Synthesis of organic halides via palladium(0) catalysis
Chen
Chen
and
Xiaofeng
Tong
*
Shanghai Key Laboratory of Functional Materials Chemistry, East China University of Science and Technology, Shanghai, 200237, China. E-mail: tongxf@ecust.edu.cn; Fax: +(86)-21-6425-3881; Tel: +(86)-21-6425-3881
First published on 14th March 2014
Abstract
This critical review provides a brief overview of recent advances in the palladium(0)-catalysed synthesis of organic halides. It is well established that very sterically hindered phosphine ligands are capable of promoting the reductive elimination of carbon halides from C–Pd(II)–X species (X = F, Cl, Br, I), which has proved to be a more common process than was previously assumed and exhibits great potential for organic halides synthesis, including aryl fluorides.
Introduction
Organic halides represent one of the most widely used starting materials in transition-metal catalysis. This is mainly due to their facile and viable oxidative addition to low valent metal centers. In this context, numerous theoretical and experimental studies have been conducted to disclose the details of oxidative addition of organic halides to Pd(0)-complexes, especially the ligand effects and the origins of different reactivities of organic halides in the formation of C–Pd(II)–X species.1 Since the oxidative addition of organic halides to Pd(0)-centers is usually thermodynamically favorable, the reverse process, the reductive elimination of C–Pd(II)–X, has been regarded as less likely to form organic halides. However, the principle of microscopic reversibility tells us that this type of reductive elimination still remains possible. This raises the fundamental question of how to realize this highly endothermic process.Despite the apparent difficulty in the reductive elimination of C–Pd(II)–X, recent developments have clearly demonstrated that this process can be a significantly more common reaction than previously assumed and has exhibited some advantages toward organic halide synthesis. The purpose of this critical article is to provide a brief overview of these developments. Besides, this article will also include some Pd(0)-catalyzed halide-atom-transfer cyclizations. The processes involving Pd(IV) complexes and other metals are not covered.2
Stoichiometric studies on C–halide reductive elimination
Although early examples of carbon halide reductive elimination started to appear in the literature about 40 years ago,3 the variant involving Pd(II) was firstly observed from the isolable Pd(II)-complex 1 by Roy and Hartwig in 2001 (Scheme 1).4 The authors disclosed that only very sterically hindered phosphine ligands were able to promote the reductive elimination of 1, leading to the formation of the corresponding aryl halides. The details about ligand screening are summarized in Scheme 1. Furthermore, a large excess of ligand was also found to be required and the highest yields were obtained when approximately 15 equiv. of P(tBu)3 were used. Interestingly, the reactivity of the halide followed the trend I > Br > Cl, strongly indicating the importance of the kinetic factors in carbon halide reductive elimination.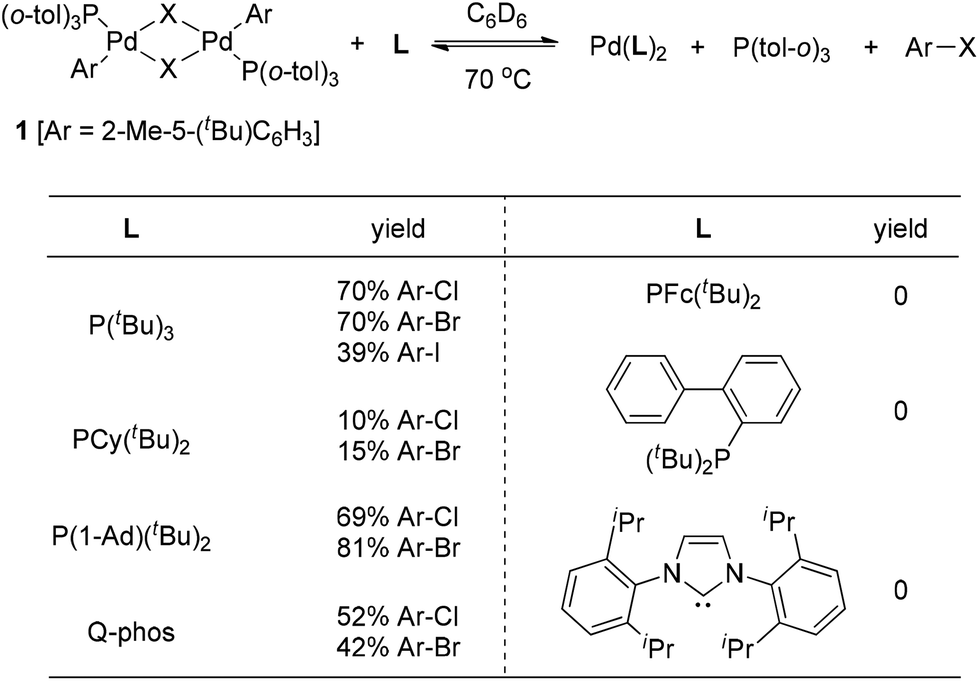 | ||
| Scheme 1 Reductive elimination of complex 1 and ligand effects. | ||
These results clearly demonstrated that very sterically hindered phosphane ligands can alter the thermodynamics so severely that aryl halide reductive elimination is favored thermodynamically. We believe that sterically hindered phosphane ligands not only create a crowded coordination environment around the Pd(II) center to facilitate the aryl halide reductive elimination, but also make the resulting Pd(0) species so crowded as to slow down the reversed aryl halide oxidative addition.
In contrast to the well-known heavier halide ligated complexes (L)nRPdX (X = Cl, Br, I), the fluoride-ligated analogues are rarely documented. The isolable and reliably characterized complex (PPh3)2(Ph)PdF was firstly reported by Grushin and co-authors in 1997.5 Treatment of (PPh3)2(Ph)PdI with AgF readily forms (PPh3)2(Ph)PdF, which has been a standard method to synthesize this class of Pd(II)–F complexes (Scheme 2).
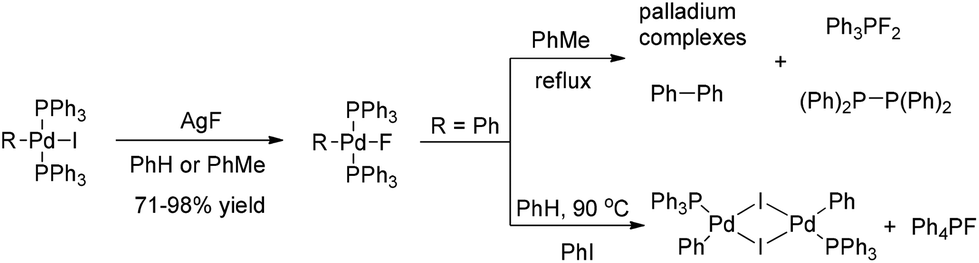 | ||
| Scheme 2 Formation and transformations of (PPh3)2RPdF. | ||
Although considerable efforts have been made by chemists, there are still few successful reports of the formation of ArF via the reductive elimination of (PPh3)2(Ar)PdF.6 For example, the thermal decomposition of (PPh3)2(Ph)PdF in refluxing toluene often results in the formation a P–F bond among other compounds such as Ph3PF2 and [(Ph)2P]2 instead of the desired reductive elimination product PhF. Even in the presence of additional PhI, (PPh3)2(Ph)PdF was found to be converted into [(PPh3)PhPdI]2 dimer and Ph4PF (Scheme 2). These results clearly demonstrate that forming an Ar–F bond via the carbon fluoride reductive elimination is extremely challenging.
In 2007, Yandulov and Tran used the complex [Pd(4-NO2-C6H4)L(μ-F)]2 [L = P(o-Tolyl)3 or P(tBu)3] to investigate the Ar–F reductive elimination.7 Again, the thermal decomposition of these two complexes indeed formed somewhat complicated mixtures of products rather than the desired ArF, even in the case of bulky ligand P(tBu)3. However, treatment of [Pd(4-NO2-C6H4)(PtBu3)(μ-F)]2 with bulkier ligand P(2-Trip-C6H4)(tBu)2 indeed led to the formation of ArF albeit in very low yield. Although the use of the strong activating nitro group might cast doubts on whether the resulting ArF arose from the aryl fluoride reductive elimination,8 this finding may provide a first step towards addressing this challenging task.
