yoon and S. K. Kim, Org. Lett., 2007, 9, 3291 CrossRef CAS PubMed
-
(a) D. W. Will, G. Breipohl, D. Langner, J. Knolle and E. Uhlmann, Tetrahedron, 1995, 51, 12069 CrossRef CAS
-
(a) F. H. Carpenter and D. T. Gish, J. Am. Chem. Soc., 1952, 74, 3818 CrossRef CAS
- A. Isidro-Llobet, J. Guasch-Camell, M. Alvarez and F. Albericio, Eur. J. Org. Chem., 2005, 3031 CrossRef CAS
- L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M. Egholm, O. Buchardt, P. E. Nielsen, J. Coull and R. H. Berg, J. Pept. Sci., 1995, 1, 175 CrossRef CAS PubMed
-
(a) F. Wojciechowski and R. H. E. Hudson, J. Org. Chem., 2008, 73, 3807 CrossRef CAS PubMed
-
(a) E. C. B. Johnson and S. B. H. Kent, Tetrahedron Lett., 2007, 48, 1795 CrossRef CAS PubMed
- J.-S. Zheng, H.-N. Chang, J. Shi and L. Liu, Sci. China: Chem., 2012, 55, 64 CrossRef CAS PubMed
- G. Leriche, L. Chisholm and A. Wagner, Bioorg. Med. Chem., 2012, 20, 571 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, 1H NMR, 13C NMR, ESI-MS and copies of NMR spectra for all products. See DOI: 10.1039/c4qo00217b |
| This journal is © the Partner Organisations 2014 |