It is well known that very sterically hindered phosphane ligands are capable of producing monomeric (L)RPdX with T-shaped geometry at the palladium center, which has been proved to be essential for some Pd-catalyzed transformations.9 Buchwald and co-workers recently disclosed that this capability of bulkier phosphine ligands is also crucial for aryl fluoride reductive elimination. Indeed, they found that monomeric complex 3 readily underwent reductive elimination to form ArF upon heating in toluene (Scheme 3).10 The yields in these reactions increased significantly when the thermolysis was conducted in the presence of additional aryl bromides. More importantly, in the latter cases, the oxidative addition complexes 2 were detected as the only phosphorus-containing products, strongly suggesting that LPd(0) was formed along with the ArF product.
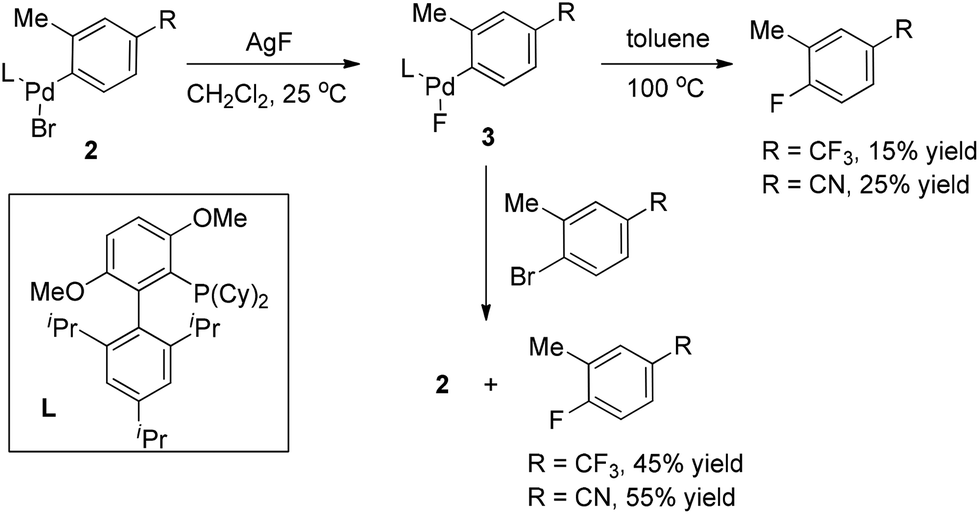 | ||
| Scheme 3 The reductive elimination of carbon fluoride. | ||
Interestingly, the [(Ph3P)2(PhCO)PdF] complex, which was formed upon carbonylation of [(Ph3P)2(Ph)PdF] with CO, rapidly underwent reductive elimination of benzoyl fluoride under CO atmosphere at room temperature (Scheme 4).5 This observation may be attributed to the formation of a σ-benzoyl-palladium bond which would create an electron-deficient metal center. In contrast, heavier halide analogues [(Ph3P)2Pd(COPh)X] (X = I, Br, Cl) cannot undergo a similar reductive elimination even at elevated temperatures (Scheme 4).11
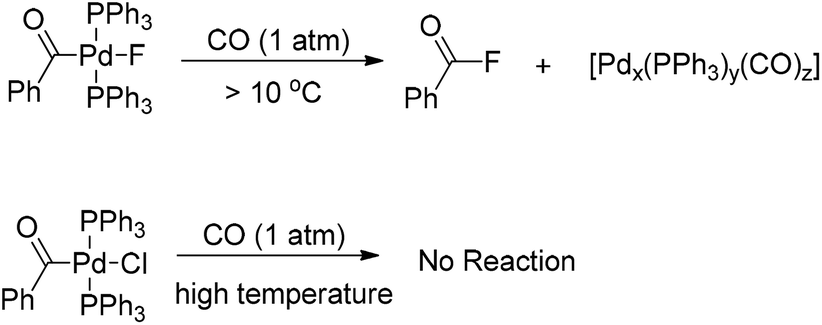 | ||
| Scheme 4 Reductive elimination of [(Ph3P)2Pd(COPh)X] (X = F or Cl). | ||
However, Quesnel and Arndtsen very recently discovered that complex [(PtBu3)(PhCO)PdCl] readily underwent reductive elimination upon treatment with CO, resulting in the nearly quantitative formation of benzoyl chloride, along with the phenyl iodide oxidative addition product [(PtBu3)(PhCO)PdI] (Scheme 5).12 The ability of P(tBu)3 to promote this transformation may arise from its size and donor ability, which both create a coordination site on the palladium atom for the π-acidic CO ligand. Accordingly, the association with a CO ligand may result in an electron-deficient palladium center. Thus, the dual steric and electronic effects of P(tBu)3 and CO facilitate the reductive elimination of benzoyl chloride. Without CO atmosphere, no reaction was observed even at elevated temperature and in the presence of excess phenyl iodide to trap any resultant (tBu)3PPd(0) species, suggesting that the reductive elimination of acid halide is neither kinetically viable nor thermodynamically favored.
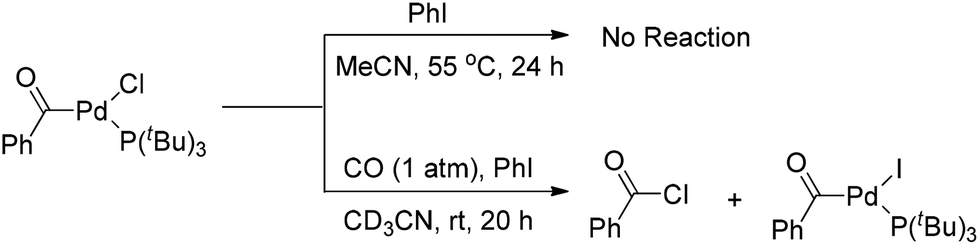 | ||
| Scheme 5 Reductive elimination of [(PtBu3)(PhCO)PdCl]. | ||
Palladium(0)-catalysed carbon halide reductive elimination
The investigations based on the well-defined LnRPdX (n = 1, 2) complexes have shed light on the carbon halide reductive elimination. Most importantly, research on the above-mentioned complexes has established that very sterically hindered phosphane ligands are the key point for this thermodynamically disfavored process. These fundamental advances have paved the way for the emergence of some novel Pd(0)-catalyzed transformations involving a carbon halide reductive elimination step in the catalytic cycle, which provide an alternative for the efficient synthesis of organic halides.Synthesis of aryl halides and acid chlorides
On the basis of their observation of the reductive elimination of complex 3 to form the ArF product, Buchwald and co-workers elegantly developed a Pd(0)-catalyzed process for direct conversion of aryl triflates into the corresponding aryl fluorides simply using CsF as the fluoride source (Fig. 1).10 Ligand tBuBrettphos proved to be most efficient for this transformation. The authors believe that the use of the sterically demanding, electron-rich biaryl monophosphine tBuBrettPhos not only promotes the reductive elimination because of its large size but also prevents the formation of dimeric [LPdAr(F)]2 complexes. In consistency with the findings described in the section on the stoichiometric version of the transformation, both of these factors appear to be critical for the reductive elimination of C–Pd(II)–X. As shown in Fig. 1, this transformation features a wide range of substrates. Importantly, some heterocyclic substrates, including flavones, indoles as well as quinolines, can be fluorinated. Fluorinated fluorescein and quinine can be synthesized using this system, demonstrating the potential of this method for the preparation of pharmaceutically relevant F-containing compounds.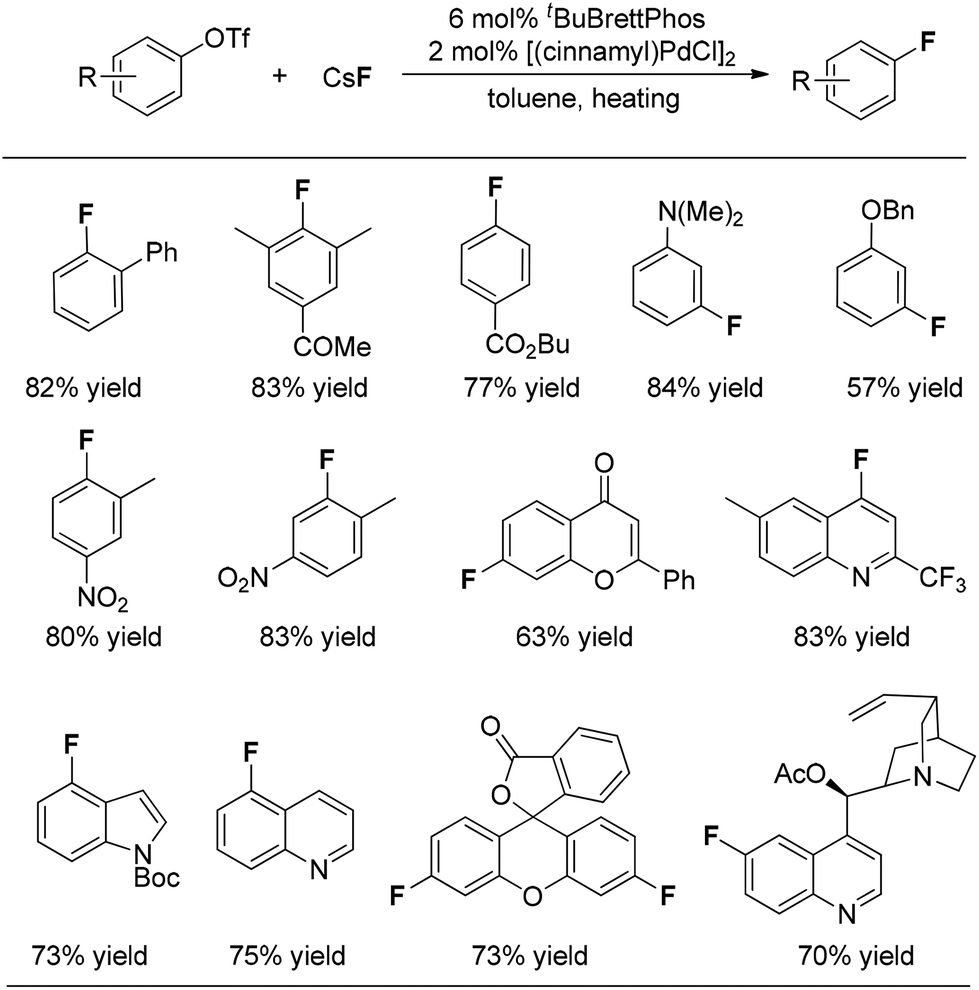 | ||
| Fig. 1 Pd(0)-catalyzed conversion of aryl triflates into the corresponding aryl fluorides. | ||
Interestingly, regioisomeric products were observed in a few cases. Although the authors did not provide a precise mechanism to explain the formation of regioisomers, they found that switching the reaction solvent from toluene to cyclohexane completely suppressed the formation of the undesired isomer. As can be seen in Fig. 2, the use of cyclohexane afforded almost exclusively the desired product.
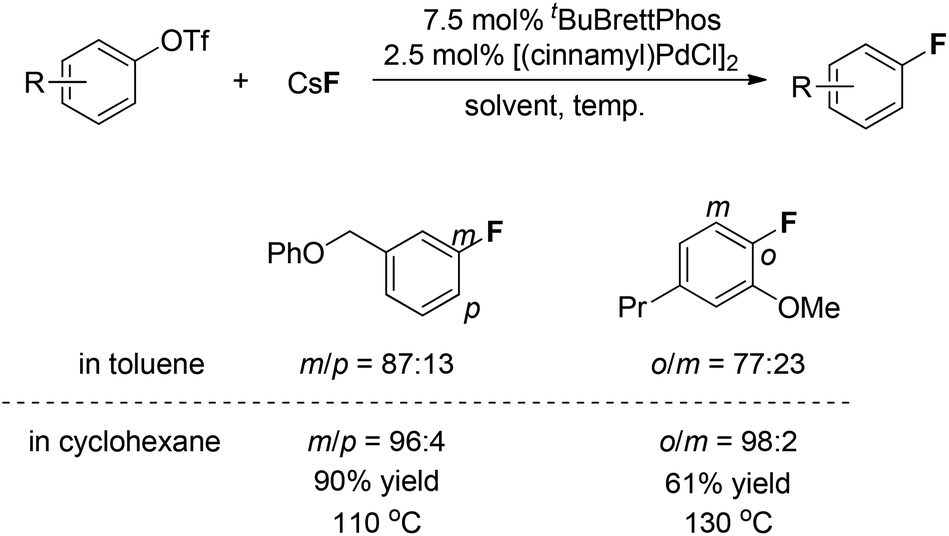 | ||
| Fig. 2 Solvent effects on the regioselectivity in Pd(0)-catalyzed fluorinations. | ||
In 2013, the Pd(0)-catalyzed fluorination of aryl triflates was significantly improved by the same group.13 Buchwald and co-workers found that complex (AdBrettPhos-Pd)2(COD) is an efficient precatalyst for the fluorination of electron-rich substrates, as well as heterocycle-containing aryl triflates, providing the corresponding aryl fluorides in synthetically useful yields and contaminated only with easily separated side products (Fig. 3). Although the precise structure of (AdBrettPhos-Pd)2(COD) has not been well established, the authors believe that this precatalyst could provide the active catalytic species without generation of reactive byproducts such as Cl−, dba, carbazole or HF which accompany the activation of other Pd sources. This represents a significant improvement for this reaction.
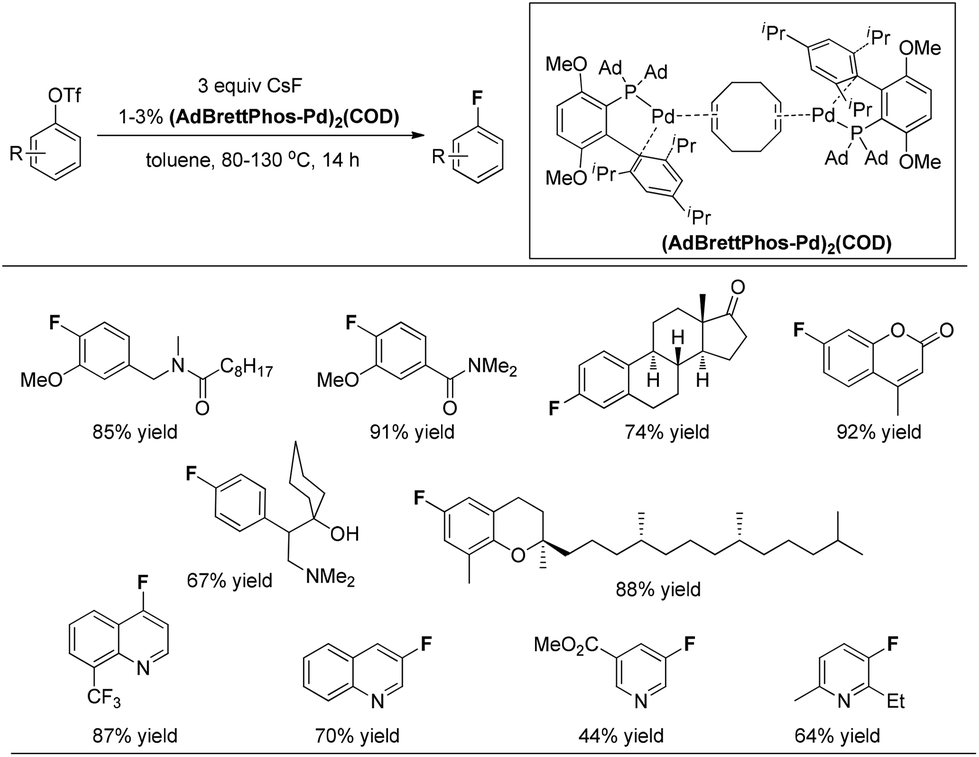 | ||
| Fig. 3 An improved catalyst system for the Pd(0)-catalyzed fluorination. | ||
Later on, Buchwald and co-workers successfully applied these reaction conditions to the conversion of aryl and vinyl triflates to bromides and chlorides.14 As highlighted in Fig. 4 (conditions A), a wide range of organic bromides and chlorides can be synthesized with the use of KBr or KCl as the halide source. PEG 3400 as the phase transfer catalyst was required to increase the solubility of KX (X = Br, Cl) while the combination of butanone and (iBu)3Al was utilised to generate the dialkylaluminum alkoxide in situ to suppress the formation of undesired by-products (Fig. 4).
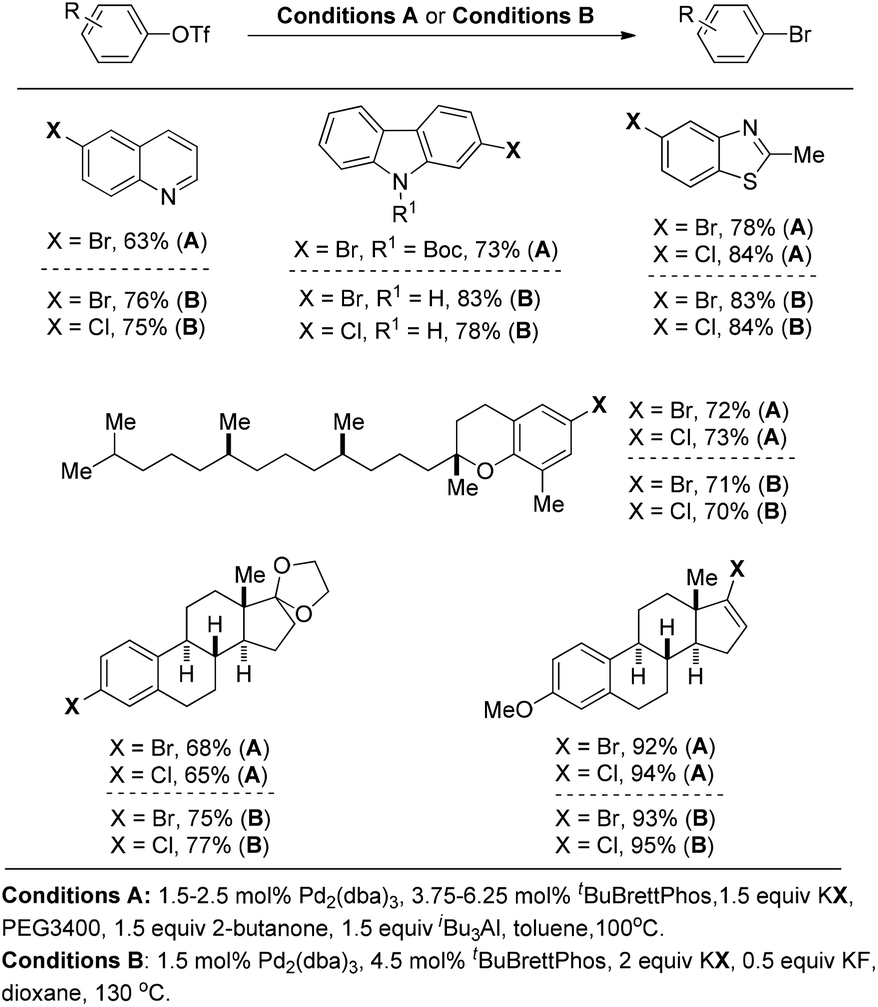 | ||
| Fig. 4 Selected examples of the Pd(0)-catalyzed conversion of aryl triflates into the corresponding aryl halides. | ||
To address the issue of the complexity of the reaction conditions, the authors developed more general and practical conditions with only KF as the additive, which greatly improved the reaction efficiency (Fig. 4, conditions B).15 Although in these cases the aryl halide reductive elimination is an endoergonic step, the overall reaction proceeds downhill.
Very recently, Arndtsen and co-workers developed the first example of a Pd(0)-catalyzed synthesis of acid chlorides from aryl iodides and CO (Scheme 6).12
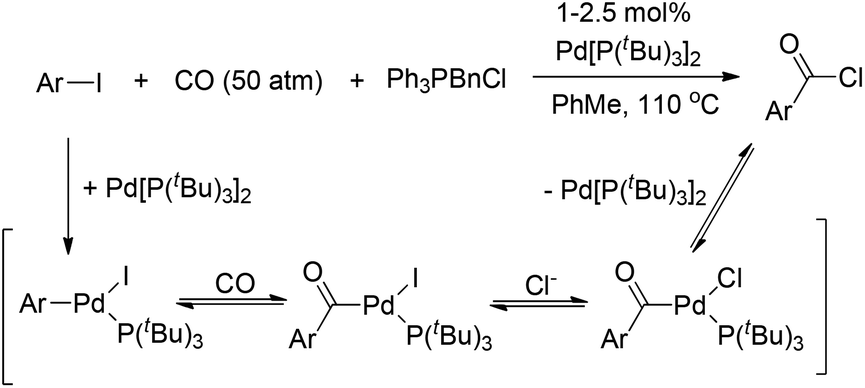 | ||
| Scheme 6 Palladium-catalyzed carbonylation approach to acid chloride synthesis. | ||
For these reaction conditions, the following points should be emphasized: (i) high reaction temperature. Due to the much higher reactivity of the product (acid chloride) than the starting material (aryl iodide), the oxidative addition of acid chlorides to Pd(0) was presumed to slow down the reaction. Thus, high reaction temperatures were required to overcome this kinetic product inhibition, resulting in a clean catalytic route to generate benzoyl chloride from phenyl iodide and CO. (ii) High CO pressure. Moderate pressure is viable and elevated CO pressure allows the isolation of these acid chloride products via simple filtration of the insoluble halide salts and precipitation of the palladium catalyst. These two issues are very different from the case of stoichiometric experiments relevant to the isolated complex [(PtBu3)(PhCO)PdCl].
When the resulting acid chloride was trapped by the amine, the so-called aminocarbonylation of the aryl iodide was smoothly realized at room temperature under CO atmosphere. As illustrated in Fig. 5, a wide range of aryl iodides and amines are suitable substrates under these reaction conditions, even including nitrogen-containing heterocycles which normally exhibit too poor nucleophilicity and coordinating ability to participate in palladium-catalyzed carbonylations.
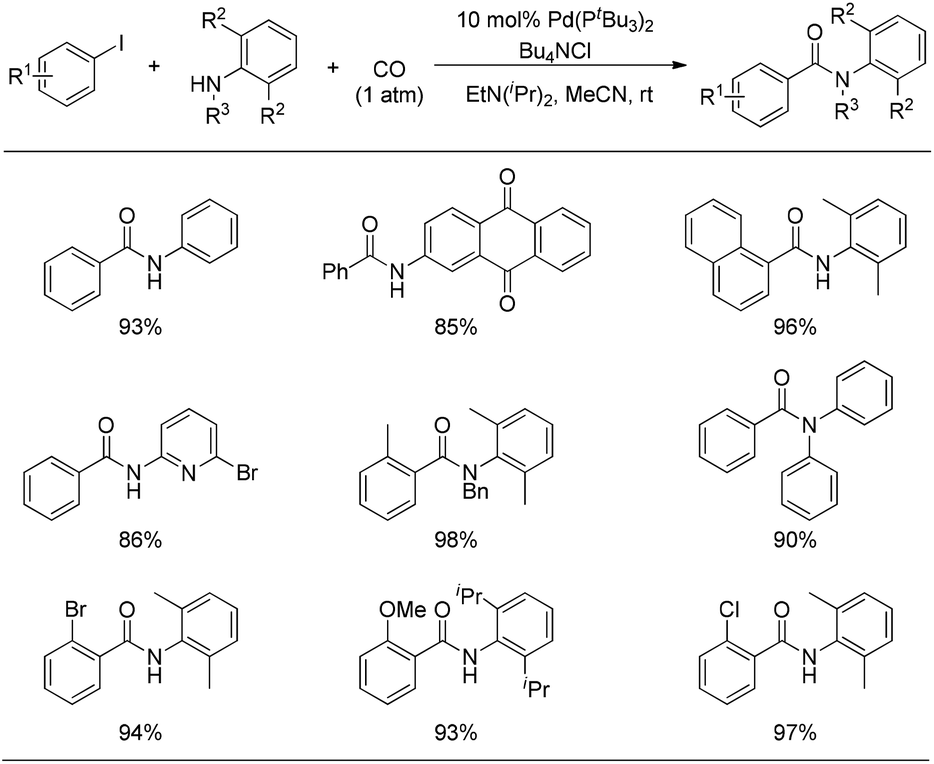 | ||
| Fig. 5 Ambient temperature and pressure palladium-catalyzed aminocarbonylations. | ||
Synthesis of alkyl iodides
In sharp contrast to the isolable and well-characterized Ln(aryl)PdX complex, Ln(alkyl)Pd(II)X complexes are less documented. The Ln(alkyl)PdX complex is believed to be highly reactive owing to the absence of stabilizing electronic interaction with metal d-orbitals.16 The fast and thermodynamically favored β-hydride elimination is the predominant process for the (alkyl)PdX species. Therefore, in the research toward the synthesis of alkyl halides via reductive elimination of aryl halides, care should be taken in the formation of a Ln(alkyl)PdX species without a β-hydride in the alkyl group.In 2011, Newman and Lautens employed Pd(Qphos)2 as the catalyst to realize carboiodination of alkenes 4.17 As illustrated in Scheme 7, insertion of an alkene into the Ar–Pd(II) bond of intermediate A, which was readily generated via oxidative addition of Pd(0) to an aryl iodide, was cleverly designed to form (alkyl)Pd(II)–I intermediate B in which the β-hydride was blocked by the R group. The authors found that only bulky ligands such as QPhos, P(tBu)3 as well as PhP(tBu)2 were able to promote the reductive elimination of intermediate B, giving the alkyl iodide in good conversion, while less bulky ligands PCy3 and P(o-tol)3 were totally inert. Again, these results clearly demonstrate the importance of very sterically hindered phosphane ligands for the reductive elimination of C–Pd(II)–X.
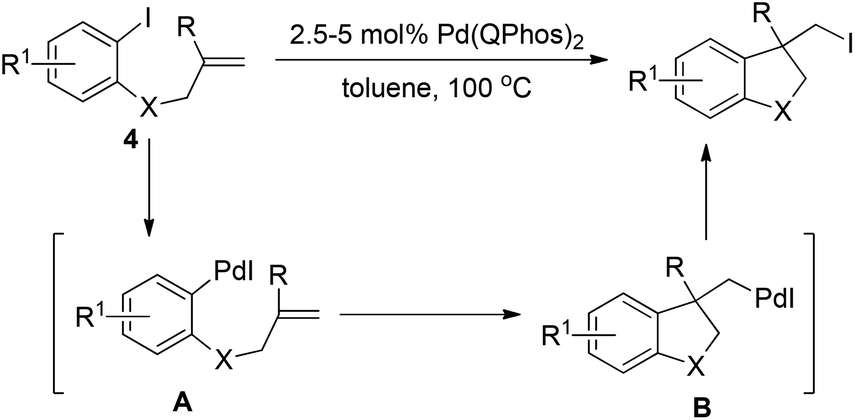 | ||
| Scheme 7 Palladium-catalyzed intramolecular carboiodination of alkene-tethered aryl iodides. | ||
Lautens and co-workers subsequently developed a novel “zipper”-type transformation of polyunsaturated aryl iodides 5, under the same conditions (Scheme 8), in which the reductive elimination of carbon iodide was firstly used as an efficient terminating step in a domino process.18
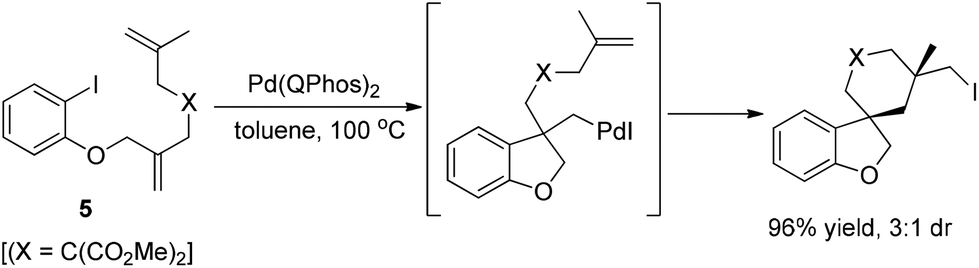 | ||
| Scheme 8 Domino synthesis of polycyclic alkyl iodide. | ||
Lautens and co-workers then used the reductive elimination of carbon iodide as the terminating step to realize the domino reaction of 2-iodo-α-methyl styrene 6 with alkynes (Scheme 9).19 This methodology allows access to polysubstituted indenes through the formation of two new carbon–carbon bonds and one new carbon–iodine bond.
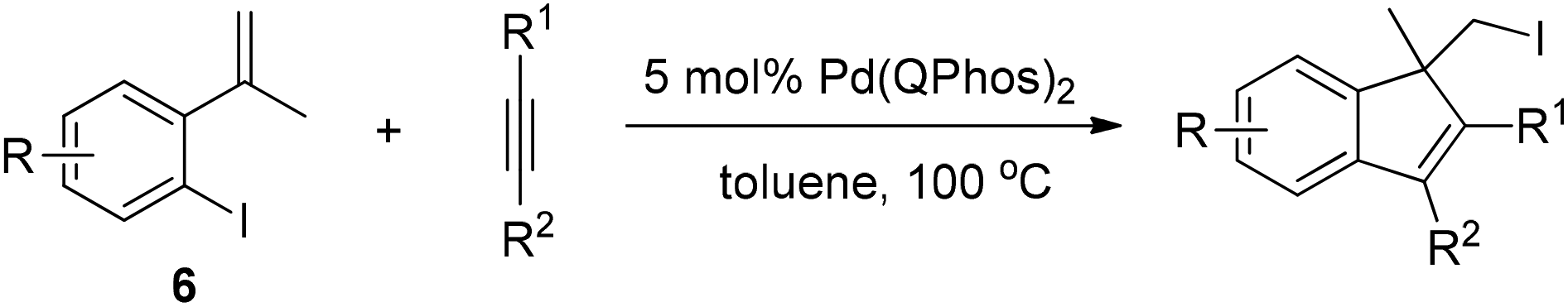 | ||
| Scheme 9 Pd(0)-catalyzed conjunctive carboiodination to indene synthesis. | ||
Almost simultaneously, Tong and co-workers developed a similar Pd(0)-catalyzed carboiodination process with the use of vinyl iodides 7 (Scheme 10).20 Unlike the very sterically hindered phosphane ligands for the above-mentioned reductive elimination event, DPPF was found to be the most efficient ligand for this transformation although a high loading (30 mol%) was required.
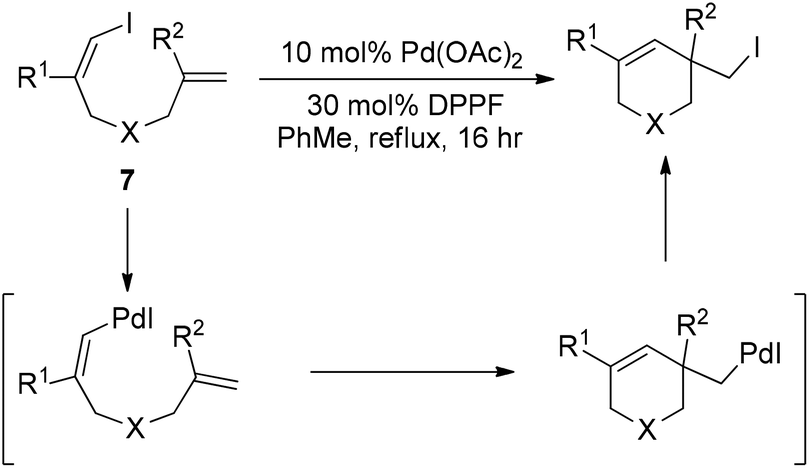 | ||
| Scheme 10 Pd(0)-catalyzed carboiodination of alkene-tethered vinyl iodides. | ||
Furthermore, the fact that no deuterium scrambling was observed in product 8a strongly supported that the reductive elimination of (alkyl)Pd(II)–X is a stereospecific process (Scheme 11). Interestingly, the deuterium scrambling was found to be strongly time-dependent, implying that the combination of Pd(OAc)2 and DPPF might be able to initiate a radical process.
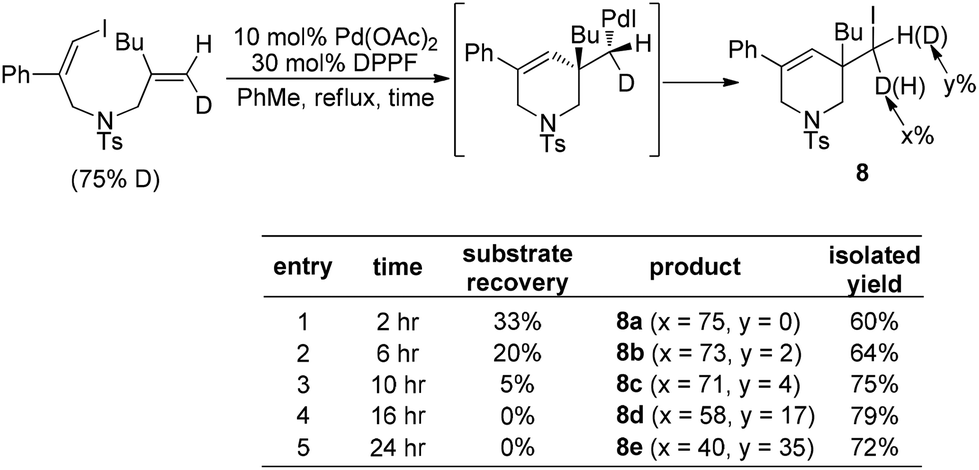 | ||
| Scheme 11 Time-dependent deuterium scrambling. | ||
Later, Lautens and co-workers successfully realized the Pd(0)-catalyzed carboiodination of aryl bromides 9 with the help of 2 equivalents of KI in dilute toluene (Scheme 12).18 Without the additive KI, no synthetically useful quantities of carbobromination product were detected, strongly implying that the alkyl halide reductive elimination may be less efficient with bromides than with iodides.
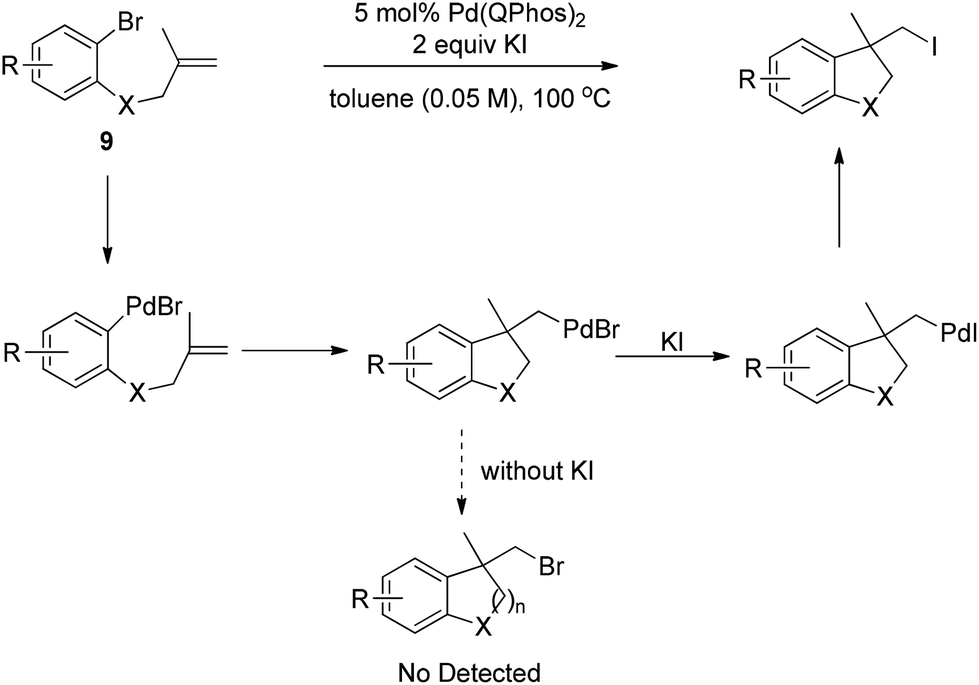 | ||
| Scheme 12 Palladium-catalyzed halogen-exchange carboiodination. | ||
Houk and co-workers employed a DFT study to investigate the mechanism of Lautens's carboiodinations.21 Their calculated data disclosed that the dissociative mechanism is favored for the alkyl halide reductive elimination step. Thus, the different reactivities of halides may arise from the different bond dissociation energies of the Pd–X bond. Indeed, the calculated bond dissociation energies of Pd–I, Pd–Br and Pd–Cl are 72.8 kcal mol−1, 81.1 kcal mol−1 and 91.9 kcal mol−1, respectively, indicating that C–Br and C–Cl reductive eliminations are required to overcome higher activation energy barriers. Their calculation also showed that the alkene insertion step is exoergonic by 10 kcal mol−1, which compensates the energy of the alkyl iodide reductive elimination and serves as an important driving force in this catalytic cycle.
Harnessing the reductive elimination of carbon halides
During their study on the Pd(0)-catalyzed transformation of gem-dibromoolefins 10, Lautens and co-workers observed that a boronic acid was necessary for conversion of the starting material (Scheme 13).22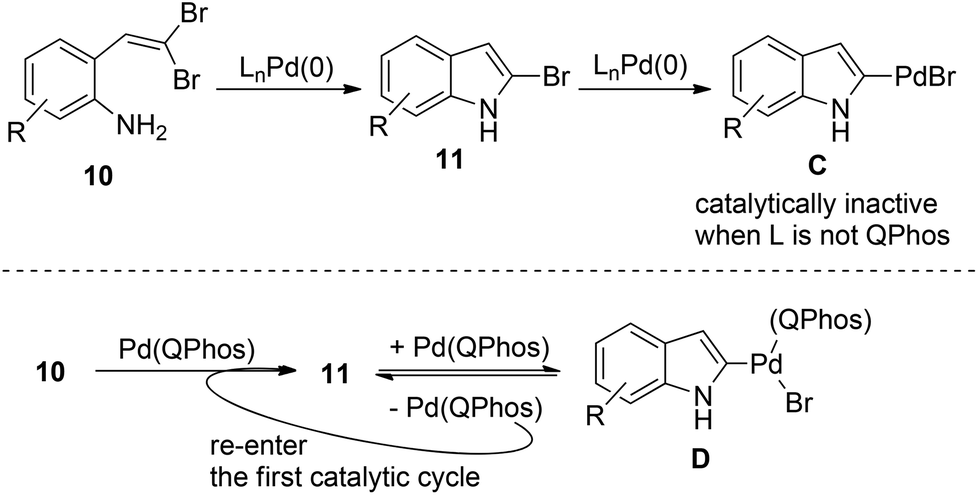 | ||
| Scheme 13 The importance of the C–Br reductive elimination in the palladium-catalyzed synthesis of 2-bromoindoles. | ||
The intermediate 11 should be formed after the first catalytic cycle relevant to the C–N bond formation. However, this product was never observed directly in the reaction and the starting material was recovered in the absence of boronic acid. These results forced the authors to suggest that the active Pd(0) catalyst was undergoing irreversible oxidative addition after the first turnover, leading to catalytically inactive Pd(II) species C, thus shutting down the whole catalytic cycle. Fortunately, P(tBu)3 was found to efficiently promote the aryl bromide reductive elimination of intermediate D, therefore releasing the catalyst to re-enter the first catalytic cycle (Scheme 13).23 This unique mechanism allows for the efficient C–N bond formation to form novel 2-bromoindoles 11 from gem-dibromoolefins 10. These findings clearly demonstrate the importance of harnessing the carbon halide reductive elimination in some palladium(0) catalysis.
The power of this concept was further highlighted by the Pd(0)-catalyzed transformation of polyhalogenated substrates 12 (Scheme 14).24 In the case of di-iodinated substrate 12, one can anticipate that the 4-iodo group would react before the more hindered 2-iodo group, which might generate intermediate E. As is the case for the reaction of compound 10, intermediate E could lead to a catalytic dead end without an additional reaction partner such as alkenes or nucleophiles. Through the carbon iodide reductive elimination of intermediate F enforced by QPhos, di-iodinated substrate 12 was readily converted into the desired product 13. The concept of harnessing the reductive elimination of carbon halide provides a general and attractive solution for commonly encountered problems associated with polyhalogenated substrates.
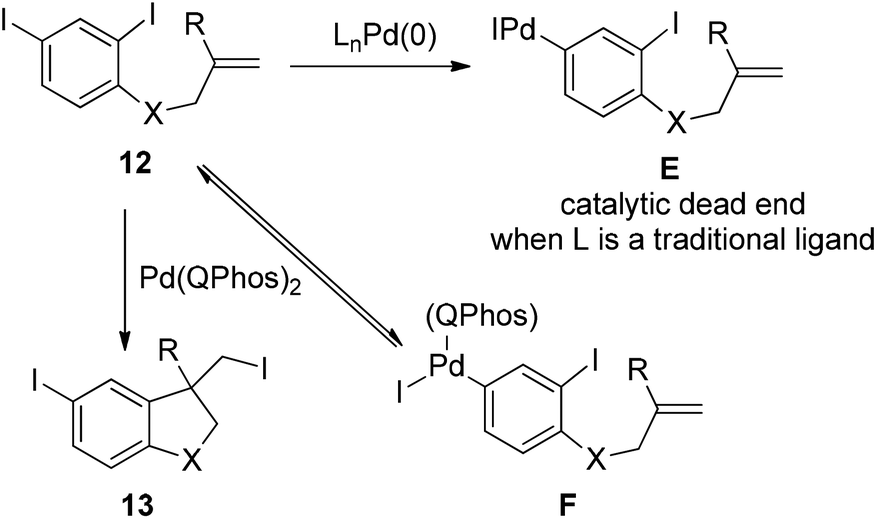 | ||
| Scheme 14 The importance of the C–I reductive elimination in palladium-catalyzed transformation of polyhalogenated substrates. | ||
Synthesis of alkyl halide via Pd(0)-catalyzed radical process
As early as 1988, Mori and co-workers reported the Pd(PPh3)4-catalyzed halide-transfer cycloisomerization of alkene-tethered α-haloester 14 (Scheme 15).25 Interestingly, no β-hydrogen elimination products were observed, which might rule out any reaction pathway involving σ-alkyl palladium(II) intermediates. In fact, the σ-alkyl palladium(II) species, which were produced by the reaction of compound 16 and Pd(OAc)2, actually led to two types of β-hydrogen elimination products 17 and 18 (Scheme 15). Thus, one can conclude that the Pd(0)-catalyzed conversion of 14 to 15 might be a free-radical chain reaction.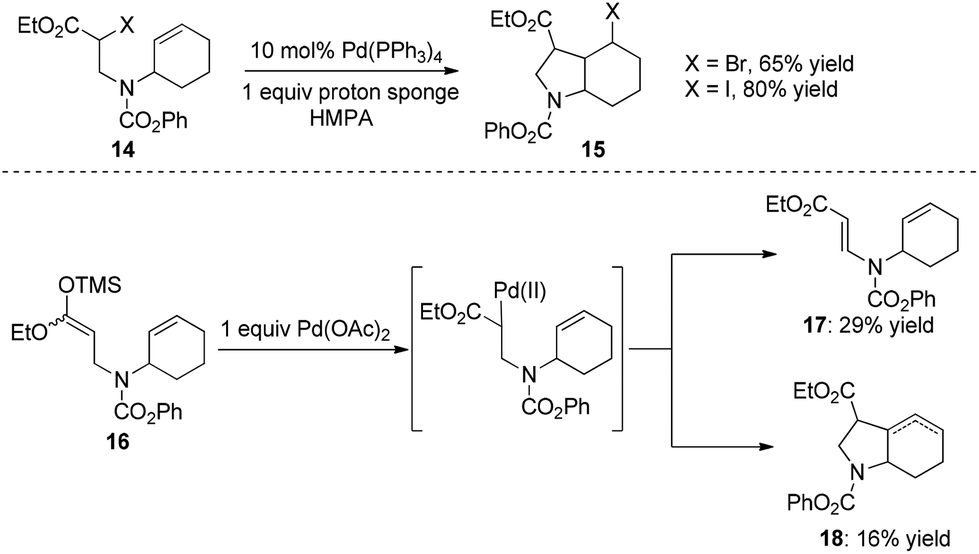 | ||
| Scheme 15 Mori's Pd(0)-catalyzed halide-transfer cyclization. | ||
The Pd(0)-initiated free-radical halide-transfer reaction was further proved by Jiang and co-workers (Scheme 16).26 They found that the combination of Pd(OAc)2 and DPPF could efficiently promote the transformation of alkene-tethered alkyl iodide 19 into product 20. The free-radical chain mechanism was solidly established on the basis of the isolation of two TEMPO radical adducts 21 and 22 (Scheme 16).
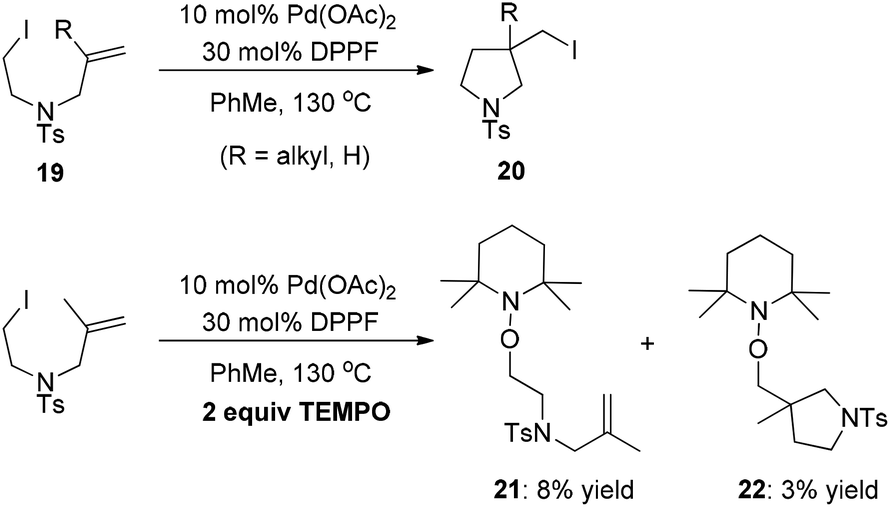 | ||
| Scheme 16 Pd(0)-catalyzed iodide-transfer cyclization. | ||
Very recently, Cook and Monks developed a Pd(PPh3)4-catalyzed zipper-type reaction of alkyl iodide 23, leading to the isolation of the diquinane 24 bearing a primary iodide group (Scheme 17).27 Their observations, such as ligand influence on stereoselectivity and compromised reactivity in the absence of the terminal olefin, strongly indicated the importance of the coordination between a putative Pd(I)I intermediate and the terminal olefin (Scheme 17). More importantly, the authors found that alkyl iodide 23 was quantitatively recovered under various conditions for some well-known radical atom-transfer cyclizations, clearly demonstrating the superiority of this Pd(0)-catalyzed transformation over traditional free radical methods.
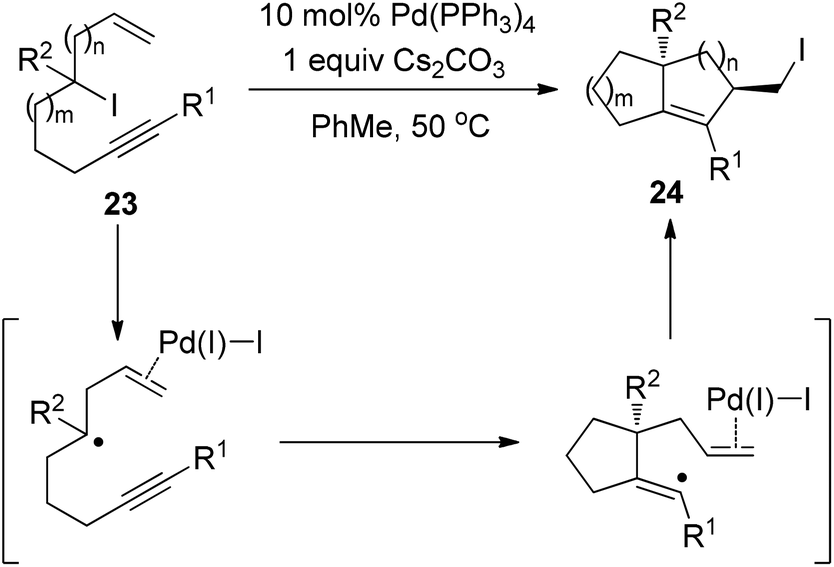 | ||
| Scheme 17 Palladium-catalyzed intramolecular iodine-transfer reactions in the presence of β-hydrogen atoms. | ||
Conclusions and outlook
This critical review provides an overview of the recent advances related to the reductive elimination of C–Pd(II)–X species. Very sterically hindered phosphane ligands have been proven to be essential for this thermodynamically disfavored process. While oxidative additions are normally widely utilized in numerous Pd-catalyzed transformations as the initial step in the catalytic cycle, there is evidence that the reverse reaction of carbon halide reductive elimination can be significantly more common than was previously assumed and has exhibited some advantages for the synthesis of organic halides, especially the synthetically challenging organofluorine compounds. It is now clear that further attention should be paid to the following points: (i) to develop the asymmetric version, (ii) to uncover a more precise mechanism for the alkyl iodide reductive elimination, (iii) to realize the more challenging alkyl-Br and alkyl-Cl reductive elimination, and (iv) to develop novel Pd-catalyzed transformations with the use of the reductive elimination of C–Pd(II)–X as the terminating step. It is our hope and expectation that these aims will be addressed in the future by chemists worldwide, resulting in more generally applicable synthetic methods.Acknowledgements
We appreciate the financial support from NSFC (no. 21272066), the Fundamental Research Funds for the Central Universities, Fok Ying-Tong Education Foundation for Young Teachers in the Higher Education Institutions of China (131011) and NCET (no. 12-0851).Notes and references
- For reviews and selected examples, see: (a) L. Xue and Z. Lin, Chem. Soc. Rev., 2010, 39, 1692 RSC; (b) G. Zeni and R. C. Larock, Chem. Rev., 2006, 106, 4644 CrossRef CAS PubMed; (c) C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664 CrossRef CAS PubMed; (d) M. Ahlquist and P.-O. Norrby, Organometallics, 2007, 26, 550 CrossRef CAS; (e) D. W. Old, J. P. Wolfe and S. L. Buchwald, J. Am. Chem. Soc., 1998, 120, 9722 CrossRef CAS; (f) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 1998, 37, 3387 CrossRef CAS; (g) A. F. Littke and G. C. Fu, Angew. Chem., Int. Ed., 2002, 41, 4176 CrossRef CAS; (h) U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366 CrossRef CAS PubMed.
- For selected reviews, see: (a) A. Vigalok, Chem.–Eur. J., 2008, 14, 5102 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (d) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC.
- J. D. Ruddick and B. L. Shaw, J. Chem. Soc. A, 1969, 2969 RSC.
- (a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232 CrossRef CAS; (b) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 13944 CrossRef CAS PubMed; (c) A. H. Roy and J. F. Hartwig, Organometallics, 2004, 23, 1533 CrossRef CAS.
- (a) S. L. Fraser, M. Yu Antipin, V. N. Khroustalyov and V. V. Grushin, J. Am. Chem. Soc., 1997, 119, 4769 CrossRef CAS; (b) M. C. Pilon and V. V. Grushin, Organometallics, 1998, 17, 1774 CrossRef CAS.
- For reviews, see: (a) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed; (b) V. V. Grushin, Chem.–Eur. J., 2002, 8, 1006 CrossRef CAS.
- D. V. Yandulov and N. T. Tran, J. Am. Chem. Soc., 2007, 129, 1342 CrossRef CAS PubMed.
- V. V. Grushin and W. J. Marshall, Organometallics, 2007, 26, 4997 CrossRef CAS.
- For reviews, see: (a) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed; (b) R. J. Lundgren, K. D. Hesp and M. Stradioto, Synlett, 2011, 2443 CAS; (c) S. M. Wong, C. M. So and F. Y. Kwong, Synlett, 2012, 1132 CAS.
- (a) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661 CrossRef CAS PubMed; (b) T. J. Maimone, P. J. Milner, T. Kinzel, Y. Zhang, M. K. Takase and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 18106 CrossRef CAS PubMed.
- P. E. Garrou and R. F. Heck, J. Am. Chem. Soc., 1976, 98, 4115 CrossRef CAS.
- J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841 CrossRef CAS PubMed.
- H. G. Lee, P. J. Milner and S. L. Buchwald, Org. Lett., 2013, 15, 5602 CrossRef CAS PubMed.
- X. Shen, A. M. Hyde and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 14076 CrossRef CAS PubMed.
- J. Pan, X. Wang, Y. Zhang and S. L. Buchwald, Org. Lett., 2011, 13, 4974 CrossRef CAS PubMed.
- For reviews, see: (a) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674 CrossRef CAS PubMed; (b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656 CrossRef CAS PubMed.
- S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2011, 133, 1778 CrossRef CAS PubMed.
- S. G. Newman, J. K. Howell, N. Nicolaus and M. Lautens, J. Am. Chem. Soc., 2011, 133, 14916 CrossRef CAS PubMed.
- X. Jia, D. A. Petrone and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 9870 CrossRef CAS PubMed.
- H. Liu, C. Li, D. Qiu and X. Tong, J. Am. Chem. Soc., 2011, 133, 6187 CrossRef CAS PubMed.
- Y. Lan, P. Liu, S. G. Newman, M. Lautens and K. N. Houk, Chem. Sci., 2012, 3, 1987 RSC.
- Y.-Q. Fang and M. Lautens, J. Org. Chem., 2008, 73, 538 CrossRef CAS PubMed.
- S. G. Newman and M. Lautens, J. Am. Chem. Soc., 2010, 132, 11416 CrossRef CAS PubMed.
- D. A. Petrone, M. Lischka and M. Lautens, Angew. Chem., Int. Ed., 2013, 52, 10635 CrossRef CAS PubMed.
- M. Mori, Y. Kubo and Y. Ban, Tetrahedron, 1988, 44, 4321 CrossRef CAS.
- H. Liu, Z. Qiao and X. Jiang, Org. Biomol. Chem., 2012, 10, 7274 CAS.
- B. M. Monks and S. P. Cook, Angew. Chem., Int. Ed., 2013, 52, 14214 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2014 |